Introduction
Renal cell carcinoma (RCC) is the most common kidney
cancer subtype. More than 100,000 cases are diagnosed worldwide per
year, and RCC accounts for approximately 2–5% of all adult
neoplastic diseases and its incidence is rising (1). RCC originates from the tubular
structures of the kidney and comprises a heterogeneous group of
malignancies. RCC can be classified into four major histological
cell types, of which clear cell RCC (ccRCC) is the most common
(75–80%) (2). Other types include
papillary (10–15%), chromophobe (5%) and collecting duct (1%) RCC.
Metastasis may be present at the time of RCC diagnosis and such
cases are resistant to radiotherapy, chemotherapy and targeted
therapy. Despite great efforts in the field of RCC therapy, RCC
remains an incurable disease and the median survival period of
patients with advanced RCC is approximately 26 months (3). Therefore, there is a compelling demand
to identify both molecular targets and novel drugs with which to
treat this disease.
Inhibition of vascular endothelial growth factor
(VEGF) receptors is a promising approach for the targeted therapy
of RCC. Antiangiogenic drugs for RCC treatment such as sunitinib,
pazopanib, sorafenib and axitinib have recently received particular
attention (3–5). Sunitinib is a small-molecule inhibitor
of multiple receptor tyrosine kinases (RTKs), that inhibits the
family of VEGF receptors (VEGFR-1, VEGFR-2 and VEGFR-3), PDGF
receptors (PDGFR-α and PDGFR-β), FLT3, the stem cell growth factor
receptor KIT and RET. The repressive effect of sunitinib on tumor
growth and invasion is mainly achieved by antagonizing the
activation of VEGFR and PDGFR, two crucial players in the
pathogenesis of RCC. Sunitinib treatment for metastasized RCC
contributes to a significantly longer progression-free survival and
overall survival. Although sunitinib has become the mainstay of RCC
treatment, the occurrence of resistance is a main obstacle
overshadowing its clinical benefit. Additional underlying
therapeutic targets for RCC include mammalian target of rapamycin
(mTOR) kinase, immune checkpoint proteins, indoleamine
2,3-dioxygenase and transforming growth factor-β (3–5).
As tumor cells may escape the inhibition of VEGF
signaling using other parallel pathways as growth and survival
signals (6), it is necessary to seek
accessory pathways to develop new treatment approaches and avoid
chemotherapeutic resistance. Tropomyosin-related kinases (Trks) are
a family of receptor tyrosine kinases consisting of 3 isoforms
(TrkA, TrkB and TrkC) that are activated by neurotrophins. In the
present study, we demonstrated that GNF-5837, an inhibitor of TrkA
and TrkB, reduced the cell viability of renal carcinoma cells via
inducing cell cycle arrest and apoptosis. Meanwhile, GNF-5837 also
suppressed the migration ability of renal carcinoma cells. Taken
together, these results identify the potential pharmacological
merit of a Trk inhibitor for the chemotherapy of RCC.
Materials and methods
Reagents
GNF-5837 and sunitinib were purchased from Selleck
Chemicals (Houston, TX, USA).
Cell culture
The human RCC cell lines 786O and Caki-2 were
purchased from the Shanghai Institute of Cell Biology (Shanghai,
China) and were cultured in Roswell Park Memorial Institute
(RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS;
Gibco/Thermo Fisher Scientific, Inc.), penicillin (100 U/ml), and
streptomycin (100 µg/ml) under a humidified atmosphere containing
5% CO2 maintained at 37°C.
MTS assay
Cell viability was determined using the CellTiter
96® AQueous One Solution Cell Proliferation
assay kit (Promega, Madison, WI, USA). Cells (5×103)
were seeded into 96-well plates and treated with GNF-5837 or other
agents for the depicted time intervals. After treatment, 10 µl MTS
(Promega) was added into each well for a 2-h incubation. The
absorbance was measured using a model ELX800 Micro Plate Reader
(Bio-Tek Instruments, Winooski, VT, USA) at 490 nm.
EdU incorporation assay
Cell proliferation was determined by incorporation
of 5-ethynyl-2′-deoxyuridine (EdU) using an EdU Cell Proliferation
Assay kit (Guangzhou RiboBio Co., Ltd., Guangzhou, China) as
previously described (7). Cells were
cultured in triplicate in 24-well plates at a density of
5×104 and treated with GNF-5837 for 48 h at 37°C, and
then 50 mM EdU was added and incubated for an additional 2 h at
37°C. The cells were fixed with 4% formaldehyde for 30 min at room
temperature and treated with 2 mg/ml glycine for 5 min. After
washing with phosphate-buffered saline (PBS), the cells were
treated with 0.5% Triton X-100 for 10 min at room temperature for
permeabilization. Apollo® reaction cocktail (100 µl) was
added to each well and incubation was carried out for 30 min in the
dark at room temperature. After washing with Triton X-100, the
cells were stained with 100 µl Hoechst 33342 for 30 min and
visualized under a fluorescence microscope (Olympus Corp., Japan)
at ×200 magnification. A total of 100 cells from the different
groups was analyzed for EdU labeling in this study.
Flow cytometric analysis
To assess cell cycle progression by flow cytometry,
786O cells (2×105) after GNF-5837 treatment were
suspended in 100 µl of PBS, and 200 µl of 95% ethanol was added.
The cells were then incubated at 4°C for 1 h, washed with PBS,
resuspended in 250 µl of 1.12% sodium citrate buffer (pH 8.4)
together with 12.5 µg of RNase, and incubated at 37°C for an
additional 30 min. The cellular DNA was then stained by 250 µl
propidium iodide (PI) for 30 min at room temperature. Red
fluorescence emitted from the PI-DNA complex was analyzed at 488
nm/600 nm (excitation/emission wavelength) using a FACScan flow
cytometer (BD LSR II). The data of relative DNA content were
analyzed by ModFit LT software package (https://www.vsh.com/products/mflt/mfFeatures.asp)
to reveal cell cycle distribution.
Apoptosis assay
786O and Caki-2 cells (2×105) were seeded
in 6-well plates and allowed to attach overnight. After GNF-5837
treatment for 48 h, cells were harvested and resuspended in 200 µl
binding buffer after washing twice with cold PBS. Then, apoptotic
cells were assessed by an Annexin-V apoptosis detection kit
(MultiSciences Biotech, Hangzhou, China) according to the
manufacturer's protocol. The cell suspension was incubated with 10
µl Annexin V-fluorescein isothiocyanate (FITC) stock solution for
30 min at 4°C in the dark, and then incubated with 5 µl PI solution
for 5 min. Cell samples were analyzed by flow cytometry (BD LSR II;
BD Biosciences, Franklin Lakes, NJ, USA) and apoptotic cell
fractions were determined.
Real-time polymerase chain reaction
(qPCR)
Total RNA was isolated using the RNAiso Plus (Takara
Biotechnology, Dalian, China), and cDNA was prepared using
Transcriptor First Strand cDNA Synthesis kit (Roche) according to
the manufacturer's instructions. Quantitative reverse transcriptase
PCR (RT-qPCR) was performed to quantify the expression of the genes
of interest using LightCycler® 480 SYBR-Green I Master
(Roche, Indianapolis, IN, USA) according to the manufacturer's
protocol on Roche light cycler version 3.5. The following thermal
profile was applied: 1 cycle at 95°C for 10 min, 40 cycles at 95°C
for 10 sec, 60°C for 30 sec, and 72°C for 15 sec. The level of
expression of each target gene was calculated using the
2−ΔΔCq method (8). The
relative amount of each mRNA was normalized to GAPDH. Each
sample was examined in triplicate. qPCR analysis was performed with
the following primers: Survivin 5′-GAGGCTGGCTTCATCCACTG-3′
(forward) and 5′-GCACTTTCTTCGCAGTTTCCTC-3′ (reverse); Bcl2
5′-TCGCCCTGTGGATGACTGA-3′ (forward) and 5′-CAGAGACAGCCAGGAGAAATC-3′
(reverse); Bax 5′-TGGCAGCTGACATGTTTTCTGAC-3′ (forward) and
5′-TCACCCAACCACCCTGGTCTT-3′ (reverse); GAPDH
5′-CTCACCGGATGCACCAATGTT-3′ (forward) and
5′-CGCGTTGCTCACAATGTTCAT-3′ (reverse). The designed primers were
synthesized by Sangon Biotech (Shanghai, China).
Western blot analysis
The cells (1×106) were washed with cold
PBS and lysed on ice in 100 µl modified radioimmunoprecipitation
assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25%
Na-deoxycholate, 150 mM NaCl, 1 mM Na3VO4,
and 1 mM NaF) containing protease inhibitors [100 µM
phenylmethylsulfonyl fluoride, 10 µM leupeptin, 10 µM pepstatin and
2 mM ethylenediaminetetraacetic acid (EDTA)]. The protein content
was measured using a BCA Protein Assay kit (Beyotime, Shanghai,
China). The proteins were separated by 10% SDS-PAGE (sodium dodecyl
sulphate-polyacrylamide gel electrophoresis) and transferred to
polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford,
MA, USA), and then blotted with the specific antibodies. Antibodies
against phosphorylated TrkA (cat. no. AP0492; dilution 1:500),
total TrkA (cat. no. A15618; dilution 1:500), phosphorylated TrkB
(cat. no. AP0423; dilution 1:500), total TrkB (cat. no. A2099;
dilution 1:500), and β-actin (cat. no. AC026; dilution 1:500) were
obtained from ABclonal Biotechnology (Wuhan, China). Antibodies
against phosphorylated Rac1 (cat. no. 2461; dilution 1:1,000),
total Rac1 (cat. no. 4651; dilution 1:1,000), phosphorylated ERK
(cat. no. 9101; dilution 1:1,000), total ERK (cat. no. 4695;
dilution 1:1,000), phosphorylated AKT (cat. no. 4060; dilution
1:1,000), and total AKT (cat. no. 9272; dilution 1:1,000) were
obtained from Cell Signaling Technology (Danvers, MA, USA).
Antibodies specific for p21 (cat. no. sc-6246; dilution 1:200),
c-Myc (cat. no. sc-40; dilution 1:200), poly(ADP-ribose)
polymerase-1 (PARP-1) (cat. no. sc-56197; dilution 1:200), and
survivin (cat. no. sc-17779; dilution 1:200) were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). The secondary
antibodies were horseradish peroxidase conjugated goat anti-rabbit
IgG (cat. no. AS014; dilution 1:2,000; ABclonal Biotechnology) and
anti-mouse IgG (cat. no. AS003; dilution 1:2,000; ABclonal
Biotechnology) antibodies. Detection of specific proteins was
carried out with an ECL chemiluminescence detection kit (Vigorous,
Beijing, China) according to the manufacturer's instructions.
Wound healing assay
786O and Caki-2 cells were seeded in 6-well plates
for the wound healing assay. When monolayer cells grew to about 80%
confluence, they were wounded by the tip of a sterile 200-µl
micropipette. Cells were washed with PBS (pH 7.4) for 3 times and
then cultured with complete medium (10% FBS) containing 20 µM
GNF-5837. The migration of the cells at the edge of the scratch was
analyzed at 24 h. Images were captured by an inverted microscope
(Olympus Corporation, Japan) at ×10.
Statistical analysis
All data in this study are displayed as means ± SD
(n≥3 repeats). Comparisons were analyzed by Student's t-test or
one-way analysis of variance (ANOVA) followed by the
Student-Newman-Keuls (SNK) test. The significance was analyzed with
SPSS 10.0 software (SPSS, Inc., Chicago, IL, USA) and a P-value
<0.05 was considered to indicate a statistically significant
result.
Results
GNF-5837 decreases the cell viability
of the RCC cell lines
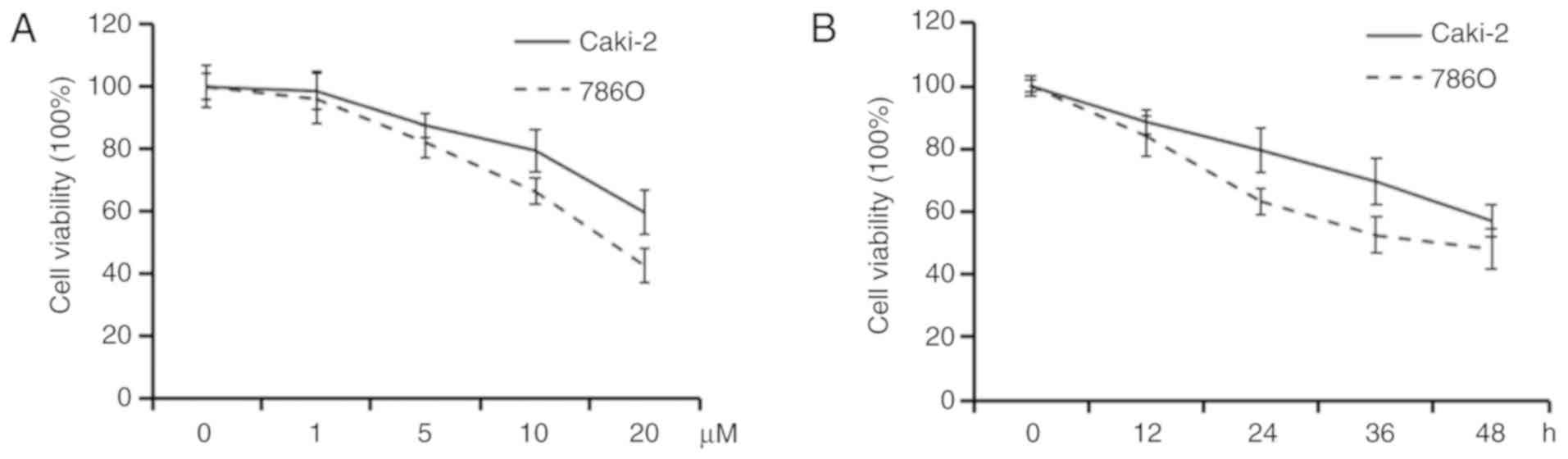
In order to evaluate the effects of a Trk inhibitor
GNF-5837 on RCC cell lines, cell viability was tested in Caki-2 and
786O cell lines by MTS assay. The 786O cell line has many
characteristics of ccRCC. Although the Caki-2 cell line was
primarily recognized as a ccRCC cell line, an accumulating amount
of data showed that it is a cell line of papillary RCC. After
exposure to elevated concentrations of GNF-5837 (0–20 µM) for 48 h,
both of the tested RCC cell lines showed a dose-dependent decrease
in cell viability (Fig. 1A). Our
results revealed that GNF-5837 at 20 µM exerted a significant
effect on cell viability. Compared with the DMSO-treated control,
the viability of the Caki-2 and 786O cells was decreased to about
57.1 and 48.2% after treatment with 20 µM GNF-5837 for 48 h,
respectively. MTS assays also showed that exposure to 20 µM
GNF-5837 resulted in marked time-dependent reduction in cell
viability (Fig. 1B).
GNF-5837 inhibits Trks and its
downstream signaling pathways
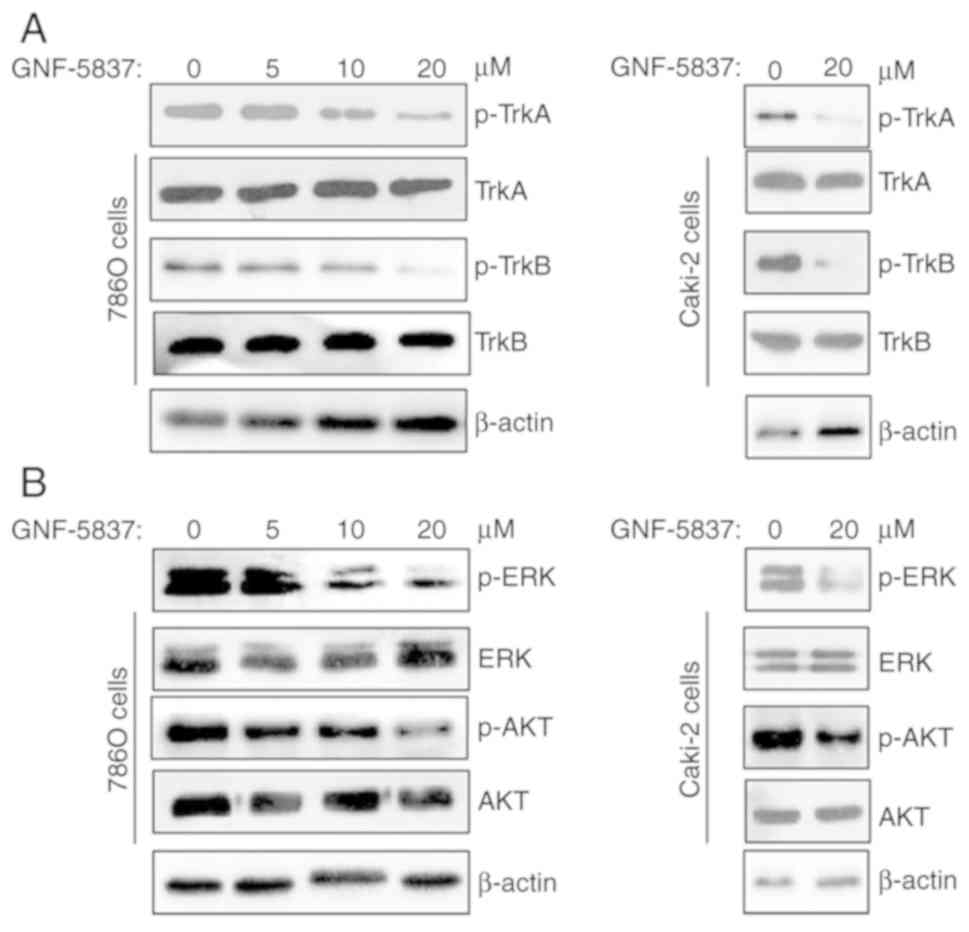
Subsequently, we evaluated the inhibition of
GNF-5837 on Trk signaling in 786O cells. As shown in Fig. 2A, GNF-5837 treatment (0–20 µM) for 48
h in 786O cells effectively decreased the phosphorylation of TrkA
and TrkB signaling in a dose-dependent manner. High concentration
of GNF-5837 (20 µM) produced similar effects in Caki-2 cells.
Activation of Trk signaling has been demonstrated to affect cell
growth, survival, and differentiation via modulating the PI3K/AKT
and MAPK/ERK signaling pathways. Thus, we further assessed the
effect of GNF-5837 treatment on Trk downstream molecules including
AKT and ERK kinases. As shown in Fig.
2B, GNF-5837 directly interrupted the phosphorylation
activation of AKT and ERK kinases in a dose-dependent manner in
786O cells. Similar results were also observed in Caki-2 cells.
Overall, these findings indicate that GNF-5837 treatment inhibits
Trk kinases and downstream signaling pathways.
GNF-5837 suppressed the cell growth of
RCC cells
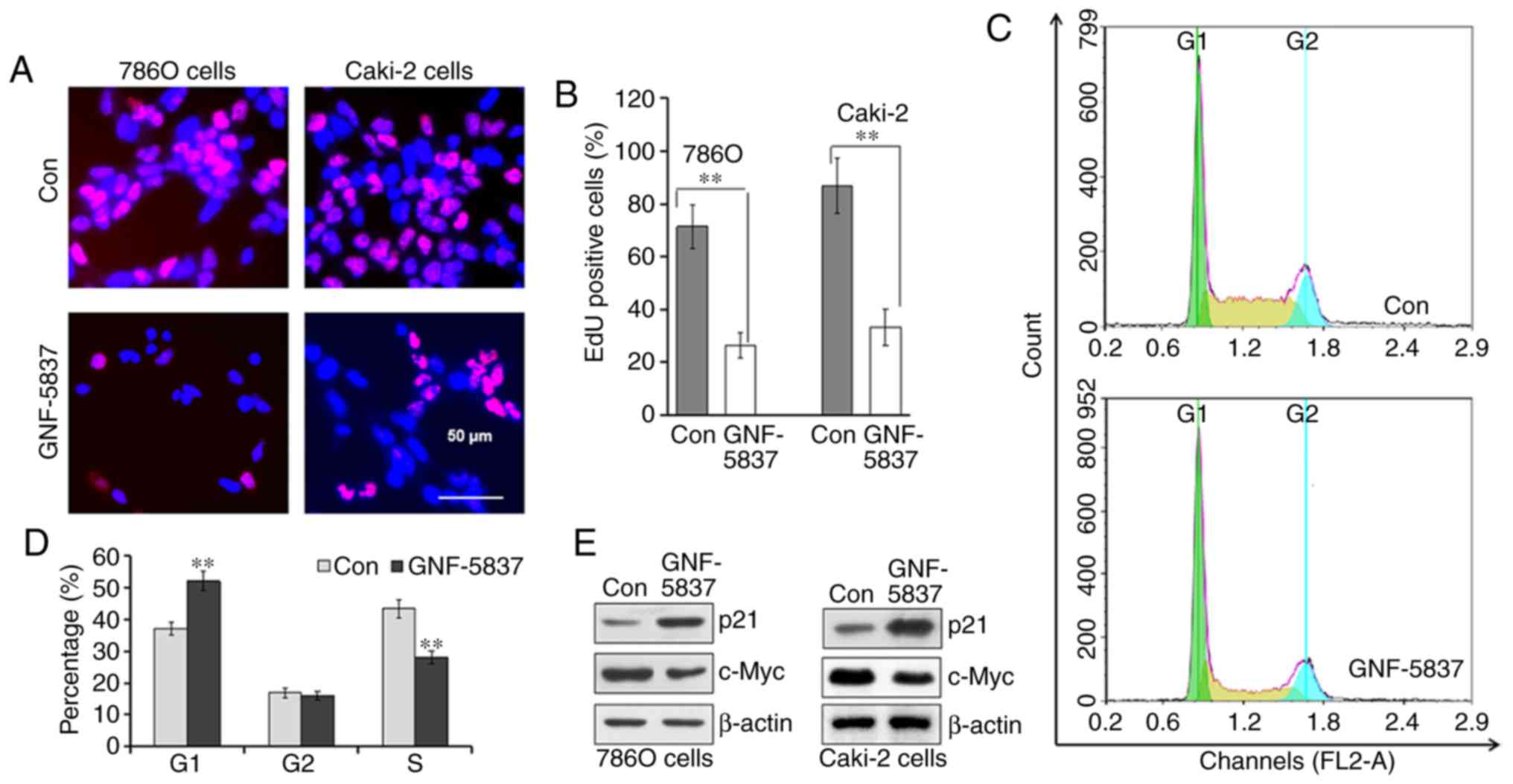
Next, we investigated the effect of GNF-5837
treatment on cell growth. Firstly, EdU incorporation assay was used
to detect DNA synthesis. After incubation with 20 µM GNF-5837 for
48 h, the number of EdU-positive 786O and Caki-2 cells was
significantly decreased (Fig. 3A).
For example, EdU-incorporating cells accounted for as much as 71.4%
of the total 786O cells in the control group, while only 26.3%
cells were positive for EdU staining after GNF-5837 treatment for
48 h (Fig. 3B). These results
indicated that the Trk inhibitor inhibited the proliferation of RCC
cells. Secondly, the cell cycle profile was analyzed using
propidium iodide staining and flow cytometry after treatment with
20 µM GNF-5837 for 48 h in 786O cells. Compared with the control,
GNF-5837 treatment significantly decreased the cell populations in
the S phase (Fig. 3C and D).
Meanwhile, GNF-5837 treatment mainly induced cell cycle arrest in
the G0/G1 phase. Coincidently, GNF-5837 treatment upregulated the
expression of p21 protein, a CDK inhibitor, in both 786O and Caki-2
cell lines (Fig. 3E). In comparison,
GNF-5837 exerted a moderate inhibition on the expression of c-Myc,
a master regulator of cell division.
GNF-5837 induces cell apoptosis
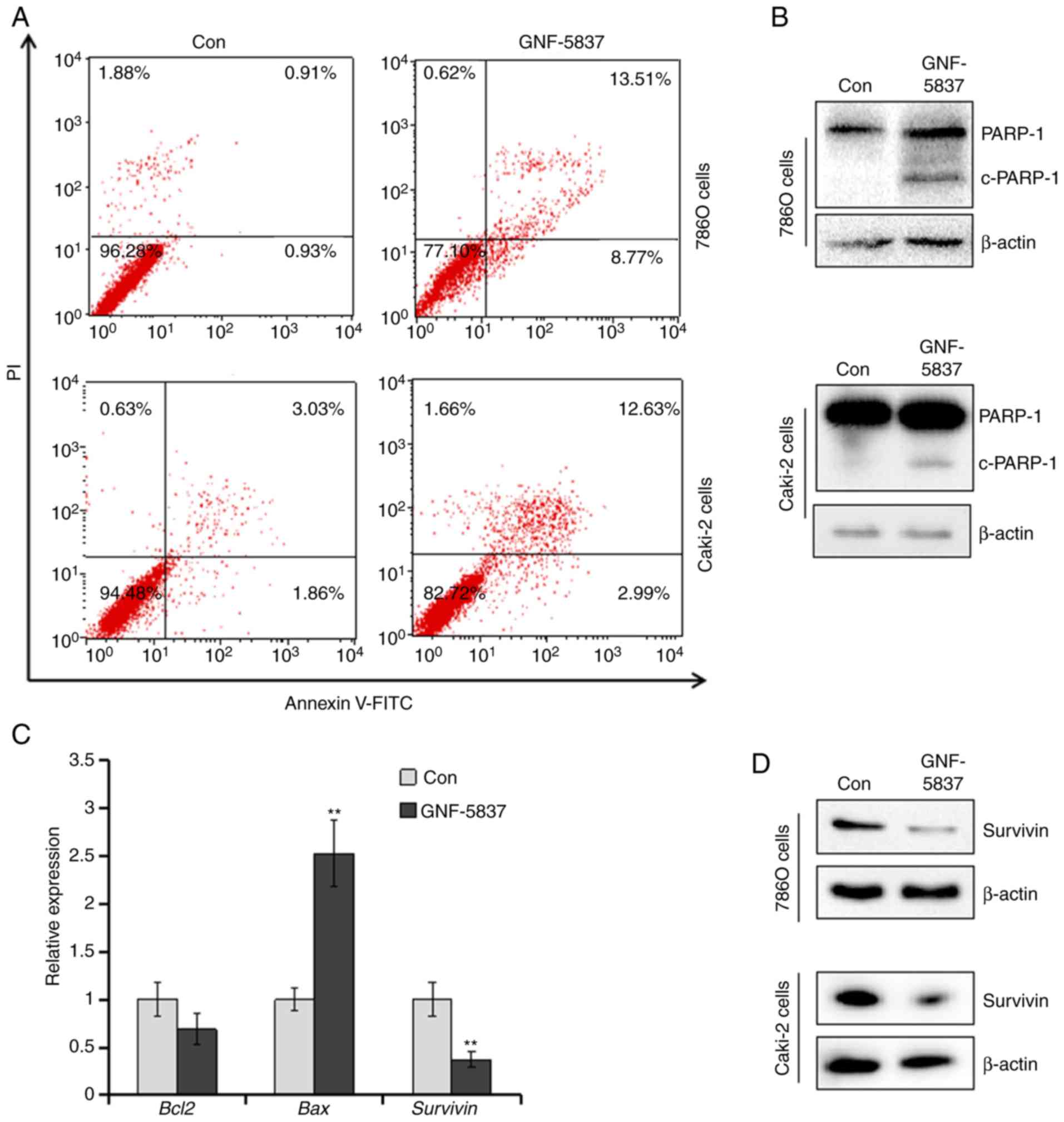
Since GNF-5837 was found to inhibit the viability
and growth of Caki-2 and 786O cells, we investigated whether
GNF-5837 induces the apoptosis of RCC cells. After incubation with
20 µM GNF-5837 for 48 h, 786O and Caki-2 cells were stained with
Annexin V-FITC and propidium iodide (PI) and analyzed by flow
cytometry. As shown in Fig. 4A,
GNF-5837 increased the apoptotic populations (Annexin
V-FITC-positive cells) to 20.6±2.1 and 15.5±2.9% in the 786O and
Caki-2 cells (P<0.01, vs. control), respectively. Next,
immunoblotting assay was performed to investigate the expression of
cleaved PARP-1, a hallmark of apoptosis. As expected, incubation
with 20 µM GNF-5837 triggered the cleavage of PARP-1 in 786O and
Caki-2 cells (Fig. 4B). Furthermore,
apoptosis-related genes were examined by qPCR assay. As shown in
Fig. 4C, the expression of
anti-apoptotic survivin gene (BIRC5) in 786O cells was
significantly downregulated by GNF-5837 treatment. Bcl2
expression was slightly altered. Conversely, the pro-apoptotic
protein Bax was significantly induced. Immunoblotting assay
further confirmed that GNF-5837 treatment suppressed the abundance
of the survivin protein (Fig.
4D).
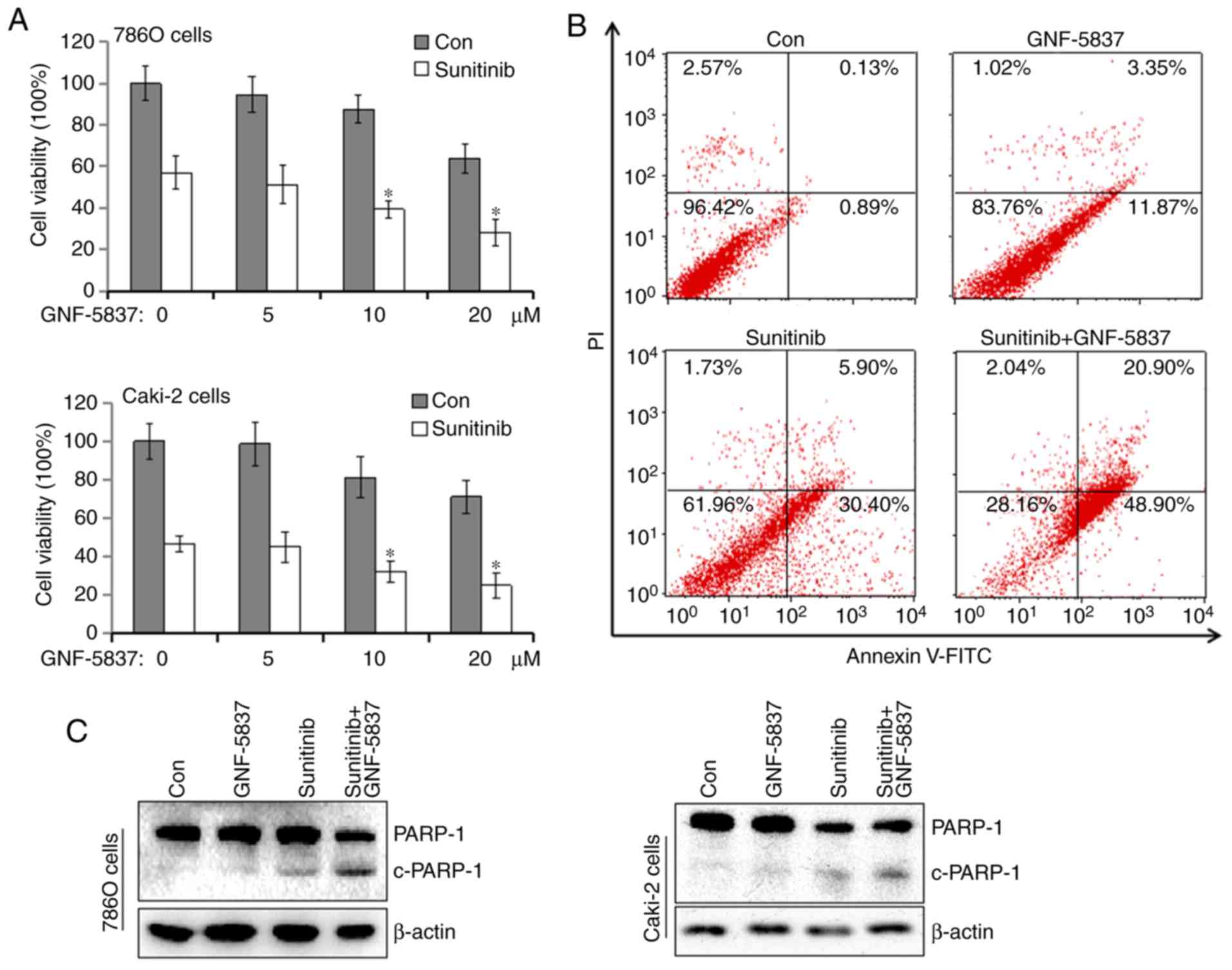
GNF-5837 potentiates sunitinib-induced
cytotoxicity
Subsequently, we estimated the combined effects of
GNF-5837 together with sunitinib, a tyrosine kinase inhibitor in
the treatment of metastatic RCC. By comparison, sunitinib is a
strong apoptosis inducer and alone can trigger a majority of cells
to undergo apoptosis. Thus, we shortened the treatment period to
reduce the toxicity of sunitinib, thereby to manifest the
cooperative effect of GNF-5837. 786O cells were exposed to 20 µM
sunitinib together with increasing concentrations of GNF-5837 for
24 h. Following treatment, MTS assay was performed. As shown in
Fig. 5A, sunitinib alone decreased
the cell viability of 786O cells by 42.9%. When the cells were
cotreated with 10 or 20 µM GNF-5837, the inhibition rate increased
to 60.7 and 68.8% following cotreatment, respectively. Similar
results were obtained for the Caki-2 cells. The results from flow
cytometry also demonstrated that cotreatment with GNF-5837 and
sunitinib greatly triggered cell apoptosis in the 786O cells
(Fig. 5B). Apoptosis occurred in
13.9±1.2, 31±5.3 and 60±8.8% of 786O cells in the GNF-5837,
sunitinib and combined treatment groups, respectively. PARP-1
cleavage in 786O and Caki-2 cells was increased after the combined
treatment (Fig. 5C). These results
indicate that there is an enhanced antitumor effect of sunitinib
and GNF-5837.
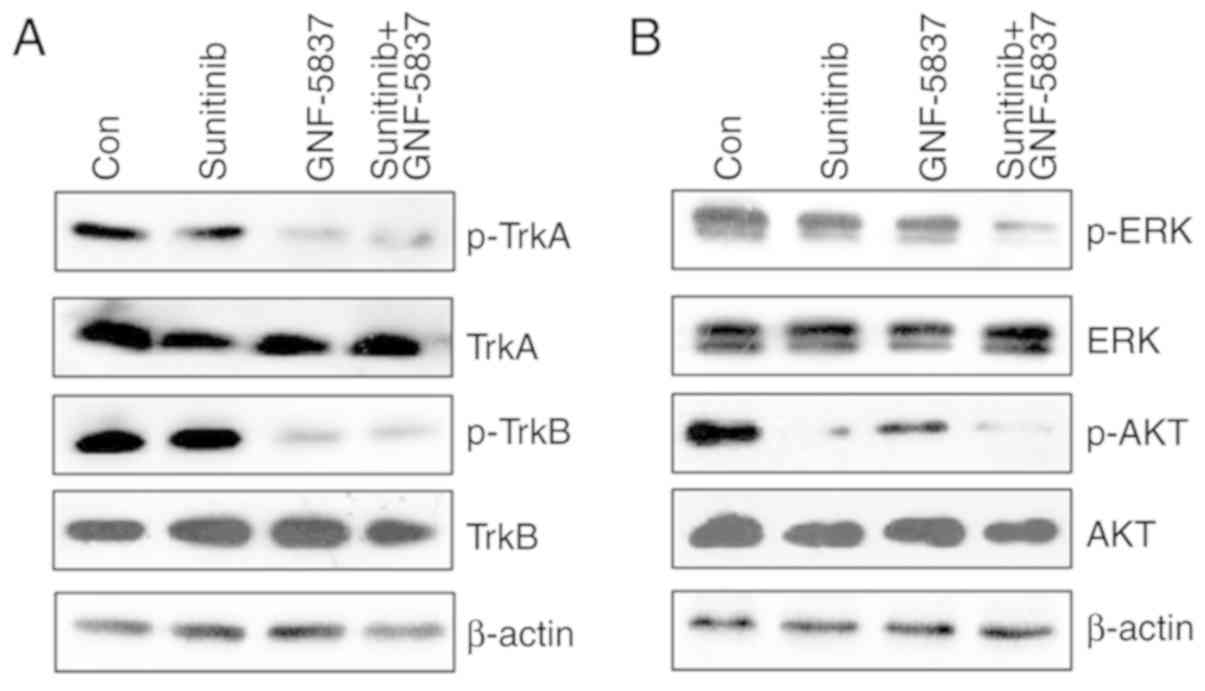
GNF-5837 cooperates with sunitinib to
inhibit the ERK cascade
To further ascertain the underlying mechanism
involving the the synergy of GNF-5837 and sunitinib, we examined
the alteration of Trk, ERK and AKT signals in 786O cells under
different circumstances. As shown in Fig.
6A, sunitinib alone did not affect the active levels of TrkA
and TrkB kinases, which were greatly suppressed upon GNF-5837
treatment. In contrast, sunitinib effectively reduced the
activities of ERK and AKT kinases (Fig.
6B). By comparison, the combined treatment with sunitinib and
GNF-5837 mainly yielded a cooperative inhibition on ERK activity.
As the ERK cascade is critical for cell growth and survival, the
synergistic action of GNF-5837 and sunitinib may exert cell
toxicity via the repression of the ERK cascade.
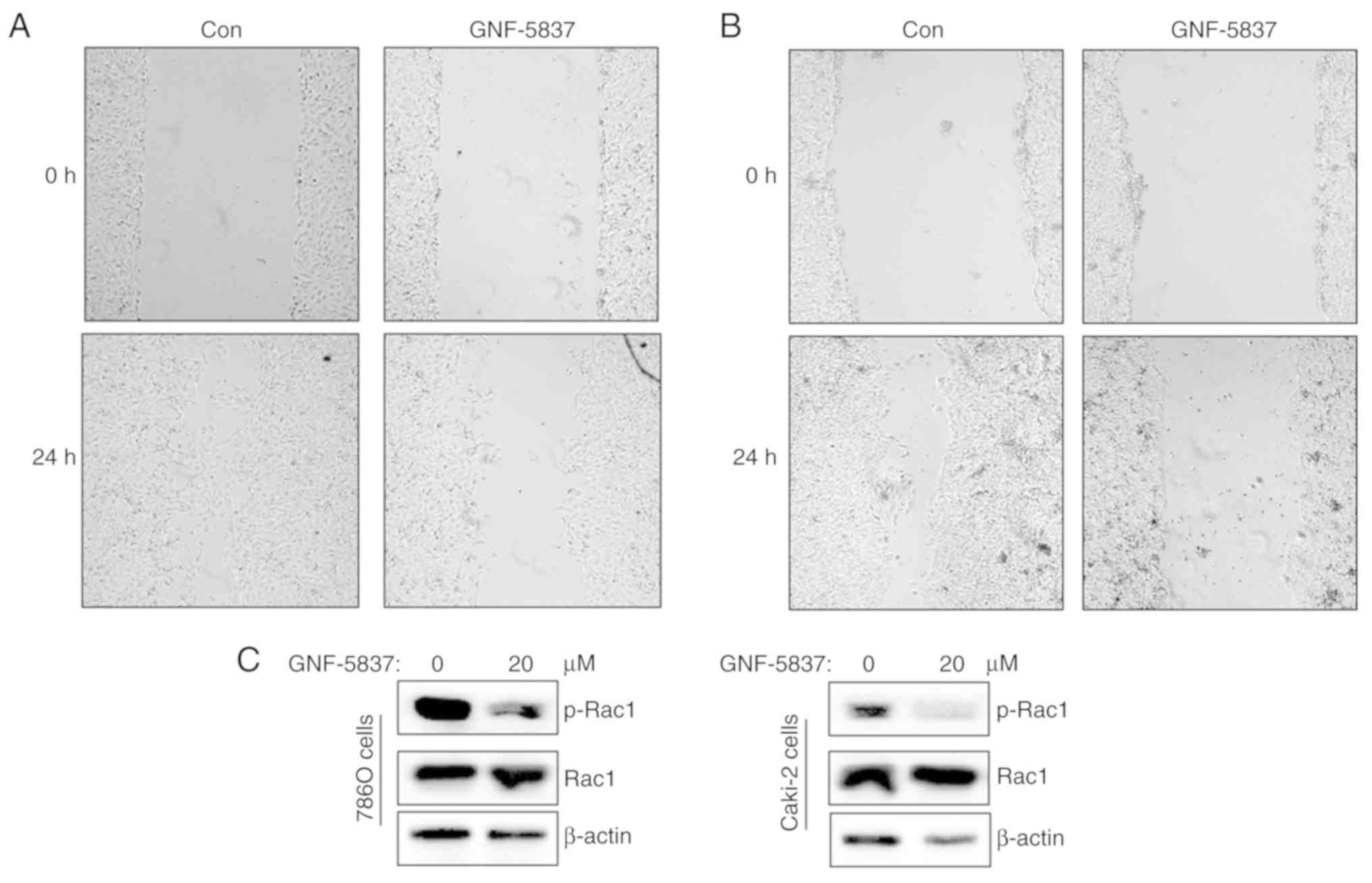
GNF-5837 suppresses cell
migration
About one-third of patients with RCC present with
metastatic disease at diagnosis. Metastatic RCCs are poorly
responsive to treatment and have a poorer prognosis.
Pharmacological modulation of tumor metastasis contributes to
improved clinical outcome. Indeed, Trk signaling combined with
neurotrophins is reported to stimulate cell migration and invasion
(9,10). Thus, we performed wound healing assay
to assess whether GNF-5837 inhibits cell migration. As shown in
Fig. 7A and B, GNF-5837 treatment was
able to reduce the migratory distance of 786O and Caki-2 cells
compared with the control group. As one small GTPase, Rac1,
mediates cytoskeleton rearrangements to facilitate synaptic
plasticity and tumor metastasis, we further investigated the effect
of GNF-5837 on Rac1 activity. As shown in Fig. 7C, GNF-5837 treatment reduced the
phosphorylation level of Rac1, accounting for the impaired cell
migration.
Discussion
RCC is a lethal and invasive tumor, and continues to
present a major therapeutic challenge. Diverse growth factors and
their corresponding kinase receptors are aberrantly expressed in
this malignancy and contribute to tumor cell growth and invasion
(11). Targeting growth factors
and/or their receptors presents a promising therapeutic strategy
for RCC. In the present study, we demonstrated that the pan-Trk
inhibitor GNF-5837 exerts its cytotoxicity on RCC cells by inducing
growth arrest and apoptosis.
Neurotrophins are growth factors that play important
roles in neurogenesis during development and regeneration. The
family of neurotrophins include nerve growth factor (NGF), brain
derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3) and
neurotrophin-4/5 (NT-4/5) (12–14). Four
mammalian neurotrophins bind to two distinct types of receptors:
Tropomyosin receptor kinase (Trk) receptors and the p75
neurotrophin receptor (p75NTR) (15,16). The
Trk receptors are high-affinity NGF receptors and transmembrane
tyrosine kinases. The Trk receptors include TrkA, TrkB and TrkC,
each of which exhibits different specificities for neurotrophins
(17,18). NGF has preferential affinity for TrkA,
BDNF and NT-4/5 for TrkB, and NT-3 for TrkC. The p75 neurotrophin
receptor (p75NTR) is the low-affinity NGF receptor and belongs to
the tumor necrosis factor (TNF) receptor superfamily (19).
Trk receptors activate and autophosphorylate
tyrosine residues in their intracellular tails, triggering various
downstream signaling pathways that involve cytoplasmic adaptors and
enzymes such as extracellularly regulated kinases (ERKs),
phosphatidyl inositol kinase 3 (PI3K) and phospholipase C (PLC)
(20,21). Binding of a neurotrophin to Trk
receptors predominantly leads to promotion of cell survival,
proliferation, and differentiation. Each neurotrophin also binds to
p75NTR and activates multiple signaling pathways including the
c-Jun N-terminal kinase (JNK) signaling cascade, which can enhance
neuronal survival or induce cell death depending on cellular
context (15,22).
Neurotrophins also participate in physiological
events outside of the nervous system, such as embryonic
development, vascularization and organ homeostasis. In addition,
increasing evidence has shown that neurotrophins and their
receptors are involved in the growth, angiogenesis, dissemination,
and drug resistance in different tumor types (10,11).
Neurotrophins and its receptors are abnormally overexpressed in
lung, pancreatic, breast, liver, skin, ovarian, thyroid, and
gastric tumors. For instance, primary and metastatic melanoma cell
lines synthesize and secrete all neurotrophins and express both
p75NTR and Trk receptors (11).
Neurotrophin and their receptors are frequently found yielding an
autocrine stimulation of tumor cell proliferation and metastasis
(12,19,20). It is
well known that NGF/TrkA as a mitogenic signal promotes cell
proliferation and invasion in breast cancer via signaling pathways
similar to those implicated in neuronal development, such as ERK
and AKT cascades (17). Additionally,
NGF was found to modulate self-renewal and plasticity of tumor stem
cells, and was found to be involved in the promotion of
epithelial-mesenchymal transition (EMT) (22). In ovarian carcinomas, NGF activates
TrkA in granulosa cells, where it functions as an indirect
angiogenic factor by enhancing vascular endothelial growth factor
(VEGF) expression, resulting in tumor cell proliferation, migration
and vasculogenesis (18). BDNF is
also known to participate in the cell division and dissemination of
tumor cells through autocrine stimulation of TrkB and p75NTR.
Aberrant BDNF/TrkB signaling is recognized as a driver and
potential target in gastrointestinal cancer (13). Functional experiments have illuminated
that BDNF administration promotes tumor cell proliferation,
invasion and migration in both gastric and bowel cancer cell lines,
as well as tumor cell metastasis in mouse models (14,16).
Multiple studies (11–13,15,18,20,22)
have highlighted the potential utility of neurotrophins and their
receptors as diagnostic and prognostic biomarkers in malignancies
of diverse origins. Both TrkA and p75NTR are increased in thyroid
cancer, and in particular TrkA expression is highly associated with
lymph node invasion, implicating its involvement in tumor
metastasis (15). NGF may serve as a
biomarker of high-grade prostate cancer. Urinary NGF levels, as
analyzed in a cohort of 115 prostate cancer patients, were found to
be significantly correlated with tumor grade (Gleason score) and
were thought to contribute to nerve infiltration in the tumor
microenvironment (23). Studies in
ovarian cancer have unveiled a dramatically increased expression of
NGF, TrkA, and p75NTR in tumor tissues (21). In gastric cancer, BDNF is
overexpressed and correlated with factors reflecting disease
progression and poor prognosis, pointing to its value as a
diagnostic and prognostic factor of clinical significance (13). Moreover, colorectal cancer patients
with higher levels of BDNF and TrkB display greatly worse overall
survival and elevated cancer metastasis (14).
However, the detailed effects of neurotrophin
signaling in RCC largely remain unclear. Comprehensive molecular
profiling has identified the abnormal activation of NGF signaling
in metastatic clear cell renal cell carcinoma (24). A cohort study of 83 clear cell RCC
tumors showed that overexpression of p75NTR, pro-BDNF, and to a
lesser extent TrkB was detected by immunohistochemistry (16). In particular, p75NTR, mainly expressed
in tumor tissues, was highly correlated with a higher Fuhrman grade
in multivariate analysis. p75NTR inhibition by silencing or
blocking antibody repressed cell survival and migration in RCC cell
lines. Meanwhile, TrkB silencing caused apoptosis, inhibited
proliferation, retarded invasion as well as improved antitumor
efficiency of sorafenib in anoikis-resistant RCC cells (25).
Trk inhibitors such as KK5101, GNF-5837, AZD6918,
GW441756 and GNF-4256, have exhibited suppressive effects on tumor
proliferation, migration, and metastasis (26–30). In
the present study, we observed that a pan-Trk inhibitor GNF-5837
exerted inhibitory effects on cell proliferation, survival and
migration in RCC cells. GNF-5837 greatly suppressed the activation
of TrkA and TrkB in 786O and Caki-2 cells, thereby leading to the
inhibition of downstream signals such as ERK and AKT kinases. As
ERK and AKT kinases are important downstream modulators of Trk
receptors, their inhibition should negatively regulate cell growth
and survival. We further observed that GNF-5837 effectively induced
G0/G1-phase arrest, increased p21 expression and downregulated
c-Myc to achieve growth inhibition. qPCR analysis revealed that the
expression of anti-apoptotic Survivin was downregulated by
GNF-5837. Conversely, the pro-apoptotic gene Bax was greatly
induced following GNF-5837 treatment. Despite that, our results
indicated that GNF-5837 is a moderate apoptosis inducer. Seemingly,
GNF-5837 mainly exerts cell toxicity via inhibiting cell growth. In
addition, neurotrophins are closely linked with cell migration of
neural cells (9,10). Herein, our results indicated that
GNF-5837 also suppressed cell migration in RCC cells. Neurotrophin
receptors have been shown to alter the expression of E-cadherin and
Twist, thereby affecting epithelial-to-mesenchymal transition (EMT)
and cell migration (31). BDNF-TrkB
was sufficient to activate the Rho GTPase proteins Rac1 and Cdc42
to modulate cytoskeleton rearrangements, leading to the
facilitation of synaptic plasticity (32). Our findings showed that GNF-5837
treatment impeded Rac1 activation, which contributed to the
suppression of cell migration. Overall, our results further shed
light on the putative effects of neurotrophin signaling in the
pathological progress of RCC. Therefore, inhibition of
neurotrophins and their receptors may be a novel potential
therapeutic strategy for RCC. Despite that, it is essential to
explore the origin of neurotrophins such as NGF and BDNF, and the
precise role of distinct receptors in RCC. In addition, the present
study was limited as it did not investigate the effects of GNF-5837
on normal renal cells.
In fact, a subset of preclinical trials have shown
that blockade of neurotrophin/receptor by neutralizing antibody,
siRNA-mediated silence, or pharmacological inhibition causes the
inhibition of growth and invasion in a variety of human tumors. For
instance, targeting NGF signaling via anti-NGF or anti-TrkA
blocking antibodies has demonstrated tumor growth inhibition,
reduced metastasis, and increased cancer cell apoptosis in breast
cancer xenografted mice (33).
Furthermore, siRNA-mediated knockdown of TrkA in breast cancer
cells suppressed cancer cell growth and cell cycle progression with
stagnation at G0/G1 phase, also increased chemo-sensitivity to
paclitaxel and reduced the incidence of lung metastasis in
xenograft models (34). Similarly,
knockdown of TrkB in human lung adenocarcinoma cancer cell lines
significantly decreased their migratory and metastatic ability
in vitro and in vivo (35).
Tumor cells may escape the antagonism of VEGF
signaling using other parallel pathways, such as neurotrophin
signaling. Herein, we provided evidence that GNF-5837 can
potentiate sunitinib-induced cytotoxicity via inhibition of TrkA
and TrkB. Despite that, we lacked in vivo tumorigenic
assessment and the combined effects on cell invasion. The combined
treatment of GNF-5837 and sunitinib mainly interrupted ERK
activity. Thus, the enhanced suppression on cell growth and
survival may be attributed to ERK inhibition. In fact, a previous
study noted that ERK signaling plays a role in the generation of
sunitinib resistance in RCC patients receiving anti-angiogenic
therapy (36). However, it is still
unclear whether there is a synergistic effect of GNF-5837 and
sunitinib. A series of experiments is needed to determine the
combination index and associated mechanisms. Meanwhile, it is also
obscure whether GNF-5837 can impede the generation of sunitinib
resistance. Further investigation is still required to explore
whether neurotrophins and their receptors are involved in sunitinib
resistance.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Zhejiang
Provincial Natural Science Foundation (grant no. LY18H160002).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
GD and TL conceived and designed the study and the
experiments. YiC acquired the funding. YiC, HW and YuC performed
the experiments. MW analyzed the data and conducted the statistical
analysis. TL wrote the manuscript. YiC and HW critically revised
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
These authors declare that they have no competing
interests.
References
|
1
|
Hsieh JJ, Purdue MP, Signoretti S, Swanton
C, Albiges L, Schmidinger M, Heng DY, Larkin J and Ficarra V: Renal
cell carcinoma. Nat Rev Dis Primers. 3:170092017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Graves A, Hessamodini H, Wong G and Lim
WH: Metastatic renal cell carcinoma: Update on epidemiology,
genetics, and therapeutic modalities. Immunotargets Ther. 2:73–90.
2013.PubMed/NCBI
|
|
3
|
Rodriguez-Vida A, Hutson TE, Bellmunt J
and Strijbos MH: New treatment options for metastatic renal cell
carcinoma. ESMO Open. 2:e0001852017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mattei J, da Silva RD, Sehrt D, Molina WR
and Kim FJ: Targeted therapy in metastatic renal carcinoma. Cancer
Lett. 343:156–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
da Silva RD, Gustafson D, Nogueira L,
Werahera PN, Molina WR and Kim FJ: Targeted therapy for metastatic
renal carcinoma: An update. J Kidney Cancer VHL. 1:63–73. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Posadas EM, Limvorasak S and Figlin RA:
Targeted therapies for renal cell carcinoma. Nat Rev Nephrol.
13:496–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Huang Q, Zhou H, Wang Y, Hu X and
Li T: Inhibition of canonical WNT/β-catenin signaling is involved
in leflunomide (LEF)-mediated cytotoxic effects on renal carcinoma
cells. Oncotarget. 7:50401–50416. 2016.PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee WD, Wang KC, Tsai YF, Chou PC, Tsai LK
and Chien CL: Subarachnoid hemorrhage promotes proliferation,
differentiation, and migration of neural stem cells via BDNF
upregulation. PLoS One. 11:e01654602016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oliveira SL, Trujillo CA, Negraes PD and
Ulrich H: Effects of ATP and NGF on proliferation and migration of
neural precursor cells. Neurochem Res. 40:1849–1857. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Truzzi F, Marconi A, Lotti R, Dallaglio K,
French LE, Hempstead BL and Pincelli C: Neurotrophins and their
receptors stimulate melanoma cell proliferation and migration. J
Invest Dermatol. 128:2031–2040. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang W, Chen J and Guo X: The role of
nerve growth factor and its receptors in tumorigenesis and cancer
pain. Biosci Trends. 8:68–74. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okugawa Y, Tanaka K, Inoue Y, Kawamura M,
Kawamoto A, Hiro J, Saigusa S, Toiyama Y, Ohi M, Uchida K, et al:
Brain-derived neurotrophic factor/tropomyosin-related kinase B
pathway in gastric cancer. Br J Cancer. 108:121–130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tanaka K, Okugawa Y, Toiyama Y, Inoue Y,
Saigusa S, Kawamura M, Araki T, Uchida K, Mohri Y and Kusunoki M:
Brain-derived neurotrophic factor (BDNF)-induced
tropomyosin-related kinase B (Trk B) signaling is a potential
therapeutic target for peritoneal carcinomatosis arising from
colorectal cancer. PLoS One. 9:e964102014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Faulkner S, Jobling P, Rowe CW, Rodrigues
Oliveira SM, Roselli S, Thorne RF, Oldmeadow C, Attia J, Jiang CC,
Zhang XD, et al: Neurotrophin receptors TrkA, p75NTR,
and sortilin are increased and targetable in thyroid cancer. Am J
Pathol. 188:229–241. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De la Cruz-Morcillo MA, Berger J,
Sánchez-Prieto R, Saada S, Naves T, Guillaudeau A, Perraud A,
Sindou P, Lacroix A, Descazeaud A, et al: p75 neurotrophin receptor
and pro-BDNF promote cell survival and migration in clear cell
renal cell carcinoma. Oncotarget. 7:34480–34497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lagadec C, Meignan S, Adriaenssens E,
Foveau B, Vanhecke E, Romon R, Toillon RA, Oxombre B, Hondermarck H
and Le Bourhis X: TrkA overexpression enhances growth and
metastasis of breast cancer cells. Oncogene. 28:1960–1970. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vera C, Tapia V, Vega M and Romero C: Role
of nerve growth factor and its TRKA receptor in normal ovarian and
epithelial ovarian cancer angiogenesis. J Ovarian Res. 7:822014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Griffin N, Faulkner S, Jobling P and
Hondermarck H: Targeting neurotrophin signaling in cancer: The
renaissance. Pharmacol Res. 135:12–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Demir IE, Tieftrunk E, Schorn S, Friess H
and Ceyhan GO: Nerve growth factor & TrkA as novel therapeutic
targets in cancer. Biochim Biophys Acta. 1866:37–50.
2016.PubMed/NCBI
|
|
21
|
Yu X, Liu Z, Hou R, Nie Y and Chen R:
Nerve growth factor and its receptors on onset and diagnosis of
ovarian cancer. Oncol Lett. 14:2864–2868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tomellini E, Touil Y, Lagadec C, Julien S,
Ostyn P, Ziental-Gelus N, Meignan S, Lengrand J, Adriaenssens E,
Polakowska R and Le Bourhis X: Nerve growth factor and proNGF
simultaneously promote symmetric self-renewal, quiescence, and
epithelial to mesenchymal transition to enlarge the breast cancer
stem cell compartment. Stem Cells. 33:342–353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liss MA, Gordon A, Morales B, Osann K,
Skarecky D, Lusch A, Zaldivar F and Ahlering TE: Urinary nerve
growth factor as an oncologic biomarker for prostate cancer
aggressiveness. Urol Oncol. 32:714–719. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghatalia P, Yang ES, Lasseigne BN, Ramaker
RC, Cooper SJ, Chen D, Sudarshan S, Wei S, Guru AS, Zhao A, et al:
Kinase gene expression profiling of metastatic clear cell renal
cell carcinoma tissue identifies potential new therapeutic targets.
PLoS One. 11:e01609242016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang P, Xing Z, Li X, Song Y, Zhao J,
Xiao Y and Xing Y: Tyrosine receptor kinase B silencing inhibits
anoikis-resistance and improves anticancer efficiency of sorafenib
in human renal cancer cells. Int J Oncol. 48:1417–1425. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fang Z, Han B, Jung KH, Lee JH, El-Damasy
AK, Gadhe CG, Kim SJ, Yan HH, Park JH, Lee JE, et al: A novel
tropomyosin-related kinase A inhibitor, KK5101 to treat pancreatic
cancer. Cancer Lett. 426:25–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Albaugh P, Fan Y, Mi Y, Sun F, Adrian F,
Li N, Jia Y, Sarkisova Y, Kreusch A, Hood T, et al: Discovery of
GNF-5837, a selective TRK inhibitor with efficacy in rodent cancer
tumor models. ACS Med Chem Lett. 3:140–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Z, Zhang Y, Tong Y, Tong J and Thiele
CJ: Trk inhibitor attenuates the BDNF/TrkB-induced protection of
neuroblastoma cells from etoposide in vitro and in vivo. Cancer
Biol Ther. 16:477–483. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Croucher JL, Iyer R, Li N, Molteni V,
Loren J, Gordon WP, Tuntland T, Liu B and Brodeur GM: TrkB
inhibition by GNF-4256 slows growth and enhances chemotherapeutic
efficacy in neuroblastoma xenografts. Cancer Chemother Pharmacol.
75:131–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bapat AA, Munoz RM, Von Hoff DD and Han H:
Blocking nerve growth factor signaling reduces the neural invasion
potential of pancreatic cancer cells. PLoS One. 11:e01655862016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kupferman ME, Jiffar T, El-Naggar A,
Yilmaz T, Zhou G, Xie T, Feng L, Wang J, Holsinger FC, Yu D and
Myers JN: TrkB induces EMT and has a key role in invasion of head
and neck squamous cell carcinoma. Oncogene. 29:2047–2059. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hedrick NG, Harward SC, Hall CE, Murakoshi
H, McNamara JO and Yasuda R: Rho GTPase complementation underlies
BDNF-dependent homo- and heterosynaptic plasticity. Nature.
538:104–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Adriaenssens E, Vanhecke E, Saule P,
Mougel A, Page A, Romon R, Nurcombe V, Le Bourhis X and Hondermarck
H: Nerve growth factor is a potential therapeutic target in breast
cancer. Cancer Res. 68:346–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang J, Wang LS, Ye SL, Luo P and Wang
BL: Blockage of tropomyosin receptor kinase a (TrkA) enhances
chemo-sensitivity in breast cancer cells and inhibits metastasis in
vivo. Int J Clin Exp Med. 8:634–641. 2015.PubMed/NCBI
|
|
35
|
Sinkevicius KW, Kriegel C, Bellaria KJ,
Lee J, Lau AN, Leeman KT, Zhou P, Beede AM, Fillmore CM, Caswell D,
et al: Neurotrophin receptor TrkB promotes lung adenocarcinoma
metastasis. Proc Natl Acad Sci USA. 111:10299–10304. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Diaz-Montero CM, Mao FJ, Barnard J, Parker
Y, Zamanian-Daryoush M, Pink JJ, Finke JH, Rini BI and Lindner DJ:
MEK inhibition abrogates sunitinib resistance in a renal cell
carcinoma patient-derived xenograft model. Br J Cancer.
115:920–928. 2016. View Article : Google Scholar : PubMed/NCBI
|