Introduction
The most common type of liver cancer is
hepatocellular carcinoma (HCC), and the prognosis of patients with
advanced HCC is poor due to acquired resistance to current
chemotherapeutic regimens through the de-regulation of signaling
pathways governing cell proliferation and survival (1). Resistance to apoptosis of HCC cells is
a critical obstacle in cancer treatment. Among the diverse
modalities inducing apoptosis in cancer cells including HCC cells,
tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), a
death receptor ligand is one of the promising anticancer agents due
to its capability to induce apoptosis selectively in cancer cells
but not in most normal cells (2).
However, most primary cancer cells show resistance to TRAIL
monotherapy. Therefore, combination therapies are required for
reduced development of drug resistance, better effectiveness, and
reduced toxicity. TRAIL combinations have been studied to induce
synergism or sensitize TRAIL-resistant cancer cells (3), and identification of effective
combination that synergize with TRAIL to kill HCC cells is needed
for a more extensive and successful application of TRAIL-based
therapies in the future.
TRAIL-induced apoptosis occurs through the binding
of TRAIL to its cognate surface receptors. Following the binding of
TRAIL to the death receptor TRAIL-R1 (DR4) and/or TRAIL-R2 (DR5),
the activated receptors recruit the adapter protein FAS-associated
death domain (FADD) and the effector capase-8, resulting in the
assembly of the death-inducing signaling complex (DISC). After
binding the DISC, caspase-8 undergoes cleavage and promotes
apoptosis by activating the downstream effector caspase-3 and the
mitochondrial apoptotic pathway (2). The cellular-FLICE inhibitory protein
(c-FLIP), which consists of two isoforms, FLIPL and
FLIPS, resembles an initiator procaspase, except in the
absence of a proteolytic domain. Following the recruitment of
c-FLIP to the DISC, this protein competes with procaspases-8 and
−10, blocking the processing and activation of these procaspases
and inhibiting DR4- and DR5-mediated cell death. Therefore, c-FLIP
hinders apoptosis by inhibiting the activation of caspase-8 and
accordingly the inhibition of c-FLIP enhances TRAIL-induced
apoptosis in cancer cells (4).
It has been shown that several cancer cell lines
including HCC cells are resistant to TRAIL (5). An overexpression of c-FLIP, an
endogenous antiapoptotic factor which inhibits procaspase-8 in DISC
complex, may represent an important mechanism for resistance to
apoptosis in cancer cells (6). In
addition, the downregulation of antiapoptotic proteins involving
c-FLIP and/or upregulation of death receptors, and the activation
of C/EBP homologous protein (CHOP) can overcome TRAIL resistance in
cancer cells (7). CHOP, which is
induced during the unfolded protein response, mediates the
transcriptional control during endoplasmic reticulum (ER)
stress-induced apoptosis (8).
c-FLIPL is a CHOP control target, and CHOP downregulates
c-FLIPL expression at the post-transcriptional level
(9).
It has been known that an interplay of autophagy and
apoptosis, which are interconnected in their signaling pathways,
greatly affects cell death during stress responses. An insufficient
activity of autophagy may trigger apoptosis due to accumulation of
aberrant proteins and defective organelles, while excessive
activity of autophagy can also lead to cell death, even in the
insufficient stimuli of apoptosis (10). Therefore, the interconnection of
signaling pathways of both autophagy and apoptosis is not
surprising, and may modulate sensitivity to anticancer drugs.
Autophagy is associated with improvement of TRAIL sensitivity of
cancer cells through both upregulation of DR5 and c-FLIP
degradation (11,12). It has been suggested that the
addition of autophagy-inducing agents to some apoptosis-inducing
therapeutic agents could be a useful therapeutic approach to
overcome resistance of cancers (13).
Nonsteroidal anti-inflammatory drugs (NSAIDs), which
are a structurally diverse group of drugs, are widely used to treat
inflammation, fever and pain, and are currently known to have
diverse effects in cancer. Celecoxib (CCB), a non-cyclooxygenase
(COX)-2 selective NSAID, exhibits therapeutic effects in tumor
cells in combination with chemotherapy and radiotherapy in
preclinical investigations (14).
Ibuprofen, a commonly used NSAID, was also found to enhance the
anticancer effect of cisplatin in lung cancer cells (15). The antitumor effects of NSAIDs seems
to be at least in part related to their autophagy-modulating
effects, and the use of NSAIDs in combination with anticancer drugs
may be helpful for the treatment of drug-resistant tumors (16).
In this study, we investigated whether NSAID could
potentiate TRAIL cytotoxicity of TRAIL-resistant HCC cells, and the
molecular mechanism underlying the combined effect of NSAID and
TRAIL on sensitization of TRAIL-resistant HCC cells to TRAIL for
reversal of TRAIL resistance by inducing autophagic cell death as
well as apoptosis.
Materials and methods
Cell culture and reagents
Hepatocellular carcinoma (HCC) cell lines (SNU-354,
SNU-423, SNU-449 and SNU-475) derived from HCC tissues of patients
were purchased from the Korea Cell Line Bank (17). TRAIL-resistant SNU-475/TR subline
was isolated from SNU-475 cells by stepwise increases in
concentrations of TRAIL, starting with 10 ng/ml and reaching 200
ng/ml. After approximately 2 month of drug exposure, SNU-475/TR
cells were able to grow in the presence of 200 ng/ml TRAIL, and
this phenotype remained stable through passages. SNU-475/TR cells
were at least 100-fold more resistant than SNU-475 cells to TRAIL.
Cells were maintained in RPMI-medium (Welgene) supplemented with
10% (v/v) heat-inactivated fetal bovine serum (FBS; Welgene), 100
U/ml penicillin and 100 µg/ml streptomycin in a 5% CO2
humidified incubator at 37°C. Recombinant human soluble TRAIL was
obtained from R&D systems. Celecoxib (CCB), 2,5-dimethyl
celecoxib (DMC), cycloheximide (CHX), 3-methyladenine (3-MA),
chloroquine (CQ), LY294002 (LY), and 4-phenylbutyric acid (4-PBA)
were purchased from Sigma-Aldrich; Merck KGaA.
Cell proliferation assay
Cell proliferation was measured by counting viable
cells using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
colorimetric dye-reduction method. Exponentially growing cells
(1×104 cells/well) were plated in a 96-well plate and
incubated in growth medium treated with the indicated concentration
of TRAIL in the absence or presence of the NSAID at 37°C. After 96
h, the medium was removed using centrifugation (500 × g for 10
min), and MTT-formazan crystals solubilized in 100 µl DMSO. The
optical density (OD) of each sample at 570 nm was measured using an
ELISA reader. The optical density of the medium was proportional to
the number of viable cells. Inhibition of proliferation was
evaluated as a percentage of control growth (no drug in the
medium). All experiments were repeated in at least two experiments
in triplicate.
Western blot analysis
Cells were lysed by RIPA cell lysis buffer (1X) with
EDTA (GenDEPOT, Inc.) and the Bradford protein (Bio-Rad
Laboratories, Inc.) assay was used for protein determination. The
protein samples from each group were resolved by 10–15% SDS-PAGE
(20 µg protein per lane) and transferred to PVDF membrane (EMD
Millipore). Membranes were blocked with 5% non-fat milk in TBST at
room temperature (RT) for 1 h, and then incubated at 4°C overnight
with the primary antibodies: Caspase-8 (dilution 1:1,000, cat. no.
9746), caspase-9 (dilution 1:1,000, cat. no. 9508), caspase-3
(dilution 1:1,000, cat. no. 9662), c-FLIP (dilution 1:1,000, cat.
no. 56343), ALDH1 (dilution 1:1,000, cat. no. 36671), Nanog
(dilution 1:1,000, cat. no. 3580), CHOP (dilution 1:1,000, cat. no.
5554), CD44 (dilution 1:1,000, cat. no. 5640), β-tubulin (dilution
1:10,000, cat. no. 2146), AMPK (dilution 1:1,000, cat. no. 2532),
phosphorylated (p)-AMPK (Thr172) (dilution 1:1,000, cat. no. 2535),
Akt (dilution 1:1,000, cat. no. 9272), p-Akt (Thr308) (dilution
1:1,000, cat. no. 9271), mTOR (dilution 1:1,000, cat. no. 2972),
p-mTOR (Ser2448) (dilution 1:1,000, cat. no. 2971), p70S6K
(dilution 1:1,000, cat. no. 9202), p-p70S6K (Thr389) (dilution
1:1,000, cat. no. 9205), 4E-BP1 (dilution 1:1,000, cat. no. 9644)
and p-4E-BP1 (Thr37/46) antibodies (dilution 1:1,000, cat. no.
2855) (all above were purchased from Cell Signaling Technology,
Inc.) ATF4 (dilution 1:1,000, cat. no. ab34034), DR5 (dilution
1:1,000, cat. no. ab16942; Abcam), PARP (dilution 1:10,000, cat.
no. sc-7150; Santa Cruz Biotechnology, Inc.), β-actin (dilution
1:10,000, cat. no. A5316; Sigma-Aldrich; Merck KGaA), LC3B
(dilution 1:5,000, cat. no. NB100-2220), p62 (dilution 1:1,000,
cat. no. NBP1-31381; Novus) CD133 (dilution 1:1,000, cat. no.
PAB12663; Abnova). Membranes were washed with TBST three times (15
min per time), incubated with horseradish peroxidase-conjugated
secondary antibody (dilution 1:10,000, cat. no. 7074; dilution
1:50,000, cat. no. 7076; Cell Signaling Technology, Inc.) and
washed thrice with TBST. Target proteins were visualized using ECL
and detected with an enhanced chemiluminescence (ECL) detection
system (NEL103E001EA, PerkinElmer Inc.), and the film (Agfa
RADIOMAT™) that detected chemiluminescence signals from Western
blot was developed by JP-33 Automatic X-Ray Film Processor (JPI
America, Inc.) in a darkroom.
Apoptosis analysis by flow
cytometry
Cell apoptosis was detected using an Annexin
V/propidium iodide (PI) apoptosis detection kit according to the
manufacturer's protocols. Cells were treated with TRAIL in the
presence or absence of CCB (or DMC). After 24 h, the cells were
harvested by trypsinization and washed in PBS. The cells were
centrifuged (400 × g for 5 min) and resuspended in 100 µl binding
buffer, and then 5 µl FITC Annexin V and 3 µl PI was added. The
cells were gently vortexed and incubated for 15 min at RT in the
dark. After that 400 µl of binding buffer was added to each tube.
The samples were analyzed using a CANTO II (BD Biosciences) flow
cytometer and the data were analyzed using FlowJo (ver.10; Tress
Star).
Fluorescence-activated cell sorting
(FACS) analysis
Expression profiles of cell surface molecules on HCC
cell lines were conducted using FITC mouse anti-human CD44
(dilution 1:50, cat. no. 555478; BD Biosciences), APC mouse
anti-human CD133 (dilution 1:100, cat. no. 130-090-854; Miltenyi
Biotec) and FITC mouse anti-human DR5 (dilution 1:10, cat. no.
MA1-19759; Invitrogen; Thermo Fisher Scientific, Inc.) monoclonal
antibodies (mAbs). Cells (5×105 cells/well) were
centrifuged at 500 × g and resuspended in 500 µl PBS. Cells were
then incubated for 1 h on ice with 5 µl of mouse IgG, anti-CD44 or
anti-CD133 mAb for 30 min. After washing with PBS, PE-conjugated
rabbit anti-mouse IgG (dilution 1:500, cat. no. IC002P; R&D
Systems) was added to the cell suspensions, incubated for 30 min on
ice, and washed with PBS. After rinsing, samples were analyzed by
flow cytometry using a FACS Calibur flow cytometer (BD
Biosciences). The data were analyzed using CellQuest software
(version 3.3; BD Biosciences). Cell surface expression of CD44 or
DR5 on HCC cells treated with or without NSAID was determined by
flow cytometry. Briefly, cells were washed once at the time of
harvesting with PBS/0.1% sodium azide and aliquoted into
polystyrene tubes. Cells were stained with FITC-labeled anti-CD44
mAb or anti-DR5 mAb. Autofluorescence and isotype (IgG2b)-matched
control Abs (dilution 1:500, cat. no. MA5-14447; BD Biosciences)
were included. Data was collected using BD FACSCanto II or BD
FACSCalibur flow cytometer (BD Biosciences) and subsequently
analyzed with FlowJo software (version 10; Tree Star).
Statistical analysis
Statistical analyses were performed using SPSS ver.
24.0 (IBM Corp.). One-way analysis of variance (ANOVA) with Tukey's
post hoc test was performed to analyze multiple groups, and
unpaired t-test was used to analyze differences in data between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
NSAID sensitizes TRAIL-resistant
CD44-overexpressing HCC cells to TRAIL
Expression of CD44 and CD133 as markers of liver
cancer stem cells can contribute to hepatocellular carcinoma (HCC)
development and poor responsiveness of HCC towards chemotherapy and
radiotherapy (18). Since high
susceptibility to TRAIL of HCC cells is associated with CD133
expression (19), we therefore
investigated whether the acquisition of resistance to TRAIL is
associated with distribution of CD44/CD133 expression in HCC cells.
When both SNU-475/TR cells exhibiting high-level resistance to
TRAIL and the parental TRAIL-sensitive SNU-475 cells were treated
with increasing doses of TRAIL, marked activation of caspases
(caspase-8, −9 and −3) and PARP, showing cleavage of caspases and
PARP, occurred in the SNU-475 cells but not in the SNU-475/TR
cells, indicating resistance of SNU-475/TR cells to TRAIL (Fig. 1A and B). Moreover, SNU-475/TR cells
exhibited relatively high basal expression of CD44 but undetectable
expression of CD133, as compared to those in SNU-475 cells, and
also the upregulated CD44 level of SNU-475/TR cells was not
modulated by TRAIL treatment (Fig.
1B), suggesting that the expression of CD44 in HCC cells might
be linked to the acquisition of the TRAIL-resistance phenotype. We
next compared cell surface expression of DR5, a major predictor of
TRAIL sensitivity, between SNU-475/TR and SNU-475 cells by FACS
analysis. As expected, the cell surface expression of DR5 in
SNU-475/TR cells was significantly decreased when compared with
that in SNU-475 cells (Fig. 1C). We
therefore evaluated the expression of both CD44 and CD133
cell-surface markers in four HCC cell lines and SNU-475/TR subline
(Fig. 1D). The surface expression
of CD44 in SNU-423, SNU-449, SNU-475/TR, SNU-423 and SNU-449 cells
was significantly higher than that in the SNU-475 and SNU353 cells
whereas the surface expression of CD133 in SNU-353, SNU-423,
SNU-449 and SNU-475/TR cells was significantly lower than in
SNU-475 cells. Indeed, we previously reported that CD133-high
SNU475 cells were more sensitive to TRAIL than CD133-low SNU-423,
SNU-449 and SNU-354 cells (19).
These results indicate that the expression ratio of CD44/CD133
plays a critical role in HCC cells for TRAIL sensitivity, and
downregulation of CD44 is required for enhancing the TRAIL
sensitivity of HCC cells.
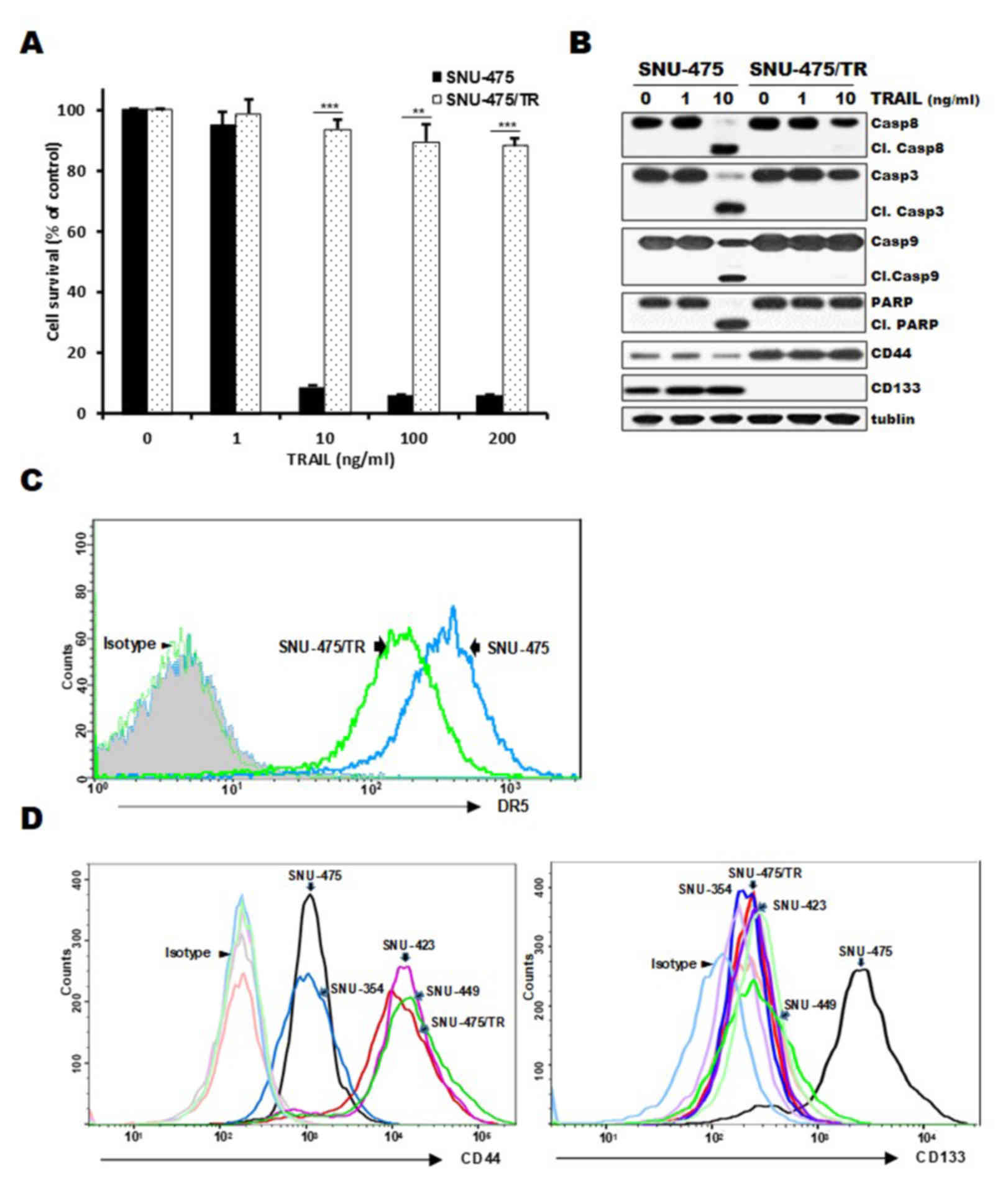 | Figure 1.Comparison of cytotoxicity, activity
of caspases and surface expression of DR5, CD44 and CD133 in
SNU-475/TR cells and other HCC cells in the absence and presence of
TRAIL. (A) SNU-475/TR cells and the parental SNU-475 cells were
treated with various concentrations of TRAIL (0. 1. 10. 100 and 200
ng/ml). Percentage of cell survival was determined after 96 h of
incubation using MTT assay. Each bar represents the mean ± SD of
triplicate experiments. **P<0.01, ***P<0.001 compared to the
SNU-475 cells. (B) SNU-475/TR and SNU-475 cells were treated with
TRAIL (1 or 10 ng/ml) for 24 h, and the level of activation by the
cleavage (Cl.) of pro-caspase-8 (Casp8), −3 (Casp3), −9 (Casp9) and
PARP and the expression of CD44/CD133 were determined by western
blot analysis. Tubulin was used as a loading control. (C) Both cell
lines were stained with anti-DR5 antibody to determine the surface
expression of DR5 by a flow cytometer. (D) Four HCC and SNU-475/TR
cell lines were stained with anti-CD133 or anti-CD44 antibody to
determine the surface expression of CD44 or CD133 by a flow
cytometer. HCC, hepatocellular carcinoma; TRAIL, TNF-related
apoptosis inducing ligand; DR4, death receptor TRAIL-R1; PARP,
poly(ADP-ribose) polymerase. |
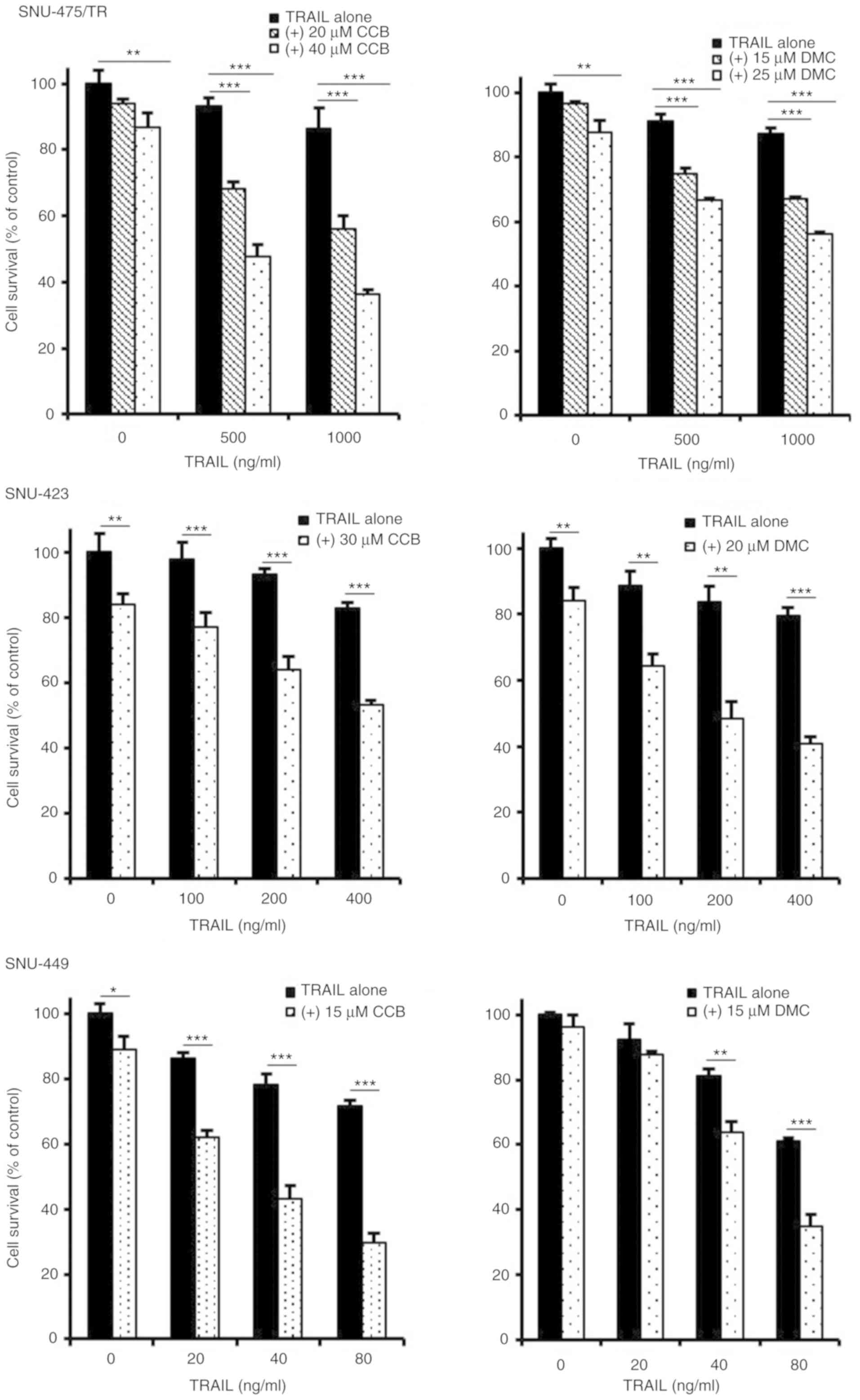
To enhance the TRAIL-mediated cytotoxicity and thus
to overcome TRAIL resistance of TRAIL-resistant HCC cells, it is
important to develop novel TRAIL sensitizing agent in combination
with TRAIL. Indeed, combining TRAIL with single-drug therapy such
as TRAIL-sorafenib combination has shown potential in colon cancer
cells (20). In the present study,
we investigated the combined effect of NSAID as TRAIL a sensitizing
agent with TRAIL for obtaining sensitization of TRAIL-resistant HCC
cells to TRAIL. To evaluate the effectiveness of NSAID as a new
modulator of TRAIL sensitivity, the changing susceptibility of
TRAIL-resistant HCC cells to TRAIL by NSAIDs such as celecoxib
(CCB) and 2,5-dimethyl celecoxib (DMC), a non-COX-2 inhibitory
analog of celecoxib was determined by MTT assay (Fig. 2). TRAIL-resistant
CD44-overexpressing SNU-475/TR, SNU-423 and SNU-449 cells were
treated with various concentrations of TRAIL in the presence or
absence of CCB or DMC. SNU-475/TR cells showed increased
susceptibility to TRAIL by combined treatment with CCB and TRAIL
than TRAIL alone, indicating potentiation of TRAIL sensitivity in
SNU-475/TR cells by CCB. We also determined whether DMC could
enhance susceptibility of SNU-475/TR cells to TRAIL. DMC
significantly potentiated TRAIL cytotoxicity in SNU-475/TR cells,
suggesting that DMC could exert TRAIL-potentiating effect,
regardless of COX-2 activity. Similar results were observed in
other TRAIL-resistant SNU-423 and SNU-449 cells co-treated with
TRAIL and CCB (or DMC). SNU-354 cells also showed that both CCB and
DMC potentiated TRAIL cytotoxicity, and another non-selective COX
inhibitor ibuprofen also potentiated TRAIL cytotoxicity in
TRAIL-resistant HCC cells (data not shown). These results strongly
suggest that NSAID could be a promising candidate for a new class
of TRAIL sensitizers in HCC cells.
NSAID modulates the expression of
DR5/c-FLIP and CSC markers through ATF4/CHOP signaling axis in
TRAIL-resistant HCC cells
Since enhancement of DR5 expression is mediated
through upregulation of ER stress-induced major transcriptional
factor CHOP (8,21), we determined whether CCB could
induce upregulation of DR5 via activation of ATF4 and CHOP, one of
the direct ATF4 target genes, in SNU-475/TR cells in comparison to
parental SNU-475 cells. CCB-induced ATF4/CHOP-dependent DR5
upregulation was observed in SNU-475/TR cells but not in SNU-475
cells (Fig. 3A, upper rows). It has
been reported that the level of c-FLIP is a key regulator of TRAIL,
and upregulation of c-FLIP induces TRAIL resistance (22). Downregulation of c-FLIP plays a
critical role in overcoming TRAIL resistance and is caused by
facilitating autophagy-mediated c-FLIP degradation, leading to
apoptosis (23). We found that
CCB-induced c-FLIPL/S downregulation in SNU-475/TR cells
with high basal level of c-FLIPL/S was more prominent
than SNU-475 cells, and concurrently the expression of CSC markers
CD44, ALDH1 and Nanog that are overexpressed in SNU-475/TR cells
was decreased by CCB (Fig. 3A,
bottom rows). To investigate the mechanisms underlying enhancement
of TRAIL-induced cytotoxicity by NSAID, modulation of DR5 cell
surface expression by NSAID in HCC cells was investigated by FACS
analysis since DR5 plays a critical role in TRAIL sensitization via
the extrinsic pathway. In SNU-475/TR cells, the cell surface
expression of DR5 was significantly increased by CCB treatment
(Fig. 3B). Next, we determined
whether NSAID could induce DR5 activation in other TRAIL-resistant
HCC cells. SNU-449 and SNU-354 cells also showed that DR5 surface
expression was increased by CCB treatment (Fig. 3C). Similar results were obtained in
DMC-treated with HCC cells. The cell surface expression of DR5 was
significantly increased in SNU-475/TR, SNU-354 and SNU-449 cells by
DMC treatment (Fig. 3D). These
results indicate that NSAID-induced DR5 activation might contribute
to sensitization of TRAIL-resistant HCC cells to TRAIL.
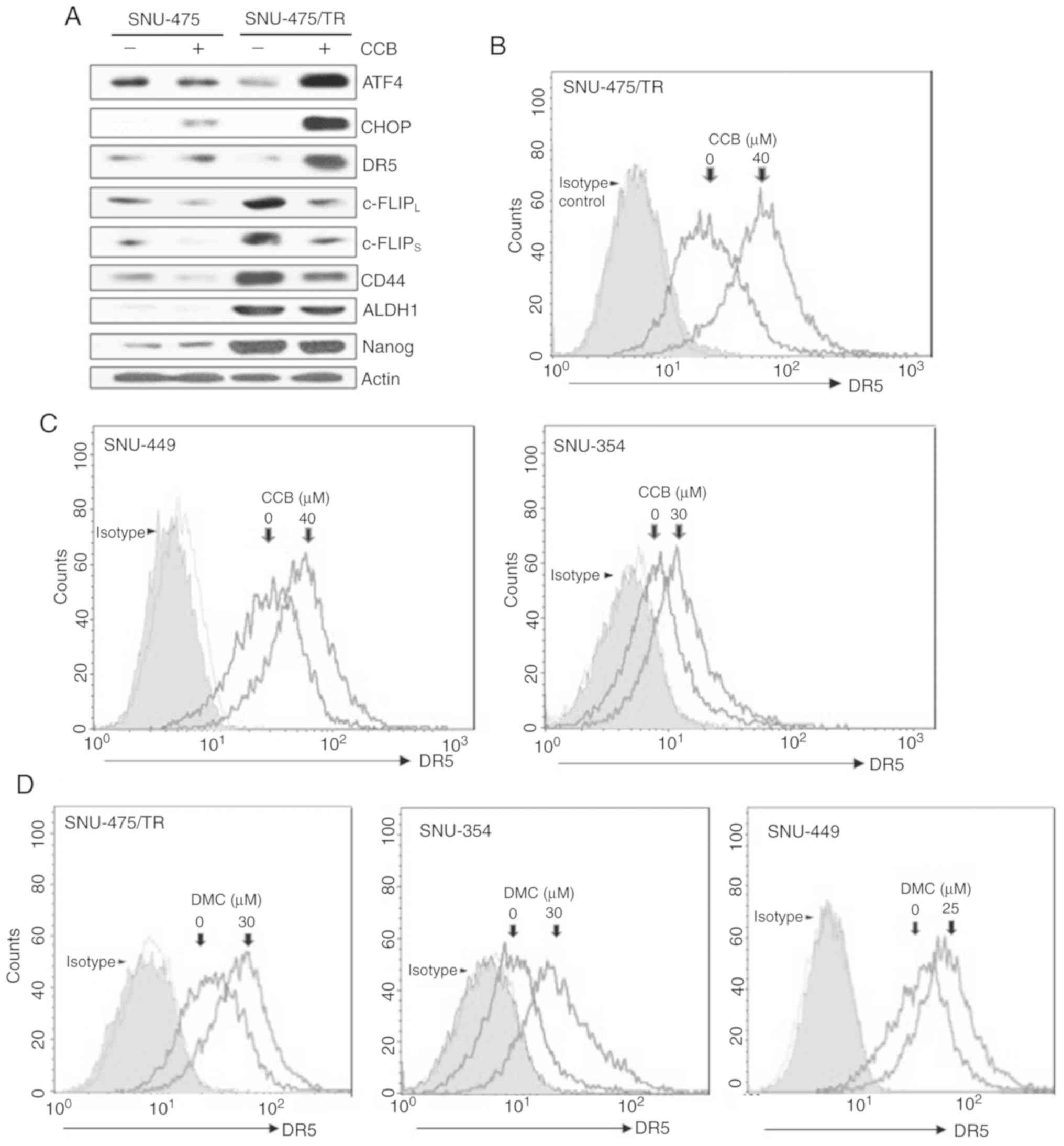 | Figure 3.CCB-induced regulation of DR5/c-FLIP
and CSC markers through the ATF/CHOP signaling axis in SNU-475/TR
cells, and the effect of NSAID on the cell surface expression of
DR5 in HCC cells. (A) The altered levels of ATF4, CHOP, DR5 (mature
form), c-FLIPL/S, CD44, ALDH1 and Nanog in SNU-475 and
SNU-475/TR cells by CCB treatment (40 µM for 24 h) were determined
by western blot analysis. Actin was used as a loading control.
(B-D) HCC cell lines were treated with the indicated dose of CCB or
DMC for 24 h. The cells were stained with control IgG or anti-DR5
antibody and subsequently labeled with PE-conjugated secondary
antibodies to determine the surface expression of DR5, and the cell
surface expression were measured by a flow cytometer. CCB,
celecoxib; DR5, death receptor TRAIL-R2; c-FLIP, cellular-FLICE
inhibitory protein; CSC, cancer stem cell; ATF, activating
transcription factor; CHOP, C/EBP homologous protein; ALDH1,
aldehyde dehydrogenase 1; NSAID, non-steroidal anti-inflammatory
drug; HCC, hepatocellular carcinoma; DMC, 2,5-dimethyl
celecoxib. |
NSAID induces degradation of c-FLIP
and upregulation of DR5 through activation of ER stress-dependent
autophagy
The protein c-FLIP was found to be degraded by
autophagy and competes with microtubule-associated protein light
chain 3 (LC3) for Atg3 binding (12,24).
LC3 is widely used to monitor autophagy. The amount of LC3
conversion (LC3-I to LC3-II) is clearly correlated with the number
of autophagosomes, and an increase in endogenous LC3-II may be
regarded as a marker for autophagy (25). In addition, ER stress-inducible
transcription factors ATF4/CHOP signaling has been shown to play
significant role in the regulation of autophagy (26). In SNU-475/TR cells, we therefore
examined whether CCB (or DMC) treatment could induce ER
stress-dependent autophagy and lead to the modulation of DR5 and
c-FLIP expression (Fig. 4A). CCB
dose-dependently upregulated both LC3-II and CHOP, resulting in
downregulation of c-FLIPL/S and upregulation of DR5.
These results indicate CCB-induced autophagic degradation of c-FLIP
and CHOP-mediated upregulation of DR5. Similarly, DMC treatment of
SNU-475/TR cells also resulted in an increase in LC3-II, CHOP and
DR5 levels and a decrease in c-FLIPL/S levels. We
further confirmed the ability of CCB to induce autophagy related to
the degradation of CD44 through ATF4/CHOP-dependent autophagy in
SNU-475/TR cells since the degradation of CD44 in cancer cells is
mediated by autophagy (27). When
SNU-475/TR cells were treated with CCB in the presence of autophagy
inhibitor, we determined whether CCB-induced modulation of DR5 and
c-FLIPL/CD44 was blocked via autophagy
inhibition-mediated ATF/CHOP inactivation (Fig. 4B). Autophagy inhibitor chloroquine
(CQ) that blocks the final steps of autophagic degradation
prevented CCB-induced upregulation of both ATF4/CHOP and DR5 and
downregulation of c-FLIPL and CD44 in SNU-475/TR cells.
Similar results were observed when SNU-475/TR cells were treated
with CCB in the presence of another autophagy inhibitor
3-methyladenine (3-MA) or LY294002 (LY) that suppresses autophagy
via inhibition of class III PI3K. CCB-induced upregulation of ATF4
and DR5 and downregulation of CD44 was significantly attenuated by
3-MA treatment in SNU-475/TR cells. In addition to LC3, degradation
of p62 is another widely used marker to monitor autophagic activity
and thus the reduced level of p62 can be used to monitor autophagic
flux (25). The CCB-induced
reduction of p62 level and subsequent downregulation of
c-FLIPL and upregulation of DR5 were blocked by LY
treatment. These results indicate that ATF4/CHOP-induced autophagy
induction might be associated with degradation/downregulation of
c-FLIP and CD44. As shown in Fig.
4C, SNU-354 cells also demonstrated that treatment of CCB or
DMC resulted in a significant increase in LC3-II and decrease in
p62 level and concurrent downregulation of c-FLIPL/S,
and upregulation of DR5 in a dose-dependent manner, which were
prevented by LY treatment, indicating that NSAID positively
regulates autophagy and TRAIL activity in HCC cells. Since
autophagy-mediated c-FLIP degradation in lung adenocarcinoma A549
cells has been reported (23), we
examined the CCB-mediated c-FLIPL degradation in HCC
cells in the presence of protein synthesis inhibitor cycloheximide
(CHX) after treatment with CCB (Fig.
4D). When the half-life of c-FLIPL protein was
determined in SNU-475/TR cells treated with or without CCB by
performing a CHX chase assay, degradation of c-FLIP protein in
CCB-treated cells was accelerated in the presence of CHX compared
with CCB-untreated cells. Similarly, the half-life of
c-FLIPL protein in SNU-449 cells was also significantly
reduced by CCB treatment. These results suggest that CCB causes
reduction of c-FLIP level in TRAIL-resistant HCC cells possibly
through autophagic degradation.
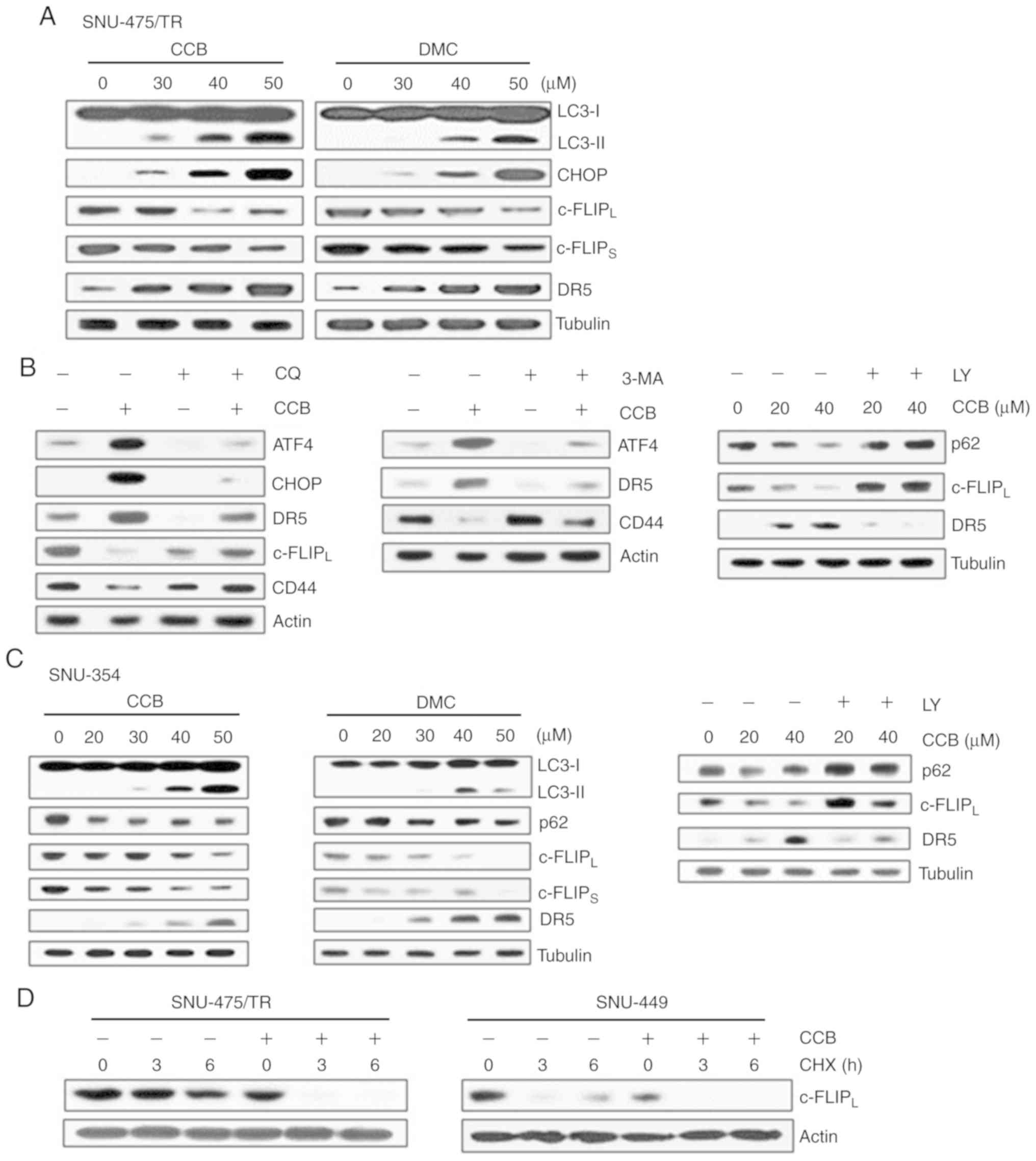 | Figure 4.Effect of NSAID on expression of
CHOP/DR5 and c-FLIP and autophagic degradation of c-FLIP in HCC
cells. (A) SNU-475/TR cells were treated with serial doses of CCB
or DMC for 24 h. (B) SNU-475/TR cells were pretreated with 5 µM
chloroquine (CQ), 10 mM 3-methyladenine (3-MA) or 5 µM LY294002
(LY) for 3 h followed by treatment of 40 µM CCB for 24 h. (C)
SNU-354 cells were treated with serial doses of CCB or DMC for 24 h
or 5 µM LY for 3 h followed with treatment of 40 µM CCB for 24 h.
Subsequently, altered levels of the indicted molecules in these
cells were determined by western blot analysis. (D) SNU-475/TR and
SNU-449 cells were treated with or without 50 µM CCB for 24 h, and
were collected at 0, 3 and 6 h following treatment with 20 µg/ml
cycloheximide (CHX) and the level of c-FLIPL was
determined by western blot analysis. TRAIL, TNF-related apoptosis
inducing ligand; NSAID, non-steroidal anti-inflammatory drug; CHOP,
C/EBP homologous protein; DR5, death receptor TRAIL-R2; c-FLIP,
cellular-FLICE inhibitory protein; HCC, hepatocellular carcinoma;
CCB, celecoxib; DMC, 2,5-dimethyl celecoxib. |
NSAID reduces CSC marker CD44 surface
expression and accelerates TRAIL-mediated downregulation of c-FLIP
and activation of caspases and PARP
Since TRAIL-resistance phenotype of SNU-475/TR cells
was closely linked to high expression of CD44, we determined
whether CCB could modulate surface expression of CD44 by FACS
analysis (Fig. 5A). SNU-475/TR
cells expressing a high level of CD44 showed that CCB significantly
reduced CD44 surface expression when compared to SNU-475 cells.
Similarly, inhibition of CD44 surface expression by DMC treatment
occurred in SNU-475/TR and SNU-449 cells. These results
demonstrated that treatment of HCC cells with DMC as well as CCB
induced reduction of CD44 surface expression and suggest that NSAID
could suppress the expression of CD44, one of the major causes of
TRAIL resistance, and overcome TRAIL resistance in HCC cells. We
next determined the effect of TRAIL and NSAID, singly and in
combination, on apoptosis induction in HCC cells. SNU-449 cells
showed an increase in the induction of apoptosis after co-treatment
with TRAIL and CCB (or DMC) compared to the individual drug
treatments (Fig. 5B). The increase
in apoptosis after the combination treatment of TRAIL and NSAID was
observed in other HCC cells (data not shown). We also investigated
the effects of NSAID on TRAIL-mediated c-FLIP expression and
caspase activity in various HCC cells. We found that NSAID
accelerated TRAIL-mediated c-FLIP downregulation, and subsequent
caspase and PARP activation in HCC cells (Fig. 5C). CCB or DMC promoted
TRAIL-mediated c-FLIPL/S downregulation and subsequent
activation of caspase-8 and −3 and PARP cleavage in SNU-449 cells,
indicating acceleration of TRAIL-mediated cell death by NSAID.
Similarly, TRAIL-mediated c-FLIPL downregulation and
caspase-8 and −3 and PARP activation were significantly accelerated
by combined treatment of TRAIL and CCB in SNU-423 and SNU-354 cells
compared to the results of TRAIL treatment alone. Our data suggest
that sensitization of TRAIL-resistant HCC cells to TRAIL by NSAID
is partially caused by degradation/downregulation of c-FLIP and
CD44 via ATF4/CHOP-induced autophagy.
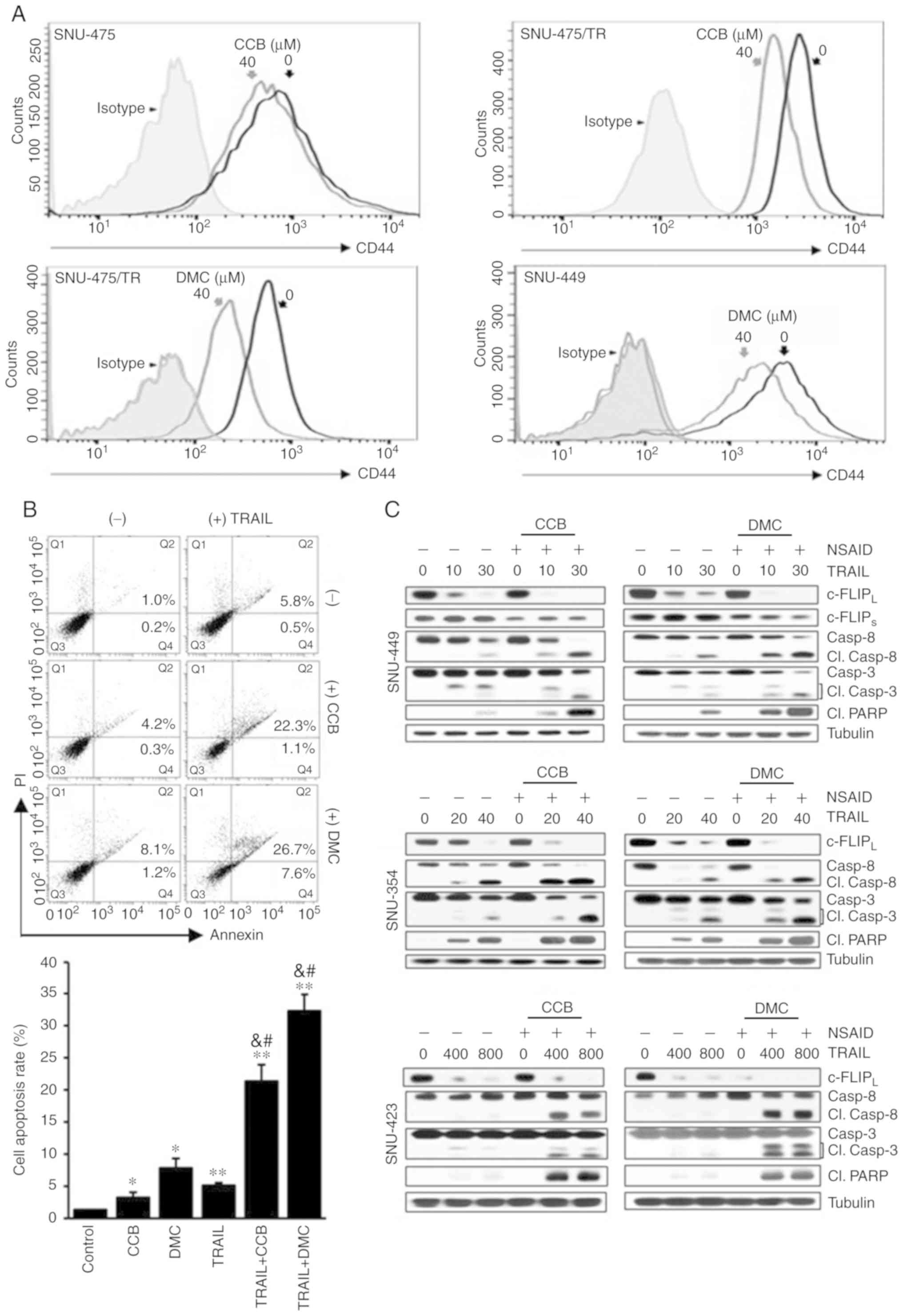 | Figure 5.Acceleration of TRAIL-mediated
caspase and PARP activities by CCB in HCC cells through inhibition
of CD44 surface expression and downregulation of c-FLIP. (A)
SNU-475, SNU-475/TR and SNU-449 cells treated with 40 µM CCB or DMC
for 24 h were stained with anti-CD44 antibody to determine the
surface expression of CD44 by a flow cytometer. (B) Apoptosis
assay. SNU-449 cells treated with TRAIL (30 ng/ml) in the presence
or absence of NSAID (20 µM CCB or DMC). Annexin V/PI double
staining by flow cytometry (upper image). Statistical analysis of
the cell apoptosis assay (lower histogram). The number of apoptotic
cells is the sum of Q2 and Q4. Data are mean ± SD, n=3. *P<0.01,
**P<0.001 vs. the control; &P<0.001 vs. TRAIL
alone; #P<0.001 vs. CCB or DMC alone, respectively.
(C) SNU-449, SNU-354 and SNU-423 cells treated with the indicated
doses of TRAIL (ng/ml) in the presence or absence of NSAID (20 µM
CCB or DMC) for 24 h. The levels of c-FLIP and activity of caspase
and PARP in these cells were determined by western blot analysis.
Cl., cleaved; TRAIL, TNF-related apoptosis inducing ligand; PARP,
poly(ADP-ribose) polymerase; CCB, celecoxib; DMC, 2,5-dimethyl
celecoxib; HCC, hepatocellular carcinoma; NSAID, non-steroidal
anti-inflammatory drug; c-FLIP, cellular-FLICE inhibitory
protein. |
NSAID activates AMPK but inhibits
Akt/mTOR signaling pathway through ER-mediated ATF4/CHOP
pathway
It has been reported that autophagy is promoted by
AMPK and inhibited by mTOR, and mTOR phosphorylates downstream
targets such as ribosomal protein S6 kinase (p70S6K) and eukaryotic
initiation factor 4E-binding protein (4E-BP1) (28). We investigated both total and
phosphorylated forms of AMPK and key proteins of Akt/mTOR signaling
pathway. Treatment of SNU-475/TR cells with CCB (or DMC) resulted
in an increase in phosphorylated AMPK (p-AMPK) and a decrease in
phosphorylated Akt (p-Akt), mTOR (p-mTOR), p70S6K (p-p70S6K) and
4E-BP1 (p-4E-BP1) without affecting total protein levels of them,
indicating that activation of AMPK phosphorylation and inhibition
of Akt/mTOR/p70S6K/4E-BP1 phosphorylation by CCB participates in
the event of CCB-induced autophagy (Fig. 6A). These results indicate the
possibility that autophagic response triggered by NSAID via AMPK
activation and inhibition of Akt/mTOR signaling may be linked to
activation of ATF/CHOP signaling in HCC cells. Since ATF4/CHOP is
the key signal for autophagy induced by ER stress, and the above
data showed that CCB-induced up-regulation of DR5 and
downregulation of c-FLIP and CD44 occurred through ATF4/CHOP
pathway, we determined whether ER stress inhibitor 4-phenylbutyric
acid (4-PBA) could modulate the CCB-induced expression of
ATF4/CHOP, DR5, c-FLIP and CD44 in SNU-475/TR cells (Fig. 6B). Our data showed that blocking ER
stress with 4-PBA suppressed CCB-induced upregulation of ATF4 and
CHOP and subsequent upregulation of DR5 and also blocked
downregulation of c-FLIP and CD44. We further determined whether
treatment of 4-PBA could block CCB-induced surface expression of
DR5 and CD44 by FACS analysis (Fig.
6C). In SNU-475/TR cells, CCB-mediated increase of DR5 and
decrease of CD44 surface expression was significantly attenuated by
4-PBA. These results indicate that CCB triggers activation of ER
stress-dependent autophagy, leading to promote upregulation of DR5
and degradation/downregulation of c-FLIP and CD44.
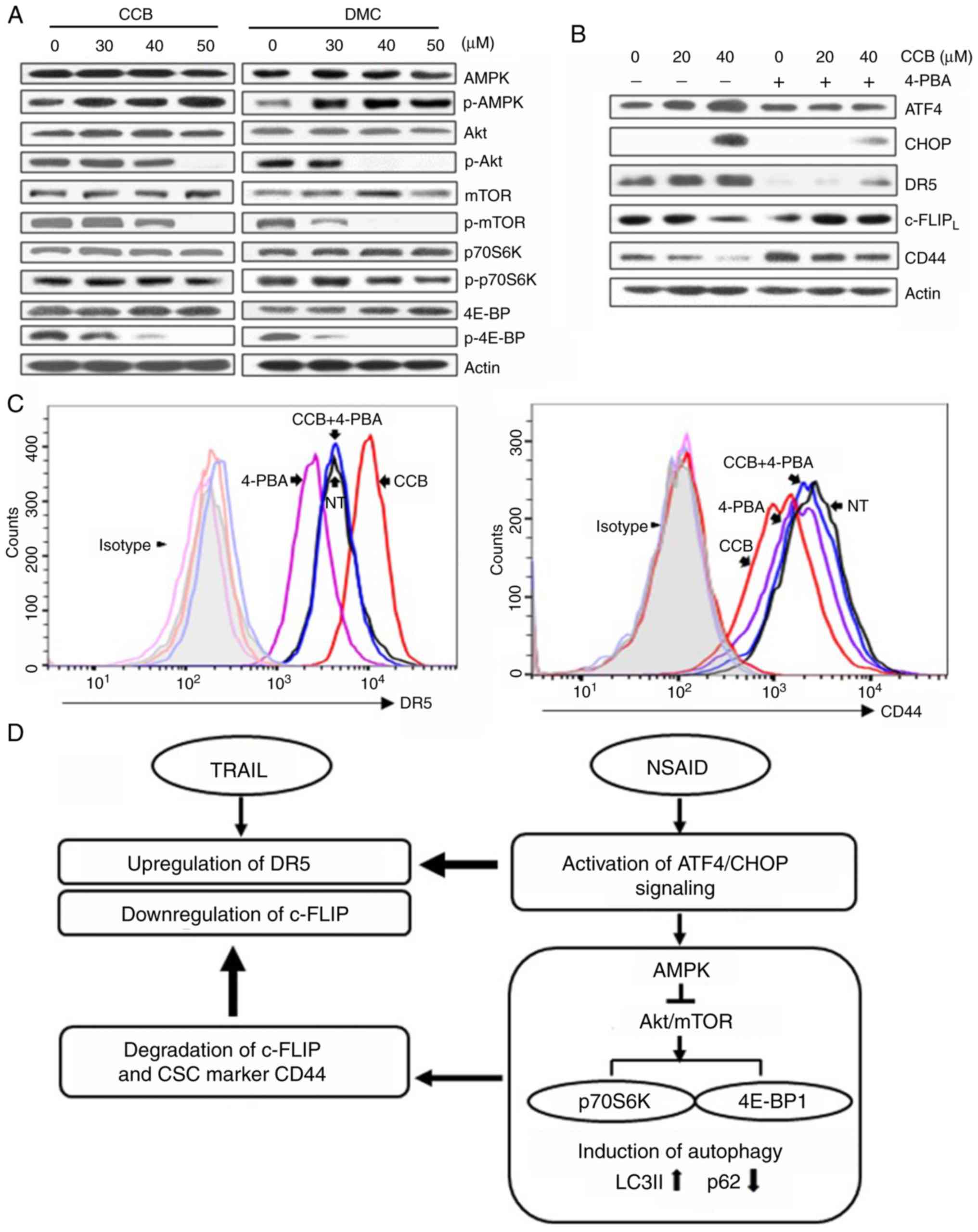 | Figure 6.Activation of AMPK and inhibition of
Akt/mTOR signaling by NSAID and inhibition of NSAID-activated
ATF4/CHOP pathway by ER stress inhibitor. (A) SNU-475/TR cells were
treated with the indicated doses of CCB or DMC for 24 h or (B) the
cells were pretreated with or without 1.5 mM 4-phenylbutyric acid
(4-PBA) for 24 h before treatment of serial doses of CCB for 24 h,
and the altered levels of indicated molecules were determined by
western blot analysis. (C) SNU-475/TR cells were additionally
treated with CCB (40 µM for 24 h) after pretreatment of 4-PBA (1.5
mM for 24 h) and stained with anti-DR5 or anti-CD44 antibody to
determine the cell surface expression of DR5 or CD44 by a flow
cytometer. (D) Proposed scheme showing how NSAID enhances TRAIL
activity. NSAID induces ER stress via activation of ATF4/CHOP
signaling, which upregulates TRAIL-mediated DR5 expression and also
induces autophagy via AMPK activation and inhibition of Akt/mTOR
signaling. NSAID-induced autophagy causes degradation of
c-FLIP/CD44 that can accelerate TRAIL-mediated c-FLIP
downregulation. NSAID, non-steroidal anti-inflammatory drug; ATF,
activating transcription factor; ER, endoplasmic reticulum; CCB,
celecoxib; DMC, 2,5-dimethyl celecoxib; DR5, death receptor
TRAIL-R2; CHOP, C/EBP homologous protein; TRAIL, TNF-related
apoptosis inducing ligand; CSC, cancer stem cell; LC3,
microtubule-associated protein light chain 3. |
In summary, we propose that the autophagic response
triggered by NSAID could be attributed to activation of ATF/CHOP
signaling, which leads to upregulation of TRAIL-mediated DR5
expression, and also NSAID-induced autophagy via AMPK activation
and inhibition of Akt/mTOR signaling. This consequently leads to
increased LC3B-II and decreased p62 levels resulting in degradation
of c-FLIP and TRAIL resistance-related CSC marker CD44, which
accelerates TRAIL-mediated c-FLIP downregulation, and reduces CSC
marker protein in TRAIL-resistant HCC cells (Fig. 6D).
Discussion
Resistance to TNF-related apoptosis inducing ligand
(TRAIL) remains a major issue in the treatment of cancer, and
overcoming resistance to TRAIL is desirable for TRAIL-based
therapy. In the present study, to sensitize TRAIL-resistant
hepatocellular carcinoma (HCC) cells to TRAIL, we examined the
cytotoxic effect of TRAIL alone and in combination with a
non-steroidal anti-inflammatory drug (NSAID) such as celecoxib
(CCB) or 2,5-dimethyl celecoxib (DMC), a non-cyclooxygenase (COX)-2
inhibitor analog of CCB in TRAIL-resistant HCC cells involving
SNU-475/TR, SNU-423 and SNU-449 cells with high levels of CD44. We
propose NSAID as a new sensitizer for TRAIL in TRAIL-resistant HCC
cells. NSAID significantly potentiated the sensitivity of
TRAIL-resistant HCC cells to TRAIL and thus could overcome TRAIL
resistance in a COX-2-independent manner.
In addition to the anti-inflammatory effects, some
NSAIDs including CCB are effective in the treatment and prevention
of cancer, and the anticancer effects of NSAIDs include their
ability to induce apoptosis (29).
Although CCB-mediated apoptosis has not been fully elucidated, it
appears to be associated with induction of endoplasmic reticulum
(ER) stress involving upregulation of CHOP and downregulation of
the anti-apoptotic protein survivin (30). It has been reported that several
signaling pathways are involved in the complex cross-talk between
apoptosis and autophagy, and autophagy has been closely linked to
ER stress/unfolded protein response pathway (31). ATF4 and CHOP have been shown to play
a significant role in the regulation of autophagy (32). Both CHOP and ATF4 are increased upon
treatment with CCB or other agents, and CCB induced CHOP and DR5
expression through an ATF4-dependent mechanism. ATF4 can directly
upregulate CHOP expression through binding to the the promoter
region of CHOP, and subsequently CHOP upregulates a number of
autophagy genes (33).
Autophagy has been known to affect the apoptotic
process, serving either a pro-survival or pro-death function
(10). Indeed, TRAIL has been shown
to induce autophagy as well as apoptosis in cancer cell lines.
TRAIL induces autophagy in lung adenocarcinoma cells and activates
a Fas-associated death domain pathway mediated by both autophagy
and apoptosis (34). In addition,
gefitinib, ginsenoside and compound K were also found to increase
DR5 expression through induction of autophagy, and inhibition of
autophagy reduced DR5 expression in human colon cancer cells
(11,35). We showed that CCB and DMC increased
ATF4 and subsequent CHOP expression in parallel with upregulation
of DR5 in TRAIL-resistant HCC cells. Treatment of SNU-475/TR cells
with NSAID resulted in a dose-dependent increase in LC3-II and
decrease in p62 levels, leading to autophagic
degradation/downregulation of c-FLIP and CD44 through activation of
the ATF/CHOP pathway, which contributes to enhance TRAIL-mediated
apoptosis in TRAIL-resistant HCC cells. Insight into the complex
network of TRAIL-induced apoptosis and autophagy contributes to the
development of novel chemosensitizers to reverse TRAIL resistance
in HCC cells. Our data showed that treatment of HCC cells with
NSAID resulted in the increased expression of DR5 and concurrent
decreased level of c-FLIP and CD44, major causes of TRAIL
resistance, leading to accelerate TRAIL-induced cell death.
Therefore, NSAIDs may represent promising chemosensitizers with
which to reverse TRAIL resistance in CD44-overexpressing
TRAIL-resistant HCC cells by inducing apoptotic and autophagic cell
death to enhance TRAIL sensitivity. We previously reported that
NSAIDs exert their autophagy-inducing effect through activation of
AMPK and inhibition of the Akt/mTOR/p70S6K/4EBP signaling axis in
CD44high K562 cells (36). Similarly, we showed that autophagic
responses triggered by CCB could be attributed to activation of the
ATF/CHOP signaling axis, which leads to induction of autophagy in
TRAIL-resistant HCC cells via AMPK activation and inhibition of
Akt/mTOR/p70S6K/4EBP1 signaling pathway.
Since cancer stem cells (CSCs), which are resistant
to anticancer therapy, are usually characterized by a dysregulation
of autophagy, it is crucial to investigate their relationship
(37). Notably, nigericin induced
autophagy and subsequently suppressed CSC properties in glioma
cells (38), and induction of
autophagy by rottlerin was found ot lead to apoptosis in breast
CSCs (39). These reports indicate
a possibility that induction of autophagy could finally suppress
stemness and lead to cell death in CSCs, and thus, autophagy is a
potential target to decrease the resistance of CSCs to anticancer
therapy. The CSC population in HCC can be responsible for
metastasis, recurrence and chemoresistance of HCC (40). In HCC, markers most frequently used
for CSC are CD44 and CD133 (18),
and particularly CD44 expression was reported to be correlated with
high HCC histologic grades, vascular invasion, and reduced survival
outcomes (41). Another potential
CSC marker is CD133 that is released from the plasma membrane to
the cytoplasm, and overexpression of CD133 constitutively activates
autophagy via inhibition of mTORC1 and mTORC2 signaling and
potentiation of autophagy flux (42). In the present study, we revealed a
pivotal role of CD44/CD133 expression for HCC resistance towards
TRAIL. CD133−/CD44high SNU475/TR cells are
significantly resistant to TRAIL compared with
CD133high/CD44− SNU475 cells. In SNU-475/TR
cells, the expression of CD44 on the cell surface was significantly
enhanced while the CD133 level was markedly reduced as compared to
those in the parental TRAIL-sensitive SNU-475 cells. Other
CD133−/CD44high HCC cells such as SNU-423 and
SNU-449 cells were also resistant to TRAIL, indicating that
upregulation of CD44 may be associated with acquisition of TRAIL
resistance in HCC cells, and thus a high ratio of CD44/CD133
relative expression is an important marker for TRAIL
resistance.
It has been reported that CD44 protein degradation
in human breast and pancreatic cancer cells can occur through the
lysosomal/autophagic pathway (43).
In addition, CD44 degradation in head and neck squamous cell
carcinoma cells was blocked by autophagy inhibitor CQ not by
pretreatment with the proteasome inhibitor MG132 (27). We also showed that CCB inhibited
cell surface expression of CD44, and pre-treatment of autophagy
inhibitor 3-MA or CQ blocked CCB-induced reduction of CD44 level in
SNU-475/TR cells, indicating CCB-induced autophagic degradation of
CD44 in HCC cells. In addition, the expression of stemness-related
makers ALDH1 and Nanog as well as CD44 in SNU-475/TR cells was
downregulated by CCB. Indeed, ALDH1high cells were found
to demonstrate higher resistance to common chemotherapeutic
reagents, and ALDH1A3 protein, one of the ALDH1 family members, was
downregulated by autophagy (44),
and the expression of Nanog as well as CD44 in gastric cancer stem
cells was also decreased by ibuprofen (45), indicating the possibility of
NSAID-induced autophagic degradation of CSC marker proteins CD44,
ALDH1 and Nanog in SNU-475/TR cells. Recently, most studies have
focused on CSCs due to their abilities to cause tumorigenicity,
drug resistance, and cancer recurrence. NSAIDs are among the most
commonly used medications worldwide and inexpensive. Here, we
showed the effectiveness of NSAIDs on TRAIL-resistant HCC cells
overexpressing CSC markers to perform effective cancer therapy. The
combination of NSAID and TRAIL resulted in a greater cytotoxicity
than TRAIL alone by NSAID-induced acceleration of TRAIL-mediated
c-FLIP downregulation and DR5 activation via ER stress
ATF4/CHOP-mediated autophagic cell death pathway, leading to
subsequent activation of caspases and PARP and finally
NSAID-mediated potentiation of TRAIL-induced cell death in
TRAIL-resistant HCC cells. Taken together, our findings highlight a
novel mechanism underlying the combination effect of NSAID and
TRAIL, and thus NSAID can be considered as a novel chemosensitizer
of TRAIL for effective treatment of HCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by Pusan National
University Research Grant, 2019.
Availability of data and materials
The datasets used during the present study are
available from the corresponding authors upon reasonable
request.
Authors' contributions
SHK and CDK conceived and designed the study. SHL,
HJM and YSL performed the experiments and the statistical analysis
and participated in the writing of the manuscript. YSL participated
in the revision of the manuscript critically for important
intellectual content. All authors approved the manuscript and agree
to be accountable for all aspects of the work in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen S, Cao Q, Wen W and Wang H: Targeted
therapy for hepatocellular carcinoma: Challenges and opportunities.
Cancer Lett. 460:1–9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang W, Wu DB, Fu SY, Chen EQ, Tang H and
Zhou TY: Insight into the role of TRAIL in liver diseases. Biomed
Pharmacother. 110:641–645. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trivedi R and Mishra DP: Trailing TRAIL
resistance: Novel targets for TRAIL sensitization in cancer cells.
Front Oncol. 5:692015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang Y, Yang X, Xu T, Kong Q, Zhang Y,
Shen Y, Wei Y, Wang G and Chang KJ: Overcoming resistance to
TRAIL-induced apoptosis in solid tumor cells by simultaneously
targeting death receptors, c-FLIP and IAPs. Int J Oncol.
49:153–163. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koehler BC, Urbanik T, Vick B, Boger RJ,
Heeger S, Galle PR, Schuchmann M and Schulze-Bergkamen H:
TRAIL-induced apoptosis of hepatocellular carcinoma cells is
augmented by targeted therapies. World J Gastroenterol.
15:5924–5935. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim S, Seo SU, Min KJ, Woo SM, Nam JO,
Kubatka P, Kim S, Park JW and Kwon TK: Garcinol enhances
TRAIL-induced apoptotic cell death through up-regulation of DR5 and
down-regulation of c-FLIP expression. Molecules. 23:16142018.
View Article : Google Scholar
|
|
7
|
Jeon MY, Min KJ, Woo SM, Seo SU, Choi YH,
Kim SH, Kim DE, Lee TJ, Kim S, Park JW and Kwon TK: Maritoclax
enhances TRAIL-induced apoptosis via CHOP-mediated ipregulation of
DR5 and miR-708-mediated downregulation of cFLIP. Molecules.
23:3030–3043. 2018. View Article : Google Scholar
|
|
8
|
Cubillos-Ruiz JR, Bettigole SE and
Glimcher LH: Tumorigenic and Immunosuppressive effects of
endoplasmic reticulum stress in cancer. Cell. 168:692–706. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noh HJ, Lee SJ, Sung EG, Song IH, Kim JY,
Woo CH, Kwon TK and Lee TJ: CHOP down-regulates cFLIP(L) expression
by promoting ubiquitin/proteasome-mediated cFLIP(L) degradation. J
Cell Biochem. 113:3692–3700. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheng J, Qin H, Zhang K, Li B and Zhang X:
Targeting autophagy in chemotherapy-resistant of hepatocellular
carcinoma. Am J Cancer Res. 8:354–365. 2018.PubMed/NCBI
|
|
11
|
Chen L, Meng Y, Sun Q, Zhang Z, Guo X,
Sheng X, Tai G, Cheng H and Zhou Y: Ginsenoside compound K
sensitizes human colon cancer cells to TRAIL-induced apoptosis via
autophagy-dependent and -independent DR5 upregulation. Cell Death
Dis. 7:e23342016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Xu X, Shi S, Wang Q, Saxton B, He W,
Gou X, Jang JH, Nyunoya T, Wang X, et al: Autophagy-mediated
degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by
combination of TRAIL and Chal-24. J Cell Biochem. 117:1136–1144.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duffy A, Le J, Sausville E and Emadi A:
Autophagy modulation: A target for cancer treatment development.
Cancer Chemother Pharmacol. 75:439–447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Chen F and Shang L: Advances in
antitumor effects of NSAIDs. Cancer Manag Res. 10:4631–4640. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Endo H, Yano M, Okumura Y and Kido H:
Ibuprofen enhances the anticancer activity of cisplatin in lung
cancer cells by inhibiting the heat shock protein 70. Cell Death
Dis. 5:e10272014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu C, Li WB, Liu JB, Lu JW and Feng JF:
Autophagy: Novel applications of nonsteroidal anti-inflammatory
drugs for primary cancer. Cancer Med. 7:471–484. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ku JL and Park JG: Biology of SNU cell
lines. Cancer Res Treat. 37:1–19. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rozeik MS, Hammam OA, Ali AI, Magdy M,
Khalil H, Anas A, Abo El Hassan AA, Rahim AA and El-Shabasy AI:
Evaluation of CD44 and CD133 as markers of liver cancer stem cells
in Egyptian patients with HCV-induced chronic liver diseases versus
hepatocellular carcinoma. Electron Physician. 9:4708–4717. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee SH, Hyun SK, Kim HB, Kang CD and Kim
SH: Potential role of CD133 expression in the susceptibility of
human liver cancer stem-like cells to TRAIL. Oncol Res. 24:495–509.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pennarun B, Kleibeuker JH, Boersma-van Ek
W, Kruyt FA, Hollema H, de Vries EG and de Jong S: Targeting FLIP
and Mcl-1 using a combination of aspirin and sorafenib sensitizes
colon cancer cells to TRAIL. J Pathol. 229:410–421. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JY, Jung KH, Morgan MJ, Kang YR, Lee
HS, Koo GB, Hong SS, Kwon SW and Kim YS: Sensitization of
TRAIL-induced cell death by 20(S)-ginsenoside Rg3 via CHOP-mediated
DR5 upregulation in human hepatocellular carcinoma cells. Mol
Cancer Ther. 12:274–285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hassanzadeh A, Farshdousti Hagh M, Alivand
MR, Akbari AAM, Shams Asenjan K, Saraei R and Solali S:
Down-regulation of intracellular anti-apoptotic proteins,
particularly c-FLIP by therapeutic agents; the novel view to
overcome resistance to TRAIL. J Cell Physiol. 233:6470–6485. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nazim UM, Jeong JK and Park SY:
Ophiopogonin B sensitizes TRAIL-induced apoptosis through
activation of autophagy flux and downregulates cellular FLICE-like
inhibitory protein. Oncotarget. 9:4161–4172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He MX and He YW: c-FLIP protects T
lymphocytes from apoptosis in the intrinsic pathway. J Immunol.
194:3444–3451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang P and Mizushima N: LC3- and
p62-based biochemical methods for the analysis of autophagy
progression in mammalian cells. Methods. 75:13–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Su M, Mei Y and Sinha S: Role of the
Crosstalk between autophagy and apoptosis in Cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nanbu T, Umemura N, Ohkoshi E, Nanbu K,
Sakagami H and Shimada J: Combined SN-38 and gefitinib treatment
promotes CD44 degradation in head and neck squamous cell carcinoma
cells. Oncol Rep. 39:367–375. 2018.PubMed/NCBI
|
|
28
|
Matter MS, Decaens T, Andersen JB and
Thorgeirsson SS: Targeting the mTOR pathway in hepatocellular
carcinoma: Current state and future trends. J Hepatol. 60:855–865.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Umar A, Steele VE, Menter DG and Hawk ET:
Mechanisms of nonsteroidal anti-inflammatory drugs in cancer
prevention. Semin Oncol. 43:65–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jendrossek V: Targeting apoptosis pathways
by Celecoxib in cancer. Cancer Lett. 332:313–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song S, Tan J, Miao Y, Li M and Zhang Q:
Crosstalk of autophagy and apoptosis: Involvement of the dual role
of autophagy under ER stress. J Cell Physiol. 232:2977–2984. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Matsumoto H, Miyazaki S, Matsuyama S,
Takeda M, Kawano M, Nakagawa H, Nishimura K and Matsuo S: Selection
of autophagy or apoptosis in cells exposed to ER-stress depends on
ATF4 expression pattern with or without CHOP expression. Biol Open.
2:1084–1090. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
B'chir W, Chaveroux C, Carraro V, Averous
J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P and
Bruhat A: Dual role for CHOP in the crosstalk between autophagy and
apoptosis to determine cell fate in response to amino acid
deprivation. Cell Signal. 26:1385–1391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Y, Zhou X, Qiao J and Bao A:
Autophagy is a regulator of TRAIL-induced apoptosis in NSCLC A549
cells. J Cell Commun Signal. 11:219–226. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen L, Meng Y, Guo X, Sheng X, Tai G,
Zhang F, Cheng H and Zhou Y: Gefitinib enhances human colon cancer
cells to TRAIL-induced apoptosis of via autophagy- and JNK-mediated
death receptors upregulation. Apoptosis. 21:1291–1301. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moon HJ, Park SY, Lee SH, Kang CD and Kim
SH: Nonsteroidal anti-inflammatory drugs sensitize
CD44-overexpressing cancer cells to Hsp90 inhibitor through
autophagy activation. Oncol Res. 27:835–847. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nazio F, Bordi M, Cianfanelli V, Locatelli
F and Cecconi F: Autophagy and cancer stem cells: Molecular
mechanisms and therapeutic applications. Cell Death Differ.
26:690–702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hegazy AM, Yamada D, Kobayashi M, Kohno S,
Ueno M, Ali MA, Ohta K, Tadokoro Y, Ino Y, Todo T, et al:
Therapeutic strategy for targeting aggressive malignant gliomas by
disrupting their energy balance. J Biol Chem. 291:21496–21509.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kumar D, Shankar S and Srivastava RK:
Rottlerin-induced autophagy leads to the apoptosis in breast cancer
stem cells: Molecular mechanisms. Mol Cancer. 12:1712013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ji J and Wang XW: Clinical implications of
cancer stem cell biology in hepatocellular carcinoma. Semin Oncol.
39:461–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Luo Y and Tan Y: Prognostic value of CD44
expression in patients with hepatocellular carcinoma:
Meta-analysis. Cancer Cell Int. 16:472016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bhattacharya S, Yin J, Winborn CS, Zhang
Q, Yue J and Chaum E: Prominin-1 is a novel regulator of autophagy
in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci.
58:2366–2387. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haakenson JK, Khokhlatchev AV, Choi YJ,
Linton SS, Zhang P, Zaki PM, Fu C, Cooper TK, Manni A, Zhu J, et
al: Lysosomal degradation of CD44 mediates ceramide
nanoliposome-induced anoikis and diminished extravasation in
metastatic carcinoma cells. J Biol Chem. 290:8632–8643. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu W, Schecker J, Würstle S, Schneider F,
Schönfelder M and Schlegel J: Aldehyde dehydrogenase 1A3 (ALDH1A3)
is regulated by autophagy in human glioblastoma cells. Cancer Lett.
417:112–123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Akrami H, Moradi B, Borzabadi Farahani D
and Mehdizadeh K: Ibuprofen reduces cell proliferation through
inhibiting Wnt/β catenin signaling pathway in gastric cancer stem
cells. Cell Biol Int. 42:949–958. 2018. View Article : Google Scholar : PubMed/NCBI
|