Introduction
Among adults, renal cell carcinoma (RCC) is the most
common type of renal tumor (1,2). There
are >10,000 deaths and 220,000 new cases diagnosed with RCC each
year worldwide (3,4). The number of deaths and diagnosed
cases accounts for ~40% oc cancers in China (5,6). Data
from the National Health Commission of the PRC suggested that RCC
incidence is increasing, with a 95% increase in growth rate over
the past 28 years (1990–2018) (7).
To date, RCC has ranked in the top three malignant types of tumors
in males (8). Recently, due to
increased incorrect dietary habits, such as high salt diet,
unfiltered water drinking and sugary drink consumption, accompanied
by increasingly advanced cancer diagnosis, the mortality of RCC has
markedly increased in China (9).
However, the 5-year survival rate is low, at 25–35% (10). Since the majority of patients are
diagnosed at middle or late stages with local or remote invasion,
the combination treatment of chemotherapy and molecular-targeted
therapy are commonly utilized (11,12).
Thus, investigating novel target molecules that serve a critical
role in tumor progression and studying their molecular mechanism
will aid in identifying novel targets for anti-RCC treatment.
Additionally, there is an urgent need to further research the
development of RCC and find more effective therapeutic methods for
patients with RCC.
Bromodomain and extraterminal domain (BET) proteins
serve a role as essential epigenome regulators for gene
transcription mediation (13,14).
Bromodomain-containing protein (BRD)2, BRD3, BRD4 and bromodomain
testis-specific protein (BRDT) are members of this family protein
(15). BRDT has been well studied,
and is involved in signaling transduction, gene transcription and
tumor progression (16,17). BRDT was reported to recognize
acetylated histones and recruit transcriptional complexes, such as
elongation factors, and other regulators to facilitate
transcription progression (18,19).
However, an increasing amount of evidence indicated that BRDT could
activate gene transcription during various diseases, such as
testicular pathologies and malignant tumors (16,20,21).
In addition, BET was demonstrated as an effective target for
lymphoma, multiple myeloma and acute myeloid leukemia, as well as
in solid tumors including lung cancer, liver cancer and ovarian
cancer, since c-Myc, Fos-related antigen 1 (FOSL1) and IL-7R
oncogenes have been reported to be act BET targets (22,23).
PLX51107, INCB054329 and I-BET151, which are currently developed
BET target treatments, were reported to be effective for cancer
therapy in clinical trials (24–29).
However, the effect and molecular mechanism of BET suppression, and
the association between BET and eIF4EBP1 have not yet been fully
elucidated in RCC. Therefore, identifying the downstream signals
that regulates its functions in RCC is urgently required, which
would provide basic clinical knowledge for generating targeted
medicine in RCC.
Eukaryotic translation initiation factor 4E binding
protein 1 (eIF4EBP1) directly interacts with eukaryotic translation
initiation factor 4E (eIF4E), which functions as an essential
limiting component to recruit 40S ribosomal subunits to the 5′ end
of mRNAs (30–32). Meanwhile, the association between
proteins and eIF4E suppresses complex assembly and inhibits gene
translation (33). eIF4EBP1 is
phosphorylated in response to UV irradiation, insulin signaling and
virus infection, and diseases including malignant tumors, leading
to the disruption of eIF4E mRNA translation (34,35).
Previous studies demonstrated that eIF4EBP1 acts as a biomarker in
ovarian cancer and hepatocellular carcinoma (36,37).
eIF4EBP1 facilitates tumorigenesis, invasion, reappearance and
multidrug resistance in various cell lines and mouse experiments
(38–40). MicroRNA-125a and microRNA-125b
inhibits ovarian cancer via repressing transcription of eIF4EBP1
(41). However, the advanced
position regulators that support eIF4EBP1 expression or eIF4EBP1
partners are limited. Thus, the present study hypothesized that
BRDT may act as a partner of eIF4EBP1, and thus, regulate eIF4EBP1
transcription.
Investigating new interaction partners of eIF4EBP1
indicates a candidate method for identifying novel drug targets for
RCC. In the present study, BRDT was identified as a new interaction
protein of eIF4EBP1in RCC cells. Furthermore, it was suggested that
BRDT could regulate eIF4EBP1 levels via interacting with its
promoter sequence, and BRDT inhibitors could regulate eIF4EBP1
transcription as well as RCC progression. Overall, the results of
the present study revealed that BRDT and eIF4EBP1 may function as a
potentially novel therapeutic targets and biomarkers.
Materials and methods
Reagents
The BRDT inhibitors PLX51107 (cat. no. S8739) and
INCB054329 (cat. no. S8753) were purchased from Selleck Chemicals.
Both were dissolved in DMSO at 10 mmol/l, stored at −80°C and
diluted in DMSO just before use. Exfect transfection reagent (cat.
no. T202) was purchased from Vazyme Biotech Co., Ltd. Polyclonal
antibody against BRDT (cat. no. NBP1-88357) was purchased from
Novus Biologicals, Ltd. c-Myc (D3N8F; cat. no. 13987) and eIF4EBP1
(53H11; cat. no. 9644) rabbit monoclonal antibodies were both
purchased from Cell Signaling Technology, Inc. Rabbit polyclonal
antibody against β-actin (cat. no. 0407-3) was purchased from
Huanan Biotechnology. DMSO was purchased from Sigma-Aldrich; Merck
KGaA. NP40 lysates and western blotting reagents were all purchased
from Beyotime Institute of Biotechnology.
Cell lines and cell treatment
293T cells and human RCC cell lines ACHN, 769-P and
768-O were all obtained from American Type Culture Collection. All
cells were maintained in DMEM (cat. no. 11995; Beijing Solarbio
Science & Technology Co., Ltd.) supplemented with 10% FBS (cat.
no. 10099-141; Gibco; Thermo Fisher Scientific Inc.) and 0.1 mg/m
penicillin-streptomycin solution at 37°C in a humidified atmosphere
containing 5% CO2.
DNA plasmid construction
The eIF4EBP1 or BRDT open reading frame (ORF) was
amplified by PCR derived from ACHN cells and inserted into a
pcDNA3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.). The
promoter region from positions −1,100 to +80 of eIF4EBP1 was
amplified and inserted into a pGL3.0-basic vector (Promega
Corporation) to generate the pGL3-eIF4EBP1 promoter plasmid.
Sulforhodamine B (SRB) assays
All cells were cultured in 96-well plates at a
density of 3×103 cells/well. Cells were then treated
with different concentrations (0.5, 1, 2, 4, 8, 10 µmol/l) of PLX
for 48 h. Cells transfected with small interfering (si) RNAs
targeting eIF4EBP1 or control siRNAs (siCon) for 36 h were
re-seeded in 96-well plates at 2×103 cells/well and
further cultured for 120 h, or re-seeded to 96-well plates at
3×103 cells/well and treated with reagents at 48 h for a
further 72 h. An SRB assay was used to count the cell number. Cells
were seeded into six-well plates and transiently transfected with
eIF4EBP1 siRNAs and control siRNAs for 48 h. The cells were then
re-seeded into 96-well plates for another 5 days and subjected to
the SRB assay. The proliferation rate of cells was assessed based
on previously reported methods (20). The IC50 was determined
using GraphPad Prism (version 5.01; GraphPad Software, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from cells or tumors tissues was extracted
following the manufacturers' protocols of the RNA extraction kit
(cat. no. P1023; Beijing Solarbio Science & Technology Co.,
Ltd.). RT of RNA and qPCR were performed using the High-Capacity
RNA-to-cDNA™ kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. All qPCR
experiments were performed in triplicate using the ChamQ SYBR Color
qPCR Master Mix (Vazyme Biotech Co., LTd.) on an ABI Prism 7500
sequence detection system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The fold-change of target gene transcription was
determined using the 2−ΔΔCq method (42). The following thermocycling
conditions were used for the qPCR: Initial denaturation at 95°C for
5 min; 40 cycles of 95°C for 10 sec and 60°C for 30 sec; and a
final step at 95°C for 15 sec, 60°C for 1 min and 95°C for 15 sec.
The following primer pairs were used for the qPCR: BRDT (GenBank
accession. no. NM-207189) forward, 5′-ATCAGACCACACCTTCACATG-3′ and
reverse, 5′-AGAGCTGTTCAGAATCACCTG-3′; eIF4EBP1 (GenBank accession.
no. NM-004095.4) forward, 5′-ACCTGTGACCAAAACACCC-3′ and reverse,
5′-TCAAACTGTGACTCTTCACCG-3′; c-myc (GenBank accession. no.
NM_001354870) forward, 5′-CCAAGCTCGTCTCAGAGAAG-3′ and reverse,
5′-GATGCACTCTGAGGCGGCGG-3′ and GAPDH (GenBank accession. no.
NM-002046.7) forward, 5′-AATCCCATCACCATCTTCCAG-3′ and reverse,
5′-AAATGAGCCCCAGCCTTC-3′. Primers were synthesized by Sangon
Biotech Co., Ltd.
Immunoprecipitation (IP) and mass
spectrophotometry (MS)
To identify new binding partners of eIF4EBP1, ACHN
cells were cultured in two 5-cm dishes and transfected with 6 µg
eIF4EBP1 plasmids and control FLAG plasmids (cat. no. 635688,
Clontech Laboratories, Inc.) using Exfect transfection reagent
according to the manufacturer's instructions. After 48 h,
transfected cells were collected for lysis with NP40 buffer at 4°C
for 2–4 h. Total protein was determined using a bicinchoninic acid
(BCA) Protein Determination kit (Beyotime Institute of
Biotechonology). Proteins were centrifuged at 12,000 × g at 4°C for
10 min for the subsequent IP assay. A total of 1 µg eIF4EBP1
antibody (cat. no. 9644; Cell Signaling Technology, Inc.) or FLAG
antibody (cat. no. 14793; Cell Signaling Technology, Inc.) per 100
µg total protein was used for IP for at least 2 h with protein A/G
beads (Santa Cruz Biotechnology, Inc.) at 4°C. Finally, beads were
collected via centrifugation at 2,000 g at 4°C for 5 min and washed
three to five times using NP40 buffer. Immunoprecipitated proteins
were analyzed by MS (Table I).
 | Table I.List of immunoprecipitated
proteins. |
Table I.
List of immunoprecipitated
proteins.
| IP: Anti-FLAG | IP:
Anti-eIF4EBP1 |
|---|
|
|
|---|
| Accession no. | Name | Accession no. | Name |
|---|
| NP-001017963.2 | Heat shock protein
90 α family class member 1 | NP-001014431 | AKT
serine/threonine kinase 1 |
| NP-002145.3 | Heat shock 70 kDa
protein 4 | NP-001158095.1 | Argonaute RISC
catalytic component 2 |
| NP-001092.1 | Actin β (ACTB) | NP-997072.2 | Bromodomain testis
associated protein |
| NP-003371.2 | Vimentin (Homo
sapiens) | NP-004086.1 | Eukaryotic
translation initiation factor 4E binding protein 1 |
| AAA91576.1 | α-tubulin | NP-001017963.2 | Heat shock protein
90 α family class member 1 |
| NP-002037.2 |
Glyceraldehyde-3-phosphate
dehydrogenase | NP-000042.3 | ATM
serine/threonine kinase |
Western blotting
Protein samples were prepared as described above.
Protein concentrations were determined using a BCA Protein
Determination kit. Equivalent amounts (100 µg) of cell lysates were
subjected to 15% SDS-PAGE and then transferred to PVDF membranes
(Beijing Solarbio Science & Technology Co., Ltd.). Following
blocking with 5% bovine serum albumin (cat. no. A602449; Sangon
Biotech Co., Ltd.) in phosphate-buffered saline (PBS) containing
0.1% Tween-20 (PBS-T) for 30 min at 25°C, the membranes were
incubated with primary antibodies (1:1,000) for 12 h at 4°C with
gentle agitation. This was followed by incubation with
HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG
(1:10,000; cat. nos. K0038M-HRP and SE134; Beijing Solarbio Science
& Technology Co., Ltd.) at room temperature for 45 min.
Finally, chemiluminescent signals were scanned and analyzed using
Amersham Imager 600 (GE Healthcare).
siRNA transfection
To efficiently knock down BRDT and eIF4EBP1, the
present study designed two BRDT siRNA
(5′-UCTGCCAAGUCGACAAACAGCUAUU-3′ and
5′-GCCAAGUCGACAAACAGCUAUUAUU-3′), two eIF4EBP1 siRNA
(5′-GAGTCACAGTTTGAGATGGACATTT-3′ and
5′-AGTCACAGTTTGAGATGGACATTTA-3) and a luciferase siRNA control
sequence (5′-CAGGCCATGACAATGCTCCTGACAA-3′) (all synthesized by
Genepharm, Inc). A total of 100 nmol/l siRNAs were transfected into
ACHN, 769-P or 768-O cells for 48 h using Exfect transfection
reagent.
Luciferase reporter assay
ACHN, 769-P and 768-O cells were cultured in 24-well
plates and transfected with pGL3-eIF4EBP1 and pRL-TK (Promega
Corporation) using Exfect transfection reagents for 24 h, and then
exposed to 8 µmol/l BET inhibitors for another 24 h. The cells were
subsequently collected for analysis according to the instructions
of the Dual Luciferase Reporter Assay system (cat. no. E1910;
Promega Corporation). The Renilla vector pRL-TK was used to
normalize the transfection efficiency.
Chromatin IP (ChIP) assay
All cells (ACHN, 769-P and 768-O) were cultured in
5-cm dishes and exposed to 8 µmol/l BET inhibitors for 24 h, then
collected for the ChIP assay according to the instructions of the
ChIP assay kit (cat. no. P2078; Beyotime Institute of
Biotechnology). Briefly, in order to crosslink proteins to DNA,
whole cells were first fixed with 2% formaldehyde for 5 min at room
temperature. Next, sonication was performed to cut the cross-linked
DNA for 1 h at 4°C five times on the Qsonica Q800R3 sonicator
(Aoran Biotechnology Co., Ltd.). For the control, 1/10 volume of
total samples were used. eIF4EBP1 antibody or IgG antibody (cat.
no. 0806-1; Hangzhou Huaan Biotechnology Co., Ltd.) was used for IP
in samples. The immunoprecipitated complexes were washed three to
five times with NP40 buffer and subjected to RT-qPCR.
Xenograft nude mouse model and
treatments
The Institutional Animal Care and Use Committee of
Sun Yat-sen University approved all animal experiments. The Model
Animal Research Center of Sun Yat-sen University provided 18
6-week-old female athymic (nu/nu) mice (weight, ~18 g). All mice
were raised in specific pathogen-free conditions and had ad
libitum access to food and water in 40–60% humidity and 10/14-h
light/dark cycle at 27°C. ACHN cells were cultured at a density of
5×105 cells/ml in serum-free DMEM. Once tumors reached a
size of ~100 mm3, all the 18 mice were randomized into
three groups (n=6 in each group) and treated with BET inhibitors
(100 mg/kg/day) via oral gavage. Tumor volumes were measured every
2 days. The mice were sacrificed by injection with 150 mg/kg
barbiturate at the end of the 14-day treatment or when the tumor
diameter nearly reached 2 cm. Respiratory arrest was validated
using a ventilator and absence of heartbeat was determined by an
electrocardiographic system to confirm death after euthanasia.
After euthanizing, the tumors were removed and weighed,
homogenized, and subjected to RT-qPCR and western blot
analysis.
Statistical analysis
All results were collected from at least three
independent repeats. Data were analyzed using SPSS 19.0 (IBM Corp.)
and presented as the mean ± SD. Statistical significance between
datasets were analyzed by one or two-way ANOVA analysis followed by
Bonferroni's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
BRDT interacts with eIF4EBP1
The present study aimed to identify new interaction
proteins of eIF4EBP1, thereby enabling further understanding the
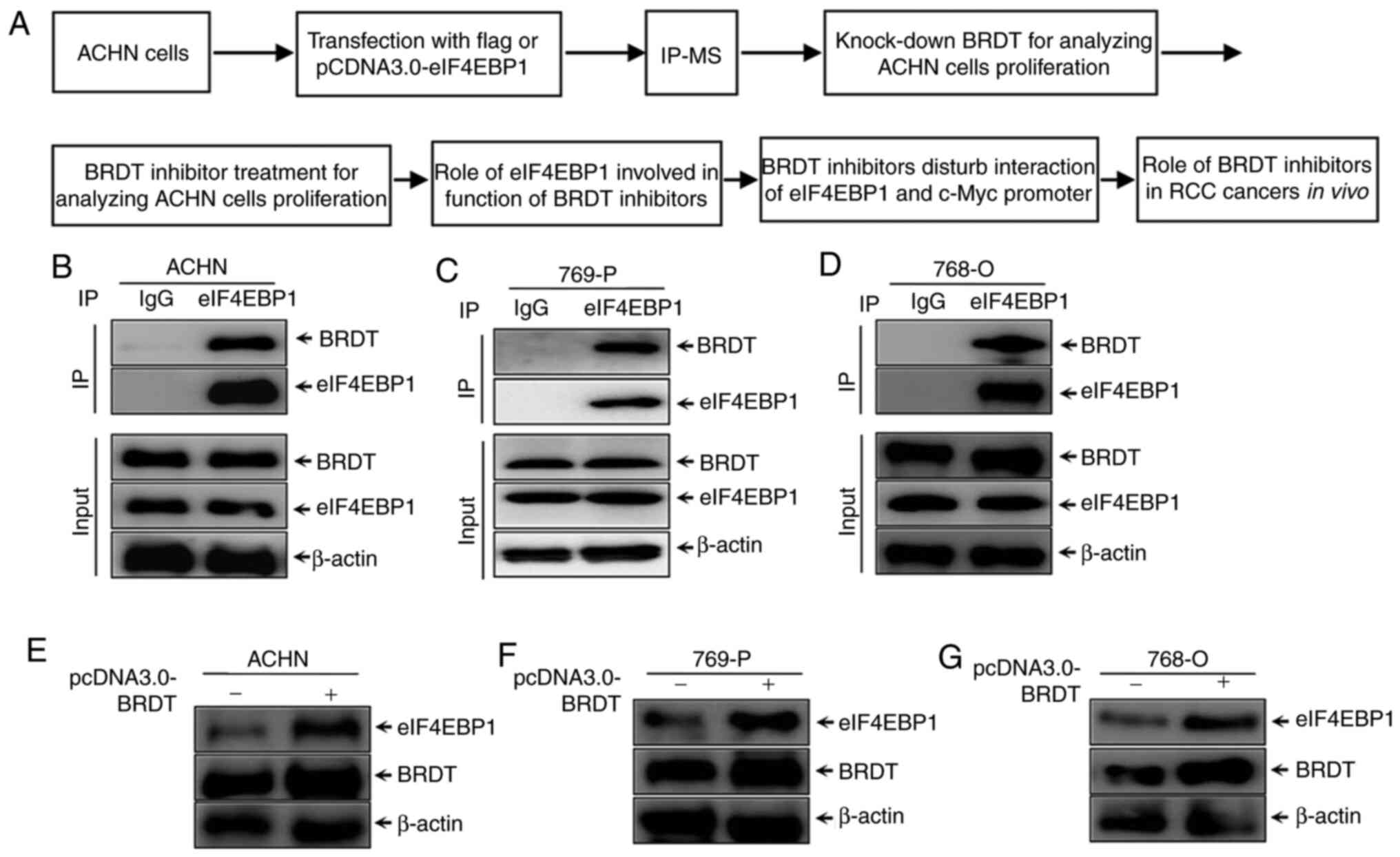
regulatory mechanisms of eIF4EBP1. The diagram of this study
process is shown in Fig. 1A. ACHN
cells were transfected with pCDNA3.0-eIF4EBP1 for IP-MS to identify
interaction partners of eIF4EBP1. Next, the function of BRDT
involved in ACHN cell proliferation was further analyzed. Finally,
the molecular mechanism of BRDT inhibitors regulated by eIF4EBP1
and the role of BRDT inhibitors in RCC cancer growth were further
analyzed. An IP assay of eIF4EBP1 in RCC cell lines was performed
in the present study. ACHN cells were transfected with eIF4EBP1
plasmid for 48 h. Proteins that were precipitated by eIF4EBP1 were
identified by MS (performed by Applied Protein Technology,
Shanghai, China). Meanwhile, samples were immunoprecipitated with
FLAG antibody as the control. The indicated identified proteins and
negative control proteins are listed in Table I. To further confirm the mass
spectrometry results and the association between eIF4EBP1 and BRDT,
co-IP was re-performed with endogenous eIF4EBP1 in ACHN cells and
769-P and 768-O whole cell lysates. IgG antibody was used as the
negative control. The results, as presented in Fig. 1B-D, demonstrated that BRDT interacts
with eIF4EBP1 endogenously in RCC cells. To further investigate the
effect of BRDT on eIF4EBP1 expression, western blotting was used to
analyze the protein levels of eIF4EBP1 following transfection of
BRDT in RCC cell lines. The results presented in Fig. 1D-F suggested that eIF4EBP1
expression was markedly increased by ectopic expression of BRDT in
RCC cell lines. Taken together, these results suggested that BRDT
was identified as a novel interaction partner of eIF4EBP1 and
increased eIF4EBP1 expression in RCC cell lines.
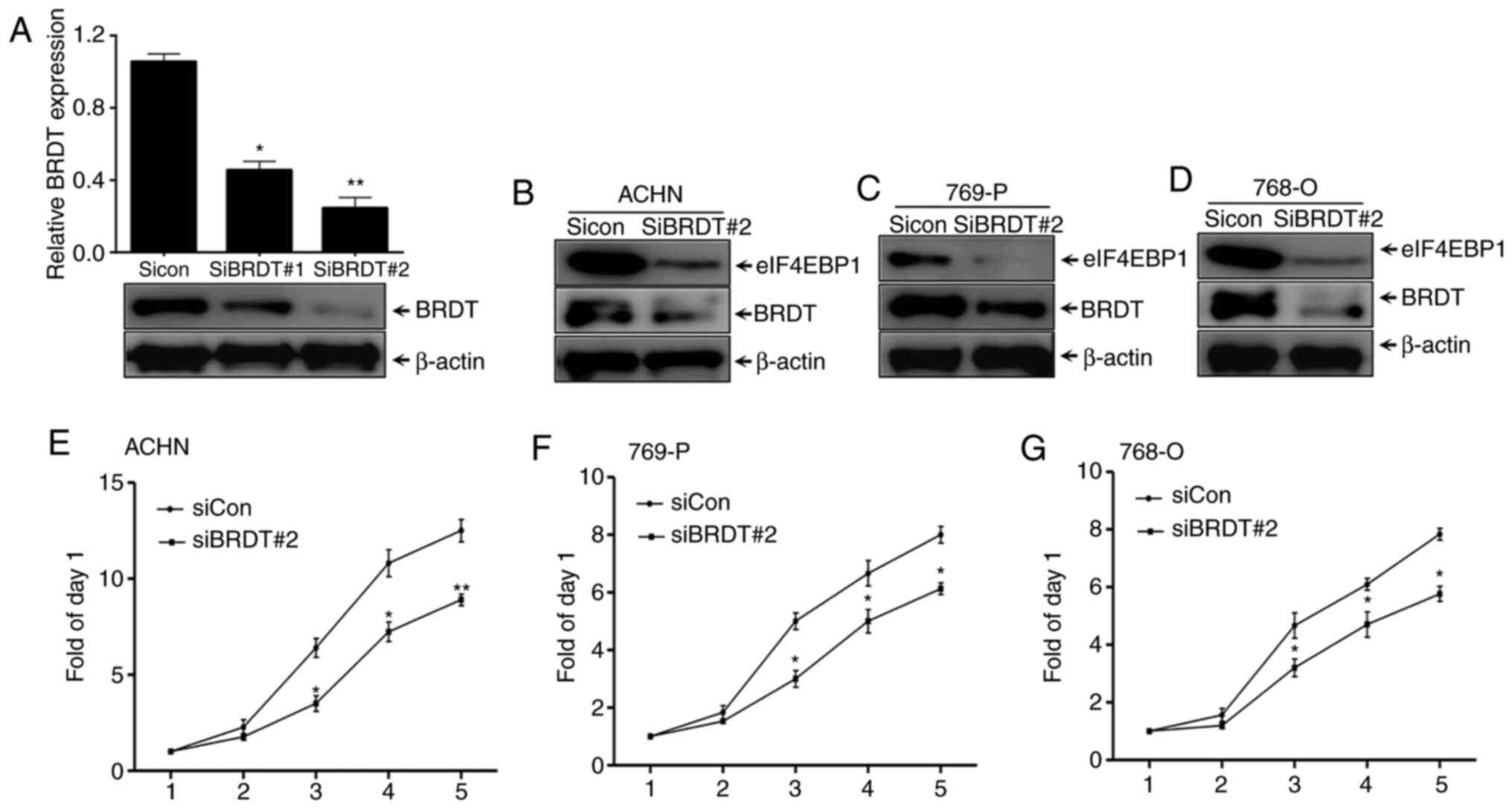
Silencing BRDT, together with
suppressed eIF4EBP1 expression, inhibits RCC proliferation
To elucidate the mechanism underlying the effects of
BRDT on eIF4EBP1 expression, the present study first assessed the
regulation of eIF4EBP1 by interfering with BRDT expression. Two
siRNAs that targeted different regions of BDRT were used, which,
along with the control siRNA, were individually transfected into
ACHN cells for 48 h. Whole cell lysates were collected for western
blotting. It was revealed that the interference efficiency of
siRNA#2 targeting BRDT was nearly 80% (Fig. 2A), indicating that BRDT was
successfully silenced. The present study also revealed that
eIF4EBP1 protein expression levels were decreased by BRDT knockdown
in the RCC cell lines, ACHN, 769-P and 768-O (Fig. 2B-D). In addition, SRB assays
suggested that knockdown of BRDT expression significantly blocked
the proliferation of ACHN, 769-P and 768-O cells compared with the
siRNA control group (Fig. 2E-G).
Overall, these data suggested that inhibition of eIF4EBP1
expression may the due to knocking down BRDT.
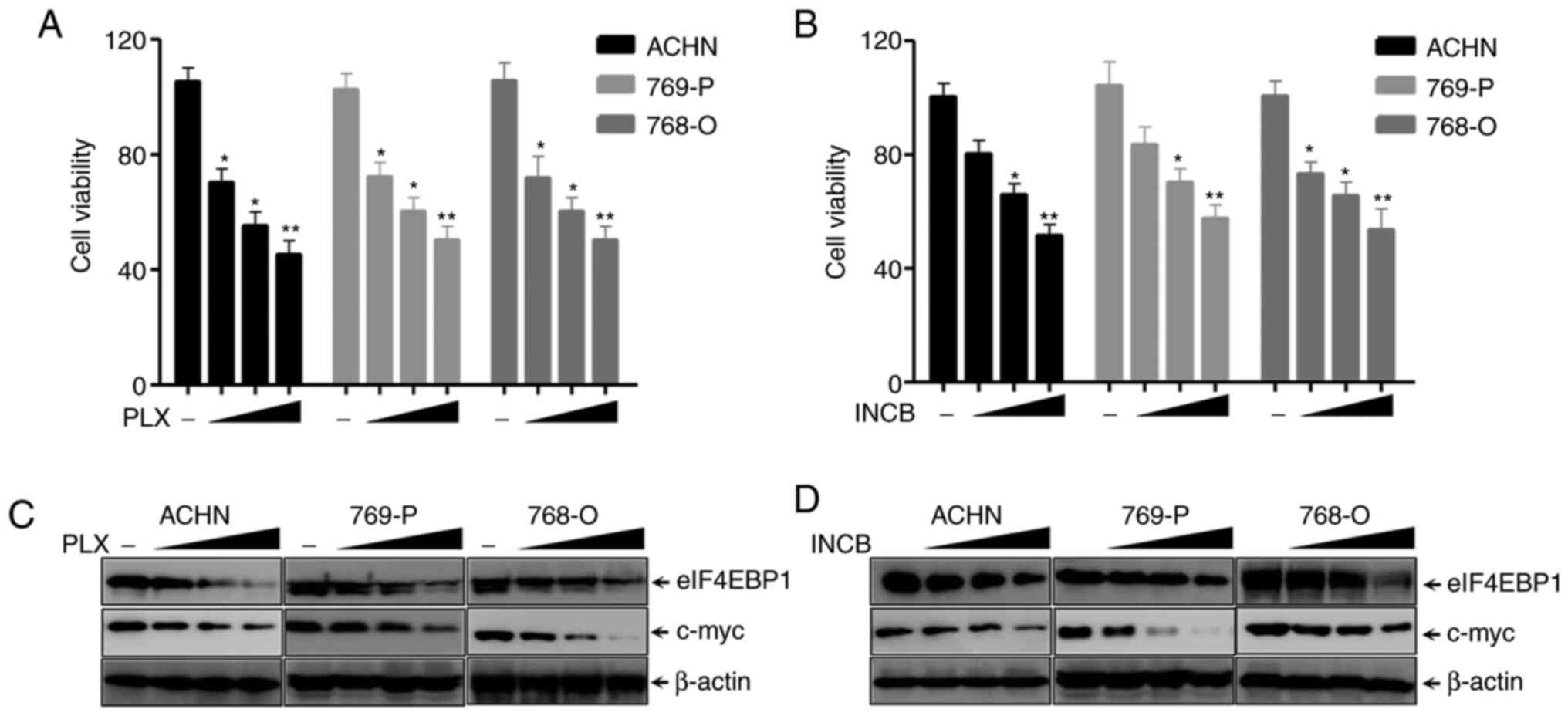
BRDT inhibitors suppress eIF4EBP1
expression and RCC proliferation
BRDT inhibitors, including PLX51107 and INCB054329,
have been demonstrated to suppress the progression of lung cancer
cells, bladder cancer cells and intestinal cancer cells (24,25).
The present study first analyzed the function of PLX51107 and
INCB054329 on various RCC cell lines. SRB assays indicated that
PLX51107 significantly inhibited the proliferation of ACHN, 769-P
and 768-O cells in a dose-dependent manner (Fig. 3A). INCB054329 also exhibited similar
results (Fig. 3B). The c-myc gene
is a downstream target of PLX51107 and INCB054329 (43–45).
The results in Fig. 3C and D show
that the BET inhibitors PLX51107 and INCB054329, markedly decreased
c-myc and eIF4EBP1 expression in a dose-dependent manner in RCC
cell lines, ACHN, 769-P and 768-O cells, which demonstrated that
eIF4EBP1 expression was significantly decreased by BRDT inhibitors
PLX51107 and INCB054329. Altogether, these results suggested that
RCC proliferation and eIF4EBP1 expression were inhibited by BET
inhibitors.
eIF4EBP1 affects the inhibitory
function of PLX51107 in RCC
The role of eIF4EBP1 in affecting the growth
inhibitory function of PLX51107 and INCB054329 in RCC cell lines,
ACHN, 769-P and 768-O cells, was further evaluated. Since the
effect of PLX51107 and INCB054329 was similar, PLX51107 was
selected for these experiments. The three cell lines were
transiently transfected with recombinant plasmids containing full
length ORFs for eIF4EBP1 or control empty vector for 48 h. The
cells were lysed with NP40 buffer and subsequently separated via
SDS-PAGE for western blotting with the indicated antibodies.
eIF4EBP1 protein levels were markedly increased in
pcDNA3.0-eIF4EBP1 groups compared with the empty vector groups,
indicating that eIF4EBP1 was successfully expressed in the three
cell lines (Fig. 4A). In addition,
SRB assay results showed that overexpression of eIF4EBP1
significantly partially attenuated the growth inhibition of
PLX51107 in the three cell lines compared with the empty vector
groups.
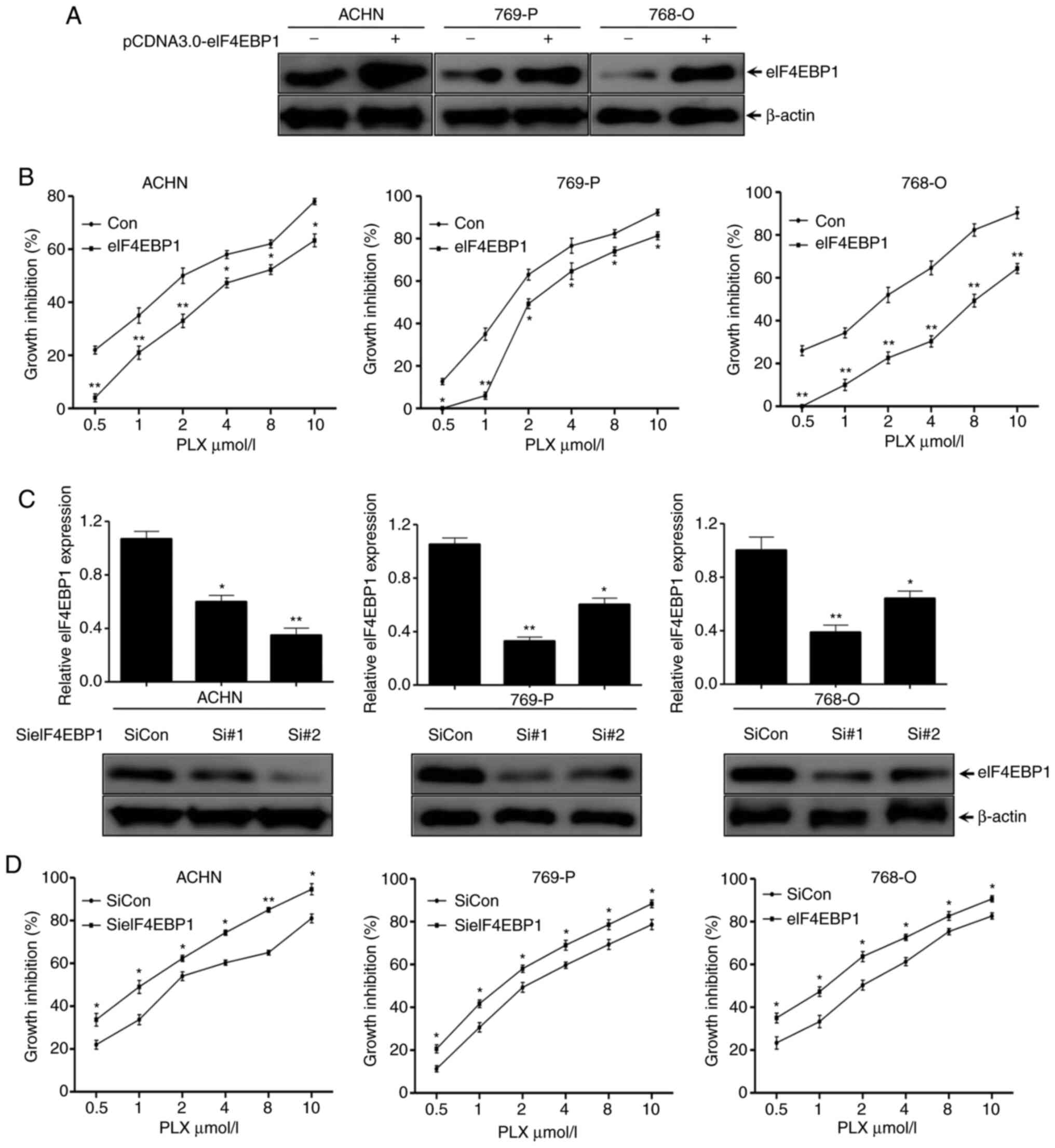 | Figure 4.eIF4EBP1 affects the inhibitory
function of PLX51107 in renal cell carcinoma. (A) ACHN, 769-P and
768-O cells were transiently transfected with pCDNA3.1-eIF4EBP1
plasmids or empty control plasmids for 48 h using Exfect
transfection reagent, and then subjected to western blotting using
the indicated antibodies. (B) The three cell lines were transfected
with the indicated plasmids as aforementioned for 24 h and then
re-cultured in 96-well plates, treated with different
concentrations of PLX51107 as indicated for another 3 days, and
subjected to an SRB assay. (C) The three cell lines were
transfected with two different siRNAs targeting two regions of
eIF4EBP1 and the control siRNAs for 48 h, and then subjected to
western blotting with the indicated antibodies. (D) The three cell
lines transfected with the indicated siRNAs of eIF4EBP1 or the
control siRNAs for 24 h were re-seeded to 96-well plates and
treated with PLX at the indicated concentrations, and then
subjected to an SRB assay. Data are representative of three
independent experiments. Bars represent the standard deviation.
*P<0.05 and **P<0.01 vs. control groups. SRB, sulforhodamine
B; siRNA, small interfering RNA; PLX, PLX51107; siCon, control
siRNA. |
To further confirm the effects of eIF4EBP1 on
regulating PLX5114 inhibitory function, two siRNAs targeting
different regions of eIF4EBP1 and control siRNA were designed. The
three cell lines were transfected with the aforementioned siRNAs
for 48 h, and then subjected to western blotting with the indicated
antibodies. The results revealed that eIF4EBP1 protein levels
significantly decreased up to 70% with the different siRNAs in
different cell lines, which was in contrast to the control siRNAs,
suggesting an efficient knockdown (Fig.
4C). Furthermore, the SRB assay suggested that the suppression
effects of PLX51107 on the growth of the cell lines increased in
eIF4EBP1-silenced cells in a dose-dependent manner (Fig. 4D). Overall, these results
demonstrated that PLX51107 suppressed the proliferation of RCC
cells via inhibition of eIF4EBP1 expression, and eIF4EBP1 affected
the inhibition of PLX51107 on RCC cells.
BRDT inhibitors decrease the
interaction between eIF4EBP1 and c-myc promoter
Due to decreased regulation of eIF4EBP1 expression
assuming an essential role in mediating the growth inhibitory
effect of PLX51107, it was hypothesized that eIF4EBP1 directly
affects c-myc expression in RCC cells. The present study first
analyzed the mRNA transcription levels of c-myc mediated by BRDT
inhibitors, PLX51107 and INCB054329. As shown in Fig. 5A and B, the c-myc mRNA transcription
levels significantly decreased upon treatment with PLX51107 and
INCB054329 at 6 and 18 h in the three cell lines. Meanwhile, the
RT-qPCR assay revealed that silencing BRDT expression also
significantly decreased c-myc mRNA transcription levels compared
with control siRNA groups (Fig.
5C), which is consistent with the results of si-eIF4EBP1
transfection (Fig. 5C). Next, the
present study assessed the promoter activity of c-myc using
dual-luciferase reporter assays. The pGL3-c-myc promoter plasmid
and the control vector pGL3.0-basic were transfected into the three
cells for 36 h, and then treated with PLX51107 and INCB054329 for
another 24 h. The Renilla plasmid, used to normalize the
transfection efficiency, was also co-transfected to the cells. As
presented in Fig. 5D, treatment
with the BRDT inhibitors, PLX51107 and INCB054329, could
significantly decrease c-myc promoter activity in the three cell
lines compared with controls, indicating that BET inhibitors
suppressed c-myc transcription. In conclusion, these data
demonstrated that BET inhibitors decreased c-myc transcription via
inhibition of BRDT and eIF4EBP1.
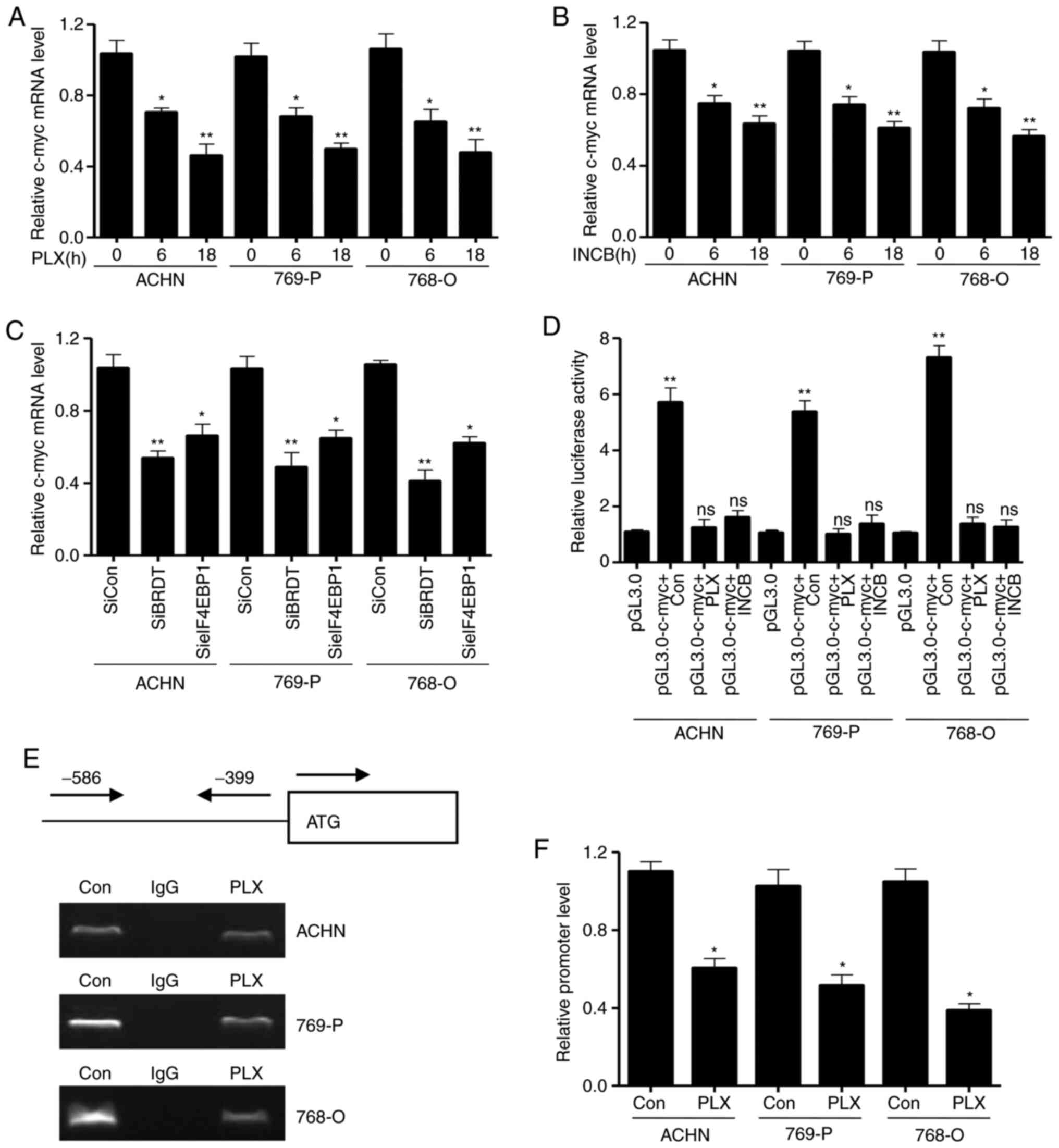 | Figure 5.Bromodomain and extraterminal domain
inhibitors decrease eIF4EBP1 associated with c-myc promoter. The
three cell lines were treated with the indicated concentrations of
(A) PLX51107 or (B) INCB054329 for different time periods (0, 6 and
18 h) and subjected to RT-qPCR. (C) The three cell lines were
transiently transfected with the indicated siRNAs or control siRNAs
for 24 h. Total RNA was extracted and subjected to RT-qPCR assay
for detecting the c-myc mRNA transcription level. (D) The three
cell lines were transiently transfected with c-myc promoter plasmid
or the control vector using Exfect reagent and treated with the
indicated concentrations of PLX51107 or INCB054329 for 36 h.
Renilla plasmids were co-transfected as the loading control.
The cells were lysed and subjected to dual luciferase reporter
assays. The three cell lines were treated with PLX51107 for 36 h,
and then subjected to chromatin immunoprecipitation using eIF4EBP1
antibody. The c-myc promoter was detected using primers designed
from −586 to −399 pre-leading regions of the start site (0 is the
ATG start site). The PCR products of c-myc promoter was analyzed by
(E) and agarose gel electrophoresis and (F) qPCR. Data are
representative of three independent experiments. Bars represent the
standard deviation. *P<0.05 and **P<0.01. RT-qPCR, reverse
transcription-quantitative PCR; PLX, PLX51107; INCB, INCB054329;
Con, control; siRNA, small interfering RNA; siCon, control siRNA;
BRDT, bromodomain testis-specific protein; eIF4EBP1, eukaryotic
translation initiation factor 4E-binding protein 1. |
To investigate the molecular mechanism underlying
how BET inhibitors inhibit c-myc promoter activity, a ChIP assay
using eIF4EBP1 antibody in the three cell lines was performed. The
three cell lines were treated with PLX51107 for 24 h. Whole cell
lysates were used for the CHIP assay. The results in Fig. 5E and F showed that the PCR products
of c-myc promoter were significantly decreased in the eIF4EBP1
antibody IP group compared with the control IgG group, implying
that PLX51107 decreased the interaction of c-myc promoter with
eIF4EBP1. These results suggested that PLX51107 inhibited eIF4EBP1
protein levels and the association of eIF4EBP1 with c-myc promoter,
and inhibited subsequent c-myc transcription.
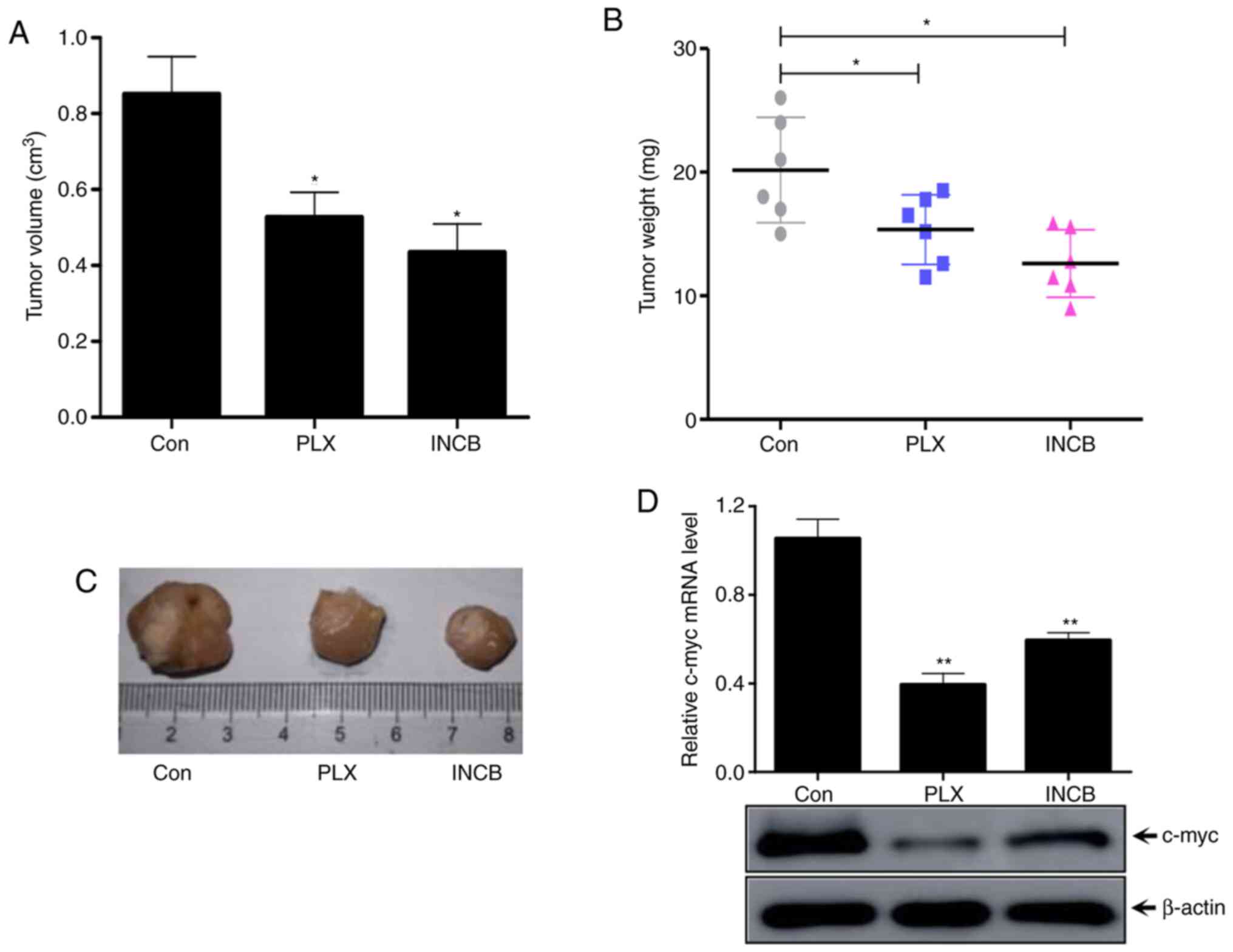
BET inhibitors suppresses ANCH tumor
progression and c-myc protein levels in vivo
To further verify the underlying mechanism and
function of BET inhibitors in RCC tumors and c-myc regulation, an
ACHN xenograft nude mouse model in vivo experiment was
established. PLX51107 (oral gavage, 100 mg/kg/day) and INCB054329
(oral gavage, 90 mg/kg/day) and negative control (oral gavage with
physiological saline) were used to treat mice for 14 days (n=6 in
each group) (24). The tumor volume
was significantly lower in both PLX51107- and INCB054329-treated
groups compared with the control group (Fig. 6A). Meanwhile, the tumor weight
significantly decreased in both PLX51107 and INCB054329 groups
compared with the control group (Fig.
6B). The images of the tumors are presented in Fig. 6C. In addition, the transcription and
protein levels of c-myc were determined by RT-qPCR and western
blotting. The results in Fig. 6C
showed that the mRNA transcription and protein levels of c-myc
in vivo tumors treated with BET inhibitors were
significantly lower compared with the control group tumors
(Fig. 6D). Taken together, these
results suggested that treatment with BRDT inhibitors PLX51107 and
INCB054329 could inhibit RCC tumor progression and decrease c-myc
expression in vivo.
Discussion
The present study revealed several results with
regard to BRDT inhibition in the progression of RCC: i)
BRDT-specific inhibitors, PLX51107 and INCB054329, inhibited the
proliferation of various RCC cell lines; ii) BRDT was identified as
a novel interaction partner of eIF4EBP1; iii) Expression of
eIF4EBP1 was directly affected by BRDT; iv) BRDT regulated the
binding of eIF4EBP1 with c-myc promoter; v) Knockdown of BRDT
protein inhibited RCC cell proliferation and vi) BRDT inhibitors
suppressed ACHN tumors growth in vivo. Targeting BET
proteins was demonstrated to inhibit tumor growth in various types
of cancer, including multiple myeloma, lymphoma, ovarian cancer,
bladder cancer, castration-resistant prostate cancer and lung
cancer (46,47). BRD4, a family of BET proteins, was
reported to function as a tumor suppressor in colon cancer and lung
cancer (48). To the best of our
knowledge, there have been no reports on the role of the BET family
in RCC cell proliferation. The present study revealed that three
RCC cell lines were sensitive to the BRDT inhibitors PLX51107 and
INCB054329.
PLX51107 and INCB054329 are small molecular
inhibitors that specifically target BRDT. PLX51107 and INCB054329
interact with the domain of BRDT that recognizes acetylated lysine
and disassociates from chromatin, thereby blocking gene
transcription (24,25), which suggests that PLX51107 and
INCB054329 may disrupt the interaction of BRDT with other
regulators to block gene transcription during chromatin remodeling.
Hence, PLX51107 and INCB054329 is thought to inhibit tumor
progression during specific conditions (49–51).
Suppression of c-Myc transcription was hypothesized to serve an
important role in BRD4-induced suppression of tumor growth in
malignant hematological diseases and solid tumors (52–54).
In lung cancer, the BET inhibitor JQ1 demonstrated antitumor
functions not only via suppression of c-myc, but also via
downregulation of FOSL1 (55). The
present study first identified that eIF4EBP1 acted as a novel
binding partner with BRDT, and it was also revealed that c-myc and
eIF4EBP1 expression was decreased by BRDT inhibitors or BRDT
knockdown. These results provided an additional mechanism for the
effects of BRDT in RCC.
In the present study, eIF4EBP1 was identified as a
novel binding partner of BRDT and c-myc was a target of eIF4EBP1.
The mRNA transcription and protein expression levels of c-myc were
inhibited by BRDT inhibitors, PLX51107 and INCB054329, or when BRDT
was silenced in RCC cell lines. This was also observed in a
xenograft mouse model. Furthermore, overexpression of eIF4EBP1
significantly attenuated BRDT inhibitor suppression on cell
proliferation, indicating that knockdown of eIF4EBP1 serves a role
in the progression of RCC. eIF4EBP1 is an important interaction
partner of eIF4E, which facilitates the restriction process during
translation initiation (56). It
was also reported to be upregulated in various types of tumors and
promote cancer growth (57). The
present study demonstrated that eIF4EBP1 downregulation
significantly improved PLX51107-induced growth suppression of RCC,
revealing a new approach for cancer therapy by co-targeting
PLX51107 and BRDT protein.
In recent decades, alternative epigenetic regulation
for gene expression has attracted an increasing amount of attention
(58,59). Targeting the downstream target of
eIF4E, eIF4F or its binding partners is emerging as a potential
treatment opportunity. The present study observed a decrease in
c-myc mRNA expression following addition of BRDT inhibitors, and a
decrease in c-myc promoter activity caused by BRDT inhibitors in a
dual luciferase reporter assay, implying a mechanism of c-myc
transcription mediation. The ChIP assay demonstrated that the
association between c-myc promoter and eIF4EBP1 was significantly
decreased by BRDT inhibitors, implying that BRDT inhibitors
decreased c-myc expression via inhibition of eIF4EBP1 protein, and
the binding of eIF4EBP1 with c-myc promoter and subsequent
transcription. There is a possibility that BRDT may also bind with
transcriptional factors or transcription regulatory proteins, or
attract multi-complexes to modified chromatin to promote
transcription (58).
In conclusion, the results of the present study
suggested that targeting BRDT by PLX51107 and INCB054329, or
BRDT-knockdown, suppressed RCC cell proliferation and tumor growth
via releasing the binding of eIF4EBP1 from the c-myc promoter and
decreasing subsequent c-myc mRNA transcription and protein
expression. These results identified a novel molecular mechanism of
BRDT-regulated eIF4EBP1 as well as c-myc in RCC tumors and
demonstrated a new method of targeting both eIF4EBP1 and c-myc to
support BRDT-targeted cancer therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Guangdong Province (grant no.
2017A030307037).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PW, NC and ZC designed the experiments. PW performed
the IP and CHIP assay. ZC designed the whole siRNA and performed
the transfection. WZ and HJ performed the plasmid construction. PW
and ZC performed the mice experiments. ZH and DP performed the dual
luciferase reporter assay. QH performed the SBR assay. PW, NC and
ZC analyzed the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All the experiments was conducted based on the
supervision and approval of the Institutional Animal Care and Use
Committee of Sun Yat-sen University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cella D, Grünwald V, Escudier B, Hammers
HJ, George S, Nathan P, Grimm MO, Rini BI, Doan J, Ivanescu C, et
al: Patient-Reported outcomes of patients with advanced renal cell
carcinoma treated with nivolumab plus ipilimumab versus sunitinib
(CheckMate 214): A randomised, phase 3 trial. Lancet Oncol.
20:297–310. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Powles T: Re: Nivolumab plus ipilimumab
versus sunitinib in advanced renal-cell carcinoma. Eur Urol.
74:679–680. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chevrier S, Levine JH, Zanotelli VR,
Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MA, Moch H,
et al: An immune atlas of clear cell renal cell carcinoma. Cell.
169:736–749. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song E, Song W, Ren M, Xing L, Ni W, Li Y,
Gong M, Zhao M, Ma X, Zhang X and An R: Identification of potential
crucial genes associated with carcinogenesis of clear cell renal
cell carcinoma. J Cell Biochem. 119:5163–5174. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Q, Ding H, He Y, Li X, Cheng Y, Xu Q,
Yang Y, Liao G, Meng X, Huang C and Li J: NLRC5 mediates cell
proliferation, migration, and invasion by regulating the
wnt/β-catenin signalling pathway in clear cell renal cell
carcinoma. Cancer Lett. 444:9–19. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Yan L, Jiao W, Ren J, Xing N,
Zhang Y, Zang Y, Wang J and Xu Z: The clinical and biological
significance of MICA in clear cell renal cell carcinoma patients.
Tumour Biol. 37:2153–2159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu H, Wang Z, Xu Q, Zhang Y, Zhai Y, Bai
J, Liu M, Hui Z and Xu N: Inhibition of STAT1 sensitizes renal cell
carcinoma cells to radiotherapy and chemotherapy. Cancer Biol Ther.
13:401–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wozniak MB, Le Calvez-Kelm F,
Abedi-Ardekani B, Byrnes G, Durand G, Carreira C, Michelon J,
Janout V, Holcatova I, Foretova L, et al: Integrative genome-wide
gene expression profiling of clear cell renal cell carcinoma in
czech republic and in the United States. PLoS One. 8:e578862013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Wang H, Ricketts CJ, Yang Y, Merino
MJ, Zhang H, Shi G, Gan H, Linehan WM, Zhu Y and Ye D: Germline
mutations of renal cancer predisposition genes and clinical
relevance in Chinese patients with sporadic, early-onset disease.
Cancer. 125:1060–1069. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghatalia P, Gordetsky J, Kuo F, Dulaimi E,
Cai KQ, Devarajan K, Bae S, Naik G, Chan TA, Uzzo R, et al:
Prognostic impact of immune gene expression signature and tumor
infiltrating immune cells in localized clear cell renal cell
carcinoma. J Immunother Cancer. 7:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pal S, Gong J, Mhatre SK, Lin SW, Surinach
A, Ogale S, Vohra R, Wallen H and George D: Real-World treatment
patterns and adverse events in metastatic renal cell carcinoma from
a large US claims database. BMC Cancer. 19:5482019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karner C, Kew K, Wakefield V, Masento N
and Edwards SJ: Targeted therapies for previously treated advanced
or metastatic renal cell carcinoma: Systematic review and network
meta-analysis. BMJ Open. 9:e246912019. View Article : Google Scholar
|
|
13
|
Suarez-Alvarez B, Morgado-Pascual JL,
Rayego-Mateos S, Rodriguez RM, Rodrigues-Diez R, Cannata-Ortiz P,
Sanz AB, Egido J, Tharaux PL, Ortiz A, et al: Inhibition of
bromodomain and extraterminal domain family proteins ameliorates
experimental renal damage. J Am Soc Nephrol. 28:504–519. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McDaniel KF, Wang L, Soltwedel T, Fidanze
SD, Hasvold LA, Liu D, Mantei RA, Pratt JK, Sheppard GS, Bui MH, et
al: Discovery of
N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide
(ABBV-075/Mivebresib), a potent and orally available bromodomain
and extraterminal domain (BET) family bromodomain inhibitor. J Med
Chem. 60:8369–8384. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Manterola M, Brown TM, Oh MY, Garyn C,
Gonzalez BJ and Wolgemuth DJ: BRDT is an essential epigenetic
regulator for proper chromatin organization, silencing of sex
chromosomes and crossover formation in male meiosis. PLoS Genet.
14:e10072092018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Rijck J, de Kogel C, Demeulemeester J,
Vets S, El Ashkar S, Malani N, Bushman FD, Landuyt B, Husson SJ,
Busschots K, et al: The BET family of proteins targets moloney
murine leukemia virus integration near transcription start sites.
Cell Rep. 5:886–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dibenedetto AJ, Guinto JB, Ebert TD, Bee
KJ, Schmidt MM and Jackman TR: Zebrafish brd2a and brd2b are
paralogous members of the bromodomain-ET (BET) family of
transcriptional coregulators that show structural and expression
divergence. BMC Dev Biol. 8:392008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang L and Wolgemuth DJ: BET protein BRDT
complexes with HDAC1, PRMT5, and TRIM28 and functions in
transcriptional repression during spermatogenesis. J Cell Biochem.
117:1429–1438. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pivot-Pajot C, Caron C, Govin J, Vion A,
Rousseaux S and Khochbin S: Acetylation-Dependent chromatin
reorganization by BRDT, a testis-specific bromodomain-containing
protein. Mol Cell Biol. 23:5354–5365. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bourova-Flin E, Chuffart F, Rousseaux S
and Khochbin S: The role of bromodomain testis-specific factor,
BRDT, in cancer: A biomarker and a possible therapeutic target.
Cell J. 19:1–8. 2017.PubMed/NCBI
|
|
21
|
Barda S, Yogev L, Paz G, Yavetz H, Lehavi
O, Hauser R, Doniger T, Breitbart H and Kleiman SE: BRDT gene
sequence in human testicular pathologies and the implication of its
single nucleotide polymorphism (rs3088232) on fertility. Andrology.
2:641–647. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suyama T, Shiraishi T, Zeng Y, Yu W,
Parekh N, Vessella RL, Luo J, Getzenberg RH and Kulkarni P:
Expression of cancer/testis antigens in prostate cancer is
associated with disease progression. Prostate. 70:1778–1787. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lagerholm S, Lagerholm S, Dutta S and Nair
P: Non-Invasive detection of c-myc p64, c-myc p67 and c-erbb-2 in
colorectal cancer. Scand J Gastroenterol. 40:1343–1350. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ozer HG, El-Gamal D, Powell B, Hing ZA,
Blachly JS, Harrington B, Mitchell S, Grieselhuber NR, Williams K,
Lai TH, et al: BRD4 profiling identifies critical chronic
lymphocytic leukemia oncogenic circuits and reveals sensitivity to
PLX51107, a novel structurally distinct BET inhibitor. Cancer
Discov. 8:458–477. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Erkes DA, Field CO, Capparelli C, Tiago M,
Purwin TJ, Chervoneva I, Berger AC, Hartsough EJ, Villanueva J and
Aplin AE: The next-generation BET inhibitor, PLX51107, delays
melanoma growth in a CD8-mediated manner. Pigment Cell Melanoma
Res. 32:687–696. 2019.PubMed/NCBI
|
|
26
|
Stubbs MC, Burn TC, Sparks R, Maduskuie T,
Diamond S, Rupar M, Wen X, Volgina A, Zolotarjova N, Waeltz P, et
al: The novel bromodomain and extraterminal domain inhibitor
INCB054329 induces vulnerabilities in myeloma cells that inform
rational combination strategies. Clin Cancer Res. 25:300–311. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wilson AJ, Stubbs M, Liu P, Ruggeri B and
Khabele D: The BET inhibitor INCB054329 reduces homologous
recombination efficiency and augments PARP inhibitor activity in
ovarian cancer. Gynecol Oncol. 149:575–584. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao Z, Yang S, Zhao H, Yang H and Jiang X:
BET inhibitor I-BET151 sensitizes GBM cells to temozolomide via
PUMA induction. Cancer Gene Ther. 27:226–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo NH, Zheng JF, Zi FM and Cheng J:
I-BET151 suppresses osteoclast formation and inflammatory cytokines
secretion by targetting BRD4 in multiple myeloma. Biosci Rep.
39:BSR201812452019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang C, Cigliano A, Jiang L, Li X, Fan B,
Pilo MG, Liu Y, Gui B, Sini M, Smith JW, et al: 4EBP1/eIF4E and
p70S6K/RPS6 axes play critical and distinct roles in
hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in
mice. Hepatology. 61:200–213. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chao MW, Wang LT, Lai CY, Yang XM, Cheng
YW, Lee KH, Pan SL and Teng CM: eIF4E binding protein 1 expression
is associated with clinical survival outcomes in colorectal cancer.
Oncotarget. 6:24092–24104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
William M, Leroux LP, Chaparro V, Lorent
J, Graber TE, M'Boutchou MN, Charpentier T, Fabié A, Dozois CM,
Stäger S, et al: eIF4E-Binding proteins 1 and 2 limit macrophage
anti-inflammatory responses through translational repression of
IL-10 and cyclooxygenase-2. J Immunol. 200:4102–4116. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bao Y, Wu X, Chen J, Hu X, Zeng F, Cheng
J, Jin H, Lin X and Chen LF: Brd4 modulates the innate immune
response through mnk2-eIF4E pathway-dependent translational control
of IkBα. Proc Natl Acad Sci USA. 114:E3993–E4001. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Batool A, Majeed ST, Aashaq S, Majeed R,
Shah G, Nazir N and Andrabi KI: Eukaryotic initiation factor 4E
(eIF4E) sequestration mediates 4E-BP1 response to rapamycin. Int J
Biol Macromol. 125:651–659. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Modrak-Wojcik A, Gorka M, Niedzwiecka K,
Zdanowski K, Zuberek J, Niedzwiecka A and Stolarski R: Eukaryotic
translation initiation is controlled by cooperativity effects
within ternary complexes of 4E-BP1, eIF4E, and the mRNA 5′ cap.
FEBS Lett. 587:3928–3934. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao W, Lam JW, Li JZ, Chen SQ, Tsang RK,
Chan JY and Wong TS: MicroRNA-138-5p controls sensitivity of
nasopharyngeal carcinoma to radiation by targeting EIF4EBP1. Oncol
Rep. 37:913–920. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cha YL, Li PD, Yuan LJ, Zhang MY, Zhang
YJ, Rao HL, Zhang HZ, Zheng XF and Wang HY: EIF4EBP1 overexpression
is associated with poor survival and disease progression in
patients with hepatocellular carcinoma. PLoS One. 10:e1174932015.
View Article : Google Scholar
|
|
38
|
Wang Z, Feng X, Molinolo AA, Martin D,
Vitale-Cross L, Nohata N, Ando M, Wahba A, Amornphimoltham P, Wu X,
et al: 4E-BP1 is a tumor suppressor protein reactivated by mTOR
inhibition in head and neck cancer. Cancer Res. 79:1438–1450. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Furic L, Rong L, Larsson O, Koumakpayi IH,
Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M,
Gaboury LA, et al: EIF4E phosphorylation promotes tumorigenesis and
is associated with prostate cancer progression. Proc Natl Acad Sci
USA. 107:14134–14139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
D'Abronzo LS and Ghosh PM: EIF4E
phosphorylation in prostate cancer. Neoplasia. 20:563–573. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee M, Kim EJ and Jeon MJ: MicroRNAs 125a
and 125b inhibit ovarian cancer cells through post-transcriptional
inactivation of EIF4EBP1. Oncotarget. 7:8726–8742. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB,
Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et
al: BET bromodomain inhibition as a therapeutic strategy to target
c-myc. Cell. 146:904–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bandopadhayay P, Bergthold G, Nguyen B,
Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R,
Masoud S, et al: BET bromodomain inhibition of MYC-amplified
medulloblastoma. Clin Cancer Res. 20:912–925. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Coudé MM, Braun T, Berrou J, Dupont M,
Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME,
et al: BET inhibitor OTX015 targets BRD2 and BRD4 and decreases
c-MYC in acute leukemia cells. Oncotarget. 6:17698–17712. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Baker EK, Taylor S, Gupte A, Sharp PP,
Walia M, Walsh NC, Zannettino AC, Chalk AM, Burns CJ and Walkley
CR: BET inhibitors induce apoptosis through a MYC independent
mechanism and synergise with CDK inhibitors to kill osteosarcoma
cells. Sci Rep. 5:101202015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shao Q, Kannan A, Lin Z, Stack BJ, Suen JY
and Gao L: BET protein inhibitor JQ1 attenuates myc-amplified MCC
tumor growth in vivo. Cancer Res. 74:7090–7102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang
Q, Lin Y, Li J, Kang T, Tao M, et al: Disrupting the interaction of
BRD4 with diacetylated twist suppresses tumorigenesis in basal-like
breast cancer. Cancer Cell. 25:210–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Takashima Y, Kikuchi E, Kikuchi J, Suzuki
M, Kikuchi H, Maeda M, Shoji T, Furuta M, Kinoshita I, Dosaka-Akita
H, et al: Bromodomain and extraterminal domain inhibition
synergizes with WEE1-inhibitor AZD1775 effect by impairing
non-homologous end joining and enhancing DNA damage in non-small
cell lung cancer. Int J Cancer. 15:1114–1124. 2019.
|
|
50
|
Wang SS, Chen G, Li SH, Pang JS, Cai KT,
Yan HB, Huang ZG and He RQ: Identification and validation of an
individualized autophagy-clinical prognostic index in bladder
cancer patients. Onco Targets Ther. 12:3695–3712. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wei W, Cao W, Zhan Z, Yan L, Xie Y and
Xiao Q: MiR-1284 suppresses gastric cancer progression by targeting
EIF4A1. Onco Targets Ther. 12:3965–3976. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xing ZY, Wang Y, Cheng L, Chen J, He XZ
and Xing W: Bromodomain-Containing protein 4 (BRD4) inhibition
sensitizes palomid 529-induced anti-renal cell carcinoma cell
activity in vitro and in vivo. Cell Physiol Biochem. 50:640–653.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liao S, Maertens O, Cichowski K and
Elledge SJ: Genetic modifiers of the BRD4-NUT dependency of NUT
midline carcinoma uncovers a synergism between BETis and CDK4/6is.
Genes Dev. 32:1188–1200. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tan Y, Wang L, Du Y, Liu X, Chen Z, Weng
X, Guo J, Chen H, Wang M and Wang X: Inhibition of BRD4 suppresses
tumor growth in prostate cancer via the enhancement of FOXO1
expression. Int J Oncol. 53:2503–2517. 2018.PubMed/NCBI
|
|
55
|
Gao Z, Yuan T, Zhou X, Ni P, Sun G, Li P,
Cheng Z and Wang X: Targeting BRD4 proteins suppresses the growth
of NSCLC through downregulation of eIF4E expression. Cancer Biol
Ther. 19:407–415. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hsu HS, Lin MH, Jang YH, Kuo TT, Liu CC
and Cheng TH: The 4E-BP1/eIF4E ratio is a determinant for rapamycin
response in esophageal cancer cells. J Thorac Cardiovasc Surg.
149:378–385. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang H, Liu Y, Ding J, Huang Y, Liu J, Liu
N, Ao Y, Hong Y, Wang L, Zhang L, et al: Targeting mTOR suppressed
colon cancer growth through 4EBP1/eIF4E/PUMA pathway. Cancer Gene
Ther. 27:448–460. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schwartz YB, Kahn TG, Stenberg P, Ohno K,
Bourgon R and Pirrotta V: Alternative epigenetic chromatin states
of polycomb target genes. PLoS Genet. 6:e10008052010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Thieffry D and Sanchez L: Alternative
epigenetic states understood in terms of specific regulatory
structures. Ann N Y Acad Sci. 981:135–153. 2002. View Article : Google Scholar : PubMed/NCBI
|