Introduction
Cutaneous neurofibromas (cNF) are present in a large
number of individuals with neurofibromatosis type 1 (NF1), a tumor
susceptibility syndrome with autosomal dominant inheritance
(1). As a hallmark symptom of
patients with NF1, cNFs typically develop at puberty and rarely
progress to malignancy. However, these benign tumors are may be a
substantial burden to the individual due to the consequent
disfigurement of the body, which is associated with significant
psychological distress (2).
Unfortunately, the available treatment options for cNF are limited
to surgical excision, which is not feasible with larger tumors or
with the presence of multiple tumors. Therefore, determining novel
methods for treating cNF is required; however, current efforts have
thus far yielded insufficient success (3).
cNFs commonly consist of Schwann cells, endothelial
cells, fibroblasts and inflammatory cells (4). Mast cells are the most abundantly
present inflammatory cells (5).
Efforts to target mast cells in order to shrink neurofibromas have
presented difficulties, as certain patients do not exhibit a
response to the inhibition of mast cell activity (6), which suggests that there may be
compensatory factors facilitating neurofibroma progression.
Furthermore, altered macrophage activity has been detected in cNF
tissues (7,8), nerve injury with coincident macrophage
invasion promotes neurofibroma formation (9) and macrophage depletion impairs tumor
maintenance in mice (10). All
these results highlight the possibility that macrophages may
infiltrate into the neurofibroma microenvironment and contribute to
tumor progression. However, the detailed mechanism underlying
macrophage accumulation requires further clarification, and this
may result in the identification of potentially viable therapeutic
targets for treating neurofibromas.
Nf1 encodes neurofibromin, which serves as the ‘off’
signal for RAS, a GTPase activating protein which activates various
receptor tyrosine kinases, such as cytokine receptors (11). Schwann cells, the primary pathogenic
cells, are capable of secreting multiple types of cytokines which
mediate macrophage recruitment (12,13).
Although active RAS primarily increases PI3K and RAF-MEK-ERK
mediated signaling in neurofibroma cells, the Hippo signaling
pathway has been validated as an important modifier in cNF
tumorigenesis (14). Yes-associated
protein 1 (YAP) and tafazzin (TAZ) are key effectors of Hippo
signaling (15). MST1 and 2 are
kinases involved in the Hippo signaling pathway which modulates the
protein expression levels and localization of YAP by upregulating
YAP degradation and phosphorylation (16). Activated MST1 and 2 promotes the
maintenance of YAP in the cytoplasm, and repressing MST1 and 2
activity enhances the nuclear translocation of YAP, where it
interacts with T-domain transcription factor (TEAD) to activate a
panel of genes (15–17). The Hippo pathway contributes to a
conducive environment for tumor growth (18) and may modulate the tumor
microenvironment (19). Regarding
the role of macrophages in neurofibromas, the association between
the Hippo pathway and macrophage accumulation requires further
study.
In the present study, it was hypothesized that Hippo
pathway effectors participated in macrophage accumulation induced
by cNF cells to contribute to the development of cNF, and that the
Hippo pathway may modulate macrophage accumulation through cytokine
secretion.
Materials and methods
Macrophage polarization
The macrophage polarization model was generated
using the human acute monocytic leukemia cell clone THP-1 (American
Type Culture Collection). A total of 3×106 THP-1 cells
were seeded in a 10 cm dish containing RPMI-1640 medium (Hyclone;
GE Healthcare Life Sciences), and 300 nmol/l phorbol 12-myristate
13-acetate was added to the cells for 48 h, followed by exposure
for 24 h to lipopolysaccharide (20 ng/ml) and interferon (IFN)-γ
(20 ng/ml) for M1-polarization. Subsequently, the cells were
propagated in Human Endothelial Serum-Free Medium (Thermo Fisher
Scientific, Inc.) with lipopolysaccharide and IFN-γ at the same
concentrations for an additional 24 h.
Cell culture and stable RNA
interference
SW10 (murine Schwann cells) and Hs 53.T cells (human
skin fibroblasts from neurofibroma patients) were grown in DMEM/F12
(Invitrogen; Thermo Fisher Scientific, Inc.) containing 10% FBS
(Invitrogen; Thermo Fisher Scientific, Inc.). THP-1 cells were
grown in RPMI-1640 medium with 10% FBS.
Cells were maintained in a humidified atmosphere
with 5% CO2 at 37°C. Two reconstructed
replication-defective lentiviruses containing specific short
hairpin RNA (shRNA) sequences targeting Nf1, CCL5 or TGF-β1, or an
shRNA control were used for SW10 and Hs 53.T cell infection;
infected cells were identified as shNf, shCCL5, shTGF-β1 and shNC
cells, respectively. shRNA sequences targeting CCR5 or TGFβ1R, or
the shRNA control were used in THP-1 cells; infected cells were
termed shCCR5, shTGFβ1R or shNC cells. The lentivirus was purchased
from Shanghai Genechem Co. Ltd. and used to transfect cells with a
viral titer >1×108 particles/ml and 0.8 µl Hitrans G
A (Shanghai Genechem Co., Ltd.). The transfected cells with a
confluence of 70–80% were considered the first generation. The
efficiency of gene knockdown was determined by western blot
analysis 3 days after transfection. The transfected cells were used
for 10 passages following transfection. To obtain conditioned
medium, the cell culture supernatant was collected by
centrifugation (167 × g, 5 min at 37°C) and filtration (0.22 µm),
followed by immediate use or storage until use at −80°C.
Antibodies and reagents
GAPDH (cat. no. 6c5) antibody was purchased from
Santa Cruz Biotechnology, Inc. Phospho-(p-)YAP (Ser127; cat. no.
13008S), YAP (cat. no. 14074), PARP/cleaved-PARP (cat. no. 9532),
Caspase 3/cleaved-Caspase 3 (cat. no. 9662), proliferating cell
nuclear antigen (PCNA; cat. no. 13110), Ki67 (cat. no. 2586), p21
(cat. no. 2947), p27 (cat. no. 3686), CDK4 (cat. no. 12790), CDK6
(cat. no. 13331), cyclin D1 (cat. no. 2978) and TGFβ receptor
(TGFβR; cat. no. 5544) antibodies were all purchased from Cell
Signaling Technology, Inc. C-C motif chemokine receptor 5 (CCR5;
cat. no. ab65850), CCL5 (cat. no. ab10394), TGF-β1 (cat. no.
ab64715), HLA-DR (cat. no. ab92511) and TAZ (cat. no. ab84927)
antibodies were purchased from Abcam. CD68 (cat. no. M087601-2)
antibody was purchased from Dako; Agilent Technologies, Inc.
XMU-MP-1 and Verteporfin (VP) were purchased from Selleck
Chemicals. The Click-iT™ plus EdU Alexa Fluor™ 488 Imaging kit
(cat. no. C10637) was purchased from Thermo Fisher Scientific, Inc.
The TGF-β1 ELISA kits (cat. no. ab119557 for SW10 cells; cat. no.
ab100647 for Hs 53.T cells) and CCL5 ELISA kits (cat. no. ab215537
for SW10 cells; cat. no. ab174446 for Hs 53.T cells) were purchased
from Abcam.
Macrophage recruitment assay
Collected conditioned medium (1 ml) was added to the
lower chambers of 24-well Transwell plates with 5-µm pore
polycarbonate membrane inserts. A total of 1×105 THP-1
cells in 0.5 ml medium were seeded into the upper chamber. After 20
h of incubation, the cells which had migrated to the lower surface
of the membrane were washed with PBS three times, fixed with 4%
paraformaldehyde for 20 min at room temperature, and stained with
0.1% crystal violet for 15 min at room temperature. Subsequently
the cells were washed with PBS three times at room temperature and
counted. The number of cells which had migrated were counted in
five randomly chosen fields using light microscopy (magnification,
×40; Olympus IX50-S8F2; Olympus Corporation).
RNA extraction and reverse
transcription-quantitative PCR (RT-q)PCR)
Total RNA from cells was extracted using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) and 1
µg total RNA was reverse transcribed using the PrimeScript™ RT
reagent kit (Takara Biotechnology Co., Ltd.) with the following
temperature protocol: 37°C for 15 min followed by 85°C for 5 sec
and held at 4°C until use. qPCRwas performed using a CFX96
real-time PCR system (Bio-Rad Laboratories, Inc.) with SYBR Green
PCR Master mix (Takara Biotechnology Co., Ltd.). The themorcycling
conditions were: 95°C for 3 min; followed by 39 cycles of 95°C for
10 sec and 55°C for 30 sec. The expression levels of the genes of
interest were quantified using the 2−ΔΔCq method
(20) and normalized to the mRNA
levels of GAPDH. The sequences of the primers used are presented in
Table SI.
Western blot analysis
The cells were washed three times in cold PBS and
then lysed using RIPA buffer containing Tris (50 mM, pH 8.0), NaCl
(150 mM), SDS (0.1%), NP40 (1%), sodium deoxycholate (0.5%) and
proteinase inhibitors [1% cocktail and 1 mM PMSF (Sigma-Aldrich;
Merck KGaA)]. Equivalent quantities of protein for each sample
(30–35 µg) were loaded on a 12% SDS-gel and resolved using
SDS-PAGE, before transferring to nitrocellulose membranes. Skimmed
milk (5%) was used to block the non-specific binding sites on the
membrane prior to incubation with the specific primary antibodies
(1:1,000 for antibodies purchased from Cell Signaling Technology
and 1:2,000 for antibodies purchased from Abcam). The membranes
were incubated with the primary antibodies overnight (>10 h) at
4°C. Subsequently, the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies; anti-mouse IgG (cat.
no. 7076) or anti-rabbit IgG (cat. no. 7074) both used at 1:200
(both from Cell Signaling Technology, Inc.) for 1 h at room
temperature. The blots were treated with luminol and hydrogen
peroxide (ECL Super Sensitive kit; DiNing), followed by
visualization using the Molecular Imagery ChemiDoc XRS System
(Bio-Rad Laboratories, Inc.).
Clinical specimens and
immunohistochemistry
To investigate macrophage infiltration in
neurofibroma tissues, 50 dermal neurofibroma samples and adjacent
tissue samples were collected from patients who underwent tumor
resection at the First Affiliated Hospital of Xi'an Jiaotong
University, between June 2010 and October 2017. Patients diagnosed
as NF1, with a cNF who received surgery were included. Patients
diagnosed as NF1 without a cNF were excluded. Approval was obtained
from the institutional review board of the First Affiliated
Hospital of Xi'an Jiaotong University prior to the collection of
samples, and consent was also obtained from the patients. The
characteristics of the recruited patients are presented in Table SII. The EnVision™ system (Dako;
Agilent Technologies, Inc.) was used for immunohistochemistry (IHC)
staining, according to the manufacturer's protocol. Tumor sections
were deparaffinized (60–65°C for 4 h), washed with xylene 3 times,
rehydrated (100% alcohol 3 times, 5 min each; 95% alcohol 2 times,
5 min each; 80% alcohol 5 min; 70% alcohol 5 min, and 50% alcohol 5
min) and subjected to heat-induced antigen retrieval. Methanol with
3% H2O2 was used to block endogenous
peroxidase and alkaline phosphatase activity. Subsequently, the
slides were incubated with primary antibodies (1:150) at 4°C
overnight, washed three times and sequentially incubated with the
EnVision secondary antibody (cat. no. GK600505; Gene Tech
Biotechnology Co., Ltd.) for 30 min at room temperature. Proteins
of interest were detected using diaminobenzidine buffer and
subsequently counterstained with hematoxylin for 3 min at room
temperature. Slides were imaged and analyzed using light microscopy
(magnification, ×40; Olympus BX51; Olympus Corporation). IHC
staining was analyzed using a scoring system, in which the staining
score was calculated by multiplying the intensity score by the
percentage score. The intensity score was defined as follows: 0,
0%; 1, <25%; 2, ≥25% and <50%; 3, ≥50% and <75%; and 4,
≥75%. The percentage score was graded as follows: 0, no staining;
1, slight staining; 2, moderate staining; and 3, strong
staining.
Cell Counting Kit-8 (CCK-8) assay
The Cell Counting Kit-8 (CCK-8) was purchased from
Dojindo Molecular Technologies, Inc. Cells (5×103 for
THP-1 cells, and 1×103 for SW10 and Hs 53.T cells) were
seeded in 96-well culture plates. After different treatments for
the indicated time, the cells were washed, followed by incubation
with serum-free medium (150 µl) containing 10% CCK-8 at 37°C for 3
h. The absorbance was measured at a wavelength of 450 nm using a
Microplate Autoreader (Bio-Tek Instruments Inc.). Independent
experiments were repeated in triplicate.
Colony formation assay
A total of 1×103 cells with the indicated
gene knocked down were seeded in 6-well plates. After ~2 weeks of
treatment with XMU-MP-1 (5 µM) or VP (5 µM), cells were washed with
PBS, fixed with 4% paraformaldehyde for 20 min, at room temperature
and stained with 0.1% crystal violet for 15 min at room
temperature. The number of colonies (≥50 cells) in each well were
counted. The colony number for each sample was defined as the
average colony number of four wells and the experiments were
performed four times.
Cell cycle analysis
Cells with the indicated treatments were washed with
cold PBS, followed by resuspension in cold 70% ethanol and stored
at −20°C for >24 h. The ethanol was removed, and the cell
pellets were washed twice with cold PBS. Prior to flow cytometry,
the cells were resuspended in PBS containing RNAse A (0.5 µg/ml)
and propidium iodide (50 µg/ml) and incubated in the dark for 30
min at room temperature. Flow cytometry was performed using a BD
FACSCalibur Flow Cytometer (BD Biosciences) and flow data was
analyzed using ModFit LT™ version 3.3 (BD Biosciences).
In vivo macrophage recruitment assay
and EdU staining
A total of 3×106 shNf1-SW10 cells were
mixed with Matrigel 1 (1:8; BD Biosciences) and subcutaneously
injected into the right thigh region of 4-week-old BALB/c nude mice
(18 mice in total, 9 male and 9 female; purchased from Beijing
Vital River Laboratory Animal Technology Co., Ltd.). The mice were
raised in the animal laboratory center of Xi'an Jiaotong University
in specific-pathogen free conditions. After 1 week, the xenografts
formed a tumor with a volume of ~0.5 cm3, and the mice
were randomly divided into three groups (6 mice/group). The mice
were injected intraperitoneally with XMU-MP-1 (1 mg/kg), VP (10
mg/kg) or 10% DMSO as a control. After 2 weeks treatment,
5×106 red fluorescent protein (RFP)-labeled THP-1 cells
were injected via the tail vein and the treatments were continued.
The RFP-labeled THP-1 cells were generated by transfecting the
cells with an RFP plasmid (Shanghai Genechem, Co. Ltd.; 2.5
µg/5×105 cells) using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. After 1 week, the mice were
sacrificed, and the tumors were obtained. For SW10 cells with the
indicated gene knocked down, 3×106 shNf1-SW10 cells were
injected subcutaneously to form the tumors. The RFP-labeled THP-1
cells (5×106) were injected 3 weeks after the
subcutaneous injection of SW10 cells, and the tumors were obtained
after 1 week. Frozen sections of tumor tissues were prepared to
analyze the THP-1 cells using fluorescence microscopy (Olympus,
IX50-S8F2; magnification, ×40). For EdU staining, after
deparaffinization, Alexa-488-azide (10 µM; cat. no. A10266; Thermo
Fisher Scientific, Inc.) was used to stain the sections on glass
slides for 10–30 min, followed by washing with PBS in Coplin jars.
Subsequently, the EdU+ cells were analyzed by
fluorescence microscopy (magnification, ×40). The animal
experiments were approved by the institutional review board of the
First Affiliated Hospital of Xi'an Jiaotong University.
Statistical analysis
A Student's t-test was used for comparisons between
two groups, using GraphPad Prism version 5.0 (GraphPad Software,
Inc.). A one-way ANOVA with a Fisher's Least Significant Difference
t-test was used to compare ≥3 groups, using SPSS Version 10 (SPSS,
Inc.). For correlation analysis, a Spearman's correlation test was
performed using SPSS. P<0.05 was considered to indicate a
statistically significant difference.
Results
Pharmacologically-activated Hippo
pathway effectors upregulate cell proliferation and macrophage
accumulation in cNF
IHC analyses was performed using anti-CD68 (specific
marker of macrophages) (21), and
anti-YAP and anti-TAZ (core effectors of the Hippo pathway) in 50
neurofibroma tissues and adjacent normal tissues. YAP and TAZ
expression was notably increased, concurrent with increased numbers
of infiltrating macrophages in cNF tissues (Fig. 1A). Further analysis of the IHC
staining showed a positive association between macrophage density
and YAP or TAZ expression in cNF tissues (Fig. S1A). Schwann cells are generally
considered to be the precursor cells of neurofibromas, although
there is some controversy regarding this (22). In addition, fibroblasts are an
important component of cNF, and ~50% of the dry weight of human cNF
is collagen (22). Thus, the
present study established a cell model of cNF by knocking down Nf1
in murine SW10 cells and Hs 53.T cells, termed shNf1-SW10 and
shNf1-Hs 53.T (Fig. S1B).
Expression of YAP and TAZ was increased and phosphorylation of YAP
was decreased in the Nf1-ablated cells, suggesting that Nf1
knockdown overactivated the core effectors of the Hippo pathway in
shNf1-SW10 and shNf1-Hs 53.T cells (Fig. S1C). Furthermore, treatment with
XMU-MP-1 enhanced the activity of YAP and TAZ in SW10 and Hs 53.T
cells, irrespective of Nf1 ablation (Fig. S1C). Preliminary studies discovered
both M1-and M2-macrophages in neurofibroma tissue, but very few
CD163+ macrophages were identified (23). Thus, M1-polarized macrophages were
induced in the present study (Fig.
S2). To investigate the potential association between
macrophage infiltration and Hippo pathway activity, an in
vitro macrophage recruitment assay was used, with conditioned
medium added to the lower chamber. Nf1-ablation in the SW10 and Hs
53.T cells resulted in improved macrophage penetration.
Furthermore, the conditioned medium collected from XMU-MP-1-treated
shNC-SW10 and shNC-Hs 53.T cells facilitated THP-1 infiltration,
whereas conditioned medium from VP (a YAP inhibitor that acts by
disturbing the YAP-TEAD interaction)-treated shNC-SW10 and shNC-Hs
53.T cells impaired THP-1 infiltration (Fig. 1B). This suggests that activated YAP
induced by XMU-MP-1 treatment or Nf1 ablation may promote THP-1
infiltration, and attenuation of YAP activity reduced THP-1
infiltration. For further confirmation, a subcutaneous tumor
xenograft model was generated in mice to examine the in vivo
effects. When the volume of tumors reached 0.5 cm3, the
mice were treated with intraperitoneal injections of XMU-MP-1 (1
mg/kg) or VP (10 mg/kg) or 10% DMSO (as a control) daily. At 1 week
prior to sacrifice, 5×106 RFP-labeled THP-1 cells were
injected via the tail vein. The results showed that XMU-MP-1
treatment significantly increased tumor weight, THP-1 infiltration
and YAP levels, which were decreased in the VP-treated mouse
xenograft tumors (Fig. 1C and D).
No difference in the body weight of mice was observed among the
three groups (Fig. S3).
Furthermore, both the colony formation assay and CCK-8 assay
confirmed the positive association between cell viability and YAP
activity in SW10 and Hs 53.T cells, as VP impaired the increase in
cell viability induced by Nf1 knockdown and XMU-MP-1 accelerated
the cell viability of Nf1-expressing cells (Fig. 1E and S1D).
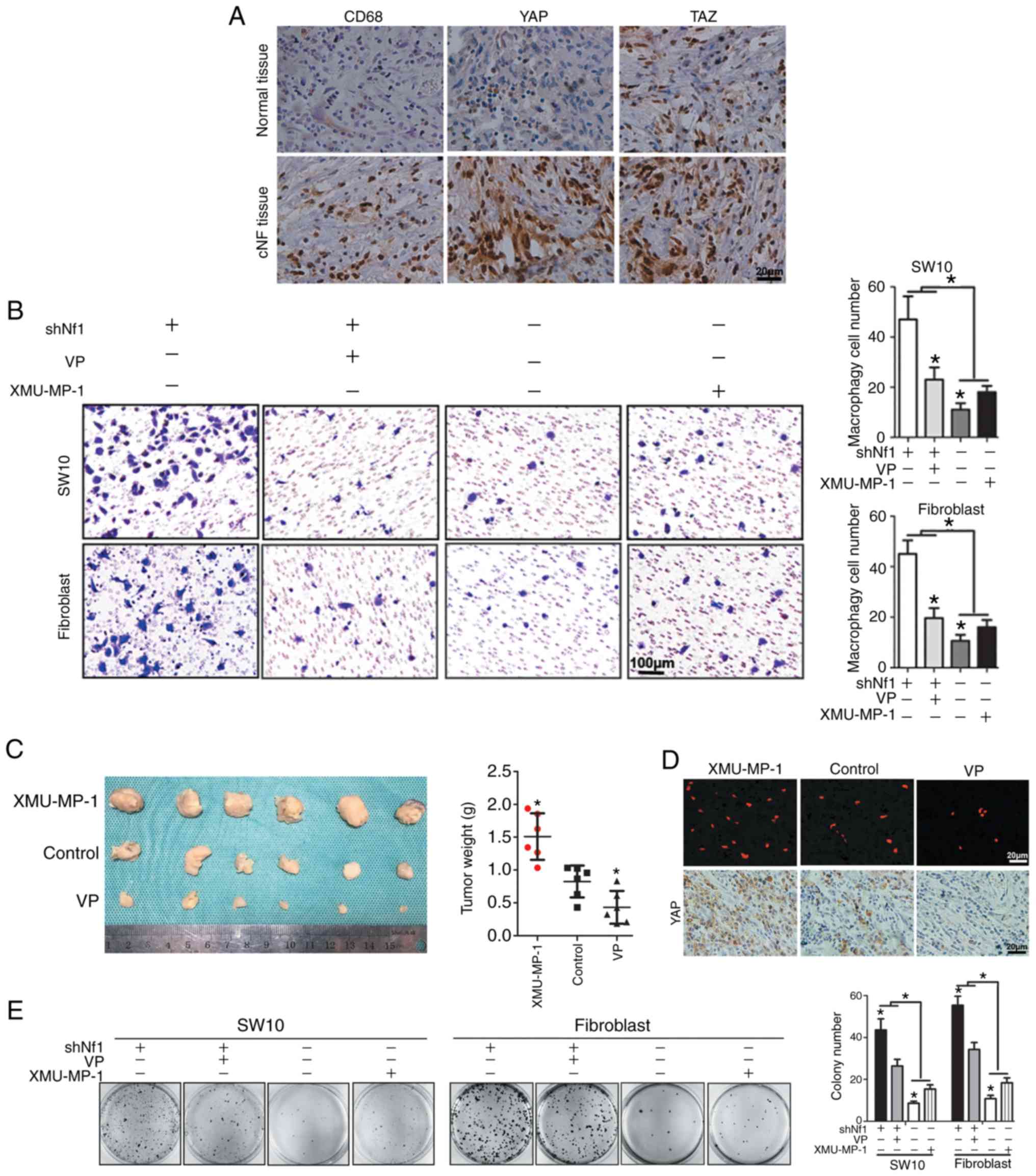 | Figure 1.Hippo pathway regulates cNF growth
and macrophage infiltration. (A) IHC staining for CD68, a marker of
macrophages, and YAP and TAZ, core effectors of the Hippo pathway,
in human cNF tissues and adjacent non-tumor tissues. Magnification,
×400. (B) VP was used to inhibit YAP activity in Nf1-ablated SW10
and Hs 53.T cells, and XMU-MP-1 was used to enhance YAP activity in
the Nf1-NC cells. Subsequently, conditioned media was collected and
used for the macrophage recruitment assay. Left, representative
images of migrated THP-1 cells. Right, quantitative analysis of the
recruited macrophages. Magnification, ×200. (C) Nude mice were
injected with shNf1-SW10 cells subcutaneously. When the volume of
the tumors reached 0.5 cm3, the mice were treated with XMU-MP-1, VP
or DMSO intraperitoneally. A total of 1 week prior to harvesting
the tumors, RFP-labeled THP-1 cells were injected via the tail vein
and tumor weights were analyzed. *P<0.05. (D) THP-1 cells were
identified by fluorescence microscopy in sections of SW10 ×enograft
tumor samples, and IHC staining for YAP expression was performed in
paraffin-embedded tumor tissues. Magnification, ×400. (E) In the
colony formation assay, shNf1-transfected or shNC-transfected SW10
and Hs 53.T cells were treated with VP and XMU-MP-1 for 2 weeks.
*P<0.05. IHC, immunohistochemistry; VP, Verteporfin; cNF,
cutaneous neurofibroma; sh, short hairpin; RFP, red fluorescent
protein; NC, negative control; Nf1, neurofibromin 1; YAP,
Yes-associated protein 1; TAZ, tafazzin. |
Taken together, the results from Fig. 1 suggest that activation of Hippo
pathway effectors facilitates SW10 and Hs 53.T cell proliferation,
and their ability to recruit macrophages in vivo and in
vitro.
The Hippo pathway modulates CCL5 and
TGF-β1 expression in cNF cells
As cell increasing viability of the YAP-activated
cNF cells, the involvement of chemokines in affecting the Hippo
pathway to mediate macrophage infiltration was investigated. Thus,
the expression of 11 chemokines were detected to investigate
altered macrophage accumulation. The results showed that CCL5 and
TGF-β1 may be involved in Hippo pathway-regulated macrophage
recruitment in Nf1-ablated SW10 and Hs 53.T cells (Fig. 2A and B). Western blotting, RT-qPCR
and ELISA confirmed that the expression and secretion of CCL5 and
TGF-β1 were upregulated when treated with XMU-MP-1 and
downregulated when treated with VP. The protein expression levels
of YAP and p-YAP, and the mRNA expression levels of YAP were not
altered by VP, as VP serves its inhibitory role by disturbing the
interaction between YAP and TEAD (Fig.
2C-E). IHC with anti-CCL5 and anti-TGF-β1 validated the robust
levels of CCL5 and TGF-β1 expression in the cNF tissues (Fig. 2F). Furthermore, the staining scores
in cNF tissues showed that both CD68 and YAP were positively
associated with CCL5 and TGF-β1 (Fig.
S4).
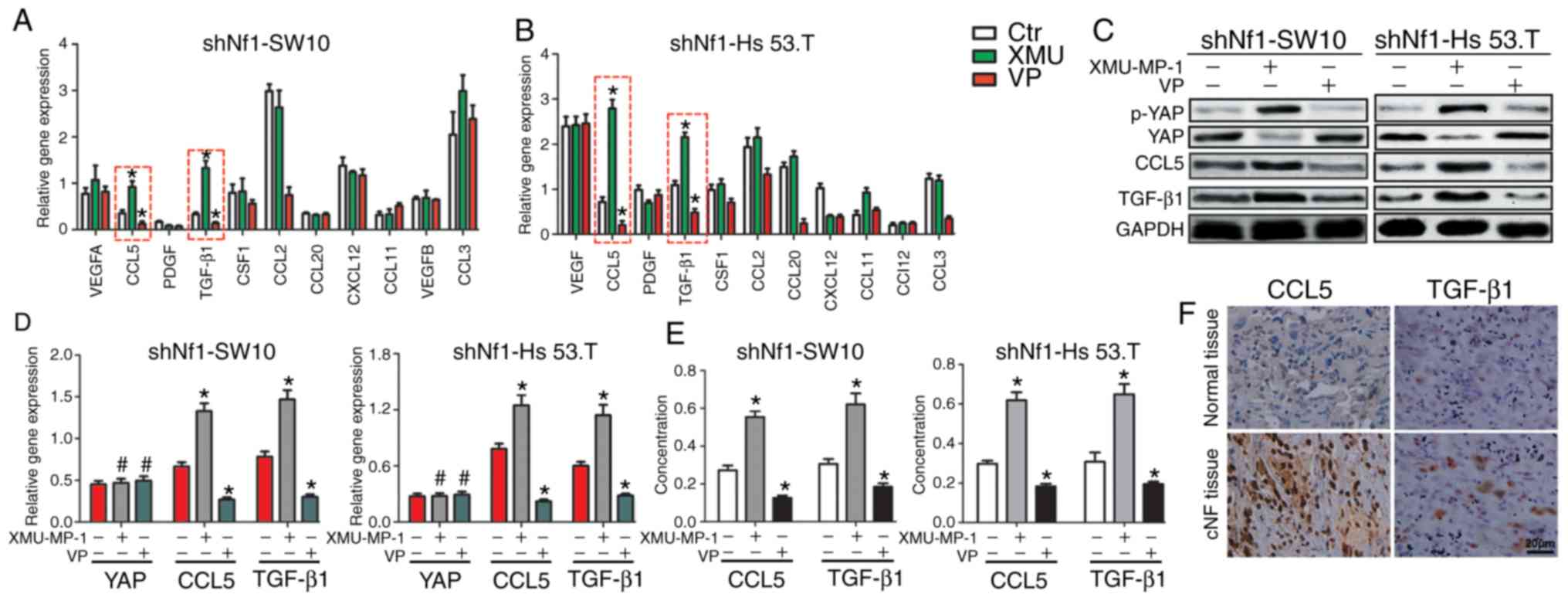 | Figure 2.The Hippo pathway regulates the
expression of CCL5 and TGF-β. (A and B) Reverse
transcription-quantitative PCR was used to profile the mRNA levels
of cytokines associated with macrophage recruitment in shNf1-SW10
and shNf1-Hs 53.T cells treated with VP, XMU-MP-1 or DMSO. (C) CCL5
and TGF-β1 were upregulated when YAP was activated and
downregulated when YAP activity was decreased at the protein level.
(D) Expression of YAP, CCL5 and TGF-β1 in shNf1-SW10 cells and
shNf1-Hs 53.T cells treated with XMU-MP-1, VP or DMSO. *P<0.05,
XMU-MP-1/VP vs. DMSO. (E) Concentration of CCL5 and TGF-β1 in the
medium of shNf1-SW10 and shNf1-Hs 53.T cells with the indicated
treatments. *P<0.05, XMU-MP-1/VP vs. DMSO. (F) Expression of
CCL5 and TGF-β1 in the patient cNF tissues and adjacent tissues.
Magnification, ×200). *P<0.05. VP, Verteporfin; sh, short
hairpin; CCL5, C-C motif chemokine ligand 5; TGF-β1, transforming
growth factor-β1; cNF, cutaneous neurofibroma; Nf1, neurofibromin
1; ctr, control. |
Taken together, Fig.
2 suggests that CCL5 and TGF-β1 may serve an important role in
altered Hippo pathway modulation of macrophage infiltration.
Knockdown of either CCL5 or TGF-β1
alone fails to significantly reduce macrophage accumulation
As CCL5 was involved in recruiting macrophages and
the CCL5 expression was altered with altered Hippo pathway
activity, it was hypothesized that the production of CCL5 by cNF
cells may recruit macrophages into the tumor microenvironment.
Therefore, shRNA targeting CCL5 was transfected into shNf1-SW10 and
shNf1-Hs 53.T cells (Fig. 3A) prior
to collecting the conditioned medium for the subsequent macrophage
recruitment assays. There was no significant difference observed in
treatment with conditioned media from the CCL5-ablated cNF cells
with or without XMU-MP-1 treatment (Fig. 3B). The failure to abolish macrophage
accumulation by CCL5 knockdown may be due to the compensatory
efficiency of TGF-β1. Therefore, CCL5-ablated cells were
transfected with shTGF-β1 to knock down TGF-β1 (Fig. 3C). Neither conditioned media from
CCL5-knockdown cells nor from TGF-β1-knockdown cells could
attenuate the macrophage accumulation. However, conditioned media
from cells with both CCL5 and TGF-β1 knocked down significantly
reduced macrophage accumulation (Fig.
3D). Furthermore, subcutaneous tumor xenografts were generated
in mice using shNf1-SW10 cells with ablation of CCL5, TGF-β1 or
both. A total of 3 weeks later, 5×106 RFP-labeled THP-1
cells were injected via the tail vein 1 week before the mice were
sacrificed. The number of recruited THP-1 cells only decreased in
xenograft tumors composed of cells with ablation of both CCL5 and
TGF-β1 (Fig. S5).
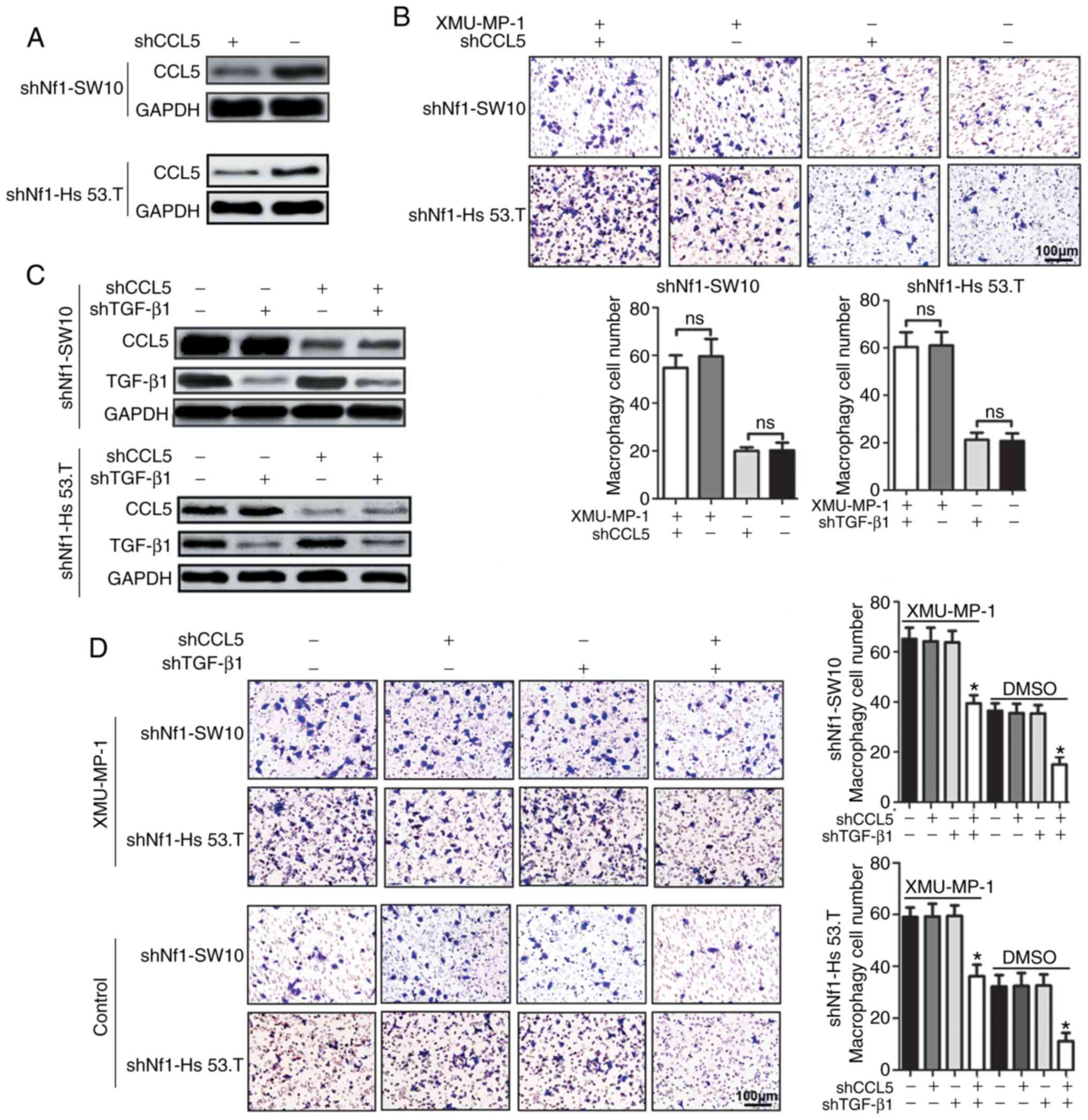 | Figure 3.CCL5 and TGF-β1 knockdown does not
reduce macrophage infiltration induced by upregulated YAP activity.
(A) Efficiency of CCL5 knockdown in shNf1-SW10 cells and shNf1-Hs
53.T cells. (B) CCL5-ablated shNf1-SW10 and shNf1-Hs 53.T cells
were treated with XMU-MP-1 or DMSO for 24 h, and the conditioned
media was collected. Conditioned media was added to the lower
chamber of the Transwell insert, and THP-1 cells passing through
the membrane were stained and analyzed. Magnification, ×200. (C)
Protein expression levels of CCL5 and TGF-β1 in shNf1-SW10 and
shNf1-Hs 53.T cells transfected with shCCL5, shTGF-β1, or both
combined. (D) Conditioned media was collected from cNF cells with
the indicated gene knocked down following treatment with XMU-MP-1
or DMSO for 24 h. Subsequently, conditioned media was added to the
lower chamber of the Transwell insert and THP-1 cells passing
through the membrane were stained and analyzed. Magnification,
×200. *P<0.05, shCCL5 + shTGF-β1 vs. shNC + shNC. shRNA, short
hairpin RNA; CCL5, C-C motif chemokine ligand 5; TGF-β1,
transforming growth factor-β1; Nf1, neurofibromin 1; ns, not
significant. |
Taken together, Fig.
3 suggests that knockdown of CCL5 or TGF-β1 alone failed to
significantly attenuate YAP-induced macrophage accumulation.
Therefore, it was hypothesized that the upregulated levels of CCL5
and TGF-β1 may exhibit additional effects on macrophages apart from
facilitating their infiltration.
Conditioned media from YAP activated
cNF cells promotes macrophage proliferation
A previous study revealed that the number of
resident macrophages increased in injured peripheral nerves where
neurofibromas had formed (24).
Therefore, a CCK-8 assay was performed, which showed that viability
of THP-1 cells was increased when grown in conditioned media from
XMU-MP-1-treated shNf1-SW10 and shNf1-Hs 53.T cells, and viability
was decreased when grown in conditioned media from VP-treated cells
(Fig. 4A). Additionally, there was
a significant increase in the protein expression levels of PCNA
induced by conditioned media from XMU-MP-1-treated cNF cells;
whereas the levels were decreased when cells were grown in
conditioned media from VP-treated cNF cells, further confirming the
increase in THP-1 cell proliferation (Fig. 4B). Conditioned media from both VP-
and XMU-MP-1-treated cNF cells did not significantly affect the
apoptotic rates of THP-1 cells (Fig.
4C). For cell cycle analysis, flow cytometry was performed, and
it was determined that the proportion of macrophages which entered
S phase and G2/M phase were increased when grown in conditioned
medium from XMU-MP-1-treated shNf1-SW10 and shNf1-Hs 53.T cell;
whereas conditioned medium from VP-treated shNf1-SW10 and shNf1-Hs
53.T cells resulted in arrest of macrophages in the G1 phase
(Figs. 4D and S6). Similarly, EdU detection in
subcutaneous tumor xenografts showed that the number of
proliferative cells (EdU+ cells) were increased in
XMU-MP-1-treated tumor sections and attenuated in VP-treated tumor
sections. Overlaying the images showed co-localized EdU+
cells and red fluorescent cells, suggesting a positive association
between macrophage cell proliferation and YAP activity. The IHC
assays for Ki67 further confirmed the increased cell proliferation
in XMU-MP-1-treated tumors and impaired cell proliferation in
VP-treated tumors (Fig. 4E).
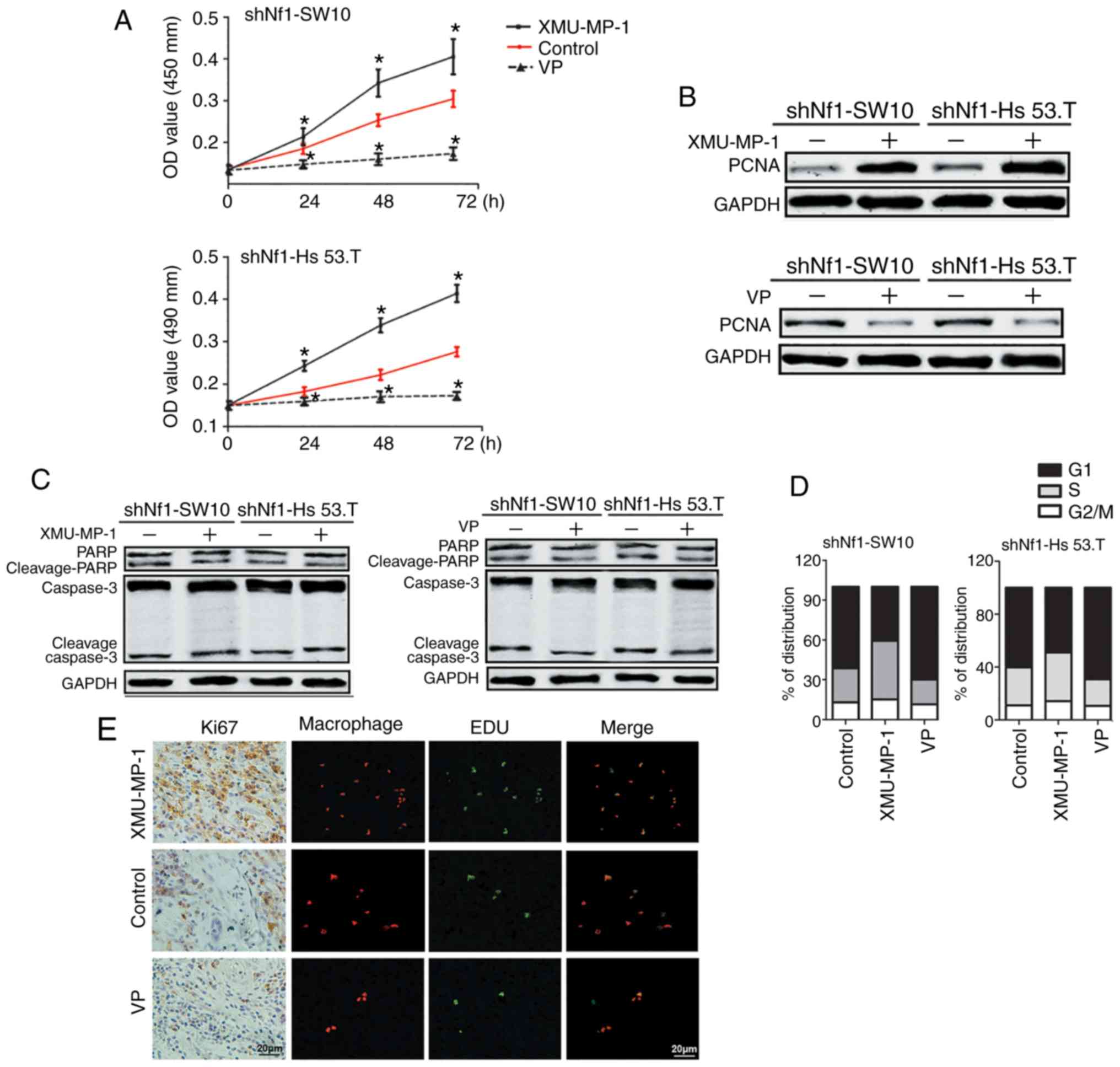 | Figure 4.Conditioned media from cNF cells with
activated YAP promotes THP-1 cell proliferation. (A) Conditioned
media was collected from XMU-MP-1-, VP- or DMSO-treated Nf1-ablated
shNf1-SW10 and shNf1-Hs 53.T cells. The conditioned media was used
to treat THP-1 cells for different periods of times, and a Cell
Counting Kit-8 assay was used to assess the cell viability of THP-1
cells. (B) Conditioned media from XMU-MP-1- or VP-treated
shNf1-SW10 or shNf1-Hs 53.T cells were collected to treat THP-1
cells. Expression of PCNA, a marker of proliferation, was assessed.
(C) Conditioned media from XMU-MP-1 or VP treated shNf1-SW10 cells
and shNf1-Hs 53.T cells were collected to treat THP-1 cells. The
protein levels of PARP, cleaved-PARP, Caspase 3 and Cleaved-Caspase
3, all markers of apoptosis, were detected in the THP-1 cells. (D)
Cell cycle distribution of these clones was detected using flow
cytometry. (E) Representative images of Ki67, RFP-labeled THP-1
cells and EdU triple staining in subcutaneous tumors from mice
treated with XMU-MP-1, VP or DMSO. Magnification, ×200. *P<0.05.
VP, Verteporfin; sh, short hairpin; cNF, cutaneous neurofibromas;
PCNA, proliferating cell nuclear antigen; medium; Nf1,
neurofibromin 1; RFP, red fluorescent protein. |
Taken together, Fig.
4 suggests that shNf1-SW10 and shNf1-Hs 53.T cells with
activated YAP facilitated THP-1 cell proliferation in an indirect
manner, which eventually contributed to macrophage accumulation in
cNFs.
Both CCL5/CCR5 and TGF-β1/TGFβ1R
suppress p21 and p27 expression
To examine the detailed mechanism involved in THP-1
proliferation with stimulation from cNF cells with altered YAP
activity, the protein levels of cell cycle-related molecules were
measured. The results showed that altered YAP activity regulated
the expression of p21 and p27, but not that of CDK4, CDK6 or cyclin
D1, in shNf1-SW10 and shNf1-Hs 53.T cells (Fig. 5A). As p21 and p27 are generally
dephosphorylated prior to entering the nucleus, where they inhibit
the cell cycle, the cytoplasmic protein and nucleoprotein levels
were assessed separately. It was demonstrated that p21 and p27
levels were higher in the cytoplasm when treated with conditioned
media from XMU-MP-1-treated shNf1-SW10 and shNf1-Hs 53.T cells, and
translocated into the nucleus when treated with conditioned media
from VP-treated shNf1-SW10 and shNf1-Hs 53.T cells (Fig. 5B). To investigate whether CCL5 and
TGF-β1 were responsible for THP-1 proliferation, THP-1 cells were
treated with conditioned media from the treated cNF cells. The
results showed that p21 and p27 levels were upregulated and PCNA
levels were downregulated following treatment with conditioned
media from CCL5- or TGF-β1-ablated shNf1-SW10 and shNf1-Hs 53.T
cells. There was a more notable alteration in the levels of p21,
p27 and PCNA in THP-1 cells stimulated with conditioned media from
both CCL5- and TGF-β1-ablated cNF cells. XMU-MP-1 activated YAP and
reduced p-YAP levels, as well as downregulating p21 and p27 levels,
and upregulating PCNA levels in the cNF cells (Figs. 5C, and S7A and B). For further confirmation,
specific shRNAs were transfected to knock down CCR5 or TGFβ1R in
THP-1 cells (Fig. 5D). Accordingly,
p21 and p27 levels were increased and PCNA levels were decreased in
both CCR5-ablated and TGFβ1R-ablated THP-1 cells when treated with
conditioned media from shNf1-SW10 and shNf1-Hs 53.T cells with or
without XMU-MP-1 treatment (Fig.
5E).
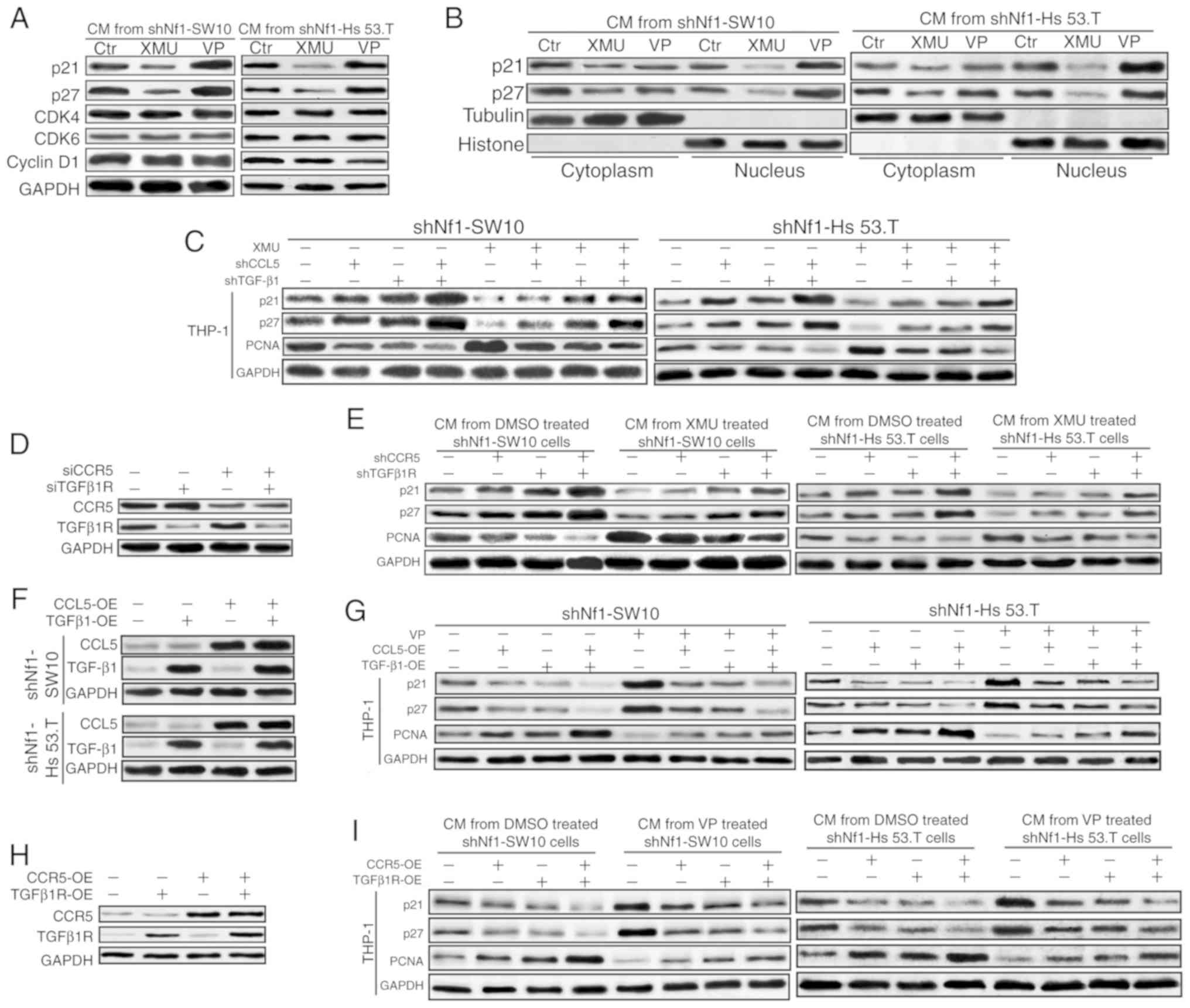 | Figure 5.CCL5 and TGF-β1 mediate the
expression of p21 and p27. (A) Nf1-ablated shNf1-SW10 and shNf1-Hs
53.T cells were treated with XMU-MP-1, VP or DMSO for 48 h before
the conditioned media was collected. Expression of p21, p27, CDK4,
CDK6 and Cyclin D1 was detected in the THP-1 cells treated with the
prepared conditioned media. (B) Nuclear and cytoplasmic proteins
were collected separately for the detection of p21 and p27 by
western blotting. (C) XMU-MP-1-treated shNf1-SW10 and shNf1-Hs 53.T
cells transfected with shCCL5, shTGF-β1 or both for 48 h prior to
collection of conditioned media. The protein levels of p21, p27,
Ki67 and PCNA in different conditioned media-treated THP-1 cells
was detected. (D) Specific shRNAs were transfected to knock down
TGFβ1R and CCR5 in THP-1 cells. (E) Conditioned media collected
from XMU-MP-1- or DMSO-treated shNf1-SW10 and shNf1-Hs 53.T cells
were used to treat THP-1 cells with knockdown of specific genes,
and the expression of p21, p27 and PCNA in the THP-1 cells was
detected. (F) Specific shRNAs targeting CCL5 or TGFβ1 were
transfected in the shNf1-SW10 and shNf1-Hs 53.T cells and the
expression of CCL5 and TGFβ1 was assessed. (G) CCL5- and/or
TGFβ1-overexpressing shNf1-SW10 and shNf1-Hs 53.T cells were
treated with VP or DMSO, and conditioned media was collected to
treat THP-1 cells. The protein levels of p21, p27 and PCNA in the
conditioned media-treated THP-1 cells were examined. (H) Specific
shRNAs against CCR5 and TGFβ1R were transfected into THP-1 cells.
(I) Conditioned media from VP- or DMSO-treated shNf1-SW10 and
shNf1-Hs 53.T cutaneous neurofibroma cells was prepared to treat
THP-1 cells. The protein levels of p21, p27 and PCNA in the
conditioned media-treated THP-1 cells was detected. CDK,
cyclin-dependent kinase; VP, Verteporfin; sh, short hairpin; PCNA,
proliferating cell nuclear antigen; CCL5, C-C motif chemokine
ligand 5; TGF-β1, transforming growth factor-β1; Nf1, neurofibromin
1. |
CCL5 and TGF-β1 were overexpressed in shNf1-SW10 and
shNf1-Hs 53.T cells, and the conditioned media were collected for
subsequent experiments (Fig. 5F).
As shown in Fig. 5G, conditioned
media from cells overexpressing CCL5 and/or TGF-β1 in the presence
or absence of VP treatment reduced p21 and p27 levels, and
increased PCNA levels in THP-1 cells. No notable alterations in YAP
or p-YAP levels were observed in these cNF cells (Figs. S7C and D), consistent with the
results in Fig. 2D. Subsequently,
CCR5 and TGFβ1R were overexpressed in THP-1 cells (Fig. 5H). Similarly, the levels of p21 and
p27 were increased and the levels of PCNA were decreased in the
CCR5- or TGFβ1R-overexpressing THP-1 cells treated with conditioned
media from VP-treated or DMSO-treated shNf1-SW10 and shNf1-Hs 53.T
cells (Fig. 5I).
Taken together, these results suggest that CCL5/CCR5
and TGF-β1/TGFβ1R may modulate the expression of p21 and p27
individually and synergistically in THP-1 cells.
Both CCL5/CCR5 and TGF-β1/TGFβ1R
promote macrophage proliferation
CCL5-ablated, TGF-β1-ablated and CCL5 +
TGF-β1-ablated shNf1-SW10 and shNf1-Hs 53.T cells were treated with
XMU-MP-1 or DMSO for 24 h, and the conditioned media was
subsequently collected. THP-1 cells were treated with the prepared
conditioned media, which validated the finding that CCL5 ablation
and/or TGF-β1 ablation could reduce the increased cell viability of
THP-1 cells induced by shNf1-SW10 and shNf1-Hs 53.T (Fig. 6A). The reduced cell viability of
CCR5-ablated and TGFβ1R-ablated THP-1 cells when treated with
conditioned media from XMU-MP-1-treated or DMSO-treated cNF cells
further indicated the importance of CCL5/CCR5 and TGF-β1/TGFβ1R in
promoting THP-1 proliferation (Fig.
6B). CCL5 or TGF-β1 knockdown prevented the YAP-activated
shNf1-SW10- and shNf1-Hs 53.T cell-induced THP-1 cells from
entering into the S and G2/M phases (Figs. 6C, and S8A and B). Furthermore, both CCR5 and
TGFβ1R knockdown arrested THP-1 cells in the G1 phase when treated
with conditioned media from shNf1-SW10 and shNf1-Hs 53.T cells,
with or without XMU-MP-1 treatment (Figs. 6D, S8C
and D).
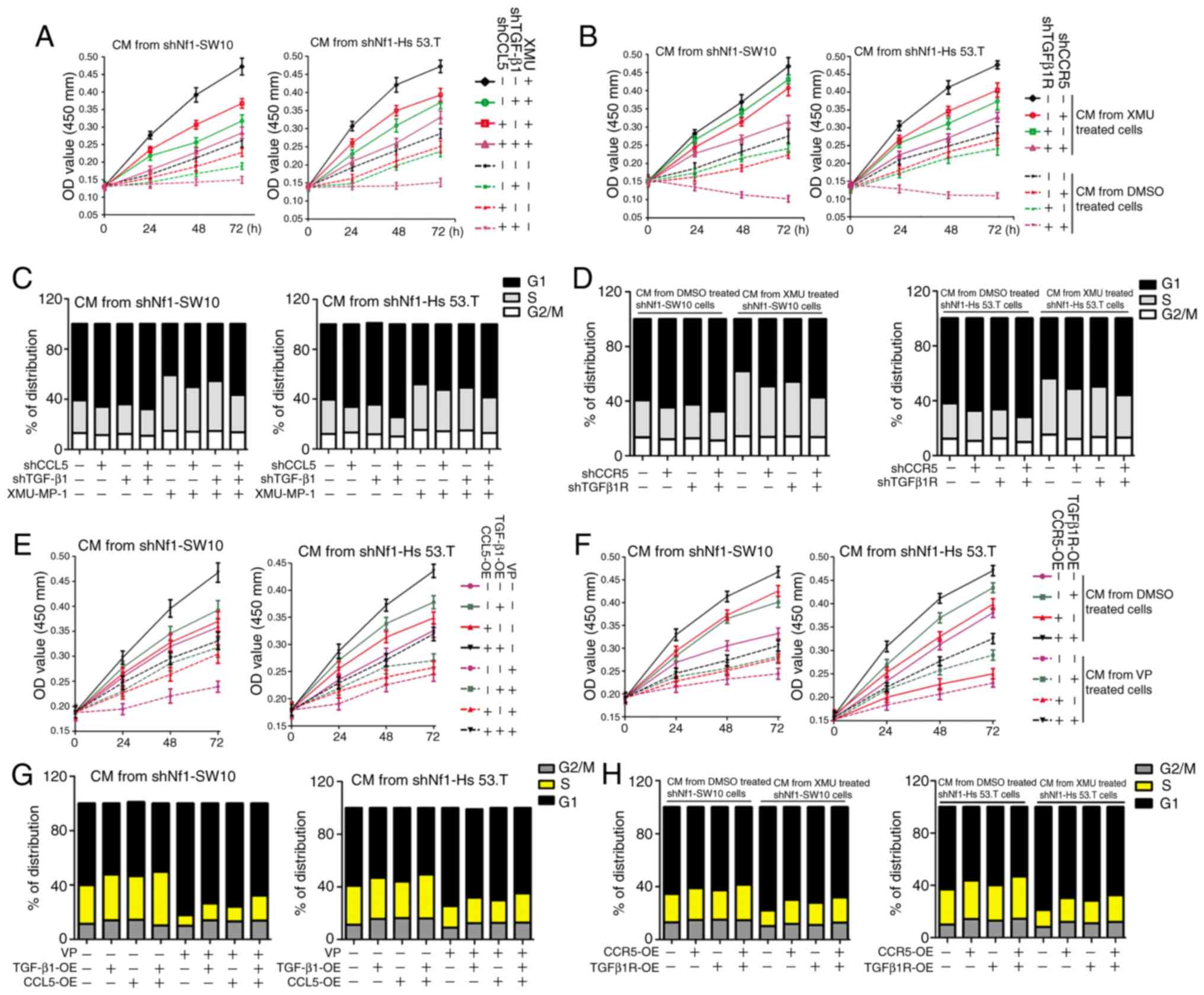 | Figure 6.CCL5/CCR5 and TGF-β1/TGFβ1R serve
important roles on the effect of cNF cells on proliferation of
THP-1 cells. (A) XMU-MP-1-treated cNF cells were transfected with
shCCL5, shTGF-β1 or both for 48 h, and conditioned media was
subsequently collected. The cell viability of THP-1 cells treated
with the conditioned media was analyzed using a CCK-8 assay. (B)
Conditioned media was collected from shNf1-SW10 and shNf1-Hs 53.T
cells treated with or without XMU-MP-1. THP-1 cells with TGFβ1R,
CCR5 or both TGFβ1R + CCR5 knocked down were treated with the
prepared conditioned media, and the cell viability was analyzed
using a CCK-8 assay. (C) Cell cycle distribution of THP-1 cells
following treatment with conditioned media from treated shNf1-SW10
and shNf1-Hs 53.T cells. (D) Cell cycle distribution in the
TGFβ1R-, CCR5- or TGFβ1R + CCR5-knockdown THP-1 cells treated with
conditioned media from XMU-MP-1-treated shNf1-SW10 and shNf1-Hs
53.T cells. (E) CCL5- or TGF-β1-overexpressing shNf1-SW10 and
shNf1-Hs 53.T cells were treated with VP or DMSO and the
conditioned media was collected. A CCK-8 assay was used to assess
the viability of THP-1 cells following treatment with different
conditioned medias. (F) THP-1 cells overexpressing CCR5 or TGFβ1R
were treated with conditioned media from VP- or DMSO-treated
shNf1-SW10 and shNf1-Hs 53.T cells. The cell viability of THP-1
cells was detected. (G) Cell cycle distribution of THP-1 cells
following treatment with conditioned media from CCL5- or
TGFβ1-overexpressing cNF cells, treated with or without VP. (H)
THP-1 cells overexpressing CCR5 and TGFβ1R prior to the treatment
with conditioned media from VP- or DMSO-treated shNf1-SW10 and
shNf1-Hs 53.T cells. Cell cycle distribution of these treated THP-1
cells were analyzed. VP, Verteporfin; sh, short hairpin; cNF,
cutaneous neurofibromas; PCNA, proliferating cell nuclear antigen;
CCL5, C-C motif chemokine ligand 5; TGF-β1, transforming growth
factor-β1; Nf1, neurofibromin 1; CCK-8, Cell Counting Kit-8. |
Similarly, VP-treated shNf1-SW10 and shNf1-Hs 53.T
cells exhibited a decreased capacity to promote proliferation of
THP-1 cells. CCL5 and/or TGF-β1 overexpression reduced this
suppression (Fig. 6E). Increased
cell viability was also observed in the CCR5- or
TGFβ1R-overexpressing THP-1 cells treated with CM from VP- or
DMSO-treated shNf1-SW10 and shNf1-Hs 53.T cells (Fig. 6F). Subsequently, cell cycle
distribution was assessed, and it was found that the proportion of
cells transitioning to the S and G2/M phases was increased when
treated with conditioned media from CCL5- and/or
TGF-β1-overexpressing shNf1-SW10 and shNf1-Hs 53.T cells, in the
presence or absence of VP (Figs.
6G, S9A and B). Accordingly,
overexpressing CCR5 or TGFβ1R also promoted THP-1 cell progression
to the S and G2/M phases, when treated with conditioned media from
VP- or DMSO-treated cNF cells (Figs.
6H, S9C and D). The results in
Fig. 6E-H suggested that YAP
reduced THP-1 cell proliferation, whereas overexpressing CCL5/CCR5
or TGF-β1/TGFβ1R, separately or concomitantly, reversed the
YAP-mediated reduction.
Taken together, Fig.
6 suggests that either CCL5/CCR5 or TGF-β1/TGFβ1R mediated cell
cycle progression of THP-1 cells and regulated THP-1 cell
proliferation synergistically.
Discussion
At present, treatment of cNF is limited to surgical
excision, which is not always curative, as the presence of numerous
or large cNF tumors are difficult to completely resect. Thus,
improving our understanding of the mechanisms underlying the
development of cNF initiation and progression may assist in the
identification of novel therapeutic targets. In the present study,
it was demonstrated that the number of macrophages was increased in
cNF tissues compared with the matching adjacent healthy tissue, and
this was accompanied by upregulated expression of YAP and TAZ,
effectors of the Hippo pathway. Additionally, it was demonstrated
that activation of the Hippo pathway effectors facilitated
macrophage accumulation by promoting both macrophage recruitment
and macrophage cell proliferation.
Clinically, patients with similar genetic
backgrounds, for example patients from the same family, were found
to exhibit different types of Cnf (14), which suggests the existence of
modifiers during neurofibroma development. A study in Nf1
heterozygous mice showed that the preferential sites for
development of neurofibromas are the cervical nerves and the
mid-thoracic nerves, flexible sites that are prone to minor but
numerous nerve injuries (25).
Similarly, the cervical nerve has also been identified as a hot
spot for development of neurofibroma formation in patients with NF1
(26). Tissue damage leads to
inflammation in order to repair the tissue, during which
inflammatory cells produce cytokines and growth factors,
stimulating factors of neoplasias (27). Mast cells and macrophages are the
two primary types of inflammatory cells in the cNF
microenvironment. Infiltrating mast cells in neurofibromas are
frequently present in an activated state, as shown by elevated
levels of local histamine and circulating serum IgE (28,29).
Macrophages in the cNF microenvironment also exhibit an
inflammatory status, shown by the presence of stronger M1
signatures than M2 signatures (23). These findings indicate that
continuous inflammation in the microenvironment may contribute to
the progression of cNF.
As mast cells are the most abundantly present type
of inflammatory cells in the cNF microenvironment (29), efforts to treat cNF by inhibiting
mast cells has been attempted. Some success has been achieved in
shrinking neurofibromas by targeting mast cells, but there remain a
number of patients who do not respond favorably to this treatment
(6). Thus, exploring complementary
factors that support neurofibroma maintenance may highlight novel
potential targets for treatment. Importantly, a clinical trial
using ketotifen (3), which
possesses antihistamine and anti-inflammatory activities, and mouse
models of cNF treated with PLX3397 (10), a dual inhibitor for both mast cells
and macrophages, indicated improved efficacy against neurofibromas
compared with inhibiting mast cells alone. Nevertheless, there were
still instances of unsatisfactory outcomes in the aforementioned
studies. Therefore, understanding the detailed mechanism involved
in macrophage accumulation may assist in identifying potential
therapeutic targets.
The critical role of Nf1 heterozygosity was first
noticed in the pioneer mouse plexiform neurofibroma model, which
showed that tumors developed in the Nf1f/− mice, but not
the Nf1f/f mice (5).
However, Nf1 heterozygosity may not always be essential, as some
Nf1f/f mice also develop neurofibromas without the
microenvironment afforded by a Nf1+/− genotype (30), suggesting the presence of
alternative signals mediating neurofibroma development. A recent
study showed that that activating Hippo pathway effectors
accelerates Nf1-associated cNF development, although altering the
Hippo pathway alone did not result in the development of tumors
(14). Furthermore, cNF also occurs
in patients with neurofibromatosis type 2 who possess mutant
neurofibromin 2, a regulator of the Hippo pathway (31). The present study showed that the
expression of YAP and TAZ was upregulated in cNF tissues, both of
which are core effectors of the Hippo pathway, and demonstrated the
positive association between YAP activity and macrophage
accumulation. These data suggest that the Hippo pathway may serve
as a modifier, although possibly not a driver, in neurofibroma
progression and modification of the tumor microenvironment.
A preliminary study showed that the Hippo pathway
modulates macrophage recruitment (32). CCL5/CCR5 and TGF-β1/TGFβR can
mediate macrophage recruitment (33,34).
The present study showed that cNF cells with activated YAP
increased macrophage accumulation by upregulating both CCL5 and
TGF-β1, and only synergistic ablation of CCL5 and TGF-β1
effectively inhibited macrophage accumulation. The possible reasons
for these results may be that other molecules not examined in the
present study contributed to macrophage accumulation.
Alternatively, cNF cells may also facilitate the
proliferation/survival of resident macrophages. The macrophages in
the tumor microenvironment included the recruited macrophages and
the resident macrophages, and the proliferation of resident
macrophages should not be overlooked during the process of
eliminating macrophages from the tumor microenvironment. The
present study revealed that repression of either CCL5 or TGF-β1
alone significantly reduced THP-1 cell viability, but did not
attenuate THP-1 cell accumulation. A possible reason for the above
results may be the increased efficiency of enhancing infiltration
and promoting proliferation of both CCL5 and TGF-β1. It was also
attempted to determine the source of macrophages in tumors, to
distinguish between infiltration/recruitment or proliferation, but
a suitable method could not be determined.
The results of the present study showed that
YAP-active neurofibroma cells regulated cell cycling in
macrophages, and this represents just one of the potential
mechanisms involved in promoting macrophage proliferation. Other
mechanisms participating in macrophage proliferation cannot be
excluded. The IHC assay with anti-Ki67 in the xenograft
subcutaneous tumors (containing macrophages and shNf1-SW10 cells)
showed that XMU-MP-1/VP treatment promoted/suppressed tumor growth.
EDU staining in the tumor tissues indicated that macrophage
proliferation was also enhanced. These results do not exclude the
potential involvement of XMU-MP-1/VP itself in promoting/reducing
macrophage proliferation. In the in vitro experiments with
conditioned media from XMU-MP-1/VP-treating shNf1-SW10/shNf1-Hs
53.T cells confirmed that YAP-activating neurofibroma cells
modulated macrophage proliferation. Taken together, these results
suggested that neurofibroma cells with activated YAP exhibited
upregulated cell growth and promoted macrophage accumulation. If
other mechanisms are also involved in neurofibroma cell-mediated
increase of macrophage proliferation, these remain to be
determined.
In conclusion, the present study demonstrated that
activation of YAP promoted proliferation of cNF cells and increased
CCL5 and TGF-β1 expression. This resulted in accumulation of
macrophages by facilitating both the recruitment and proliferation
of macrophages. Reversal of macrophage accumulation caused by
treatment with a YAP inhibitor suggested that YAP inhibition may
serve as a potential therapeutic avenue for treatment of cNF, via
impairment of both tumor cell proliferation and macrophage
accumulation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the general
project of major research plan for social development of Shaanxi
province (grant no. 2018SF-250).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
MS designed the experiments, performed the data
analyses and wrote the manuscript. LH performed the in vivo
analysis. HBZ and JJ performed the cellular experiments and
assisted with the mice experiments. HKZ performed the
immunohistochemistry assays. All authors participated in the
discussion and revision of the manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All procedures performed involving human
participants were in accordance with the ethical standards of the
Institutional Review Board of The First Affiliated Hospital of the
Xi'an Jiaotong University. The present study adhered to the
guidelines of the 1964 Helsinki declaration and its later
amendments or comparable ethical standards. Informed consent was
obtained from adult patients, or from their parents/guardians for
patients younger than 18 years old. The animal experiments were
approved by the Institutional Review Board of The First Affiliated
Hospital of Xi'an Jiaotong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Serra E, Puig S, Otero D, Gaona A, Kruyer
H, Ars E, Estivill X and Lázaro C: Confirmation of a double-hit
model for the NF1 gene in benign neurofibromas. Am J Hum Genet.
61:512–519. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Granström S, Langenbruch A, Augustin M and
Mautner VF: Psychological burden in adult neurofibromatosis type 1
patients: Impact of disease visibility on body image. Dermatology.
224:160–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riccardi VM: Ketotifen suppression of NF1
neurofibroma growth over 30 years. Am J Med Genet A. 167:1570–1577.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gutmann DH, Ferner RE, Listernick RH, Korf
BR, Wolters PL and Johnson KJ: Neurofibromatosis type 1. Nat Rev
Dis Primers. 3:170042017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu Y, Ghosh P, Charnay P, Burns DK and
Parada LF: Neurofibromas in NF1: Schwann cell origin and role of
tumor environment. Science. 296:920–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Demestre M, Herzberg J, Holtkamp N, Hagel
C, Reuss D, Friedrich RE, Kluwe L, Von Deimling A, Mautner VF and
Kurtz A: Imatinib mesylate (Glivec) inhibits Schwann cell viability
and reduces the size of human plexiform neurofibroma in a xenograft
model. J Neurooncol. 98:11–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takata M, Imai T and Hirone T:
Factor-XIIIa-positive cells in normal peripheral nerves and
cutaneous neurofibromas of type-1 neurofibromatosis. Am J
Dermatopathol. 16:37–43. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park SJ, Sawitzki B, Kluwe L, Mautner VF,
Holtkamp N and Kurtz A: Serum biomarkers for neurofibromatosis type
1 and early detection of malignant peripheral nerve-sheath tumors.
BMC Med. 11:1092013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rizvi TA, Huang Y, Sidani A, Atit R,
Largaespada DA, Boissy RE and Ratner N: A novel cytokine pathway
suppresses glial cell melanogenesis after injury to adult nerve. J
Neurosci. 22:9831–9840. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prada CE, Jousma E, Rizvi TA, Wu J, Dunn
RS, Mayes DA, Cancelas JA, Dombi E, Kim MO, West BL, et al:
Neurofibroma-associated macrophages play roles in tumor growth and
response to pharmacological inhibition. Acta Neuropathol.
125:159–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Donovan S, Shannon KM and Bollag G: GTPase
activating proteins: Critical regulators of intracellular
signaling. Biochim Biophys Acta. 1602:23–45. 2002.PubMed/NCBI
|
|
12
|
Dineen SP, Lynn KD, Holloway SE, Miller
AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB and
Brekken RA: Vascular endothelial growth factor receptor 2 mediates
macrophage infiltration into orthotopic pancreatic tumors in mice.
Cancer Res. 68:4340–4346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Keepers TR, Gross LK and Obrig TG:
Monocyte chemoattractant protein 1, macrophage inflammatory protein
1 alpha, and RANTES recruit macrophages to the kidney in a mouse
model of hemolytic-uremic syndrome. Infect Immun. 75:1229–1236.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Z, Mo J, Brosseau JP, Shipman T, Wang
Y, Liao CP, Cooper JM, Allaway RJ, Gosline SJC, Guinney J, et al:
Spatiotemporal loss of NF1 in Schwann cell lineage leads to
different types of cutaneous neurofibroma susceptible to
modification by the hippo pathway. Cancer Discov. 9:114–129. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rawat SJ, Araiza-Olivera D, Arias-Romero
LE, Villamar-Cruz O, Prudnikova TY, Roder H and Chernoff J: H-ras
inhibits the hippo pathway by promoting Mst1/Mst2
heterodimerization. Curr Biol. 26:1556–1563. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kwon Y, Vinayagam A, Sun X, Dephoure N,
Gygi SP, Hong P and Perrimon N: The Hippo signaling pathway
interactome. Science. 342:737–740. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gérard C and Goldbeter A: The balance
between cell cycle arrest and cell proliferation: Control by the
extracellular matrix and by contact inhibition. Interface Focus.
4:201300752014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murakami S, Shahbazian D, Surana R, Zhang
W, Chen H, Graham GT, White SM, Weiner LM and Yi C: Yes-associated
protein mediates immune reprogramming in pancreatic ductal
adenocarcinoma. Oncogene. 36:1232–1244. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim W, Khan SK, Liu Y, Xu R, Park O, He Y,
Cha B, Gao B and Yang Y: Hepatic Hippo signaling inhibits
protumoural microenvironment to suppress hepatocellular carcinoma.
Gut. 67:1692–1703. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi GP and Lindholt JS: Mast cells in
abdominal aortic aneurysms. Curr Vasc Pharmacol. 11:314–326. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brosseau JP, Pichard DC, Legius EH,
Wolkenstein P, Lavker RM, Blakeley JO, Riccardi VM, Verma SK,
Brownell I and Le LQ: The biology of cutaneous neurofibromas:
Consensus recommendations for setting research priorities.
Neurology. 91((2 Suppl 1)): S14–S20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi K, Komurov K, Fletcher JS, Jousma E,
Cancelas JA, Wu J and Ratner N: An inflammatory gene signature
distinguishes neurofibroma Schwann cells and macrophages from cells
in the normal peripheral nervous system. Sci Rep. 7:433152017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Müller M, Wacker K, Getts D, Ringelstein
EB and Kiefer R: Further evidence for a crucial role of resident
endoneurial macrophages in peripheral nerve disorders: Lessons from
acrylamide-induced neuropathy. Glia. 56:1005–1016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ribeiro S, Napoli I, White IJ, Parrinello
S, Flanagan AM, Suter U, Parada LF and Lloyd AC: Injury signals
cooperate with Nf1 loss to relieve the tumor-suppressive
environment of adult peripheral nerve. Cell Rep. 5:126–136. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen R, Ibrahim C, Friedrich RE,
Westphal M, Schuhmann M and Mautner VF: Growth behavior of
plexiform neurofibromas after surgery. Genet Med. 15:691–697. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Geller M, Ribeiro MG, Araújo AP, de
Oliveira LJ and Nunes FP: Serum IgE levels in neurofibromatosis 1.
Int J Immunogenet. 33:111–115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kamide R, Nomura N and Niimura M:
Characterization of mast cells residing in cutaneous neurofibromas.
Dermatologica. 179 (Suppl 1):S1241989. View Article : Google Scholar
|
|
30
|
Wu J, Williams JP, Rizvi TA, Kordich JJ,
Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA and Ratner N:
Plexiform and dermal neurofibromas and pigmentation are caused by
Nf1 loss in desert hedgehog-expressing cells. Cancer Cell.
13:105–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Evans DG: Neurofibromatosis type 2 (NF2):
A clinical and molecular review. Orphanet J Rare Dis. 4:162009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai
X, Ji X, Ji F, Gong XG, Li L, et al: Single tumor-initiating cells
evade immune clearance by recruiting type II macrophages. Genes
Dev. 31:247–259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brauß TF, Winslow S, Lampe S, Scholz A,
Weigert A, Dehne N, von Stedingk K, Schmid T and Brüne B: The
RNA-binding protein HuR inhibits expression of CCL5 and limits
recruitment of macrophages into tumors. Mol Carcinog. 56:2620–2629.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin S, Zheng JH, Xia ZH, Qian J, Deng CL
and Yang SL: CTHRC1 promotes wound repair by increasing M2
macrophages via regulating the TGF-β and notch pathways. Biomed
Pharmacother. 113:1085942019. View Article : Google Scholar : PubMed/NCBI
|














