Introduction
Esophageal squamous cell carcinoma (ESCC), the
predominant histological subtype of esophageal carcinoma, is one of
the most refractory cancer types with a high mortality rate in the
world (1,2). Although various treatment regimens have
been adopted to improve curability, the overall 5-year survival
rate for patients with ESCC over the past 30 years is still poor at
~20% (2). At present, surgical
treatment combined with radiotherapy and chemotherapy is the main
clinical therapeutic method of ESCC, and cisplatin (CDDP) is still
used as one of the pivotal chemotherapeutic drugs in the front-line
therapeutic regimen, playing an irreplaceable role in prevention of
recurrence and metastasis of ESCC (3). However, satisfactory chemotherapeutic
effects have rarely been achieved, as CDDP-based therapies always
just obtain initial chemotherapeutic success and eventually induce
CDDP resistance in ESCC (4,5).
It has been proposed that several molecular
mechanisms can drive chemoresistance to CDDP, which may be unique
to different types of cancer (4,5). Prominent
CDDP-resistance mechanisms involve various biological regulatory
processes, including blockade of DNA damage response signal,
activation of DNA repair pathways, epithelial-mesenchymal
transition (EMT), thiols and metallothionein-mediated
detoxification, and efflux of CDDP from cancer cells (4). Despite the progress in identifying
mechanisms responsible for resistance to CDDP, the underlying
mechanisms of resistance in ESCC are not entirely understood and
the clinical treatment of CDDP-resistant ESCC remains a critical
obstacle. Therefore, the present study aimed to further elucidate
mechanisms behind CDDP resistance in ESCC and novel biomarkers,
which may be used to predict prognosis and chemosensitivity.
Accumulating evidence has suggested that
CDDP-induced nuclear DNA damage alone is not enough to illustrate
its high-level of effectiveness or cytotoxicity (6,7). Several
recent studies have demonstrated that, independent of CDDP-DNA
adduct generation, the mechanism of CDDP-stimulated production of
reactive oxygen species (ROS) and subsequent oxidative damage of
the DNA base also have a crucial involvement in CDDP cytotoxicity
(7–9).
One of the best characterized oxidative DNA lesions is
7,8-dihydro-8-oxoguanine, which can induce G:C to T:A transversion
mutations. The mutations are recognized and repaired by mutY
homolog (MUTYH) (10,11). MUTYH is a key component of the
system of base excision repair (BER), which is able to remove the
oxidized bases paired erroneously, thereby ensuring the maintenance
of DNA integrity (10,11). It has been confirmed by experimental
evidence that MUTYH has an important role in various cancer
types; it was reported that modification of the tissue expression
of MUTYH is associated with the elevated risk of development
of various cancer types, such as colorectal cancer, lung cancer,
esophageal carcinoma, thyroid cancer and head and neck cancer
(12–17). Mutations in MUTYH reduce BER
effectiveness and predisposition to cancer (17,18).
Recent research also suggests that patients with colorectal cancer
have lower MUTYH expression in cancer tissues compared with
normal tissues (19). Meanwhile, the
MUTYH gene was found to present reduced expression in more
advanced stages of colorectal cancer, and lower MUTYH
expression may contribute to worse patient prognosis (12). In the present study, the association
of MUTYH with CDDP resistance in ESCC cells was analyzed.
The present data demonstrated that downregulation of MUTYH
induced the EMT process by regulating both the transcription and
degradation of Twist, which eventually contributed to resistance to
CDDP.
Materials and methods
Establishment of a CDDP-resistant ESCC
cell line
The human ESCC cell line EC109 was obtained from The
Cell Bank of the Chinese Academy of Sciences and was maintained in
RPMI-1640 medium (HyClone; GE Healthcare Life Sciences)
supplemented with 10% FBS (HyClone; GE Healthcare Life Sciences).
The CDDP-resistant cell line EC109/CDDP was selected by culturing
EC109 cells in increasing concentrations of CDDP (Selleck
Chemicals) starting from 0.1 µM to a final concentration of 5.0 µM.
The cells were maintained in a humidified incubator with 5%
CO2 at 37°C.
Cell viability and cell death
analysis
The inhibitory effect of CDDP on cell viability in
EC109 and EC109/CDDP cells was determined by an MTT (Sigma-Aldrich;
Merck KGaA) colorimetric assay using a Thermo Fisher Multiskan
microplate reader (Thermo Fisher Scientific, Inc.).
Apoptosis induced by CDDP in EC109 and EC109/CDDP
cells was tested using an Annexin V-FITC/Propidium Iodide Apoptosis
Detection kit, according to the manufacturer's protocol (BD
Biosciences). A flow cytometer (FACSCaliber; BD Biosciences) was
used for fluorescence quantification.
Proteasome activity assay
The proteasome substrates Suc-LLVY-AMC
(N-succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin), Bz-LLE-AMC
(benzyloxycarbonyl-L-leucyl-leucyl-glutamyl-methylcoumarylamide)
and Bz-VGR-AMC (Bz-Val-Gly-Arg-7-amino-4-methylcoumarin) from Enzo
Life Sciences, Inc., were used to detect activities of
chymotrypsin-like (ChT-L), trypsin-like and peptidyl-glutamyl
peptide-hydrolyzing (PGPH) of proteasomes in cells, respectively.
Whole cell lysates were extracted with lysis buffer [50 mM Tris-HCl
(pH 8.0), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40 and 2 mM DTT], and
incubated at 37°C for 40 min with the substrates in the assay
buffer. Proteasome activities were determined by measuring
fluorescent activities of the substrates using a Mithras LB-940
reader (Berthold Technologies).
Transwell Matrigel invasion assay
Cell invasion was measured by a Transwell Matrigel
invasion assay using Growth Factor Reduced Matrigel Invasion
Chambers from BD Biosciences. In total, 0.5×106 cells
were resuspended in RPMI-1640 medium supplemented with 1% FBS serum
into the upper chamber with Matrigel matrix added (BD Biosciences).
RPMI-1640 medium supplemented with 10% serum, serving as a
chemoattractant, was added to the bottom well. After 48 or 72 h,
cells that migrated to the bottom chamber were stained with Giemsa
(Bejing Solarbio Science & Technology Co., Ltd.) and were
imaged under a bright-field microscope (Nikon Corp.; Magnification,
×200). The stained cells were counted using ImageJ software
(National Institutes of Health).
Wound healing assay
A wound healing assay was performed to examine cell
migration. When cells grew to approximately 95% confluence in
6-well plates, a scratch (wound) was introduced using a pipette
tip. Then, the cells were washed three times with PBS to remove
detached cells and cultured continually for 24 or 48 h. The wound
was imaged under a bright-field microscope (Nikon Corp.;
magnification, ×100) at 0, 24 and 48 h. The wound gaps were
digitally quantified using ImageJ software (National Institutes of
Health). The equation for the ‘relative wound area’ is: Relative
wound area=wound area of different group/wound area of EC109 cells
at 0 h.
ROS measurement
The production of ROS was monitored using the
fluorescent dye hydroethidine 2′,7′-dichlorodihydrofluorescein
diacetate [H(2)DCFDA; Sigma-Aldrich; Merck KGaA]. After treatment
as indicated, cells were incubated with 10 µM H(2)DCFDA at 37°C for
30 min. Then, the cells were washed with PBS and subjected to ROS
measurement by flow cytometry. In the experiments, to test the
scavenging effect of the antioxidant N-acetylcysteine (NAC;
Sigma-Aldrich; Merck KGaA) on ROS generation, cells were treated
with 2 mM NAC for 24 h before ROS measurement.
Western blot assay
A western blot assay was performed, as previously
described (20). Whole cell lysates
were prepared with RIPA buffer, according to the manufacturer's
protocol (Beyotime Institute of Biotechnology). Proteins were
quantified using a bicinchoninic acid protein assay (Beyotime
Institute of Biotechnology). Blots were incubated with primary
antibodies against MUTYH (cat. no. 19650-1-AP; rabbit polyclonal
antibody; dilution 1:500), E-cadherin (cat. no. 60335-1-lg; mouse
monoclonal antibody; dilution 1:2,000), vimentin (cat. no.
60330-1-lg; mouse monoclonal antibody; dilution 1:2,000), Twist
(cat. no. 25465-1-AP; rabbit polyclonal antibody; dilution 1:500)
and ubiquitin (cat. no. 10201-1-AP; rabbit polyclonal antibody;
dilution 1:500) (all from ProteinTech Group, Inc.) overnight at
4°C, followed by incubation with appropriate peroxidase-conjugated
secondary antibodies (anti-mouse IgG (H+L) peroxidase-labeled
polyclonal antibody, cat. no. 074-1806; dilution 1:5,000;
anti-rabbit IgG (H+L) peroxidase-labeled polyclonal antibody; cat.
no. 074-1506; dilution 1:5,000; purchased from KPL, Inc.; SeraCare
Life Sciences, Inc.). GAPDH (cat. no. sc-47724; mouse monoclonal
antibody; dilution 1:3,000; Santa Cruz Biotechnology, Inc.) served
as a protein loading control. Immunocomplexes were visualized using
chemiluminescence (EMD Millipore) and exposed to X-ray film.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was prepared using an RNAiso Plus Kit
(Takara Bio, Inc.). cDNA was synthesized using a PrimeScript RT
Reagent Kit (Takara Bio, Inc.) and RT-qPCR was carried out using
the Eppendorf RT-qPCR System (Mastercycler ep realplex; Eppendorf).
The PCR reaction conditions for all assays were as follows: 95°C
for 30 sec, followed by 40 cycles of amplifcation (95°C for 5 sec,
58°C for 30 sec and 72°C for 30 sec). Changes in the mRNA levels
were normalized to the level of GAPDH and calculated using the
2−ΔΔCq method (21). The
primer sequences are summarized in Table
I.
 | Table I.Primers for the RT-qPCR analysis. |
Table I.
Primers for the RT-qPCR analysis.
| Gene name | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| MUTYH |
TGCCACGTACAGCAGAGAC |
CAAAGGCGATAGAGGCAATGG |
| CDH1 |
ATTTTTCCCTCGACACCCGAT |
TCCCAGGCGTAGACCAAGA |
| CDH2 |
TTTGATGGAGGTCTCCTAACACC |
ACGTTTAACACGTTGGAAATGTG |
| VIM |
AGTCCACTGAGTACCGGAGAC |
CATTTCACGCATCTGGCGTTC |
| TWIST1 |
GCCGGAGACCTAGATGTCATT |
TTTTAGTTATCCAGCTCCAGAGTC |
| SNAI2 |
GCGCTCCTTCCTGGTCAAGA |
CGCCCAGGCTCACATATTCC |
| SNAI1 |
AGCGAGCTGCAGGACTCTAA |
ATCTCCGGAGGTGGGATGG |
| MMP7 |
GAGTGAGCTACAGTGGGAACA |
CTATGACGCGGGAGTTTAACAT |
| MMP9 |
TTCCAAACCTTTGAGGGCGA |
GCAAAGGCGTCGTCAATCAC |
| CLDN1 |
CTGTCATTGGGGGTGCGATA |
GACTGGGGTCATAGGGTCAT |
| GAPDH |
TGGTCACCAGGGCTGCTT |
AGCTTCCCGTTCTCAGCCTT |
Transient transfection of plasmids and
small interfering RNAs (siRNAs)
The pcDNA3.1-MUTYH plasmid was constructed to
induce overexpression of MUTYH (Shanghai Integrated Biotech
Solutions Co., Ltd.). Empty vector pcDNA3.1 served as a control.
Knockdown of MUTYH was performed by transiently transfecting
siRNA duplex oligonucleotides targeting MUTYH (BioSune). The
MUTYH siRNA sequences were as follows: Sense,
5′-GGAGGCAGAAGCAUGCUAATT-3′ and antisense,
5′-UUAGCAUGCUUCUGCCUCCTT-3′. The scramble siRNA sequences were as
follows: Sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. Transfection of the overexpression
plasmid or siRNA was performed using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
Statistical analysis
Data are presented as the mean ± SD and were
analyzed using GraphPad Prism software (GraphPad Software, Inc.).
The statistical significance of mean differences between the
treated and control groups was determined using a two-tailed
Student's unpaired t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Evaluation of the CDDP resistance of
the established CDDP-resistant ESCC cell line
The aim of the present study was to test the
sensitivity of the established EC109/CDDP cells and the parental
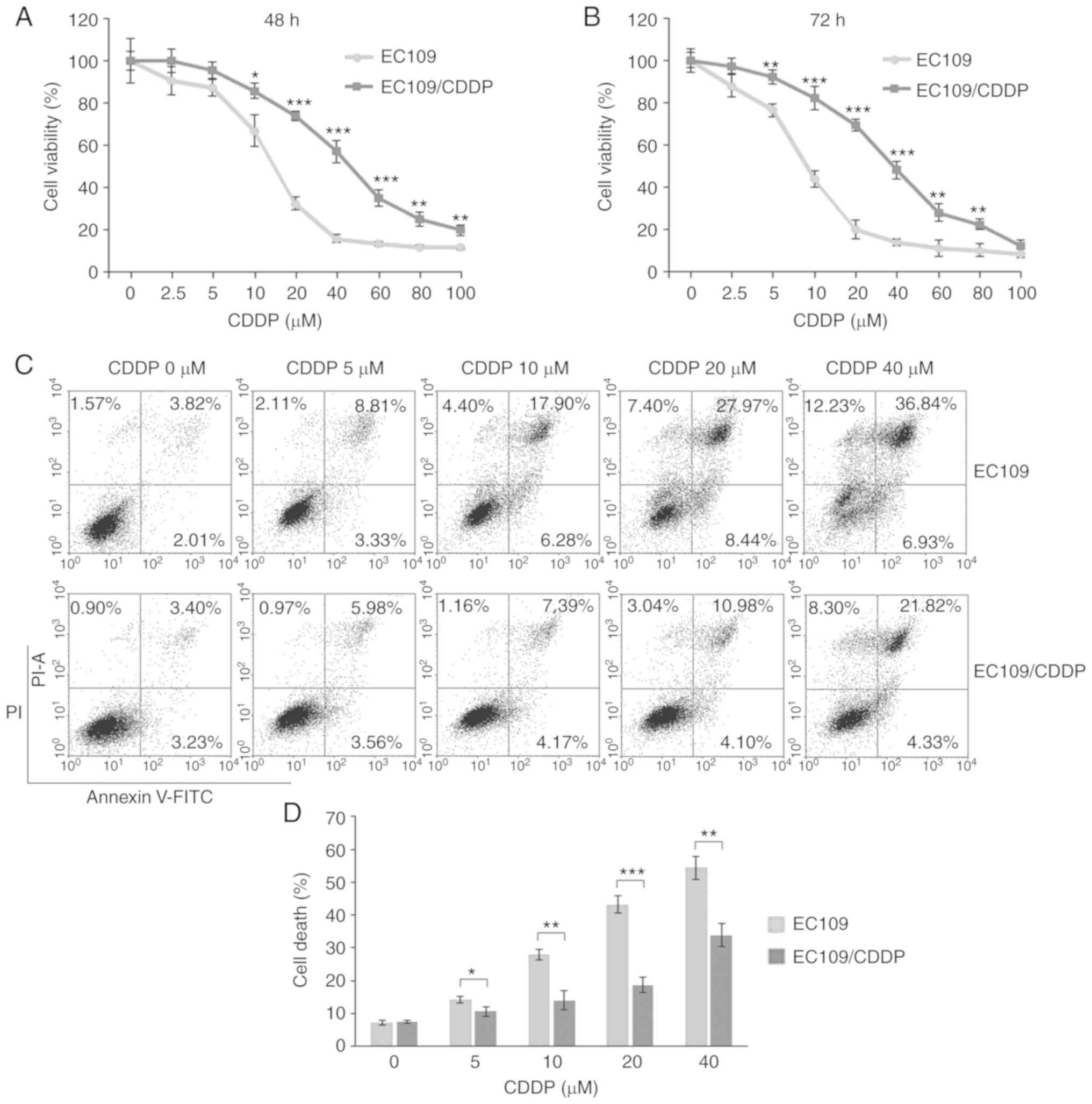
EC109 cells to CDDP. As shown in Fig.
1A, EC109/CDDP cells showed markedly higher cell viability than
EC109 after 48 h treatment with CDDP at concentrations of 2.5–100
µM and the concentrations that induced cellular death at 50%
(IC50) were calculated. EC109/CDDP cells were resistant
to CDDP, with a relatively marginally increased IC50
value at ~50 µM compared with ~10 µM in EC109 cells. The resistant
index (RI) of EC109/CDDP cells for CDDP was ~5 at 48 h. Fig. 1B demonstrates that the CDDP-resistance
of EC109/CDDP cells was more prominent at 72 h of CDDP incubation,
with an RI at ~7. Flow cytometry was performed to detect the
apoptotic cells exposed to CDDP in EC109/CDDP cells. The present
results demonstrated that CDDP at 5, 10, 20 and 40 µM caused
significant increases in the proportion of apoptotic EC109 cells,
~14.25, 28.58, 43.81 and 56.00%, respectively. By comparison, the
apoptotic EC109/CDDP cell fractions were 10.51, 12.72, 18.12 and
34.45% respectively, showing a significant reduction (Fig. 1C and D). Therefore, the present data
demonstrated that EC109/CDDP cells were resistant to CDDP when
compared with parental EC109 cells.
Downregulation of MUTYH contributes to
CDDP resistance in ESCC cells
In the process of identifying potential targets
involved in CDDP resistance, MUTYH, the crucial gene for
excision repair of oxidized bases, was noted for its pronounced
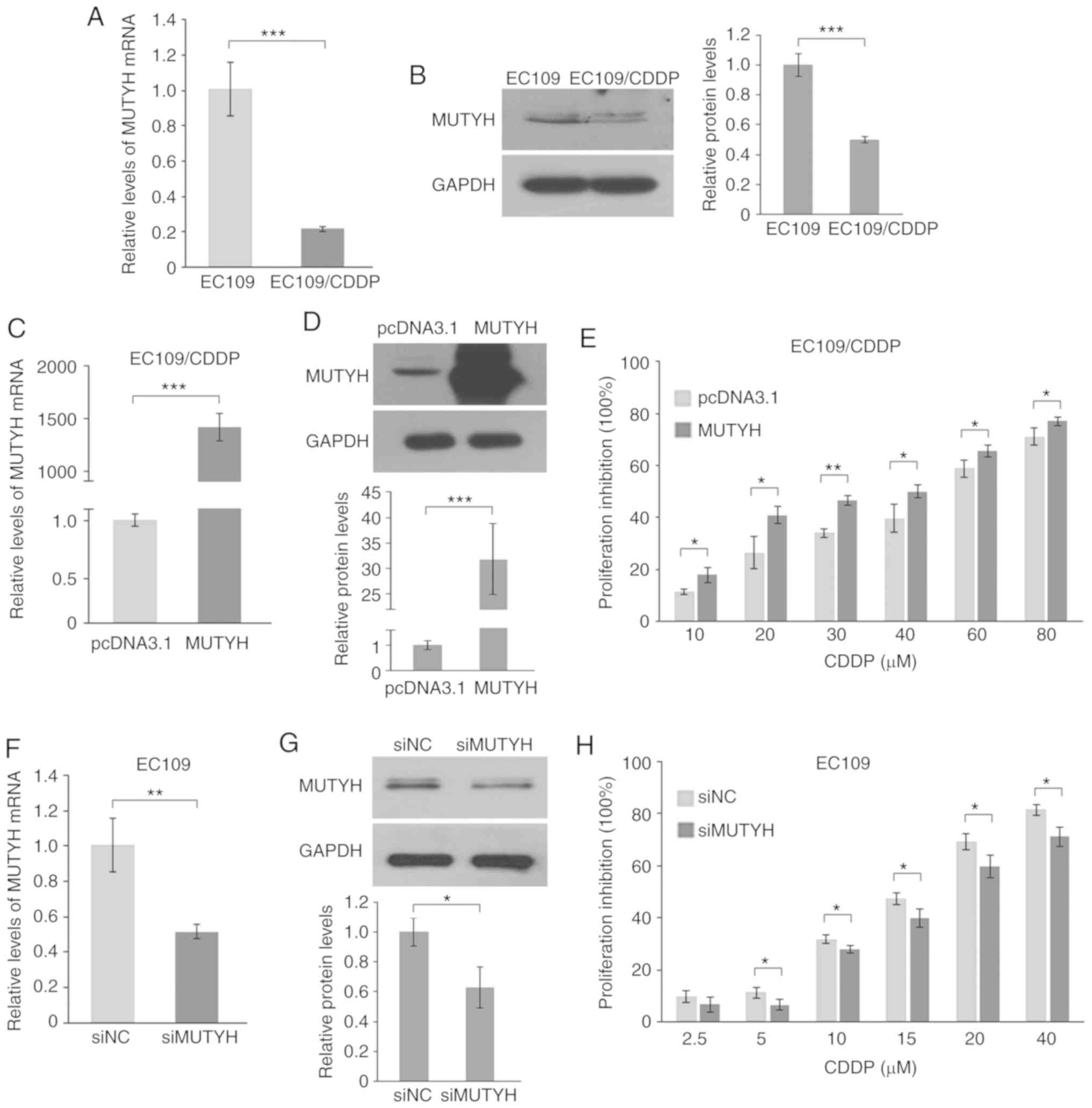
downregulation in the resistant cells. As shown in Fig. 2A, RT-qPCR analysis demonstrated that
the mRNA expression of MUTYH decreased ~5-fold in resistant
EC109/CDDP cells compared with that noted in the parental cells.
Similar to the mRNA level, the resistant cells presented a
significant decrease in the corresponding protein level of MUTYH
(Fig. 2B).
To further investigate the effect of MUTYH in
CDDP resistance, MUTYH was either overexpressed with a MUTYH
expression plasmid in resistant cells or ablated using siRNA in
parental cells, and the cell proliferation in response to CDDP was
determined. The present results demonstrated that the MUTYH
expression level was significantly enhanced by transfecting cells
with a MUTYH expression plasmid (Fig. 2C and D), and ectopic expression of
MUTYH significantly re-sensitized resistant cells to CDDP treatment
at concentrations of 10–80 µM for 48 h (Fig. 2E). Conversely, it was observed that
the MUTYH-targeted siRNA reduced the expression of
MUTYH from the basal level in sensitive EC109 cells
(Fig. 2F and G), and the result of
the cell viability assay demonstrated that knockdown of
MUTYH in sensitive EC109 cells resulted in an attenuated
response to CDDP (Fig. 2H).
Therefore, these data demonstrated that downregulation of
MUTYH, at least in part, contributes to the CDDP-resistant
phenotype.
CDDP-resistant ESCC cells exhibit an
EMT-like phenotypic change
Based on the significant effect of MUTYH on
CDDP-resistance, the signaling mechanisms underlying
MUTYH-mediated CDDP resistance in resistant cells were
investigating. By comparing the differential phenotype between the
resistant and parental cells, it was observed that EMT was markedly
enhanced in resistant cells, which may be associated with
MUTYH downregulation. EMT plays an important role in various
cancer-associated biological events, which can contribute to
invasion, metastasis as well as drug resistance of cancer (22,23). As
shown in Fig. 3A, in contrast to
parental cells, EC109/CDDP cells displayed morphological
characteristics of EMT, as observed by the transition from
cobblestone-like cells into slender spindle-shaped cells with
increased detachment.
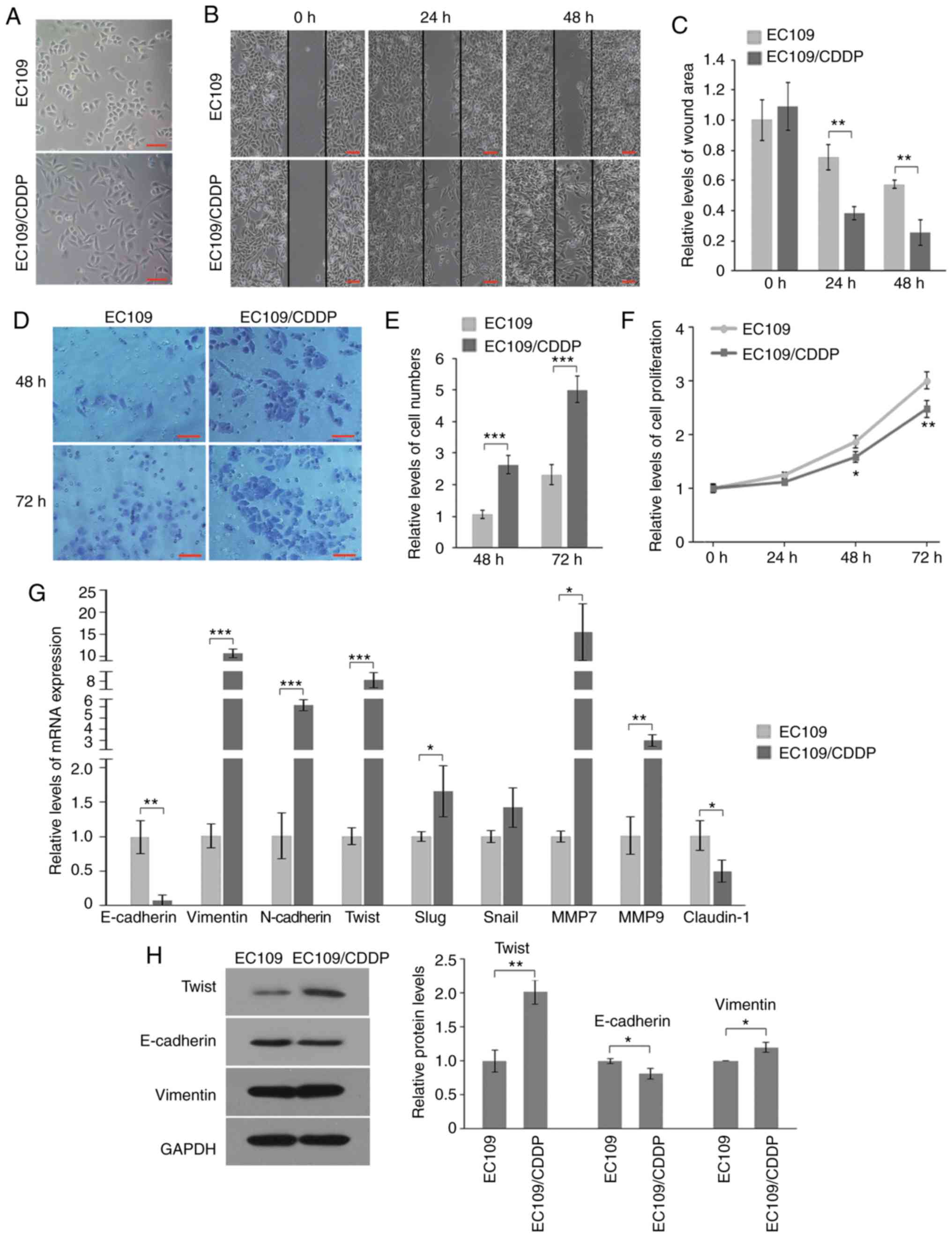 | Figure 3.Resistant cells display an EMT
phenotype. (A) Morphology of EC109 and EC109/CDDP cells (scale bar,
100 µm). (B and C) Representative images (B) and quantification
analysis (C) of the wound-healing assay of EC109 and EC109/CDDP
cells (scale bar, 100 µm). (D and E) Representative images (D) and
quantification analysis (E) of invasive behavior of EC109 and
EC109/CDDP cells (scale bar, 100 µm). (F) Cell proliferation of
EC109 and EC109/CDDP cells was determined by the MTT assay after
24, 48 or 72 h. (G and H) RT-qPCR (G) and western blotting (H)
analysis of mRNA and protein levels of various EMT-associated
markers, respectively. Protein quantification for Twist, E-cadherin
and vimentin was shown. In C, E-H: *P<0.05, **P<0.01 and
***P<0.001 vs. the EC109 cells, respectively. EMT,
epithelial-mesenchymal transition; CDDP, cisplatin. |
Enhanced capacities of migration and invasion are
common along with resistance acquisition in cancer cells. Wound
healing assays showed that EC109/CDDP cells displayed significantly
higher migratory potential, as demonstrated by more rapid and
complete wound healing than parental cells (Fig. 3B). Wound areas in parental cells were
~2- and ~2.5-fold at 24 and 48 h, respectively, as large as those
in resistant cells (Fig. 3C).
Additionally, Transwell invasion analysis revealed that the
invasiveness through Matrigel was also significantly increased in
the resistant cells, 2–3-fold compared with that noted in the
parental cells (Fig. 3D and E). To
eliminate the interference of cell proliferation on migration and
invasion, proliferation rates of the two cell lines were also
compared. The present results showed that the proliferation rate of
resistant cells decreased ~13, ~28 and ~52% at 24, 48 and 72 h,
respectively, compared with the parental cells (Fig. 3F). Therefore, the enhanced wound
healing and invasiveness through Matrigel of the EC109/CDDP cell
line were not caused by proliferation, which further demonstrated
its increased migratory and invasive capacities.
Moreover, the expressions of various genes
associated with migration and invasion in both cells were detected.
The results of RT-qPCR showed that epithelial marker E-cadherin
(encoded by CDH1 gene) was significantly downregulated
(~14-fold) while mesenchymal markers N-cadherin (encoded by
CDH2 gene) and vimentin (encoded by VIM gene) were
significantly upregulated (~10-fold and ~6-fold, respectively) in
the resistant cells compared with levels in the parental cells
(Fig. 3G). Notably, Twist (encoded by
TWIST1 gene), a key transcription factor of EMT that
transcriptionally regulates the expression of E-cadherin,
N-cadherin and vimentin, was significantly enhanced in the
EC109/CDDP cells, ~8-fold increase in resistant cells compared with
parental cells (Fig. 3G). In
contrast, the upregulation of Slug (encoded by SNAI2 gene)
and Snail (encoded by SNAI1 gene), other transcription
factors of EMT, was only ~1.6-fold and 1.4-fold, respectively
(Fig. 3G), much lower than Twist
(~8-fold). Expression changes in Twist, E-cadherin and vimentin
were further validated; the protein levels were similar, as
determined by western blotting (Fig.
3H). In addition, matrix metalloproteinase (MMP)7 and MMP9
(encoded by MMP7 and MMP9 genes, respectively), two
core MMPs, are positively correlated with invasion and were also
markedly upregulated in CDDP-resistant cells (Fig. 3G). Expression of Claudin1 (encoded by
CLDN1 gene), another important epithelial-related factor,
was also markedly decreased in CDDP-resistant cells (Fig. 3G). Taken together, these data
demonstrated an EMT phenotypic conversion in CDDP-resistant
EC109/CDDP cells, which was largely in a Twist-dependent
manner.
Downregulation of MUTYH contributes to
EMT via enhancement of Twist in resistant ESCC cells
Subsequently, it was further assessed whether there
was an association between MUTYH downregulation and
induction of EMT in resistant cells. The functional involvement of
MUTYH in EMT was studied by overexpressing MUTYH in
resistant cells. As shown in Fig. 4A,
RT-qPCR results showed that ectopic expression of MUTYH
prominently suppressed the mRNA level of Twist in EC109/CDDP cells
by ~5-fold, suggesting a role of MUTYH in the
transcriptional regulation of Twist. Correspondingly, cells
transfected with the MUTYH expression plasmid demonstrated a
significant induction of E-cadherin (~1.4-fold) and Claudin-1
(~2.5-fold), and a pronounced suppression of N-cadherin (~5-fold),
vimentin (~5-fold), MMP7 (~3-fold) and MMP9 (~17-fold). Similar to
the mRNA levels, restoration of MUTYH expression in
resistant cells significantly elevated the protein level of
E-cadherin and caused a reduction in the protein expression of
vimentin and Twist to different levels (Fig. 4B).
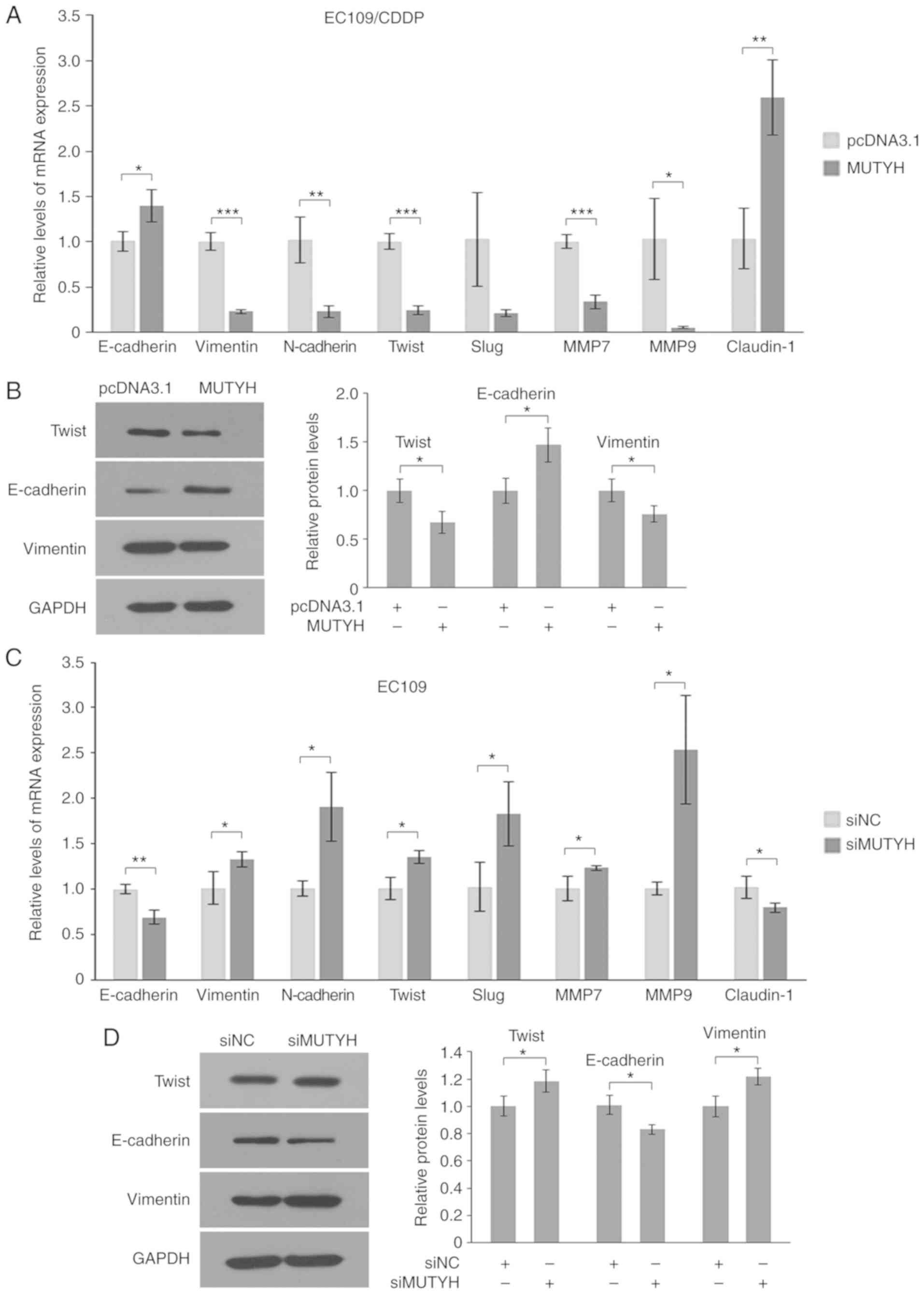 | Figure 4.MUTYH downregulation
contributes to promotion of Twist expression and EMT. (A and B)
Effect of MUTYH overexpression (MUTYH) on Twist and
other EMT-associated markers in EC109/CDDP cells. RT-qPCR (A) and
western blot analysis (B) of mRNA and protein levels, respectively.
Protein quantification for Twist, E-cadherin and vimentin is shown.
Results are the mean ± SD of three independent experiments.
*P<0.05, **P<0.01 and ***P<0.001 vs. the EC109/CDDP cells
transfected with pcDNA3.1, respectively. (C and D) Effect of
MUTYH knockdown on Twist and other EMT-associated markers in
EC109 cells. RT-qPCR (C) and western blot analysis (D) of mRNA and
protein levels, respectively. Protein quantification for Twist,
E-cadherin and vimentin is shown. Results are the mean ± SD of
three independent experiments. *P<0.05 and **P<0.01 vs. the
EC109 cells transfected with siNC, respectively. MTUYH, MutY
homolog; CDDP, cisplatin; siMUTYH, MUTYH siRNA; siNC,
negative control siRNA. |
Furthermore, it was analyzed whether knockdown of
MUTYH led to induction of EMT in the parental cells. As
shown in Fig. 4C, following
MUTYH knockdown, the parental cells presented an EMT
phenotypic conversion as demonstrated by induction of Twist,
N-cadherin, vimentin, MMP7 and MMP9, as well as the inhibition of
E-cadherin and Claudin-1. Concordantly, the expression changes of
these genes in response to depletion of MUTYH were further
confirmed by their protein levels, as observed in Fig. 4D. Therefore, these data suggested that
downregulation of MUTYH contributed to induction of EMT by
enhancing Twist and subsequently regulating its downstream targets,
including E-cadherin, N-cadherin and vimentin in CDDP-resistant
EC109/CDDP cells.
Downregulation of MUTYH blocks Twist
degradation via reduced ROS and proteasome activity in resistant
cells
Due to the essential role of Twist in EMT mediated
by downregulation of MUTYH, we aimed to ascertain whether
MUTYH affects the degradation of Twist in resistant cells.
As MUTYH is associated with ROS and previous studies have
found that ROS exert a regulatory effect on proteasome activity
(24,25), the present study aimed to investigate
whether MUTYH affects the proteasomal degradation of Twist,
which also contributed to an increase in Twist in EC109/CDDP cells.
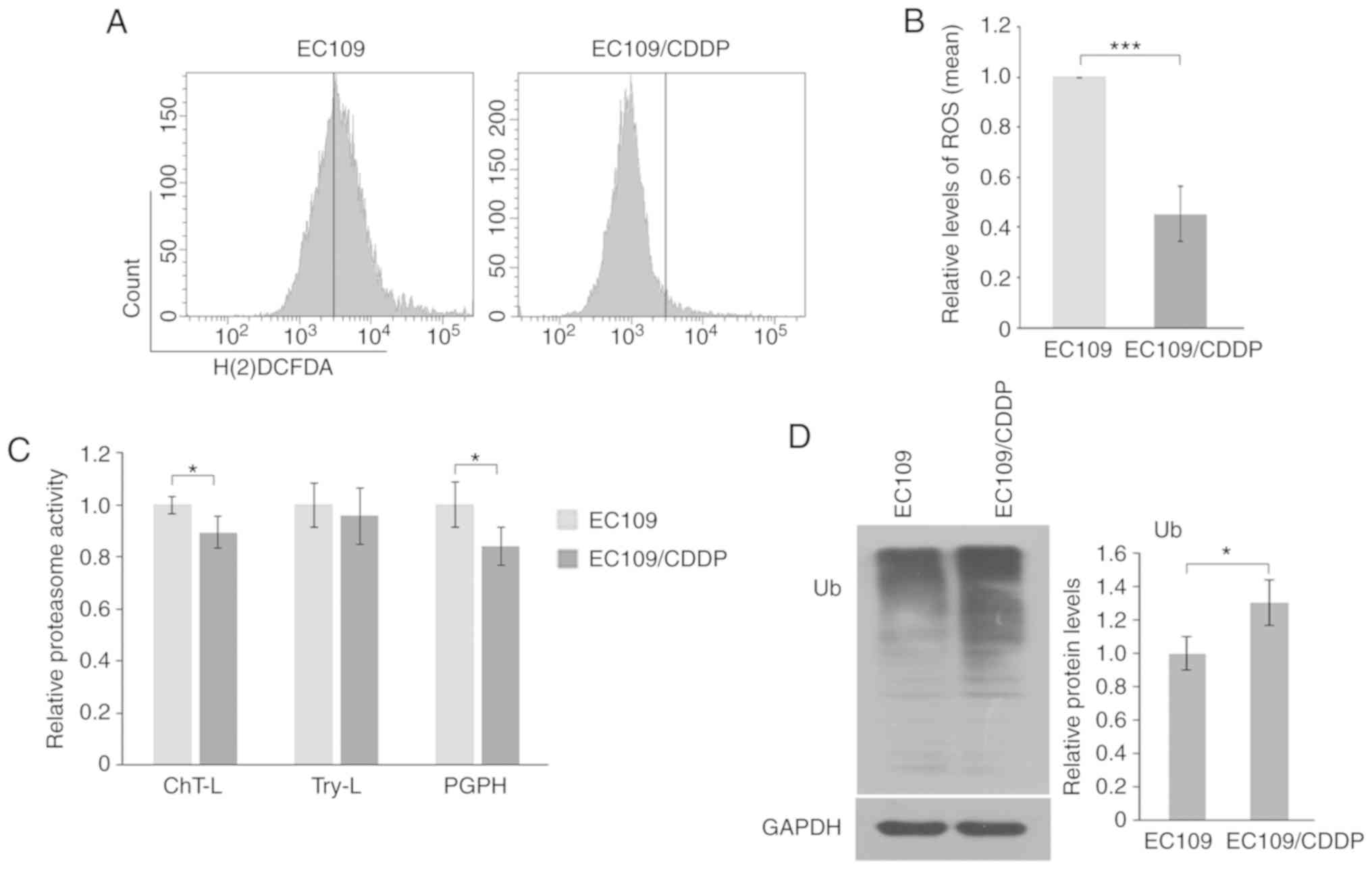
The results in Fig. 5A and B
demonstrated lower levels of ROS in resistant cells compared with
parental cells. Additionally, a lower level of proteasome activity
was identified, as demonstrated by reduced ChT-L and PGPH
activities (Fig. 5C) as well as
increased ubiquitinated proteins (Fig.
5D) in EC109/CDDP cells, suggesting that a blockade of
proteasomal degradation of Twist occurred in resistant cells.
The effect of MUTYH on ROS and proteasome
activity was additionally investigated. As shown in Fig. 6A and B, ectopic expression of
MUTYH in resistant cells led to an increase in the level of
ROS. Furthermore, as a consequence of MUTYH overexpression,
the resistant cells demonstrated downregulation of ubiquitinated
proteins, suggesting that MUTYH suppression attenuated
proteasomal degradation of proteins, including Twist (Fig. 6C).
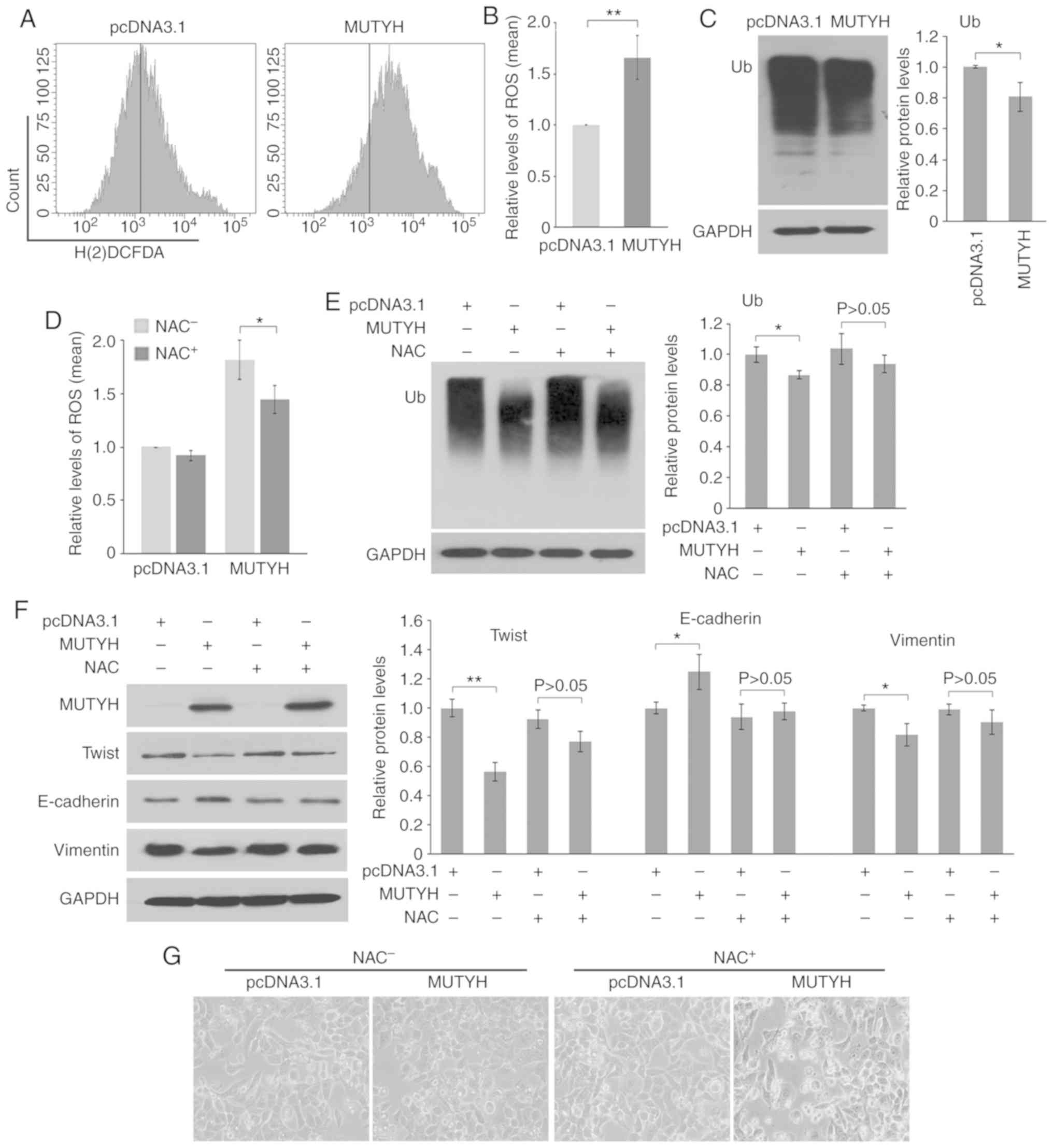 | Figure 6.Downregulation of MUTYH
attenuates Twist degradation via ROS in resistant cells. (A and B)
MUTYH overexpression (MUTYH) elevated ROS generation
in EC109/CDDP cells as detected by flow cytometry. (C) Western blot
analysis of polyubiquitinated proteins (Ub) in response to
MUTYH overexpression in EC109/CDDP cells. GAPDH served as
the loading control. Protein quantification for Ub is shown. In B
and C, the results are the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 vs. the EC109/CDDP cells
transfected with pcDNA3.1, respectively. (D) The antioxidant NAC
reduced ROS generation induced by MUTYH overexpression in
EC109/CDDP cells. Results are the mean ± SD of three independent
experiments. *P<0.05 vs. the NAC-untreated (NAC−)
EC109/CDDP cells transfected with MUTYH. (E) Addition of NAC
significantly rescued downregulation of polyubiquitinated proteins
induced by MUTYH overexpression in EC109/CDDP cells. (F and
G) Impact of NAC on EMT under conditions where MUTYH was
overexpressed in resistant cells. Western blot analysis of Twist,
E-cadherin and vimentin. (F) Morphologic analysis. (G) In E and F,
protein quantification for Ub, Twist, E-cadherin and vimentin is
shown. Results are the mean ± SD of three independent experiments.
*P<0.05, **P<0.01 vs. the EC109/CDDP cells transfected with
pcDNA3.1, respectively. P>0.05 vs. the EC109/CDDP cells
simultaneously treated with NAC and pcDNA3.1 transfection.
MTUYH, MutY homolog; ROS, reactive oxygen species; NAC,
N-acetylcysteine; CDDP, cisplatin; EMT,
epithelial-mesenchymal transition. |
To determine whether MUTYH is involved in EMT
via modulation of ROS, NAC, an antioxidant agent, was used. As
shown in Fig. 6D, NAC significantly
reduced the amount of ROS induced by overexpression of
MUTYH. Furthermore, following the blocking of ROS generation
by NAC, decreases in ubiquitinated proteins (Fig. 6E) and Twist (Fig. 6F) as well as changes of EMT markers
(Fig. 6F) in response to
overexpression of MUTYH, were predominantly rescued under
conditions where ROS was blocked by NAC in EC109/CDDP cells.
Correspondingly, ectopic expression of MUTYH led to fewer
morphological characteristics of EMT in resistant cells while this
reversing effect was markedly abolished when scavenging of ROS
occurred from the addition of NAC (Fig.
6G). Therefore, the present data demonstrated that
downregulation of MUTYH was able to reduce the degradation
of Twist through ROS, which could function together with the
increase in Twist transcription to accelerate EMT.
Discussion
MutY homolog (MTUYH) was initially thought to
play a prominent role in the BER system of oxidized DNA bases in
response to reactive oxygen species (ROS). Notably, recent studies
have identified that MTUYH is associated with tumorigenesis
or development, including in esophageal carcinoma. In the present
study, the exact role of MTUYH in the cisplatin (CDDP) resistance
of esophageal squamous cell carcinoma (ESCC) was further
investigated. Based on the establishment of a CDDP-resistant cell
model EC109/CDDP, MUTYH was initially identified to be
downregulated in resistant ESCC cells. It was identified for the
first time, to the best of our knowledge, that downregulation of
MUTYH contributed to CDDP resistance of ESCC. Further
experiments revealed that downregulation of MUTYH stimulated
EMT by enhancing both mRNA and protein levels of Twist in
CDDP-resistant EC109/CDDP cells; moreover, downregulation of
MUTYH contributed to low ROS levels and subsequently
inhibited proteasome activity, which in turn led to suppression of
Twist degradation. The present data suggested that, as a
consequence of these events, a low level of MUTYH might, at
least partially, confer Twist-mediated EMT and drug resistance.
Although CDDP alone or in combination with other
chemotherapeutic drugs is recommended as an effective front-line
strategy to treat advanced ESCC, occurrence of resistance to CDDP
is the main challenge in chemotherapy. The CDDP-resistance
mechanisms are multi-factorial and may be unique to different types
of cancer (4,5). Further understanding concerning the
CDDP-resistance mechanisms at a molecular level remains a critical
goal for cancer therapy. In the present study, a CDDP-resistant
ESCC cell line EC109/CDDP was established from parental EC109
cells. The CDDP-resistant cell line was characterized with
decreased sensitivity to CDDP and increased EMT phenotypes compared
with parental cells. Notably, a significant downregulation of
MUTYH was observed in resistant cells. The causal
association between MUTYH downregulation and CDDP-resistance
acquisition was demonstrated by transfecting an expression vector
or RNA interference. Overexpression of MUTYH restored CDDP
sensitivity, while MUTYH inhibition made cells resistant to
CDDP. To the best of our knowledge, this is the first study to
demonstrate that downregulation of MUTYH contributed to
acquisition of CDDP-resistance in ESCC.
Epithelial-mesenchymal transition (EMT) has been now
widely recognized as a vital process that contributes to cancer
progression and drug resistance (22,23). It
has been identified that EMT is executed by a relatively small
number of master regulators, mainly including the transcription
factors Twist, Snail and Slug (22,23).
Accumulating evidence has demonstrated that EMT activation confers
multi-drug-resistance to cancer cells (22,23,26).
EMT-induced multidrug resistance involves various mechanisms,
including a lower level of proliferation, increased expression of
anti-apoptotic proteins as well as ATP-binding cassette
transporters (23,26,27). In
the present study, EC109/CDDP cells underwent EMT, as characterized
by the changes in cell morphology and the expression of EMT
markers, displaying upregulation of two mesenchymal markers,
N-cadherin and Vimentin, and reduction of an epithelial marker
E-cadherin. Notably, the master regulator of EMT, Twist, also
presented a marked increase in expression in CDDP-resistant cells,
demonstrating that the occurrence of EMT in EC109/CDDP cells was
largely in a Twist-dependent manner. Based on these events
mentioned, it was hypothesized that downregulation of MUTYH
was associated with Twist-mediated EMT and in turn conferred CDDP
resistance to ESCC. In support of this hypothesis, MUTYH
suppression enhanced the Twist level and stimulated cells to
acquire a higher potential for EMT in parental cells; conversely,
restoring MUTYH in CDDP-resistant cells reduced the Twist
level and inhibited the EMT process.
Moreover, in addition to transcriptionally
regulating Twist, it was also observed that MUTYH affected
the degradation of Twist. Twist is a labile protein, rapidly
polyubiquitinated and degraded via the ubiquitin-proteasome pathway
(UPP) (28). UPP is regarded as the
primary cytosolic proteolytic machinery for protein degradation
(29,30). In the process of UPP, multiple
ubiquitin molecules firstly tag substrate proteins, which are then
degraded by the 26S proteasome. Therefore, the UPP is a major
pathway responsible for the protein quality control mechanism
(29,30). Changes in UPP capacity of degrading
proteins can occur in response to cellular environmental insults,
including oxidative stress (24,25). It
has been demonstrated that exposure to various forms of oxidative
stress leads to increased intracellular protein degradation, which
results from the elevation of the rate of ubiquitin conjugation by
increasing substrate availability and enhancement of activities of
ubiquitin-conjugating enzymes (24,25,31),
thereby elevating ubiquitination and degradation capacities. In the
present study, it was determined that compared with parental cells,
resistant E109/CDDP cells presented a lower level of ROS and
reduced activity of UPP, which led to the hypothesis that
MUTYH modulates ROS and UPP. The present data demonstrated
that MUTYH downregulation contributed to reduced ROS and UPP
activity, thereby leading to an inhibition of Twist degradation.
The addition of an antioxidant agent NAC significantly abolished
the effect of MUTYH on UPP and the EMT phenotype, further
validating this hypothesis. Nevertheless, the underlying mechanisms
of these signaling pathways require further investigation.
In summary, the present study provided novel insight
into the role of MUTYH in the acquisition of resistance to
CDDP in ESCC cells and suggested a novel mechanism in that
downregulation of MUTYH conferred EMT-mediated drug
resistance via an increased level of Twist. Regulation of the
signaling response may provide a practical therapeutic strategy to
strengthen the effect of CDDP-based chemotherapy in ESCC cells.
Acknowledgements
Not applicable
Funding
This study was supported by the National Natural
Science Foundation of China (81603140), Shandong Provincial Natural
Science Foundation, China (2015GSF118037, ZR2017BH108,
ZR2017PH033), Jinan Science and Technology Development Program
(201704087) and the Jining Key Research and Development Plan
(2018SMNS001).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
CP, YL and YG designed the research. YG, YL, YJ, SW,
DG, LZ and ZL performed the experiments. FK, NL and SW contributed
new reagents and analytic tools in conducting the experiments. YG
and YJ analyzed the data. YG and YL wrote the paper. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
Murphy G, McCormack V, Abedi-Ardekani B,
Arnold M, Camargo MC, Dar NA, Dawsey SM, Etemadi A, Fitzgerald RC,
Fleischer DE, et al: International cancer seminars: A focus on
esophageal squamous cell carcinoma. Ann Oncol. 28:2086–2093. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu J, Wei Z, Zhang J, Hu W, Ma Z and Liu
Q: Which factors are associated with extremely short-term survival
after surgery in patients with esophageal squamous cell carcinoma?
Asia Pac J Clin Oncol. 12:308–313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baba Y, Watanabe M, Yoshida N and Baba H:
Neoadjuvant treatment for esophageal squamous cell carcinoma. World
J Gastrointest Oncol. 6:121–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kartalou M and Essigmann JM: Mechanisms of
resistance to cisplatin. Mutat Res. 478:23–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Koning P, Neijt JP, Jennekens FG and
Gispen WH: Evaluation of cis-diamminedichloroplatinum (II)
(cisplatin) neurotoxicity in rats. Toxicol Appl Pharmacol.
89:81–87. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marullo R, Werner E, Degtyareva N, Moore
B, Altavilla G, Ramalingam SS and Doetsch PW: Cisplatin induces a
mitochondrial-ROS response that contributes to cytotoxicity
depending on mitochondrial redox status and bioenergetic functions.
PLoS One. 8:e811622013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Santos NA, Catão CS, Martins NM, Curti C,
Bianchi ML and Santos AC: Cisplatin-induced nephrotoxicity is
associated with oxidative stress, redox state unbalance, impairment
of energetic metabolism and apoptosis in rat kidney mitochondria.
Arch Toxicol. 81:495–504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang Y, Guo C, Vasko MR and Kelley MR:
Implications of apurinic/ apyrimidinic endonuclease in reactive
oxygen signaling response after cisplatin treatment of dorsal root
ganglion neurons. Cancer Res. 68:6425–6434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Markkanen E, Dorn J and Hübscher U: MUTYH
DNA glycosylase: The rationale for removing undamaged bases from
the DNA. Front Genet. 4:182013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruggieri V, Pin E, Russo MT, Barone F,
Degan P, Sanchez M, Quaia M, Minoprio A, Turco E, Mazzei F, et al:
Loss of MUTYH function in human cells leads to accumulation of
oxidative damage and genetic instability. Oncogene. 32:4500–4508.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nascimento EFR, Ribeiro ML, Magro DO,
Carvalho J, Kanno DT, Martinez CAR and Coy CSR: Tissue expresion of
the genes MUTYH and OGG1 in patients with sporadic colorectal
cancer. Arq Bras Cir Dig. 30:98–102. 2017.(In English, Portuguese).
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong J, Wang X, Yu Y, Yan X and Cui JW:
Association of base excision repair gene polymorphisms with the
response to chemotherapy in advanced non-small cell lung cancer.
Chin Med J (Engl). 131:1904–1908. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Santos LS, Branco SC, Silva SN, Azevedo
AP, Gil OM, Manita I, Ferreira TC, Limbert E, Rueff J and Gaspar
JF: Polymorphisms in base excision repair genes and thyroid cancer
risk. Oncol Rep. 28:1859–1868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kong F, Han XY, Luan Y, Qi TG, Sun C, Wang
J, Hou HY, Jiang YH, Zhao JJ and Cheng GH: MUTYH Association with
esophageal adenocarcinoma in a Han Chinese population. Asian Pac J
Cancer Prev. 14:6411–6413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sliwinski T, Markiewicz L, Rusin P,
Pietruszewska W, Olszewski J, Morawiec-Sztandera A, Mlynarski W and
Majsterek I: Polymorphisms of the DNA base excision repair gene
MUTYH in head and neck cancer. Exp Oncol. 31:57–59. 2009.PubMed/NCBI
|
|
17
|
Przybylowska K, Kabzinski J, Sygut A,
Dziki L, Dziki A and Majsterek I: An association selected
polymorphisms of XRCC1, OGG1 and MUTYH gene and the level of
efficiency oxidative DNA damage repair with a risk of colorectal
cancer. Mutat Res. 745-746:6–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Al-Tassan N, Chmiel NH, Maynard J, Fleming
N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS,
Sampson JR and Cheadle JP: Inherited variants of MYH associated
with somatic G:C->T:A mutations in colorectal tumors. Nat Genet.
30:227–232. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Slyskova J, Naccarati A, Pardini B,
Polakova V, Vodickova L, Smerhovsky Z, Levy M, Lipska L, Liska V
and Vodicka P: Differences in nucleotide excision repair capacity
between newly diagnosed colorectal cancer patients and healthy
controls. Mutagenesis. 27:225–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu YQ, Wang SK, Xu QQ, Yuan HQ, Guo YX,
Wang Q, Kong F, Lin ZM, Sun DQ, Wang RM and Lou HX:
Acetyl-11-keto-β-boswellic acid suppresses docetaxel-resistant
prostate cancer cells in vitro and in vivo by blocking Akt and
Stat3 signaling, thus suppressing chemoresistant stem cell-like
properties. Acta Pharmacol Sin. 40:689–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith BN and Bhowmick NA: Role of EMT in
metastasis and therapy resistance. J Clin Med. 5:E172016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shang F and Taylor A: Ubiquitin–proteasome
pathway and cellular responses to oxidative stress. Free Radic Biol
Med. 51:5–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dudek EJ, Shang F, Valverde P, Liu Q,
Hobbs M and Taylor A: Selectivity of the ubiquitin pathway for
oxidatively modified proteins: Relevance to protein precipitation
diseases. FASEB J. 19:1707–1709. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Díaz VM, Viñas-Castells R and García de
Herreros A: Regulation of the protein stability of EMT
transcription factors. Cell Adh Migr. 8:418–428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldberg AL: Protein degradation and
protection against misfolded or damaged proteins. Nature.
426:895–899. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Glickman MH and Ciechanover A: The
ubiquitin–proteasome proteolytic pathway: Destruction for the sake
of construction. Physiol Rev. 82:373–428. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shang F, Gong X and Taylor A: Activity of
ubiquitin-dependent pathway in response to oxidative stress.
Ubiquitin-activating enzyme is transiently upregulated. J Biol
Chem. 272:23086–23093. 1997. View Article : Google Scholar : PubMed/NCBI
|