Introduction
A number of DNA-damaging chemotherapeutic agents
used for the treatment of adult T-cell leukemia-lymphoma (ATL)
result in cell death diametrically by inducing DNA damage. Examples
include etoposide and anthracyclines, which are currently the most
common treatments used in ATL (1,2). DNA
damage causes cell cycle arrest and cell death. Therefore, drug
resistance represents a challenge in the clinical treatment of
leukemia, as strengthened DNA damage repair (DDR) serves a crucial
role in the resistance of ATL cells to chemotherapy (3–5). Thus,
targeting DNA repair pathways may be an effective strategy for
eradicating leukemia cells resistant to chemotherapy.
An intact DDR system is critical for maintaining
genomic stability and cell proliferation (6). Although in eukaryotes, DNA double-strand
breaks (DSBs) are repaired by non-homologous end joining (NHEJ)
mechanisms or homologous recombination (HR) (2,7), HR has
high fidelity. Abnormal DSB repair induces various types of
chromosomal aberrations, including deletions (loss of
heterozygosity), aneuploidy and chromosomal translocations-events
which are particularly relevant in carcinogenesis (6).
DNA repair protein RAD51 homolog 1 (RAD51) is a
central protein in the homologous recombination (HR) repair pathway
(8). The role of RAD51 is to
recognize the homologous sequence and facilitate homologous pairing
and DNA strand exchange, as well as to complete DNA replication
using homologous sequences as a template (9,10). A
previous study revealed that RAD51 is overexpressed in several
tumors (11). The overexpression of
RAD51 is also associated with increased tumor metastasis (12), high tumor grade (13), treatment resistance (14) and poor overall survival rate (15). Knockout of RAD51 disrupts the HR
pathway and causes embryonic death in vertebrates, whereas high
expression of RAD51 increases HR efficiency and results in tumor
resistance (16). Therefore, the
focus of the present study was RAD51 as a treatment target, in
order to determine whether the recovery of its normal expression
level would reduce tumor resistance, and even increase its
sensitivity to DNA-damaging drugs.
Imatinib is a platelet-derived growth factor
receptor tyrosine kinase, which is an inhibitor of proto-oncogenes
c-KIT and c-ABL. Previous studies have demonstrated that imatinib
(Gleevec) reduces RAD51 protein repression (17,18).
Synthetic lethality via targeting the DDR pathways and HR defects
has had clinical success with breast cancer type 1 susceptibility
protein (BRCA1) functional loss and poly(ADP-ribose) polymerase
inhibition (PARPi) (19). Direct
inhibition of RAD51 with RNAi and small molecule inhibitors, such
as halenaquinone (20), B02 (21), RI-1 (22) and IBR2 (23) aims to reduce the expression of RAD51.
Indirect inhibition is predominantly achieved by functional damage
to RAD51 protein recombinase activity or interference with RAD51
protein-protein interactions, such as with tyrosine receptor kinase
inhibitors (TKIs) (24), histone
deacetylase inhibitors (HDACis) (25)
and methylamine Pterin (26). Novel
treatments targeted at RAD51 combined with PARPi have been widely
reported in combination with traditional cancer therapies (27,28). Thus,
the present study aimed to expand this innovative treatment to
other cancer types, particularly hematological malignancies.
In the present study, RAD51 was knocked down by
small hairpin (sh)RNA in Jurkat cells, one of the highest
RAD51-expressing ATL cell lines. Cell proliferation and apoptosis
under etoposide treatment was evaluated in cell culture. In
addition, DDR efficiency and apoptosis was examined following RAD51
silencing in response to etoposide in combination with imatinib
treatment.
Materials and methods
Cell lines
293T/17, Namalwa (human Burkitt lymphoma cell),
Karpas-299 (human anaplastic large cell lymphoma cells), Daudi
(human Burkitt lymphoma cells), HL-60 (human myeloid leukemia
cells), Su-DHL-4 (human diffuse large B-cell lymphoma cells),
Kasumi-1 (human acute myeloid leukemia cells), HEL (human
erythroleukemia leukemia cells), K562 (human chronic myeloid
leukemia cells), THP1 (human monocyte leukemia cells) and Jurkat
(human T lymphocytic leukemia cells) cell lines were purchased from
the Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai,
China). The lentivirus packaging cell line 293T/17 was maintained
in Dulbecco's modified Eagle's medium (DMEM; Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA). The remaining hematological tumor
cells were cultured in Iscove's modified Dulbecco's medium (IMDM)
or RPMI-1640 medium (Hyclone; GE Healthcare Life Sciences). Cell
cultures were supplemented with 0.1 U/ml streptomycin, 0.1 U/µl
penicillin and 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cells were cultured at
37°C in a humidified atmosphere of 5% CO2.
Gene knockdown
shRNA targeting RAD51 was cloned into the
pLVX-shRNA1 vector (Clontech Laboratories, Inc., Mountainview, CA,
USA) to create shRAD51 and scrambled negative control (SCR)
vectors. The RAD51 interference sequences were as follows:
shRNA-RAD51-1, 5′-GAAGCTATGTTCGCCATTA-3′; shRNA-RAD51-2,
5′-GCCAACGATGTGAAGAAATT-3′; shRNA-RAD51-3,
5′-AAGCTATGTTCGCCATTAA-3′; shRNA-RAD51-4,
5′-GCAGTGATGTCCTGGATAA-3′; SCR, 5′-GTTCTCCGAACGTGTCACGT-3′. 293T/17
cells with shRAD51 or SCR and lentivirus packaging plasmids were
co-transfected at a ratio of 4:3:2 using calcium phosphate
precipitation to produce lentiviral particles. Transduction was
performed in the presence of 5 µg/ml polybrene. Subsequent
experiments were performed at 72 h post-transduction.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from cell lines using TaKaRa
MiniBEST Universal RNA Extraction (Takara Biotechnology Co., Ltd.,
Dalian, China) according to the manufacturer's instructions.
First-strand cDNA was synthesized in a 10 µl reaction volume with
5X prime Script RT Master mix (Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. Relative mRNA
expression of RAD51 and GADPH was assessed by qPCR on an Applied
Biosystems 7500 Fast Real-Time PCR system (Thermo Fisher
Scientific, Inc.) with SYBR Premix Ex Taq™ (Takara Biotechnology
Co., Ltd.) according to the manufacturer's instructions. The
following primers were used: GAPDH forward,
5′-CTCTGATTTGGTCGTATTGGG-3′ and reverse,
5′-TGGAAGATGGTGATGGGATT-3′; RAD51 forward,
5′-GCCACCGCCCTTTACAGAACA-3′ and reverse,
5′-TGGGATCAGCAGCAAACATCG-3′. Data were analyzed using the
2−ΔΔCq method (29), where
ΔCq=(Cq target gene-Cq GAPDH).
Cell apoptosis analysis
Apoptosis assays were performed using a BD
Bioscience Annexin V-allophycocyanin (APC) staining kit according
to the manufacturer's protocols (BD Biosciences, Franklin Lakes,
NJ, USA). Cells were seeded in 24-well plates at a density of
1×105 cells/ml and cultured in a medium with etoposide
(20 µM) or PBS for 4 h at 37°C. Then, the cells were washed with
PBS three times and incubated for 48 h at 37°C. Cells
(5–10×105) were harvested, washed with PBS three times,
and stained with Annexin V-APC and propidium iodide for 15 min at
room temperature. Apoptotic cells were detected by flow
cytometry.
Colony-forming ability assay and
cytotoxicity test
For the clonogenic assay, soft agar culture was
performed with 1.2% agarose. After 10 days, the number of colonies
was counted using an inverted microscope, and the proliferation
ability of the cells was observed. For the cytotoxicity test, cells
were collected and the cell concentration was adjusted to
105/ml; 100 µl cell suspension was seeded in each well
of a 96-well plate and different concentrations of imatinib were
added and cultured at 37°C for 48 h; 10 µl Cell Counting Kit
(CCK)-8 reagent was then added and incubated for a further 4 h. The
absorbance at 450 nm was measured with a microplate reader.
Measurement of DNA repair
capacity
HR, NHEJ reporter cassettes and pDsRed-N1 as
internal controls were kindly provided by Dr Zhiyong Mao from the
School of Life Science and Technology of Tongji University. NHEJ or
HR reporter cassettes were linearized by I-SceI endonuclease and
purified using Monarch PCR & DNA Cleanup kit (cat. no. T1030S;
New England BioLabs, Inc., Ipswich, MA, USA). Cells were
transfected with 0.5 µg NHEJ reporter construct or 2 µg HR reporter
construct, and 0.2 µg of pDsRed-N1 as internal control.
Transfections were performed using an Amaxa Nucleofector
(Walkersville, MD, USA). Cells were analyzed by
fluorescence-activated cell sorting at 72 h post-transfection, as
previously described (30).
Immunoblotting and
immunofluorescence
Cells were collected and lysed in
radioimmunoprecipitation assay lysis buffer (EpiZyme Biotech,
Shanghai, China) lysis buffer for protein extraction. Protein
concentration was determined by a bicinchonic acid protein assay
and the supernatant was used for western blotting. Proteins were
separated by SDS-PAGE (10% gel) and transferred to polyvinylidene
difluoride membranes, and membranes were blocked in 5% non-fat dry
milk. Proteins were first incubated with anti-RAD1 (1:1,000; cat.
no. ab133534; Abcam, Cambridge, MA, USA) overnight at 4°C with
gentle rotation, and then incubated with horseradish
peroxidase-conjugated anti-rabbit IgG secondary antibody (cat. no.
7074S; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA)
at room temperature. Signal development was performed using an
enhanced chemiluminescence kit (EpiZyme Biotech).
For immunofluorescence analysis, cells grown on
coverslips were fixed in 4% paraformaldehyde, permeabilized with
PBS containing 0.1% Triton X-100 (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), and blocked with 5% bovine serum albumin (BSA)
for 30 min at room temperature. Rabbit anti-human phosphorylated
(γ-) H2A histone family member X (H2AX; 1:1,000; cat. no. ab11174;
Abcam) was diluted in 5% BSA and applied at 4°C overnight.
Following rinsing with PBS three times and incubating for 1 h with
donkey anti-rabbit secondary antibody (cat. no. 20308-1, 1:1,000;
Biotium, Hayward, CA, USA) at room temperature, the slides were
washed three times in PBS and the cell nuclei were stained with
DAPI (1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.,
Carlsbad, CA, USA) for 10 min at room temperature. Images were
captured with a Leica TCS SP2 confocal fluorescence microscope
(×20; Leica Microsystems GmbH, Wetzlar, Germany).
Statistical analysis
All statistical analyses were performed using Prism
6.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean ± standard deviation. All quantitative
experiments were conducted with a minimum of three independent
experiments. For data analysis, two-tailed t-test or one-way
analysis of variance followed by Tukey's post-hoc test were used.
P<0.05 was considered to indicate a statistically significant
difference. Flow cytometry data were analyzed with FCS Express 6
Flow software (De Novo Software, Glendale, CA, USA) and protein
expression was quantified using the ImageQuant R 4.2A software (GE
Healthcare Life Sciences).
Results
RAD51 is overexpressed in leukemia
cells
To determine the expression of RAD51 in leukemia,
the mRNA expression of RAD51 was measured in several cell lines by
qPCR. Kasumi, Namalwa, Karpas 299, HL-60, Daudi, HEL, THP1, Su-4,
K562 and Jurkat cells exhibited markedly higher RAD51 expression
compared with the Kasumi cells, which were used as a control. The
Jurkat cell line exhibited the highest level of RAD51 mRNA, which
was ~7.25 times that of Kasumi cells (Fig. 1A). The Kasumi cell line was chosen as
a control because it exhibited the lowest RAD51 expression of all
tumor cell lines tested. Immunoblot analysis demonstrated that
Jurkat cells exhibited markedly higher RAD51 protein expression
compared with the other four cell lines (Fig. 1B and C). Consequently, Jurkat cells
were selected for subsequent experiments.
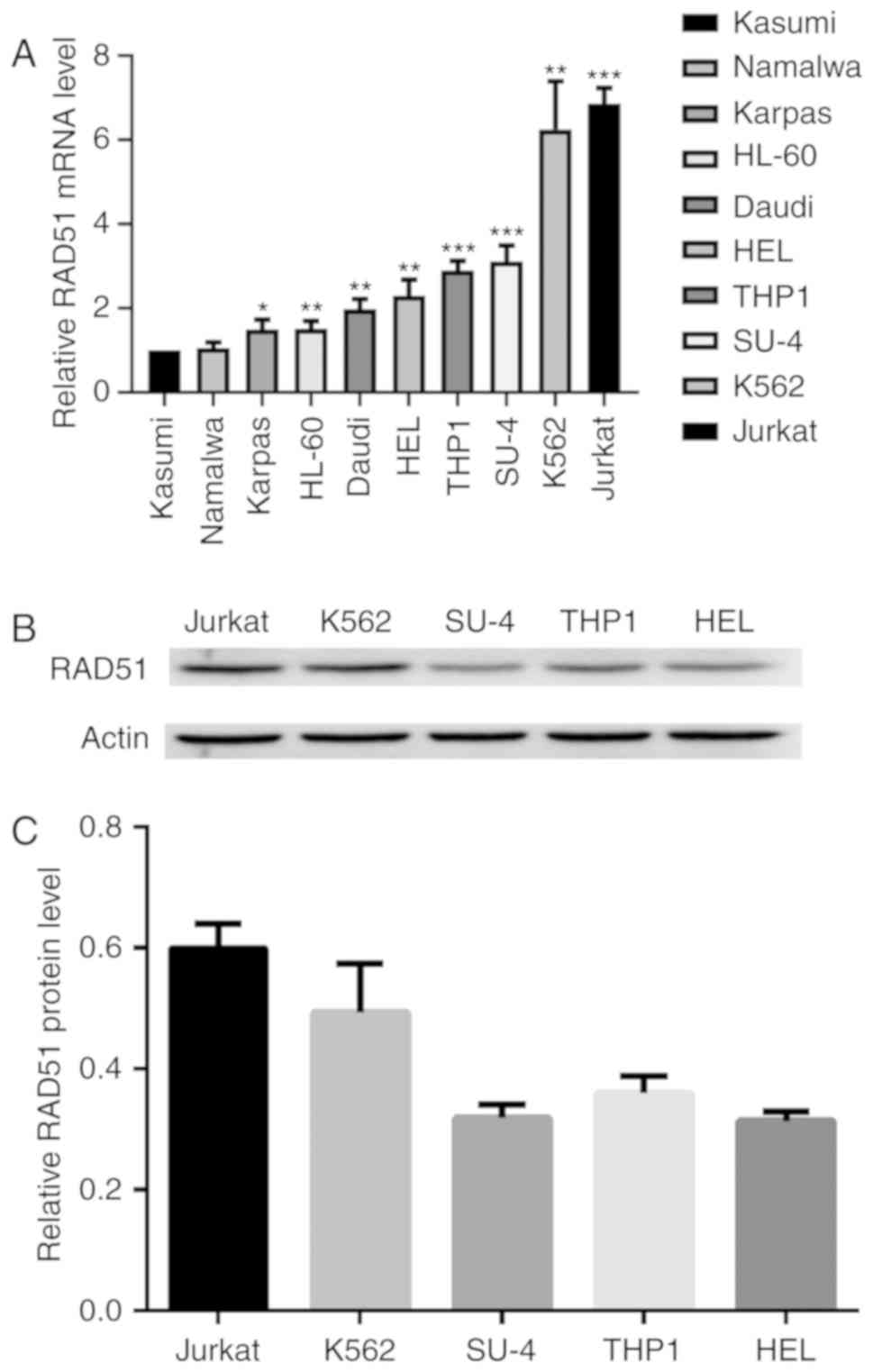 | Figure 1.RAD51 expression is increased in
leukemia cells. (A) RAD51 mRNA expression in Kasumi, Namalwa,
Karpas, HL-60, Daudi, HEL, THP1, Su-DHL-4, K562, Jurkat cells was
analyzed by reverse transcription-quantitative polymerase chain
reaction relative to GAPDH as the internal control. *P<0.05,
**P<0.01 ***P<0.001 vs. Kasumi. (B) Protein expression levels
of RAD51 and β-actin in the cell lysates of Jurkat, K562, Su-DHL-4,
THP1 and HEL cells was determined by immunoblotting. (C) RAD51
protein expression was quantified by densitometric analysis and
normalized to GAPDH expression. The normalized RAD51 expression
levels of Jurkat, K562, Su-DHL-4, THP1 and HEL are shown.
Experiments were repeated at least three times. RAD51, DNA repair
protein RAD51 homolog 1. |
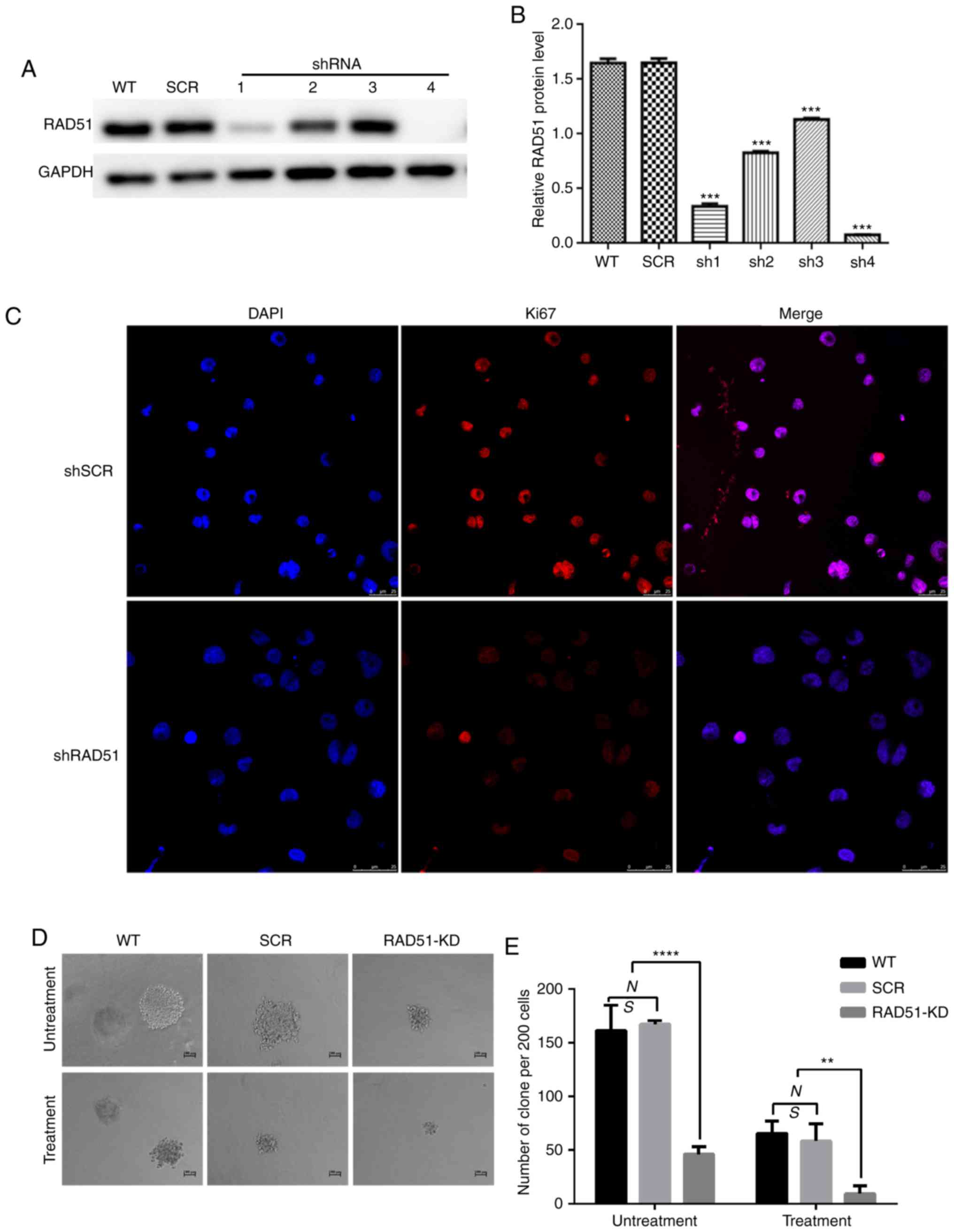
ShRNA-mediated down regulation of
RAD51 in leukemia cells
In order to determine whether the expression of
RAD51 in infected Jurkat cells was decreased, and whether the four
interferon fragments of RAD51 were designed to effectively degrade
RAD51 mRNA, Jurkat cells were infected with lentivirus expressing
shRNA targeting RAD51 (shRAD51-KD) or normal control (shRNA-SCR).
As shown in Fig. 2A and B, RAD51-sh4
exerted a prominent interference effect. Consequently, RAD51-sh4
was selected for subsequent experiments.
RAD51 knockdown reduces cell
proliferation and induces apoptosis of Jurkat cells
To elucidate the role of RAD51 in Jurkat cell DNA
damage, RAD51 was downregulated with shRNA and the colony-forming
ability and apoptotic rate was assessed following etoposide
treatment. Ki67 is a well-known cell proliferation activity marker
protein that reflects the proliferative activity of tumor cells
(31). As shown in Fig. 2C, the results revealed that the
expression of Ki67 in RAD51-KD cells was notably lower compared
with that in SCR. Similarly, shRNA-mediated downregulation of RAD51
reduced the colony-forming ability of Jurkat cells by 3-fold under
normal growth conditions, and further decreased the colony-forming
ability of Jurkat cells by 4-fold following etoposide treatment
(Fig. 2E). Furthermore, the colony
size of Jurkat cells with RAD51 shRNA was markedly smaller compared
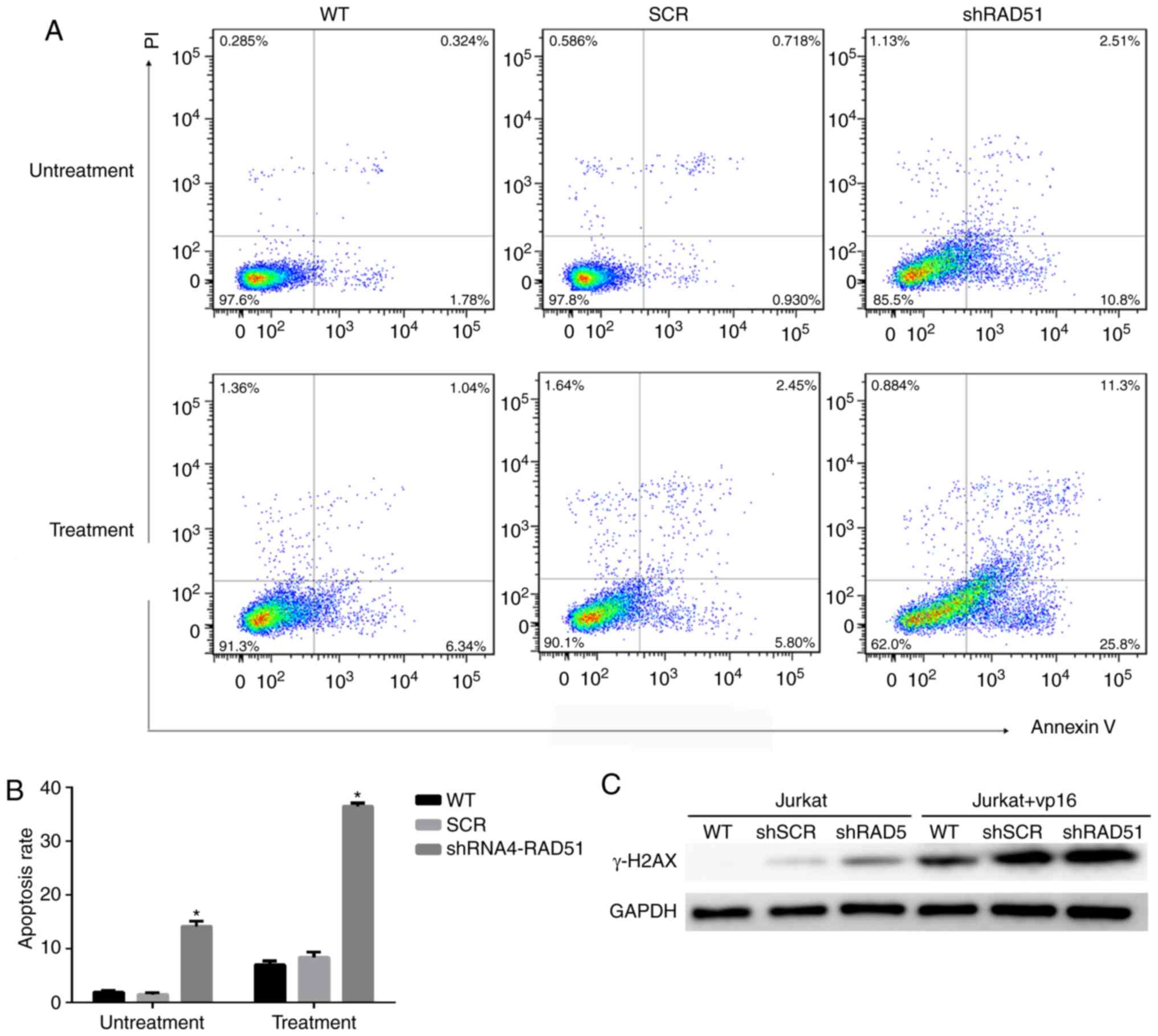
with that of cells in the WT and control SCR groups (Fig. 2D). The apoptotic rate was examined by
Annexin V staining coupled with flow cytometry. As shown in
Fig. 3A and B, the mean apoptotic
rate of Jurkat cells in the RAD51KD group was 14.12%, compared with
1.92 and 1.47% in the two negative control groups. Following
etoposide treatment, the apoptotic rate of RAD51KD group was
36.42%, compared with 6.98 and 8.38% in the control groups,
respectively (P<0.05). Taken together, these results
demonstrated that RAD51 silencing results in reduced cell viability
and an increased apoptotic rate under normal growth conditions with
etoposide treatment.
Downregulation of RAD51 alters the
efficiency of DDR in Jurkat cells
To investigate whether the increase in Jurkat cell
apoptosis was associated with a decrease in DDR function, the
expression of H2A histone family member X (H2AX) and the efficiency
of DDR was measured in a quantitative manner in Jurkat cells. The
phosphorylation of H2AX (γ-H2AX), is a sign of DNA DSBs (32). Therefore, γ-H2AX protein expression
was detected in cells treated with or without etoposide. The
expression of γ-H2AX was higher in Jurkat cells transduced with
RAD51 shRNA compared with that with control shRNA, whereas γ-H2AX
was notably higher in Jurkat cells transduced with RAD51 shRNA
following etoposide treatment (Fig.
3C), indicating that RAD51 was indispensable for the repair of
DSBs in Jurkat cells. Furthermore, fluorescent reporter constructs
were used in which a functional GFP gene was reconstituted
following an NHEJ or HR event (Fig.
4A). Notably, the results demonstrated that inhibition of RAD51
by shRNA reduced the efficiency of HR, but increased that of NHEJ
(P<0.05; Fig. 4B).
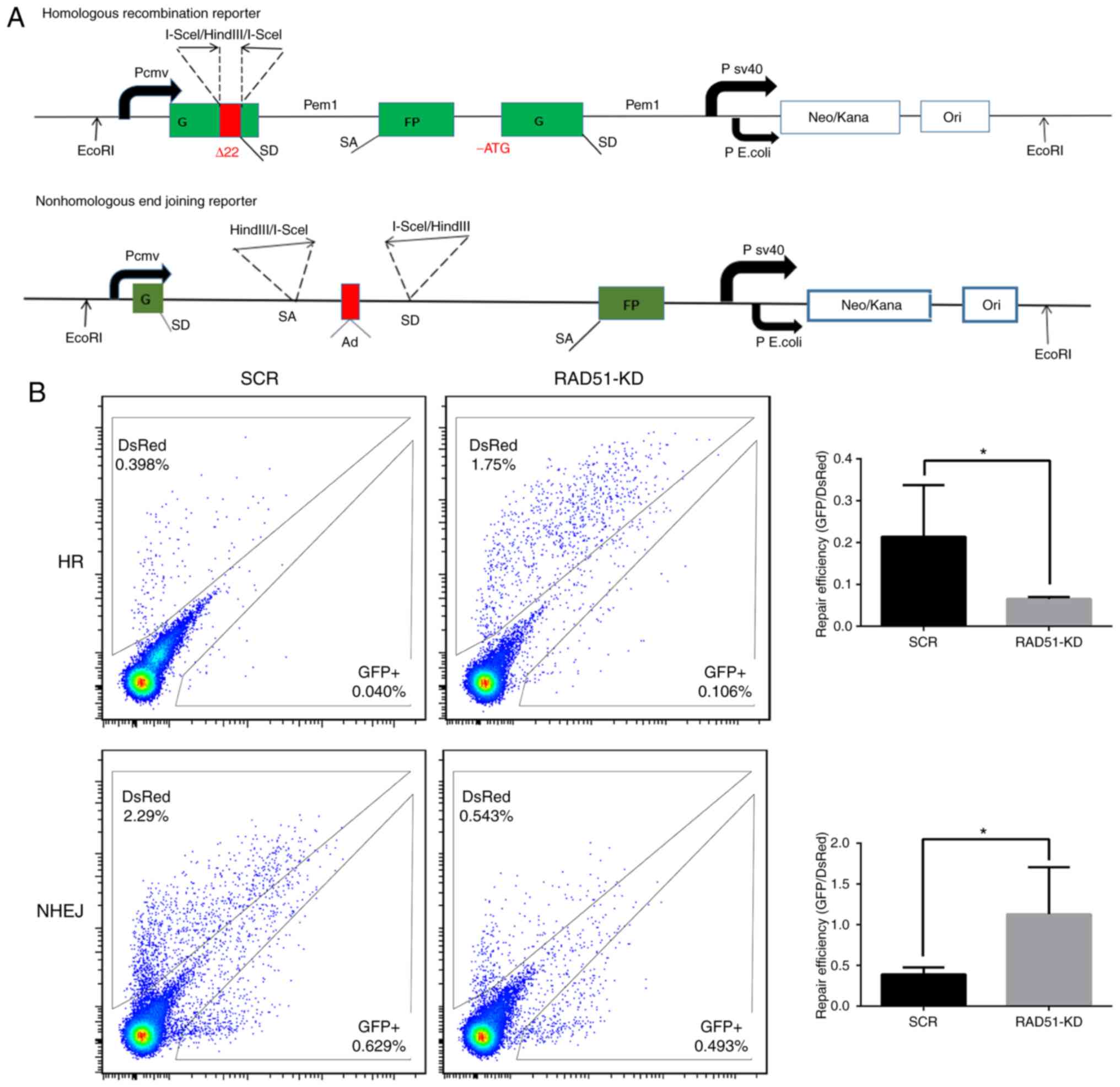 | Figure 4.Inhibition of RAD51 impairs DNA
repair in Jurkat cells. (A) Fluorescent reporter constructs were
used to measure NHEJ or HR events. (B) Analysis of HR and NHEJ in
SCR and shRAD51 Jurkat cells. Flow cytometric analysis results
(left panel) show the gating for the analysis of GFP+
and DsRed+ cells using cells transfected with GFP or
DsRed expression vectors, as well as cells transfected with a
negative control plasmid to exclude auto-fluorescent cells. The
ratio of GFP+ to DsRed+ cells (right panel),
which was used as a measure of repair efficiency, was also
presented. Experiments were repeated at least three times.
*P<0.05. RAD51, DNA repair protein RAD51 homolog 1; NHEJ,
non-homologous end joining; HR, homologous recombination; GFP,
green fluorescent protein; sh, small hairpin RNA; SCR, scramble;
KD, knockdown. |
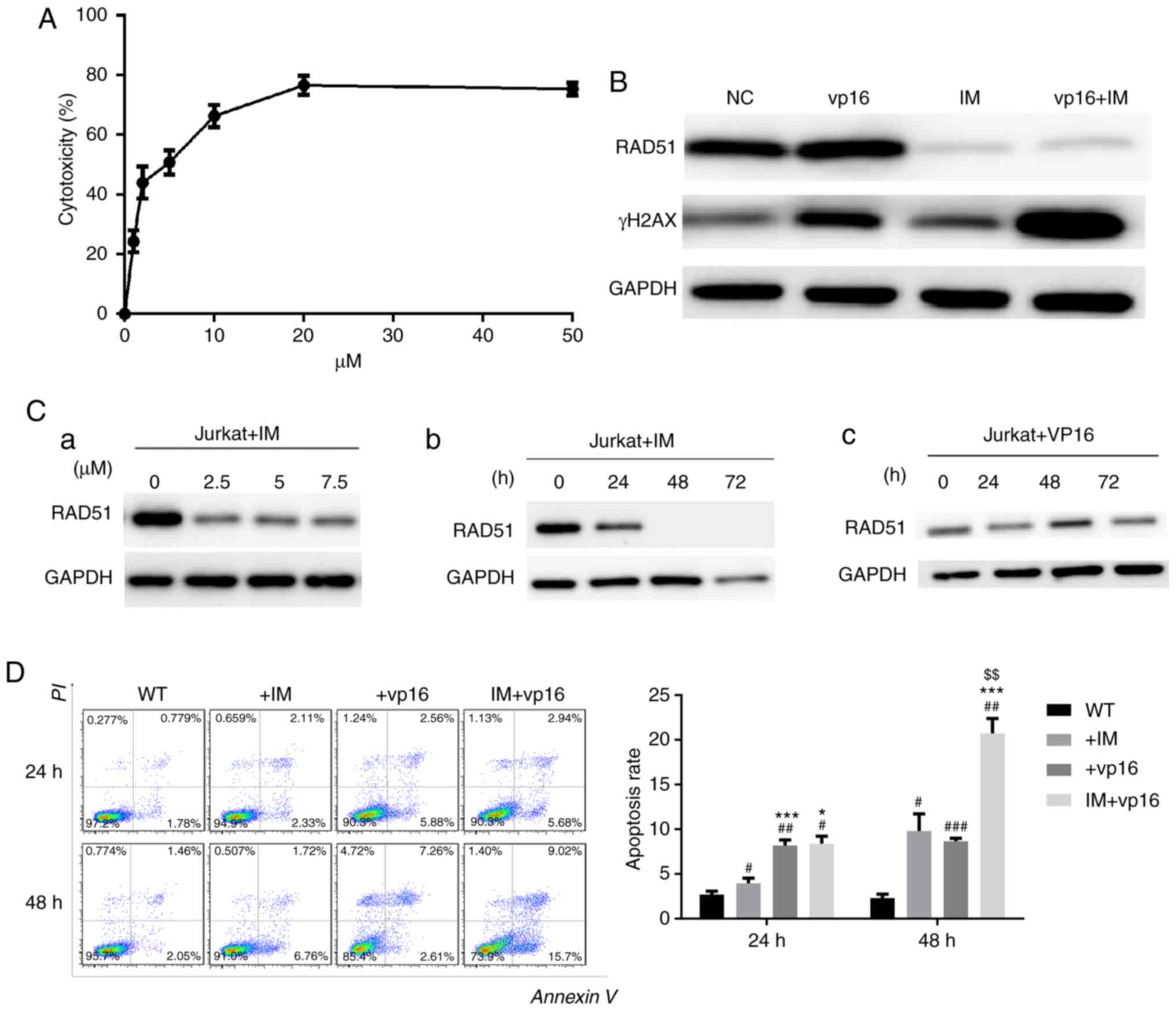
Imatinib inhibits the expression of
RAD51 and enhances chemosensitivity in Jurkat cells
To test the toxicity of imatinib on Jurkat cells,
CCK-8 assays were performed. Next, DSB levels in Jurkat cells
following RAD51 knockdown were determined, as well as the apoptotic
rate in cells treated with or without etoposide. The results
demonstrated that the IC50 of imatinib toxicity to
Jurkat cells was ~5 µM, and the toxic effect of imatinib on Jurkat
cells at 20 µM stabilized (Fig.
5A).
Using the IC50 of imatinib obtained from
the experiment described above, imatinib reduced RAD51 protein
expression, and this reduction in RAD51 was dose- and
time-dependent (Fig. 5C-a and -b). In
contrast, etoposide (VP16), which was used as a reference, did not
reduce RAD51 expression, but increased it at 48 h, which may be
associated with DDR initiation (Fig.
5C-c). The expression of γ-H2AX demonstrated that the
combination therapy group had the most severe DSB injury, i.e., the
weakest ability to repair DNA damage (Fig. 5B). Similarly, apoptosis evaluation
indicated that combination therapy markedly increased Jurkat cell
apoptosis. These results suggested that imatinib may specifically
reduce RAD51 protein (Fig. 5D) in a
dose- and time-dependent manner, and this downregulation of RAD51
by imatinib triggers apoptosis in Jurkat cells following etoposide
treatment.
Discussion
In the present study, it was demonstrated that RAD51
was overexpressed in Jurkat cells, whereas RAD51 downregulation by
shRNA and imatinib decreased cell viability and increased apoptosis
in Jurkat cells treated with etoposide. Furthermore, RAD51
downregulation resulted in impaired DNA repair capacity,
accompanied by decreased HR. Furthermore, downregulation of RAD51
by imatinib markedly increased the rate of apoptosis in combination
with etoposide treatment. These results suggested that targeting
RAD51 may be a potential strategy for the sensitization of ATL to
DNA damage-based chemotherapy.
RAD51 is a central protein in HR repair pathways,
which serves a vital role in the pathogenesis and drug resistance
of leukemia (33,34). Although several tumor cells have been
found to overexpress RAD51 (11,15), the
underlying reason remains unclear. An increasing number of studies
have demonstrated that mammalian RAD51 proteins are linked directly
or indirectly to a number of other proteins, such as anti-tumor
factors serine-protein kinase ATM (35), p53 (36), BRCA1/BRCA2 (27,28),
proto-oncogene c-Abl (37) and
SUMO-conjugating enzyme UBC9 (38).
This prompted us to investigate the role of RAD51 in tumor
development.
In the present study, downregulation of RAD51 by
shRNA and imatinib reduced the efficiency of HR repair and
increased chemosensitivity and apoptosis in ATL. These results
suggested that RAD51 has the potential to become a novel target for
the clinical treatment of acute leukemia, with the hope of
increasing ATL patient survival rate.
Etoposide is a topoisomerase II (TOP2) inhibitor
which is widely used as an anticancer drug. It increases the
expression of the TOP2 cleavage complex and thus increases
TOP2-mediated chromosome DNA breakage (39,40). In
the present study, cells were treated with etoposide at a
concentration of 20 µM for 4 h with etoposide to induce DNA damage
(41), and all the analyses were
performed 48 h later. The results demonstrated that downregulation
of RAD51 reduced cell proliferation and increased the rate of
apoptosis after etoposide treatment-induced DNA damage. This result
may be related to DDR capacity, particularly HR abnormalities. The
result of the subsequent experiments demonstrated showed that HR
pathway repair in Jurkat cells was attenuated by RAD51 knockdown ed
the rate of apoptosis following DNA damage. This result may be
related to DDR capacity, particularly HR abnormalities. The result
of the subsequent experiments demonstrated showed that HR pathway
repair in Jurkat cells was attenuated by RAD51 knockdown and
switched to the NHEJ pathway. This alteration resulted in an
increase in apoptosis resulting from incomplete repair of Jurkat
cells. A limitation of the present study was that whether key
proteins in the NHEJ pathway were upregulated was not determined,
so changes in the repair pathways could not be further
confirmed.
Several small molecule RAD51 inhibitors have been
developed (42,43). However, they lack specificity for
RAD51 and are available only for in vitro studies of RAD51
activity. As shRNA is not currently applied in clinical treatment,
inhibition of RAD51 with imatinib was also used in the current
study (24). Imatinib is the
first-line therapy for chronic myelocytic leukemia. It has been
reported that imatinib treatment reduces the expression of RAD51
and is closely associated with reduced HR in tumor cell lines with
different p53 states (18). Treatment
of tumor cells with imatinib enhances sensitivity (24), but this effect does not occur in
normal fibroblasts. In irradiated tumors, mitomycin, gemcitabine
combined with imatinib decreases tumor cell proliferation. This
synergistic effect was also demonstrated in vivo using a PC3
mouse tumor model: Combination of imatinib and radiotherapy alone
significantly delayed tumor growth, at least partially due to a
decrease in RAD51 expression (24).
The results of the present study demonstrated that imatinib reduced
RAD51 protein in ATL cells in a dose- and time-dependent manner.
Therefore, the combined treatment of imatinib and chemotherapeutic
drugs may be useful for the treatment of hematological tumors.
Imatinib reduced the expression of RAD51, but the exact mechanism
of how imatinib reduces RAD51 expression has not been fully
elucidated; this requires further investigation in future
experiments. For more far-reaching mechanisms, it will be necessary
to determine the DNA damage response caused by RAD51
overexpression.
In conclusion, the RAD51 protein is key to HR repair
pathways and was involved in the occurrence and drug resistance of
leukemia. Increased expression of RAD51 recombination protein in
various tumors is a common phenomenon (11). Acute leukemia is a malignancy with
poor treatment outcomes (3). Although
RNAi technology targets gene activity by silencing and has very
high specificity, the clinical application of siRNA is currently
limited by its off-target effects and short life span. The
limitations of this study include the lack of data from peripheral
blood samples and a non-cancerous cell line. In the present study,
no normal peripheral blood samples or non-cancerous cell lines were
used as negative controls; therefore, the experimental results can
only indicate that RAD51 may serve an important role in blood
cancer cell lines.
In the present experiment, RAD51 knockdown decreased
the repair efficiency of Jurkat cells and increased their
chemosensitivity, ultimately leading to cell apoptosis. Based on
these results, RAD51 appears to be promising as a novel target for
the clinical treatment of leukemia, and it may improve the survival
of leukemia patients.
Acknowledgements
Not applicable.
Funding
This study was supported by Ministry of Science and
Technology of China (grant no. 2016YFE0107200) and the National
Natural Science Foundation of China (grant no. 81770151).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY, XT and ZF designed the research and performed
experiments. WY, ZL, WZ, JZ and AL collected the samples, and
participated in the collection and analysis of data. XT wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rich T, Allen R and Wyllie AH: Defying
death after DNA damage. Nature. 407:777–783. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nature Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Linker C, Damon L, Ries C and Navarro W:
Intensified and shortened cyclical chemotherapy for adult acute
lymphoblastic leukemia. J Clin Oncol. 20:2464–2471. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Grady S, Finn SP, Cuffe S, Richard DJ,
O'Byrne KJ and Barr MP: The role of DNA repair pathways in
cisplatin resistant lung cancer. Cancer Treat Rev. 40:1161–1170.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hijiya N, Thomson B, Isakoff MS, Silverman
LB, Steinherz PG, Borowitz MJ, Kadota R, Cooper T, Shen V, Dahl G,
et al: Phase 2 trial of clofarabine in combination with etoposide
and cyclophosphamide in pediatric patients with refractory or
relapsed acute lymphoblastic leukemia. Blood. 118:6043–6049. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoeijmakers JH: Genome maintenance
mechanisms for preventing cancer. Nature. 411:366–374. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kanaar R, Hoeijmakers JH and van Gent DC:
Molecular mechanisms of DNA double strand break repair. Trends Cell
Biol. 8:483–489. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arnaudeau C, Helleday T and Jenssen D: The
RAD51 protein supports homologous recombination by an exchange
mechanism in mammalian cells. J Mol Biol. 289:1231–1238. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogawa T, Yu X, Shinohara A and Egelman EH:
Similarity of the yeast RAD51 filament to the bacterial RecA
filament. Science. 259:1896–1899. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suwaki N, Klare K and Tarsounas M: RAD51
paralogs: Roles in DNA damage signalling, recombinational repair
and tumorigenesis. Semin Cell Dev Biol. 22:898–905. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Raderschall E, Stout K, Freier S, Suckow
V, Schweiger S and Haaf T: Elevated levels of Rad51 recombination
protein in tumor cells. Cancer Res. 62:219–225. 2002.PubMed/NCBI
|
|
12
|
Wiegmans AP, Al-Ejeh F, Chee N, Yap PY,
Gorski JJ, Da Silva L, Bolderson E, Chenevix-Trench G, Anderson R,
Simpson PT, et al: Rad51 supports triple negative breast cancer
metastasis. Oncotarget. 5:3261–3272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maacke H, Opitz S, Jost K, Hamdorf W,
Henning W, Krüger S, Feller AC, Lopens A, Diedrich K, Schwinger E
and Stürzbecher HW: Over-expression of wild-type Rad51 correlates
with histological grading of invasive ductal breast cancer. Int J
Cancer. 88:907–913. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Short SC, Giampieri S, Worku M,
Alcaide-German M, Sioftanos G, Bourne S, Lio KI, Shaked-Rabi M and
Martindale C: Rad51 inhibition is an effective means of targeting
DNA repair in glioma models and CD133+ tumor-derived
cells. Neuro Oncol. 13:487–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barbano R, Copetti M, Perrone G, Pazienza
V, Muscarella LA, Balsamo T, Storlazzi CT, Ripoli M, Rinaldi M,
Valori VM, et al: High RAD51 mRNA expression characterize estrogen
receptor-positive/progesteron receptor-negative breast cancer and
is associated with patient's outcome. Int J Cancer. 129:536–545.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Richardson C: RAD51, genomic stability,
and tumorigenesis. Cancer Lett. 218:127–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kubler HR, van Randenborgh H, Treiber U,
Wutzler S, Battistel C, Lehmer A, Lehmer A, Wagenpfeil S, Hartung R
and Paul R: In vitro cytotoxic effects of imatinib in combination
with anticancer drugs in human prostate cancer cell lines.
Prostate. 63:385–394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ishida T, Takizawa Y, Kainuma T, Inoue J,
Mikawa T, Shibata T, Suzuki H, Tashiro S and Kurumizaka H: DIDS, a
chemical compound that inhibits RAD51-mediated homologous pairing
and strand exchange. Nucleic Acids Res. 37:3367–3376. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takaku M, Kainuma T, Ishida-Takaku T,
Ishigami S, Suzuki H, Tashiro S, van Soest RW, Nakao Y and
Kurumizaka H: Halenaquinone, a chemical compound that specifically
inhibits the secondary DNA binding of RAD51. Genes Cell.
16:427–436. 2011. View Article : Google Scholar
|
|
21
|
Huang F, Motlekar NA, Burgwin CM, Napper
AD, Diamond SL and Mazin AV: Identification of specific inhibitors
of human RAD51 recombinase using high-throughput screening. ACS
Chem Biol. 6:628–635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Budke B, Kalin JH, Pawlowski M,
Zelivianskaia AS, Wu M, Kozikowski AP and Connell PP: An optimized
RAD51 inhibitor that disrupts homologous recombination without
requiring Michael acceptor reactivity. J Med Chem. 56:254–263.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G,
Qiu XL, Chen CF, Hu CM, Goldblatt E, et al: A novel small molecule
RAD51 inactivator overcomes imatinib-resistance in chronic myeloid
leukaemia. EMBO Mol Med. 5:353–365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choudhury A, Zhao H, Jalali F, Al Rashid
S, Ran J, Supiot S, Kiltie AE and Bristow RG: Targeting homologous
recombination using imatinib results in enhanced tumor cell
chemosensitivity and radiosensitivity. Mol Cancer Ther. 8:203–213.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kachhap SK, Rosmus N, Collis SJ,
Kortenhorst MS, Wissing MD, Hedayati M, Shabbeer S, Mendonca J,
Deangelis J, Marchionni L, et al: Downregulation of homologous
recombination DNA repair genes by HDAC inhibition in prostate
cancer is mediated through the E2F1 transcription factor. PLoS One.
5:e112082010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Du LQ, Du XQ, Bai JQ, Wang Y, Yang QS,
Wang XC, Zhao P, Wang H, Liu Q and Fan FY: Methotrexate-mediated
inhibition of RAD51 expression and homologous recombination in
cancer cells. J Cancer Res Clin Oncol. 138:811–818. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scully R, Chen J, Plug A, Xiao Y, Weaver
D, Feunteun J, Ashley T and Livingston DM: Association of BRCA1
with Rad51 in mitotic and meiotic cells. Cell. 88:265–275. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sharan SK, Morimatsu M, Albrecht U, Lim
DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P and Bradley A:
Embryonic lethality and radiation hypersensitivity mediated by
Rad51 in mice lacking Brca2. Nature. 386:804–810. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seluanov A, Mao Z and Gorbunova V:
Analysis of DNA double-strand break (DSB) repair in mammalian
cells. J Vis Exp. 8:20022010.
|
|
31
|
Scholzen T and Gerdes J: The Ki-67
protein: From the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maacke H, Jost K, Opitz S, Miska S, Yuan
Y, Hasselbach L, Lüttges J, Kalthoff H and Stürzbecher HW: DNA
repair and recombination factor Rad51 is over-expressed in human
pancreatic adenocarcinoma. Oncogene. 19:2791–2795. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Henning W and Stürzbecher HW: Homologous
recombination and cell cycle checkpoints: Rad51 in tumour
progression and therapy resistance. Toxicology. 193:91–109. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen G, Yuan SS, Liu W, Xu Y, Trujillo K,
Song B, Cong F, Goff SP, Wu Y, Arlinghaus R, et al:
Radiation-induced assembly of Rad51 and Rad52 recombination complex
requires ATM and c-Abl. J Biol Chem. 274:12748–12752. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stürzbecher HW, Donzelmann B, Henning W,
Knippschild U and Buchhop S: P53 is linked directly to homologous
recombination processes via RAD51/RecA protein interaction. EMBO J.
15:1992–2002. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan ZM, Huang Y, Ishiko T, Nakada S,
Utsugisawa T, Kharbanda S, Wang R, Sung P, Shinohara A,
Weichselbaum R and Kufe D: Regulation of Rad51 function by c-Abl in
response to DNA damage. J Biol Chem. 273:3799–3802. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saitoh H, Pizzi MD and Wang J:
Perturbation of SUMOlation enzyme Ubc9 by distinct domain within
nucleoporin RanBP2/Nup358. J Biol Chem. 277:4755–4763. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nitiss JL: Targeting DNA topoisomerase II
in cancer chemotherapy. Nat Rev Cancer. 9:338–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu
YJ, Yen TJ, Chiang CW and Chan NL: Structural basis of type II
topoisomerase inhibition by the anticancer drug etoposide. Science.
333:459–462. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Muslimovic A, Nystrom S, Gao Y and
Hammarsten O: Numerical analysis of etoposide induced DNA breaks.
PLoS One. 4:e58592009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Z, Sun B, Clewell RA, Adeleye Y,
Andersen ME and Zhang Q: Dose-response modeling of
etoposide-induced DNA damage response. Toxicol Sci. 137:371–384.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, He Y and Luo Y: Crystal structure of
an archaeal Rad51 homologue in complex with a metatungstate
inhibitor. Biochemistry. 48:6805–6810. 2009. View Article : Google Scholar : PubMed/NCBI
|