Introduction
Nasopharyngeal carcinoma (NPC), considered a rare
tumour, has a unique geographical distribution and has the highest
prevalence in Southeast Asia, such as southeastern China, including
Guangdong and Hong Kong, and in other regions (India and Thailand)
(1). Distant metastasis is a leading
cause of treatment failure in patients with nasopharyngeal
carcinoma, in which more than 70% of them have locoregionally
advanced disease (2). Even after
undergoing radical treatment, ~30–40% of patients with
locoregionally advanced NPC ultimately develop distant metastasis
(3). The present
tumour-node-metastasis (TNM) system was suggested to have some
limitations in predicting which patients will develop distant
metastasis because it is entirely based on anatomical information
(4).
Consequently, increasing the number of biomarkers
has been researched to improve the prognosis and treatment
efficiency of nasopharyngeal carcinoma, such as Epstein-Barr virus
DNA (EBV DNA), lactate dehydrogenase (LDH), VEGF, and distant
metastasis gene signature (DMGN) (5–8). However,
new biomarkers that reflect tumour heterogeneity should be studied
to determine their clinical roles and to guide personalized therapy
(9). MicroRNAs (miRNAs) have been
revealed to suppress or promote many cancers, such as miRNA-195,
miR-BART6-3p, microRNA-150, microRNA-29c, and microRNA-29b, which
are potential biomarkers for diagnosis, prognosis, and personalized
treatment (10–14). However, the molecular mechanism of
miRNAs in nasopharyngeal carcinoma has not been completely
established (15).
In the present study, the microarray data of
GSE32960 (16) and GSE12452 (17) from the Gene Expression Omnibus (GEO)
dataset were applied to identify differentially expressed genes
(DEGs) via integrative bioinformatics approaches. The search tools
of the starBase v2.0, DAVID, STRING, and GEO databases were used to
identify the core differentially expressed miRNAs (DEMs) and
differentially expressed mRNAs (DEmRNAs), as well as DEM-DEmRNA
interactions. In total, 46 DEMs and 2,956 DEmRNAs were identified
as being aberrantly expressed. In the first analyses, four
significant miRNAs were revealed to be associated with overall
survival (OS), disease-free survival (DFS), and distant
metastasis-free survival (DMFS) in NPC via a series of
bioinformatics analyses. Notably, the risk score of four miRNAs was
a greatly effective prognostic factor. Finally, a regular
enrichment analysis was performed for key differentially expressed
genes (DEGs) that participated in some vital pathways related to
cancer pathogenesis, such as the focal adhesion, PI3K/Akt, p53, and
mTOR signalling pathways. The present study aimed to identify key
genes associated with the prognosis of NPC and to provide a
theoretical basis for future molecular mechanisms.
Materials and methods
miRNA and mRNA expression data and
pre-processing
The gene expression profile data (GSE32960 and
GSE12452) were downloaded from the NCBI Gene Expression Omnibus
(GEO) database. However, the detailed clinical data presented in
Table SIV was obtained from the
researchers. The GSE32960 dataset contained the microRNA profile of
312 paraffin-embedded NPC specimens and 18 normal nasopharyngeal
tissues (16). The aim of the
researchers of this study was to evaluate whether microRNAs can
predict the survival and efficacy of concurrent chemotherapy in
nasopharyngeal carcinoma (NPC) patients. The GSE12452 dataset
contained the mRNA expression profile of 31 nasopharyngeal
carcinomas and 10 normal healthy nasopharyngeal tissue specimens.
The mRNA expression levels were measured for essentially all human
genes and all latent Epstein-Barr virus (EBV) genes in
nasopharyngeal carcinoma tissue samples and normal ones. The aim of
the authors of this study was to analyse data for differential gene
expression between nasopharyngeal carcinoma tissue samples and
normal nasopharyngeal tissues and for correlations with levels of
viral gene expression (17).
Statistically significant DEMs between NPC samples and normal
samples were obtained with the cut-off criteria of adjust P-value
[false discovery rate (FDR)] <0.01 and fold change (FC) >2.5.
Likewise, with a cut-off criteria of adjust P-value (FDR) <0.05
and a fold change (FC) >1.5, statistically significant mRNAs
expressing differentially were also obtained. R software (version
3.5.1, http://www.r-project.org/) and MEV
(version 4.9.0, http://mev.tm4.org/) were applied to
the significance analysis of differentially expressed genes.
Construction of weighted gene
co-expression network
First, with the use of the systems biology method,
the weighted gene co-expression network, a scale-free network from
gene expression data was constructed (18). Next, a soft-thresholding power
(soft-threshold, β=3) was selected in accordance with standard
scale-free networks, with which a hierarchical clustering tree was
produced using the WGCNA package (19). Then, the correlations between the 30
modules and clinical traits and P-values were assessed using R
functions in the WGCNA package. Subsequently, the adjacency was
transformed into a topological overlap matrix (TOM). In addition,
an average linkage hierarchical clustering was performed on the
basis of the TOM-based dissimilarity measure. Finally, a minimum
size (gene group) of 10 for the gene dendrogram and a cut-line of
0.25 for the module dendrogram were selected.
Risk score
miRNAs that were associated significantly with DMFS
were selected to construct a miRNA signature with the risk-score
method. The risk score of each patient was the sum of the
multiplication of the log-transformed normalized expression value
and the regression coefficient of each gene. The risk score was
used for survival analysis.
Combination of differentially
expressed mRNAs with target gene prediction of DEMs
To increase the accuracy of our prediction, a Venn
plot was generated to obtain the common genes of differentially
expressed mRNAs (fold change >1.5) and the potential target
genes of miRNAs (http://bioinformatics.psb.ugent.be/webtools/Venn/).
The potential target genes of four hub DEMs (hsa-miR-142-3p,
hsa-miR-150, hsa-miR-29b, and hsa-miR-29c) were analysed by using
online tool starBase v2.0 (http://starbase,sysu.edu.cn/starbase2/index.php)
(20).
Gene ontology (GO) analysis and
pathway enrichment analysis
To further clarify the mechanism of our 127 mRNAs of
interest, they were uploaded to the Database for Annotation,
Visualization, and Integrated Discovery (DAVID, http://david.ncifcrf.gov/) for GO functional
annotation and biological pathway analysis (21). P<0.05 was used as the cut-off
criterion. The biological significance of mRNAs was explored by GO
term enrichment analysis, including molecular function (MF),
biological process (BP), cellular component (CC), and biological
pathway (BPA). The results of the aforementioned functional
enrichment analysis were visualized via the package (‘ggplot2’) of
R software (version 3.5.1) and GraphPad Prism (version 5.0;
GraphPad Software).
PPI network and miRNA-mRNA correlation
network analysis
The Search Tool for the Retrieval of Interacting
Genes (STRING) database (https://string-db.org/) provides PPI information of
mRNAs regarding the predicted and experimental interactions of
proteins (22). Cytoscape (version
3.6.0) software was used for the construction of the PPI network
under the interaction information of 101 interaction genes
(23). Superimposing four hub miRNAs
with 127 mRNAs, a miRNA-mRNA correlation network was constructed
utilizing Cytoscape software.
Statistical analysis
Statistical analysis was performed using SPSS 16.0
and R software 3.5.1. Graphs were generated using GraphPad Prism
5.0. Student's t-test was used to evaluate the statistical
significance of the difference in the means between groups with a
stringency of P<0.05, which was considered to be significant.
The χ2 test was used to calculate the association
between two categorical variables. Kaplan-Meier survival analysis
and univariate/multivariate Cox regression analysis were used to
assess the expression levels of DEMs and prognostic
characteristics. Receiver operating characteristic (ROC) curves
were used to compare the specificity and sensitivity for the
prediction of survival by the risk score, TNM stage, T stage, N
stage, and sex of NPC patients. All P-values were two-sided.
Results
Identification of DEMs
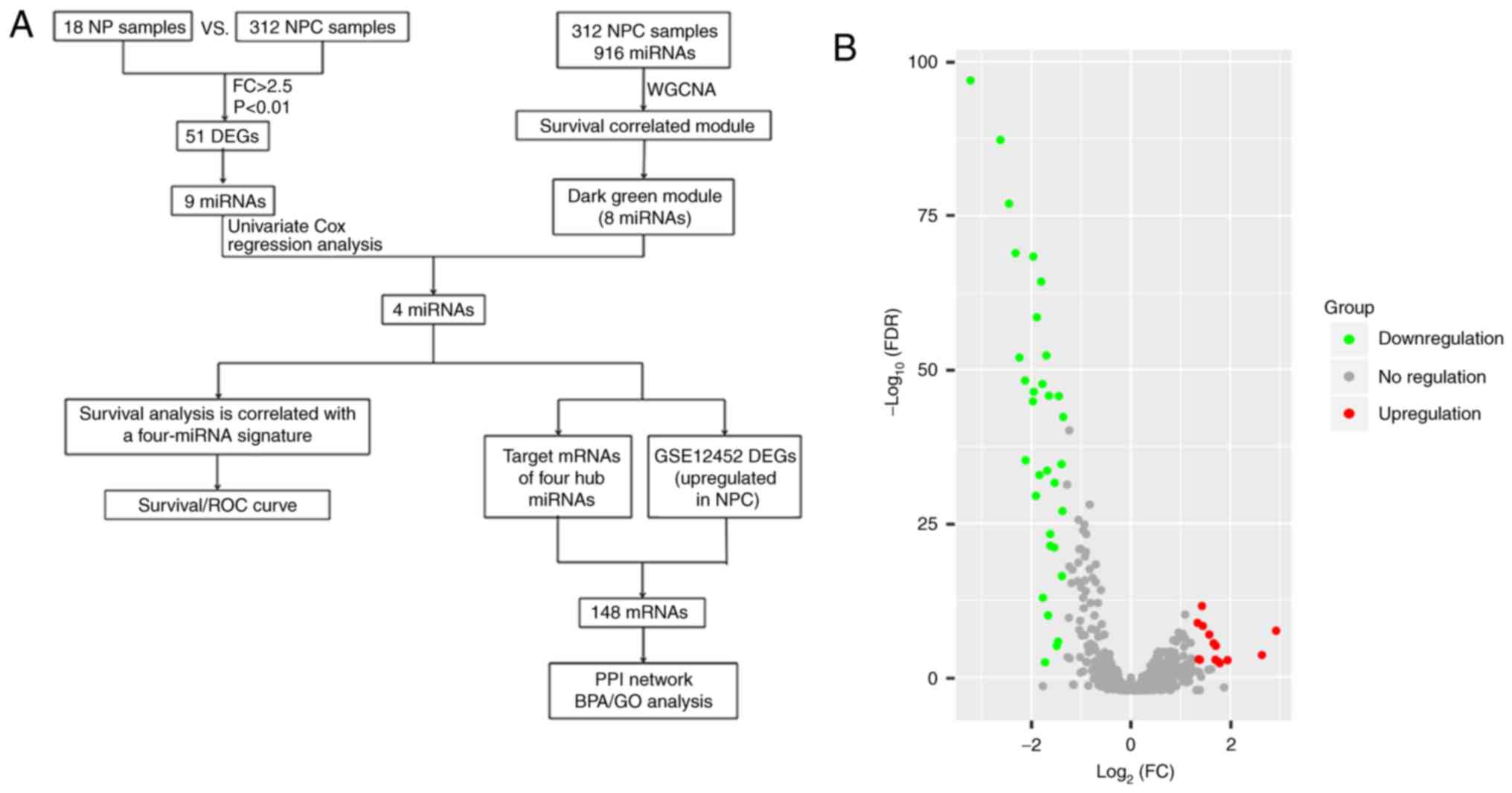
The brief work-flow of this study is presented in
Fig. 1A. The microarray data of
GSE32960, including 312 NPC tissue samples and 18 normal tissue
samples, were obtained from the NCBI-GEO database. After applying
the cut-off criteria of adjust P-value (FDR) <0.01 and fold
change >2.5, a total of 46 DEMs were considered statistically
significant between NPC tissues and normal tissues (Table SI). The results of 32 significantly
downregulated miRNAs and 14 significantly upregulated miRNAs are
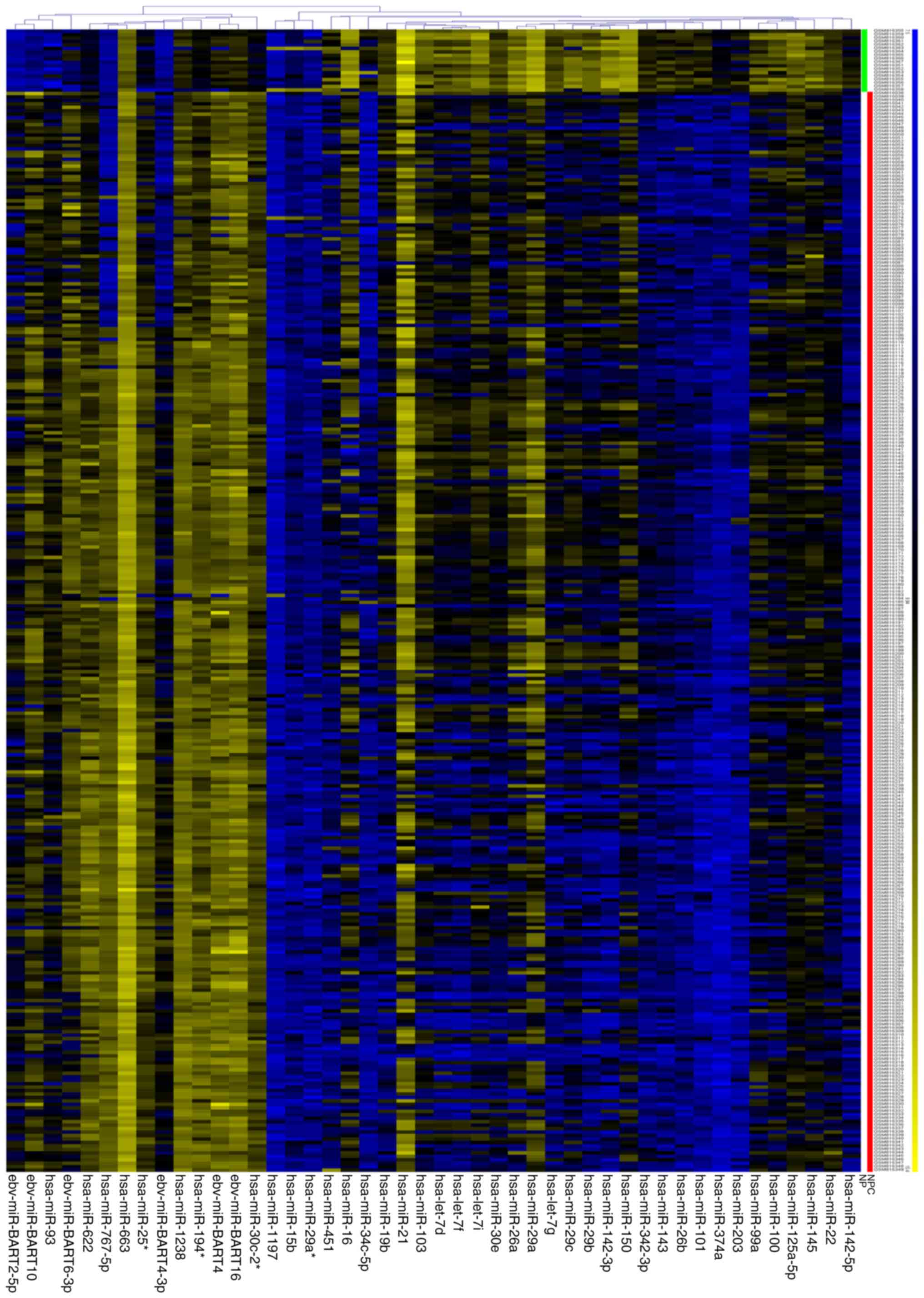
displayed in the volcano plot (Fig.
1B). A heat map of these 46 DEMs is presented in Fig. 2.
Construction of a weighted
co-expression network and identification of key modules
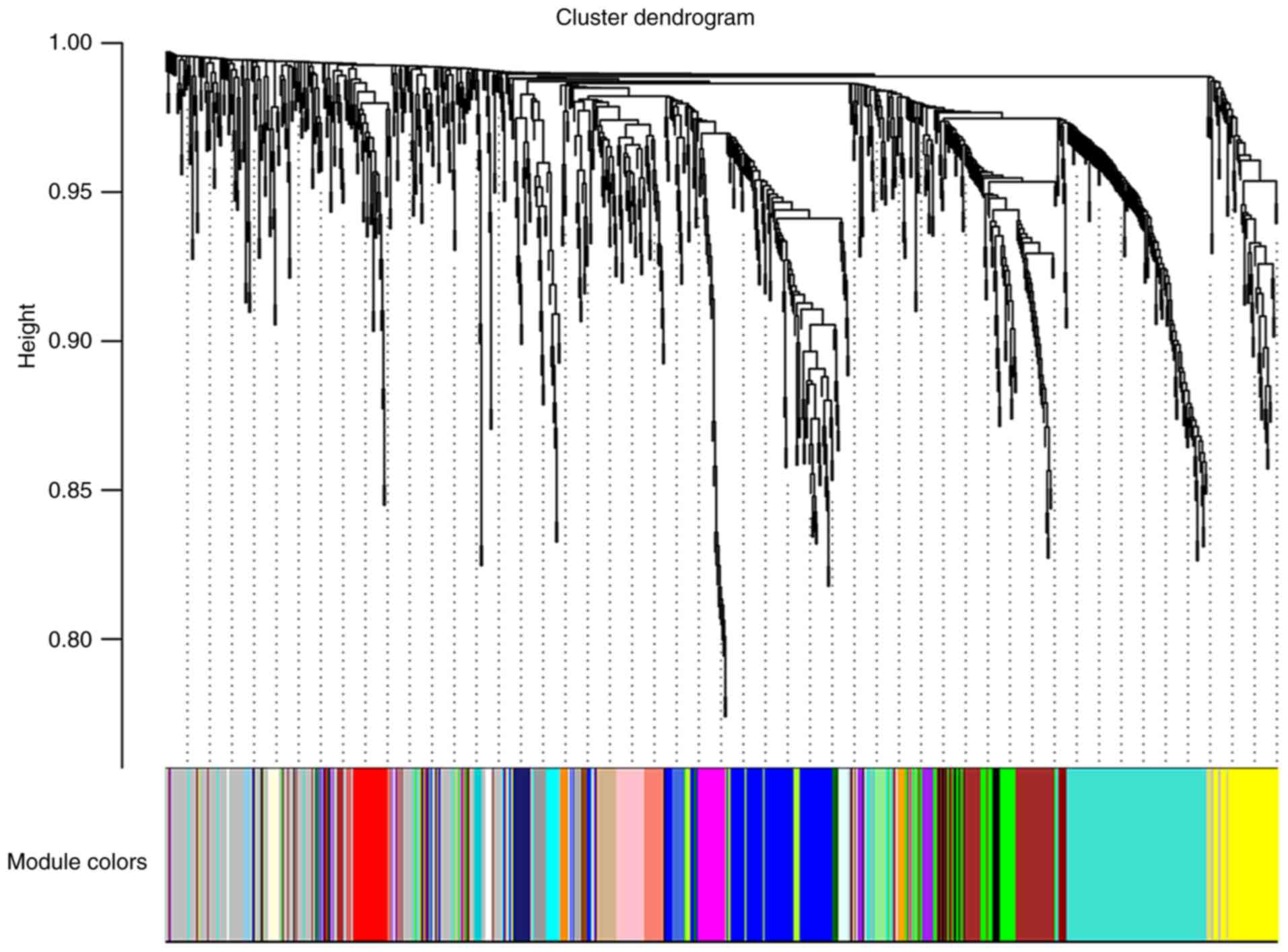
First, the WGCNA package was used to cluster the
miRNA expression of 312 cases of nasopharyngeal carcinoma in
GSE32960 (Fig. S1). The results
revealed that only the expression of GSM816316 was abnormal, which
is considered an outlier sample, and thus, GSM816316 was excluded
from our analysis. To ensure a scale-free network, a soft-threshold
β=3 was selected to produce a hierarchical clustering tree using a
WGCNA package as the soft-thresholding power and then a total of 30
modules were identified (Figs. 3 and
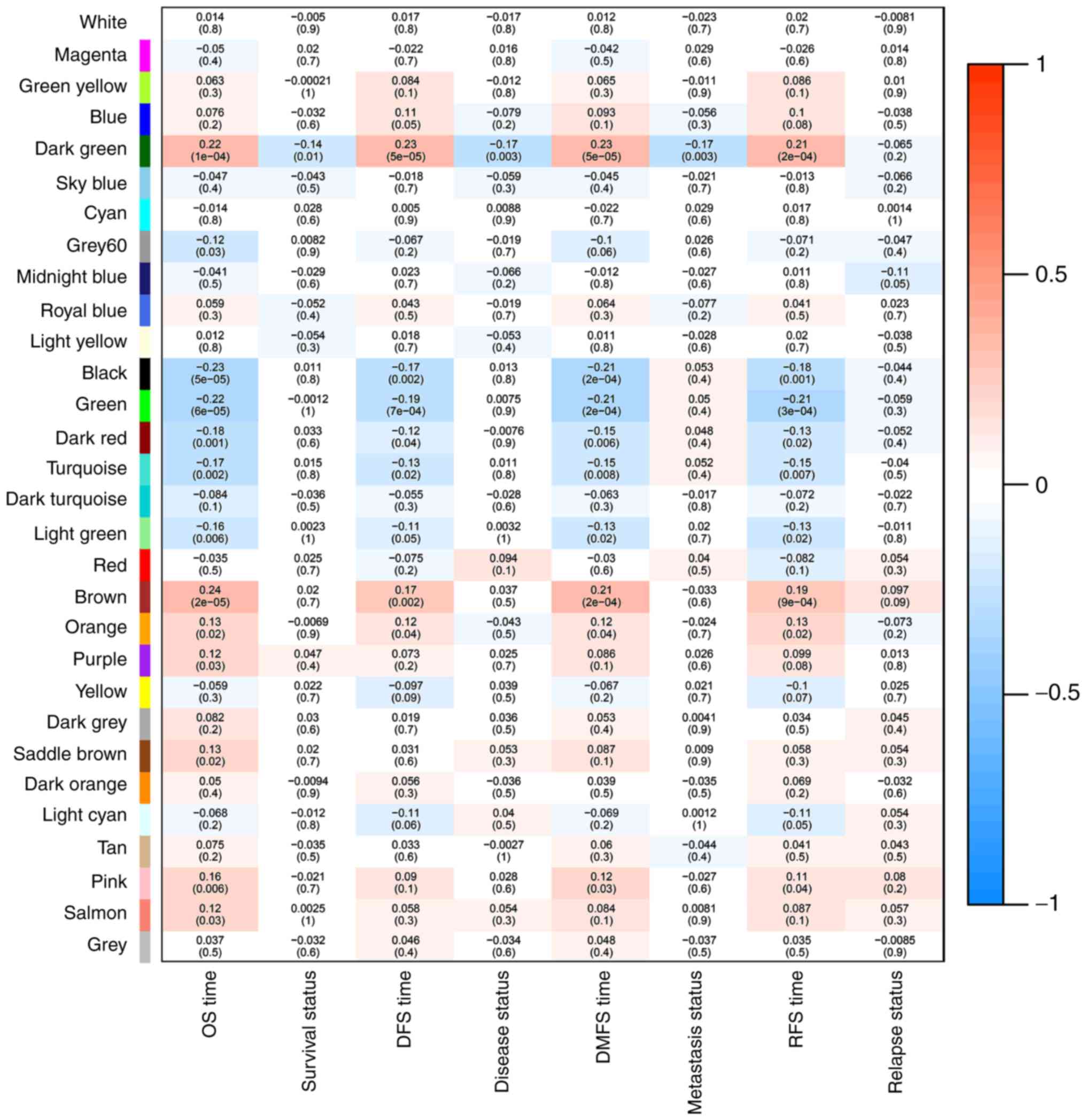
S2). Then, a co-expression network
of the associations between clinical traits and these modules was
constructed using data from GSE32960, including 311 NPC samples
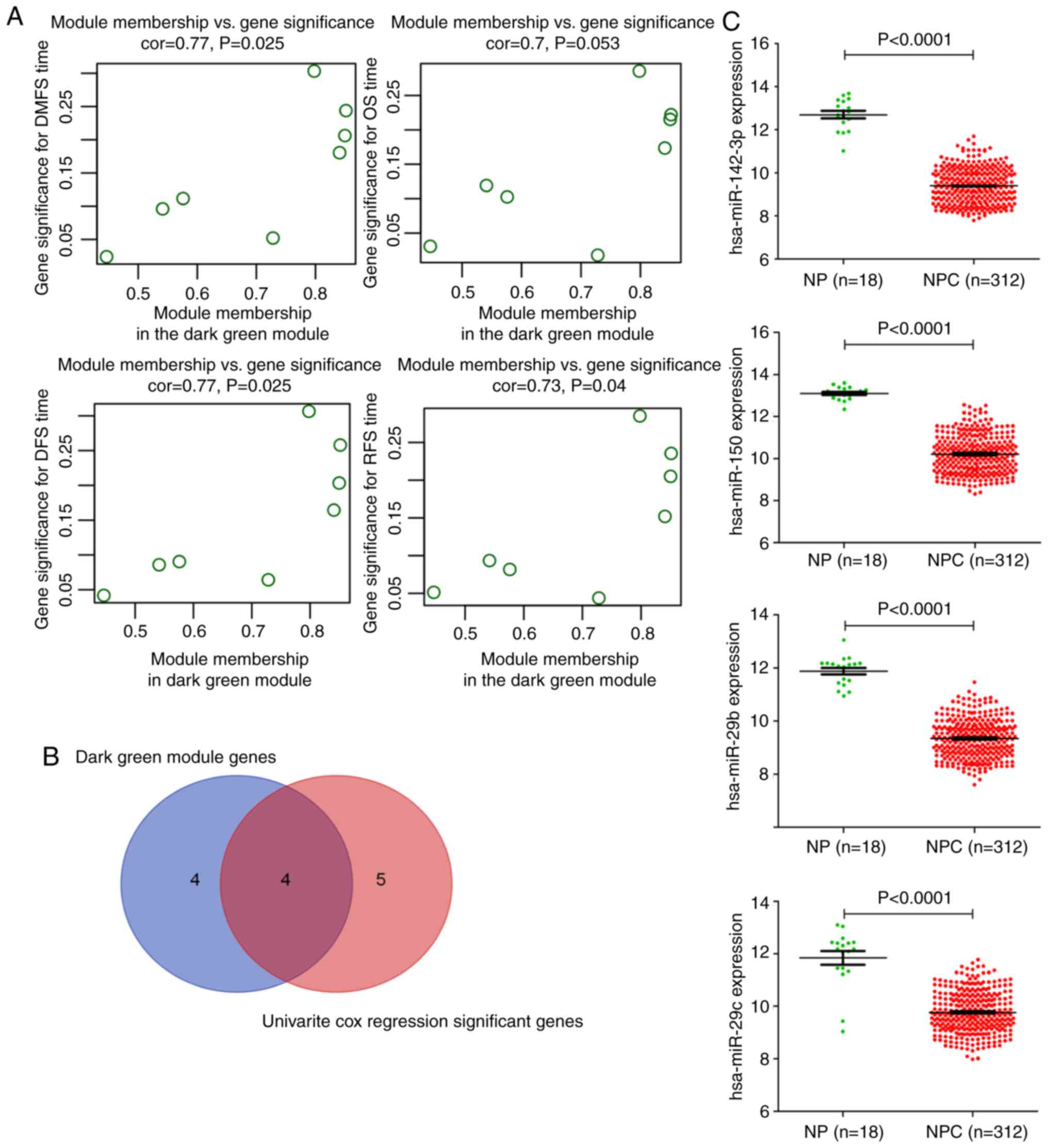
associated with complete clinical data (Fig. 4). Notably, the dark green module was
most significantly associated with survival status, such as DMFS,
RFS, DFS, and OS in NPC patients. Thus, the dark green module that
is most relevant to survival status was defined as a
sur-module.
Identification of hub miRNAs
The 8 miRNAs belonging to the dark green module are
listed in Table SII. The correlation
of the 8 miRNAs of the dark green module and the survival time of
patients is presented in Fig. 5A. To
determine which genes were associated with distant metastasis-free
survival, univariate Cox regression analysis of the DEMs
individually was performed (Table
SIII). Additionally, 9 miRNAs were independently significantly
related to DMFS (P<0.05). Finally, the overlapping four miRNAs
(hsa-miR-142-3p, hsa-miR-150, hsa-miR-29b, and hsa-miR-29c) were
obtained as our candidate miRNAs from the two methods
aforementioned (Fig. 5B). As a
result, these four miRNAs were identified as potential prognostic
molecules. Then, all of the tissue data was applied to determine
the four hub miRNA expression levels between 312 NPC tissues and 18
normal tissues, and Fig. 5C revealed
that compared with the normal tissues, the hub miRNAs were
significantly decreased (P<0.001). The detailed clinical data is
presented in Table SIV. Moreover, in
the present study, it was determined that the four miRNAs were not
only significantly downregulated but positively associated with
DFS, of which miR-29c and miR-142-3p were positively associated
with DMFS (Figs. S3–S5).
Feasibility analysis of miRNA as a
prognostic factor for survival
We obtained a formula to calculate the risk score
for every patient from the expression values of the four hub
miRNAs, weighted by the regression coefficient (24,25).
Risk score=−(0.6×expression value of
hsa-miR-142-3p)-(0.37×expression value of
hsa-miR-150)-(0.42×expression value of
hsa-miR-29b)-(0.66×expression value of miR-29c).
With this risk score formula, the 312 NPC patients
were divided into low-risk or high-risk groups with the median risk
score (−19.78) as the cut-off. Furthermore, the survival difference
between the two groups of patients was plotted (Fig. 6A). Notably, compared with patients
with low-risk scores of the four miRNAs, patients with high-risk
scores had a shorter DMFS [hazard ratio (HR) 2.276, 95% CI,
1.403–3.692; P<0.0009], OS (2.119, 95% CI, 1.337–3.357;
P<0.0014), DFS (2.183, 95% CI, 1.457–3.271; P<0.0002), and
RFS (1.768, 95% CI, 0.975–3.207; P<0.0606). The relative
clinical characteristics are presented in Table SV. To better understand which of the
groupings were critical in the development of clinical outcome,
univariate and multivariable Cox regression analyses were performed
using a forward conditional method in view of the results of the
univariate analysis. With this aforementioned analysis, it was
clearly observed that the risk scores of the four miRNAs and T
stage were independent prognostic factors for DFS, DMFS, RFS, and
OS in patients with nasopharyngeal carcinoma (Tables SVI and SVII). Patients in the high-risk group had
significantly higher risk scores, T-stage, and TNM stages than the
low-risk group. Furthermore, to compare the sensitivity and
specificity of prediction, ROC analysis was performed, and the risk
score of four miRNAs exhibited a better prediction for survival
than TNM stage, T stage, N stage and sex with regard to DMFS, OS,
RFS, and DFS in patients with nasopharyngeal carcinoma (Fig. 6B). Coincidentally, the AUCs of the
risk score in DMFS and DFS were both 0.69 (95% CI, 0.63–0.75;
P<0.0001). Similarly, the AUCs of the risk score in RFS and OS
were both 0.68 (95% CI, 0.62–0.74; P<0.0001). Thus, the risk
score of the four miRNAs was an effective prognostic factor.
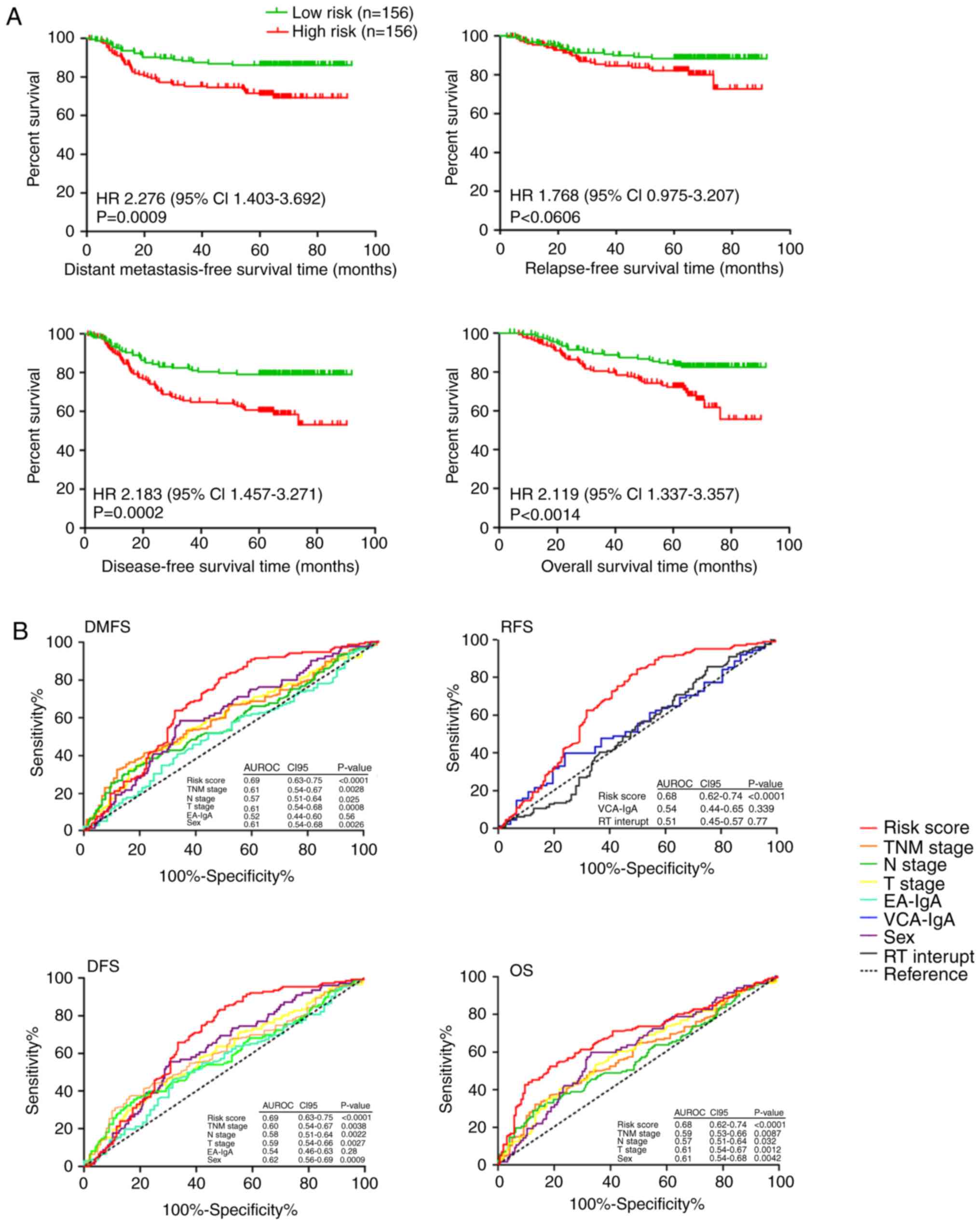 | Figure 6.Feasibility analysis of miRNA as a
survival prognostic factor. (A) Kaplan-Meier curves of DMFS, RFS,
DFS, and OS according to the risk score of the four-miRNA signature
in NPC patients. A Kaplan-Meier curve was drawn by GraphPad Prism
(version 5.0). (B) Comparisons of the sensitivity and specificity
for prediction of survival by the risk score of the four-microRNA
signature, TNM stage, T stage, N stage, EA-IgA, VCA-IgA, sex or RT
interrupt in 312 NPC patients. Survival ROC curve was drawn by
GraphPad Prism (version 5.0). The HRs and P-values were calculated
through an adjusted multivariate Cox regression analysis, including
risk score (high risk vs. low risk), sex, age (≥45 years vs. <45
years), AJCC7 N stage (stage 2–3 vs. 0–1), AJCC7 T stage (stage
III–IV vs. I–II), AJCC7 TNM stage (stage III–IV vs. I–II),
concurrent chemotherapy (Yes vs. No), EA-IgA (≥1:40 vs. 1:10-1:20
vs. <1:10), RT boosting (Yes vs. No), RT interrupt (0 day vs.
>1 days), sex, VCA-IgA (≥1:640 vs. 1:80-1:320 vs.<1:80), and
WHO type (undifferentiated non-keratinizing vs. differentiated
non-keratinizing vs. keratinizing squamous cell) as covariates for
each analysis. miRNA, microRNA; DMFS, distant metastasis-free
survival; OS, overall survival; RFS, relapse-free survival; DFS,
disease-free survival; NPC, nasopharyngeal carcinoma; TNM,
tumour-node-metastasis; VCA-IgA, viral capsid
antigen-immunoglobulin A; EA-IgA, early antigen-immunoglobulin A;
RT, radiotherapy; ROC, receiver operating characteristic; HR,
hazard ratio. |
Combination of differentially
expressed mRNAs with predicted targets of hub miRNAs
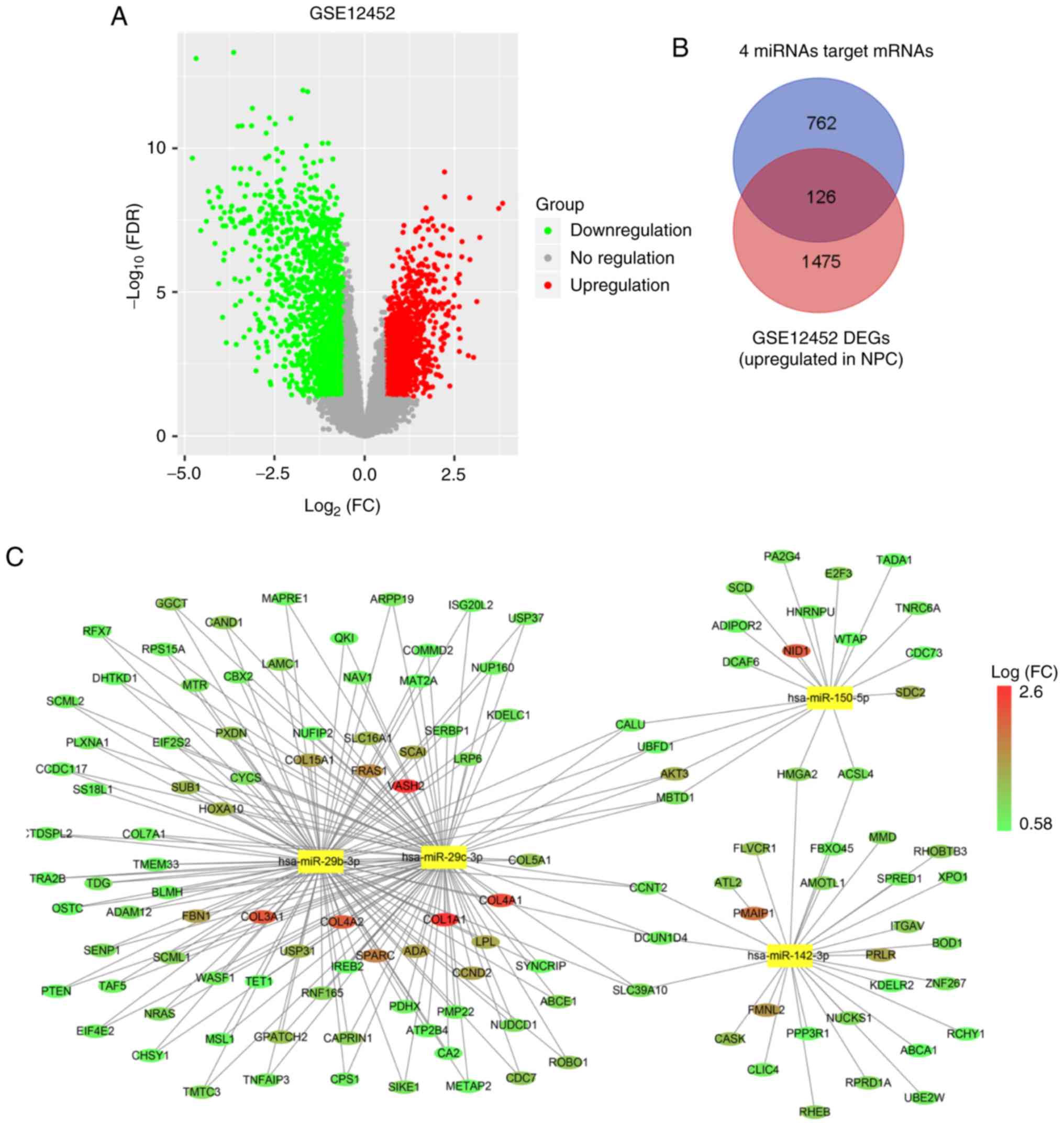
Bioinformatics analysis was applied to explore the
potential correlation between miRNAs and mRNA expression profiles.
The expression profile of GSE12452, including 31 NPC tissue samples
and 10 normal tissue samples, was also obtained from the NCBI-GEO
database. First, with the differential expression analysis, 2,956
mRNAs were identified as being aberrantly expressed (fold change
>1.5, adjust P-value (FDR) <0.05). There are two reasons for
screening differentially expressed genes. One is that our main
focus is on miRNAs, thus the screening of the differentially
expressed miRNAs is more stringent. Another reason is that the
regulation of mRNAs is complicated. Thus, the screening criteria
for mRNAs was lowered in order to avoid missing genes that are not
obviously altered by miRNA targeting. Of these, 1,601 mRNAs were
upregulated and 1,355 mRNAs were downregulated in NPC tissues
compared with normal tissues. The results of 1,355 downregulated
mRNAs and 1,601 upregulated mRNAs are displayed in the volcano plot
(Fig. 7A). Second, we predicted
possible target genes of four hub miRNAs. The online target
prediction tool starBase v2.0 was used to predict the target genes
of hsa-miR-142-3p, hsa-miR-150, hsa-miR-29b, and hsa-miR-29c. The
results revealed that 888 protein-coding genes were associated with
the four hub miRNAs to generate 1,502 miRNA-mRNA target pairs.
Finally, the 127 shared genes were obtained from 888 target genes
of hub genes and 1,601 upregulated mRNAs as aforementioned
(Fig. 7B; Table SVIII). As a result, 127 mRNAs were
identified for the next analysis.
The construction of the miRNA-mRNA
correlation network
After merging the target genes of 4 miRNAs with 127
mRNAs, the miRNA-mRNA correlation network was constructed (Fig. 7C). In this network, circular nodes
represent mRNAs, and rectangle nodes represent miRNAs. The size of
the nodes is equal to the number of miRNAs corresponding to the
mRNA, and the colour of the nodes represents the fold-change to
mRNA.
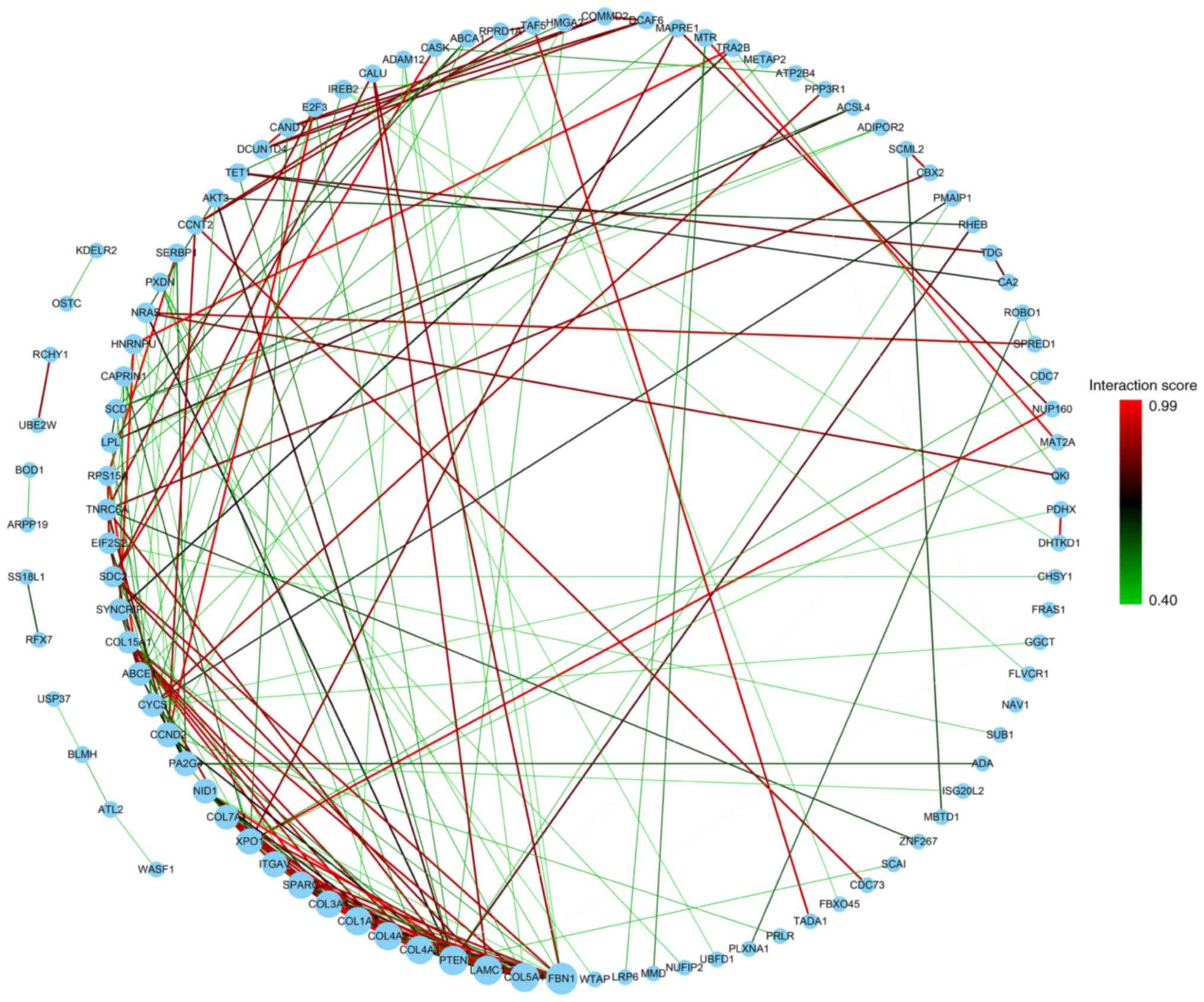
Gene ontology (GO) and pathway
analysis
Functional interactions and related pathway analysis
were performed on 127 mRNAs screened in the previous step to find a
mechanism that affects patient survival. First, the screened 127
mRNAs were input into the String website for analysis, and the
interaction information of 101 genes was obtained. Then, the
previous information was imported into Cytoscape software. The
number of interactions of each gene was considered as the size of
its node, and the comprehensive score of each interaction was
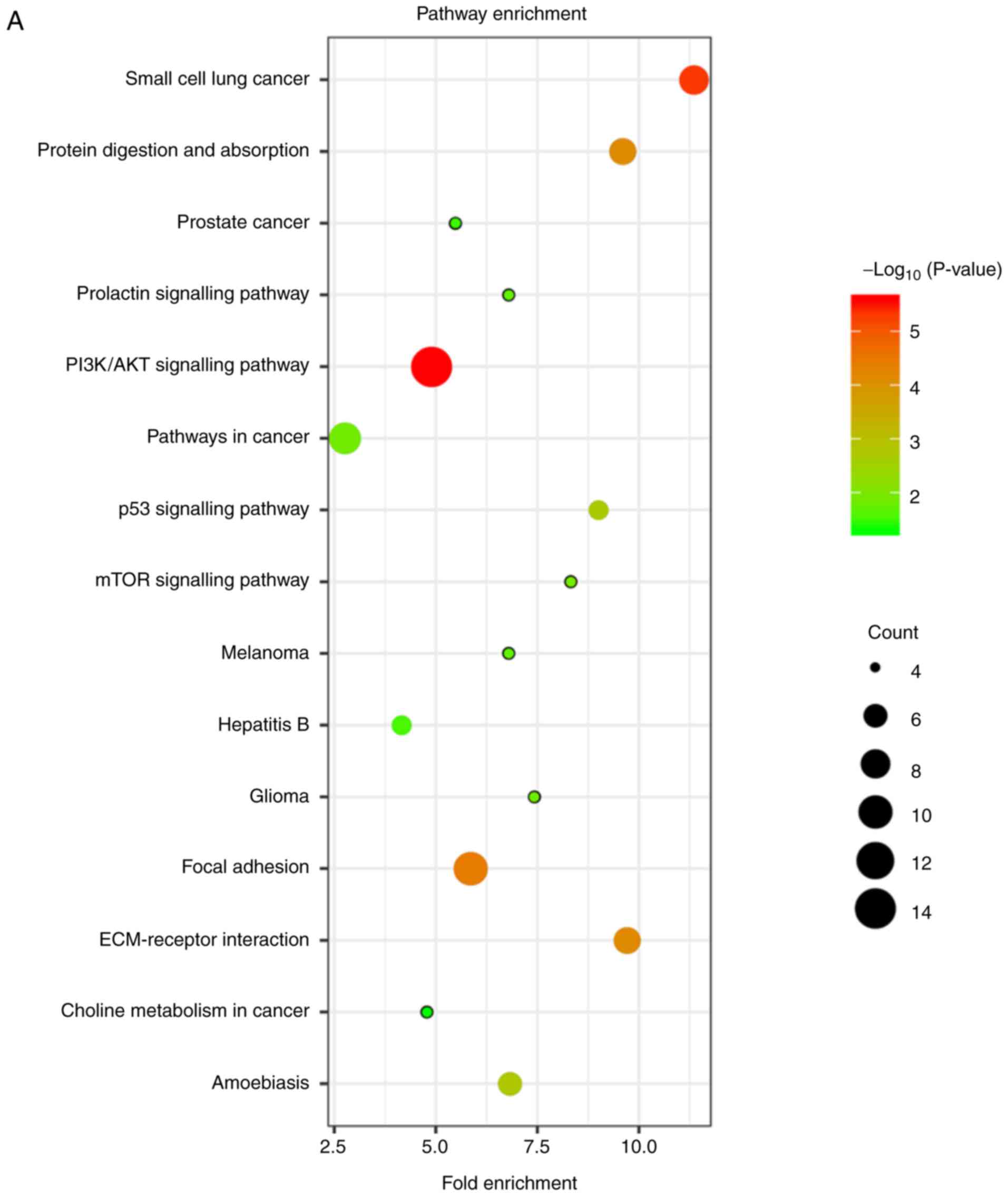
distinguished by colour (Fig. 8). BP,
CC, MF and biological pathway analyses of these 127 mRNAs are
presented in Fig. 9A and B. The GO
analysis indicated that the genes were mostly enriched in the
extracellular matrix organization, blood vessel development,
platelet-derived growth factor binding, and extracellular matrix
(Table SIX). Biological pathways
were mainly enriched in the focal adhesion, PI3K/Akt, p53, mTOR,
and ECM-receptor interaction signalling pathways (Table SX).
Discussion
Prognostic assessment is a crucial part of
appropriate treatment choices. In recent years, miRNAs have been
reported in the initiation and development of many cancers and are
potential biomarkers for diagnosis, prognosis, and personalized
treatment (26,27). However, miRNAs as a new biomarker
still need to be identified to guide individual treatment for
patients. One of the biggest challenges in developing miRNA-based
therapeutics is to identify the best miRNA candidates or miRNA
targets for each disease type (28).
Another major challenge is radioresistance in NPC (29). Hence, to improve the prognosis and
clinical treatment of NPC patients, it is urgent to identify
crucial prognostic biomarkers. However, by combining miRNAs and
mRNAs, the functional properties related to NPC pathogenesis cannot
be determined. Therefore, miRNA expression profiles were analysed
in NPC samples relative to normal samples to reveal the potential
role of miRNAs in the prognosis of NPC (Figs. 1 and 2).
In the present study, 46 DEMs were identified by
analysing the GSE32960 data. In addition, a co-expression network
of the associations between clinical traits and the modules was
constructed using this dataset, including 312 NPC samples (Figs. 3 and 4).
It was revealed that the dark green module was most significantly
associated with survival status, such as DMFS, RFS, DFS and OS, by
WGCNA and univariate Cox regression analyses. Furthermore, to
better understand which of the 46 differentially expressed miRNAs
was critical in the development of clinical outcome, univariate Cox
regression analysis was performed. In the present study, it was
also revealed that four hub miRNAs (hsa-miR-142-3p, hsa-miR-150,
hsa-miR-29b, and hsa-miR-29c) were significantly downregulated and
positively associated with DFS. Of them, hsa-miR-29c and
hsa-miR-142-3p were positively associated with DMFS by Kaplan-Meier
survival analysis (Figs. 5 and
S3-S5). Subsequently, a four-miRNA
signature was constructed to predict the prognosis of NPC patients.
The risk score of the four miRNAs revealed a better prediction of
survival than did TNM stage, T stage, N stage and sex alone with
regard to DMFS, OS, RFS, and DFS (Fig.
6). Clinically, NPC is a unique malignancy that is highly
invasive and metastatic. It was confirmed that ~50–60% of patients
developed distant metastases during the process of the disease
(30). Patients with loco-regionally
advanced NPC (stages III and IV) were reported to have a 5-year
survival rate of only 40% despite treatment with standard RT. In
contrast, the great majority of patients died from distant
recurrences (31). It has been
reported in the literature that mir-29c is downregulated in
nasopharyngeal carcinoma and targets a variety of mRNAs, such as
extracellular matrix proteins involved in cell migration and
metastasis. Increased accumulation of mRNAs encoding proteins may
contribute to the invasion and metastasis of NPC (32). Therefore, a decrease in the expression
of mir-29c in NPC cells may contribute to its aggressive
characteristics. Recently, a study indicated that miR-142-3p, a key
suppressive regulator, was epigenetically silenced by DNMT1 and
suppressed NPC cell metastasis and EMT by targeting ZEB2 (33). Therefore, miR-142-3p may be a
potential prognostic marker and therapeutic target to fight against
the metastasis of NPC. One study in NPC revealed that miR-150 can
modulate the EMT course in NPC/HK-1 cells and lead to cell invasion
(34). In addition, miR-29a/b may
contribute to the increase in migration and invasion of S18 cells,
a type of nasopharyngeal carcinoma cell (35). Furthermore, miRNAs emerging as
important modulators in biological pathways can regulate target
gene expression and play a key role in tumourigenesis through
translational repression or mRNA degradation, indicating that these
miRNAs are candidates for clinical applications in the treatment of
cancer (36–39). The function and mechanism of the four
miRNAs in NPC pathogenesis have not been presented thoroughly.
Further investigation into their functions and partners may provide
us with more targets and strategies for therapy. Moreover,
differentially expressed mRNA data (GSE32960) were integrated with
predicted miRNA targets to increase the accuracy of target
prediction. Therefore, bioinformatics analysis was applied to
explore the potential correlation between miRNAs and mRNA
expression profiles. The target genes of downregulated miRNAs in
NPC should be upregulated. Accordingly, the 127 shared genes were
obtained from 888 targets genes of hub genes and 1,601 upregulated
mRNAs as aforementioned. The 127 mRNAs were then identified by
superimposing differentially expressed mRNAs and target genes of
miRNAs, which were used for subsequent gene functional enrichment
analysis (Fig. 7). Increasing
evidence has confirmed that miRNAs play an important role in
regulating the expression of protein-coding genes (40–42). To
understand the potential functional roles of miRNAs, GO and BP
analyses were performed. The network revealed that AKT3, PTEN,
FBN1, LAMC1, COL5A1, COL1A1, COL4A1, and other genes may play an
important role in the interaction (Fig.
8). In particular, the results revealed that their target genes
were significantly associated with the focal adhesion, PI3K/Akt,
p53, and mTOR signalling pathways (Fig.
9A). In the present study, PTEN, a type of tumour suppressor
and a negative regulator of PI3K/Akt-dependent cellular survival,
has been implicated in several cancer progressions and was revealed
to be involved in p53 signalling (43–46). The
aforementioned results indicated a possible role of PTEN in p53 and
its related signalling pathways in the dysregulation of miRNAs
during NPC pathogenesis (47–49). Of course, miRNAs play a role in tumour
suppression in other pathways, and we confirmed that the tumour
suppressor miR-216b inhibited the KRAS-related AKT and ERK pathways
(50). Therefore, we conclude that
the results of our gene functional enrichment analysis are reliably
consistent with the present studies.
In conclusion, we successfully identified a
four-miRNA signature using an integrated bioinformatics analysis
for predicting the prognosis of patients with NPC and then analysed
their target genes along with potential biological signalling
pathways in the development and progression of NPC. GO and BP
analyses enabled the identification of possible associations
between miRNAs and protein-coding genes and revealed the potential
roles of miRNAs in NPC pathogenesis. Additionally, the greatest
advantage in the application of miRNA biology in the clinical
management of patients with NPC is its ability to target multiple
genes. However, the regulatory roles of the four miRNAs related to
p53 signalling or other vital signalling pathways in the genesis
and development mechanism of NPC and the detailed regulatory
mechanisms still require further study before this four-miRNA
signature can be successfully applied clinically. It is our sincere
hope that the present study may help promote future individualized
treatment of NPC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
We are deeply grateful to all donors who
participated in this study.
Funding
The present study was supported by the National Key
Research and Development Program and the National Natural Science
Foundations of China (2017YFC1200204, 31670171 and 81728011) and
the Innovation Foundations for Graduates of Central South
University (2018zzts821).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding authors on request.
Authors' contributions
SZ conceived, designed, and performed the
statistical analysis and wrote the paper. WY and SL participated in
analyzing the data, performing the statistical analysis and
drafting the manuscript. LY, XZ and PC helped to analyze the data.
YX, LL, WD and SX provisioned suggestions in figure preparation and
critically revised the manuscript. JL conceived and supervised the
study. All authors have read and approved the final version of the
manuscript and agree to be accountable for all aspects of the work
in ensuring that questions related to the accuracy or integrity of
any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GO
|
Gene Ontology
|
|
NPC
|
nasopharyngeal carcinoma
|
|
miRNAs
|
microRNAs
|
|
GEO
|
Gene Expression Omnibus
|
|
WGCNA
|
weighted gene co-expression network
analysis
|
|
DEMs
|
differentially expressed miRNAs
|
|
PPI
|
protein-protein interaction
|
|
MF
|
molecular function
|
|
BP
|
biological process
|
|
CC
|
cellular component
|
|
BPA
|
biological pathway analysis
|
|
ROC
|
receiver operating characteristic
|
|
DMFS
|
distant metastasis-free survival
|
|
OS
|
overall survival
|
|
DFS
|
disease-free survival
|
|
DEGs
|
differentially expressed genes
|
|
TNM
|
tumor-node-metastasis
|
|
FDR
|
false discovery rate
|
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pan JJ, Ng WT, Zong JF, Lee SW, Choi HC,
Chan LL, Lin SJ, Guo QJ, Sze HC, Chen YB, et al: Prognostic
nomogram for refining the prognostication of the proposed 8th
edition of the AJCC/UICC staging system for nasopharyngeal cancer
in the era of intensity-modulated radiotherapy. Cancer.
122:3307–3315. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hui EP, Leung SF, Au JS, Zee B, Tung S,
Chua D, Sze WM, Law CK, Leung TW and Chan AT: Lung metastasis alone
in nasopharyngeal carcinoma: A relatively favorable prognostic
group. A study by the Hong Kong nasopharyngeal carcinoma study
group. Cancer. 101:300–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ng WT, Yuen KT, Au KH, Chan OS and Lee AW:
Staging of nasopharyngeal carcinoma-the past, the present and the
future. Oral Oncol. 50:549–554. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin JC, Chen KY, Wang WY, Jan JS, Liang
WM, Tsai CS and Wei YH: Detection of Epstein-Barr virus DNA in the
peripheral-blood cells of patients with nasopharyngeal carcinoma:
Relationship to distant metastasis and survival. J Clin Oncol.
19:2607–2615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou GQ, Tang LL, Mao YP, Chen L, Li WF,
Sun Y, Liu LZ, Li L, Lin AH and Ma J: Baseline serum lactate
dehydrogenase levels for patients treated with intensity-modulated
radiotherapy for nasopharyngeal carcinoma: A predictor of poor
prognosis and subsequent liver metastasis. Int J Radiat Oncol Biol
Phys. 82:e359–e365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lv X, Xiang YQ, Cao SM, Qian CN, Li NW,
Guo L, Mai HQ, Chen QY, Huang PY, Luo D, et al: Prospective
validation of the prognostic value of elevated serum vascular
endothelial growth factor in patients with nasopharyngeal
carcinoma: More distant metastases and shorter overall survival
after treatment. Head Neck. 33:780–785. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang XR, Li YQ, Liang SB, Jiang W, Liu F,
Ge WX, Tang LL, Mao YP, He QM, Yang XJ, et al: Development and
validation of a gene expression-based signature to predict distant
metastasis in locoregionally advanced nasopharyngeal carcinoma: A
retrospective, multicentre, cohort study. Lancet Oncol. 19:382–393.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goretti E, Wagner DR and Devaux Y: miRNAs
as biomarkers of myocardial infarction: A step forward towards
personalized medicine? Trends Mol Med. 20:716–725. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deng Z, Wang Y, Fang X, Yan F, Pan H, Gu
L, Xie C, Li Y, Hu Y, Cao Y and Tang Z: Research on miRNA-195 and
target gene CDK6 in oral verrucous carcinoma. Cancer Gene Ther.
24:282–288. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He B, Li W, Wu Y, Wei F, Gong Z, Bo H,
Wang Y, Li X, Xiang B, Guo C, et al: Epstein-Barr virus-encoded
miR-BART6-3p inhibits cancer cell metastasis and invasion by
targeting long non-coding RNA LOC553103. Cell Death Dis.
7:e23532016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watanabe A, Tagawa H, Yamashita J, Teshima
K, Nara M, Iwamoto K, Kume M, Kameoka Y, Takahashi N, Nakagawa T,
et al: The role of microRNA-150 as a tumor suppressor in malignant
lymphoma. Leukemia. 25:1324–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saito Y, Suzuki H, Imaeda H, Matsuzaki J,
Hirata K, Tsugawa H, Hibino S, Kanai Y, Saito H and Hibi T: The
tumor suppressor microRNA-29c is downregulated and restored by
celecoxib in human gastric cancer cells. Int J Cancer.
132:1751–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fang JH, Zhou HC, Zeng C, Yang J, Liu Y,
Huang X, Zhang JP, Guan XY and Zhuang SM: MicroRNA-29b suppresses
tumor angiogenesis, invasion, and metastasis by regulating matrix
metalloproteinase 2 expression. Hepatology. 54:1729–1740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee KT, Tan JK, Lam AK and Gan SY:
MicroRNAs serving as potential biomarkers and therapeutic targets
in nasopharyngeal carcinoma: A critical review. Crit Rev Oncol
Hematol. 103:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu N, Chen NY, Cui RX, Li WF, Li Y, Wei
RR, Zhang MY, Sun Y, Huang BJ, Chen M, et al: Prognostic value of a
microRNA signature in nasopharyngeal carcinoma: A microRNA
expression analysis. Lancet Oncol. 13:633–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sengupta S, den Boon JA, Chen IH, Newton
MA, Dahl DB, Chen M, Cheng YJ, Westra WH, Chen CJ, Hildesheim A, et
al: Genome-wide expression profiling reveals EBV-associated
inhibition of MHC class I expression in nasopharyngeal carcinoma.
Cancer Res. 66:7999–8006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Horvath S and Dong J: Geometric
interpretation of gene coexpression network analysis. PLoS Comput
Biol. 4:e10001172008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang JH, Li JH, Shao P, Zhou H, Chen YQ
and Qu LH: starBase: A database for exploring microRNA-mRNA
interaction maps from Argonaute CLIP-Seq and Degradome-Seq data.
Nucleic Acids Res. 39((Database Issue)): D202–D209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dennis G, Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43((Database Issue)): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu SL, Chen HY, Chang GC, Chen CY, Chen
HW, Singh S, Cheng CL, Yu CJ, Lee YC, Chen HS, et al: MicroRNA
signature predicts survival and relapse in lung cancer. Cancer
Cell. 13:48–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lossos IS, Czerwinski DK, Alizadeh AA,
Wechser MA, Tibshirani R, Botstein D and Levy R: Prediction of
survival in diffuse large-B-cell lymphoma based on the expression
of six genes. N Engl J Med. 350:1828–1837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ji J, Shi J, Budhu A, Yu Z, Forgues M,
Roessler S, Ambs S, Chen Y, Meltzer PS, Croce CM, et al: MicroRNA
expression, survival, and response to interferon in liver cancer. N
Engl J Med. 361:1437–1447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Z and Rana TM: Therapeutic targeting of
microRNAs: Current status and future challenges. Nat Rev Drug
Discov. 13:622–638. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qu JQ, Yi HM, Ye X, Zhu JF, Yi H, Li LN,
Xiao T, Yuan L, Li JY, Wang YY, et al: miRNA-203 reduces
nasopharyngeal carcinoma radioresistance by targeting IL8/AKT
signaling. Mol Cancer Ther. 14:2653–2664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cvitkovic E, Bachouchi M, Boussen H,
Busson P, Rousselet G, Mahjoubi R, Flores P, Tursz T, Armand JP and
Azli N: Leukemoid reaction, bone marrow invasion, fever of unknown
origin, and metastatic pattern in the natural history of advanced
undifferentiated carcinoma of nasopharyngeal type: A review of 255
consecutive cases. J Clin Oncol. 11:2434–2442. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Teo PM, Kwan WH, Lee WY, Leung SF and
Johnson PJ: Prognosticators determining survival subsequent to
distant metastasis from nasopharyngeal carcinoma. Cancer.
77:2423–2431. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sengupta S, den Boon JA, Chen IH, Newton
MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B and
Ahlquist P: MicroRNA 29c is down-regulated in nasopharyngeal
carcinomas, up-regulating mRNAs encoding extracellular matrix
proteins. Proc Natl Acad Sci USA. 105:5874–5878. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, He Q, Wen X, Hong X, Yang X, Tang X,
Zhang P, Lei Y, Sun Y, Zhang J, et al: EZH2-DNMT1-mediated
epigenetic silencing of miR-142-3p promotes metastasis through
targeting ZEB2 in nasopharyngeal carcinoma. Cell Death Differ.
26:1089–1106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yue PY, Ha WY, Lau CC, Cheung FM, Lee AW,
Ng WT, Ngan RK, Yau CC, Kwong DL, Lung HL, et al: MicroRNA
profiling study reveals miR-150 in association with metastasis in
nasopharyngeal carcinoma. Sci Rep. 7:120122017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qiu F, Sun R, Deng N, Guo T, Cao Y, Yu Y,
Wang X, Zou B, Zhang S, Jing T, et al: miR-29a/b enhances cell
migration and invasion in nasopharyngeal carcinoma progression by
regulating SPARC and COL3A1 gene expression. PLoS One.
10:e01209692015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng Z, Qu JQ, Yi HM, Ye X, Huang W, Xiao
T, Li JY, Wang YY, Feng J, Zhu JF, et al: miR-125b regulates
proliferation and apoptosis of nasopharyngeal carcinoma by
targeting A20/NF-κB signaling pathway. Cell Death Dis. 8:e28552017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tan G, Tang X and Tang F: The role of
microRNAs in nasopharyngeal carcinoma. Tumour Biol. 36:69–79. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cho WC: MicroRNAs in cancer-from research
to therapy. Biochim Biophys Acta. 1805:209–217. 2010.PubMed/NCBI
|
|
39
|
Zuo LL, Zhang J, Liu LZ, Zhou Q, Du SJ,
Xin SY, Ning ZP, Yang J, Yu HB, Yue WX, et al: Cadherin 6 is
activated by Epstein-Barr virus LMP1 to mediate EMT and metastasis
as an interplay node of multiple pathways in nasopharyngeal
carcinoma. Oncogenesis. 6:4022017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin CW, Li XR, Zhang Y, Hu G, Guo YH, Zhou
JY, Du J, Lv L, Gao K, Zhang Y and Deng H: TAp63 suppress
metastasis via miR-133b in colon cancer cells. Br J Cancer.
110:2310–2320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu H, Lu J, Zuo L, Yan Q, Yu Z, Li X,
Huang J, Zhao L, Tang H, Luo Z, et al: Epstein-Barr virus
downregulates microRNA 203 through the oncoprotein latent membrane
protein 1: A contribution to increased tumor incidence in
epithelial cells. J Virol. 86:3088–3099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Peng G, Liao Y and Shen C: miRNA-429
inhibits astrocytoma proliferation and invasion by targeting BMI1.
Pathol Oncol Res. 23:369–376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mayo LD and Donner DB: The PTEN, Mdm2, p53
tumor suppressor-oncoprotein network. Trends Biochem Sci.
27:462–467. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stambolic V, MacPherson D, Sas D, Lin Y,
Snow B, Jang Y, Benchimol S and Mak TW: Regulation of PTEN
transcription by p53. Mol Cell. 8:317–325. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Freeman DJ, Li AG, Wei G, Li HH, Kertesz
N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et
al: PTEN tumor suppressor regulates p53 protein levels and activity
through phosphatase-dependent and -independent mechanisms. Cancer
Cell. 3:117–130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jung SH, Hwang HJ, Kang D, Park HA, Lee
HC, Jeong D, Lee K, Park HJ, Ko YG and Lee JS: mTOR kinase leads to
PTEN-loss-induced cellular senescence by phosphorylating p53.
Oncogene. 38:1639–1650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Andreozzi M, Quagliata L, Gsponer JR, Ruiz
C, Vuaroqueaux V, Eppenberger-Castori S, Tornillo L and Terracciano
LM: VEGFA gene locus analysis across 80 human tumour types reveals
gene amplification in several neoplastic entities. Angiogenesis.
17:519–527. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen HX, Xu XX, Tan BZ, Zhang Z and Zhou
XD: MicroRNA-29b inhibits angiogenesis by targeting VEGFA through
the MAPK/ERK and PI3K/Akt signaling pathways in endometrial
carcinoma. Cell Physiol Biochem. 41:933–946. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cheng JZ, Chen JJ, Xue K, Wang ZG and Yu
D: Clinicopathologic and prognostic significance of VEGF, JAK2 and
STAT3 in patients with nasopharyngeal carcinoma. Cancer Cell Int.
18:1102018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng M, Tang H, Zhou Y, Zhou M, Xiong W,
Zheng Y, Ye Q, Zeng X, Liao Q, Guo X, et al: miR-216b suppresses
tumor growth and invasion by targeting KRAS in nasopharyngeal
carcinoma. J Cell Sci. 124:2997–3005. 2011. View Article : Google Scholar : PubMed/NCBI
|