Introduction
Diffuse large B-cell lymphoma (DLBCL) is an
aggressive form of B-cell lymphoma, which is the most common type
of non-Hodgkins lymphoma (NHL), and comprises ~30% of all NHL
neoplasms. Over half of all DLBCLs are curable using standard
chemotherapy (cyclophosphamide, doxorubicin, vincristine and
prednisone) combined with rituximab (R-CHOP), while approximately
one third of cases will relapse or become refractory following
first line therapy (1,2). The two primary subtypes of DLBCL, termed
germinal center B cell (GCB) and activated B cell (ABC) have been
identified via gene expression profiling studies (3). The distinct genetic characteristics of
these two subtypes suggest that they arise from different stages of
lymphoid differentiation, with the GCB subtype deriving from the
lymphoid cells residing in the germinal center, and the ABC subtype
from B cells at a plasmablastic stage. This results in different
clinical features and prognoses, where following R-CHOP combination
treatment, the prognosis of patients with the ABC subtype is poorer
than those with the GCB subtype, who possess a 3-year
progression-free survival (PFS) rate of 40 vs. 75%, respectively
(4). The pathogenic hallmark of ABC
DLBCL is the constitutive activation of the NF-κB pathway, which is
often caused by mutations occurring following the upstream
activation of the B-cell receptor (BCR) and Toll-like receptors
(TLR) pathways (5). These activating
mutations were identified in subsets of patients with ABC DLBCL,
and include mutations in CD79B, caspase recruitment
domain-containing protein 11 (CARD11) in the BCR pathway (6,7) and
myeloid differentiation primary response gene 88 (MYD88) in the TLR
pathway (8).
The human TLRs, first reported in the 1990s, are
functionally involved in both innate immunity and the initiation of
adaptive immune responses (9). MYD88
is a key adaptor molecule for the majority of TLR and IL-1 receptor
(IL-1R) signaling cascades. The MYD88 protein contains both a
Toll/interleukin-1 receptor (TIR) domain that can bind to the TIR
domain of TLRs, and a death domain (DD), which provides a docking
site for IL-1R-associated kinases (IRAKs). It has been demonstrated
that six MYD88 molecules, four IRAK4 and four IRAK1 or IRAK2
molecules are able to associate via their DDs to form a
high-molecular-weight (HMW) (>180 kDa) complex, the myddosome.
The myddosome platform is essential for the activation of the
nuclear factor kappa-light-chain-enhancer of activated B cells
(NF-κB) pathway via a cascade of phosphorylation events that
originate from the phosphorylation of IRAK4, the most upstream
kinase in this cascade (10,11). Highly recurrent somatic mutations in
MYD88 were first demonstrated in ABC DLBCL by Ngo et al
(8), with a specific point mutation
(L265P) occurring most frequently; L265P was observed in ~29% of
ABC DLBCL cases, but rarely in GCB DLBCL. The high prevalence of
MYD88 L265P in patients with Waldenstrom macroglobulinemia (WM) has
also been reported in previous publications, with an observed
mutation frequency rate of 87% (observed in 1,324 of 1,520 patients
with WM, from 25 publications) (12).
In addition, MYD88 L265P has also been identified in other types of
B-cell neoplasm, with mutation frequency rates in monoclonal
gammopathy of undetermined significance of the IgM class (IgM MGUS;
52%), primary DLBCL of the central nervous system (CNS; 70%),
cutaneous DLBCL of leg-type (54%) and testicular DLBCL (74%)
(12). Consistent with previous
studies, the majority of these subtypes of DLBCL are of ABC origin.
Ngo et al (8) further
demonstrated that MYD88 L265P was a gain-of-function driver
mutation, which promoted ABC DLBCL cell survival by assembling a
myddosome complex and the phosphorylation of IRAK kinases; this
resulted in constitutive NF-κB activation, type I interferon (IFN)
signaling and IL-6/IL-10-engaged autocrine activation of the
JAK-STAT 3 pathway (8).
In ABC DLBCL cells, interactions between MYD88
L265P-mutated and wild-type (WT) TIR domains enhance MyD88
oligomerization, which serves a key role in myddosome complex
formation, resulting in the recruitment of IRAKs and induced NF-κB
signaling activation (13). ST2825, a
synthetic peptidomimetic compound, interferes with the association
between MYD88 proteins, potentially by targeting the interface
between the TIR domains (14).
Although MYD88 L265P is essential in promoting the survival of ABC
DLBCL cells, the therapeutic strategies for targeting MYD88 remain
largely undetermined. In the present study, the ability of ST2825
to disrupt MYD88 oligomerization-induced myddosome assembly was
investigated in ABC DLBCL cells, in addition to the subsequent
ability to inhibit NF-κB signaling and tumor cell survival.
Materials and methods
Reagents
ST2825 was purchased from MedChemExpress. The
Bruton's tyrosine kinase (BTK) inhibitor ibrutinib, and B-cell
lymphoma-2 (BCL-2) inhibitor ABT-199 were purchased from Selleck
Chemicals. All drugs were dissolved in 100% dimethyl sulfoxide
(DMSO). For all samples in all of the experiments, the final DMSO
concentrations were diluted to 0.1% with cell culture media,
including the vehicle controls.
Cell lines and cell culture
SU-DHL-4, OCI-LY10 and TMD8 cell lines were
purchased from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. The MYD88 L265P mutation of each cell
line was identified using Sanger sequencing. The cells were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). The HEK293T cell line
was cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc.) with 10% FBS. All cell lines were
cultured at 37°C in a 5% CO2 incubator.
Evaluation of cell viability and
apoptosis
Cell viability was assessed using WST-1 reagent
(Roche Diagnostics) as instructed by the manufacturer. Briefly,
~2×104 cells/well were seeded into 96-well plates and
treated with either the vehicle (DMSO) or ST2825 at serial
concentrations for 24, 48 or 72 h. After treatment, 10 µl WST-1
reagent was added to each well, followed by a 4-h incubation at
37°C. Cell viability was calculated by measuring the absorbance at
440 nm using a 96-well plate reader, and the data were normalized
to that of the vehicle-treated cells. For drug combination
experiments, cell viability was determined 72 h after treatment
with the indicated drugs. Flow cytometric analysis of apoptosis was
performed using an Annexin V-FITC Apoptosis Detection kit
(Sigma-Aldrich; Merck KGaA) 48 h after ST2825 treatment (5 or 10
µM) according to the manufacturer's protocol; the results were
acquired using a flow cytometer (BD Influx; BD Biosciences) and
analyzed using BD FACSuite software version 1.0.6. For drug
combination experiments, the apoptotic cell populations were
analyzed 48 h after treatment with the indicated drugs. The nuclear
morphology of the apoptotic cells was further examined using DAPI
staining. Cells were collected and coated onto slides after washing
with PBS three times. The cells were then fixed in 4%
paraformaldehyde at room temperature for 15 min and then
permeabilized with 0.1% Triton X-100 at 37°C for 5 min. The cells
were subsequently stained with 5 µg/ml DAPI in a dark room at 37°C
for 15 min. After three washes with PBS, the cells were visualized
using a fluorescence microscope (Eclipse 80i; Nikon Corporation)
under a 40× oil immersion objective.
Western blot analysis
Western blot analysis was performed as previously
described (15). Cells were lysed
with RIPA lysis buffer supplemented with proteinase and phosphatase
inhibitors (Cell Signaling Technology, Inc.) at 4°C for 20 min. The
supernatants were collected after centrifugation at 13,000 × g for
15 min (4°C), and the protein concentration was determined using a
bicinchoninic acid (BCA) protein assay kit (Thermo Fisher
Scientific, Inc.). Equal amounts of protein extract (30 µg per
lane) were loaded for separation onto a 10% SDS-PAGE gel and
transferred to PVDF membranes (EMD Millipore), before blocking in
5% non-fat milk/TBST at room temperature for 1 h. Then, the
membranes were incubated overnight at 4°C with primary antibodies
(all purchased from Cell Signaling Technology, Inc., and used at a
dilution of 1:1,000) against MYD88 (cat. no. 4823S), BTK (cat. no.
8547S), phospho-BTK (cat. no. 5082S), inhibitor of NF-κB (IκB; cat.
no. 4812S) and phospho-IκB (cat. no. 2859S), followed by incubation
with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary
antibody (cat. no. sc-2030; Santa Cruz Biotechnology, Inc.;
1:5,000) for 1 h at room temperature. The β-actin antibody (cat.
no. ab8227; Abcam) was used at a 1:1,000 dilution. Detection was
performed using an enhanced chemiluminescence substrate (ECL; EMD
Millipore), and the signals were visualized and analyzed using the
Bio-Rad Gel Imaging System and the Cool Imager™ workstation II
(Viagene, Biotech, Inc.).
MyD88 protein aggregation in DLBCL cell lines
(OCI-LY10 and TMD8) was analyzed using fractionated cell lysates.
Following overnight treatment with DMSO or 10 µM ST2825,
5×106 cells were incubated in each well of a 6-well
plate and then collected and lysed using cell lysis buffer
(Beyotime Institute of Biotechnology) supplemented with PMSF and
Benzonase (Sigma-Aldrich; Merck KGaA). The protein concentration
was determined using a BCA protein assay kit (Thermo Fisher
Scientific, Inc.) and equal amounts of cell lysates (60 µg) were
then fractionated by centrifugation (16,000 × g, 10 min); the
supernatant fractions were regarded as whole cell lysates (WCL) and
the pellet fractions as insoluble HMW aggregates. The pellets were
then washed with PBS and dissolved by boiling in SDS sample buffer
with shaking. Western blotting was conducted using the
aforementioned anti-MYD88 (Cell Signaling Technology, Inc.) and
anti-β-actin (Abcam) antibodies and goat anti-rabbit HRP-conjugated
secondary antibody at the indicated dilutions.
Co-immunoprecipitation
Cells from each line were lysed using cell lysis
buffer for western and immunoprecipitation (Beyotime Institute of
Biotechnology) supplemented with 1 mM PMSF. The samples were
centrifuged at 13,000 × g for 15 min, and the protein concentration
was determined using a BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Then protein A/G coated magnetic beads (Bio-Rad
Laboratories, Inc.) were incubated with 5 µg IP antibodies against
MYD88 or BTK (Cell Signaling Technology, Inc.) at room temperature
with rotation for 10 min. After washing with PBS-T, the
antibody-conjugated beads were added into the antigen-containing
lysate, and rotated for 1 h at room temperature. Immunoprecipitates
were then washed three times with PBS-T, separated on a magnetic
stand and eluted using Laemmli loading buffer for further
immunoblotting analysis.
Confocal microscopy
HEK293T cells were seeded into 12-well plates
(2×105 cells/well) 24 h before transfection to achieve a
confluency of 50–70%. Cells were then transiently transfected with
2 µg mCitrine-TIR WT or mCitrine-TIR L265P plasmids using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. After 24 h, the cells
were treated with DMSO or 10 µM ST2825 and incubated for a further
48 h. Following incubation, the cells were observed using a laser
scanning Nikon A1R confocal microscope under a 40× oil immersion
objective (numerical aperture, 1.4). An excitation of 488 and
520–570 nm emission were used for visualization. The plasmids for
mCitrine-TIR WT or L265P mutant were provided by Dr Roman Jerala
(13).
NF-κB reporter assays
CMV-Renilla luciferase lentivirus (Cignal
Lenti CMV Renilla Control; Qiagen, Inc.) was used as an internal
control for normalization, and NF-κB reporter lentivirus with
firefly luciferase expression (Cignal Lenti NF-κB Reporter; Qiagen,
Inc.) was used as the experimental reporter. The two lentiviruses
were used to co-transfect the OCI-LY10 and TMD8 cells. Briefly,
OCI-LY10 and TMD8 cells were resuspended in 0.5 ml
lentivirus-containing medium (containing 250 µl Cignal Lenti NF-κB
Reporter and 250 µl Cignal Lenti CMV Renilla Control) at a
concentration of 1×106 cells/ml in a 24-well tissue
culture plate in the presence of polybrene (8 µg/ml). The plates
were centrifuged at 800 × g for 90 min at 32°C. The cells were then
washed and resuspended in fresh medium for an additional 72 h. Then
puromycin and hygromycin were used together for selection. For the
drug experiments, cells were subsequently seeded at a density of
4×105 cells/ml and treated for 12 h with the vehicle,
ST2825, ibrutinib or a combination of ST2825 and ibrutinib at the
indicated concentrations. Luciferase activity was measured in
96-well plates using the Dual-Luciferase Reporter Assay system
(Promega Corporation) according to the manufacturer's protocol.
Cytokine measurement
The two ABC DLBCL cell lines were cultured in fresh
media and treated for 24 h with 5 or 20 µM ST2825 or DMSO. The
concentrations of IL-10 and IFN-β in the culture supernatants were
measured by ELISA (cat. no. DY217B-05 and DY814-05, respectively;
R&D Systems, Inc.) according to the manufacturer's protocol.
All experiments were performed in triplicate.
Synergism analysis
Synergism of the drug combinations (including ST2825
combined with ibrutinib or ABT-199) was evaluated using CalcuSyn
software (Premier Biosoft International), which is based on the
median-effect principle applied by Chou and Talalay (16). The combined effects and combination
index (CI) for each dose combination were calculated and are
presented as a heat map. Drug synergism, addition and antagonism
were defined by CI values of <1.0, 1.0 and >1.0,
respectively.
Statistical analysis
Statistical analysis of the experimental data was
performed using GraphPad Prism software version 6 (GraphPad
Software, Inc.), and the data are presented as the mean ± standard
deviation, unless otherwise indicated. The unpaired t-test was used
to assess the statistical significance of the differences between
two groups, and one-way analysis of variance with Dunnett's or
Tukey's test was used for those between multiple groups. P<0.05
was considered to indicate a statistically significant
difference.
Results
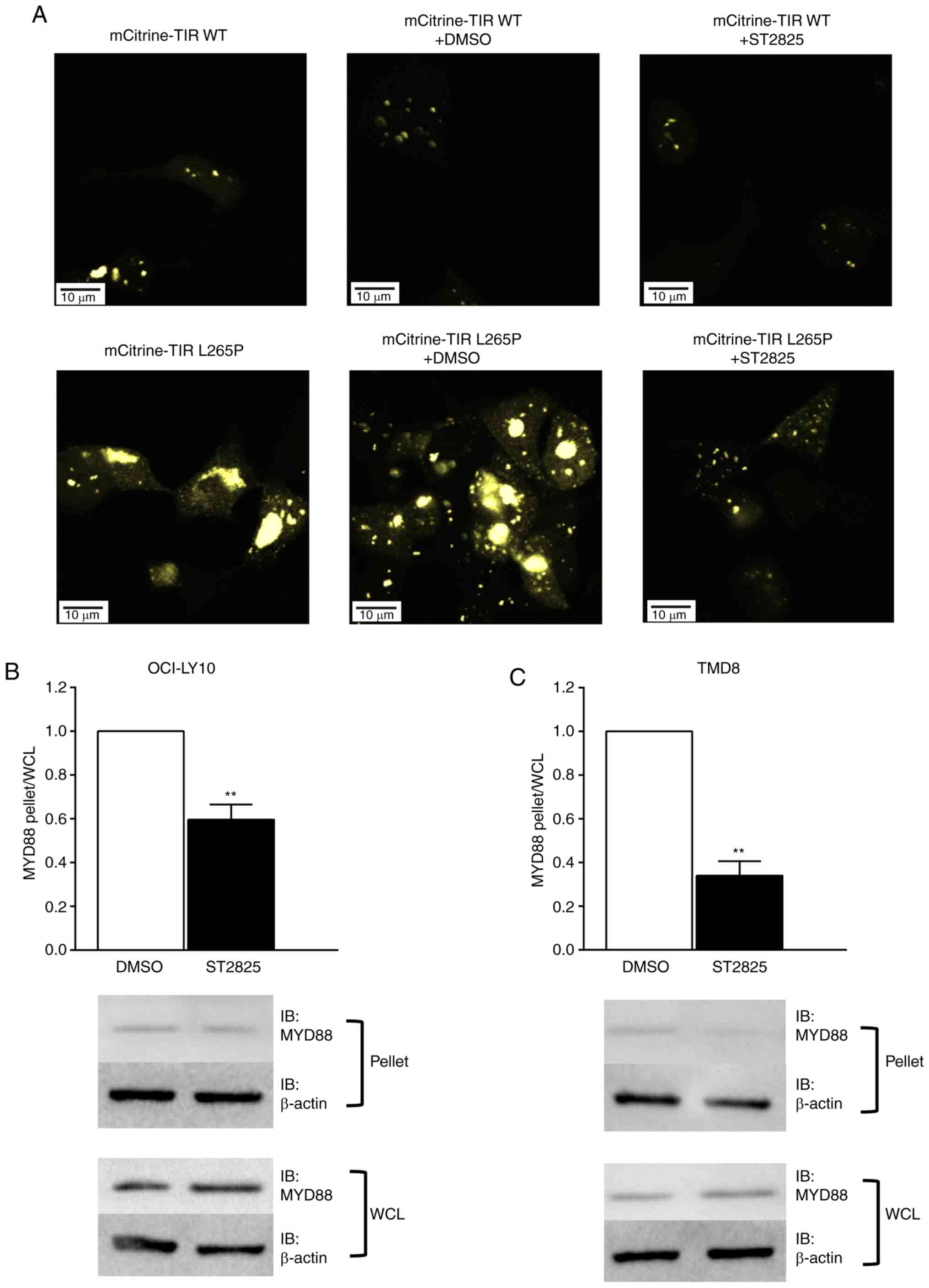
MYD88 oligomerization augmented via
the L265P-mutated TIR domain is blocked by ST2825
To determine whether oligomerization of the MyD88
TIR-L265P mutants could be blocked by ST2825, MyD88 TIR WT or
mutant linked to the fluorescent protein mCitrine were expressed in
HEK293T cells. Consistent with the results of a previous report
(13), mCitrine-TIR mutants were
strongly aggregated, whilst this was not the case for the WT
mCitrine TIR. Furthermore, the aggregation of mCitrine-TIR mutants
was markedly inhibited following treatment with ST2825 (Fig. 1A).
The HMW fraction was separated from the cellular
lysates via centrifugation, and western blotting was used to
determine the presence of MyD88 aggregates in the pelleted HMW
fractions. MyD88 aggregation was detected in the HMW fraction in
DLBCL cell lines with the L265P mutation (OCI-LY10 and TMD8). The
changes in MyD88 aggregation following treatment with ST2825 were
then investigated by comparing the ratios of endogenous MyD88
levels between pelleted HMW fractions and whole cell lysates.
Fig. 1B and C illustrate a
significantly lower pellet:lysate ratio in the ST2825-treated cells
compared with the DMSO treated cells, thus indicating that the
degree of MyD88 aggregation was significantly decreased following
ST2825 treatment. A previous study indicated that the formation of
MYD88-mutant-containing aggregates coincided with that of myddosome
complexes (13). According to these
data, it may be that augmented MyD88 oligomerization is blocked by
ST2825 in ABC DLBCL cells, resulting in the disruption of myddosome
assembly.
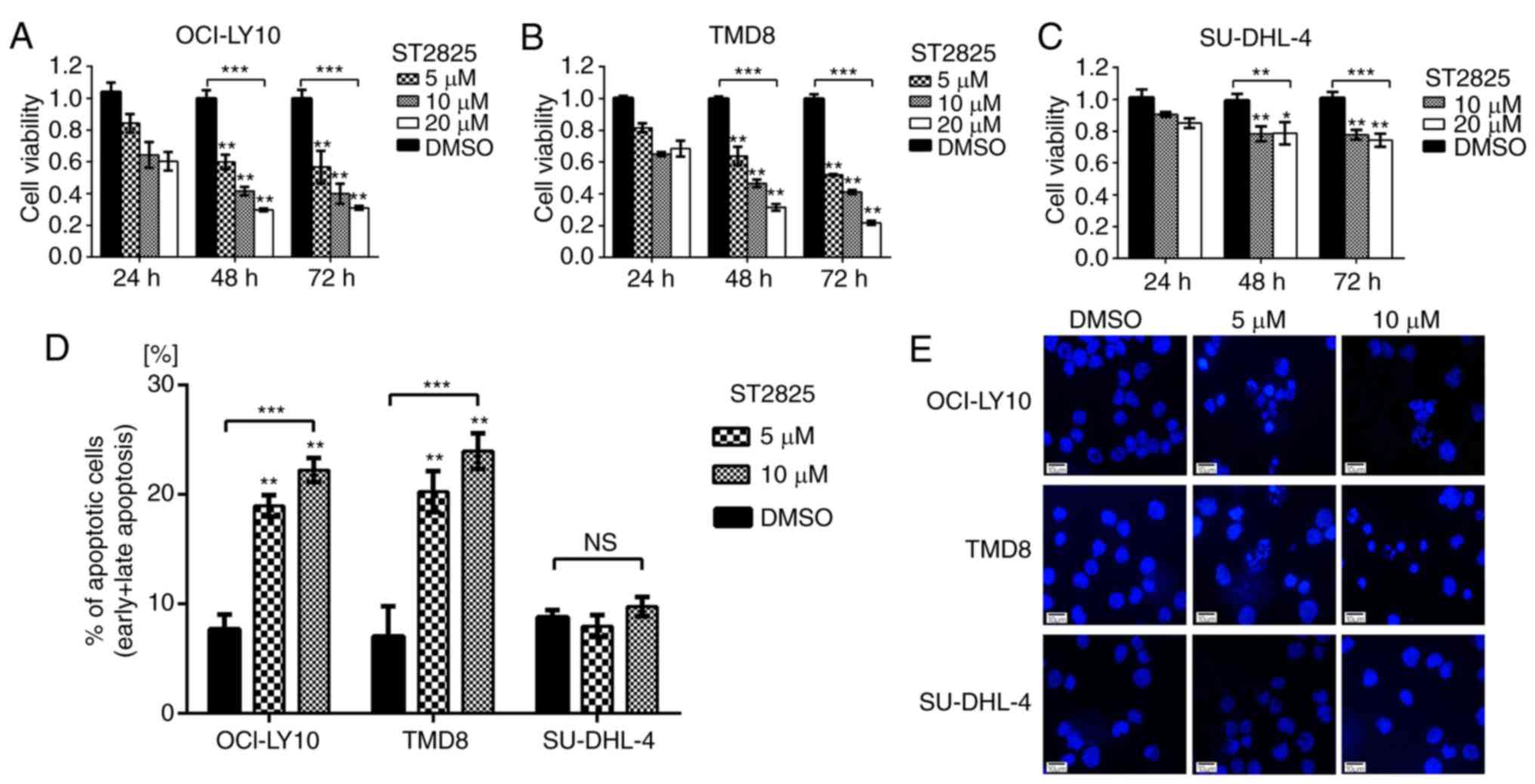
Disruption of myddosome assembly
inhibits cell survival and promotes apoptosis in ABC DLBCL cells
with the L265P mutation
WST-1 assays were used to evaluate the impact of
ST2825-induced myddosome assembly disruption on tumor cell
survival. As presented in Fig. 2,
ST2825 exerted greater cytotoxicity on MYD88-mutated (OCI-LY10 and
TMD8) vs. WT (SU-DHL-4) B-cells. Compared with the DMSO control
groups, the cell viability of OCI-LY10 cells treated with ST2825
for 72 h was decreased in a concentration-dependent manner, with
~50% inhibition at 5 µM ST2825, ~60% at 10 µM and ~70% at 20 µM
(P<0.01; Fig. 2A). Similar results
were observed in TMD8 cells (P<0.01; Fig. 2B). However, the growth inhibition
effects of ST2825 on SU-DHL-4 cells were only ~20-25% at 10 or 20
µM after 72 h of treatment, though these results were still
significant. Flow cytometry revealed that the percentage of
apoptotic cells (including early and late apoptotic cells) was
significantly increased in OCI-LY10 and TMD8 cells 48 h after
treatment with ST2825 (P<0.01; Figs.
2D and S1), while no significant
difference was observed in SU-DHL-4 cells. Consistent with these
results, the characteristic apoptotic morphological changes
(condensed and fragmented nuclear) were observed by DAPI staining
in OCI-LY10 and TMD8 cells treated with ST2825, while there were no
obvious morphological changes in SU-DHL-4 cells (Fig. 2E).
Activated MYD88 signaling pathway
driven by the L265P mutation is inhibited by ST2825
Since the disruption of myddosome assembly inhibited
survival and induced apoptosis in MYD88-L265P DLBCL cells, the
present study investigated the underlying molecular mechanism
responsible for this effect. Gain-of-function driver mutation L265P
engages the NF-κB pathway by facilitating the phosphorylation and
degradation of IκB proteins (8).
Treatment of ABC DLBCL cell lines with ST2825 resulted in decreased
ratios of phosho-/total IκB in both OCI-LY10 and TMD8 cells
(Fig. 3A). To further investigate
NF-κB activity, NF-κB reporter assays were performed. ST2825
decreased transcription of the NF-κB-dependent luciferase reporter
in OCI-LY10 and TMD8 cells (Fig. 3B).
In addition to the NF-κB pathway, JAK-STAT3 signaling was also
demonstrated to contribute to cell survival in ABC DLBCL. MYD88
L265P mutations promote JAK-STAT3 signaling via the increased
production of IL-6 and IL-10 in ABC DLBCL tumors. Furthermore, the
activation of MYD88 signaling in MYD88-L265P DLBCL cells can induce
IFN-β production, which may be involved in the immune modulation of
the ABC DLBCL cell microenvironment (8). The present study demonstrated that
treatment with ST2825 significantly reduced the secretion of IL-10
and IFN-β (Fig. 3C and D), while such
inhibition was not observed for IL-6 secretion (data not
shown).
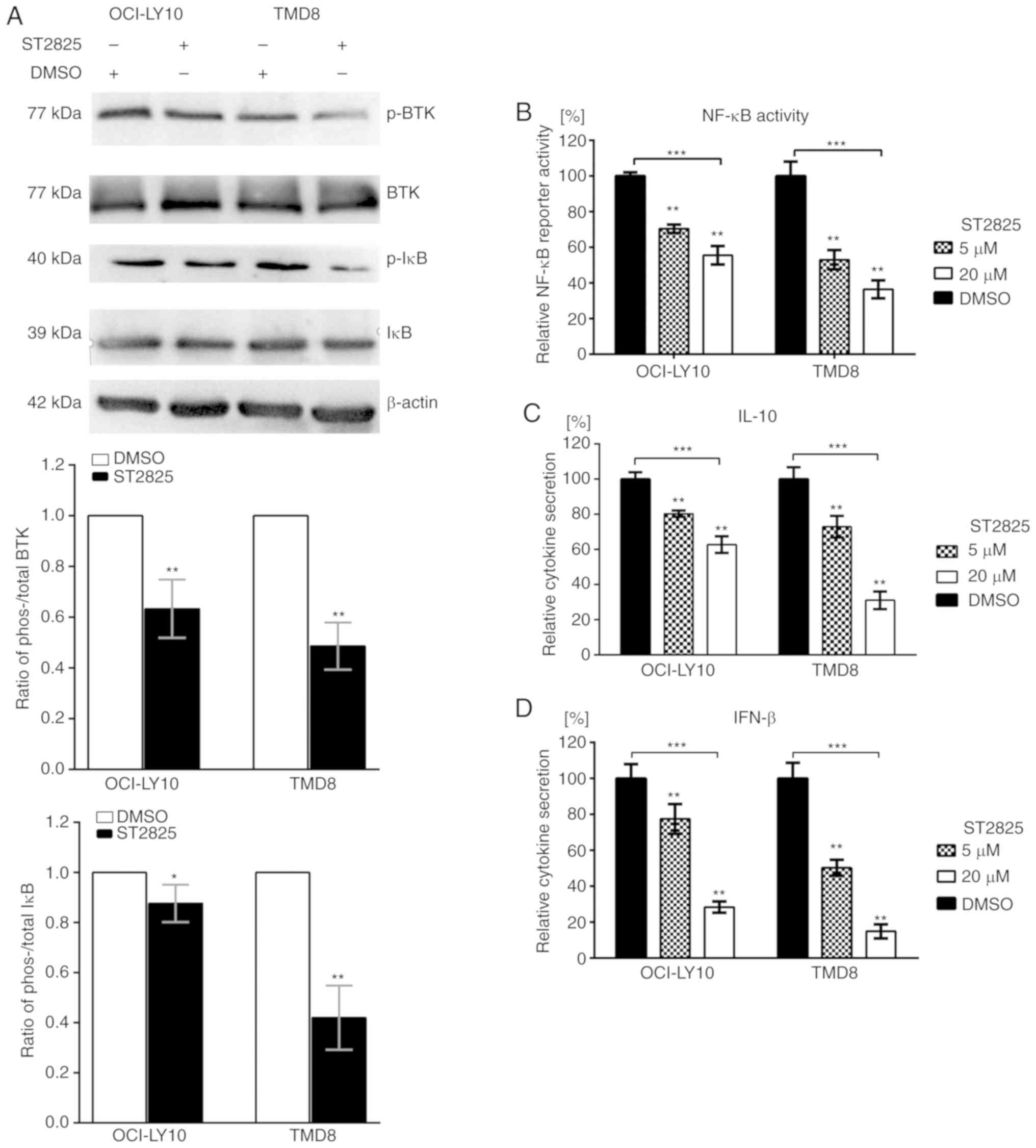 | Figure 3.Effects of ST2825 on the MYD88
signaling pathway in ABC DLBCL cells with the MYD88 L265P mutation.
(A) ABC DLBCL lines (OCI-LY10 and TMD8) were treated with 10 µM
ST2825 or DMSO for 6 h. The protein expression levels of
phosphorylated BTK and IκB, total BTK and IκB, and β-actin were
determined via western blotting. Upper: Western blot results are
representative of three independent experiments; lower: Both the
phosphorylated and total BTK (or IκB) expression levels were
normalized to the β-actin control, and presented as a ratio of the
phospho/total BTK (or IκB). (B) NF-κB-dependent luciferase activity
in ABC DLBCL lines treated with ST2825 or DMSO for 24 h. Data are
presented as the mean ± standard error of the mean from three
independent experiments. (C) Secretion of IL-10 from ABC DLBCL
cells treated for 24 h with ST2825 or DMSO. (D) Secretion of IFN-β
from ABC DLBCL cells treated for 24 h with ST2825 or DMSO.
Statistical analysis by one-way ANOVA with Dunnett's post hoc test.
Data are presented as the mean ± standard deviation from
experiments with three replicates. *P<0.05; **P<0.01;
***P<0.001. MYD88, myeloid differentiation primary response gene
88; DLBCA, diffuse large B-cell lymphoma; ABC, activated B cell;
BTK, Bruton's tyrosine kinase; IĸB, inhibitor of nuclear factor
kappa B kinase; IFN, interferon; p-, phosphorylated. |
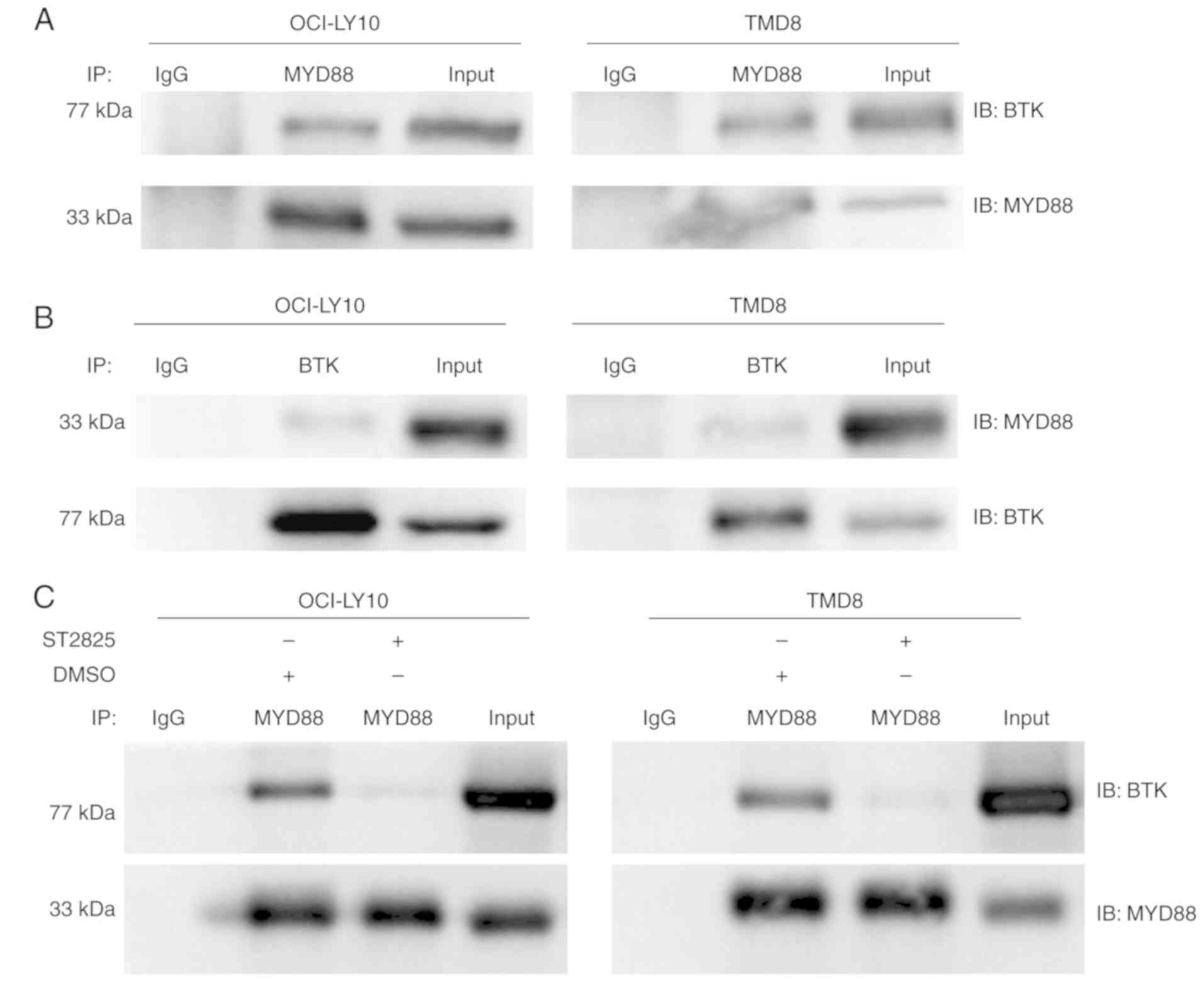
BTK is associated with MYD88, which is
altered following treatment with ST2825 in MYD88 L265P-expressing
ABC DLBCL cells
Chronically active BCR signaling, responsible for
the constitutive activation of the NF-κB pathway, is important for
cell survival in ABC DLBCL. The BCR signaling component BTK serves
a key role in triggering NF-κB activation in the majority of ABC
DLBCLs (6,17). According to the sequencing results of
155 ABC DLBCL biopsy samples, Ngo et al (8) identified that the most common mutations
were found in MYD88, CD79B/A, A20 and CARD11. These mutations
partially overlapped, and the overlap between the MYD88 L265P and
CD79B/A mutations had the highest frequency. Among cases with a
MYD88 L265P mutation, 34% had a simultaneous CD79B/A mutation,
which was reversed among cases with a CD79B/A mutation; even more
had a simultaneous MYD88 L265P mutation (43%). These data indicate
that there may be crosstalk between chronically-active BCR and
MYD88 signaling (8). In a previous
study, co-immunoprecipitation experiments revealed that in HEK293
cells, ectopically expressed BTK associated with MYD88 (18). In addition, it was also reported that
BTK interacted with MYD88 in lipopolysaccharide-stimulated
macrophages to promote the activation of MYD88-dependent pathways
(19). In the present study,
co-immunoprecipitation experiments where performed to investigate
whether endogenous MYD88 interacts with BTK in ABC DLBCL cells,
with activation of the MYD88 signaling pathway driven by the L265P
mutation. Robust MYD88 co-immunoprecipitation with BTK was observed
in L265P-expressing ABC DLBCL cells (OCI-LY10 and TMD8) (Fig. 4A). This binding between BTK and MYD88
was also confirmed through reverse pull-down studies (Fig. 4B). It was further demonstrated that
the binding of BTK with MYD88 was inhibited following treatment
with ST2825 in OCI-LY10 and TMD8 cells (Fig. 4C). In addition, treatment with ST2825
decreased the ratio of phos-/total BTK expression in OCI-LY10 and
TMD8 ABC DLBCL cell lines (Fig.
3A).
Disruption of myddosome assembly
synergizes with BTK inhibition to enhance ABC DLBCL cell death
MYD88 and BCR signaling often converge at IκB,
leading to constitutive activation of the NF-κB pathway in ABC
DLBCL cells. In addition, a recent study demonstrated that MYD88,
the BCR and TLR9 form a multi-protein supercomplex, which drives
pro-survival-associated NF-κB signaling in ibrutinib-responsive
DLBCL cells (20). Furthermore, the
present study identified the direct interaction between BTK and
MYD88 in ABC DLBCL cell lines. Thus, the potential dual inhibition
of MYD88 and BTK signaling and its synergistic effects on ABC DLBCL
cell death were investigated. The combination of ST2825 and the BTK
inhibitor ibrutinib promoted OCI-LY10 and TMD8 cell death (Figs. 5A and B, S2A and B). The inhibitory effect for each
combination at different doses was visualized using a heat map.
Analysis of the CI indicated that ST2825 and ibrutinib were
synergistic at most doses, though greater synergistic effects were
observed at higher doses of ST2825 (≥1.3 µM) for both OCI-LY10 and
TMD8 cells (Figs. 5C and S2C). The enhanced cytotoxicity was
associated with stronger inhibition of NF-κB activity in tumor
cells treated by dual inhibition, compared with that of each drug
alone (Figs. 5D and S2D).
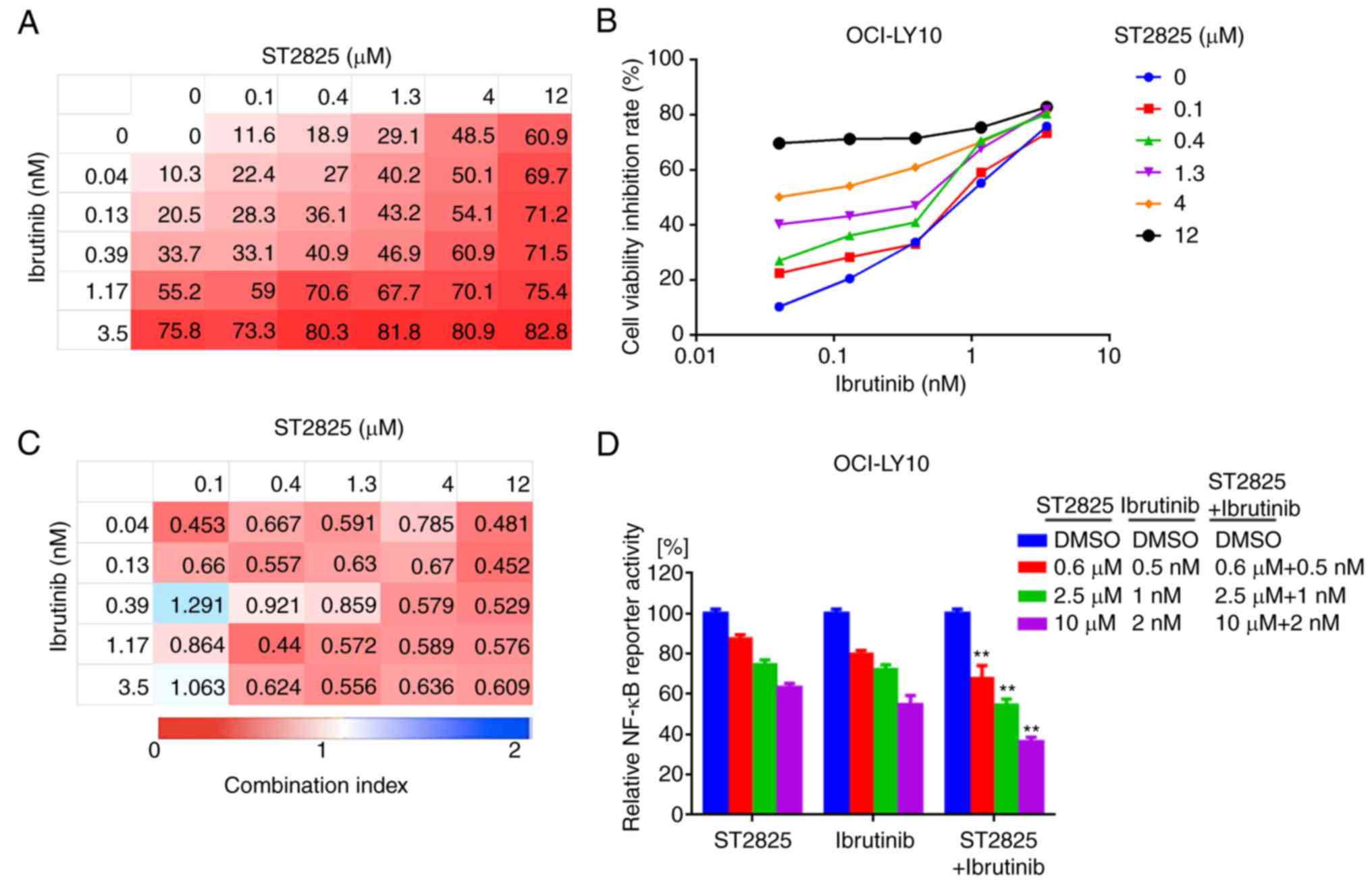 | Figure 5.Synergistic effects of BTK inhibitor
and myddosome assembly inhibitor on ABC DLBCL cells. (A, B)
OCI-LY10 cells were treated for 72 h with ibrutinib, ST2825 or
both, at the indicated doses, followed by a WST-1 assay. Inhibition
at varying dosimetry values for the inhibitor of BTK (ibrutinib)
and MYD88 (ST2825) in OCI-LY10 cells is depicted with (A) heat maps
and (B) line graphs. (C) Synergism was evaluated via CI analysis,
and the CI values of OCI-LY10 cells at varying dosimetry values for
ibrutinib and ST2825 are demonstrated via heat maps. (D) Relative
NF-κB luciferase activity was measured, following treatment of the
OCI-LY10 cells for 12 h with the indicated concentrations of
ibrutinib, ST2825 or both. Statistical analysis by one-way ANOVA
and Tukey's post hoc test. Data are presented as the mean ±
standard error of the mean from three independent experiments.
**P<0.01. BTK, Bruton's tyrosine kinase; ABC, activated B cell;
DLBCA, diffuse large B-cell lymphoma; MYD88, myeloid
differentiation primary response gene 88; CI, combination index;
NF-κB, nuclear factor kappa-light-chain-enhancer of activated B
cells. |
Disruption of myddosome assembly
synergizes with BCL-2 inhibition to enhance ABC DLBCL cell
death
BCL-2 upregulation is prevalent in B-cell lymphoma
and promotes tumor cell survival by blocking apoptosis (21,22). In
addition, high BCL-2 expression levels were associated with poor
clinical outcome in patients with ABC DLBCL (23). Given that BCL-2 serves a key role in
the underlying oncogenic mechanism of B-cell lymphoma,
BCL-2-targeted therapy has developed rapidly in recent years
(24). In the present study,
combination treatment with ST2825 and the BCL-2 inhibitor ABT-199
enhanced OCI-LY10 and TMD8 cell death (Figs. 6A and B, S3A and B). Analysis of the CI indicated
that ST2825 and ibrutinib were synergistic at higher doses of
ST2825 (≥1.3 µM) and ABT-199 (≥0.5 µM) in OCI-LY10 cells (Fig. 5C). Similar results were revealed in
TMD8 cells (Fig. S3C). This
combination treatment also consistently promoted apoptosis, as
indicated by the increased number of apoptotic cells (including
early and late apoptotic cells) detected by flow cytometry,
compared with treatment with the individual drugs alone (Figs. 6D, S3D
and S4).
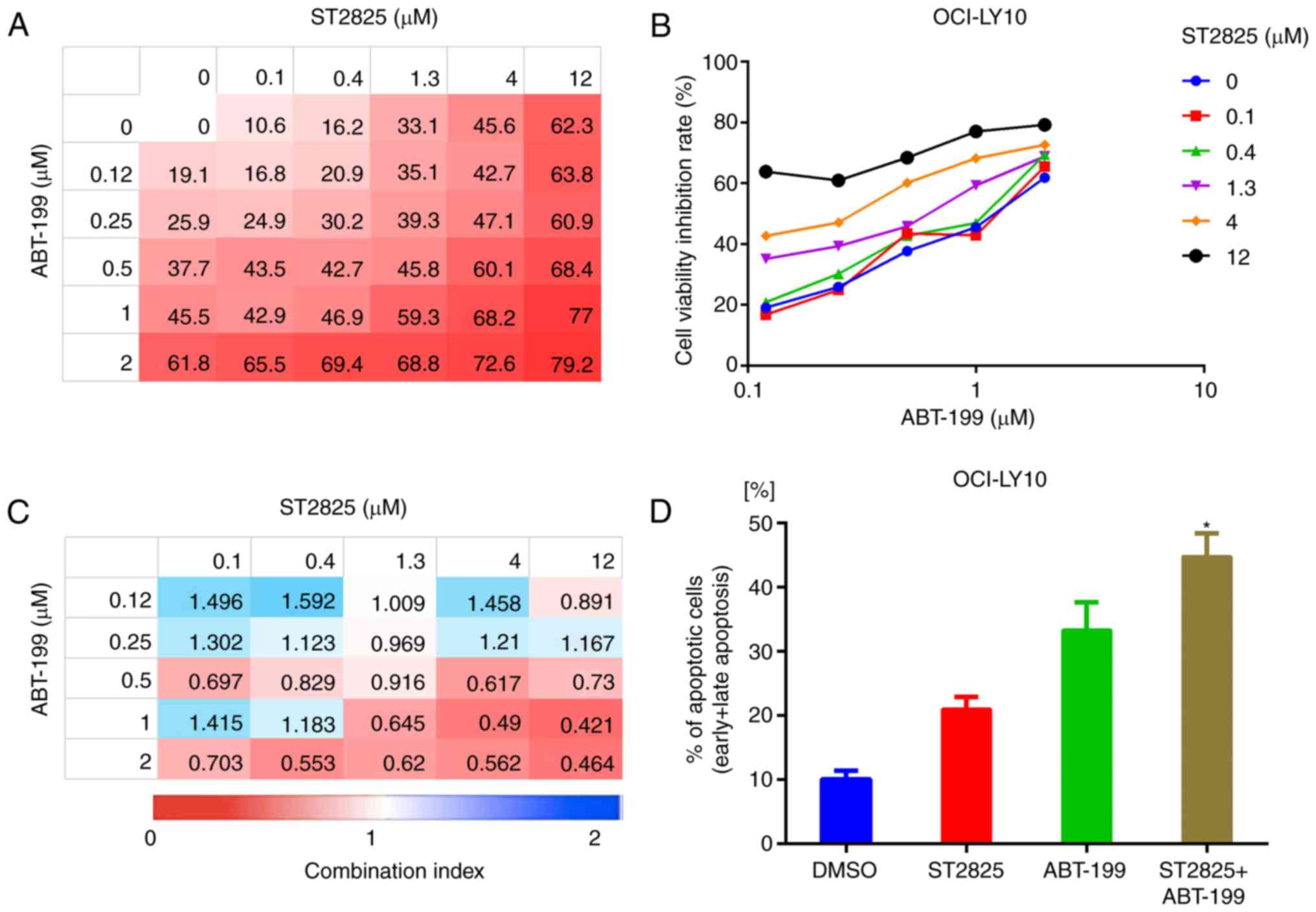 | Figure 6.Synergistic effect of BCL-2 inhibitor
and myddosome assembly inhibitor on ABC DLBCL cells. (A, B)
OCI-LY10 cells were treated for 72 h with ABT-199, ST2825 or both,
at the indicated doses, followed by a WST-1 assay. Inhibition at
varying dosimetries for the inhibitor of BCL-2 (ABT-199) and MYD88
(ST2825) in OCI-LY10 cells is depicted with (A) heat maps and (B)
line graphs. (C) Synergism was evaluated via CI analysis, and the
CI values of OCI-LY10 cells at varying dosimetry values for ABT-199
and ST2825 are demonstrated via heat maps. (D) Apoptotic cell
population (annexin V-positive) of OCI-LY10 cells analyzed by flow
cytometry, following treatment for 48 h with the vehicle, ST2825
(10 µM), ABT-199 (1 µM) or combination treatment. Statistical
analysis by one-way ANOVA with Tukey's post hoc test. Data are
presented as the mean ± standard deviation *P<0.05. BCL-2,
B-cell lymphoma-2; ABC, activated B cell; DLBCA, diffuse large
B-cell lymphoma; MYD88, myeloid differentiation primary response
gene 88; CI, combination index. |
Discussion
Recurrent MYD88 L265P mutations have been
demonstrated in numerous types of B-cell neoplasm, including IgM
MGUS, WM, DLBCL, mucosa-associated lymphoid tissue lymphoma and
chronic lymphocytic leukemia (12).
MYD88 L265P has been demonstrated to be the most frequently
occurring mutation in the ABC subtype of DLBCL (20–40%), whereas it
is rarely observed in the GCB subtype (8,17,25,26). A
study in Spain reported that the MYD88 L265P mutation was
significantly associated with inferior PFS and overall survival in
patients with DLBCL (P<0.01), and was identified as a
significant risk factor for mortality (hazard ratio, 2.4) via
multivariate Cox regression analysis (27). More recently, two studies identified
four or five subtypes of DLBCL based on distinct genetic features
and pathogenetic mechanisms, via comprehensive genetic analyses.
The results of both studies demonstrated that the subtype
associated with more frequent mutations in MYD88 indicated a less
favorable patient outcome (25,26).
MYD88 L265P was identified as an oncogenic driver
mutation, with enhanced phosphorylation of IRAK kinases and
activation of the NF-κB and JAK-STAT3 signaling pathways, resulting
in the promotion of cell survival in numerous B-cell neoplasms,
including ABC DLBCL (8,12,28).
Furthermore, mice occasionally developed ABC-DLBCL-like clonal
lymphomas following conditional expression of MYD88 L265P in B
cells specifically (29,30). MYD88 is a central adaptor to the TLRs/
IL-1R signaling pathway, which contains three distinct domains: A
C-terminal TIR domain, an N-terminal DD and a short linker region
in-between (31). During normal
immune responses, TLRs are primarily stimulated by foreign ligands
such as the chemical components of infecting microbes. Following
ligand binding, MYD88 is recruited to the activated receptor,
resulting in heterodimerization with the receptor and
homodimerization with another MyD88 molecule via TIR-TIR
interactions (32,33). These MYD88 oligomers further recruit
the IRAKs via DDs, promoting myddosome formation and triggering
subsequent downstream NF-κB and JAK-STAT3 signaling. The MYD88
L265P mutation, located in the TIR domain, reinforces dimerization
between the mutant and WT TIR domains and augments MYD88
oligomerization. In contrast to native MYD88 oligomers, those
containing L265P mutants can trigger myddosome assembly and
constitutive activation of NF-κB signaling in the absence of
external stimuli (13,34). The disruptive power of the proline
residue potentially contributes to augmented oligomerization of
MYD88 mutant TIR domains. The leucine-to-proline substitution at
position 265 in the β-sheets (at the hydrophobic core of MYD88)
would disrupt the secondary structure of the TIR domain, thus
stabilizing the core of the TIR domain dimer interface (12,35). The
BB-loop, interacting with the αE-helix, is critical for MYD88
TIR-TIR interactions (35,36). ST2825, a synthetic peptidomimetic that
interacts with the BB-loop, competently inhibits MYD88 WT TIR-TIR
interactions (14). Furthermore, it
was observed that ST2825 was able to disrupt the aggregation of
MYD88 mutant TIR domains in the present study, thus resulting in
decreased cell viability and increased apoptosis of MYD88
L265P-expressing ABC DLBCL cells.
The NF-κB transcription factor family (which can be
activated by the MYD88 L265P mutation) serves a crucial role in ABC
DLBCL cell survival (8,37). IκBα has been regarded as a critical
component of the NF-κB signaling pathway. The present study
demonstrated that IκBα phosphorylation was blocked by inhibiting
MYD88 oligomerization in L265P-expressing ABC DLBCL cells treated
with ST2825. Consistent with these results, decreased NF-κB
activity was observed following ST2825 treatment using a luciferase
NF-κB reporter assay. Furthermore, the present study also revealed
that ST2825 decreased the secretion of IL-10 and IFN-β (the
production of which are mediated by MYD88), which may be involved
in the immune modulation of the ABC DLBCL microenvironment
(8).
BTK, a mediator from BCR activity to NF-κB, serves
an essential role in chronically-active BCR signaling, which
supports tumor cell survival in ABC DLBCL cells (6,17). As
reported by Yang et al (38),
it was also observed that MYD88 interacted with BTK in L265P ABC
DLBCL cell lines, and that the binding between MYD88 and BTK was
abrogated by ST2825. Furthermore, the present study demonstrated
that the use of ST2825 inhibited BTK activity in MYD88
L265P-expressing ABC DLBCL cell lines. Collectively, the present
study demonstrated that when bound to BTK, MYD88 L265P could
influence the activity of the former. Given the association between
MYD88 and BTK, and their critical oncogenic roles in
L265P-expressing ABC DLBCL cells, the present study investigated
the effects of the dual inhibition of MYD88 and BTK. The data
revealed that the combined use of ST2825 and ibrutinib resulted in
synergistic killing effects, which were associated with enhanced
inhibition of NF-κB activity.
Based on the pathways regulated by MYD88 L265P
activity, there are several therapeutic strategies that have been
designed to halt this specific oncogenic process. The targets of
these therapies include BTK, IRAK1/4, JAK and myddosome assembly.
The BTK inhibitor ibrutinib, the only U.S. Food and Drug
Administration approved drug capable of influencing the L265P
driven pathway (potentially by abrogating the MYD88-BTK complex),
inhibits the survival of L265P-expressing cell lines, which is
consistent with the observations of the present study. A phase 1/2
clinical trial demonstrated that ibrutinib produced total responses
in 37% of 38 patients with ABC DLBCL (17). The patients with concurrent MYD88 and
BCR component mutations appeared to be more sensitive to ibrutinib,
while those with MYD88 mutations independent of chronically-active
BCR signaling exhibited resistance to ibrutinib. This indicates
that novel drugs that are more specific to MYD88 signaling require
further investigation. A number of IRAK inhibitors have
demonstrated convincing abilities to inhibit the growth of
L265P-expressing cell lines in pre-clinical models (39–41).
However, MYD88 dimerization may present a more favorable target
considering its proximity to the origin of hyperactivity,
particularly as IRAKs are not involved in all MyD88-dependent
signaling. Mini-peptides that have been designed to compete with
MYD88 TIR domain interactions disrupted myddosome signaling and
blocked WM cell proliferation (42,43).
Notably, ST2825 is a synthetic compound, created to mimic a portion
in the BB-loop of the MYD88 TIR domain, which can inhibit MYD88
dimerization (14). In the present
study, the use of ST2825 disrupted myddosome assembly and
contributed to growth inhibition, as well as attenuating NF-κB
activity in L265P ABC DLBCL cell lines.
The MYD88 L265P mutation frequently occurs alongside
other genetic events in ABC-DLBCLs, such as CD79B mutations
(44–46), loss of the tumor suppressor TNFAIP3 or
the overexpression of BCL-2 (30,34). It
has also been revealed that to induce lymphoma formation, MYD88
L265P may need to occur in addition to other genetic alterations,
while MYD88 mutations alone are potentially not sufficient. It has
been reported that MYD88 L265P and CD79B mutations cooperate to
block peripheral deletions and promote spontaneous plasmablast
differentiation in B cells (46). In
addition, TNFAIP3 loss enhances NF-κB signaling driven by MYD88
L265P, and contributes to ibrutinib resistance in lymphoma cell
lines. Furthermore, MYD88 mutations cooperate with BCL-2
overexpression to promote self-reactive B cell accumulation and
lymphomagenesis in vivo (30,34). These
findings provide a framework for the rational design of combination
therapy with dual inhibitors, targeting multiple signaling
pathways. The present study demonstrated that the dual inhibition
of MYD88 oligomerization (ST2825) and BTK activity (ibrutinib)
results in synergistic cell death by decreasing NF-κB activity. It
was also observed that combined treatment targeting both the
myddosome (ST2825) and BCL-2 (ABT-199) lead to synergistic toxicity
and increased apoptosis in ABC DLBCL cell lines.
To the best of our knowledge, the present study is
the first to report that myddosome assembly is disrupted by the
synthetic small-molecule compound ST2825 in MYD88 L265P ABC DLBCL
cells, resulting in proliferation inhibition, increased apoptosis
and decreased NF-κB activity. The interplay between the MYD88
mutant and BTK further confirms that oncogenic MYD88 L265P
cooperates with BCR signaling to promote tumor cell survival. It
was subsequently revealed that combined ST2825, either with the BTK
inhibitor ibrutinib, or the BCL-2 inhibitor ABT-199, resulted in
synergistic cell death in ABC DLBCL cells. Therefore, targeting
myddosome assembly may be a promising therapeutic strategy for
MYD88-mutated ABC DLBCL. Future studies may focus on developing
more ST2825-like synthetic peptidomimetic compounds with improved
efficacy and safety.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Roman Jerala and
Mateja M. Keber for supplying the plasmids and for their helpful
discussion.
Funding
The present work was supported by the Chongqing
Research Program of Basic Research and Frontier Technology (grant
no. cstc2017jcyjA0632), Chongqing Health and Family Planning
Commission Grant (grant no. zy201402109) to XW, and the Science and
Technology Research Program of Chongqing Municipal Education
Commission (grant no. KJ1600225) to Jing Hu.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article (and its Supplementary
files).
Authors' contributions
XW designed the research studies, performed the
experiments, analyzed the data and wrote the manuscript. YT and NSH
performed the experiments. JH and FZZ analyzed data and critically
revised the manuscript. ZLH and MG collected and analyzed the data.
WLF supervised the experimental work and critically revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
NHL
|
non-Hodgkins lymphoma
|
|
GCB
|
germinal center B cell
|
|
ABC
|
activated B cell
|
|
PFS
|
progression-free survival
|
|
BCR
|
B-cell receptor
|
|
TLR
|
toll-like receptors
|
|
CARD11
|
caspase recruitment domain-containing
protein 11
|
|
NF-κB
|
nuclear factor-kappa B
|
|
MYD88
|
myeloid differentiation primary
response gene 88
|
|
IL-1R
|
interleukin-1 receptor
|
|
TIR
|
Toll/interleukin-1 receptor
|
|
DD
|
death domain
|
|
HMW
|
high-molecular-weight
|
|
WM
|
Waldenstrom macroglobulinemia
|
|
WT
|
wild-type
|
|
BTK
|
Bruton's tyrosine kinase
|
|
BCL-2
|
B-cell lymphoma-2
|
|
IκB
|
inhibitor of NF-κB
|
|
CI
|
Combination index
|
References
|
1
|
Teras LR, DeSantis CE, Cerhan JR, Morton
LM, Jemal A and Flowers CR: 2016 US lymphoid malignancy statistics
by World Health Organization subtypes. CA Cancer J Clin.
66:443–459. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alizadeh AA, Eisen MB, Davis RE, Ma C,
Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al:
Distinct types of diffuse large B-cell lymphoma identified by gene
expression profiling. Nature. 403:503–511. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pfreundschuh M, Kuhnt E, Trümper L,
Osterborg A, Trneny M, Shepherd L, Gill DS, Walewski J, Pettengell
R, Jaeger U, et al: CHOP-like chemotherapy with or without
rituximab in young patients with good-prognosis diffuse
large-B-cell lymphoma: 6-year results of an open-label randomised
study of the MabThera International Trial (MInT) Group. Lancet
Oncol. 12:1013–1022. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rawlings DJ, Schwartz MA, Jackson SW and
Meyer-Bahlburg A: Integration of B cell responses through Toll-like
receptors and antigen receptors. Nat Rev Immunol. 12:282–294. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davis RE, Ngo VN, Lenz G, Tolar P, Young
RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al:
Chronic active B-cell-receptor signalling in diffuse large B-cell
lymphoma. Nature. 463:88–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lenz G, Davis RE, Ngo VN, Lam L, George
TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al: Oncogenic
CARD11 mutations in human diffuse large B cell lymphoma. Science.
319:1676–1679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ngo VN, Young RM, Schmitz R, Jhavar S,
Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al:
Oncogenically active MYD88 mutations in human lymphoma. Nature.
470:115–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ntoufa S, Vilia MG, Stamatopoulos K, Ghia
P and Muzio M: Toll-like receptors signaling: A complex network for
NF-κB activation in B-cell lymphoid malignancies. Semin Cancer
Biol. 39:15–25. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin SC, Lo YC and Wu H: Helical assembly
in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature.
465:885–890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu X, Li W, Deng Q, Li L, His ED, Young
KH, Zhang M and Li Y: MYD88 L265P mutation in lymphoid
malignancies. Cancer Res. 78:2457–2462. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Avbelj M, Wolz OO, Fekonja O, Benčina M,
Repič M, Mavri J, Krüger J, Schärfe C, Delmiro Garcia M, Panter G,
et al: Activation of lymphoma-associated MyD88 mutations via
allostery-induced TIR-domain oligomerization. Blood. 124:3896–3904.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Loiarro M, Capolunghi F, Fantò N, Gallo G,
Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, Ruggiero V
and Sette C: Pivotal advance: Inhibition of MyD88 dimerization and
recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound.
J Leukoc Biol. 82:801–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H, Huang Z, Gao M, Huang N, Luo Z, Shen
H, Wang X, Wang T, Hu J and Feng W: Inhibition of YAP suppresses
CML cell proliferation and enhances efficacy of imatinib in vitro
and in vivo. J Exp Clin Cancer Res. 35:1342016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilson WH, Young RM, Schmitz R, Yang Y,
Pittaluga S, Wright G, Lih CJ, Williams PM, Shaffer AL, Gerecitano
J, et al: Targeting B cell receptor signaling with ibrutinib in
diffuse large B cell lymphoma. Nat Med. 21:922–926. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jefferies CA, Doyle S, Brunner C, Dunne A,
Brint E, Wietek C, Walch E, Wirth T and O'Neill LA: Bruton's
tyrosine kinase is a Toll/interleukin-1 receptor domain-binding
protein that participates in nuclear factor kappaB activation by
Toll-like receptor 4. J Biol Chem. 278:26258–26264. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P,
Yao H and Cao X: Intracellular MHC class II molecules promote
TLR-triggered innate immune responses by maintaining activation of
the kinase Btk. Nat Immunol. 12:416–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Phelan JD, Young RM, Webster DE, Roulland
S, Wright GW, Kasbekar M, Shaffer AL III, Ceribelli M, Wang JQ,
Schmitz R, et al: A multiprotein supercomplex controlling oncogenic
signalling in lymphoma. Nature. 560:387–391. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lenz G, Wright GW, Emre NC, Kohlhammer H,
Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al:
Molecular subtypes of diffuse large B-cell lymphoma arise by
distinct genetic pathways. Proc Natl Acad Sci USA. 105:13520–13525.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Iqbal J, Neppalli VT, Wright G, Dave BJ,
Horsman DE, Rosenwald A, Lynch J, Hans CP, Weisenburger DD, Greiner
TC, et al: BCL2 expression is a prognostic marker for the activated
B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol.
24:961–968. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davids MS: Targeting BCL-2 in B-cell
lymphomas. Blood. 130:1081–1088. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chapuy B, Stewart C, Dunford AJ, Kim J,
Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et
al: Molecular subtypes of diffuse large B cell lymphoma are
associated with distinct pathogenic mechanisms and outcomes. Nat
Med. 24:679–690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmitz R, Wright GW, Huang DW, Johnson
CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer
AL, et al: Genetics and pathogenesis of diffuse large B-cell
lymphoma. N Engl J Med. 378:1396–1407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fernández-Rodríguez C, Bellosillo B,
García-García M, Sánchez-González B, Gimeno E, Vela MC, Serrano S,
Besses C and Salar A: MYD88 (L265P) mutation is an independent
prognostic factor for outcome in patients with diffuse large B-cell
lymphoma. Leukemia. 28:2104–2106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weber ANR, Cardona Gloria Y, Çınar Ö,
Reinhardt HC, Pezzutto A and Wolz OO: Oncogenic MYD88 mutations in
lymphoma: Novel insights and therapeutic possibilities. Cancer
Immunol Immunother. 67:1797–1807. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Janz S: Mouse model of
MYD88L265P-dependent DLBCL. Blood. 127:2660–2661. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Knittel G, Liedgens P, Korovkina D, Seeger
JM, Al-Baldawi Y, Al-Maarri M, Fritz C, Vlantis K, Bezhanova S,
Scheel AH, et al: B-cell-specific conditional expression of
Myd88p.L252P leads to the development of diffuse large B-cell
lymphoma in mice. Blood. 127:2732–2741. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hardiman G, Rock FL, Balasubramanian S,
Kastelein RA and Bazan JF: Molecular characterization and modular
analysis of human MyD88. Oncogene. 13:2467–2475. 1996.PubMed/NCBI
|
|
32
|
Fekonja O, Benčina M and Jerala R:
Toll/interleukin-1 receptor domain dimers as the platform for
activation and enhanced inhibition of Toll-like receptor signaling.
J Biol Chem. 287:30993–31002. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jing X, Tian Z, Gao P, Xiao H, Qi X, Yu Y,
Ding X, Yang L and Zong L: HBsAg/β2GPI activates the NF-κB pathway
via the TLR4/MyD88/IkBα axis in hepatocellular carcinoma. Oncol
Rep. 40:1035–1045. 2018.PubMed/NCBI
|
|
34
|
Wang JQ, Jeelall YS, Beutler B, Horikawa K
and Goodnow CC: Consequences of the recurrent MYD88(L265P) somatic
mutation for B cell tolerance. J Exp Med. 211:413–426. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vyncke L, Bovijn C, Pauwels E, Van Acker
T, Ruyssinck E, Burg E, Tavernier J and Peelman F: Reconstructing
the TIR side of the myddosome: A paradigm for TIR-TIR interactions.
Structure. 24:437–447. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohnishi H, Tochio H, Kato Z, Orii KE, Li
A, Kimura T, Hiroaki H, Kondo N and Shirakawa M: Structural basis
for the multiple interactions of the MyD88 TIR domain in TLR4
signaling. Proc Natl Acad Sci USA. 106:10260–10265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pasqualucci L and Zhang B: Genetic drivers
of NF-κB deregulation in diffuse large B-cell lymphoma. Semin
Cancer Biol. 39:26–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang G, Zhou Y, Liu X, Xu L, Cao Y,
Manning RJ, Patterson CJ, Buhrlage SJ, Gray N, Tai YT, et al: A
mutation in MYD88 (L265P) supports the survival of
lymphoplasmacytic cells by activation of Bruton tyrosine kinase in
Waldenström macroglobulinemia. Blood. 122:1222–1232. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kelly PN, Romero DL, Yang Y, Shaffer AL
III, Chaudhary D, Robinson S, Miao W, Rui L, Westlin WF, Kapeller R
and Staudt LM: Selective interleukin-1 receptor-associated kinase 4
inhibitors for the treatment of autoimmune disorders and lymphoid
malignancy. J Exp Med. 212:2189–2201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Markovtsov VV, Lamagna C, Chan M, Yi S,
Young C, Frances R, Siu S, Braselmann S, Li H, Singh R, et al:
Potential role for R191, potent and selective IRAK4 kinase
inhibitor, in treatment of hematologic malignancies. AACR Abstract.
2016.
|
|
41
|
Ni H, Shirazi F, Baladandayuthapani V, Lin
H, Kuiatse I, Wang H, Jones RJ, Berkova Z, Hitoshi Y, Ansell SM, et
al: Targeting myddosome signaling in Waldenström's
macroglobulinemia with the interleukin-1 receptor-associated kinase
1/4 inhibitor R191. Clin Cancer Res. 24:6408–6420. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao
Y, Sheehy P, Manning RJ, Patterson CJ, Tripsas C, et al: MYD88
L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J
Med. 367:826–833. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu X, Hunter ZR, Xu L, Chen J, Chen JG,
Tsakmaklis N, Patterson CJ, Castillo JJ, Buhrlage S, Gray N, et al:
Targeting myddosome assembly in Waldenström macroglobulinaemia. Br
J Haematol. 177:808–813. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim Y, Ju H, Kim DH, Yoo HY, Kim SJ, Kim
WS and Ko YH: CD79B and MYD88 mutations in diffuse large B-cell
lymphoma. Hum Pathol. 45:556–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yamada S, Ishida Y, Matsuno A and Yamazaki
K: Primary diffuse large B-cell lymphomas of central nervous system
exhibit remarkably high prevalence of oncogenic MYD88 and CD79B
mutations. Leuk Lymphoma. 56:2141–2145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang JQ, Jeelall YS, Humburg P, Batchelor
EL, Kaya SM, Yoo HM, Goodnow CC and Horikawa K: Synergistic
cooperation and crosstalk between MYD88L265P and
mutations that dysregulate CD79B and surface IgM. J Exp Med.
214:2759–2776. 2017. View Article : Google Scholar : PubMed/NCBI
|