Introduction
Hepatocellular carcinoma (HCC) is the fifth most
prevalent cancer worldwide, with a vast majority of patients having
intermediate to advanced stage disease upon diagnosis. Accordingly,
the recommended therapy for such patients includes transarterial
chemoembolization, molecular-targeted therapy, or checkpoint
immunotherapy (1). However,
systemic chemotherapies have demonstrated limited efficacy in the
treatment of advanced HCC (2).
Indeed, one study showed that FOLFOX4 (fluorouracil, leucovorin and
oxaliplatin) chemotherapy for patients with advanced HCC exhibited
marginal benefits (3). A plethora
of mechanisms have explained the chemoresistance of HCC, such as
P-glycoprotein (4), cancer cell
stemness (5), DNA repair (6), evasion of apoptosis (7) and autophagy (8).
Mitophagy is a selective form of autophagy that
removes malfunctioning or damaged mitochondria and thus maintains
cellular homeostasis. Mitophagy is associated with many
neurodegenerative disorders and cancer (9). For instance, the dysfunction caused by
mutations in phosphatase and tensin homolog-induced putative kinase
1 (PINK1) can impair mitophagy, while accumulation of dysregulated
mitochondria results in neuron apoptosis, which may account for
Parkinson's disease (10). On the
other hand, mitophagy can facilitate cancer cell survival by
instantly clearing damaged mitochondria (11).
Mitophagy involves three main stages (9): Mitochondrial fission, autophagosome
assembly, and fusion with lysosomes to degrade damaged
mitochondria. The first step involves the scission of mitochondria
controlled mainly by dynamin-related protein 1 (DRP1) (12), which functions to fragment large
mitochondria into smaller ones (13). Given that DRP1-dependent
mitochondrial fission is critical for mitophagy, we hypothesized
that targeting DRP1 may influence cancer cell resistance to
treatment.
Here, we showed that i) cisplatin induced mitophagy
in surviving HCC cells by activating DRP1; ii) DRP1 inhibitor
Mdivi-1 increased the apoptosis of cisplatin-treated HCC cells by
targeting mitophagy; iii) Mdivi-1 downregulated Bcl-xL and
upregulated Bax, thereby facilitating cytochrome c leakage
from the damaged mitochondria into the cytosol; iv) Mdivi-1 acted
synergistically with cisplatin to augment HCC apoptosis in
vivo. Therefore, it was demonstrated that targeting
DRP1-mediated mitophagy could be a potential approach toward
enhancing chemotherapeutic efficacy for HCC, tipping the balance in
favor of cancer cell apoptosis.
Materials and methods
Reagents
Cisplatin and Mdivi-1 were purchased from MedChem
Express, while DMSO was obtained from Sigma-Aldrich/Merck KGaA.
Primary antibodies against LC3B (cat. no. 3868; dilution 1:1,000;
Cell Signaling Technology), phospho-Ser616-DRP1 (cat. no. 3455;
dilution 1:1,000; Cell Signaling Technology), HSP60 (cat. no.
12165; dilution 1:1,000; Cell Signaling Technology), cleaved
caspase-3 (cat. no. 9661; dilution 1:1,000; Cell Signaling
Technology), Bax (cat. no. 5023; dilution 1:1,000; Cell Signaling
Technology) and Bcl-xL (cat. no. 2764; dilution 1:1,000; Cell
Signaling Technology), phospho-Ser139-histone H2AX (γ-H2AX) (cat.
no. ab2893; dilution 1:1,000; Abcam), DRP1 (cat. no. ab184274;
dilution 1:1,000; Abcam), cytochrome c (cat. no. ab133504;
dilution 1:1,000; Abcam) and CoxIV (cat. no. ab202554; dilution
1:1,000; Abcam), TOM20 (cat. no. sc-17764; dilution 1:200; Santa
Cruz Biotechnology, Inc.) and β-actin (cat. no. AA132; dilution
1:1,000; Beyotime Biotechnology, China) were used for western blot
analysis.
Cells and cell culture
HCC cell lines MHCC97H (Liver Cancer Institute,
Zhongshan Hospital, Fudan University, Shanghai, China) and Huh7
(obtained from the Japanese Cancer Research Resources Bank, Tokyo,
Japan) were cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Thermo Fisher Scientific, Inc.). Cells
were incubated at 37°C in a 5% CO2 atmosphere. After
reaching 70–80% confluency, the cells were subjected to subsequent
experiments.
Western blotting
To obtain total proteins, cells were lysed by
radioimmunoprecipitation assay (RIPA) buffer (Beyotime
Biotechnology) supplemented with protease inhibitor cocktail (Weao
Biotechnology). Mitochondrial protein extraction was performed
using a mitochondrial isolation kit (Beyotime Biotechnology)
according to the manufacturer's protocol. Protein concentration was
determined using an enhanced BCA Protein assay kit (Beyotime
Biotechnology). Proteins (30 µg per lane) were loaded onto 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) gels and then separated. Thereafter, the gels were
transferred to polyvinylidene difluoride (PVDF) membranes
(Millipore). After being blocked with 5% bovine serum albumin (BSA)
for 1 h, the membranes were incubated with the primary antibodies
at 2–4°C overnight. Next, the membranes were incubated with the
appropriate secondary antibodies (anti-mouse IgG HRP-linked
antibody, dilution 1:4,000; cat. no. 7076, Cell Signaling
Technology; anti-biotin D5A7 rabbit monoclonal antibody, HRP
conjugate, dilution 1:4,000; cat. no. 5571, Cell Signaling
Technology) for about 1 h at room temperature. After being washed
three times, the membranes were visualized using High-Signal ECL
Substrate (Tanon).
Immunofluorescence staining
Cells or tissues were fixed with paraformaldehyde
for 20 min and then permeabilized with 0.3% Triton X-200 (Beyotime
Biotechnology) for 10 min to enhance specimen permeability.
Thereafter, the specimens were blocked with 10% BSA (Roche) and
incubated with primary antibodies (LC3B, cat. no. 3868, dilution
1:100, Cell Signaling Technology; TOM20, cat. no. sc-17764,
dilution 1:100, Santa Cruz Biotechnology, Inc.) at 2–4°C for 16 h.
Next, samples were incubated with the appropriate
fluorescence-conjugated secondary antibodies (FITC-conjugated goat
anti-mouse IgG secondary antibody, F-2761, dilution 1:1,000, Thermo
Fisher Scientific, Inc; TRITC-conjugated goat anti-rabbit IgG
antibody, T-2769, Thermo Fisher Scientific, Inc.). Hoechst (Thermo
Fisher Scientific, Inc.) was used to stain the nuclei. Processed
cells or tissues were viewed using a fluorescence microscope
(Olympus, magnification, ×200). All images were analyzed using
software Image-Pro Plus 6 (Media Cybernetics, Inc.).
Flow cytometry to detect apoptosis and
mitochondrial reactive oxygen species (ROS)
Apoptosis rates were measured using the Dead Cell
Apoptosis kit (Thermo Fisher Scientific, Inc.). Briefly, cells were
harvested and incubated with Annexin V and propidium iodide (PI)
based on the protocol outlined in the kit. Thereafter, the cells
were washed and then loaded onto a flow cytometer (BD Biosciences)
and FlowJo software (Tree Star), according to the instructions.
Mitochondrial ROS levels were measured using Mitosox
(Yeasen). Briefly, cells were incubated in a culture medium
containing Mitosox (2 µM) for 10 min and then washed using warm
PBS. Next, the mitochondrial ROS level was determined using flow
cytometry.
JC-1 to probe mitochondrial membrane
potential
Changes in mitochondrial membrane potential were
measured using JC-1 staining. Briefly, cells
(5×105/well) in a 6-well plate were incubated with JC-1
(1 µg/ml) in culture medium at 37°C for 10 min. After the culture
medium containing JC-1 was removed, samples were washed with PBS
and measured using a microplate spectrophotometer and a
fluorescence microscope (magnification, ×200; Olympus Corporation)
to detect relative levels of red J-aggregates (intact mitochondria,
excitation/emission: 585 nm and 590 nm) and green J-monomers
(uncoupled mitochondria, excitation/emission: 514 and 529 nm).
Changes in the red/green fluorescence intensity ratio were used to
assess mitochondrial depolarization. A decrease in red fluorescence
indicated loss of mitochondrial membrane potential.
Mitophagy detection
Cells (5×105/cm2) in a 6-well
plate were fixed with 4% paraformaldehyde for 20 min and then
permeabilized with 0.2% Triton X-100 for 10 min. Thereafter, the
samples were incubated with primary antibody against TOM20 (cat.
no. sc-17764; dilution 1:100; Santa Cruz Biotechnology, Inc.) or
LC3B (cat. no. 3868; dilution 1:100; Cell Signaling Technology) for
1 h at room temperature. Next, they were incubated with the
appropriate secondary antibodies (FITC-conjugated goat anti-mouse
IgG secondary antibody, F-2761, dilution ratio: 1:1,000, Thermo
Fisher Scientific, Inc; TRITC-conjugated goat anti-rabbit IgG
antibody, T-2769, Thermo Fisher Scientific, Inc.) for 1 h at room
temperature and counterstained with Hoechst. Specimens were viewed
using a fluorescence microscope (Olympus America Inc.;
magnification, ×200). TOM20 antibody was used to mark the
mitochondrial outer membrane, while LC3B antibody was used to label
the autophagic vesicles. Mitophagy was quantified through the
co-localization of these two molecules as previously described
(14).
Animal experiments
Animal experiments were approved by the Committee on
Animal Research (Zhongshan Hospital, Fudan University, Shanghai,
China). All animal experiments were performed following the
guidelines formulated by Shanghai Medical Experimental Animal Care
Commission.
Twelve BALB/c nude mice (4–6 weeks old, male, body
weight 18–20 g) were purchased from Shanghai SLAC Laboratory Animal
Co., Ltd., and housed in animal rooms with a 10-h light/14-h dark
cycle and at a constant temperature (22–27°C) and a relative
humidity of 40–60% under specific pathogen-free conditions, and
with unlimited water and standard laboratory chow. A suspension of
2×107 MHCC97H cells was subcutaneously injected into the
right flank of each BALB/c nude mice (18–20 g, 4–6 weeks old).
After the tumor size reached 10 mm in diameter, mice bearing tumors
were randomly divided into three groups and received a drug (100
µl) via peritoneal cavity injections every 3 days: control group
(10% DMSO, n=4), cisplatin group (2.5 mg/kg, n=4), and combination
treatment group (2.5 mg/kg cisplatin and 50 mg/kg Mdivi-1, n=4).
Such treatment was continued for 2 weeks. Mice were sacrificed by
cervical dislocation at 48 h after the last treatment, and tumor
xenografts were harvested for further experiments.
The Cancer Genome Atlas (TCGA)
DRP1 expression levels in HCC and normal liver
tissue were compared using the UALCAN website (http://ualcan.path.uab.edu/). Survival analysis based
on the target gene in the TCGA database of patients with HCC
(n=371) (TCGA data portal, http://cancergenome.nih.gov/) was conducted using
Kaplan-Meier analysis at the KMplot website (http://kmplot.com) according to the threshold
expression value automatically set by the website.
Statistical analysis
Data are expressed as means ± standard deviations
from three independent experiments and were analyzed using Graphpad
Prism 7 (GraphPad Software, Inc.). Comparisons between two samples
or among three groups were performed using unpaired Student's
t-test or one-way analysis of variance (ANOVA, Bonferroni post hoc
test). A two-sided P-value of <0.05 was considered statistically
significant.
Results
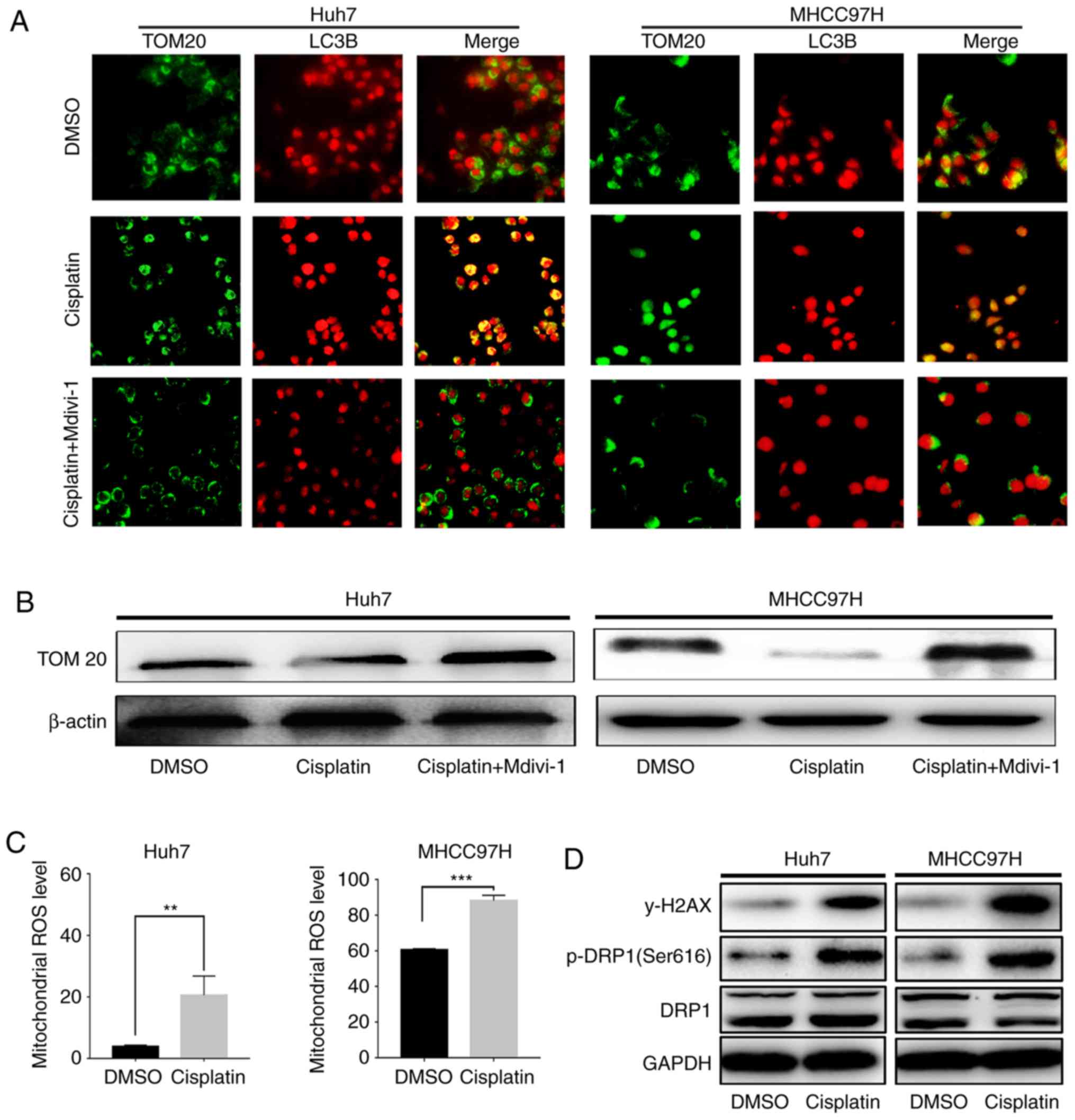
Cisplatin increases mitophagy in
surviving HCC cells by activating DRP1
After treating HCC cells (MHCC97H and Huh7) with
cisplatin (at 6 and 4 µg/ml, respectively) for 24 h, a significant
increase in mitophagy was observed, as indicated by increased TOM20
and LC3 co-localization (Fig.
1A).
Mitophagy typically occurs when ROS cause
mitochondrial damage (9). Upon
severe cisplatin-induced DNA damage (Fig. 1D), an increase in mitochondrial ROS
among the surviving HCC cells (Fig.
1C) and a marked increase in DRP1 phosphorylation at Ser616
(p-DRP1, Fig. 1D) were observed.
More importantly, DRP1-specific inhibitor Mdivi-1 (50 nM)
significantly suppressed cisplatin-induced mitophagy (Fig. 1A). Meanwhile, the expression of
TOM20 in mitochondria, which is used to identify mitochondrial
turnover, was measured in these cells. Accordingly, a significant
decrease in HCC cell TOM20 expression was observed after cisplatin
treatment (Fig. 1B), indicating
accelerated mitochondrial degradation, including mitophagy
degradation, after cisplatin treatment. Moreover, Mdivi-1 reversed
the cisplatin-induced decrease in TM20 (Fig. 1B), suggesting that inhibition of
cisplatin-induced mitophagy can decrease mitochondrial degradation
by targeting DRP1. The aforementioned data demonstrated that
cisplatin treatment induces mitophagy while inhibition of DRP1
activity can significantly reduce mitophagy. These results suggest
that HCC cells survive chemotherapy by activating DRP1-mediated
mitophagy, which may be trigged by chemotherapy-induced DNA damage
and an increase in mitochondrial ROS. This indicates that
DRP1-mediated mitophagy protects HCC cells against chemotherapy
insult.
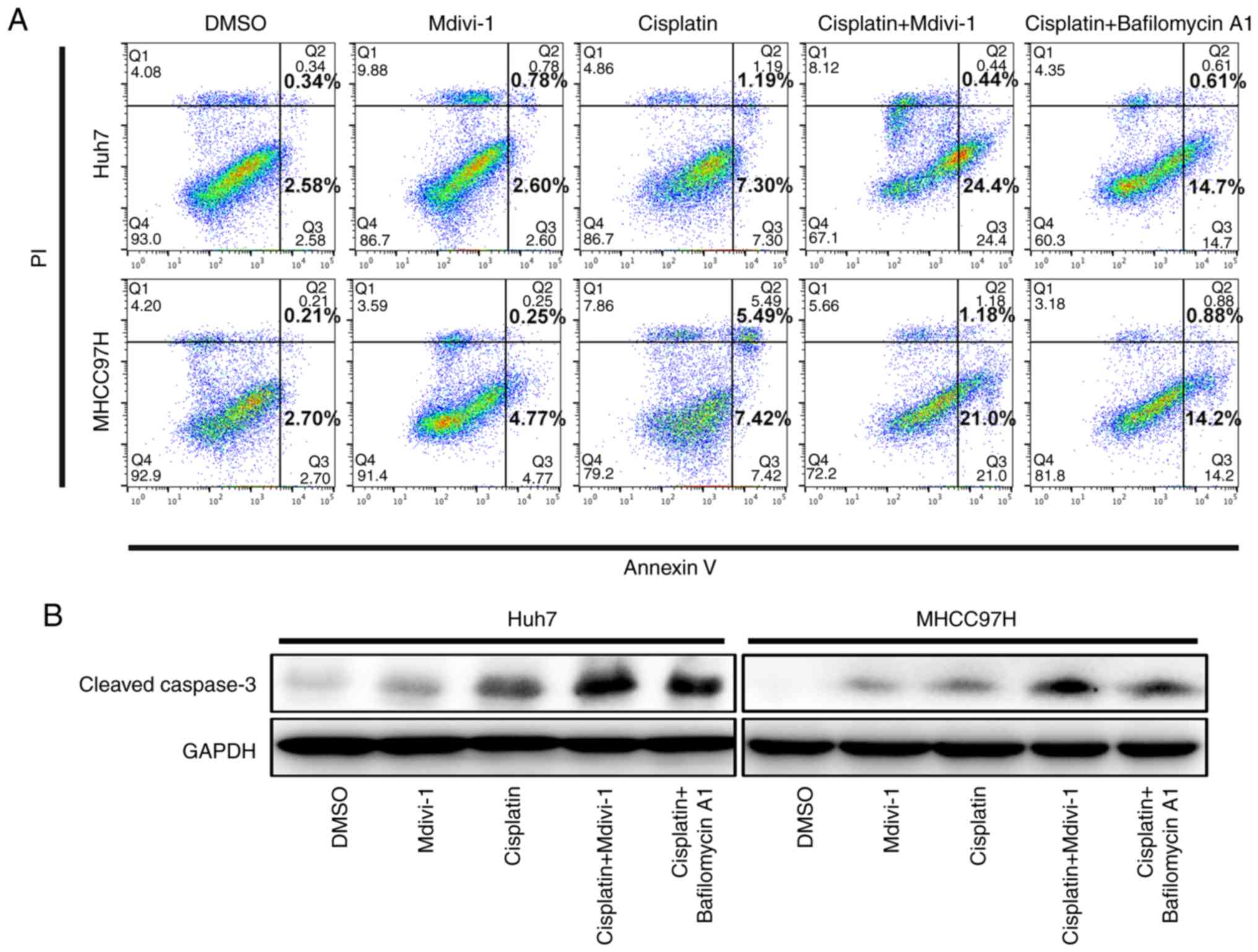
Inhibition of mitophagy by DRP1
inhibitor leads to increased apoptosis of cisplatin-treated HCC
cells
To validate whether targeting mitophagy could
influence the response of HCC cells to cisplatin treatment, HCC
cells were subjected to different treatments. Firstly, Mdivi-1
alone did not induce the apoptosis of HCC cells (Fig. 2A). However,
cisplatin+Mdivi-1-treated HCC cells exhibited significantly higher
apoptosis compared to those treated with cisplatin alone (Fig. 2A), implicating that targeting
DRP1-mediated mitophagy promotes apoptosis in cisplatin-treated HCC
cells. Cisplatin-treated Huh7 and MHCC97H cells had an apoptosis
rate of 8.5 and 12.9%, respectively. Interestingly, following
combination treatment, the apoptosis rates of both cell lines
increased markedly to 24.8 and 22.2%, respectively. In addition,
HCC cell apoptosis was significantly higher with cisplatin and
bafilomycin A1 (10 nM, a lysosomal inhibitor) treatment than with
cisplatin treatment alone, implying that blockade of autolysosomes,
including the inhibition of the lysosomal degradation of
mitochondria during mitophagy, facilitates apoptosis of
cisplatin-treated HCC cells. Furthermore, the aforementioned
findings were confirmed by western blotting for cleaved caspase-3
(Fig. 2B). Hence, our results
indicate that inhibition of DRP1-mediated mitophagy increases the
susceptibility of cisplatin-treated HCC cells to apoptosis rather
than directly causing apoptosis.
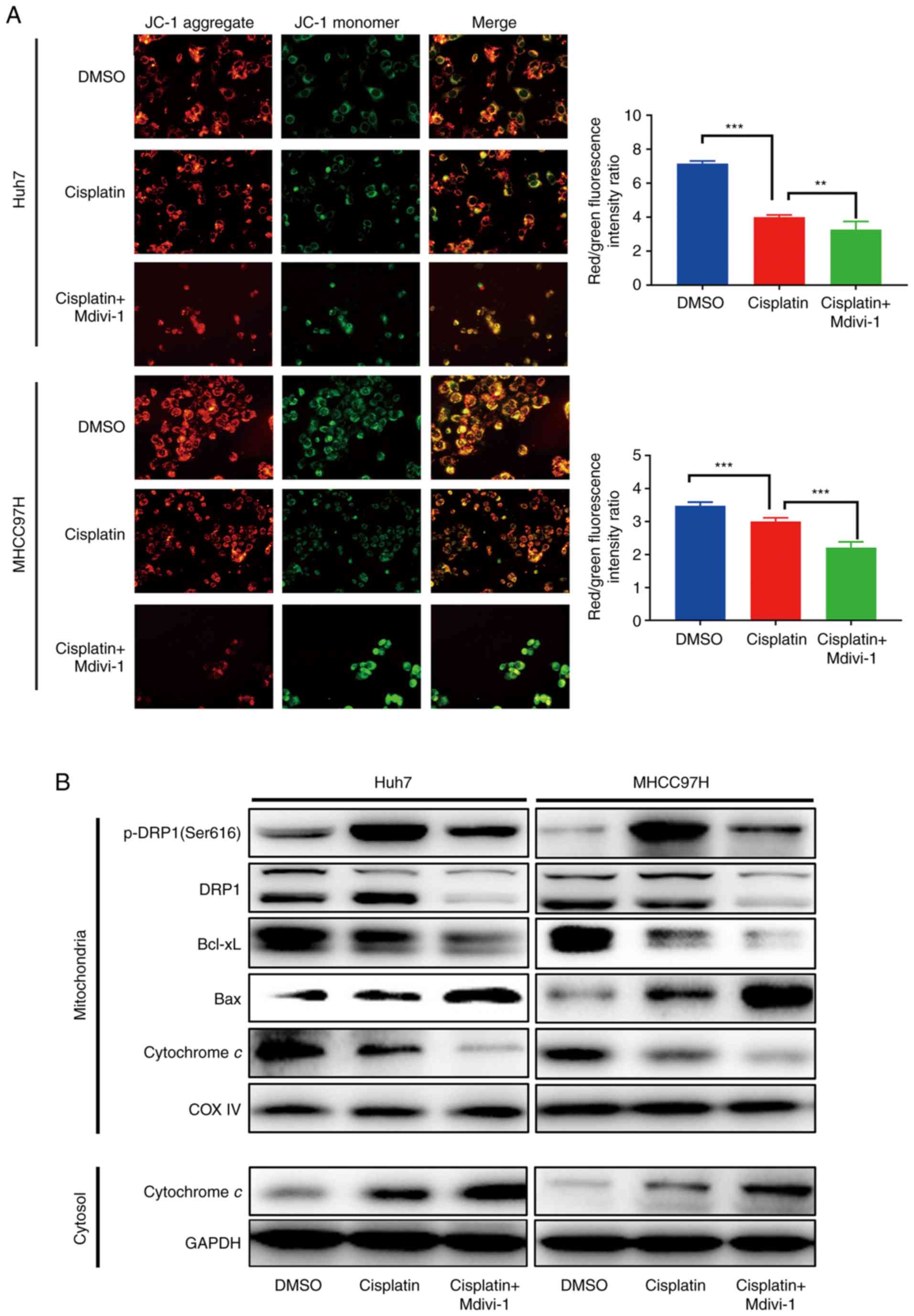
Disruption in mitophagy decreases the
mitochondrial membrane potential, subsequently releasing cytochrome
c from the damaged mitochondria
Combination treatment with cisplatin and Mdivi-1
caused more mitochondrial damage in both MHCC97H and Huh7 cell
lines, as indicated by the decrease in mitochondrial membrane
potential using JC-1 aggregate/JC-1 monomer fluorescence intensity
ratio and JC-1 staining (Fig. 3A).
More importantly, cisplatin and Mdivi-1 treatment of HCC cells
induced considerable leakage of cytochrome c into the
cytosol (Fig. 3B). This suggests
that inhibition of DRP1-mediated mitophagy using Mdivi-1 could
promote cytochrome c release from mitochondria into the
cytosol by decreasing the mitochondrial membrane potential.
Among the major proteins that maintain mitochondrial
membrane potential, Bcl-xL and Bax are considered crucial given
that they can counteract each other by competing for
voltage-dependent anion channel (VDAC) in mitochondria. More
specifically, Bax can increase VDAC activity and helps with the
formation of permeability transition pores, whereas Bcl-xL
inactivates VDAC (15–17). Although other molecules, such as
Bid/Bik, function to maintain integrity, they do not influence the
mitochondrial potential (15).
Initially, Bax, Bcl-xL, and cytochrome c expression was
measured at four time points (3, 6, 12, and 24 h). Our initial
results showed that the expression of the aforementioned proteins
started to change between 12 and 24 h after the treatment (data not
shown). Therefore, Bax, Bcl-xL and cytochrome c expression
in HCC cells was analyzed 24 h after treatment. Notably,
mitochondrial Bax expression was markedly higher in HCC cells
receiving combination treatment than those receiving cisplatin
treatment alone. Accordingly, HCC cells exposed to the combination
treatment had much less mitochondrial Bcl-xL. Meanwhile, cytochrome
c was significantly increased in the cytosol (Fig. 3B).
These results suggest that targeting DRP1-mediated
mitophagy using Mdivi-1 downregulates Bcl-xL and upregulates Bax,
which decreases the mitochondrial membrane potential leading to
increased mitochondrial membrane permeability and subsequent
release of cytochrome c from damaged mitochondria into the
cytosol, thereby accelerating cisplatin-induced apoptosis in HCC
cells.
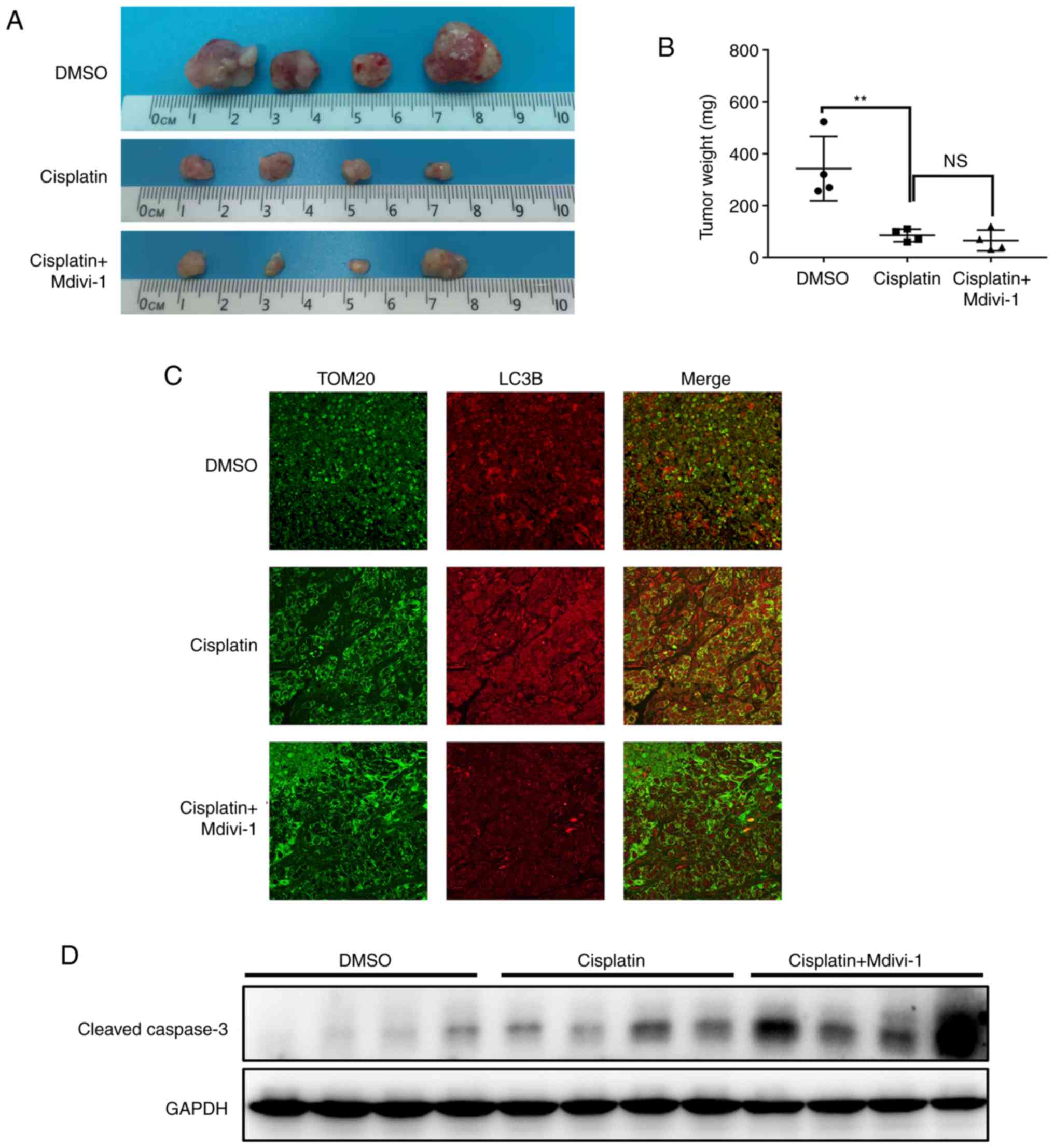
Mdivi-1 exacerbates cisplatin-induced
HCC apoptosis in vivo
We examined whether targeting mitophagy could
promote cisplatin-induced HCC apoptosis in vivo. After
establishing a mouse xenograft model by subcutaneously injecting
MHCC97H cells, mice were randomly divided into three groups
receiving different treatments: the control group treated with
DMSO, the cisplatin group, and the combination treatment group
(cisplatin and Mdivi-1). Accordingly, tumor growth was markedly
lower in the cisplatin group than that noted in the control group
as evidenced by tumor size and weight. Despite the lack of a
significant difference in tumor reduction between the cisplatin
group and combination treatment group, the addition of Mdivi-1 to
cisplatin treatment further inhibited tumor growth (Fig. 4A and B), suggesting a synergistical
effect in the combined use of Mdivi-1 and cisplatin. Moreover,
cisplatin induced mitophagy in HCC in vivo, which was
significantly blocked by the DRP1 inhibitor Mdivi-1 (Fig. 4C). Consistent with a markedly
inhibitory effect on tumor growth, the expression of apoptotic
marker cleaved caspase-3 was significantly increased in the
combination treatment group (Fig.
4D). These data suggest that Mdivi-1 acts synergistically with
cisplatin to suppress HCC xenograft growth in vivo through
the blockade of DRP1-mediated mitophagy and an increase in
apoptosis.
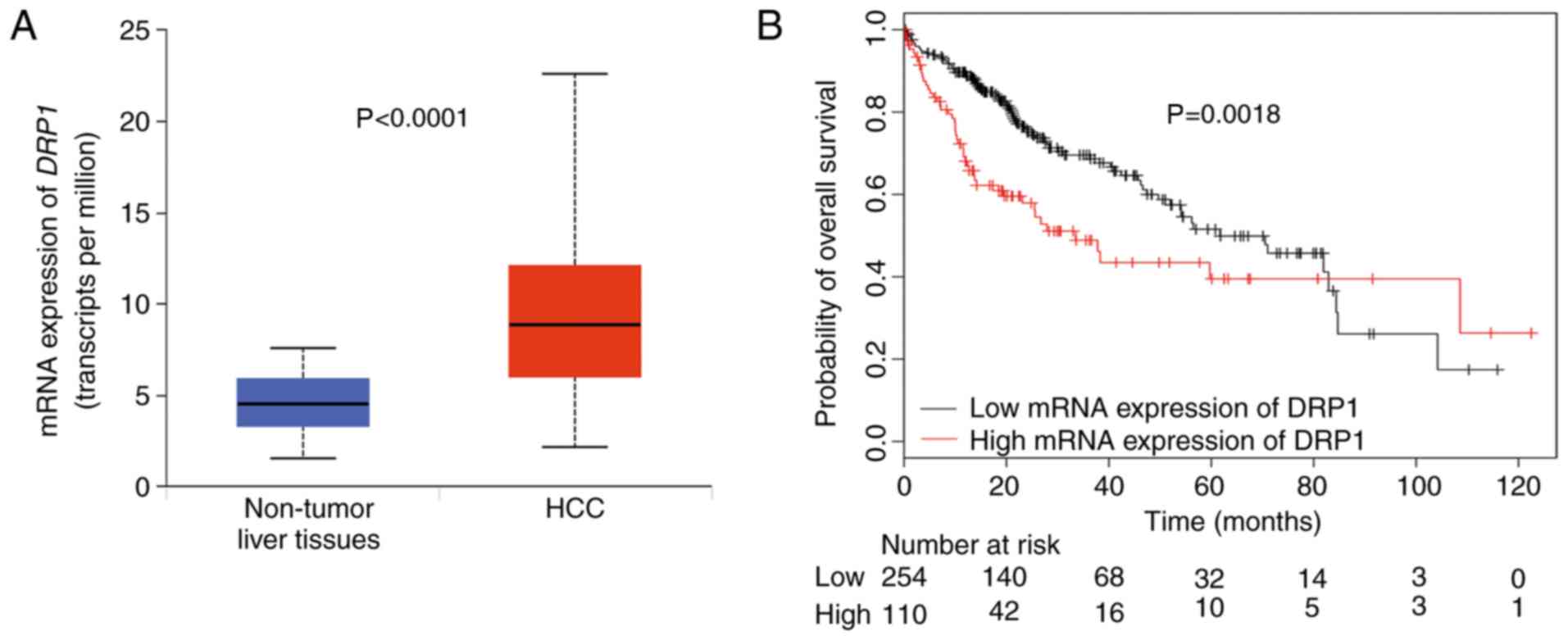
Negative correlation between DRP1
expression and survival among patients with HCC
As shown in Fig. 5A,
DRP1 mRNA expression was markedly increased in the HCC than
in the non-tumoral liver tissues. Moreover, patients with HCC
having high DRP1 mRNA expression had worse overall survival
compared to those with low DRP1 mRNA expression (Fig. 5B). Therefore, these results suggest
that high DRP1 expression may serve as an independent
predictor for poor HCC prognosis.
Discussion
The principal finding of the present study was that
targeting dynamin-related protein 1 (DRP1)-mediated mitophagy by
combining cisplatin with Mdivi-1 (a specific DRP1 inhibitor) could
aggravate the cisplatin-induced apoptosis of HCC cells, providing a
novel strategy for boosting the efficacy of hepatocellular
carcinoma (HCC) chemotherapy. Furthermore, our results revealed
that targeting DRP1-mediated mitophagy could reduce the
mitochondrial membrane potential by upregulating Bax and
downregulating Bcl-xL, which increased mitochondrial membrane
permeability and cytochrome c release from damaged
mitochondria, thereby augmenting cisplatin-induced apoptosis of HCC
cells. Therefore, the clinical implication of our findings is that
targeting mitophagy could potentiate the effects of chemotherapy
against HCC.
Mitophagy is a vital mechanism for maintaining
cellular homeostasis through the selective degradation of damaged
or malfunctioning mitochondria. This mechanism promotes cellular
survival by preventing the release of apoptotic factors, such as,
cytochrome c and apoptosis-inducing factors, into the
cytosol (18). Mitophagy, however,
could be exploited by tumor cells as an adaptive stress response
that helps them survive under stressful conditions, such as hypoxia
and ischemia, resulting in treatment resistance. Indeed, reports
have shown that inhibition of FUNDC1-mediated mitophagy during
cardiac ischemia–reperfusion injury promoted cellular apoptosis
(19). Anticancer treatment,
including chemotherapy, can cause mitochondrial damage (20). Accordingly, damaged mitochondria, if
not recycled or cleared through mitophagy, can increase cancer cell
susceptibility to death (18).
Analogous to the increase in mitophagy among cardiomyocytes
suffering from ischemia, we showed that HCC cells survive cisplatin
exposure by activating DRP1-mediated mitophagy, suggesting that
mitophagy protects HCC cells against chemotherapy.
DRP1, which initiates mitochondrial dynamics, plays
a critical role in mitophagy (9).
Although other DRP1-independent pathways lead to mitophagy
(21), our study showed that DRP1
activation initiates chemotherapy-induced mitophagy. DRP1 is
activated under cellular stress, including ROS, ATP deficiency, and
calcium overloading (22). The
present study showed that DRP1 activation during
chemotherapy-induced mitophagy might be triggered by
cisplatin-induced DNA damage and mitochondrial ROS. Given that
cisplatin can bind not only mitochondrial DNA but also
mitochondrial molecules (23,24),
it could perhaps directly damage mitochondrial DNA and trigger a
burst of mitochondrial ROS, which results in the activation of DRP1
and mitophagy. DRP1 inhibitor Mdivi-1 specifically targets DRP1
GTPase to block DRP1 activity (25). The present study showed that
Mdivi-1-induced disruption of mitophagy exacerbated the apoptotic
response of HCC cells to cisplatin treatment, suggesting the
therapeutic potential of targeting mitophagy in antitumor
treatments. Additionally, based on the TCGA database, we found that
DRP1 was a promising predictor for outcomes of patients with
HCC.
Mechanistically, during the blockade of
cisplatin-induced mitophagy by Mdivi-1, we observed more Bax and
less Bcl-xL in the mitochondria, which resulted in a decrease in
the mitochondrial membrane potential, an increase in mitochondrial
permeability and cytochrome c release from damaged
mitochondria into the cytosol, and the initiation of the apoptotic
cascade. Moreover, evidence suggests the interaction between Bcl-xL
and DRP1. Accordingly, Li et al reported that while Bcl-xL
can activate DRP1 and thus mobilize mitochondria to facilitate
synapse formation, the depletion of DRP1 may hamper this process
(26,27).
This study has some limitations. First, despite
highlighting the importance of DRP1 in cisplatin-induced mitophagy,
we cannot be certain whether other molecules involved in mitophagy,
such as PINK1, Nix, and BNIP3, can hold the same role. Second, only
cisplatin was used herein. Apart from cisplatin, doxorubicin has
been commonly used in clinical settings. However, there is a
possibility that doxorubicin-induced mitophagy may not be dependent
on DRP1 activation. Third, we did not assess the changes in DRP-1
and p-DRP1 after Mdivi-1 treatment, thus making it impossible to
determine whether Mdivi-1 worked by inhibiting the GTPase activity
of p-DRP1 or by altering protein expression. Fourth, Bordt et
al suggested that Mdivi-1 not only impairs DPR1 GTPase activity
but also modulates mitochondrial ROS production (28). Therefore, it is possible that
Mdivi-1 could exert its pro-apoptotic effects via other mechanisms.
Fifth, considering the small number of mice in each group (n=4),
the conclusions presented herein may suffer from low power.
Finally, although the apoptosis rate of MHCC97H cells treated with
cisplatin alone was not apparent, animal experiments showed that
cisplatin can significantly suppress MHCC97H cell growth. This
inconsistency may be due to differences in cisplatin concentrations
or MHCC97H responses to cisplatin in vitro and in
vivo.
In conclusion, the present study demonstrated that
suppression of cisplatin-mediated mitophagy increases cell
apoptosis in HCC via DRP1 inhibition, providing a preclinical proof
of concept for combination therapy targeting mitophagy to
potentiate chemotherapy.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81272723).
Availability of data and materials
The data that support the findings of this study are
available from the corresponding author upon reasonable
request.
Authors' contributions
MM and RXC designed the experiments. MM, XHL, HHL
and RZ performed the experiments and conducted data collection and
analyses. MM and RXC wrote the paper. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
No human patients were enrolled in the present
study. The animal experiments were approved by the Committee on
Animal Research (Zhongshan Hospital, Fudan University, Shanghai,
China). All animal experiments were performed following the
guidelines formulated by Shanghai Medical Experimental Animal Care
Commission.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
European Association for the Study of the
Liver. Electronic address, . simpleeasloffice@easloffice.eu;
European Association for the Study of the Liver: EASL clinical
practice guidelines: Management of hepatocellular carcinoma. J
Hepatol. 69:182–236. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nowak AK, Chow PK and Findlay M: Systemic
therapy for advanced hepatocellular carcinoma: A review. Eur J
Cancer. 40:1474–1484. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qin S, Bai Y, Lim HY, Thongprasert S, Chao
Y, Fan J, Yang TS, Bhudhisawasdi V, Kang WK, Zhou Y, et al:
Randomized, multicenter, open-label study of oxaliplatin plus
fluorouracil/leucovorin versus doxorubicin as palliative
chemotherapy in patients with advanced hepatocellular carcinoma
from Asia. J Clin Oncol. 31:3501–3508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J, Duan B, Guo Y, Zhou R, Sun J, Bie B,
Yang S, Huang C, Yang J and Li Z: Baicalein sensitizes
hepatocellular carcinoma cells to 5-FU and epirubicin by activating
apoptosis and ameliorating P-glycoprotein activity. Biomed
Pharmacother. 98:806–812. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee TK, Castilho A, Cheung VC, Tang KH, Ma
S and Ng IO: CD24(+) liver tumor-initiating cells drive
self-renewal and tumor initiation through STAT3-mediated NANOG
regulation. Cell Stem Cell. 9:50–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roos WP, Thomas AD and Kaina B: DNA damage
and the balance between survival and death in cancer biology. Nat
Rev Cancer. 16:20–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lo SJ, Fan LC, Tsai YF, Lin KY, Huang HL,
Wang TH, Liu H, Chen TC, Huang SF, Chang CJ, et al: A novel
interaction of nucleophosmin with BCL2-associated X protein
regulating death evasion and drug sensitivity in human hepatoma
cells. Hepatology. 57:1893–1905. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ding ZB, Hui B, Shi YH, Zhou J, Peng YF,
Gu CY, Yang H, Shi GM, Ke AW, Wang XY, et al: Autophagy activation
in hepatocellular carcinoma contributes to the tolerance of
oxaliplatin via reactive oxygen species modulation. Clin Cancer
Res. 17:6229–6238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vives-Bauza C, Zhou C, Huang Y, Cui M, de
Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of Parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:378–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eisner V, Picard M and Hajnóczky G:
Mitochondrial dynamics in adaptive and maladaptive cellular stress
responses. Nat Cell Biol. 20:755–765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roy M, Reddy PH, Iijima M and Sesaki H:
Mitochondrial division and fusion in metabolism. Curr Opin Cell
Biol. 33:111–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fang EF, Palikaras K, Sun N, Fivenson EM,
Spangler RD, Kerr JS, Cordonnier SA, Hou Y, Dombi E, Kassahun H, et
al: In vitro and in vivo detection of mitophagy in human cells,
C. elegans, and mice. J Vis Exp. 22:2017.
|
|
15
|
Shimizu S and Tsujimoto Y: Proapoptotic
BH3-only Bcl-2 family members induce cytochrome c release,
but not mitochondrial membrane potential loss, and do not directly
modulate voltage-dependent anion channel activity. Proc Natl Acad
Sci USA. 97:577–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shimizu S, Narita M and Tsujimoto Y and
Tsujimoto Y: Bcl-2 family proteins regulate the release of
apoptogenic cytochrome c by the mitochondrial channel VDAC.
Nature. 399:483–487. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vander Heiden MG, Chandel NS, Williamson
EK, Schumacker PT and Thompson CB: Bcl-xL regulates the membrane
potential and volume homeostasis of mitochondria. Cell. 91:627–637.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubli DA and Gustafsson AB: Mitochondria
and mitophagy: The yin and yang of cell death control. Circ Res.
111:1208–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Debatin KM, Poncet D and Kroemer G:
Chemotherapy: Targeting the mitochondrial cell death pathway.
Oncogene. 21:8786–8803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Graef M: A dividing matter:
Drp1/Dnm1-independent mitophagy. J Cell Biol. 215:599–601. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lima AR, Santos L, Correia M, Soares P,
Sobrinho-Simões M, Melo M and Máximo V: Dynamin-related protein 1
at the crossroads of cancer. Genes (Basel). 9:E1152018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marullo R, Werner E, Degtyareva N, Moore
B, Altavilla G, Ramalingam SS and Doetsch PW: Cisplatin induces a
mitochondrial-ROS response that contributes to cytotoxicity
depending on mitochondrial redox status and bioenergetic functions.
PLoS One. 8:e811622013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cassidy-Stone A, Chipuk JE, Ingerman E,
Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR
and Nunnari J: Chemical inhibition of the mitochondrial division
dynamin reveals its role in Bax/Bak-dependent mitochondrial outer
membrane permeabilization. Dev Cell. 14:193–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Alavian KN, Lazrove E, Mehta N,
Jones A, Zhang P, Licznerski P, Graham M, Uo T, Guo J, et al: A
Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics
during endocytosis. Nat Cell Biol. 15:773–785. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Chen Y, Jones AF, Sanger RH, Collis
LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, et al:
Bcl-xL induces Drp1-dependent synapse formation in cultured
hippocampal neurons. Proc Natl Acad Sci USA. 105:2169–2174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bordt EA, Clerc P, Roelofs BA, Saladino
AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, et
al: The putative Drp1 inhibitor mdivi-1 is a reversible
mitochondrial complex I inhibitor that modulates reactive oxygen
species. Dev Cell. 40:583–594.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|