Introduction
Glioblastoma (GBM) is the most common and highly
malignant glial tumor (Grade IV - World Health Organization).
Complete surgical resection and adjuvant treatments (chemotherapy
and radiotherapy) improve patient survival, but the prognosis for
adult patients with GBM remains poor, with a median survival of
15–18 months (1–3). Temozolomide (TMZ) has been the most
widely used drug treatment for patients with GBM (4), and MGMT promoter methylation
has been considered to be a predictor for chemotherapeutic response
to alkylating and methylating agents (5,6).
However, there is a possibility that other DNA repair pathways may
also promote GBM resistance to TMZ-induced base lesions (7). N7-methyl-guanine
(N7-methyl-G) and N3-methyl-adenine (N3-methyl-A)
adducts comprise >80% of TMZ-induced DNA lesions and are
processed through base excision repair (BER), which is a
multiprotein mechanism that is initiated by several damage-specific
glycosylases (8). Therefore,
resistance to TMZ can be caused by an efficient repair process via
BER (9–11), although other alternative processes
may also occur.
Information on the mechanisms involved in tumor
resistance has the potential to provide the fundamental basis for
novel therapeutic strategies. In this context, an approach that has
been extensively investigated is synthetic lethality, which may
occur when two gene functions are compromised due to the
simultaneous loss/or mutations in both genes, which can lead to
cell death (12). Over two decades,
this approach has been studied to explore the potential of applying
PARP inhibition to cancer therapy (12–14),
and the PARP-BRCA interaction provides the first successful example
of clinical application in patients with breast/ovarian cancer
(15–17). The increased susceptibility of
breast cancer cells to PARP inhibitor (PARPi) is thought to result
from the association between PARP-1, BER and homologous
recombination (HR) repair pathways (17). Furthermore, the sensitivity to PARPi
has also been reported in cells that present other genetic
alterations affecting HR, including mutations in the phosphatase
and tensin homologue deleted on chromosome ten (PTEN) gene
(18) and ataxia telangiectasia
mutated (ATM) deficiency (19).
PTEN is a tumor-suppressor gene, which appears to play a
role in astrocytomas (20). Changes
in the PTEN gene, including the loss of heterozygosity,
mutation and methylation, have been identified in at least 60% of
GBMs (21). In addition to the
phosphatase activity that is attributed to PTEN (22), it has been demonstrated that this
protein is associated with the centromere, specifically interacting
with the CENP-C region, becoming a critical controller of the
dynamic organization of the centromere and promoting genomic
stability. This can also lead to defects in double-strand break
(DSB) repair when PTEN is absent, suggesting its role in the
HR pathway (23). The authors of
the aforementioned study also demonstrated that PTEN acts at the
chromatin level, influencing the remodeling of the region
encompassing the RAD51 promoter, thereby controlling
RAD51 transcription by E2F-1. However, conflicting and
limited results have been indicated regarding PTEN influence
on the expression of RAD51 and its paralogs, as well as on
HR efficiency (24). In glioma
cells, it has previously been demonstrated that a disruption of HR
components (RAD51 and BRCA2) sensitizes these cells
to alkylating agents, and it has been demonstrated that the
inhibitor of PARP-1 (olaparib) increases cell death (25).
The genetic heterogeneity of GBM tumors is well
recognized in the literature, but whether PTEN and MGMT status
influence GBM response to treatment with TMZ combined with a PARP-1
inhibitor, has not yet been fully determined. The genetic changes
present in tumor cells may contribute to drug sensitivity;
therefore, it is relevant to investigate molecular signatures that
represent potential predictive markers of susceptibility to
therapies for patients with GBM.
The present study hypothesized that PARP-1
inhibition may increase the cytotoxicity of TMZ-induced lesions in
GBM cells due to the role of the enzyme in damage responses and
multiple DNA repair pathways. Therefore, the present study
investigated whether these mechanisms can be influenced by MGMT and
PTEN status. While it is well established that MGMT is the
main factor that leads to GBM resistance to TMZ, the impact of
PTEN deficiency in repair pathways, and the consequences of
PARP-1 inhibition and PTEN silencing (or deficiency), in
terms of synthetic lethality, was also evaluated in TMZ-treated GBM
cells.
In the present study, GBM cell lines presenting
different PTEN status and MGMT activity were used, and the
results of combined treatments (TMZ plus PARPi - NU1025) were
analyzed. The results demonstrated the effectiveness of these
treatments in sensitizing TMZ-resistant and -sensitive cells,
independent of MGMT activity. However, PARP-1 inhibition was unable
to sensitize U87MG TMZ-sensitive cells, either as a single
treatment, or in the TMZ-combined treatment. The cellular responses
to TMZ/NU1025 in TMZ-resistant cells involved antiproliferative
activity, G2/M arrest, DSBs and the induction of apoptosis.
Regarding the influence of PTEN status on drug-treated
cells, the results of the current study indicated that
PTEN-silenced LN18 cells did not exhibit sensitization to
PARPi tested alone, indicating an absence of synthetic lethality.
Furthermore, the responses to the combined treatment (TMZ plus
PARPi) were also independent of PTEN status. PARPi combined
with TMZ treatment (during three days) caused a strong reduction in
cell viability at 20 days, in contrast to cells treated with TMZ
alone. Therefore, the combination of PARPi with TMZ was revealed to
be a promising strategy that can be used to overcome TMZ-resistance
in GBM cells, and these effects are independent of MGMT and
PTEN.
Materials and methods
Cell lines and culture
T98G (CRL-1690; glioblastoma), LN18 (CRL-2610;
glioblastoma) and U87MG (HTB-14™; glioblastoma of unknown origin)
cell lines were purchased from American Type Culture Collection,
and U251MG (glioblastoma) was provided by Guido Lenz Department of
Biophysics, Federal University of Rio Grande do Sul (UFRGS - Porto
Alegre, RS, Brazil) (26). All cell
lines were authenticated (STR profiling method) and evaluated for
mycoplasma contamination prior to the experiments. The cell lines
differ regarding the proficiency for the TP53 gene, and the
activity of the MGMT repair enzyme (T98G and LN18 are TP53
deficient with high MGMT activity; U87MG is TP53 proficient
and lacks MGMT activity; U251MG is TP53 deficient with no
MGMT activity). T98G, U251MG and U87MG are PTEN-mutated but
LN18 is PTEN wild-type (27,28).
T98G and LN18 cells, which are MGMT proficient, are resistant to
TMZ treatment, indicating IC50 values >500 µM, unlike
U87MG and U251MG cells, which are sensitive (IC50 values
<50 µM) (27). Therefore, T98G
and LN18 are referred to as resistant cells, whereas U87MG and
U251MG are referred to as sensitive to TMZ in the present
study.
Cells were kept frozen in liquid nitrogen. After
thawing, the cells were cultured in HAM F10/DMEM (1:1) medium
(Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS
(Sigma-Aldrich; Merck KGaA), penicillin (100 U/ml; Sigma-Aldrich;
Merck KGaA), and streptomycin (100 mg/ml; Sigma-Aldrich; Merck
KGaA) at 37°C in a humidified 5% CO2 incubator.
Cell treatment with TMZ and PARP-1
inhibitor
For TMZ treatment (TEMODAL- Shering-Plough Corp.),
the concentrations used in the present study were based on previous
results (9,27); those selected were above the values
of IC50 calculated for the cell lines, although previous
reports show that concentrations of 10–25 µM are equivalent to
those found in the spinal fluid of patients after treatment
(29,30). Therefore, TMZ-resistant cells (T98G
and LN18; MGMT-proficient) were treated with 100 and 200 µM of TMZ,
while TMZ-sensitive cells (U87MG and U251MG; MGMT-deficient) were
treated with 10 µM.
For PARP-1 inhibition, the NU1025 agent
(Sigma-Aldrich; Merck KGaA) was used in the current study, since it
has been successfully used by several authors (31–35).
Two concentrations of NU1025 (NU-100 and 200 µM) were added 20 min
prior to TMZ treatment. MGMT inhibition was achieved using 30 µM of
O6-BG inhibitor (Sigma-Aldrich; Merck KGaA) 1 h prior to TMZ
treatment. All drugs remained in cell cultures until subsequent
experimentation. To test the PARP-1 inhibition efficiency by NU1025
agent, the cells were treated with H2O2 (20
mM) for 10 min following NU1025 incubation, and were subsequently
evaluated using immunofluorescent detection for poly-ADP-ribose
(PAR) polymers.
Small interfering (si)RNA
transfection
LN18 cells were transfected with PTEN
siRNA (cat. no. sc-29459; Santa Cruz Biotechnology, Inc.) and a
non-specific siRNA control (cat. no. sc-37007; Santa Cruz
Biotechnology, Inc.) at a final concentration of 100 nM with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. A control siRNA was used
as a negative control, consisting of a scrambled sequence (20–25
nucleotides) that does not target any known genes in the target
cells. The efficiency of LN18 siRNA transfected cells was confirmed
using western blot analysis. Treatment with NU1025 and TMZ was
performed 72 h after transfection, and the experiments were
repeated three times.
Protein isolation and western blot
analysis
Cells were lysed in 200 µl of the RIPA buffer
reagent (Thermo Fisher Scientific, Inc.) supplemented with Halt™
Protease Inhibitor Cocktail kit (Thermo Fisher Scientific, Inc.).
Protein concentration was determined using BCA Protein Assay
reagents (Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Proteins (30 µg) were separated by
electrophoresis in NuPAGE 4–12% Bis-Tris gel (Thermo Fisher
Scientific, Inc.) and blotted onto a PVDF membrane (Thermo Fisher
Scientific, Inc.). Samples were incubated in blocking buffer before
the addition of the primary antibody. The immuno-detection was
accomplished using a WesternBreeze Chemiluminescent kit (Thermo
Fisher Scientific, Inc.). The antibodies used were as follows:
anti-mouse PTEN (cat. no. 9556; dilution 1:1,000), anti-rabbit
phospho-AKT(ser473) (cat. no. 9271; dilution 1:500),
anti-rabbit AKT (cat. no. 9272; dilution 1:1,000), anti-rabbit MGMT
(cat. no. 2739; dilution 1:1,000), and anti-rabbit β-actin (cat.
no. 4967; dilution 1:2,000) or anti-rabbit β-tubulin (cat. no.
2146; dilution 1:1,000), which were used as endogenous controls for
normalization. All antibodies were purchased from Cell Signaling
Technology, Inc. The chemiluminescence detection was performed
using the ImageQuant LAS 500 (GE Healthcare Life Sciences) and
quantified using Gel-Pro Analyzer 4.0 software (Media Cybernetics,
Inc).
Cell proliferation assay
GBM cells (T98G, LN18, U87MG and U251MG) were seeded
(2,000 cells/well) in 12-well plates and incubated at 37°C. After
24 h, cells were treated with TMZ and NU1025, and cell viability
was evaluated after 7 days. Cells were subsequently washed with PBS
followed by incubation with XTT reagent kit as recommended by the
manufacturer's protocol (Roche Molecular Diagnostics).
Clonogenic assay
A clonogenic assay was performed according to
Franken et al (36). After
seeding triplicates of T98G and LN18 cells in 6-well plates (1,000
cells/well), drug treatments were performed. Approximately 10 days
after treatment, cells were washed in PBS, fixed (methanol) and
stained with Giemsa (20 min at room temperature). The colonies with
>50 cells were counted using a stereomicroscope at 16×
magnification (Carl Zeiss).
Cell cycle analysis
After TMZ and NU1025 treatment, T98G and LN18 cells
were washed with PBS and fixed in 70% ethanol, stained for 15 min
(37°C) with a solution containing propidium iodide (PI) (5 µg/ml)
and RNase (50 µg/ml) and analyzed in a Guava EasyCyte Mini System
(Merck KGaA), according to the manufacturer's protocol. Percentages
of cells undergoing G0/G1, S, or G2/M phase were collected on days
one and three after treatment and analyzed using Guava Personal
Cell Analysis system (Merck KGaA).
Apoptosis assays and Annexin-V
staining
Apoptosis detection was evaluated at 3 and 5 days
following TMZ and NU1025 treatment. Apoptosis induction was
measured using the Guava Nexin reagent (Merck KGaA), according to
manufacturer's protocol. The samples were processed using flow
cytometry and analyzed using Guava Personal Cell Analysis system
(Merck KGaA).
Flow cytometry for γH2AX and PAR
analysis
For γH2AX and PAR (poly-ADP-ribose) immunostaining,
cells were fixed with paraformaldehyde (3%) and permeabilized
(Triton-X 0.5%). Cells were then incubated with either primary
rabbit monoclonal antibody to γH2AX(Ser-139) (cat. no.
sc101696; Santa Cruz Biotechnology, Inc.), or anti-mouse pADPr
(product code ab14459; Abcam), both diluted (1:400) and incubated
for 1 h at 37°C. Cells were then incubated with Alexa
Fluor® 488 anti-rabbit IgG (cat. no. A21441; Invitrogen;
Thermo Fisher Scientific, Inc.) or Alexa Fluor® 594
anti-mouse IgG (cat. no. A21201; Invitrogen; Thermo Fisher
Scientific, Inc.; dilution 1:400) for 30 min at 37°C. The
percentage of positive cells was calculated using the Guava
Personal Cell Analysis system (Merck KGaA).
RNA isolation and reverse
transcription-quantitative (RT-q)PCR by PCR array
The transcriptional profiles were analyzed for a set
of DNA repair genes and evaluated using RT-qPCR with a customized
TaqMan® Assay Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Cells were collected at 6 and 24 h
post-treatment, and total RNA was isolated using illustra RNAspin
Mini (GE Healthcare). RNA integrity was performed using a RNA 6000
Nano kit (Agilent Technologies, Inc.) and Bioanalyzer 2100 (Agilent
Technologies, Inc.), following the manufacturer's protocol. The
first-strand complementary DNA (cDNA) was synthesized from 1 µg of
each RNA sample using the SuperScript® VILO™ Master Mix
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. A cDNA pool of three independent
experiments was used for the screening of gene expression profiles.
The reactions were prepared using TaqMan® Fast Universal
PCR Master Mix (Applied Biosystems). The TaqMan® Assay
plate used was customized by Thermo Fisher Scientific, Inc., and
contained 21 genes (DNA repair pathways) and two reference genes
(Table SI). All plates were run on
QuantStudio 3 Real-Time PCR Systems (Applied Biosystems) and
analyzed using QuantStudio™ Design & Analysis Software1.3.1
(Applied Biosystems) using the 2−ΔΔCq method (37). TBP and HPRT1 genes
were used as endogenous controls.
Statistical analysis
Statistical analysis was performed using the
SigmaStat software (version 3.5; Jandel Scientific Software). A
one-way ANOVA, followed by Holm-Sidak multiple comparison tests,
was used to establish whether significant differences existed
between the groups. P<0.05 was considered to indicate a
statistically significant difference. All experiments were
independently performed at least three times and the results are
expressed as the mean ± standard deviation. Results obtained for
gene expression (cDNA pool of three independent experiments) are
expressed as fold-change values.
Results
PARPi potentiates TMZ-induced
cytotoxicity in GBM TMZ-resistant cell lines
To confirm specific genetic alterations in each cell
line, MGMT, PTEN, and AKT protein expression was analyzed (Fig. S1A). Additionally, the
p(ser473)AKT was evaluated to confirm its activation due
to PTEN deficiency in T98G and U87MG cells. To validate PARP-1
inhibition by NU1025, T98G cells were treated with
H2O2 (20 mM) prior to NU1025 treatment (100
or 200 µM), and PAR levels were assessed using flow cytometry.
Following a 10 min incubation, a significant increase in PARP-1
activity (PAR detection) was detected in
H2O2-treated cells, while as expected, NU1025
treatment markedly decreased PAR polymers in response to
H2O2 treatment (Fig. S1B). Cells were then treated with
TMZ (200 µM) and NU1025 (100 or 200 µM). As detected using a XTT
assay, which was performed after 7 days of continuous treatment,
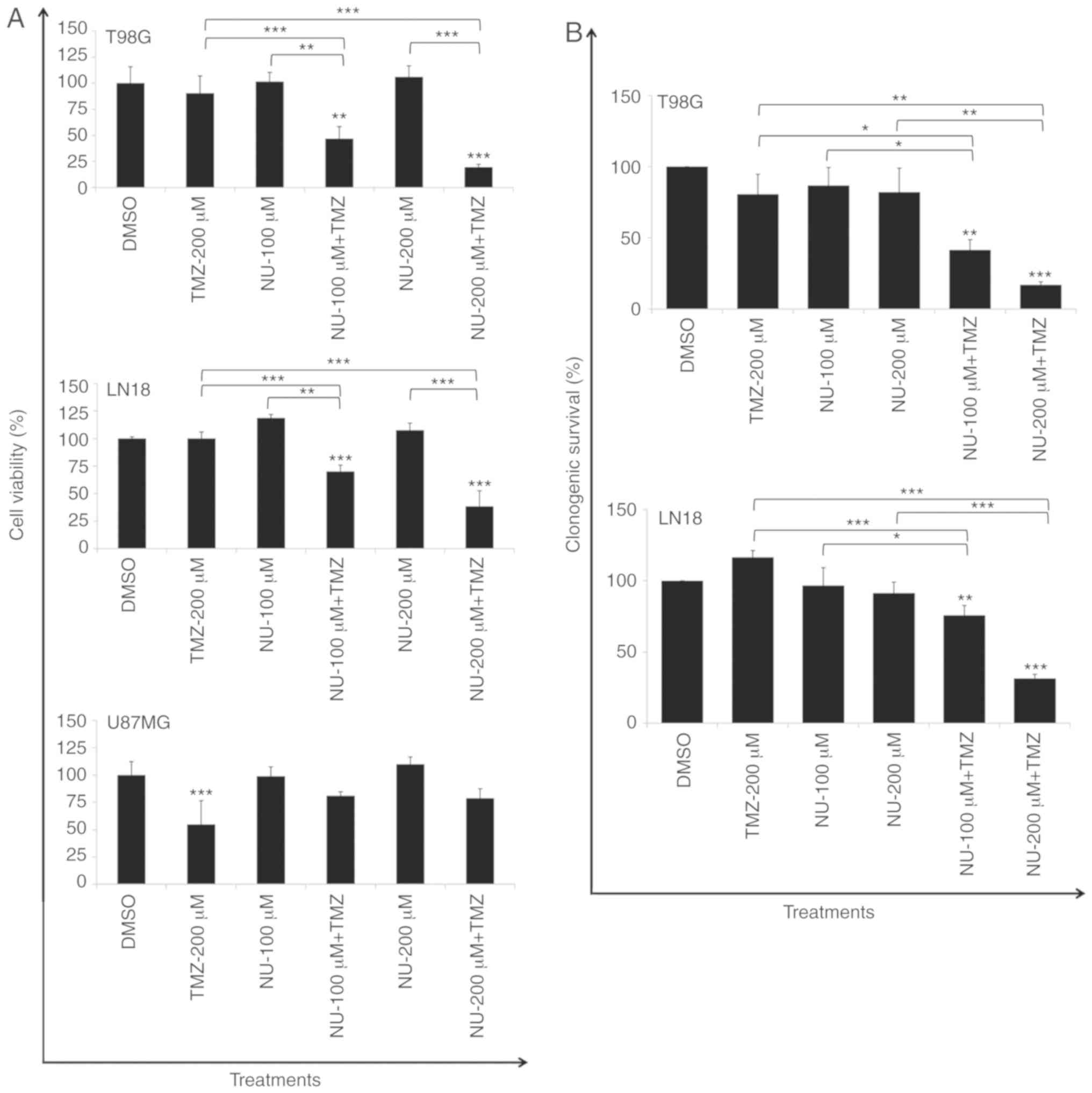
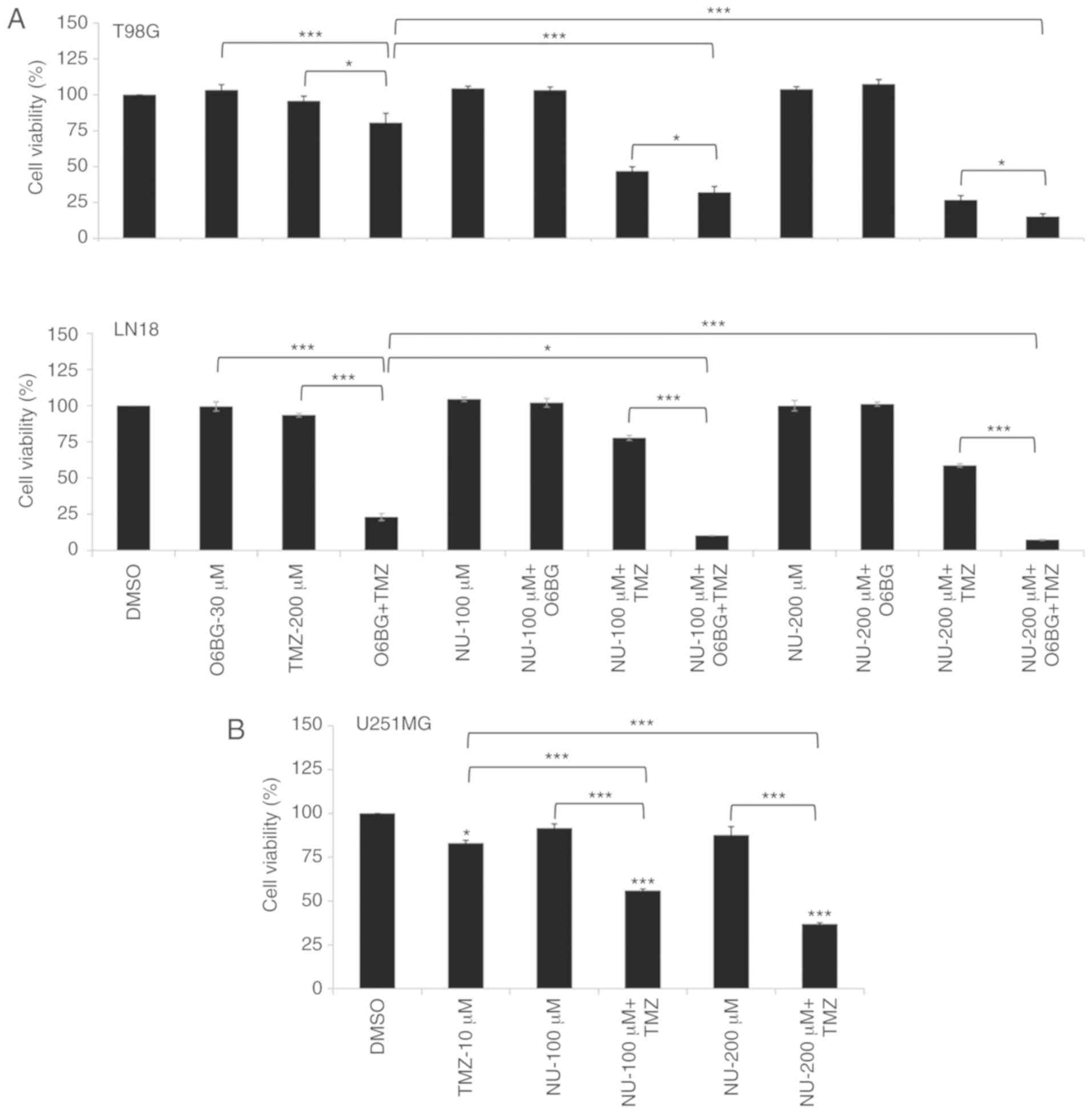
the drug combination caused a significant reduction in cell
viability in T98G (PTEN-mutated) and LN18 cells
(PTEN-wild type), which are TMZ-resistant (MGMT proficient
cells), and this was independent to the PTEN status
(Fig. 1A). Survival rates were
reduced ~4.7- and 3.7-fold following the combined treatment (NU-200
µM + TMZ-200 µM) for T98G and LN18 cells, respectively, compared
with a TMZ single treatment, as evaluated at 10 days after
treatment (Fig. 1B). Similar
experiments were performed in TMZ-sensitive U87MG cells
(PTEN-mutated), which did not present MGMT, to analyze their
sensitivity to the combined treatment. A marked reduction in cell
viability was observed in cells treated with TMZ (10 µM, single
treatment), and a significant difference was not indicated between
TMZ alone and TMZ combined to NU1025 (Fig. 1A).
NU1025 (single treatment) did not demonstrate a
cytotoxic effect in PTEN proficient (LN18) and deficient
cells (T98G and U87MG), indicating that PTEN status may not
contribute to the synthetic lethality promoted by PARPi in the
absence of DNA damage, which is induced by TMZ in GBM cells.
Combined treatment of PARPi and TMZ
induces G2/M blockage and apoptosis
It has been previously reported that BER
intervention by APE1 depletion in T98G cells caused G2/M arrest,
DSBs and apoptosis induction in response to TMZ treatment (10). The present study investigated
whether the inhibition of PARP-1 could cause similar effects
considering its role in the BER pathway. Whether differences in the
PTEN status could lead to different drug responses was therefore
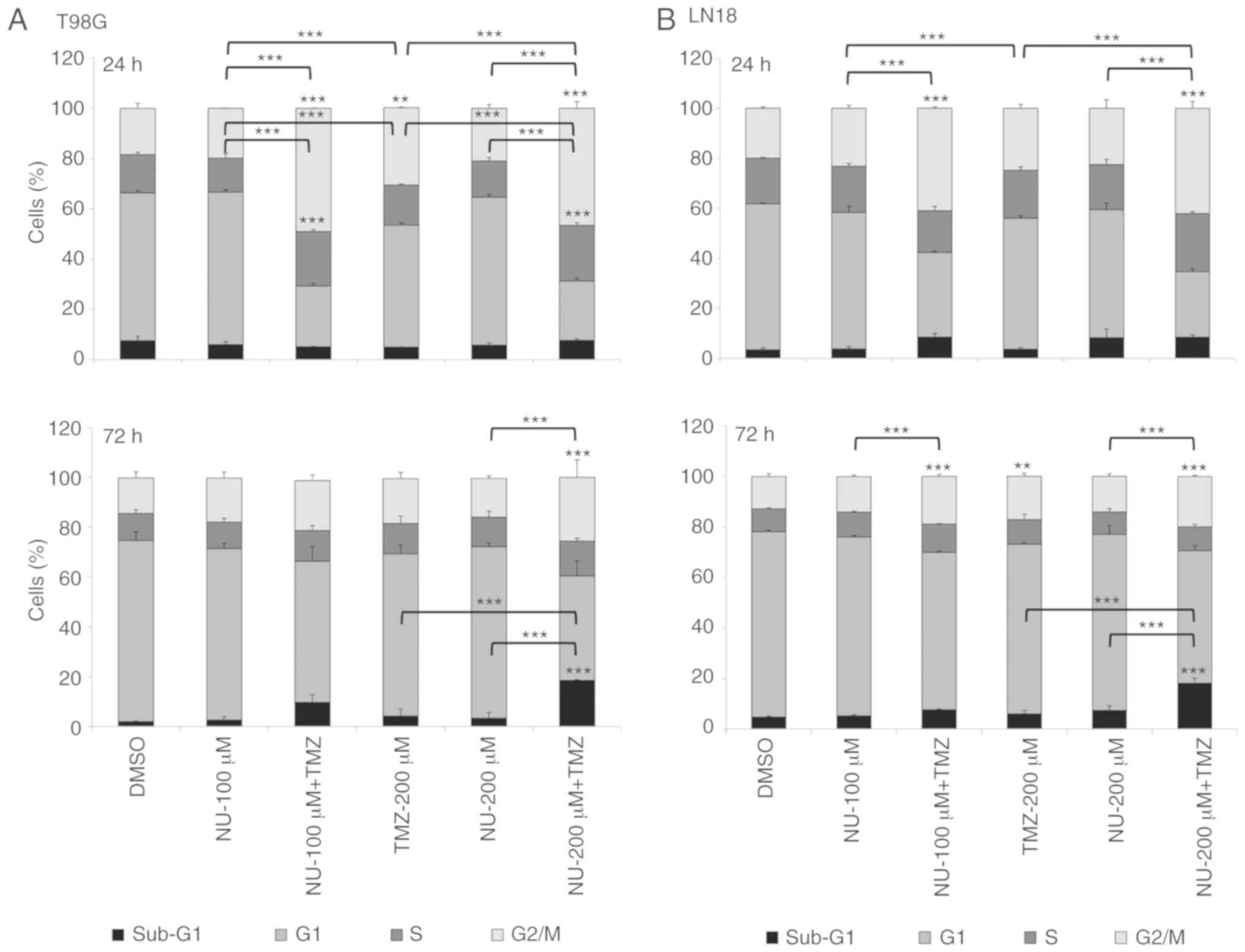
assessed. T98G and LN18 cells treated with TMZ (200 µM) plus NU1025
(100 or 200 µM) indicated a significant G2/M block at 24 h after
treatment compared with single-treatments (TMZ or NU1025) and
control (DMSO) cells. G2/M arrest was maintained after 72 h,
compared with NU1025-treated and control cells, either in T98G
(NU-200 µM + TMZ) and LN18 cells (NU-100 or 200 µM + TMZ).
Furthermore, only T98G cells treated with NU1025 + TMZ also
demonstrated a significant increase in the proportion of S-phase
cells observed at 24 h, compared with single-treatments and the
control. T98G and LN18 cell lines also revealed a significant
increase in the sub-G1 content (DNA fragmentation) following
treatment with TMZ plus NU1025 (200 µM) in cells collected after 72
h, compared with single drug-treatments or the control group
(Fig. 2A and B). The cell cycle
blockade may have occurred as a result of DSBs generated by the
combined treatment, as evaluated by the detection of γH2AX-positive
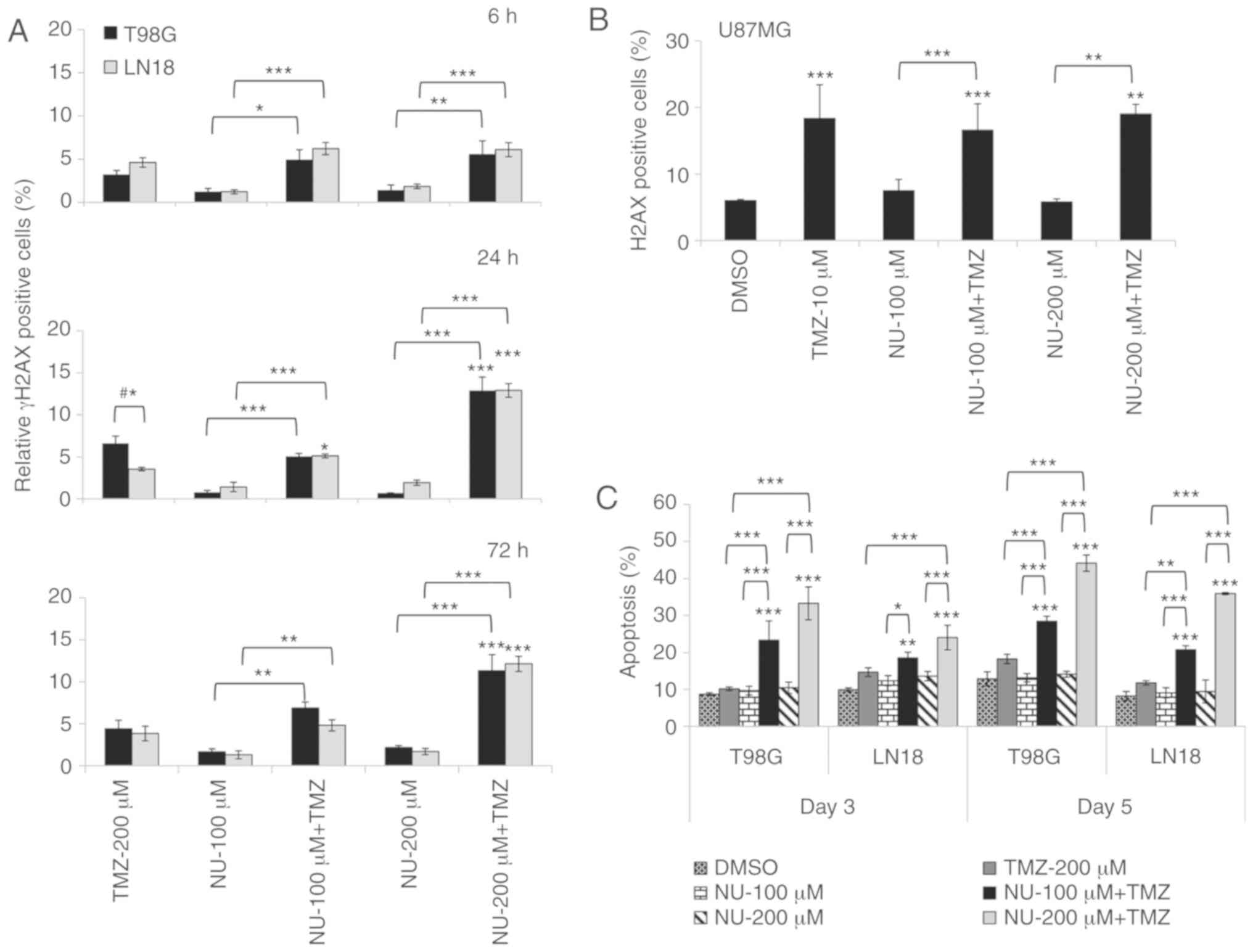
cells (6, 24 and 72 h), which were demonstrated to be increased. In
these experiments, γH2AX-positive cells were calculated (average of
relative values), as the experimental vs. the control (DMSO), in
order to compare the responses between cell lines, taking into
account their genetic background, including the PTEN status.
The results indicated that DSB induction was similar in both cell
lines (T98G and LN18), except for TMZ single treatment in T98G
cells, which presented a greater percentage of γH2AX-positive cells
detected at 24 h (Fig. 3A). These
data indicated that differences in the PTEN status did not
significantly affect the induction of DSBs. The amount of
γH2AX-positive cells increased from 6 to 72 h following the
combined treatments, and the reduction in cell viability may be a
consequence of a decreased DNA repair that is promoted by PARP
inhibition. In U87MG cells, the γH2AX induction (Fig. 3B) was likely due to the TMZ
single-treatment, confirming the lack of response to the combined
treatment, as demonstrated by the results of the cell viability
assay.
Furthermore, the results of the present study
indicated that the responses to DNA damage observed for the
combined treatment was different when comparing the two cell lines,
since T98G indicated higher levels of apoptosis induction than LN18
cells. This may be due to LN18 being more efficient in DSB repair
than T98G cells (Fig. 3C). NU1025
single treatment did not induce changes in the cell cycle kinetics,
and did not cause any effect on the induction of DSBs and
apoptosis, corroborating the results of cell viability and
clonogenic survival.
PTEN silencing did not affect the
efficacy of PARPi plus TMZ-treatment
Considering the possible involvement of PTEN in the
HR pathway (18), experiments were
performed in LN18 PTEN-silenced cells to demonstrate the
lack of synthetic lethality, since this phenomenon was not observed
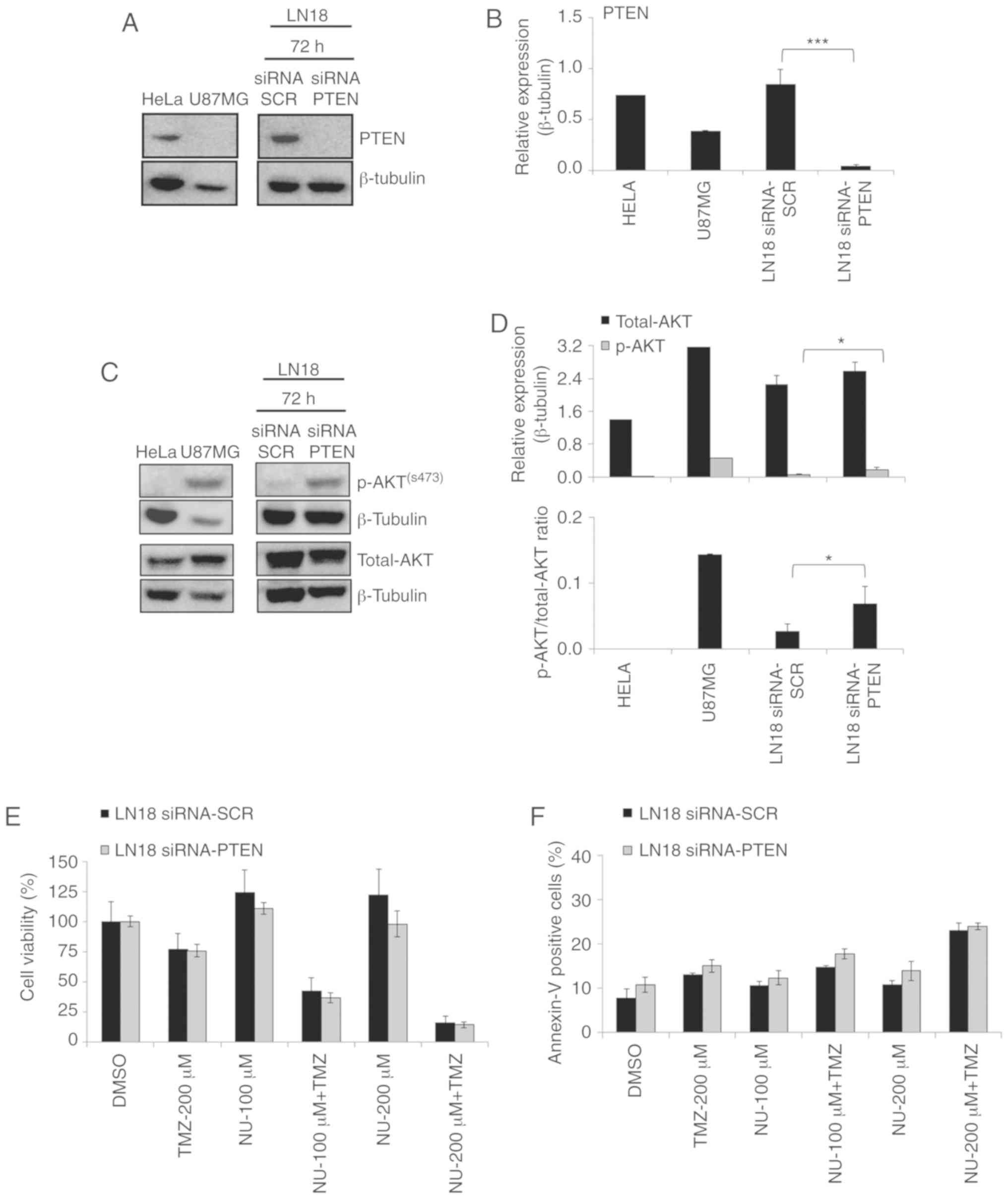
in PTEN-deficient cells (T98G, U251, and U87MG). siRNA PTEN
LN18 cells indicated a substantial decrease in gene expression
(94.7% of knockdown; data not shown), as well as in PTEN protein
level (Fig. 4A and B). The
total-AKT expression was not significantly affected by the
PTEN silencing, but an increase in p-AKT expression was
observed (Fig. 4C and D). This
indicates that PTEN downregulation promotes the modulation of the
PI3K/AKT signaling pathway, and this is supported by the increase
in the p-AKT/total-AKT ratio in PTEN-silenced cells (Fig. 4D). However, in these cells,
PTEN downregulation did not significantly influence the
responses to PARPi tested as a single agent, and in a combination
with TMZ, compared with siSCR cells (transfection control), as
evaluated using cell viability (Fig.
4E) and Annexin assays (Fig.
4F). These results indicated that PTEN does not
contribute to the occurrence of synthetic lethality in GBM cells,
and is not crucial for the cytotoxicity induced by the combined
treatment (TMZ + NU1025) in LN18 TMZ-resistant cells.
MGMT repair does not influence the
efficiency of PARPi plus TMZ treatment
To evaluate whether the effects of TMZ plus NU1025
combined treatment are dependent on MGMT activity, the O6-BG agent
was used to inhibit the activity of the enzyme in T98G and LN18
cells. A significant reduction in cell viability was observed in
the TMZ/O6-BG and TMZ/O6-BG/NU (100 and 200 µM) treated cells. The
results indicated that the inhibition of MGMT activity (O6-BG) did
not significantly counteract the effectiveness of PARP inhibition
in TMZ-treated cells, since it also contributed to a possible
additive effect on cell sensitization (Fig. 5A). Additionally, to test whether
MGMT repair influenced the response to PARPi plus TMZ treatment,
experiments were performed in U251MG cells, which are deficient for
MGMT activity (27), and also carry
TP53 and PTEN mutations (28). The results obtained for U251MG cells
revealed that MGMT activity does not affect cellular responses to
drug treatments, and corroborated the results obtained in T98G and
LN18 cells under MGMT inhibition (Fig.
5B). However, the absence of a response observed for U87MG
cells suggested the influence of other genetic alteration, since
these cells also lack MGMT activity.
PARPi treatment overcomes TMZ-induced
resistance
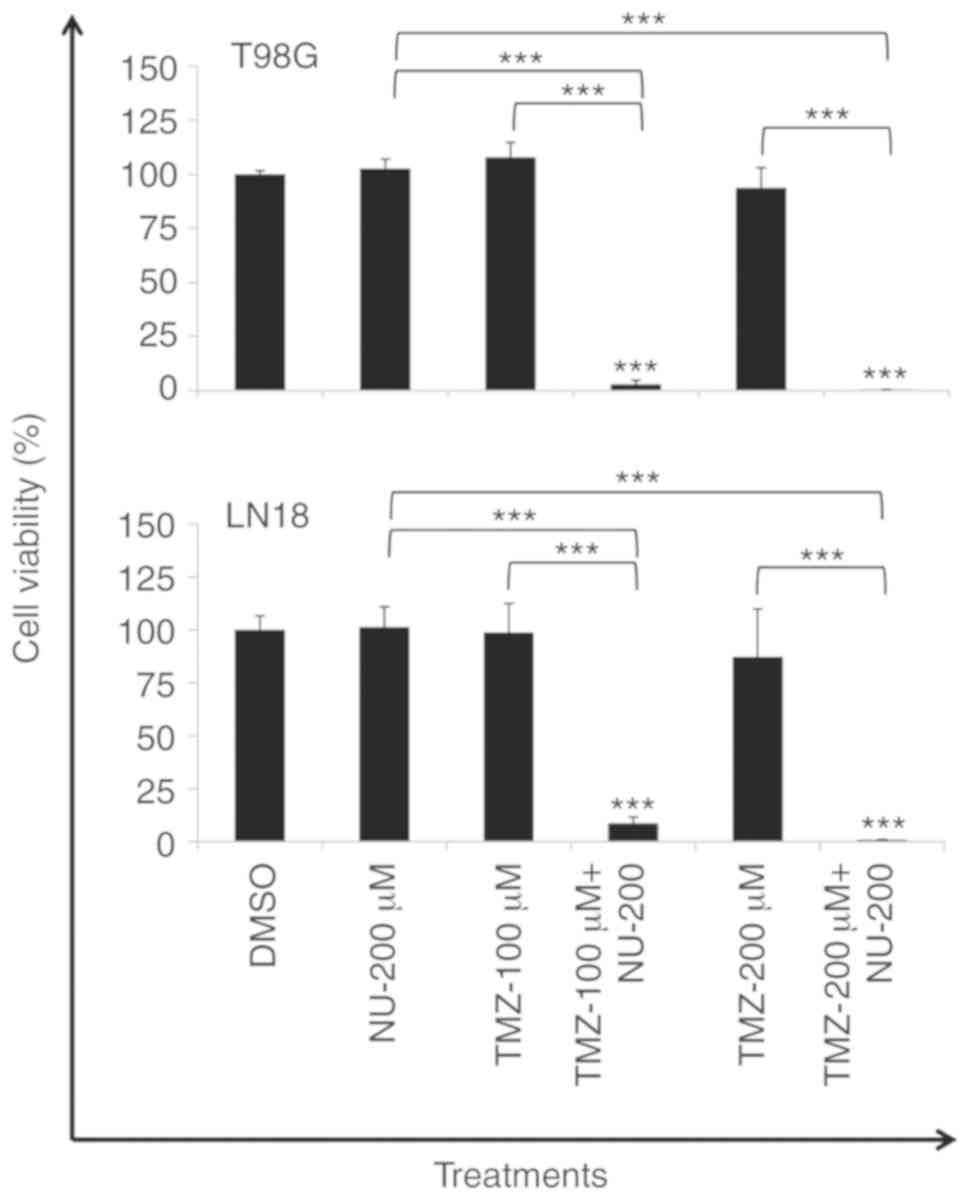
Cell viability was also determined following 20 days
of recovery time after three consecutive days of TMZ (100 and 200
µM) treatment, administered alone or in combination with NU1025
(200 µM), to evaluate the occurrence of resistance to the combined
treatment. TMZ treatment was unable to reduce cell viability
following 20 days. However, a period of three consecutive days of
combined treatment (TMZ plus NU-200 µM) was efficient to
significantly decrease the viability in T98G and LN18 cell lines
(Fig. 6), demonstrating the absence
of resistance under the conditions tested, and indicating that the
induced-lethality was independent to the PTEN status. Taken
together, these results demonstrated the effectiveness of the drug
combination to sensitize TMZ-resistant tumor cells.
Transcript expression of GBM cells
exposed to TMZ plus PARPi treatment
To determine the influence of the PARP-1 inhibitor
on the recruitment of repair mechanisms in response to TMZ
treatment, a gene set of DNA repair genes was selected to evaluate
transcriptional expression profiles (6 and 24 h) in LN18 and T98G
cell lines. BER (APEX1, PARP1, FEN1, LIG1, and
XRCC1), HR (BRCA1, RAD51, RAD51B/C/D
and LIG4), non-homologous end joining (NHEJ; PRKDC
and XRCC5/6), nucleotide excision repair (NER; XPA,
XPC, and XRCC4), mismatch repair (MMR; MSH2/3)
and MGMT genes were evaluated in the current study. In
addition, transcriptional expression of HR genes was compared
between the two cell lines, which possess a different PTEN
status. To establish differential transcript expression,
fold-change (Log2) values >|1.3| were considered to be the cut
off.
In the clustering analysis, the two cell lines
presented a distinct damage response, however, each cell line was
clustered according to the time-point (6 or 24 h), except for T98G
NU1025-treated cells evaluated after 24 h (Fig. 7). Regarding DNA repair pathways in
T98G PTEN-deficient cells, BER (PARP1, FEN1, and
LIG1) and HR (BRCA1 and RAD51B/C) genes were
indicated to be slightly induced by TMZ (24 h), while the effect of
PARP-1 inhibition by NU1025 abolished these responses in the
combined treatment (Fig. 7;
Table SII). However, LN18
PTEN-proficient cells did not exhibit changes in expression
in response to treatments (Fig. 7;
Table SIII). An upregulation of
RAD51 paralogs (B and C) was indicated
following a single treatment of TMZ in T98G PTEN-deficient
cells, but the same result was not revealed in LN18
PTEN-proficient cells, indicating that there is no
correlation between PTEN status and the expression of these
genes, which are associated with the HR pathway.
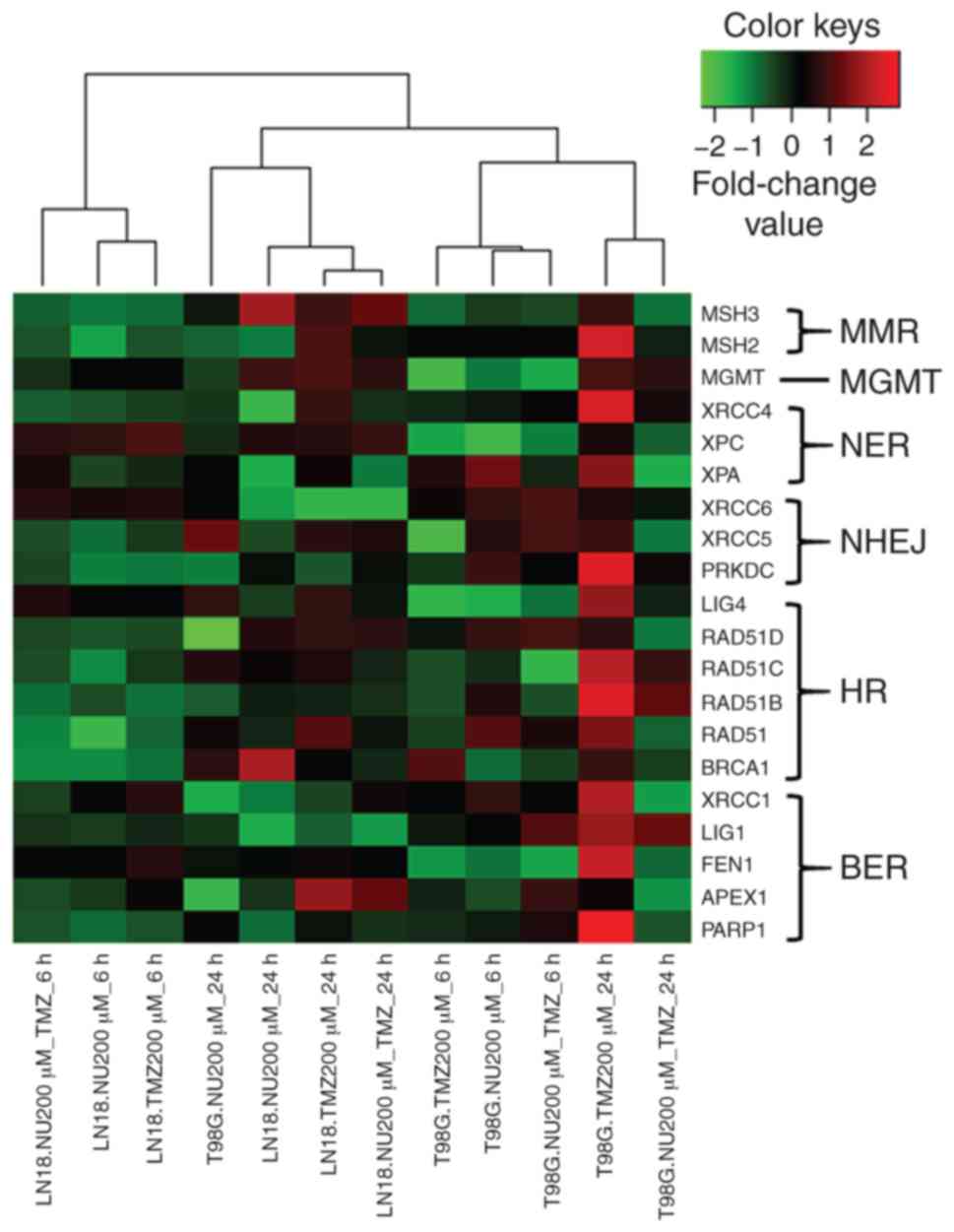 | Figure 7.GBM cell lines showing gene
expression profiles in response to PARPi and TMZ combination.
Hierarchical clustering illustrating a graphical representation of
transcript expression profiles for a gene set encompassing the
major repair pathways (BER, NER, MGMT, MMR, HR and NHEJ), as
analyzed in LN18 and T98G cells treated with TMZ (200 µM) and
NU1025 (200 µM) for 6 and 24 h. The genes and samples were grouped
by using Pearson's correlation, according to the similarity of
expression level. Upregulated and downregulated genes are
represented in red and green colors, respectively; black represents
a lack of differential expression. Variations in the color
intensity represent different levels of transcript expression. GBM,
glioblastoma; TMZ, temozolomide; BER, base excision repair; NER,
nucleotide excision repair; MGMT, O6-methylguanine DNA
methyltransferase; MMR, mismatch repair; HR, homologous
recombination; NHEJ, non-homologous end joining. |
Discussion
Despite a number of studies concerning the use of
poly-ADP-ribose polymerase inhibitors (PARPi) as a strategy for
cancer treatment (38–40), information on the mechanisms and
genetic factors influencing the responses to PARPi and temozolomide
(TMZ) combination are still largely undetermined, especially in
glioblastoma (GBM). To study this approach and the mechanisms
associated with drug responses, four cell lines that present
different genetic alterations regarding MGMT activity and
PTEN genes were analyzed in the current study.
The results indicated that the combined treatment
(TMZ plus NU1025) was effective in reducing the cell viability and
clonogenic survival rates presented by TMZ-resistant cells (T98G
and LN18, which are MGMT-proficient). The potentiating effects of
the TMZ + PARPi combination treatment occurred independently to
MGMT activity, as observed in U251MG TMZ-sensitive (MGMT-deficient)
and MGMT-inhibited cells (T98G and LN18 cell lines). The drug
combination caused a G2/M arrest due to the generation of
double-strand breaks (DSBs) (possibly via the conversion of
N-methylpurine lesions, which were not removed due to repair
intervention by PARPi), leading to the induction of apoptosis in
T98G and LN18 cells. In these cells, increased amount of unrepaired
DNA damage was caused by the inhibition of PARP-1 by NU1025,
leading to cell death following drug treatments.
However, these responses were not observed in
TMZ-sensitive U87MG cells, in which MGMT enzyme activity is absent,
in contrast to the results reported by Tentori et al
(41), using another PARPi (GPI
15427). In the present study, the decrease observed in cell
viability in U87MG cells occurred as a consequence of TMZ-induced
DSBs in the combined treatments. Erice et al (42), demonstrated that MGMT-proficient
melanoma cells, strongly responded to the combination of TMZ and
PARPi. For GBM, a number of studies have reported conflicting
results regarding markers of response to PARP inhibitors combined
with TMZ, including MGMT, PARP-1 or PTEN expression (41–44),
emphasizing the importance of these studies regarding the influence
of genetic alterations in drug responses. LN18 (PTEN-proficient)
and T98G (PTEN-mutant) cells present high MGMT activity,
demonstrating high resistance to TMZ, even at concentrations of
100–200 µM, and they were sensitized only by the combined
treatment. On the other hand, U87MG and U251MG cells
(MGMT-deficient; PTEN-mutant) were more sensitive to TMZ (10
µM), and none of them were affected by PARPi tested alone. However,
only U251MG cells were sensitized upon the combined treatment.
Thus, regardless of TMZ concentration, PTEN-deficient cells
presented different responses to the TMZ and PARPi combined
treatment. The current results in U87MG cells suggest a possible
influence of additional genetic alterations, since MGMT activity
and the PTEN status did not interfere in the efficiency of
the combined treatment (PARPi + TMZ). Alterations in TP53
may also cause impact on drug responses and may distinguish these
cells (U87MG is wild-type while T98G, LN18 and U251MG are mutant
for this gene). It has been demonstrated that radiation-induced
changes in transcript profiles of GBM cell lines are dependent on
the functional status of TP53 (45). It has also been reported that
TP53 influences the differential sensitivity of GBM cell
lines to combined treatments, including topoisomerase I inhibitor
topotecan, radiation therapy, and PARPi (such as NU1025) (35). Zaky et al (46) revealed that TP53 regulated
APE1 transcriptional expression in colorectal adenocarcinoma
tumor cells, which was also supported by a study performed by
Poletto and colleagues (47) in
normal and transformed human fibroblasts. In a previous study, the
impact of APE1 gene silencing was assessed in terms of the
TMZ response, and while U87MG cells did not exhibit sensitization
to TMZ treatment, T98G was significantly sensitized by APE1
downregulation (10). It has been
indicated that APE1/CHK2 signaling is associated with the
coordination of DSB repair, facilitating homologous recombination
(HR); however, where there is low APE1 expression, non-homologous
end joining (NHEJ) becomes the prevalent repair pathway (48). The authors showed that U87MG cells
presented a low basal APE1 protein expression, and on the basis of
this information (48), it can be
suggested that the error-prone NHEJ might be a predominant repair
pathway in these cells, explaining the lack of responses to PARP-1
inhibition in the present study. Therefore, when studying different
cell lines, determining the genetic background of each GBM cell
line is critical to understand the responses to PARP-1
inhibition.
In the present study, the influence of PTEN
status was studied in T98G (PTEN-mutated) and LN18
(PTEN-wild type) cell lines, and these cells presented
similar responses regarding G2/M blockade and DSB induction caused
by the combined treatments, regardless of the PTEN status
and AKT activation. T98G cells (PTEN-deficient) demonstrated a
reduction in cell viability and induction of apoptosis that were
slightly more pronounced than LN18 cells (PTEN-proficient) in
response to the combined treatment, suggesting some survival
pathway or differential damage tolerance between the cell lines,
which may have been due to different inherent genetic alterations
in these tumor cells. PTEN has been a subject of controversial
studies regarding its role in HR efficiency (24). Thus, PTEN knockdown
experiments by siRNA were conducted to confirm whether the lack of
this gene function could contribute to the occurrence of synthetic
lethality, or interfere in the combined treatment, considering the
possible involvement of PTEN in the HR pathway (18). PTEN gene silencing in LN18
did not modify the cellular responses in terms of viability and
apoptosis induction, suggesting that this gene did not cause any
impact in the responses of the two cell lines to the NU1025 single-
or combined treatment (TMZ + NU1025), although the relevance of
PTEN roles had been recognized regarding its participation in the
PI3K-AKT signaling pathway, and regulation of the G2/M checkpoint,
which is associated with genomic instability in tumor cells
(49,50). Conflicting conclusions have been
reported regarding the role of PTEN in drug responses. This
may be due to the fact that cancer cells, which have been developed
within the context of a genotype with a PTEN mutation,
accumulate secondary genetic aberrations, differing from cells in
which PTEN has been experimentally removed (24,51).
This may explain the responses to drug treatments that are observed
in PTEN-silenced LN18 cells.
The transcript expression profiles of a gene set,
whose genes represent the major repair pathways (BER, NER, MGMT,
MMR, HR and NHEJ), were also examined, since multiple DNA repair
pathways can affect the cellular sensitivity to damage caused by
methylating agents (52,53). The results revealed that following a
24-h treatment (after one cell doubling), T98G cells treated with
TMZ exhibited a number of upregulated genes, including PARP1,
FEN1 and LIG1 (BER), BRCA1, RAD51B/C (HR),
PRKDC (NHEJ) and XRCC4 (NER), while most genes were
downregulated or unmodulated at 6 h after treatment. LN18 cells did
not exhibit changes in expression profiles for the whole gene set.
In other studies, the expression of HR genes (BRCA1, BRCA2,
RAD51 and FANCD2) were also markedly increased in U251MG
and A172 cells at 48 h after TMZ treatment, but only BRCA1
was induced in U87MG cells, suggesting a more effective DNA repair
capacity in U251MG and A172 than U87MG cells (54). The expression of RAD51 and
its paralogs did not change in LN18 PTEN-proficient cells,
which was different to the effect in T98G PTEN-deficient
cells, which presented an upregulation of RAD51B/C. In a
previous study, U251MG cells (PTEN-mutated) exposed to
ionizing radiation (IR) indicated RAD51 foci, suggesting a
functional HR (48). This finding
indicated a negligible PTEN influence on RAD51 transcript
expression in GBM cells. These results also revealed that cell
responses to TMZ treatment may differ among cell lines depending on
their genetic background. Furthermore, for transcript gene
expression, the hierarchical cluster indicated that the cell lines
with differences in PTEN status (T98G and LN18) were grouped
separately, according to time-point (6 or 24 h). These observations
reinforce the relevance of the genetic background inherent to each
cell line and indicated that cell responses to drug treatment
cannot be driven by a single genetic alteration, including
PTEN status.
Regarding the PARPi effect on gene expression
profiles, NU1025 was effective in inducing a downregulation of the
majority of genes in TMZ-treated T98G cells. Since PARP-1 and
PARylation serve an important role in the orchestration of the
early steps of DNA damage responses, damage signaling and
cross-talk with DNA repair pathways (52,55,56),
the alterations in transcript expression may be associated with the
slight sensitization achieved in T98G cells in response to the
combined treatment (TMZ + NU1025), compared with LN18 cells.
In the present study, the survival rates following
20 days (recovery time following cell treatment with TMZ plus PARPi
along 3 consecutive days) were close to zero in both cell lines,
strongly indicating the efficiency of PARP-1 inhibition in cell
death, regardless of PTEN status. PARPi in TMZ-treated cells
may act to avoid the activation of DNA repair mechanisms, which, as
suggested by Ströbel et al (48) and Nagel et al (57), can lead to an adaptive response to
TMZ.
In conclusion, the present study highlights the
therapeutic potential of PARPi and TMZ combination for GBM
treatment, demonstrating that TMZ-resistant GBM cells, due to the
high MGMT activity (known as the main predictor of TMZ response in
GBM), can be sensitized by the combination of PARPi with TMZ
treatment. The results revealed that PTEN gene status and
MGMT activity are not genetic predictive markers for the responses
to TMZ/PARPi combination in GBM, even though the results with U87MG
TMZ-sensitive cells, which were not sensitized to the combined
treatment, suggest that other genetic alterations (inherent to the
genetic background) may influence the success of the combined
treatment. Additionally, PTEN did not influence the expression of
HR genes, and its downregulation or deficiency did not sensitize
GBM cells to the PARPi single-treatment used in the present study,
indicating an absence of synthetic lethality.
Supplementary Material
Supporting Data
Acknowledgements
The authors are indebted to Mr Luiz A. Costa Jr.
for the technical assistance, and Natalia C.S. Moreira for helping
with the identification of the cell lines. Our special gratitude to
Professor Dr Aguinaldo L. Simões and Mrs Maria do Carmo T. Canas
for performing authentication of the GBM cell lines.
Funding
We acknowledge the financial support from São Paulo
Research Foundation (FAPESP, Brazil, grant nos: 2013/12033-0,
2016/17862-3 and 2013/09352-7), National Council for Scientific and
Technological Development (CNPq, Brazil) and Coordination for the
Improvement of Higher Education Personnel (CAPES, Brazil).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
APM carried out the experimental design,
performance of the experiments and analyses of all samples,
interpreted the data and wrote the manuscript; SCGL assisted in
various experiments. PRDVG participated in the experimental design,
data interpretation and revised the manuscript. DJX conducted the
qPCR array experiments and data analysis. ETSH supervised the
research work and experimental design, participated in the data
collection and interpretation and manuscript preparation. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Authors' information
Elza Tiemi Sakamoto-Hojo, ORCID:
0000-0002-1383-3314.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 17
(Suppl 4):iv1–iv62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thakkar JP, Dolecek TA, Horbinski C,
Ostrom QT, Lightner DD, Barnholtz-Sloan JS and Villano JL:
Epidemiologic and molecular prognostic review of glioblastoma.
Cancer Epidemiol Biomarkers Prev. 23:1985–1996. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wick W, Osswald M, Wick A and Winkler F:
Treatment of glioblastoma in adults. Ther Adv Neurol Disorder.
11:17562864187904522018.
|
|
4
|
Dresemann G: Temozolomide in malignant
glioma. OncoTargets Ther. 3:139–146. 2010. View Article : Google Scholar
|
|
5
|
Mutter N and Stupp R: Temozolomide: A
milestone in neuro-oncology and beyond? Expert Rev Anticancer Ther.
6:1187–1204. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cabrini G, Fabbri E, Lo Nigro C, Dechecchi
MC and Gambari R: Regulation of expression of
O6-methylguanine-DNA methyltransferase and the treatment
of glioblastoma (Review). Int J Oncol. 47:417–428. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshimoto K, Mizoguchi M, Hata N, Murata
H, Hatae R, Amano T, Nakamizo A and Sasaki T: Complex DNA repair
pathways as possible therapeutic targets to overcome temozolomide
resistance in glioblastoma. Front Oncol. 2:1862012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wallace SS, Murphy DL and Sweasy JB: Base
excision repair and cancer. Cancer Lett. 327:73–89. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Montaldi AP and Sakamoto-Hojo ET:
Methoxyamine sensitizes the resistant glioblastoma T98G cell line
to the alkylating agent temozolomide. Clin Exp Med. 13:279–288.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Montaldi AP, Godoy PR and Sakamoto-Hojo
ET: APE1/REF-1 down-regulation enhances the cytotoxic effects of
temozolomide in a resistant glioblastoma cell line. Mutat Res Genet
Toxicol Environ Mutagen. 793:19–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu L and Gerson SL: Therapeutic impact of
methoxyamine: Blocking repair of abasic sites in the base excision
repair pathway. Curr Opin Investig Drugs. 5:623–627.
2004.PubMed/NCBI
|
|
12
|
Lord CJ, Tutt AN and Ashworth A: Synthetic
lethality and cancer therapy: Lessons learned from the development
of PARP inhibitors. Annu Rev Med. 66:455–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hartwell LH, Szankasi P, Roberts CJ,
Murray AW and Friend SH: Integrating genetic approaches into the
discovery of anticancer drugs. Science. 278:1064–1068. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhattacharjee S and Nandi S: DNA damage
response and cancer therapeutics through the lens of the fanconi
anemia DNA repair pathway. Cell Commun Signal. 15:412017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Helleday T: The underlying mechanism for
the PARP and BRCA synthetic lethality: Clearing up the
misunderstandings. Mol Oncol. 5:387–393. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mendes-Pereira AM, Martin SA, Brough R,
McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ and Ashworth A:
Synthetic lethal targeting of PTEN mutant cells with PARP
inhibitors. EMBO Mol Med. 1:315–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weston VJ, Oldreive CE, Skowronska A,
Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns
P, et al: The PARP inhibitor olaparib induces significant killing
of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood.
116:4578–4587. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Cancer genome atlas research network: Integrated genomic analysis
identifies clinically relevant subtypes of glioblastoma
characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1.
Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen WH, Balajee AS, Wang J, Wu H, Eng C,
Pandolfi PP and Yin Y: Essential role for nuclear PTEN in
maintaining chromosomal integrity. Cell. 128:157–170. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lester A, Rapkins R, Nixdorf S, Khasraw M
and McDonald K: Combining PARP inhibitors with radiation therapy
for the treatment of glioblastoma: Is PTEN predictive of response?
Clin Transl Oncol. 19:273–278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quiros S, Roos WP and Kaina B: Rad51 and
BRCA2-New molecular targets for sensitizing glioma cells to
alkylating anticancer drugs. PLoS One. 6:e271832011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Filippi-Chiela EC, Thomé MP, Bueno e Silva
MM, Pelegrini AL, Ledur PF, Garicochea B, Zamin LL and Lenz G:
Resveratrol abrogates the temozolomide-induced G2 arrest leading to
mitotic catastrophe and reinforces the temozolomide-induced
senescence in glioma cells. BMC Cancer. 13:1472013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hermisson M, Klumpp A, Wick W, Wischhusen
J, Nagel G, Roos W, Kaina B and Weller M: O6-methylguanine DNA
methyltransferase and p53 status predict temozolomide sensitivity
in human malignant glioma cells. J Neurochem. 96:766–776. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishii N, Maier D, Merlo A, Tada M,
Sawamura Y, Diserens AC and Van Meir EG: Frequent co-alterations of
TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human
glioma cell lines. Brain Pathol. 9:469–479. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ostermann S, Csajka C, Buclin T, Leyvraz
S, Lejeune F, Decosterd LA and Stupp R: Plasma and cerebrospinal
fluid population pharmacokinetics of temozolomide in malignant
glioma patients. Clin Cancer Res. 10:3728–3736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patel M, McCully C, Godwin K and Balis FM:
Plasma and cerebrospinal fluid pharmacokinetics of intravenous
temozolomide in non-human primates. J Neurooncol. 61:203–207. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cimmino G, Pepe S, Laus G, Chianese M,
Prece D, Penitente R and Quesada P: Poly(ADPR)polymerase-1
signalling of the DNA damage induced by DNA topoisomerase I poison
in D54(p53wt) and U251(p53mut) glioblastoma cell lines. Pharmacol
Res. 55:49–56. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sabisz M, Wesierska-Gadek J and
Skladanowski A: Increased cytotoxicity of an unusual DNA
topoisomerase II inhibitor compound C-1305 toward HeLa cells with
downregulated PARP-1 activity results from re-activation of the p53
pathway and modulation of mitotic checkpoints. Biochem Pharmacol.
79:1387–1397. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, Stevens MF, Laughton CA,
Madhusudan S and Bradshaw TD: Acquired resistance to temozolomide
in glioma cell lines: Molecular mechanisms and potential
translational applications. Oncology. 78:103–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cieślar-Pobuda A, Saenko Y and
Rzeszowska-Wolny J: PARP-1 inhibition induces a late increase in
the level of reactive oxygen species in cells after ionizing
radiation. Mutat Res. 732:9–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sabbatino F, Fusciello C, Somma D, Pacelli
R, Poudel R, Pepin D, Leonardi A, Carlomagno C, Della Vittoria
Scarpati G, Ferrone S and Pepe S: Effect of p53 activity on the
sensitivity of human glioblastoma cells to PARP-1 inhibitor in
combination with topoisomerase I inhibitor or radiation. Cytometry
A. 85:953–961. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Atkins RJ, Ng W, Stylli SS, Hovens CM and
Kaye AH: Repair mechanisms help glioblastoma resist treatment. J
Clin Neurosci. 22:14–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sandhu SK, Yap TA and de Bono JS:
Poly(ADP-ribose) polymerase inhibitors in cancer treatment: A
clinical perspective. Eur J Cancer. 46:9–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Majuelos-Melguizo J, Rodríguez MI,
López-Jiménez L, Rodríguez-Vargas JM, Martí Martín-Consuegra JM,
Serrano-Sáenz S, Gavard J, de Almodóvar JM and Oliver FJ: PARP
targeting counteracts gliomagenesis through induction of mitotic
catastrophe and aggravation of deficiency in homologous
recombination in PTEN-mutant glioma. Oncotarget. 6:4790–4803. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tentori L, Ricci-Vitiani L, Muzi A,
Ciccarone F, Pelacchi F, Calabrese R, Runci D, Pallini R, Caiafa P
and Graziani G: Pharmacological inhibition of poly(ADP-ribose)
polymerase-1 modulates resistance of human glioblastoma stem cells
to temozolomide. BMC Cancer. 14:1512014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Erice O, Smith MP, White R, Goicoechea I,
Barriuso J, Jones C, Margison GP, Acosta JC, Wellbrock C and
Arozarena I: MGMT expression predicts PARP-mediated resistance to
temozolomide. Mol Cancer Ther. 14:1236–1246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gupta A, Yang Q, Pandita RK, Hunt CR,
Xiang T, Misri S, Zeng S, Pagan J, Jeffery J, Puc J, et al: Cell
cycle checkpoint defects contribute to genomic instability in PTEN
deficient cells independent of DNA DSB repair. Cell Cycle.
8:2198–2210. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Balvers RK, Lamfers ML, Kloezeman JJ,
Kleijn A, Berghauser Pont LM, Dirven CM and Leenstra S: ABT-888
enhances cytotoxic effects of temozolomide independent of MGMT
status in serum free cultured glioma cells. J Transl Med.
13:742015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Godoy PR, Mello SS, Magalhães DA, Donaires
FS, Nicolucci P, Donadi EA, Passos GA and Sakamoto-Hojo ET:
Ionizing radiation-induced gene expression changes in TP53
proficient and deficient glioblastoma cell lines. Mutat Res.
756:46–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zaky A, Busso C, Izumi T, Chattopadhyay R,
Bassiouny A, Mitra S and Bhakat KK: Regulation of the human
AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53
in response to DNA damage. Nucleic Acids Res. 36:1555–1566. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Poletto M, Legrand AJ, Fletcher SC and
Dianov GL: P53 coordinates base excision repair to prevent genomic
instability. Nucleic Acids Res. 44:3165–3175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ströbel T, Madlener S, Tuna S, Vose S,
Lagerweij T, Wurdinger T, Vierlinger K, Wöhrer A, Price BD, Demple
B, et al: Ape1 guides DNA repair pathway choice that is associated
with drug tolerance in glioblastoma. Sci Rep. 7:96742017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ming M and He YY: PTEN in DNA damage
repair. Cancer Lett. 319:125–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mukherjee A and Karmakar P: Attenuation of
PTEN perturbs genomic stability via activation of Akt and
down-regulation of Rad51 in human embryonic kidney cells. Mol
Carcinog. 52:611–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hunt CR, Gupta A, Horikoshi N and Pandita
TK: Does PTEN loss impair DNA double-strand break repair by
homologous recombination? Clin Cancer Res. 18:920–922. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fu D, Calvo JA and Samson LD: Balancing
repair and tolerance of DNA damage caused by alkylating agents. Nat
Rev Cancer. 12:104–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schuhwerk H, Atteya R, Siniuk K and Wang
ZQ: PARPing for balance in the homeostasis of
poly(ADP-ribosyl)ation. Semin Cell Dev Biol. 63:81–91. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chai KM, Wang CY, Liaw HJ, Fang KM, Yang
CS and Tzeng SF: Downregulation of BRCA1-BRCA2-containing complex
subunit 3 sensitizes glioma cells to temozolomide. Oncotarget.
5:10901–10915. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ray Chaudhuri A and Nussenzweig A: The
multifaceted roles of PARP1 in DNA repair and chromatin remodelling
(Review). Nat Rev Mol Cell Biol. 18:610–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Polo SE and Jackson SP: Dynamics of DNA
damage response proteins at DNA breaks: A focus on protein
modifications. Genes Dev. 25:409–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nagel ZD, Kitange GJ, Gupta SK, Joughin
BA, Chaim IA, Mazzucato P, Lauffenburger DA, Sarkaria JN and Samson
LD: DNA repair capacity in multiple pathways predicts
chemoresistance in glioblastoma multiforme. Cancer Res. 77:198–206.
2017. View Article : Google Scholar : PubMed/NCBI
|