Introduction
Acute lymphoblastic leukemia (ALL) is a
heterogeneous hematopoietic malignancy, characterized by the
expansion of lymphoid progenitor cells. Massive proliferation of
cells, extensive infiltration, and inhibition of normal
hematopoiesis are the main clinical manifestations of ALL. It is
the most common neoplasm in pediatric patients. Approximately 85%
of ALLs are of the B-cell lineage; the rest are of T-cell lineage
(1). T-cell ALL (T-ALL) presents as
an uncontrolled malignant accumulation of T-cell progenitors
(1,2). T-ALL cases can occur at any age, while
the majority of cases occur between 2 and 5 years of age (3). Current treatments focus on intensive
chemotherapy, targeted therapy, and bone marrow transplantation
(4). Although 80% of cases
initially achieve remission (5),
most patients show relapse or the cancers resist chemotherapy,
resulting in a poor prognosis of patients (1). Therefore, efficient and low-toxicity
antitumor compounds are crucial to ameliorate the prognosis of
patients with T-ALL.
Currently, several treatment approaches methods for
T-ALL are in practice; these include inducing apoptosis, inhibiting
proliferation, causing cell cycle arrest and inducing
differentiation (6,7). Chinese herbs have been widely used to
treat cancers as they are cost-effective and have positive
outcomes. More than half antitumor compounds in use today are
herbaceous (8).
Gambogic acid (GA) is a natural medicinal compound
that is extracted from the traditional Chinese herb Garcinia
hanburyi. Its molecular formula is C38H44O8, and it has a
molecular weight of 628.75 g/mol. GA is a potential antineoplastic
drug against several solid tumors and hematological malignancies
(9–11), including hepatocellular (12), malignant (13), breast carcinoma (14), and chronic myeloid leukemia
(15). GA has been shown to affect
the physiological function and signaling pathways of several tumor
cell lines (16,17). Activation of cell apoptosis has been
suggested to be the main mechanism underlying the repressive effect
of GA on solid tumors. The related molecular mechanisms possibly
include inhibition of telomerase activity or obstructing
transferrin receptor internalization (9,10). In
addition, it has been found that GA can inhibit multiple cell
signaling pathways, including the Wnt/β-catenin pathway, and
transcription factors, including nuclear factor-kappa B, activator
protein 1, NRF2, PPAR-γ, tumor necrosis factor-α, sonic hedgehog,
and nitric oxide synthase (18,19).
However, the potential effect of GA on T-ALL is not fully
understood, and the underlying molecular mechanisms require further
investigation.
β-catenin, a transcriptional coactivator of the
Wnt/β-catenin signaling pathway, is degraded by proteasomes when
cell function is normal. Once the pathway is activated, β-catenin
constantly accumulates in the cytoplasm and then translocates to
the nucleus. In the nucleus, β-catenin interfaces with
high-mobility group transcription factors belonging to the T-cell
factor/lymphoid enhancer factor family to activate downstream
target genes, including c-Myc, Axin2, and CyclinD1.
All of these mechanisms are attractive therapeutic targets
(20–23).
The canonical Wnt pathway (Wnt/β-catenin pathway) is
involved in the regulation of various physiological and
pathological processes during early-and late-stage embryonic
development (24) and in
carcinogenesis (25,26). Different types of tumors cause
aberrance of the Wnt/β-catenin pathway. Previous studies have shown
that Wnt/β-catenin pathway plays a crucial role in various
leukemias (27–29). This pathway is closely associated
with self-renewal, overexpression and deletion of mutations in
hematopoietic stem cell (HSC). These factors all play important
roles in leukemogenesis, drug resistance, and tumor relapse
(30). Furthermore, it has been
demonstrated that Wnt/β-catenin signaling is involved in the
development of T cells and often the deregulation of T-ALL
(31,32). Thus, Wnt/β-catenin signaling is a
worthy target for improving T-ALL therapy results.
In the current study, we investigated the antitumor
effects of GA using T-ALL cell lines and patient samples. The study
was expanded to examine the possible mechanisms causing the
observed effects of GA. GA demonstrated marked potency in inducing
apoptosis, regulating cell cycle arrest, inhibiting proliferation,
and inducing autophagy in T-ALL cells. Further investigations
revealed that the Wnt/β-catenin signaling pathway plays an
important role in the inhibitory function of GA. Thus, GA is a
potential antitumor adjuvant agent.
Materials and methods
Cell culture
The human ALL lines Jurkat and Molt-4 were obtained
from the cell bank of the Chinese Academy of Science (Shanghai,
China). Jurkat cells and Molt-4 cells were routinely cultured in
RPMI-1640 (Gibco) medium supplemented with 10% fetal bovine serum
(Gibco) with 1% glutamine-penicillin (Beyotime). Cells were
cultured at 37°C with 5% CO2.
T-ALL patients and healthy samples
lymphocyte cells isolation
Following institutional guidelines, blood samples
were obtained following informed consent from six patients with
T-ALL and six healthy patients. Patients were recruited from the
Zhejiang Provincial People's Hospital in November and December,
2018. Lymphocytes were isolated from EDTA-treated blood by density
gradient centrifugation using Lymphocyte Separation Medium (TBD) as
per the manufacturer's instructions. Peripheral anti-coagulated
blood (4 ml) was mixed with calcium-magnesium-free
phosphate-buffered saline (PBS) (ratio 1:1). Samples were layered
onto 5-ml lymphocyte separation medium and centrifuged at 300 × g
for 25 min at room temperature. The interface layer was centrifuged
at 50 × g for another 10 min at room temperature, and the pellet
was collected. The enriched cells were cultured in RPMI-1640 medium
supplemented with 20% fetal bovine serum.
The relevant ethics approval was granted by the
Zhejiang Provincial People's Hospital Ethics Committee.
Gambogic acid preparation
GA with a purity of >98% was purchased from
Chengdu Must Bio-Technology Co. Ltd. It was dissolved in dimethyl
sulfoxide and then stored at −20°C for further analyses.
Cell viability assay using Cell
Counting Kit-8
Cell viability after GA treatment was determined
using a Cell Counting Kit-8 (CCK-8) assay (Beyotime). Cells were
seeded in 96-well plates (1×105 cells/well) and treated
with different concentrations (1.2, 2.4 and 4.8 µM) of GA, and
controls were treated with 0.1% DMSO. The plates were incubated for
24, 48 and 72 h. After incubation, 20 µl of CCK-8 solution was
added to each well. The plates were further incubated at 37°C in a
CO2 incubator for 3 h. Optical density was analyzed
using a microplate reader at 450 nm.
Cycle analysis
The cell cycle distribution of Jurkat cells after GA
treatment (at various concentrations) were analyzed using a Cell
Cycle Staining Kit (LianKe) following the manufacturer's
instructions. Jurkat cells were suspended and mixed in DNA staining
solution (50 µg/ml RNase A for 20 min at room temperature, followed
by 50 µg/ml propidium iodide for 10 min in the dark). After the 30
min treatment, distribution of cell cycle stages were detected by
flow cytometry (Becton Dickinson). In the present analysis, each
phase of the cell cycle was recorded as a percentage.
Apoptosis analysis
GA-treated Jurkat cells (at various concentrations)
were suspended in 2-ml PBS to obtain a cell density of
1–5×106/ml. The cells were tested using a PI Apoptosis
Detection Kit (MultiSciences Biotech Co.) according to the
manufacturer's protocol. Briefly, 10-µl PI was added to the
suspension, incubated for 20 min at room temperature, and
maintained in the dark. The percentage of apoptotic cells was
examined by flow cytometry. The results were analyzed using Novo
Express software.
Immunocytochemistry
Jurkat cells were treated with various
concentrations of GA and immersed overnight. Coverslips were
treated with 0.2-mg/ml poly-L-lysine for 4 h prior to placing them
on coverslips. After 1.5 h of incubation, the cells were placed
firmly on the coverslips. These were fixed with 4% PFA (Beyotime)
for 10 min at room temperature. Samples were fixed in 0.1% Triton
X-100 (Beyotime) and 5% BSA prior to incubation. The primary
antibody against p-GSK3β S9 (1:200, Cell Signaling Technology, cat.
no. 5558) was added, and the specimens were incubated overnight at
4°C. The next day, the cells were washed with PBS three times and
the appropriate fluorescent secondary antibody Alexa Fluor 488 was
added (1:1,000, Beyotime, cat. no. A0423). The samples were
incubated at room temperature for 1 h. Cells were stained with DAPI
(Beyotime) to allow visualization of the nucleus. Finally, images
were obtained using a fluorescence microscope (Olympus).
Monodansylcadaverine staining
Jurkat cells were plated in 6-well plates and
treated with different concentrations (1.2, 2.4 and 4.8 µM) of GA,
and controls were treated with 0.1% DMSO. After treatment, the
cells were collected and suspended with PBS at a density of
1.5×106 cells/ml, followed by co-staining with
monodansylcadaverine (MDC) plus DAPI for another 30 min at 37°C.
Approximately 10–20 µl of the cell suspension was placed onto a
glass slide and covered with a cover glass. The MDC staining was
examined and photographed using a fluorescence microscope.
Western blot analysis
Lysed cells in RIPA lysis buffer (Beyotime) were
maintained on ice for 30 min and centrifuged at 14,000 × g for 10
min at 4°C. The protein concentration in the supernatant was
verified by BCA Protein Assay (Thermo Scientific Inc). Total
proteins were resolved by 10% SDS-polyacrylamide gel
electrophoresis and transferred onto PVDF membranes. The membranes
were blocked in 5% de-fatted milk for 1 h at room temperature and
then incubated with primary antibodies at 4°C overnight. The
matched secondary antibody (1:3,000, Beyotime, cat. nos. A0208,
A0192) was incubated for 2 h at room temperature. Protein levels
were detected using an enhanced chemiluminescence reagent and
quantified by ImageJ software. The antibodies used in the study
were as follows: Anti-CyclinB (1:1,000, Abcam, cat. no. ab32053),
anti-CDK1 (1:500, Beyotime, cat. no. AF0111), anti-Beclin1
(1:1,000, Thermo, cat. no. MA5-15825), and anti-LC3-II (1:1,500,
Sigma, cat. no. 610832); anti-Bax (1:1,000, Abcam, cat. no.
ab32503), anti-Bcl-2 (1:1,000, Abcam, ab182858), anti-cleaved
caspase3 (1:1,000, Cell Signaling Technology, cat. no. 9661S),
anti-cleaved PARP (1:1,000, Abcam, cat. no. ab191217) anti-p-GSK3β
S9 (1:1,000, Cell Signaling Technology, cat. no. 5558), and
anti-Atg7 (1:1,000, Cell Signaling Technology, cat. no. 8558s);
anti-β-catenin (1:1,000, Cell Signaling Technology, cat. no.
19807S), anti-c-Myc (1:1,000, Cell Signaling Technology, cat. no.
2278s), and anti-CyclinD1 (1:1,000, Abcam, cat. no. ab134175); and
anti-β-actin (1:3,000, Beyotime, cat. no. AF5001).
Cell transfection
Jurkat cells were seeded in 6-well plates and
transfected using Lipofectamine 3000 (Invitrogen) with recombinant
β-catenin and blank plasmids. The procedure was performed in
accordance with the manufacturer's instructions. After 24 h, the
cells were treated with GA (2.5 µM). Observations were made after
48 h using an inverted fluorescence microscope. The expression of
marker proteins in stably transfected cells were analyzed by
western blot analysis.
Statistical analysis
Data are presented as mean ± standard deviation.
Data were analyzed using SPSS21.0. The data were collected from at
least three independent experiments. Significance of the difference
between two groups means of data sets was determined using the
Student's t-test. Multiple comparisons test was analyzed by one-way
ANOVA (Dunnett's correction). Results with P<0.05 and P<0.01
were considered statistically significant.
Results
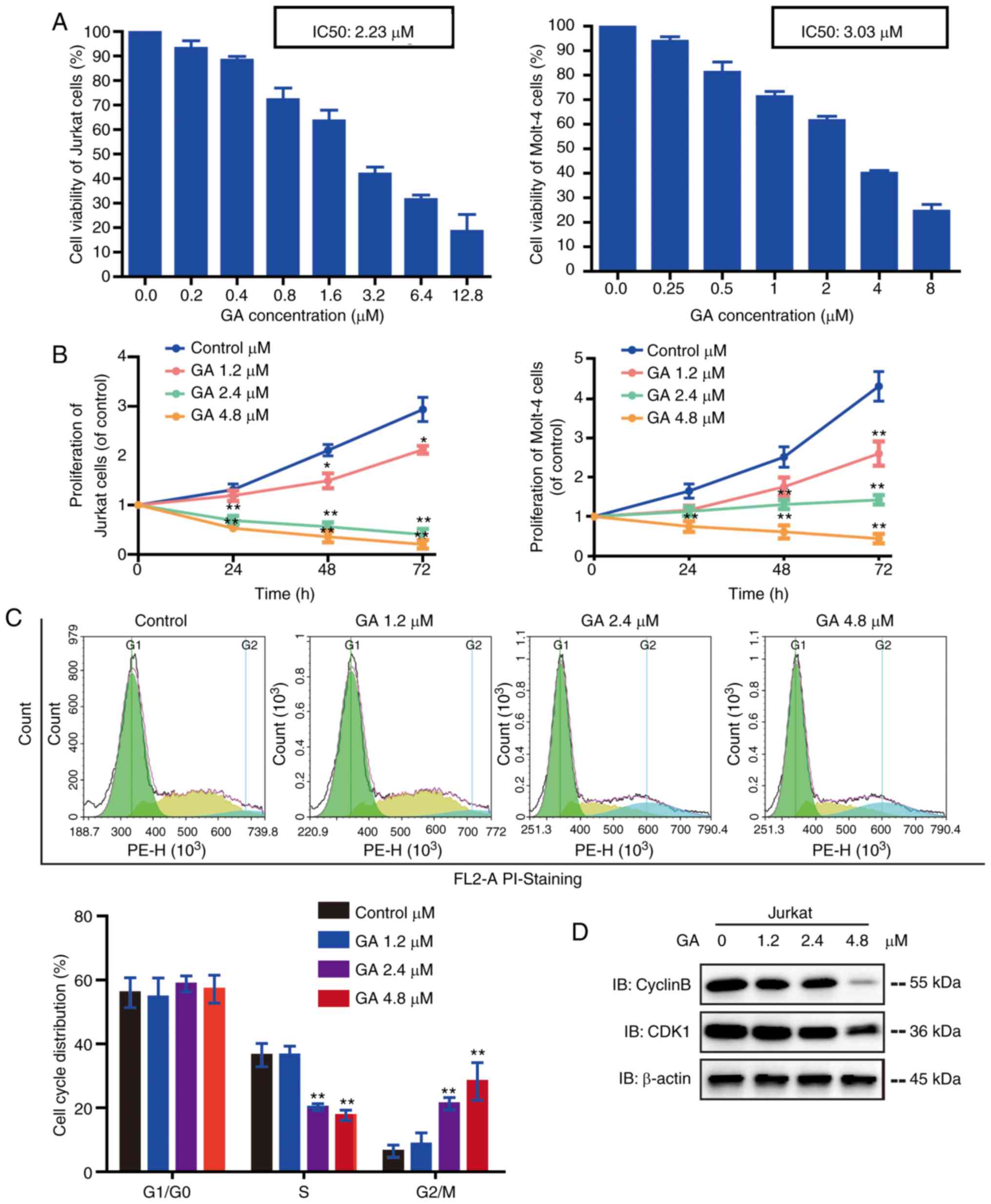
GA inhibits the proliferation of human
leukemia cell lines
To detect the anti-proliferative effects of GA in
vitro, the human leukemia cell lines Jurkat and Molt-4 cells
were exposed to increasing concentrations of GA. GA reduced the
viability of Jurkat and Molt-4 cells in a dose-dependent manner
(Fig. 1A-B). The IC50 values of GA
for Jurkat and Molt-4 were 2.23 and 3.03 µM, respectively. Jurkat
and Molt-4 cells were treated with GA (1.2, 2.4 and 4.8 µM) for 0,
24, 48 and 72 h. At the higher concentrations (2.4 and 4.8 µM), GA
inhibited the growth of cells significantly, and the inhibition
rate increased with time. These results suggested that GA
cytotoxicity is dose- and time-dependent in leukemia cells.
GA induces G2/M phase cell cycle
arrest in Jurkat cells
To test whether GA could induce Jurkat cell cycle
arrest, cell cycle distribution was performed. The results
demonstrated that the exposure of cells to GA at concentrations of
2.4 and 4.8 µM led to a remarkable increase in G2/M phase, with a
corresponding decrease in the S phase compared with exposure of
cells to control or 1.2-µM GA (Fig.
1C). To determine the underlying mechanisms causing G2/M phase
arrest in Jurkat cells, we examined the effect of GA on the
expression of CyclinB and CDK1 (key regulators of G2/M transition).
Western blot results revealed that the protein levels of CyclinB
and CDK1 were significantly decreased by GA (Fig. 1D). Taken together, these results
indicate that GA may inhibit the proliferation of Jurkat cells by
inducing cell cycle arrest at the G2/M phase.
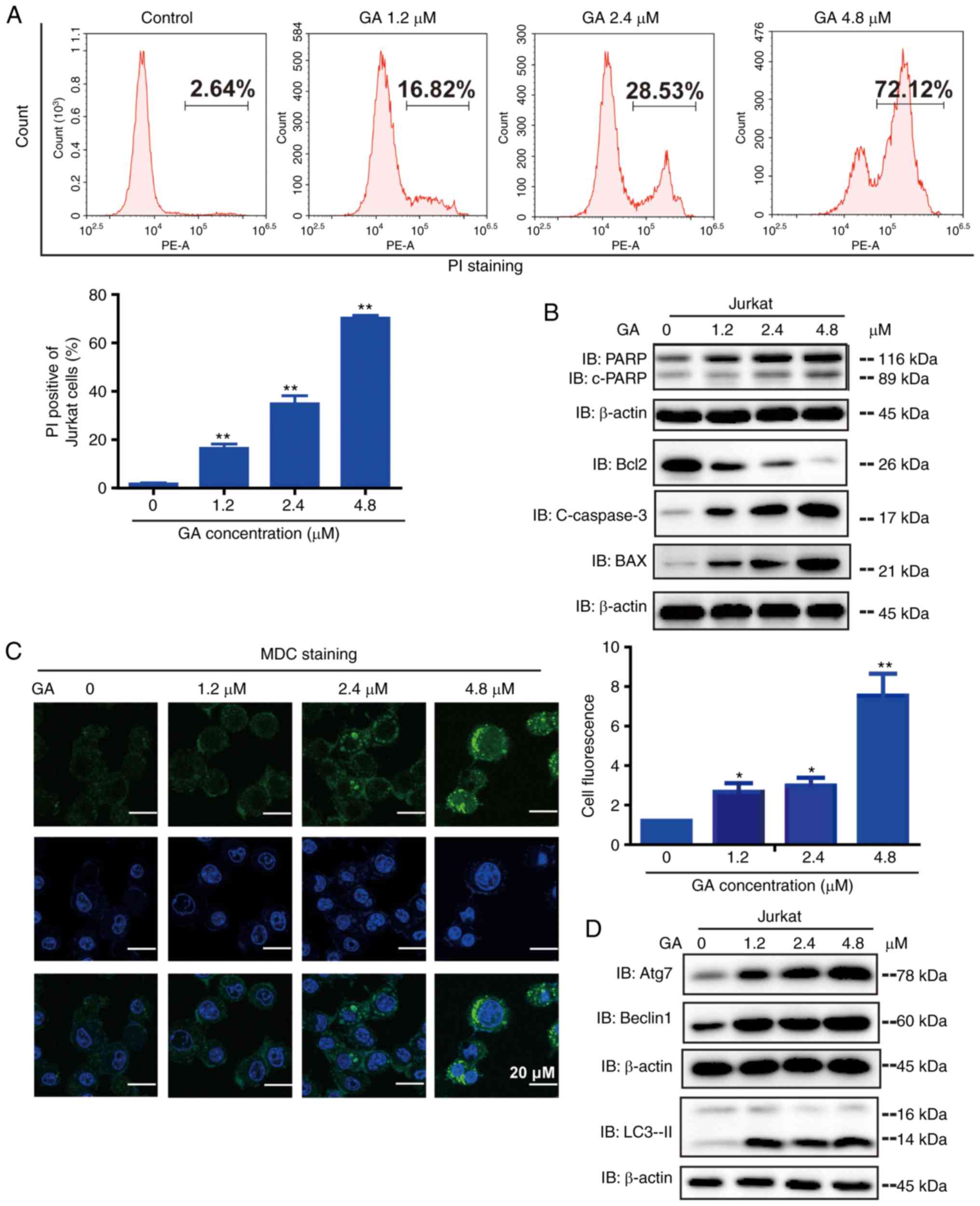
GA induces apoptosis of Jurkat
cells
It is known that apoptosis is programmed cell death.
Thus, we examined whether GA induces apoptosis in cancer cells.
Using a flow cytometric analysis the presence of apoptosis in
GA-treated cells was identified. GA treatment induced significant
apoptosis in Jurkat cells in a dose-dependent manner (Fig. 2A). Apoptosis was detected by western
blot analysis (Fig. 2B). It was
noted that GA treatment upregulated the expression of c-PARP,
c-Caspase-3, and Bax but downregulated the expression of Bcl-2
compared with control treatment. Overall, these results
demonstrated that GA upregulated the expression of pro-apoptotic
proteins, thus causing apoptosis in Jurkat cells.
GA induced autophagy in Jurkat
cells
We observed whether autophagic vacuoles were formed
when Jurkat cells were treated with GA by MDC staining (Fig. 2C). The number of MDC-positive
fluorescent points in GA-treated cells was markedly greater than
that in the control group. These results demonstrated that the
inhibitory effect of GA on cell viability is related to the
induction of autophagy. To investigate whether GA regulated the
process of autophagy, the protein expression of autophagy-related
markers were assessed by western blot analysis (Fig. 2D). Significant upregulation of Atg7,
Beclin-1, and LC3-II occurred in GA-treated groups. These results
suggest that GA may encourage autophagy of Jurkat cells.
GA regulates Wnt/β-catenin signaling
in Jurkat cells
To identify other mechanisms involved in the
inhibitory effect of GA, we focused on the Wnt pathway (Fig. 3A). The protein levels of β-catenin,
as well as downstream proteins CyclinD1 and c-Myc, were
significantly decreased in a dose-dependent manner after GA
treatment. GSK3β, a highly effective kinase within the β-catenin
destruction complex, phosphorylated β-catenin and promoted its
destruction by proteasomes. Therefore, GA suppressed the expression
of p-GSK3β S9 and subsequently promoted β-catenin degradation to
reduce β-catenin nuclear import. Immunostaining results showed that
after GA treatment (Fig. 3B), the
expression of p-GSK3β S9 decreased. Our findings indicated that GA
inhibits the growth of Jurkat cells by regulating the Wnt/β-catenin
pathway.
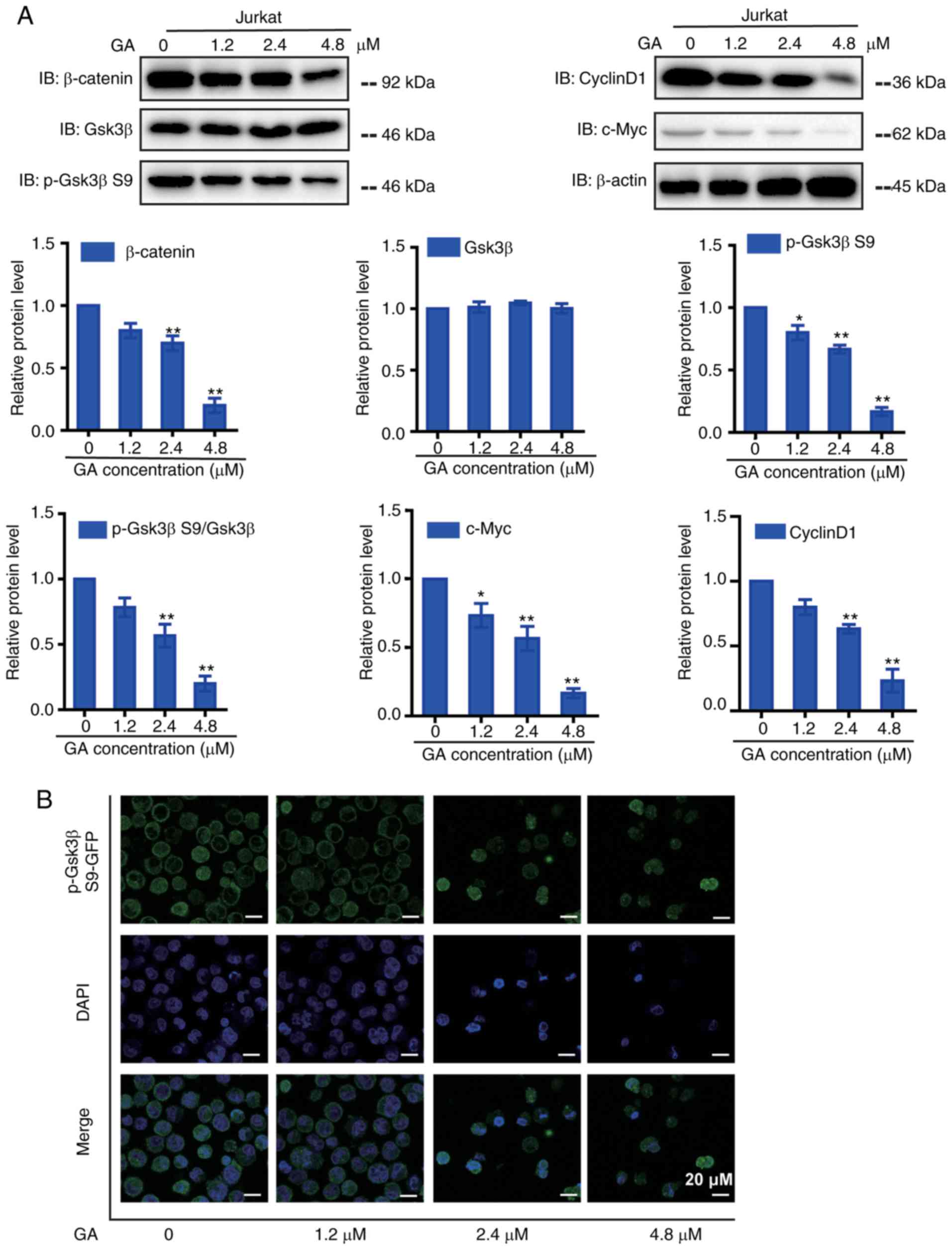 | Figure 3.GA treatment down-regulated
Wnt/β-catenin signaling pathways in Jurkat cells. (A) Western blot
detected the expression of Wnt signaling pathway proteins in Jurkat
cells after GA treatment (β-catenin, p-GSK3β S9, GSK3β, CyclinD1,
c-Myc), Corresponding statistical chart of β-catenin, p-GSK3β,
GSK3β, p-GSK3β S9/GSK3β, CyclinD1 and c-Myc is also shown. (B)
Single immunocytochemistry of p-GSK3β S9 (green) in Jurkat cells
treated with GA at indicated concentrations for 24 h. *P<0.05
and **P<0.01 compared with control, controls were treated with
0.1% DMSO. Bar scale: 20 µm. |
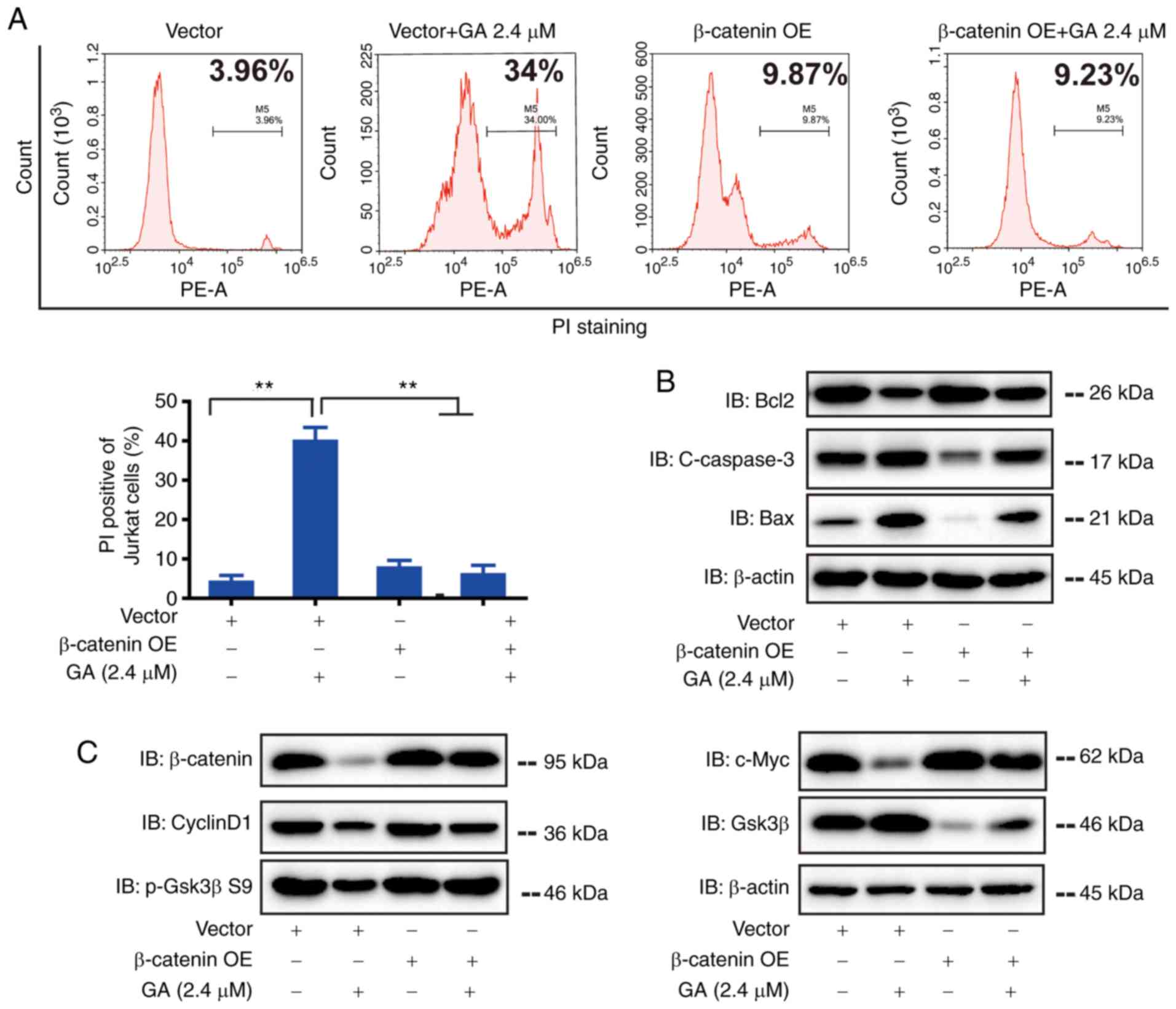
Overexpression of β-catenin inhibited the apoptosis
of Jurkat cells. To further confirm the role of β-catenin in
GA-induced regulation of Jurkat cell proliferation and apoptosis,
we transfected the cells with plasmid-carrying β-catenin. We
established that the overexpression of β-catenin protects Jurkat
cells from apoptosis. Flow cytometry results showed that after
β-catenin plasmid transfection, a proportion of the apoptosis
(induced by GA) recovered (Fig.
4A). Western blot results revealed that the transfection of
β-catenin plasmid reversed the increase of Bax and c-caspase-3 and
decreased Bcl-2 induced by GA (2.4 µM) (Fig. 4B). These results confirmed the
hypothesis that the overexpression of β-catenin significantly
inhibited the apoptosis of Jurkat cells. After transfection of
β-catenin plasmid into GA-treated Jurkat cells, the protein levels
of β-catenin, CyclinD1, GSK-3β, p-GSK3β S9, and c-Myc were measured
using western blot analysis (Fig.
4C). The results showed that the expression of GSK3β
significantly decreased in GA-treated Jurkat cells transfected with
β-catenin compared with that in cells transfected with the negative
control. In the negative control group, the expression of
β-catenin, p-GSK3β S9, CyclinD1, and c-Myc are significantly
increased.
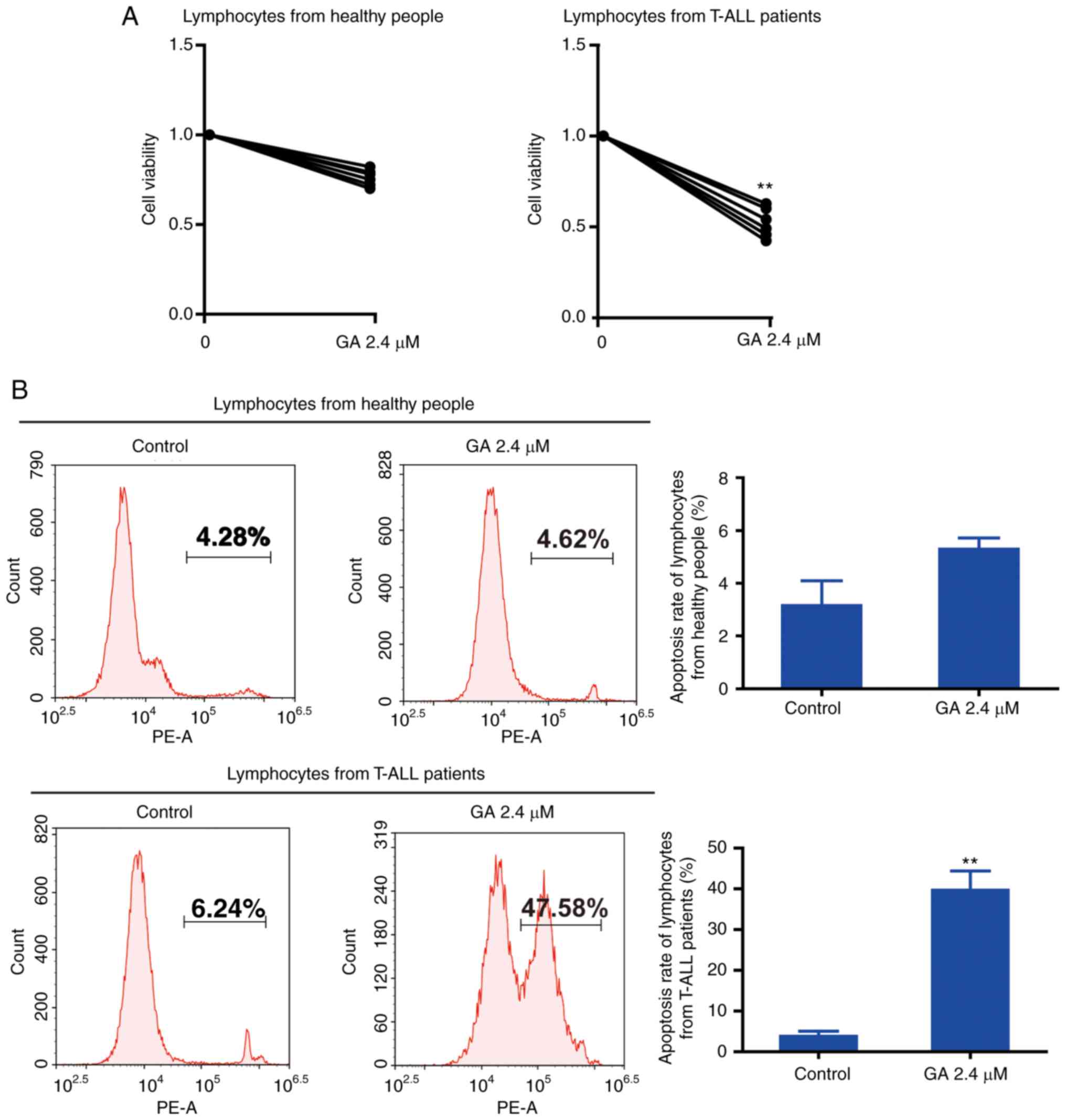
Effects of GA on healthy people and
T-ALL patients
GA toxicity was then examined. The results showed
that GA is toxic to Jurkat cells. To investigate the potential of
GA for cancer therapy, we evaluated the effects of GA on lymphocyte
cells from healthy individuals and patients with T-ALL. Apoptotic
assays revealed that cell viability of healthy individuals
decreased slightly when treated with GA (Fig. 5). Conversely, GA treatment induced
significant apoptosis in lymphocyte cells from patients with
T-ALL.
Discussion
T-ALL is one of the most common types of aggressive
hematological malignancies in children, and has high heterogeneity
(33). The current treatment
options for ALL include chemotherapy (such as VDLP or VILP regimen)
and allogeneic bone marrow transplantation. However, the side
effects of myelosuppression and central nervous system issues are
still severe (34) leading to a
poor prognosis (35). Drug
resistance and inevitable recurrence also contribute to the poor
prognosis of patients (36).
Therefore, there is a need to explore new therapeutic targets for
T-ALL treatment. GA, obtained from G. hanburryi, exhibits
cytotoxic abilities in several solid tumors both in vitro
and in vivo. The assumed mechanism of GA in treating solid
tumors primarily involves activating apoptotic pathways (9,10).
However, the exact role and possible molecular mechanisms
underlying GA activity against T-ALL are still poorly understood
and require extensive studies. In the current study, we evaluated
the potential antileukemic effect of GA on T-ALL cells. We found
that GA suppressed cell proliferation, affected cell cycle
distribution, induced apoptosis, and activated autophagy in T-ALL
cells. We identified that the antileukemic effect of GA on T-ALL
cells may involve regulating the Wnt/β-catenin signaling pathway by
inactivating p-GSK3β S9 and activating GSK3β. These combined
activities promote the degradation of the β-catenin protein.
Apoptosis induction plays an essential role in the
antitumor strategies of various therapies. Previous findings have
emphasized the fact that traditional Chinese medicines can
strengthen the activation of caspase-independent cell apoptosis
pathways (37). Flow cytometry
analysis revealed that GA induces apoptosis in T-ALL cells.
c-Caspase3 is known to activate other caspases, playing a critical
role in the execution of the mitochondrial apoptotic pathway. In
the present research, GA increased the expression levels of
c-caspase 3 in T-ALL cells, indicating that the mitochondrial
apoptosis pathway is affected by GA. Bax and Bcl2 are critical
regulators of apoptosis, however, Bax is a proapoptotic protein,
and Bcl2 is anti-apoptotic protein (38). GA reduced the expression of Bcl2 and
increased that of Bax. Therefore, GA's ability to regulate the
balance between Bax and Bcl2 provides evidence that GA induces
apoptosis in T-ALL cells. The ability of GA to induce apoptosis is
one of the mechanisms contributing to the anticancer effects of GA
in T-ALL.
The existing literature shows that T-ALL stem cells
are highly dependent on the Wnt/β-catenin signaling pathway
(39). Downstream target proteins
of Wnt/β-catenin signaling play major roles in neoplastic
transformation, including cell cycling (c-Myc and CyclinD1), tumor
growth, cell invasion, and tumor metastasis (40). Our flow cytometry results show that
GA disturbs T-ALL cell cycle and induces G2/M arrest. Cell cycle
arrest is closely related to cycle-regulating proteins (41). CyclinBl and CDKl are critical
regulators of cell cycle progression, specifically in the G2/M
transition (42). In our
experiment, GA regulated the expression levels of CyclinD1,
CyclinBl, and CDKl proteins. The ability of GA to induce cell cycle
arrest highlights its antineoplastic role. This demonstrates
another mechanism that GA uses to induce anticancer effects on
T-ALL.
The Wnt/β-catenin signaling pathway plays an
important role in the self-renewal of HSCs, leukemia progression,
and BCR-ABL kinase inhibitor resistance (43–46).
In the Wnt signaling pathway, GSK-3β binds to several proteins and
undergoes phosphorylation, thereby inducing β-catenin degradation.
GSK-3β has been regarded as a tumor suppressor as it negatively
regulates the Wnt/β-catenin signaling pathway (47). It can regulate catalytic kinase
activity through phosphorylation of different serine/threonine
residue sites. Phosphorylation at tyrosine 216 significantly
increases the enzymatic activity of GSK3β, whereas phosphorylation
at serine 9 decreases enzyme activity (48). Phosphorylation at S9 is the main
mechanism causing GSK3β inactivation (49). In the present study, we revealed
that the p-GSK3β S9 level decreases but the catalytic activity of
GSK3β increases by GA treatment. Preliminary results demonstrated
that decreased β-catenin protein levels in T-ALL cells may result
from GSK3β activation. β-catenin overexpression is associated with
neoplasm progression, cell proliferation, and cell survival
(50). To evaluate our theories, we
overexpressed β-catenin and verified the results using western blot
analysis. The degree of proliferation and apoptosis in T-ALL cells
after GA treatment showed that the inhibitory effect of GA on T-ALL
cells was reversed. Overall, our results demonstrated that GA can
suppress growth of T-ALL cells by inactivating the Wnt/β-catenin
signaling pathway.
It has been reported that the Wnt/β-catenin
signaling pathway is aberrantly upregulated during the occurrence
and development of cancers. Thus, mature blood cells lack Wnt
signaling (30). However, leukemia
cells are highly prolific with high Wnt/β-catenin signaling. The
deficiency of Wnt ligands is associated with low levels of
β-catenin as destruction complexes target β-catenin for
degradation. Our research found that GA could exert inhibitory
effects on T-ALL cell lines and lymphocyte cells from patients with
ALL. These results suggest that GA is more sensitive to lymphocyte
cells from patients with T-ALL.
Autophagy is a highly conservative and protective
process of cell recycling and degeneration of damaged functional
proteins and organelles through lysosomal degradation and digestion
(51,52). Autophagy plays an essential role in
normal cell stabilization and tumorigenesis, drug resistance, and
other pathophysiological processes (53–55).
Currently, there is no uniform classification criterion for
autophagy. Autophagy is regulated by autophagy-related genes. LC3
acts as an autophagosomal marker for monitoring autophagy (56). Beclin1 and Atg7 also belong to
autophagy markers as they participate in the formation and
maturation of autophagosomes (57).
MDC is localized and mainly restricted to the lysosomal membrane
(it is also used to assess autophagy activity). Recent research has
shown that autophagy and the Wnt/β-catenin pathway are closely
related. Autophagy negatively regulates Wnt signaling (58). The tumor inhibitor GABARAPL1
suppresses the Wnt/β-catenin pathway by activating autophagy
(59). In our study,
autophagy-related proteins (LC3-II, Beclin1, and Atg7) were
upregulated, and MDC fluorescence intensity increased significantly
after GA treatment. This suggests that GA induces autophagy in
T-ALL cells. Activation of autophagy can inhibit the Wnt/β-catenin
pathway, further encouraging cell damage. However, the specific
mechanisms require further research.
In summary, our study demonstrates that GA exhibits
effective antileukemia characteristics in T-ALL cells. The
proliferation, apoptosis, and autophagy regulation effects of GA on
T-ALL cells seem to result from downregulating Wnt/β-catenin
signaling and upregulating GSK3β activity, thereby inducing
β-catenin degradation. Collectively, our experimental results
indicate that GA is a promising antileukemia agent for the
treatment of T-ALL.
Acknowledgements
Not applicable
Funding
This study was supported by grants from the National
Natural Science Foundation of China (no. 81570198); National
Science and Technology Major Project of China (no.
SQ2017ZX09030110); Medical and Health Science and Technology
Project of Zhejiang Province (grant no. 2017KY209).
Availability of data and materials
All relevant raw data in our trial will be made
freely available to any researchers who wish to use them for
non-commercial purposes.
Authors' contributions
YL, YW, TTW designed the study. JD, DK, GY, QZ
performed the clinic trials. DK and GY analyzed the data. TTW and
JD wrote the paper, and were responsible for the images. All
authors approved the final version of the manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. Ethics approval was granted by the Zhejiang Provincial
People's Hospital Ethics Committee. Informed consent was obtained
from the subjects included in the present study.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to
declare.
References
|
1
|
Belmonte M, Hoofd C, Weng AP and Giambra
V: Targeting leukemia stem cells: Which pathways drive self-renewal
activity in T-cell acute lymphoblastic leukemia? Curr Oncol.
23:34–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Vlierberghe P and Ferrando A: The
molecular basis of T cell acute lymphoblastic leukemia. J Clin
Invest. 122:3398–3406. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Coustan-Smith E, Song G, Clark C, Key L,
Liu P, Mehrpooya M, Stow P, Su X, Shurtleff S, Pui CH, et al: New
markers for minimal residual disease detection in acute
lymphoblastic leukemia. Blood. 117:6267–6276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pui CH, Carroll WL, Meshinchi S and Arceci
RJ: Biology, risk stratification, and therapy of pediatric acute
leukemias: An update. J Clin Oncol. 29:5512011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pui CH, Robison LL and Look AT: Acute
lymphoblastic leukaemia. Lancet. 371:1030–1043. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cailleteau C, Micallef L, Lepage C, Cardot
PJ, Beneytout JL, Liagre B and Battu S: Investigating the
relationship between cell cycle stage and diosgenin-induced
megakaryocytic differentiation of HEL cells using sedimentation
field-flow fractionation. Anal Bioanal Chem. 398:1273–1283. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leger DY, Liagre B and Beneytout JL: Role
of MAPKs and NF-kappaB in diosgenin-induced megakaryocytic
differentiation and subsequent apoptosis in HEL cells. Int J Oncol.
28:201–207. 2006.PubMed/NCBI
|
|
8
|
Cragg GM and Newman DJ: Nature: A vital
source of leads for anticancer drug development. Phytochem Rev.
8:313–331. 2009. View Article : Google Scholar
|
|
9
|
Yu J, Guo QL, You QD, Lin SS, Li Z, Gu HY,
Zhang HW, Tan Z and Wang X: Repression of telomerase reverse
transcriptase mRNA and hTERT promoter by gambogic acid in human
gastric carcinoma cells. Cancer Chemother Pharmacol. 58:434–443.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kasibhatla S, Jessen KA, Maliartchouk S,
Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma
N, et al: A role for transferrin receptor in triggering apoptosis
when targeted with gambogic acid. Proc Natl Acad Sci USA.
102:12095–12100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao L, Guo QL, You QD, Wu ZQ and Gu HY:
Gambogic acid induces apoptosis and regulates expressions of Bax
and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol
Pharm Bull. 27:998–1003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Duan D, Zhang B, Yao J, Liu Y, Sun J, Ge
C, Peng S and Fang J: Gambogic acid induces apoptosis in
hepatocellular carcinoma SMMC-7721 cells by targeting cytosolic
thioredoxin reductase. Free Radic Biol Med. 69:15–25. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Liu Y, Wang L, He J, Zhang H, Chen
X, Li Y, Yang J and Tao J: Gambogic acid induces apoptosis by
regulating the expression of Bax and Bcl-2 and enhancing caspase-3
activity in human malignant melanoma A375 cells. Int J Dermatol.
48:186–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li C, Qi Q, Lu N, Dai Q, Li F, Wang X, You
Q and Guo Q: Gambogic acid promotes apoptosis and resistance to
metastatic potential in MDA-MB-231 human breast carcinoma cells.
Biochem Cell Biol. 90:718–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi X, Chen X, Li X, Lan X, Zhao C, Liu S,
Huang H, Liu N, Liao S, Song W, et al: Gambogic acid induces
apoptosis in imatinib-resistant chronic myeloid leukemia cells via
inducing proteasome inhibition and caspase-dependent Bcr-Abl
downregulation. Clin Cancer Res. 20:151–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chantarasriwong O, Batova A, Chavasiri W
and Theodorakis EA: Chemistry and biology of the caged Garcinia
xanthones. Chemistry. 16:9944–9962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kashyap D, Mondal R, Tuli HS, Kumar G and
Sharma AK: Molecular targets of gambogic acid in cancer: Recent
trends and advancements. Tumour Biol. 37:12915–12925. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo H, Vong CT, Chen H, Gao Y, Lyu P, Qiu
L, Zhao M, Liu Q, Cheng Z, Zou J, et al: Naturally occurring
anti-cancer compounds: Shining from Chinese herbal medicine. Chin
Med. 14:482019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Teimouri M, Junaid M, Saleem S, Khan A and
Ali A: In-vitro analysis of selective nutraceuticals binding to
human transcription factors through computer aided molecular
docking predictions. Bioinformation. 12:354–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rubinfeld B, Albert I, Porfiri E, Fiol C,
Munemitsu S and Polakis P: Binding of GSK3beta to the
APC-beta-catenin complex and regulation of complex assembly.
Science. 272:1023–1026. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li VS, Ng SS, Boersema PJ, Low TY,
Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi
T and Clevers H: Wnt signaling through inhibition of β-catenin
degradation in an intact axin1 complex. Cell. 149:1245–1256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tetsu O and McCormick F: Beta-Catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cadigan KM and Nusse R: Wnt signaling: A
common theme in animal development. Genes Dev. 11:3286–3305. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nejak-Bowen KN and Monga SP: Beta-catenin
signaling, liver regeneration and hepatocellular cancer: Sorting
the good from the bad. Semin Cancer Biol. 21:44–58. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abrahamsson AE, Geron I, Gotlib J, Dao KH,
Barroga CF, Newton IG, Giles FJ, Durocher J, Creusot RS, Karimi M,
et al: Glycogen synthase kinase 3beta missplicing contributes to
leukemia stem cell generation. Proc Natl Acad Sci USA.
106:3925–3929. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Banerji V, Frumm SM, Ross KN, Li LS,
Schinzel AC, Hahn CK, Kakoza RM, Chow KT, Ross L, Alexe G, et al:
The intersection of genetic and chemical genomic screens identifies
GSK-3α as a target in human acute myeloid leukemia. J Clin Invest.
122:935–947. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Z, Smith KS, Murphy M, Piloto O,
Somervaille TC and Cleary ML: Glycogen synthase kinase 3 in MLL
leukaemia maintenance and targeted therapy. Nature. 455:1205–1209.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Staal FJ, Famili F, Garcia Perez L and
Pike-Overzet K: Aberrant Wnt signaling in leukemia. Cancers
(Basel). 8:782016. View Article : Google Scholar
|
|
31
|
Weerkamp F, van Dongen JJ and Staal FJ:
Notch and Wnt signaling in T-lymphocyte development and acute
lymphoblastic leukemia. Leukemia. 20:1197–1205. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weng AP, Millholland JM, Yashiro-Ohtani Y,
Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H,
Tobias J, et al: A: c-Myc is an important direct target of Notch1
in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev.
20:2096–2109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koch U and Radtke F: Mechanisms of T cell
development and transformation. Annu Rev Cell Dev Biol. 27:539–562.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
den Hoed MA, Pluijm SM, te Winkel ML, de
Groot-Kruseman HA, Fiocco M, Hoogerbrugge P, Leeuw JA, Bruin MC,
van der Sluis IM, Bresters D, et al: Aggravated bone density
decline following symptomatic osteonecrosis in children with acute
lymphoblastic leukemia. Haematologica. 100:1564–1570. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sutton R, Shaw PJ, Venn NC, Law T,
Dissanayake A, Kilo T, Haber M, Norris MD, Fraser C, Alvaro F, et
al: Persistent MRD before and after allogeneic BMT predicts relapse
in children with acute lymphoblastic leukaemia. Br J Haematol.
168:395–404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bleckmann K and Schrappe M: Advances in
therapy for Philadelphia-positive acute lymphoblastic leukaemia of
childhood and adolescence. Br J Haematol. 172:855–869. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X,
Ren X, An Y, Wu Y, Sun W, et al: DHA inhibits proliferation and
induces ferroptosis of leukemia cells through autophagy dependent
degradation of ferritin. Free Radic Biol Med. 131:356–369. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Y, Wang W, Xu J, Li L, Dong Q, Shi Q,
Zuo G, Zhou L, Weng Y, Tang M, et al: Dihydroartemisinin inhibits
tumor growth of human osteosarcoma cells by suppressing
Wnt/β-catenin signaling. Oncol Rep. 30:1723–1730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giambra V, Jenkins CE, Lam SH, Hoofd C,
Belmonte M, Wang X, Gusscott S, Gracias D and Weng AP: Leukemia
stem cells in T-ALL require active Hif1α and Wnt signaling. Blood.
125:3917–3927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gottardi CJ and Gumbiner BM: Distinct
molecular forms of beta-catenin are targeted to adhesive or
transcriptional complexes. J Cell Biol. 167:339–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gao S, Li X, Ding X, Jiang L and Yang Q:
Huaier extract restrains the proliferative potential of
endocrine-resistant breast cancer cells through increased ATM by
suppressing miR-203. Sci Rep. 7:73132017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gavet O and Pines J: Progressive
activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell.
18:533–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Y, Krivtsov AV, Sinha AU, North TE,
Goessling W, Feng Z, Zon LI and Armstrong SA: The Wnt/beta-catenin
pathway is required for the development of leukemia stem cells in
AML. Science. 327:1650–1653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Heidel FH, Bullinger L, Feng Z, Wang Z,
Neff TA, Stein L, Kalaitzidis D, Lane SW and Armstrong SA: Genetic
and pharmacologic inhibition of β-catenin targets
imatinib-resistant leukemia stem cells in CML. Cell Stem Cell.
10:412–424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hamilton A, Helgason GV, Schemionek M,
Zhang B, Myssina S, Allan EK, Nicolini FE, Müller-Tidow C, Bhatia
R, Brunton VG, et al: Chronic myeloid leukemia stem cells are not
dependent on Bcr-Abl kinase activity for their survival. Blood.
119:1501–1510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Perrotti D, Jamieson C, Goldman J and
Skorski T: Chronic myeloid leukemia: Mechanisms of blastic
transformation. J Clin Invest. 120:2254–2264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sparks AB, Morin PJ, Vogelstein B and
Kinzler KW: Mutational analysis of the APC/beta-catenin/Tcf pathway
in colorectal cancer. Cancer Res. 58:1130–1134. 1998.PubMed/NCBI
|
|
48
|
Beurel E, Grieco SF and Jope RS: Glycogen
synthase kinase-3 (GSK3): Regulation, actions, and diseases.
Pharmacol Ther. 148:114–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Frame S, Cohen P and Biondi RM: A common
phosphate binding site explains the unique substrate specificity of
GSK3 and Its Inactivation by Phosphorylation. Mol Cell.
7:1321–1327. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huber BE and Thorgeirsson SS: Analysis of
c-myc expression in a human hepatoma cell line. Cancer Res.
47:3414–3420. 1987.PubMed/NCBI
|
|
51
|
Hönscheid P, Datta K and Muders MH:
Autophagy: Detection, regulation and its role in cancer and therapy
response. Int J Radiat Biol. 90:628–635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schneider JL and Cuervo AM: Autophagy and
human disease: Emerging themes. Curr Opin Genet Dev. 26:16–23.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ghavami S, Gupta S, Ambrose E, Hnatowich
M, Freed DH and Dixon IM: Autophagy and heart disease: Implications
for cardiac ischemia-reperfusion damage. Curr Mol Med. 14:616–629.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hashimoto D, Bläuer M, Hirota M, Ikonen
NH, Sand J and Laukkarinen J: Autophagy is needed for the growth of
pancreatic adenocarcinoma and has a cytoprotective effect against
anticancer drugs. Eur J Cancer. 50:1382–1390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ryter SW and Choi AMK: Autophagy in lung
disease pathogenesis and therapeutics. Redox Biol. 4:215–225. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kimura S, Fujita N, Noda T and Yoshimori
T: Monitoring autophagy in mammalian cultured cells through the
dynamics of LC3. Methods Enzymol. 452:1–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liu G, Liu J, Pian L, Gui S and Lu B:
α-lipoic acid protects against carbon tetrachloride-induced liver
cirrhosis through the suppression of the TGF-β/Smad3 pathway and
autophagy Mol Med Rep. 19:841–850. 2019.PubMed/NCBI
|
|
58
|
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T,
Fu W, Zhang J, Wu W, Zhang X and Chen YG: Autophagy negatively
regulates Wnt signalling by promoting Dishevelled degradation. Nat
Cell Biol. 12:781–790. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang Y, Wang F, Han L, Wu Y, Li S, Yang
X, Wang Y, Ren F, Zhai Y, Wang D, et al: GABARAPL1 Negatively
regulates Wnt/β-catenin signaling by mediating Dvl2 degradation
through the autophagy pathway. Cell Physiol Biochem. 27:503–512.
2011. View Article : Google Scholar : PubMed/NCBI
|