Colorectal cancer (CRC) is the fourth most common
type of malignancy after breast, lung and prostate cancer,
accounting for 49,190 deaths annually in the USA alone (1). Numerous risk factors have been
identified, such as age, a family history of CRC, ethnic background
(individuals of African descent), a high carbohydrate diet, poor
physical activity, obesity and metabolic syndrome, smoking and
alcohol abuse (2–5). Originating from epithelial cells at the
base of intestinal crypts, the current model of carcinogenesis is
that of the adenoma-carcinoma sequence, first described in the
1990s by Bert Vogelstein and Kenneth Kinzler [Fearon and Vogelstein
(6) and Kinzler and Vogelstein
(7)]. This model proposed a
sequential transformation of the normal colorectal epithelium to an
adenoma that could further transform into an invasive and
metastatic tumor (carcinoma). Mutations in key regions begin to
aggregate, turning normal mucosa to an early adenoma and, after a
certain point of no return, the accumulated genetic alterations
transform it to a carcinoma. Chromosomal instability,
microsatellite instability, CpG island methylation and activating
oncogenic mutations in genes such as adenomatous polyposis
coli (APC), K-ras and p53 are found to
play a key role in this sequence (7–9).
However, as it was found thereafter, the Vogelstein
model could explain 90–95% of CRC cases. The remaining 5–10% of
cases were found to be germline-inherited cancers, such as familial
adenomatous polyposis (FAP) and hereditary non-polyposis colorectal
cancer (HNPCC). Notably, 2–3% of all CRC cases are associated with
pre-existing inflammation and are referred to as colitis-associated
cancer (CAC) (10). In these cases,
the activation of nuclear factor (NF)-κB signaling in
tumor-associated macrophages (TAMs) leads to the indirect
activation of signal transducer and activator of transcription
(STAT)3 in pre-malignant intestinal epithelial cells (IECs)
(11,12). Even though epidemiologic studies have
witnessed a shift towards younger age groups over the past decade,
the age group most commonly affected remains that of the
middle-aged (>50 years of age) (13), a finding closely related to the
Vogelstein model (the accumulation of mutations) (14,15).
Moreover, although CRC is not considered a
sex-related malignancy per se, sex differences in incidence rates
do exist (16–21). As far as the male population is
considered, cancer incidence exhibits two peaks; the first one
appears before the age of 35 and the second after the age of 55. On
the other hand, in the female population, there is a single peak
trend, between 35 and 54 years of age (22,23).
Taking into consideration that physical activity performed before
or after cancer diagnosis is related to a reduced mortality risk
among CRC survivors (24) and is
therefore recommended, along with the high prevalence of the
use/misuse and abuse of anabolic agents with hormonal activity,
such as testosterone, dihydrotestosterone (DHT), finasteride,
insulin, insulin-like growth factor-1 (IGF-1) and growth hormone
(GH) in the sports community over the past decades (25), a great concern of any possible
carcinogenic properties or synergistic effects of the anabolic
agents with the already well-studied and identified CRC risk
factors has emerged (5). Nonetheless,
the data are not consistent: An increasing body of evidence
indicates that adequate levels of vitamin D, structurally related
to a number of anabolic agents, can indeed protect against
carcinogenesis via genomic and non-genomic mechanisms. In addition,
the general population experiences uncontrolled multi-chemical
exposure from several different sources at doses around or well
below regulatory limits (pesticides, food additives, lifestyle
products components) (5,15,26) that
can contribute to genotoxicity, endocrine disruption, target organ
toxicity (3,4,27–29) by affecting systemic mechanistic
pathways, such as oxidative stress and cell aging (14,30–32).
These, along with the finding that human colorectal adenocarcinomas
express specific steroid hormone receptors (33–40), has
sparked the interest of the scientific community to unveil any
possible pathogenetic mechanisms. Nonetheless, an increasing body
of evidence indicates that adequate levels of vitamin D,
structurally related to a number of anabolic agents, can indeed
protect against carcinogenesis via genomic and non-genomic
mechanisms.
An androgen is considered any molecule capable of
inducing and maintaining the male phenotype in an organism (male
primary and secondary sexual characteristics and fertility) and
taking part in the universal outgrowth of the musculoskeletal
system and the anabolic shift of the metabolic status (41). Generally, the androgen-producing
endocrine glands are able to synthesize five androgens via a sole
pathway: Testosterone, dehydroepiandrosterone sulfate (DHEAS),
dehydroepiandrosterone (DHEA), androstenedione and androstenediol,
the latter of which has both androgenic and estrogenic properties.
The molecules that prevail in this category are testosterone (the
principal androgen in mammals) and DHT (potent metabolite of
testosterones). In fact, they are the only androgens with direct
androgenic activity. Other molecules, such as DHEA, due to their
inferior potency, have received less attention. In an adult male
organism, testosterone is primarily produced by Leydig cells in the
testes. In addition, the extra-gonadal synthesis of testosterone
and DHT by the adrenal testosterone precursor, DHEA, also occurs
(42). Although adrenal androgens
represent a minor fraction of the circulating testosterone for an
adult male with an intact androgen biosynthesis cascade, they can
be the main androgens in a female or a pre-puberty male (43). In the majority of cases, the classical
mode of action of androgen superfamily is mediated by the androgen
receptor.
Anabolic androogenic steroids (AAS) are used in the
treatment of several disorders, such as hypogonadism, cachexia of
various etiologies, hypercalcemia, hypercalciuria, in oncology as a
supportive treatment and other chronic diseases (44). Since the early 1930s, AAS have been
extensively used by amateur and professional athletes and the
general public for the improvement of their physical condition and
athletic performance (45–49). When used for ergogenic or recreational
purposes, the dose levels are usually 5ement of physical condition
and athletic performance (47,50,51).
At such supraphysiological levels, AAS can cause a number of severe
side-effects, including liver dysfunction, renal disorders,
cardiotoxicity and potentially, stroke (52). In addition, anti-androgen therapy is
also relatively common, having a wide variety of applications
ranging from severe conditions (such as the treatment of prostate
cancer and polycystic ovary syndrome) to more benign or even
aesthetic conditions (such as acne and male pattern hair-loss).
Thus, given their wide use in modern society, it is reasonable to
scrutinize whether misbalanced androgen levels may possibly have a
direct or indirect connection with CRC (41). In the following paragraphs, the
current data regarding androgens and CRC will be presented
according to the clinical significance of the studied molecule.
Testosterone, being the most clinically important
androgen, has attracted scientific interest from as early as 1986.
At that time, studies advocated that androgens played a protective
role against CRC. In detail, Izbicki et al (1986) conducted
experiments on 40 male rats. They found that chemical castration
increased colonic tumor incidence, while testosterone
administration following surgical castration produced a borderline
statistically significant reduction in tumor incidence
(P<0.053), particularly in the right colon (22). In recent years, hypotestosteronemia
(defined as levels of testosterone <11 nmol/l or 320 ng/dl) was
found to contribute to the development of CRC (53). Further data indicating the protective
role of testosterone came from studies on patients who received
androgen deprivation treatment for prostate cancer. In detail, it
was found that the group with the higher risk of developing CRC was
that of the orchiectomized male patients followed by patients
receiving gonadotropin-releasing hormone (GnRH) agonist therapy
(particularly if the treatment was prolonged) (54). On the contrary, there is evidence to
suggest that androgens may act as promoters of colon carcinogenesis
(2,55–57).
Experiments using genetically modified mice, found that in
orchiectomized males, the tumor load was lower when there was no
administration of male hormone replacement therapy (2). Other researchers have indicated that, at
an early stage, androgens may play an active role in the transition
from adenoma to carcinoma (6). In
contrast to both previous statements, a large study on 4,165 males
aged 70–88 years demonstrated that increased testosterone levels
were associated neither with an increased nor with a decreased risk
of colon cancer risk (58). The same
conclusion was achieved by a prospective study of 8,771 males and
females from the general population of Denmark, who were
followed-up for >30 years (59).
Whichever the case may be, androgens can be related to CRC either
through direct mechanisms (mediated through androgen receptors),
indirect mechanisms (smoking and alcohol habits, metabolic syndrome
and type 2 diabetes mellitus, altered gut microbiota and increased
stress hormones) or even a combination of both (60). Worthy of mention is the fact that
regardless of the nature of the study (in vitro, in vivo or
epidemiological), a common ground has yet to be found given the
opposing data derived from these studies. Thus, in order to reach a
consensus on the possible carcinogenetic properties of
testosterones and the circumstances under which they appear, and to
provide the grounds for a safe comparison between studies, research
teams will have to adopt a common methodology as the basis of their
experiments (27).
Despite being less extensively studied, opposing
results have been obtained on this less potent molecule as well. In
an observational study on 170 individuals, the plasma levels of
dehydroepiandrosterone sulfate were shown to be inversely
associated with the risk of developing colon cancer (with a
borderline statistical significance) (61). Moreover, another study proposed that
DHEA strongly blunts serum deprivation-induced apoptosis. The
anti-apoptotic effects of DHEA have been found to be completely
reversed by testosterone through the blockade of DHEA receptors,
thereby antagonizing its actions (62).
There are namely two modes of action, direct action
and indirect action. These are discussed below.
Steroid hormones, and thus androgens, exert their
effects mainly through interactions with specific receptor proteins
(37,56). The presence of ARs in human colonic
tumors was first shown by Alford et al (34). The gene encoding the AR is located on
the X chromosome. It contains two polymorphic trinucleotide repeat
segments that encode polyglutamine (CAG) and polyglycine (GGC)
(normally, ranging from 6 to 39 repeats). Surprisingly, only the
number of CAG repeats has been found to be associated with
misbalanced androgen levels. In fact, studies have demonstrated an
inverse association between the number of repeats and the risk of
prostate cancer (63,64). According to Hoque et al, the
number of these repeats is gradually reduced in prostate cancers
(65). Existing data advocate that
fewer CAG repeats result in a higher transcription activity of AR,
a finding positively associated with prostate cancer (66,67). In
parallel, a greater number of repeats are associated with increased
serum androgen levels, indicating a protective role of these
against CRC (61,66).
The above-mentioned results are in discordance with
a following larger scale study of 1,798 CRC cases and 1,810
controls. In that study, the implication of the AR CAG
repeat polymorphism in colorectal cancer prognosis was investigated
for the first time. Of note, no association was found between the
above-mentioned polymorphism and CRC overall or the
disease-specific survival rate. As outlined by the authors, the
genotyping error rate calculated from the duplicated samples was
relatively high for the AR CAG repeat polymorphism (69). A genotyping error occurs when the
genotype of an individual observed in the laboratory does not
correspond to the individual's true genotype (70). The causes could be categorized as a
variation of the DNA sequence, the low quantity or quality of DNA,
biochemical artifacts (low quality reagents, poor equipment
precision or reliability, Taq polymerase errors, the lack of
specificity, electrophoresis artifact) and human error (sample
manipulation, experimental error, data handling) (71). Since the main cause of genotyping
error is human error (71), Rudolph
et al concluded that further studies are required to extract
solid results (69).
It should be pointed out that the trans-activating
function of AR is dependent on its ligands, the androgens. The
receptor participation in controlling cellular differentiation and
proliferation in hormone-dependent tissues does not always occur in
the same manner (75). Catalano et
al shed some light onto this phenomenon. They mentioned two
isoforms of the AR; androgen receptor A (AR-A) (87 kDa) and
androgen receptor B (AR-B) (110 kDa). In the healthy colonic
mucosa, both receptors are present; however, in the neoplastic
colonic mucosa, only AR-A could be detected. The loss of expression
of AR-B and the continuous expression of AR-A was proposed to
indicate a loss of cell differentiation (76).
Another category of AR was also discovered; the
membrane androgen receptors (mARs). Normally, the actions of the
androgens are mediated through intracellular receptors (iARs). mARs
seem to mediate opposite actions than iARs, inducing tumor
regression (77). Furthermore, the
affinity and selectivity of mARs differ among specific androgens
(78). Through this receptor,
testosterone exerts pro-apoptotic effects in both prostate and
colon cancer cells. In human colon cancer cell lines, the
activation of the ligand-bound androgen receptor suppresses the
transcription of β-catenin. Consequently, there is a
decreased expression of β-catenin target oncogenes,
including cyclin D1 (79).
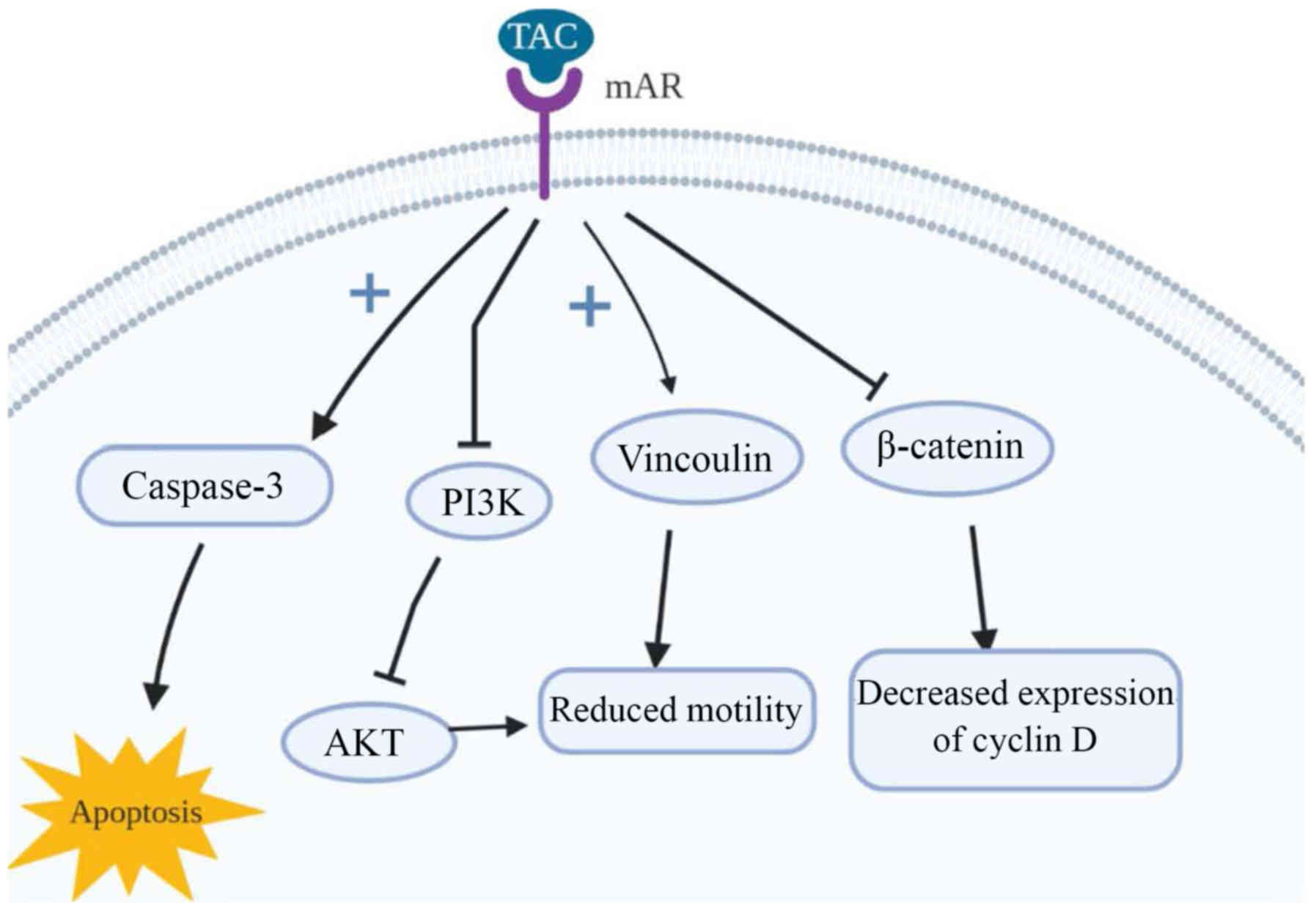
The long-term activation of mAR by
testosterone-albumin conjugated (TAC) treatment has also been
linked to the dephosphorylation of protein Kinase B (Akt) both
in vitro and in vivo (77). It is closely connected with the
invasiveness of colon cancer cells in response to a variety of
stimuli [heregulin, P21 (RAC1) activated kinase 1 (PAK1), Sprouty-2
etc.] (80–82), a finding supported through the
examination of the upstream regulators of Akt, in particular PI3K.
Upon long-term TAC treatment, it is dephosphorylated, leading to
reduced cell motility in colon cancer and, consequently,
invasiveness. Of note, although testosterone seems to induce p-Akt
downregulation when it binds to mARs, iAR bondage induces p-Akt
upregulation, even within the same cells. The main target of mAR
activation that may regulate cell motility is thought to be
vinculin; a cytoskeletal protein which links integrin adhesion
molecules to actin. The inhibition or silencing of vinculin via the
phosphorylation by specific inhibitors, as is the case with
activation of mARs, largely reverses both actin reorganization and
the inhibition of migration (77). An
illustrated representation of the mode of action of mAR is
presented in Fig. 1.
Upstream in the AR activation pathway, the role of
co-activator-associated arginine methyltransferase 1 (CARM1) is
crucial. CARM1 is a protein with arginine-specific histone
methyltransferase activity. It primarily binds to the histone and
p160 co-activators, leading to the activation of nuclear
receptors, ARs included. Thus, it promotes nuclear receptor
activity and acts as a molecular switch for gene-specific
transcription factors including p53, NF-ĸB, lymphoid
enhancer-binding factor 1 (LEF1)/transcription factor 4 (TCF-4) and
E2Fs (83,84). Taking the above into consideration, it
is clear that the role of CARM1 is of utmost importance for cell
proliferation and survival (85,86). Kim
et al reported the overexpression of CARM1 in CRC specimens,
but a weak expression in normal mucosal cells. They demonstrated
that CARM1 inhibits the p53 response and instead promotes the NF-ĸB
response in Caco2 cells (87).
However, the mechanisms involved remain to be fully clarified
(87,88).
Decreased levels of androgens seem to lead to a net
increase in stress hormone levels, such as cortisol, which affect
the tumor environment (89). The
involvement of the innate immune system in the development of CRC
has been demonstrated as well. The neutrophil count is reduced, as
found by Chuang et al in castrated males, although it can be
restored to normal levels through androgen supplementation
(90). Androgens also seem to affect
the production and excretion of biliary steroids and bile,
compounds that are suggested to act as co-carcinogens (17,38).
Lastly, an induction of insulin resistance following androgen
deprivation therapy (91) has been
linked to increased risk of developing CRC (92). The connection between insulin and CRC
will be further discussed.
Synthetic anabolic agents are categorized into two
categories: Anabolic-androgenic steroids (AAS) and selective
androgen receptor modulators (SARMs) (93). The AAS molecules that have thus far
been approved as therapeutic agents are testosterone,
nortestosterone, dihydrochlormethyltestosterone (DHCMT), metenolon,
metandienone, methyltestosterone, oxandrolone, fluoxymesterone,
stanozolol, formestane, 5on, metandieno (94). SARMS are non-steroidal alternatives to
AAS with the selective activation of the AR in either muscle tissue
or bones (93).
A number of adverse effects of these compounds have
been described when used either as medicine or as doping agents. As
far as AAS's relation to cancer is concerned, a positive
association with hepatocellular carcinoma (95), renal cancer (96–98), soft
tissue carcinoma (99),
adenocarcinoma (100,101) and lymphosarcoma (102) has been found, along with a case
report hypothesizing their involvement in leiomyosarcoma (103). SARMs have also been linked to
prostate cancer (104). However, to
the best of our knowledge, there are currently no data available in
the current literature associating any of these compounds with
CRC.
Insulin is one of the principal anabolic hormones in
the majority of animals since it regulates the metabolism of almost
all key energy points in favor of their synthesis and storage. The
target substances of insulin k are namely carbohydrates, lipids and
proteins. Acting on adipocytes, hepatocytes and muscle cells,
induces and maintains to a certain extent, an anabolic state which
is described by the synthesis of carbohydrates, fatty acids and
proteins, while reducing their degradation. Although a basic
requirement is the prior balance between the circulating levels of
the target substances and the intracellular ones, for a short
period of time, it can overcome the concentration gradient of a
substance and induce its endocytosis (105). The role of insulin in CRC was first
introduced by the observation that obesity was associated with an
increased risk of developing CRC in males. Subsequently,
hyperinsulinemia and insulin resistance were linked to obesity and
CRC development (106). In a large
epidemiological study of almost 25,000 patients with type 2
diabetes mellitus (107) and in a
large meta-analysis of 16 studies (108), a direct association between
long-term insulin therapy and type 2 diabetes mellitus with an
increased risk of developing CRC was found. It has also been
established that CRC survivors with excess amounts of blood insulin
have a greater risk of recurrence (109). By contrast, a large registry-based
study in Connecticut that included 9,395 patients with CRC
(110) and a smaller Norwegian study
of 1,194 hospitalized patients with CRC (111), failed to find an association between
diabetes and CRC-specific death. It must be stated though, that the
latter studies included patients with metastatic CRC as well.
To date, two isoforms of IR have been described,
differing at the short exon 11 (encodes 12 amino acids). The
absence of exon 11 transcripts the IR-A (short isoform), while its
presence the IR-B (long isoform) (115). Existing evidence supports that the
two IR isoforms play different biological roles. IR-A mostly exerts
mitogenic effects and IR-B modulates cell metabolism (116). Abbruzzese et al found a
strong IR expression in adenomas and low-grade adenocarcinomas
(117). The expression of IR-A has
been found in cells that have lost their differentiation, a finding
which is in accordance with the presence of this receptor in cancer
(118,119).
On the other hand, VIRs are thought to contribute to
neovascularization following abduction by elevated insulin levels.
VIRs are frequently found in CRC, particularly in left-sided CRCs,
and they are significantly associated with tumor invasiveness
(120).
From another point of view, insulin resistance in
vascular endothelial cells can promote tumor formation, possibly
through mechanisms involving chronic inflammation (122). This resistance is characteristic of
endothelial dysfunction in obesity and type 2 diabetes (123–126)
and was found to promote tumor development. By contrast, there was
almost no effect of insulin signaling on intestinal carcinogenesis
through epithelial receptors (122).
In order to fully understand the role of insulin in
the tumor cascade, special reference must be made to extracellular
vesicles (EVs). These are vesicles found in the extracellular space
of various cell types. They can be found under normal and
pathological conditions (127,128).
EVs package biologically active content (including proteins, mRNA
and miRNA), which they further transfer to the recipient cells. Due
to their action they are considered as mediators of signaling
cascades (129,130). EVs are known to mediate various
biological cascades relative to cancer, such as the activation of
Wnt signaling and the activation of PI3K/Akt signaling (129).
IGF is a hormone that serves as the mediator of
growth hormone (GH)-stimulated somatic growth, as well as a
mediator of GH-independent anabolic responses in a number of cells
and tissues, while it is also associated with mitogenesis, cell
survival and differentiation (135–142).
In detail, IGF-1 promotes cell cycle progression and inhibits
apoptosis either by triggering other growth factors or by
interacting with pathways which have an established role in
carcinogenesis and cancer promotion (142). Ma et al stated that there is
an increase in IGF-1 levels in patients with CRC (143), while other studies have advocated
the overexpression of IGF-R, as well (144–147).
In accordance with this, several studies have linked elevated
plasma IGF-1 and IGF-1R downstream signaling to an enhanced risk of
colorectal neoplasia and a poor survival (148–152).
Furthermore, Peters et al found that IGF-1 was closely
related to the expression of the proliferation marker Ki-67
(153). Ki-67 is a nuclear protein
that is active only in dividing cells and absent in cells locked in
G0 phase. This is a logical outcome when taking into consideration
that IGF-1 can stimulate the expression of cyclin D1, a molecule
that accelerates the progression of the cell cycle from G1 to S
phase (154). However, there is no
prognostic relevance of Ki-67 in CRC, regardless of the stage of
disease (155,156). Through a reverse line of thought,
octreotide, a molecule that lowers the IGF-1 concentration, has
been shown to attenuate the growth rate of tumor cells in
vivo (157).
A family of six circulating proteins termed
insulin-like growth factor binding proteins (IGFBPs) has been found
to interfere with the action of IGF. Thus, their involvement in CRC
must be investigated, as well. They act either as tumor suppressors
by limiting IGFs activity (158) or
as inhibitors of cancer growth through IGF-independent mechanisms
(159). High IGFBP2 plasma levels
were found by Liou et al to be independently associated with
a reduced overall survival (OS) of patients with CRC (160). By contrast, IGFBP3 has been shown to
be inversely associated with CRC, as its plasma levels were found
to be lower in those patients (143). Notably, IGFBP-3 can either oppose or
enhance the biologic action of IGF-I through direct bondage to
IGF-I or indirectly to IGF-R (161).
In a prospective cohort study of 210 patients with
CRC, IGF-1 expression was shown to be closely associated with tumor
size and the depth of invasion. However, it was stated that a shift
of investigation towards the IGF-1/IGFBP-3 ratio is warranted, as
it better describes the biological effects of IGF-1 (152). A nested case control study of males
in the Physician's Health Study demonstrated an increased risk of
CRC in subjects with high IGF-I levels. The risk was decreased when
high IGFBP-3 levels were measured (143). A study of 460 patients was carried
out to further examine the association between IGF-I and the
IGF-I/IGFBP-3 ratio with colorectal adenomatous polyps. A
statistically significant positive association was found, with
greater odds ratio when the case group was limited to advanced
adenomas. This finding indicates a possible stimulation of
non-advanced adenomas towards advanced adenomas (112).
Closer attention must be paid to the IGF-R, as it
has been stated that it contributes to resistance to cytotoxic
(162), radiation (163) and targeted therapies (164–166).
Indeed, the silencing of the receptor increases the intracellular
drug concentration (such as oxaliplatin and vincristine); an effect
mediated via the PI3K/Akt pathway (167). A progressive increase in
IGF-1R expression occurs in normal colonic mucosa, while it
transits to adenomatous, as well as in the transition from
adenomatous to carcinomatous tissue (147). Peters et al have confirmed a
strong expression of the IGF-1 receptor in >99% of all CRC cell
lines of their experiments (153).
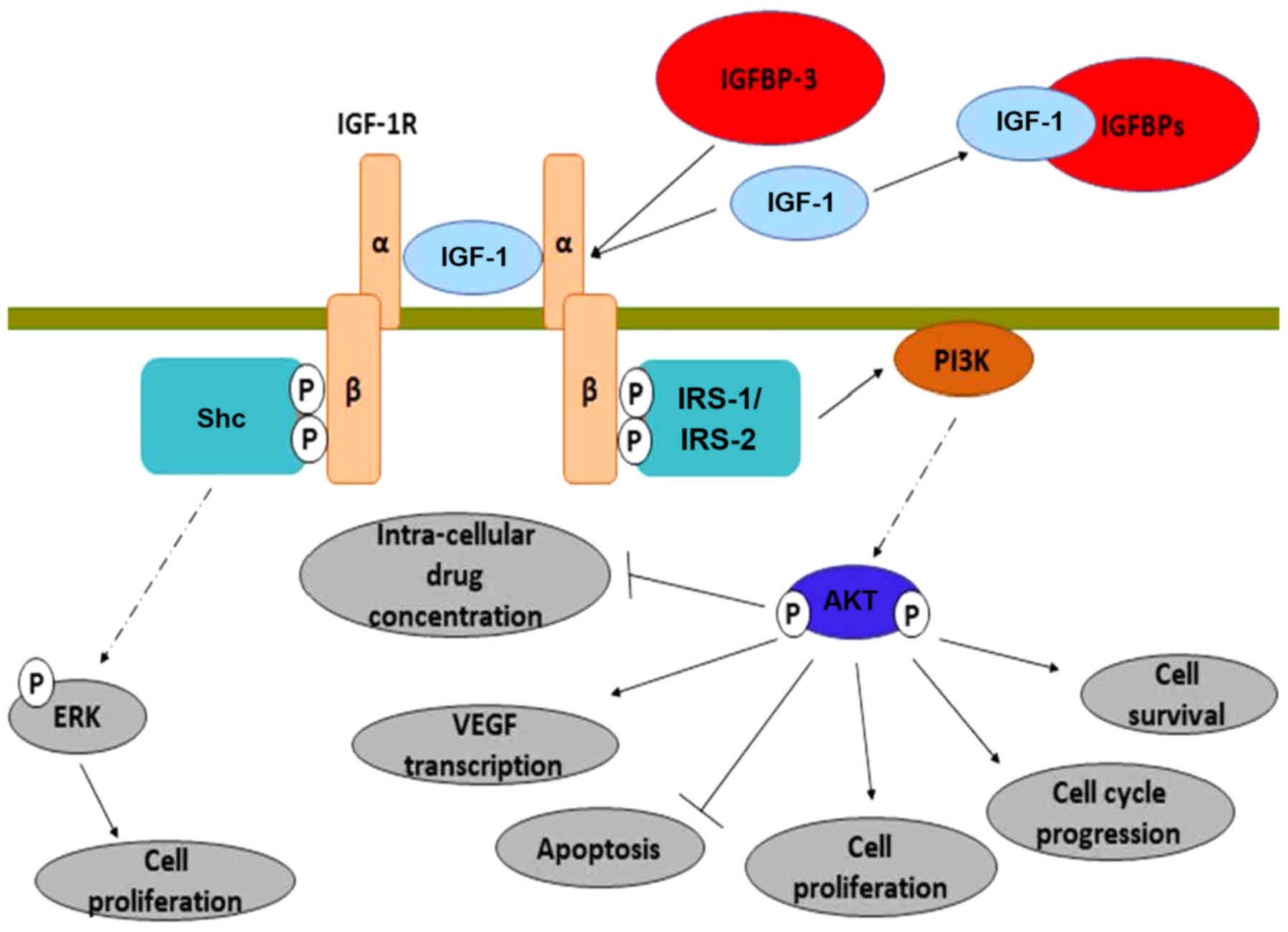
Following its activation, IGF-R induces multiple
intracellular mechanisms, as shown in Fig. 2. It induces the transcription of the
vascular endothelial growth factor (VEGF) gene (168,169),
upregulates the anti-apoptotic protein, Bcl-xL (170) and inhibits the action of β-catenin
(through PI3K/Akt activation) (171). From another perspective, Nahor et
al demonstrated that the tumor-suppression genes p63 and
p73 inhibit the IGF-1R promoter, reducing the endogenous
IGF-1R levels in a dose-dependent manner. Through this mechanism,
it was proposed that they control colon cancer proliferation
(172).
The pro-oncogenic activities of IGF-1R are solely
mediated through its proximal downstream effectors: Insulin
receptor substrate 1 (IRS-1) and 2 (IRS-2) (173,174).
IRS-1 expression appears to be inversely associated with CRC
differentiation. However, it may be upregulated in both primary and
metastatic human CRC, a finding that has not been observed in
normal colonic epithelium (175). It
has been further supported that the upregulation of IRS-1 can occur
directly from androgens (64). IRS-2
mRNA and protein expression have a positive association with the
transition from normal colorectal epithelium to adenoma and
adenocarcinoma. Furthermore, IRS-2 overexpression promotes
the invasiveness of CRC cells. It activates the oncogenic PI3K/Akt
pathway and at the same time reduces cell adhesion (176). Finally, IRS-1 and IRS-2
polymorphisms have been independently associated with the risk of
developing CRC in a direct manner (177).
The above-mentioned findings are further supported
by the action of NT157 in murine and human CRC cells. NT157 is a
molecule that, through bondage to an allosteric site of the IGF-1R,
it induces a conformational change. As a result, the receptor is
dissociated from IRS1 and IRS2 proteins. Consequently, IGF-1R
stronger interacts with the adaptor protein Shc, leading to an
enhanced activation of extracellular signal-regulated kinase (ERK).
Indeed, experiments have confirmed that NT157 activates ERK1/2,
without activating Akt (178).
Vitamin D regulates cellular functions, such as
differentiation and proliferation in normal and malignant tissues.
It also regulates cell adhesion in tumor cells and modifies tumor
angiogenesis, invasion and metastasis along with decreasing
oxidative DNA damage (179). Vitamin
D deficiency has been associated with various cancer types
(180,181).
Vitamin D first attracted scientific attention
after an inverse association was observed between solar UV-B
exposure and CRC incidence in both genres (182). As it has been well-established, UV-B
radiation is essential for the production of vitamin D3, which
after two steps becomes 1,25-(OH)2-vitamin D
(calcitriol), the most active component (183,184).
The study by Boscoe and Schymura on 3.1 million
individuals from the northern part of the USA supported that low
levels of vitamin D can induce the progression of CRC, although no
association was found with disease onset. Their proposal was based
upon a higher death rate which occurred during the winter months
(when levels of vitamin D are markedly reduced) (182). Feskanich et al found an
inverse association of 25-OH-vitamin D and CRC in observation in
the female population, although only in areas where high levels of
UV-B are available (185). In fact,
levels of 25-OH-vitamin D >20 ng/ml have been advocated to
provide protection against CRC (186). If levels of 25-OH-vitamin D are
>82 ng/ml, then it is estimated that the cancer incidence is
decreased by 50% (187). However, no
association has been found between 1,25-OH2-vitamin D
and CRC (185); a finding disputing
the results of two previous studies (188,189).
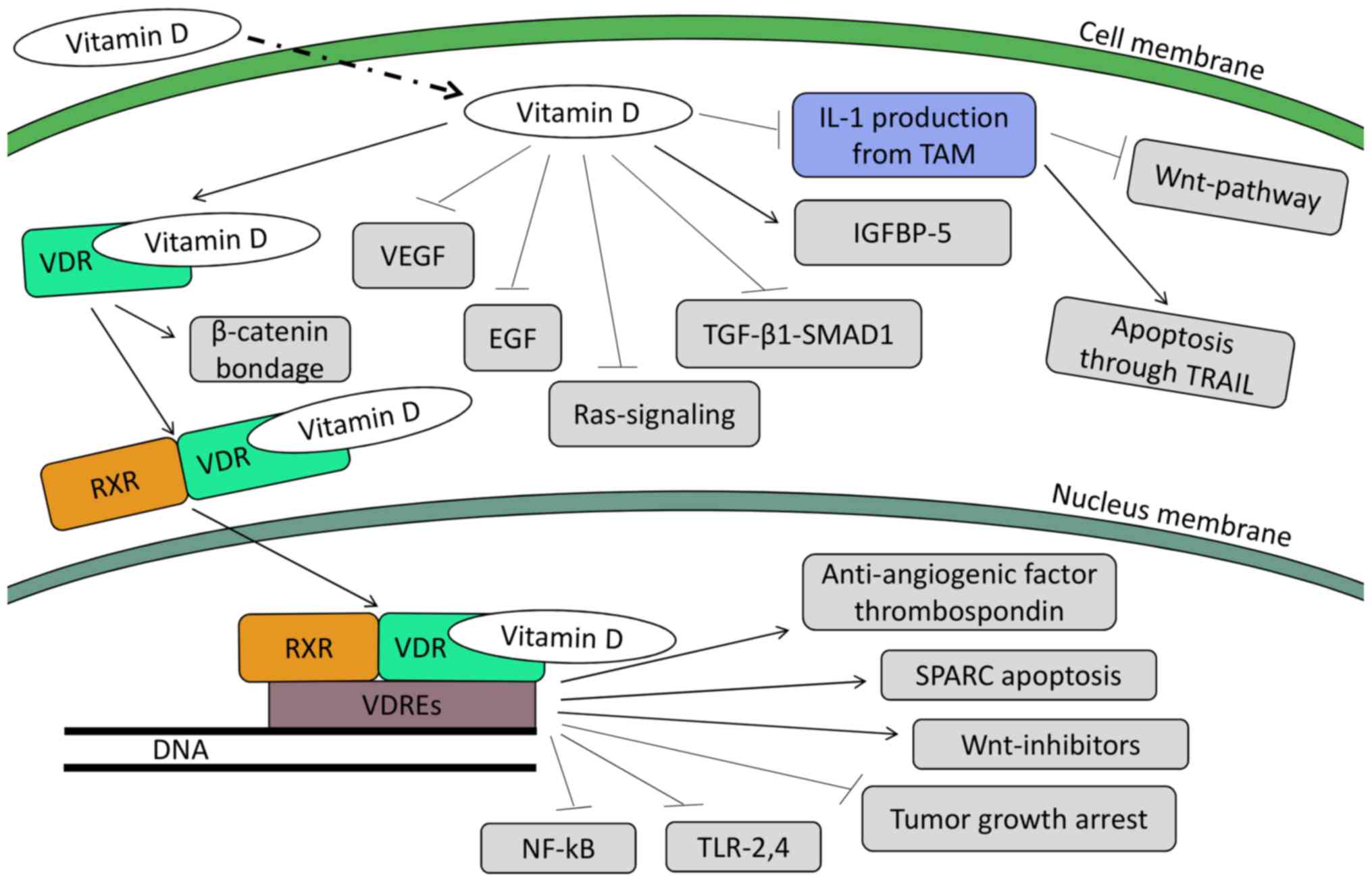
Further action of vitamin D in human colon tumor
cells leads to the upregulation of the potent anti-angiogenic
factor, thrombospondin 1 (199). The
upregulation of the transcription of the Wnt-inhibitors, the
DICKKOPF-1 and DICKKOPF-4 genes, has also been linked
with the action of calcitriol (200). There are data to suggest that
calcitriol regulates apoptosis as well (201). Following treatment of colorectal
cell lines with 1,25-OH2-vitamin D, it was found that
apoptosis was triggered through secreted protein acidic and rich in
cysteine (SPARC)-induced VDR synthesis (202). In another cell line, treatment with
calcitriol induced the mRNA expression of the pro-apoptotic
protein, G0S2 (G0/G1 switch gene) (203). Furthermore, WAF1 and KIP1 were found
to be up-regulated, leading to cell-cycle arrest in G1 phase
(201,204,205).
Other studies have demonstrated the effects of
calcitriol on tumor growth. Through the induction of
cyclin-dependent kinase inhibitors, such as p21, p27 and cystatin D
and the inhibition of pro-proliferative genes, including
c-my and cyclin D1, tumor growth is halted (198). NF-κB is another target family
of genes (fundamental for the cancer cell survival) which are
downregulated by vitamin D (206).
Finally, the reduction of colon cancer cell lines has been
suggested following the decreased expression of Toll-like
receptor (TLR)2 and 4 on human monocytes (207).
Apart from the genomic action through the nuclear
receptor, calcitriol can bind to membrane receptors in certain
tissues (including the intestines), leading to non-genomic,
non-nuclear actions (208–211). It has been well documented that in
CRC, APC mutation is by far one of the most common, allowing
various downstream pro-oncogenic pathways to upshift. One of these
pathways is the Wnt pathway, where β-catenin acts as a
transcriptional co-regulator, cooperating with transcription
factors of the T-cell factor (TCF) family to determine gene
expression (212,213). Pendás-Franco et al described
the protective action of calcitriol in colon cancer cells,
according to which it induces VDR to bind with β-catenin and
restrain it from translocating to the nucleus and inducing the
expression of pro-carcinogenic genes (200). Furthermore, vitamin D-related
compounds have been found to induce the production of IGFBP-5.
These molecules bind both with IGF-1 and IGF-2, suppressing the
stimulating effect of these molecules (214).
Furthermore, vitamin D seems to be able to
reprogram the tumor-associated macrophages (TAMs) in a manner that
halts their tumor-promoting actions (215), probably by inhibiting STAT1
activity. As a result, there is no production of interleukin (IL)-1
from the latter, rendering the tumor colon cells sensitive to
apoptosis through the TRAIL pathway (216). In parallel, due to the lack of
IL-1β expression, the Wnt pathway is deactivated (215).
Further observations of a crosstalk between vitamin
D and TGF-β1/SMAD1 signaling in the growth inhibition of human
colon cancer-derived cells has shown that this interaction halts
tumor growth by blocking the expression of cell cycle proteins and
inhibiting the action of cyclins D1, D2, D3 and E (217). It has also been found to inhibit
mitogenic Ras signaling, as well as the epidermal growth factor
(EGF) (218–220). Furthermore, Ben-Shoshan et al
exhibited an inhibition of vascular endothelial growth factor
(VEGF) in colon cancer cell lines by vitamin D (221).
Last but not least, the association of vitamin D
with calcium must be examined. It seems they exert a synergistic
effect in reducing CRC incidence. This was first described in
Apcmin mouse models by Harris and Go (222) and later on by Lappe et al who
carried a clinical trial on post-menopausal women in Nebraska. They
concluded that although calcium alone reduced the all-cancer
incidence by 44%, when accompanied by vitamin D the reduction
reached 77% (223).
Human growth hormone (hGH) or somatotropin, is a
peptide-hormone secreted mainly by somatotropic cells within the
lateral wings of the anterior pituitary. After entering the
bloodstream it reaches its target organs (namely the liver,
muscles, bones and adipose tissue) binding to its receptor [growth
hormone receptor (GHR)] and thus inducing its anabolic properties
through the activation of the mitogen activated protein kinase
(MAPK)/ERK and JAK/STAT pathways (224,225).
It has been well documented that GH plays a key role in
longitudinal growth during childhood, while maintaining various
important metabolic functions throughout life (promoting lipolysis,
protein synthesis and gluconeogenesis, while reducing glucose
uptake from the liver) (226). Of
note though, GH action is slightly more sophisticated. Apart from
its direct action, an indirect one through the production of IGF-1
also takes place, representing an important part of GH physiology.
In fact, a potent stimulus of IGF-1 production is the GH per se.
Moreover, the tissues producing IGF-1 (the liver 75% and the
peripheral tissues) are indeed the target organs of GH (227). However, it has been proven that GH
is not only synthesized in the pituitary, but also in various other
tissues, such as the large intestine, prostate and breast (228,229).
In this case however, GH lacks the endocrine potential and its
action is mainly restricted to an autocrine or paracrine manner
(230).
Due to its proliferative properties, GH has
attracted reasonable attention for its carcinogenic potential. In
fact, various studies have demonstrated that GH is indeed able to
create a favorable microenvironment for tumor cells. In detail,
GH overexpression is linked to an increased risk of
malignancies (231), while its
downregulation is linked to a carcinoprotective state. As for CRC
risk per se, GH has been proven to act as a tumor promoter in colon
tissue by suppressing p53 (232),
phosphatase and tensin homolog (PTEN) and APC
(8), while it has also been proven
that colon cancer cells overexpress GHR (232). In fact, upregulated GH has been
exhibited to increase ERK phosphorylation and to decrease
APC expression (232). It is
known that a decreased APC expression promotes the nuclear
accumulation of β-catenin, which in turn increases Wnt signaling
through the activation of pro-proliferative genes (233,234).
Thus, even though it is difficult to estimate the exact
concentration for any given age above which GH poses its
carcinogenic effect on colon cells, there is enough evidence
supporting that the ability of GH to change the microenvironment of
tumor cells can have a synergistic effect with other CRC risk
factors (such as intestinal dysbiosis, smoking etc.), shifting the
balance towards tumor survival and proliferation (235).
After 30 years of intense research on CRC and its
biological behavior, only few facts have withstood the test of
time, all revealing the impressive complexity and diversity of this
entity. In fact, this is the case for the association between CRC
and anabolic substances. Driven by epidemiological observations on
both sexes, a time pattern of CRC onset has been found. However,
studies thus far have failed to reach a consensus regarding the
direct connection between androgens and the risk of developing CRC,
as some studies have indicated a negative effect, while others have
pointed out a neutral or even a protective one (Table I). In the case of androgens, the
androgen receptor (the principal mediator of their action) has been
proven to be altered in CRC in contrast to healthy individuals.
Moreover, anabolic substances are also put to scrutiny given the
intense presence of such substances in patients with CRC (namely
IGF-1). However, no direct or well-studied indirect mode of action
on CRC pathogenesis has been found for both classes. Thus, more
studies are needed that will focus on both epidemiologic data (that
will try to investigate how the use of anabolic agents, androgens
included, alters CRC statistics) and the elucidation of molecular
pathways implicated in CRC, in order to allow the extraction of
solid conclusions. In addition, as passive everyday life exposure
to hazardous chemicals could affect traditional clinical risk
factors and act synergistically, the patterns of living and
consumers' trends should also be taken into consideration when
evaluating CRC.
Not applicable.
No funding was received.
Not applicable.
JT conceived and designed the study. TK and TKN
researched the literature, performed analysis of data and drafted
the manuscript. DAS, CT and AT made substantial contributions to
the conception of the study and critically revised the article for
important intellectual content. MS, JS, TMS made substantial
contributions to the design of the study and critically revised the
article for important intellectual content. All authors have read
and approved the final manuscript.
Not applicable.
Not applicable.
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
|
1
|
Roshan MH, Tambo A and Pace NP: The role
of testosterone in colorectal carcinoma: Pathomechanisms and open
questions. EPMA J. 7:222016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amos-Landgraf JM, Heijmans J, Wielenga MC,
Dunkin E, Krentz KJ, Clipson L, Ederveen AG, Groothuis PG,
Mosselman S, Muncan V, et al: Sex disparity in colonic
adenomagenesis involves promotion by male hormones, not protection
by female hormones. Proc Natl Acad Sci USA. 111:16514–16519. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Docea AO, Goumenou M, Calina D, Arsene AL,
Dragoi CM, Gofita E, Pisoschi CG, Zlatian O, Stivaktakis PD,
Nikolouzakis TK, et al: Adverse and hormetic effects in rats
exposed for 12 months to low dose mixture of 13 chemicals: RLRS
part III. Toxicol Lett. 310:70–91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsatsakis AM, Docea AO, Calina D, Buga AM,
Zlatian O, Gutnikov S, Kostoff RN and Aschner M: Hormetic
Neurobehavioral effects of low dose toxic chemical mixtures in
real-life risk simulation (RLRS) in rats. Food Chem Toxicol.
125:141–149. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsatsakis AM, Kouretas D, Tzatzarakis MN,
Stivaktakis P, Tsarouhas K, Golokhvast KS, Rakitskii VN, Tutelyan
VA, Hernandez AF, Rezaee R, et al: Simulating real-life exposures
to uncover possible risks to human health: A proposed consensus for
a novel methodological approach. Hum Exp Toxicol. 36:554–564. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kinzler KW and Vogelstein B: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nikolouzakis TK, Vassilopoulou L,
Fragkiadaki P, Mariolis Sapsakos T, Papadakis GZ, Spandidos DA,
Tsatsakis AM and Tsiaoussis J: Improving diagnosis, prognosis and
prediction by using biomarkers in CRC patients (Review). Oncol Rep.
39:2455–2472. 2018.PubMed/NCBI
|
|
10
|
Terzic J, Grivennikov S, Karin E and Karin
M: Inflammation and colon cancer. Gastroenterology.
138:2101–2114.e5. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Greten FR, Eckmann L, Greten TF, Park JM,
Li ZW, Egan LJ, Kagnoff MF and Karin M: IKKbeta links inflammation
and tumorigenesis in a mouse model of colitis-associated cancer.
Cell. 118:285–296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L, et al: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chlebowski RT, Wactawski-Wende J,
Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, Rosenberg CA,
Taylor VM, Harris R, Chen C, et al Women's Health Initiative
Investigators, : Estrogen plus progestin and colorectal cancer in
postmenopausal women. N Engl J Med. 350:991–1004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsoukalas D, Fragkiadaki P, Docea AO,
Alegakis AK, Sarandi E, Thanasoula M, Spandidos DA, Tsatsakis A,
Razgonova MP and Calina D: Discovery of potent telomerase
activators: Unfolding new therapeutic and anti-aging perspectives.
Mol Med Rep. 20:3701–3708. 2019.PubMed/NCBI
|
|
15
|
Tsatsakis AM, Docea AO and Tsitsimpikou C:
New challenges in risk assessment of chemicals when simulating real
exposure scenarios; simultaneous multi-chemicals' low dose
exposure. Food Chem Toxicol. 96:174–176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Potter JD and McMichael AJ: Large bowel
cancer in women in relation to reproductive and hormonal factors: A
case-control study. J Natl Cancer Inst. 71:703–709. 1983.PubMed/NCBI
|
|
17
|
McMichael AJ and Potter JD: Reproduction,
endogenous and exogenous sex hormones, and colon cancer: A review
and hypothesis. J Natl Cancer Inst. 65:1201–1207. 1980.PubMed/NCBI
|
|
18
|
Young JL Jr, Asire AJ and Polalack ES:
SEER Program: Cancer incidence and mortality in the United States:
1973–1976. US Department of Health, Education, and Welfare, NCI.
671978.
|
|
19
|
Burbank F: Patterns in cancer mortality in
the United States: 1950–1967. Natl Cancer Inst Monogr. 71:1–594.
1971.PubMed/NCBI
|
|
20
|
Haenszel W and Correa P: Cancer of the
colon and rectum and adenomatous polyps. A review of epidemiologic
findings. Cancer. 28:14–24. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Howell MA: The association between
colorectal cancer and breast cancer. J Chronic Dis. 29:243–261.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Izbicki JR, Wambach G, Hamilton SR,
Harnisch E, Hogenschurz R, Izbicki W and Kusche J: Androgen
receptors in experimentally induced colon carcinogenesis. J Cancer
Res Clin Oncol. 112:39–46. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nikolouzakis TK, Stivaktakis PD, Apalaki
P, Kalliantasi K, Sapsakos TM, Spandidos DA, Tsatsakis A, Souglakos
J and Tsiaoussis J: Effect of systemic treatment on the micronuclei
frequency in the peripheral blood of patients with metastatic
colorectal cancer. Oncol Lett. 17:2703–2712. 2019.PubMed/NCBI
|
|
24
|
Qiu S, Jiang C and Zhou L: Physical
activity and mortality in patients with colorectal cancer: a
meta-analysis of prospective cohort studies. Eur J Cancer Prev.
April 5–2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gabrielsen JS, Najari BB, Alukal JP and
Eisenberg ML: Trends in Testosterone Prescription and Public Health
Concerns. Urol Clin North Am. 43:261–271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Georgiadis N, Tsarouhas K, Tsitsimpikou C,
Vardavas A, Rezaee R, Germanakis I, Tsatsakis A, Stagos D and
Kouretas D: Pesticides and cardiotoxicity. Where do we stand?
Toxicol Appl Pharmacol. 353:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Margina D, Nițulescu G, Ungurianu A,
Mesnage R, Goumenou M, Sarigiannis DA, Aschner M, Spandidos DA,
Renieri EA, Hernández AF and Tsatsakis A: Overview of the effects
of chemical mixtures with endocrine disrupting activity in the
context of real life risk simulation (RLRS): an integrative
approach (Review). World Acad Sci J (In press).
|
|
28
|
Veremchuk LV, Tsarouhas K, Vitkina TI,
Mineeva EE, Gvozdenko TA, Antonyuk MV, Rakitskii VN, Sidletskaya
KA, Tsatsakis AM and Golokhvast KS: Impact evaluation of
environmental factors on respiratory function of asthma patients
living in urban territory. Environ Pollut. 235:489–496. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zafiropoulos A, Tsarouhas K, Tsitsimpikou
C, Fragkiadaki P, Germanakis I, Tsardi M, Maravgakis G,
Goutzourelas N, Vasilaki F, Kouretas D, et al: Cardiotoxicity in
rabbits after a low-level exposure to diazinon, propoxur, and
chlorpyrifos. Hum Exp Toxicol. 33:1241–1252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsatsakis A, Docea AO, Constantin C,
Calina D, Zlatian O, Nikolouzakis TK, Stivaktakis PD, Kalogeraki A,
Liesivuori J, Tzanakakis G, et al: Genotoxic, cytotoxic, and
cytopathological effects in rats exposed for 18 months to a mixture
of 13 chemicals in doses below NOAEL levels. Toxicol Lett. Sep
12–2019.(Epub ahead of print). View Article : Google Scholar :
|
|
31
|
Ozcagli E, Kara M, Kotil T, Fragkiadaki P,
Tzatzarakis MN, Tsitsimpikou C, Stivaktakis PD, Tsoukalas D,
Spandidos DA, Tsatsakis AM, et al: Stanozolol administration
combined with exercise leads to decreased telomerase activity
possibly associated with liver aging. Int J Mol Med. 42:405–413.
2018.PubMed/NCBI
|
|
32
|
Kadıoğlu E, Taçoy G, Özçağlı E, Okyay K,
Akboğa MK, Çengel A and Şardaş S: The role of oxidative DNA damage
and GSTM1, GSTT1, and hOGG1 gene polymorphisms in coronary artery
disease risk. Anatol J Cardiol. 16:931–938. 2016.PubMed/NCBI
|
|
33
|
McClendon JE, Appleby D, Claudon DB,
Donegan WL and DeCosse JJ: Colonic neoplasms: Tissue estrogen
receptor and carcinoembryonic antigen. Arch Surg. 112:240–241.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alford TC, Do HM, Geelhoed GW, Tsangaris
NT and Lippman ME: Steroid hormone receptors in human colon
cancers. Cancer. 43:980–984. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sica V, Contieri E, Nola E, Bova R,
Papaleo G and Puca GA: Estrogen and progesterone binding proteins
in human colorectal cancer. A preliminary characterization of
estradiol receptor. Tumori. 67:307–314. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Odagiri E, Jibiki K, Demura R, Shinozaki
H, Nakamura S, Demura H and Suzuki H: Steroid receptors and the
distribution of IR-carcinoembryonic antigen in colonic cancer. Dis
Colon Rectum. 27:787–791. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jacobson HL: Present status of steroid
hormone receptor in large bowel cancer. Prog Cancer Res Ther.
29:3671984.
|
|
38
|
Izbicki JR, Schmitz R, Hoppen HO, Izbicki
W and Troidl H: Effects of steroid hormone therapy on primarily
xenotransplanted human colorectal adenocarcinomas. J Cancer Res
Clin Oncol. 108:345–350. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wobbes T, Beex LVAM and Koenders AMJ:
Estrogen and progestin receptors in colonic cancer? Dis Colon
Rectum. 27:591–592. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bucci L, Salfi R, Meraviglia F and Delric
G: Hormonal receptors in colorectal cancers (abstract). Second
European Conference on Clinical Oncology and Cancer Nursing.
8:41–98. 1983.
|
|
41
|
Handelsman DJ: Androgen Physiology,
Pharmacology and AbuseEndotext [Internet]. Feingold KR, Anawalt B,
Boyce A, et al: MDText.com, Inc.; South Dartmouth, MA: 2000
|
|
42
|
Hiort O, Holterhus PM and Nitsche EM:
Physiology and pathophysiology of androgen action. Baillieres Clin
Endocrinol Metab. 12:115–132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guyton AC and Hall JE: Textbook of medical
physiology11th. Elsevier Saunders; Philladelphia, PA: 2006
|
|
44
|
Brenu EW, McNaughton L and
Marshall-Gradisnik SM: Is there a potential immune dysfunction with
anabolic androgenic steroid use?: A review. Mini Rev Med Chem.
11:438–445. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsarouhas K, Kioukia-Fougia N, Papalexis
P, Tsatsakis A, Kouretas D, Bacopoulou F and Tsitsimpikou C: Use of
nutritional supplements contaminated with banned doping substances
by recreational adolescent athletes in Athens, Greece. Food Chem
Toxicol. 115:447–450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsitsimpikou C, Chrisostomou N, Papalexis
P, Tsarouhas K, Tsatsakis A and Jamurtas A: The use of nutritional
supplements among recreational athletes in Athens, Greece. Int J
Sport Nutr Exerc Metab. 21:377–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vasilaki F, Tsitsimpikou C, Tsarouhas K,
Germanakis I, Tzardi M, Kavvalakis M, Ozcagli E, Kouretas D and
Tsatsakis AM: Cardiotoxicity in rabbits after long-term nandrolone
decanoate administration. Toxicol Lett. 241:143–151. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Baggish AL, Weiner RB, Kanayama G, Hudson
JI, Picard MH, Hutter AM Jr and Pope HG Jr: Long-term
anabolic-androgenic steroid use is associated with left ventricular
dysfunction. Circ Heart Fail. 3:472–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tsitsimpikou C, Tsarouhas K, Spandidos DA
and Tsatsakis AM: Detection of stanozolol in the urine of athletes
at a pg level: The possibility of passive exposure. Biomed Rep.
5:665–666. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sattler FR, Jaque SV, Schroeder ET, Olson
C, Dube MP, Martinez C, Briggs W, Horton R and Azen S: Effects of
pharmacological doses of nandrolone decanoate and progressive
resistance training in immunodeficient patients infected with human
immunodeficiency virus. J Clin Endocrinol Metab. 84:1268–1276.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Santos MA, Oliveira CV and Silva AS:
Adverse cardiovascular effects from the use of anabolic-androgenic
steroids as ergogenic resources. Subst Use Misuse. 49:1132–1137.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bonetti A, Tirelli F, Catapano A, Dazzi D,
Dei Cas A, Solito F, Ceda G, Reverberi C, Monica C, Pipitone S, et
al: Side effects of anabolic androgenic steroids abuse. Int J
Sports Med. 29:679–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gould DC and Petty R: The male menopause:
does it exist? For: Some men need investigation and testosterone
treatment. West J Med. 173:76–78. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gillessen S, Templeton A, Marra G, Kuo YF,
Valtorta E and Shahinian VB: Risk of colorectal cancer in men on
long-term androgen deprivation therapy for prostate cancer. J Natl
Cancer Inst. 102:1760–1770. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Izbicki JR, Schmitz R, Kamran D and
Izbicki W: Androgens as promoters of colon carcinogenesis. Cancer
Detect Prev. 6:355–362. 1983.PubMed/NCBI
|
|
56
|
Mehta RG, Fricks CM and Moon RC: Androgen
receptors in chemically-induced colon carcinogenesis. Cancer. 45
(Suppl):1085–1089. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Moon RC and Fricks CM: Influence of
gonadal hormones and age on 1,2-dimethylhydrazine-induced colon
carcinogenesis. Cancer. 40 (Suppl):2502–2508. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hyde Z, Flicker L, McCaul KA, Almeida OP,
Hankey GJ, Chubb SA and Yeap BB: Associations between testosterone
levels and incident prostate, lung, and colorectal cancer. A
population-based study. Cancer Epidemiol Biomarkers Prev.
21:1319–1329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Orsted DD, Nordestgaard BG and Bojesen SE:
Plasma testosterone in the general population, cancer prognosis and
cancer risk: A prospective cohort study. Ann Oncol. 25:712–718.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Koliarakis I, Psaroulaki A, Nikolouzakis
TK, Kokkinakis M, Sgantzos MN, Goulielmos G, Androutsopoulos VP,
Tsatsakis A and Tsiaoussis J: Intestinal microbiota and colorectal
cancer: a new aspect of research. J BUON. 23:1216–1234.
2018.PubMed/NCBI
|
|
61
|
Alberg AJ, Gordon GB, Hoffman SC, Comstock
GW and Helzlsouer KJ: Serum dehydroepiandrosterone and
dehydroepiandrosterone sulfate and the subsequent risk of
developing colon cancer. Cancer Epidemiol Biomarkers Prev.
9:517–521. 2000.PubMed/NCBI
|
|
62
|
Anagnostopoulou V, Pediaditakis I,
Alkahtani S, Alarifi SA, Schmidt EM, Lang F, Gravanis A,
Charalampopoulos I and Stournaras C: Differential effects of
dehydroepiandrosterone and testosterone in prostate and colon
cancer cell apoptosis: The role of nerve growth factor (NGF)
receptors. Endocrinology. 154:2446–2456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ferro P, Catalano MG, Raineri M, Reato G,
dell'Eva R, Risio M, Foà R, Fortunati N and Pfeffer U: Somatic
alterations of the androgen receptor CAG repeat in human colon
cancer delineate a novel mutation pathway independent of
microsatellite instability. Cancer Genet Cytogenet. 123:35–40.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Slattery ML, Sweeney C, Murtaugh M, Ma KN,
Wolff RK, Potter JD, Caan BJ and Samowitz W: Associations between
ERalpha, ERbeta, and AR genotypes and colon and rectal cancer.
Cancer Epidemiol Biomarkers Prev. 14:2936–2942. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hoque A, Albanes D, Lippman SM, Spitz MR,
Taylor PR, Klein EA, Thompson IM, Goodman P, Stanford JL, Crowley
JJ, et al: Molecular epidemiologic studies within the Selenium and
Vitamin E Cancer Prevention Trial (SELECT). Cancer Causes Control.
12:627–633. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Krithivas K, Yurgalevitch SM, Mohr BA,
Wilcox CJ, Batter SJ, Brown M, Longcope C, McKinlay JB and Kantoff
PW: Evidence that the CAG repeat in the androgen receptor gene is
associated with the age-related decline in serum androgen levels in
men. J Endocrinol. 162:137–142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ding D, Xu L, Menon M, Reddy GP and
Barrack ER: Effect of a short CAG (glutamine) repeat on human
androgen receptor function. Prostate. 58:23–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Westberg L, Baghaei F, Rosmond R,
Hellstrand M, Landén M, Jansson M, Holm G, Björntorp P and Eriksson
E: Polymorphisms of the androgen receptor gene and the estrogen
receptor beta gene are associated with androgen levels in women. J
Clin Endocrinol Metab. 86:2562–2568. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rudolph A, Shi H, Försti A, Hoffmeister M,
Sainz J, Jansen L, Hemminki K, Brenner H and Chang-Claude J: Repeat
polymorphisms in ESR2 and AR and colorectal cancer risk and
prognosis: Results from a German population-based case-control
study. BMC Cancer. 14:8172014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bonin A, Bellemain E, Bronken Eidesen P,
Pompanon F, Brochmann C and Taberlet P: How to track and assess
genotyping errors in population genetics studies. Mol Ecol.
13:3261–3273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Pompanon F, Bonin A, Bellemain E and
Taberlet P: Genotyping errors: Causes, consequences and solutions.
Nat Rev Genet. 6:847–859. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang R, Wang G, Song Y, Wang F, Zhu B,
Tang Q, Liu Z, Chen Y, Zhang Q, Muhammad S, et al: Polymorphic CAG
Repeat and Protein Expression of Androgen Receptor Gene in
Colorectal Cancer. Mol Cancer Ther. 14:1066–1074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chamberlain NL, Driver ED and Miesfeld RL:
The length and location of CAG trinucleotide repeats in the
androgen receptor N-terminal domain affect transactivation
function. Nucleic Acids Res. 22:3181–3186. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Choong CS, Kemppainen JA, Zhou ZX and
Wilson EM: Reduced androgen receptor gene expression with first
exon CAG repeat expansion. Mol Endocrinol. 10:1527–1535. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ferro P, Catalano MG, Dell'Eva R,
Fortunati N and Pfeffer U: The androgen receptor CAG repeat: A
modifier of carcinogenesis? Mol Cell Endocrinol. 193:109–120. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Catalano MG, Pfeffer U, Raineri M, Ferro
P, Curto A, Capuzzi P, Corno F, Berta L and Fortunati N: Altered
expression of androgen-receptor isoforms in human colon-cancer
tissues. Int J Cancer. 86:325–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gu S, Papadopoulou N, Nasir O, Föller M,
Alevizopoulos K, Lang F and Stournaras C: Activation of membrane
androgen receptors in colon cancer inhibits the prosurvival signals
Akt/bad in vitro and in vivo and blocks migration via
vinculin/actin signaling. Mol Med. 17:48–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gu S, Papadopoulou N, Gehring EM, Nasir O,
Dimas K, Bhavsar SK, Föller M, Alevizopoulos K, Lang F and
Stournaras C: Functional membrane androgen receptors in colon
tumors trigger pro-apoptotic responses in vitro and reduce
drastically tumor incidence in vivo. Mol Cancer. 8:1142009.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chesire DR and Isaacs WB: Ligand-dependent
inhibition of beta-catenin/TCF signaling by androgen receptor.
Oncogene. 21:8453–8469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yoshioka T, Nishikawa Y, Ito R, Kawamata
M, Doi Y, Yamamoto Y, Yoshida M, Omori Y, Kotanagi H, Masuko T, et
al: Significance of integrin αvβ5 and erbB3 in enhanced cell
migration and liver metastasis of colon carcinomas stimulated by
hepatocyte-derived heregulin. Cancer Sci. 101:2011–2018. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Holgren C, Dougherty U, Edwin F, Cerasi D,
Taylor I, Fichera A, Joseph L, Bissonnette M and Khare S: Sprouty-2
controls c-Met expression and metastatic potential of colon cancer
cells: Sprouty/c-Met upregulation in human colonic adenocarcinomas.
Oncogene. 29:5241–5253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Huynh N, Liu KH, Baldwin GS and He H:
P21-activated kinase 1 stimulates colon cancer cell growth and
migration/invasion via ERK- and AKT-dependent pathways. Biochim
Biophys Acta. 1803:1106–1113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Teyssier C, Ou CY, Khetchoumian K, Losson
R and Stallcup MR: Transcriptional intermediary factor 1alpha
mediates physical interaction and functional synergy between the
coactivator-associated arginine methyltransferase 1 and
glucocorticoid receptor-interacting protein 1 nuclear receptor
coactivators. Mol Endocrinol. 20:1276–1286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chen D, Ma H, Hong H, Koh SS, Huang SM,
Schurter BT, Aswad DW and Stallcup MR: Regulation of transcription
by a protein methyltransferase. Science. 284:2174–2177. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Koh SS, Li H, Lee YH, Widelitz RB, Chuong
CM and Stallcup MR: Synergistic coactivator function by
coactivator-associated arginine methyltransferase (CARM) 1 and
beta-catenin with two different classes of DNA-binding
transcriptional activators. J Biol Chem. 277:26031–26035. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
El Messaoudi S, Fabbrizio E, Rodriguez C,
Chuchana P, Fauquier L, Cheng D, Theillet C, Vandel L, Bedford MT
and Sardet C: Coactivator-associated arginine methyltransferase 1
(CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl
Acad Sci USA. 103:13351–13356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kim YR, Lee BK, Park RY, Nguyen NT, Bae
JA, Kwon DD and Jung C: Differential CARM1 expression in prostate
and colorectal cancers. BMC Cancer. 10:1972010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ilboudo S, Fouche E, Rizzati V, Toé AM,
Gamet-Payrastre L and Guissou PI: In vitro impact of five
pesticides alone or in combination on human intestinal cell line
Caco-2. Toxicol Rep. 1:474–489. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Carroll RE, Goodlad RA, Poole AJ, Tyner
AL, Robey RB, Swanson SM and Unterman TG: Reduced susceptibility to
azoxymethane-induced aberrant crypt foci formation and colon cancer
in growth hormone deficient rats. Growth Horm IGF Res. 19:447–456.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chuang KH, Altuwaijri S, Li G, Lai JJ, Chu
CY, Lai KP, Lin HY, Hsu JW, Keng P, Wu MC, et al: Neutropenia with
impaired host defense against microbial infection in mice lacking
androgen receptor. J Exp Med. 206:1181–1199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mårin P, Krotkiewski M and Björntorp P:
Androgen treatment of middle-aged, obese men: Effects on
metabolism, muscle and adipose tissues. Eur J Med. 1:329–336.
1992.PubMed/NCBI
|
|
92
|
Lin JH and Giovannucci E: Sex hormones and
colorectal cancer: What have we learned so far? J Natl Cancer Inst.
102:1746–1747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Thevis M and Schänzer W: Synthetic
anabolic agents: Steroids and nonsteroidal selective androgen
receptor modulatorsDoping in Sports: Biochemical Principles,
Effects and Analysis. Handbook of Experimental Pharmacology. Thieme
D and Hemmersbach P: 195. Springer; Berlin, Heidelberg: pp. 99–126.
2010, View Article : Google Scholar
|
|
94
|
Joseph JF and Parr MK: Synthetic androgens
as designer supplements. Curr Neuropharmacol. 13:89–100. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Watanabe S and Kobayashi Y: Exogenous
hormones and human cancer. Jpn J Clin Oncol. 23:1–13.
1993.PubMed/NCBI
|
|
96
|
Rosner F and Khan MT: Renal cell carcinoma
following prolonged testosterone therapy. Arch Intern Med.
152:426–429. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Martorana G, Concetti S, Manferrari F and
Creti S: Anabolic steroid abuse and renal cell carcinoma. J Urol.
162:2089. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bryden AAG, Rothwell PJN and O'Reilly PH:
Anabolic steroid abuse and renal-cell carcinoma. Lancet.
346:1306–1307. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zahm SH and Fraumeni JF Jr: The
epidemiology of soft tissue sarcoma. Semin Oncol. 24:504–514.
1997.PubMed/NCBI
|
|
100
|
Nourbakhsh M, Golestani A, Zahrai M,
Modarressi MH, Malekpour Z and Karami-Tehrani F: Androgens
stimulate telomerase expression, activity and phosphorylation in
ovarian adenocarcinoma cells. Mol Cell Endocrinol. 330:10–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
McKeown-Eyssen G: Epidemiology of
colorectal cancer revisited: Are serum triglycerides and/or plasma
glucose associated with risk? Cancer Epidemiol Biomarkers Prev.
3:687–695. 1994.PubMed/NCBI
|
|
102
|
Bronson FH and Matherne CM: Exposure to
anabolic-androgenic steroids shortens life span of male mice. Med
Sci Sports Exerc. 29:615–619. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Froehner M, Fischer R, Leike S, Hakenberg
OW, Noack B and Wirth MP: Intratesticular leiomyosarcoma in a young
man after high dose doping with oral-turinabol: A case report.
Cancer. 86:1571–1575. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chacon A and Monga M: Medical management
of benign prostatic hyperplasia. Geriatr Nephrol Urol. 9:39–48.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dimitriadis G, Mitrou P, Lambadiari V,
Maratou E and Raptis SA: Insulin effects in muscle and adipose
tissue. Diabetes Res Clin Pract. 93 (Suppl 1):S52–S59. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Giovannucci E and Michaud D: The role of
obesity and related metabolic disturbances in cancers of the colon,
prostate, and pancreas. Gastroenterology. 132:2208–2225. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yang YX, Hennessy S and Lewis JD: Insulin
therapy and colorectal cancer risk among type 2 diabetes mellitus
patients. Gastroenterology. 127:1044–1050. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Larsson SC, Orsini N, Brismar K and Wolk
A: Diabetes mellitus and risk of bladder cancer: A meta-analysis.
Diabetologia. 49:2819–2823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Flood A, Mai V, Pfeiffer R, Kahle L,
Remaley AT, Lanza E and Schatzkin A: Elevated serum concentrations
of insulin and glucose increase risk of recurrent colorectal
adenomas. Gastroenterology. 133:1423–1429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Polednak AP: Comorbid diabetes mellitus
and risk of death after diagnosis of colorectal cancer: A
population-based study. Cancer Detect Prev. 30:466–472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Jullumstrø E, Kollind M, Lydersen S and
Edna TH: Diabetes mellitus and outcomes of colorectal cancer. Acta
Oncol. 48:361–367. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Schoen RE, Weissfeld JL, Kuller LH, Thaete
FL, Evans RW, Hayes RB and Rosen CJ: Insulin-like growth factor-I
and insulin are associated with the presence and advancement of
adenomatous polyps. Gastroenterology. 129:464–475. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
LeRoith D, Baserga R, Helman L and Roberts
CT Jr: Insulin-like growth factors and cancer. Ann Intern Med.
122:54–59. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sandhu MS, Dunger DB and Giovannucci EL:
Insulin, insulin-like growth factor-I (IGF-I), IGF binding
proteins, their biologic interactions, and colorectal cancer. J
Natl Cancer Inst. 94:972–980. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Siddle K: Signalling by insulin and IGF
receptors: Supporting acts and new players. J Mol Endocrinol.
47:R1–R10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mosthaf L, Grako K, Dull TJ, Coussens L,
Ullrich A and McClain DA: Functionally distinct insulin receptors
generated by tissue-specific alternative splicing. EMBO J.
9:2409–2413. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Abbruzzese C, Diodoro MG, Sperduti I,
Mileo AM, Pattaro G, De Salvo L, Cosimelli M, Perrotti N and Paggi
MG: Detection of phosphorylated insulin receptor in colorectal
adenoma and adenocarcinoma: Implications for prognosis and clinical
outcome. J Cell Physiol. 230:562–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Frasca F, Pandini G, Scalia P, Sciacca L,
Mineo R, Costantino A, Goldfine ID, Belfiore A and Vigneri R:
Insulin receptor isoform A, a newly recognized, high-affinity
insulin-like growth factor II receptor in fetal and cancer cells.
Mol Cell Biol. 19:3278–3288. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Kosaki A and Webster NJ: Effect of
dexamethasone on the alternative splicing of the insulin receptor
mRNA and insulin action in HepG2 hepatoma cells. J Biol Chem.
268:21990–21996. 1993.PubMed/NCBI
|
|
120
|
Heckl SM, Pellinghaus M, Krüger S,
Bosselmann C, Wilhelm F, Behrens HM, Schreiber S and Röcken C:
Epithelial insulin receptor expression-prognostic relevance in
colorectal cancer. Oncotarget. 9:37497–37508. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Morcavallo A, Genua M, Palummo A,
Kletvikova E, Jiracek J, Brzozowski AM, Iozzo RV, Belfiore A and
Morrione A: Insulin and insulin-like growth factor II
differentially regulate endocytic sorting and stability of insulin
receptor isoform A. J Biol Chem. 287:11422–11436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wang X, Häring MF, Rathjen T, Lockhart SM,
Sørensen D, Ussar S, Rasmussen LM, Bertagnolli MM, Kahn CR and
Rask-Madsen C: Insulin resistance in vascular endothelial cells
promotes intestinal tumour formation. Oncogene. 36:4987–4996. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Laakso M, Edelman SV, Brechtel G and Baron
AD: Impaired insulin-mediated skeletal muscle blood flow in
patients with NIDDM. Diabetes. 41:1076–1083. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Steinberg HO, Chaker H, Leaming R, Johnson
A, Brechtel G and Baron AD: Obesity/insulin resistance is
associated with endothelial dysfunction. Implications for the
syndrome of insulin resistance. J Clin Invest. 97:2601–2610. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Rask-Madsen C, Ihlemann N, Krarup T,
Christiansen E, Kober L, Nervil Kistorp C and Torp-Pedersen C:
Insulin therapy improves insulin-stimulated endothelial function in
patients with type 2 diabetes and ischemic heart disease. Diabetes.
50:2611–2618. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Tabit CE, Shenouda SM, Holbrook M,
Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM,
Gokce N, et al: Protein kinase C-β contributes to impaired
endothelial insulin signaling in humans with diabetes mellitus.
Circulation. 127:86–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Simpson RJ, Lim JW, Moritz RL and
Mathivanan S: Exosomes: Proteomic insights and diagnostic
potential. Expert Rev Proteomics. 6:267–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gangoda L and Mathivanan S: Cortactin
enhances exosome secretion without altering cargo. J Cell Biol.
214:129–131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Gangoda L, Boukouris S, Liem M, Kalra H
and Mathivanan S: Extracellular vesicles including exosomes are
mediators of signal transduction: Are they protective or
pathogenic? Proteomics. 15:260–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Silantyev AS, Falzone L, Libra M, Gurina
OI, Kardashova KS, Nikolouzakis TK, Nosyrev AE, Sutton CW, Mitsias
PD and Tsatsakis A: Current and Future Trends on Diagnosis and
Prognosis of Glioblastoma: From Molecular Biology to Proteomics.
Cells. 8:82019. View Article : Google Scholar
|
|
131
|
Taniguchi CM, Emanuelli B and Kahn CR:
Critical nodes in signalling pathways: Insights into insulin
action. Nat Rev Mol Cell Biol. 7:85–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Huang XF and Chen JZ: Obesity, the
PI3K/Akt signal pathway and colon cancer. Obes Rev. 10:610–616.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Matsuzaki H, Daitoku H, Hatta M, Tanaka K
and Fukamizu A: Insulin-induced phosphorylation of FKHR (Foxo1)
targets to proteasomal degradation. Proc Natl Acad Sci USA.
100:11285–11290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liem M, Ang CS and Mathivanan S: Insulin
Mediated Activation of PI3K/Akt Signalling Pathway Modifies the
Proteomic Cargo of Extracellular Vesicles. Proteomics. 17:172017.
View Article : Google Scholar
|
|
135
|
Baserga R: The insulin-like growth factor
I receptor: A key to tumor growth? Cancer Res. 55:249–252.
1995.PubMed/NCBI
|
|
136
|
Párrizas M and LeRoith D: Insulin-like
growth factor-1 inhibition of apoptosis is associated with
increased expression of the bcl-xL gene product. Endocrinology.
138:1355–1358. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wang L, Ma W, Markovich R, Lee WL and Wang
PH: Insulin-like growth factor I modulates induction of apoptotic
signaling in H9C2 cardiac muscle cells. Endocrinology.
139:1354–1360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Remacle-Bonnet MM, Garrouste FL, Heller S,
André F, Marvaldi JL and Pommier GJ: Insulin-like growth factor-I
protects colon cancer cells from death factor-induced apoptosis by
potentiating tumor necrosis factor alpha-induced mitogen-activated
protein kinase and nuclear factor kappaB signaling pathways. Cancer
Res. 60:2007–2017. 2000.PubMed/NCBI
|
|
139
|
Liu B, Fang M, Lu Y, Mendelsohn J and Fan
Z: Fibroblast growth factor and insulin-like growth factor
differentially modulate the apoptosis and G1 arrest induced by
anti-epidermal growth factor receptor monoclonal antibody.
Oncogene. 20:1913–1922. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ryan PD and Goss PE: The emerging role of
the insulin-like growth factor pathway as a therapeutic target in
cancer. Oncologist. 13:16–24. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Pollak M: The insulin and insulin-like
growth factor receptor family in neoplasia: An update. Nat Rev
Cancer. 12:159–169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Islam MA, Hooiveld GJEJ, van den Berg JHJ,
van der Velpen V, Murk AJ, Rietjens IMCM and van Leeuwen FXR: Soy
supplementation: Impact on gene expression in different tissues of
ovariectomized rats and evaluation of the rat model to predict
(post)menopausal health effect. Toxicol Rep. 5:1087–1097. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ma J, Pollak MN, Giovannucci E, Chan JM,
Tao Y, Hennekens CH and Stampfer MJ: Prospective study of
colorectal cancer risk in men and plasma levels of insulin-like
growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer
Inst. 91:620–625. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lahm H, Suardet L, Laurent PL, Fischer JR,
Ceyhan A, Givel JC and Odartchenko N: Growth regulation and
co-stimulation of human colorectal cancer cell lines by
insulin-like growth factor I, II and transforming growth factor
alpha. Br J Cancer. 65:341–346. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Guo YS, Narayan S, Yallampalli C and Singh
P: Characterization of insulinlike growth factor I receptors in
human colon cancer. Gastroenterology. 102:1101–1108. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Freier S, Weiss O, Eran M, Flyvbjerg A,
Dahan R, Nephesh I, Safra T, Shiloni E and Raz I: Expression of the
insulin-like growth factors and their receptors in adenocarcinoma
of the colon. Gut. 44:704–708. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hakam A, Yeatman TJ, Lu L, Mora L, Marcet
G, Nicosia SV, Karl RC and Coppola D: Expression of insulin-like
growth factor-1 receptor in human colorectal cancer. Hum Pathol.
30:1128–1133. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lee J, Jain A, Kim P, Lee T, Kuller A,
Princen F, In-GuDo, Kim SH, Park JO, Park YS, et al: Activated cMET
and IGF1R-driven PI3K signaling predicts poor survival in
colorectal cancers independent of KRAS mutational status. PLoS One.
9:e1035512014. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Soubry A, Il'yasova D, Sedjo R, Wang F,
Byers T, Rosen C, Yashin A, Ukraintseva S, Haffner S and D'Agostino
R Jr: Increase in circulating levels of IGF-1 and IGF-1/IGFBP-3
molar ratio over a decade is associated with colorectal adenomatous
polyps. Int J Cancer. 131:512–517. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ollberding NJ, Cheng I, Wilkens LR,
Henderson BE, Pollak MN, Kolonel LN and Le Marchand L: Genetic
variants, prediagnostic circulating levels of insulin-like growth
factors, insulin, and glucose and the risk of colorectal cancer:
The Multiethnic Cohort study. Cancer Epidemiol Biomarkers Prev.
21:810–820. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Giovannucci E: Insulin, insulin-like
growth factors and colon cancer: A review of the evidence. J Nutr.
131 (Suppl):3109S–3120S. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Shiratsuchi I, Akagi Y, Kawahara A,
Kinugasa T, Romeo K, Yoshida T, Ryu Y, Gotanda Y, Kage M and
Shirouzu K: Expression of IGF-1 and IGF-1R and their relation to
clinicopathological factors in colorectal cancer. Anticancer Res.
31:2541–2545. 2011.PubMed/NCBI
|
|
153
|
Peters G, Gongoll S, Langner C, Mengel M,
Piso P, Klempnauer J, Rüschoff J, Kreipe H and von Wasielewski R:
IGF-1R, IGF-1 and IGF-2 expression as potential prognostic and
predictive markers in colorectal-cancer. Virchows Arch.
443:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Furlanetto RW, Harwell SE and Frick KK:
Insulin-like growth factor-I induces cyclin-D1 expression in MG63
human osteosarcoma cells in vitro. Mol Endocrinol. 8:510–517. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Sahin AA, Ro JY, Brown RW, Ordonez NG,
Cleary KR, el-Naggar AK, Wilson P and Ayala AG: Assessment of
Ki-67-derived tumor proliferative activity in colorectal
adenocarcinomas. Mod Pathol. 7:17–22. 1994.PubMed/NCBI
|
|
156
|
Kubota Y, Petras RE, Easley KA, Bauer TW,
Tubbs RR and Fazio VW: Ki-67-determined growth fraction versus
standard staging and grading parameters in colorectal carcinoma. A
multivariate analysis. Cancer. 70:2602–2609. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Cascinu S, Del Ferro E, Grianti C, Ligi M,
Ghiselli R, Foglietti G, Saba V, Lungarotti F and Catalano G:
Inhibition of tumor cell kinetics and serum insulin growth factor I
levels by octreotide in colorectal cancer patients.
Gastroenterology. 113:767–772. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Bowers LW, Rossi EL, O'Flanagan CH,
deGraffenried LA and Hursting SD: The Role of the Insulin/IGF
System in Cancer: Lessons Learned from Clinical Trials and the
Energy Balance-Cancer Link. Front Endocrinol (Lausanne). 6:772015.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Firth SM and Baxter RC: Cellular actions
of the insulin-like growth factor binding proteins. Endocr Rev.
23:824–854. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Liou JM, Shun CT, Liang JT, Chiu HM, Chen
MJ, Chen CC, Wang HP, Wu MS and Lin JT: Plasma insulin-like growth
factor-binding protein-2 levels as diagnostic and prognostic
biomarker of colorectal cancer. J Clin Endocrinol Metab.
95:1717–1725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Jones JI and Clemmons DR: Insulin-like
growth factors and their binding proteins: Biological actions.
Endocr Rev. 16:3–34. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Héron-Milhavet L and LeRoith D:
Insulin-like growth factor I induces MDM2-dependent degradation of
p53 via the p38 MAPK pathway in response to DNA damage. J Biol
Chem. 277:15600–15606. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Peretz S, Jensen R, Baserga R and Glazer
PM: ATM-dependent expression of the insulin-like growth factor-I
receptor in a pathway regulating radiation response. Proc Natl Acad
Sci USA. 98:1676–1681. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Huang F, Xu LA and Khambata-Ford S:
Correlation between gene expression of IGF-1R pathway markers and
cetuximab benefit in metastatic colorectal cancer. Clin Cancer Res.
18:1156–1166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Jones HE, Goddard L, Gee JM, Hiscox S,
Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE and
Nicholson RI: Insulin-like growth factor-I receptor signalling and
acquired resistance to gefitinib (ZD1839; Iressa) in human breast
and prostate cancer cells. Endocr Relat Cancer. 11:793–814. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Albanell J and Baselga J: Unraveling
resistance to trastuzumab (Herceptin): Insulin-like growth factor-I
receptor, a new suspect. J Natl Cancer Inst. 93:1830–1832. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Shen K, Cui D, Sun L, Lu Y, Han M and Liu
J: Inhibition of IGF-IR increases chemosensitivity in human
colorectal cancer cells through MRP-2 promoter suppression. J Cell
Biochem. 113:2086–2097. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Warren RS, Yuan H, Matli MR, Ferrara N and
Donner DB: Induction of vascular endothelial growth factor by
insulin-like growth factor 1 in colorectal carcinoma. J Biol Chem.
271:29483–29488. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Akagi Y, Liu W, Zebrowski B, Xie K and
Ellis LM: Regulation of vascular endothelial growth factor
expression in human colon cancer by insulin-like growth factor-I.
Cancer Res. 58:4008–4014. 1998.PubMed/NCBI
|
|
170
|
Sekharam M, Zhao H, Sun M, Fang Q, Zhang
Q, Yuan Z, Dan HC, Boulware D, Cheng JQ and Coppola D: Insulin-like
growth factor 1 receptor enhances invasion and induces resistance
to apoptosis of colon cancer cells through the Akt/Bcl-x(L)
pathway. Cancer Res. 63:7708–7716. 2003.PubMed/NCBI
|
|
171
|
Zhang QY, Wang L, Song ZY and Qu XJ:
Knockdown of type I insulin-like growth factor receptor inhibits
human colorectal cancer cell growth and downstream PI3K/Akt,
WNT/β-catenin signal pathways. Biomed Pharmacother. 73:12–18. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Nahor I, Abramovitch S, Engeland K and
Werner H: The p53-family members p63 and p73 inhibit insulin-like
growth factor-I receptor gene expression in colon cancer cells.
Growth Horm IGF Res. 15:388–396. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Ramocki NM, Wilkins HR, Magness ST,
Simmons JG, Scull BP, Lee GH, McNaughton KK and Lund PK: Insulin
receptor substrate-1 deficiency promotes apoptosis in the putative
intestinal crypt stem cell region, limits Apcmin/+ tumors, and
regulates Sox9. Endocrinology. 149:261–267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Chan BT and Lee AV: Insulin receptor
substrates (IRSs) and breast tumorigenesis. J Mammary Gland Biol
Neoplasia. 13:415–422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Esposito DL, Aru F, Lattanzio R, Morgano
A, Abbondanza M, Malekzadeh R, Bishehsari F, Valanzano R, Russo A,
Piantelli M, et al: The insulin receptor substrate 1 (IRS1) in
intestinal epithelial differentiation and in colorectal cancer.
PLoS One. 7:e361902012. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Day E, Poulogiannis G, McCaughan F,
Mulholland S, Arends MJ, Ibrahim AE and Dear PH: IRS2 is a
candidate driver oncogene on 13q34 in colorectal cancer. Int J Exp
Pathol. 94:203–211. 2013.PubMed/NCBI
|
|
177
|
Slattery ML, Samowitz W, Curtin K, Ma KN,
Hoffman M, Caan B and Neuhausen S: Associations among IRS1, IRS2,
IGF1, and IGFBP3 genetic polymorphisms and colorectal cancer.
Cancer Epidemiol Biomarkers Prev. 13:1206–1214. 2004.PubMed/NCBI
|
|
178
|
Sanchez-Lopez E, Flashner-Abramson E,
Shalapour S, Zhong Z, Taniguchi K, Levitzki A and Karin M:
Targeting colorectal cancer via its microenvironment by inhibiting
IGF-1 receptor-insulin receptor substrate and STAT3 signaling.
Oncogene. 35:2634–2644. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Krajewski W, Dzięgała M, Kołodziej A,
Dembowski J and Zdrojowy R: Vitamin D and urological cancers. Cent
European J Urol. 69:139–147. 2016.PubMed/NCBI
|
|
180
|
Lefkowitz ES and Garland CF: Sunlight,
vitamin D, and ovarian cancer mortality rates in US women. Int J
Epidemiol. 23:1133–1136. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Uitterlinden AG, Fang Y, Van Meurs JB,
Pols HA and Van Leeuwen JP: Genetics and biology of vitamin D
receptor polymorphisms. Gene. 338:143–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Boscoe FP and Schymura MJ: Solar
ultraviolet-B exposure and cancer incidence and mortality in the
United States, 1993–2002. BMC Cancer. 6:2642006. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Bishop JE, Collins ED, Okamura WH and
Norman AW: Profile of ligand specificity of the vitamin D binding
protein for 1 alpha,25-dihydroxyvitamin D3 and its analogs. J Bone
Miner Res. 9:1277–1288. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Bikle D: Vitamin D: Production,
Metabolism, and Mechanisms of ActionEndotext [Internet]. Feingold
KR, Anawalt B, Boyce A, et al: MDText.com, Inc.; South Dartmouth,
MA: 2000
|
|
185
|
Feskanich D, Ma J, Fuchs CS, Kirkner GJ,
Hankinson SE, Hollis BW and Giovannucci EL: Plasma vitamin D
metabolites and risk of colorectal cancer in women. Cancer
Epidemiol Biomarkers Prev. 13:1502–1508. 2004.PubMed/NCBI
|
|
186
|
Braun MM, Helzlsouer KJ, Hollis BW and
Comstock GW: Colon cancer and serum vitamin D metabolite levels
10–17 years prior to diagnosis. Am J Epidemiol. 142:608–611. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Gorham ED, Garland CF, Garland FC, Grant
WB, Mohr SB, Lipkin M, Newmark HL, Giovannucci E, Wei M and Holick
MF: Vitamin D and prevention of colorectal cancer. J Steroid
Biochem Mol Biol. 97:179–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Garland C, Shekelle RB, Barrett-Connor E,
Criqui MH, Rossof AH and Paul O: Dietary vitamin D and calcium and
risk of colorectal cancer: A 19-year prospective study in men.
Lancet. 1:307–309. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Garland CF and Garland FC: Do sunlight and
vitamin D reduce the likelihood of colon cancer? Int J Epidemiol.
9:227–231. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Belleli A, Shany S, Levy J, Guberman R and
Lamprecht SA: A protective role of 1,25-dihydroxyvitamin D3 in
chemically induced rat colon carcinogenesis. Carcinogenesis.
13:2293–2298. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Comer PF, Clark TD and Glauert HP: Effect
of dietary vitamin D3 (cholecalciferol) on colon carcinogenesis
induced by 1,2-dimethylhydrazine in male Fischer 344 rats. Nutr
Cancer. 19:113–124. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Thomas MG, Tebbutt S and Williamson RC:
Vitamin D and its metabolites inhibit cell proliferation in human
rectal mucosa and a colon cancer cell line. Gut. 33:1660–1663.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Brehier A and Thomasset M: Human colon
cell line HT-29: Characterisation of 1,25-dihydroxyvitamin D3
receptor and induction of differentiation by the hormone. J Steroid
Biochem. 29:265–270. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Cross HS, Farsoudi KH and Peterlik M:
Growth inhibition of human colon adenocarcinoma-derived Caco-2
cells by 1,25-dihydroxyvitamin D3 and two synthetic analogs:
Relation to in vitro hypercalcemic potential. Naunyn Schmiedebergs
Arch Pharmacol. 347:105–110. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Vandewalle B, Adenis A, Hornez L,
Revillion F and Lefebvre J: 1,25-dihydroxyvitamin D3 receptors in
normal and malignant human colorectal tissues. Cancer Lett.
86:67–73. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Zhao X and Feldman D: Regulation of
vitamin D receptor abundance and responsiveness during
differentiation of HT-29 human colon cancer cells. Endocrinology.
132:1808–1814. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Carlberg C and Dunlop TW: An integrated
biological approach to nuclear receptor signaling in physiological
control and disease. Crit Rev Eukaryot Gene Expr. 16:1–22. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Padi SK, Zhang Q, Rustum YM, Morrison C
and Guo B: MicroRNA-627 mediates the epigenetic mechanisms of
vitamin D to suppress proliferation of human colorectal cancer
cells and growth of xenograft tumors in mice. Gastroenterology.
145:437–446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Fernandez-Garcia NI, Palmer HG, Garcia M,
Gonzalez-Martin A, del Rio M, Barettino D, Volpert O, Muñoz A and
Jimenez B: 1alpha,25-Dihydroxyvitamin D3 regulates the expression
of Id1 and Id2 genes and the angiogenic phenotype of human colon
carcinoma cells. Oncogene. 24:6533–6544. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Pendás-Franco N, Aguilera O, Pereira F,
González-Sancho JM and Muñoz A: Vitamin D and Wnt/β-catenin pathway
in colon cancer: Role and regulation of DICKKOPF genes. Anticancer
Res 28 (5A). 2613–2623. 2008.
|
|
201
|
Ylikomi T, Laaksi I, Lou YR, Martikainen
P, Miettinen S, Pennanen P, Purmonen S, Syvälä H, Vienonen A and
Tuohimaa P: Antiproliferative action of vitamin D. Vitam Horm.
64:357–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Taghizadeh F, Tang MJ and Tai IT:
Synergism between vitamin D and secreted protein acidic and rich in
cysteine-induced apoptosis and growth inhibition results in
increased susceptibility of therapy-resistant colorectal cancer
cells to chemotherapy. Mol Cancer Ther. 6:309–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Pálmer HG, Sánchez-Carbayo M,
Ordóñez-Morán P, Larriba MJ, Cordón-Cardó C and Muñoz A: Genetic
signatures of differentiation induced by 1alpha,25-dihydroxyvitamin
D3 in human colon cancer cells. Cancer Res. 63:7799–7806.
2003.PubMed/NCBI
|
|
204
|
Jensen SS, Madsen MW, Lukas J, Binderup L
and Bartek J: Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3)
on the G(1)-S phase-controlling machinery. Mol Endocrinol.
15:1370–1380. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Scaglione-Sewell BA, Bissonnette M,
Skarosi S, Abraham C and Brasitus TA: A vitamin D3 analog induces a
G1-phase arrest in CaCo-2 cells by inhibiting cdk2 and cdk6: Roles
of cyclin E, p21Waf1, and p27Kip1. Endocrinology. 141:3931–3939.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Liu W, Chen Y, Golan MA, Annunziata ML, Du
J, Dougherty U, Kong J, Musch M, Huang Y, Pekow J, et al:
Intestinal epithelial vitamin D receptor signaling inhibits
experimental colitis. J Clin Invest. 123:3983–3996. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Murillo G, Nagpal V, Tiwari N, Benya RV
and Mehta RG: Actions of vitamin D are mediated by the TLR4 pathway
in inflammation-induced colon cancer. J Steroid Biochem Mol Biol.
121:403–407. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Kim KE and Brasitus TA: The role of
vitamin D in normal and pathologic processes in the colon. Curr
Opin Gastroenterol. 17:72–77. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Revelli A, Massobrio M and Tesarik J:
Nongenomic effects of 1α,25-dihydroxyvitamin D(3). Trends
Endocrinol Metab. 9:419–427. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Norman AW, Song X, Zanello L, Bula C and
Okamura WH: Rapid and genomic biological responses are mediated by
different shapes of the agonist steroid hormone, 1α,25(OH)2vitamin
D3. Steroids. 64:120–128. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Sitrin MD, Bissonnette M, Bolt MJ, Wali R,
Khare S, Scaglione-Sewell B, Skarosi S and Brasitus TA: Rapid
effects of 1,25(OH)2 vitamin D3 on signal transduction systems in
colonic cells. Steroids. 64:137–142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Jeanes A, Gottardi CJ and Yap AS:
Cadherins and cancer: How does cadherin dysfunction promote tumor
progression? Oncogene. 27:6920–6929. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Rozen F, Yang XF, Huynh H and Pollak M:
Antiproliferative action of vitamin D-related compounds and
insulin-like growth factor-binding protein 5 accumulation. J Natl
Cancer Inst. 89:652–656. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Kaler P, Augenlicht L and Klampfer L:
Macrophage-derived IL-1beta stimulates Wnt signaling and growth of
colon cancer cells: A crosstalk interrupted by vitamin D3.
Oncogene. 28:3892–3902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Kaler P, Galea V, Augenlicht L and
Klampfer L: Tumor associated macrophages protect colon cancer cells
from TRAIL-induced apoptosis through IL-1beta-dependent
stabilization of Snail in tumor cells. PLoS One. 5:e117002010.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Chen A, Davis BH, Sitrin MD, Brasitus TA
and Bissonnette M: Transforming growth factor-beta 1 signaling
contributes to Caco-2 cell growth inhibition induced by
1,25(OH)(2)D(3). Am J Physiol Gastrointest Liver Physiol.
283:G864–G874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa
M, Watanabe M, Kashiwagi K, Toriyabe T, Kawabata M, Miyazono K and
Kato S: Convergence of transforming growth factor-beta and vitamin
D signaling pathways on SMAD transcriptional coactivators. Science.
283:1317–1321. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Deeb KK, Trump DL and Johnson CS: Vitamin
D signalling pathways in cancer: Potential for anticancer
therapeutics. Nat Rev Cancer. 7:684–700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Tong WM, Hofer H, Ellinger A, Peterlik M
and Cross HS: Mechanism of antimitogenic action of vitamin D in
human colon carcinoma cells: Relevance for suppression of epidermal
growth factor-stimulated cell growth. Oncol Res. 11:77–84.
1999.PubMed/NCBI
|
|
221
|
Ben-Shoshan M, Amir S, Dang DT, Dang LH,
Weisman Y and Mabjeesh NJ: 1alpha,25-dihydroxyvitamin D3
(Calcitriol) inhibits hypoxia-inducible factor-1/vascular
endothelial growth factor pathway in human cancer cells. Mol Cancer
Ther. 6:1433–1439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Harris DM and Go VL: Vitamin D and colon
carcinogenesis. J Nutr. 134 (Suppl):3463S–3471S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Lappe JM, Travers-Gustafson D, Davies KM,
Recker RR and Heaney RP: Vitamin D and calcium supplementation
reduces cancer risk: Results of a randomized trial. Am J Clin Nutr.
85:1586–1591. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Binder G, Wittekindt N and Ranke MB:
Noonan Syndrome: Genetics and Responsiveness to Growth Hormone
Therapy. Horm Res Paediatr. 67:45–49. 2007. View Article : Google Scholar
|
|
225
|
Campbell GS: Growth-hormone signal
transduction. J Pediatr. 131:S42–S44. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Savage MO, Blum WF, Ranke MB, Postel-Vinay
MC, Cotterill AM, Hall K, Chatelain PG, Preece MA and Rosenfeld RG:
Clinical features and endocrine status in patients with growth
hormone insensitivity (Laron syndrome). J Clin Endocrinol Metab.
77:1465–1471. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Gunawardane K, Hansen TK, Muller N,
Christiansen JS and Jorgensen JOL: Normal Physiology of Growth
Hormone in AdultsEndotext [Internet]. Feingold KR, Anawalt B and
Boyce A: MDText.com, Inc.; South Dartmouth, MA: 2000
|
|
228
|
Slater MD and Murphy CR: Co-expression of
interleukin-6 and human growth hormone in apparently normal
prostate biopsies that ultimately progress to prostate cancer using
low pH, high temperature antigen retrieval. J Mol Histol. 37:37–41.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Waters MJ and Conway-Campbell BL: The
oncogenic potential of autocrine human growth hormone in breast
cancer. Proc Natl Acad Sci USA. 101:14992–14993. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Brooks AJ and Waters MJ: The growth
hormone receptor: Mechanism of activation and clinical
implications. Nat Rev Endocrinol. 6:515–525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Brown-Borg HM and Bartke A: GH and IGF1:
Roles in energy metabolism of long-living GH mutant mice. J
Gerontol A Biol Sci Med Sci. 67:652–660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Chesnokova V, Zonis S, Zhou C, Recouvreux
MV, Ben-Shlomo A, Araki T, Barrett R, Workman M, Wawrowsky K,
Ljubimov VA, et al: Growth hormone is permissive for neoplastic
colon growth. Proc Natl Acad Sci USA. 113:E3250–E3259. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Koliarakis I, Messaritakis I, Nikolouzakis
TK, Hamilos G, Souglakos J and Tsiaoussis J: Oral Bacteria and
Intestinal Dysbiosis in Colorectal Cancer. Int J Mol Sci.
20:41462019. View Article : Google Scholar :
|