Introduction
Cell-cycle regulatory proteins such as CDKs,
cyclins, and CDK inhibitors regulate the progression through
G1, S, G2, and M phases of the cell cycle
(1). However, cancer cells exhibit
dysregulated expression of these proteins, and hence, they continue
to divide by undergoing uncontrolled cell cycles (2). Targeting cell-cycle regulators to
hinder cancer cell proliferation is a promising anti-cancer
strategy. Given the crucial role of CDK complexes in cell cycle
regulation, several drugs targeting CDKs have been developed and
tested in clinical trials (3–7). In
addition, activators of CDKs, such as serine/threonine-protein
kinase PLK1 (Plk1), Aurora kinase and Cdc25 phosphatases
(Cdc25A/B/C) are attractive targets for the development of
anticancer drugs, and a number of specific inhibitors targeting
these proteins have been studied and tested in clinical trials
(8–12).
Mitotic entry and progression are regulated by the
activity of the CDK1-cyclin B complex and controlled by multiple
feedback loops (13–15). The regulatory molecules myelin
transcription factor 1 (Myt1) and Wee1-like protein kinase (Wee1)
inhibit the activity of the CDK1-cyclin B complex during the
G2 phase by phosphorylating CDK1 at Tyr15 and Thr14,
while the Cdc25 phosphatase family activates the complex by
dephosphorylation of these sites (16,17).
In addition, several feedback loops, such as Plk1-dependent
positive feedback and Bora-Aurora-Plk1 feedback, indirectly
regulate the activity of the CDK1-cyclin B complex (15).
DNA damage leads to the activation of checkpoint
kinases such as ATM kinases (targeting Ser residues) and ATR
kinases (targeting Ser/Thr), which further activate p53 and
downstream effector kinases, such as serine/threonine-protein
kinase Chk1 (Chk1) and serine/threonine-protein kinase Chk2 (Chk2)
(18). DNA damage during the
G2 phase evokes the DNA damage response, thereby
inhibiting Plk1 and Cdc25, leading to cell cycle arrest at the
G2/M checkpoint, irrespective of the p53 status
(18). Entry into the M phase is
prevented through inhibitory phosphorylation of CDK1 by Cdc25 or by
sequestration of cyclin B outside the nucleus (18,19).
During G2 arrest in response to DNA damage, p53, p21,
and APC/Ccdh1 (the anaphase-promoting complex/cyclosome
and its coactivator Cdh1) are required to permanently exit the cell
cycle (19–21). In cancer cell lines with
functionally compromised p21, the cells are unable to exit the cell
cycle (21,22).
Mitotic arrest can also induce DNA damage response
(23). Microtubule-targeting
agents, which are commonly used as anticancer drugs, disrupt
microtubule dynamics and arrest cells in the G2/M phases
(24–26). This may activate the DNA damage
response, eventually activating p53 and p21, and inducing apoptosis
in cancer cells (27,28).
The present study screened a chemical compound
library and identified CCL113, a novel sulfonamide, as a candidate
anticancer drug. The present study elucidated the effects of CCL113
on cell cycle progression using cancerous and noncancerous cell
lines and found that CCL113 selectively induced mitosis and
apoptosis in cancer cells.
Materials and methods
Cell lines
Cervical cancer-derived HeLa cells were obtained
from the American Type Culture Collection. Monkey-kidney-derived
Vero, liver-cancer-derived HepG2, and normal human fibroblasts,
TIG-1-20 cells were obtained from the National Institutes of
Biomedical Innovation, Health and Nutrition, Japan. Cells were
grown in DMEM (Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS
(Thermo Fisher Scientific, Inc.) and antibiotics (penicillin, 100
U/ml; streptomycin, 100 µg/ml and amphotericin B, 0.25 µg/ml) at
37°C and 5% CO2. The HeLa-Fucci and Vero-Fucci cell
lines were established as previously described (29).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from HeLa cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Double-stranded
cDNA was synthesized from 20 mg total RNA using Ready-to-Go
You-Prime First-Strand Beads (GE Healthcare) and Oligo(dT) primers
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
instructions. qPCR was performed using a LightCycler FastStart DNA
Master SYBR Green 1 Kit (Roche Diagnostics GmbH) according to the
manufacturer's protocol. The transcript levels of Cdc25 (forward,
5′-AGCGAAGATGATGACGGATT-3′ and reverse,
5′-GCAGAGATGAAGAGCCAAAGA-3′) were estimated from the respective
standard curves and normalized to the expression of GAPDH (forward,
5′-CATCTCTGCCCCCTCTGCTGA-3′ and reverse,
5′-GGATGACCTTGCCCACAGCCT-3′) determined in corresponding samples.
The following thermocycling conditions were used for the qPCR:
Initial denaturation for 3 min at 95°C, followed by 40 cycles of
denaturation at 94°C for 30 sec, annealing at 60°C for 15 sec and
elongation at 72°C for 30 sec. The experiments were performed in
triplicate.
Chemical compounds
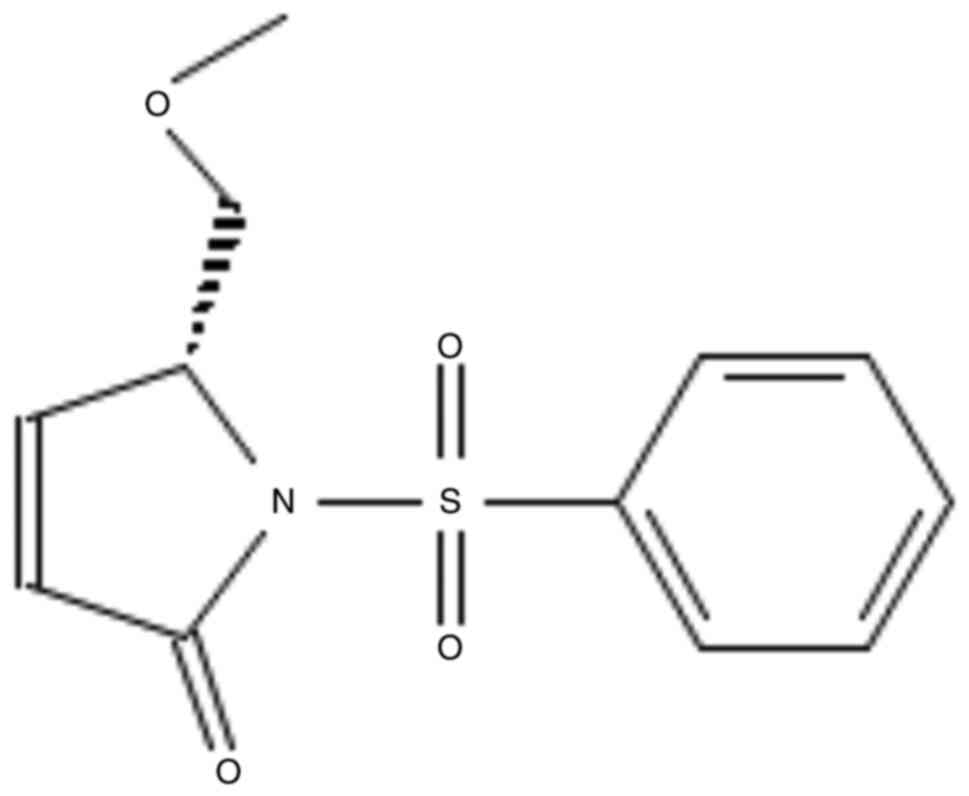
(R)-5-(methoxymethyl)-1-(phenylsulfonyl)-1,5-dihydro-2H-pyrrol-2-one
(CCL113) was provided by the Chiba Chemical Library (Fig. 1). It was synthesized from
d-pyroglutamic acid, and the schematic diagram is shown in
Supplementary Fig. S1 (30). CCL113 was dissolved in DMSO
(FUJIFILM Wako Pure Chemical Corporation) to obtain a 20-mM stock
solution. Nocodazole (Sigma-Aldrich; Merck KGaA) was dissolved in
DMSO at 100 µg/ml. DMSO was used as the vehicle control.
Cell viability
Cells (2×103 cells/well) were seeded in
96-well plates. After 24 h of culture, cells were treated with
increasing concentrations (1, 5, 10, 20, 30, and 40 µM) of CCL113
and incubated for 24 h. DMSO (<0.05%) was used as the vehicle
control, based on a previous report (31). Cell viability was determined using
the CellTiter-Glo Luminescent Cell Viability Assay (Promega
Corporation). Luminescent signals were measured using a Wallac 1420
multilabel counter (PerkinElmer, Inc.) and presented as the
proportional viability (%) by comparing the viability of
CCL113-treated cells with the DMSO-treated cells.
Staining and imaging of cultured
cells
For phase contrast microscopy, HeLa, Vero, HepG2,
and TIG-1-20 cells were treated with DMSO or 10 µM CCL113 for 8 and
24 h. Following treatment with 10 µM CCL113 for 8 and 24 h, the
cells were stained with Hoechst 33342 (Thermo Fisher Scientific,
Inc.) and incubated for 5 min at 37°C. Subsequently, the cells were
visualized at ×100 magnification with a UV-2A filter (excitation
wavelength 365 nm; emission wavelength 400 nm) on a Nikon Eclipse
TE2000-U microscope (Nikon Corporation). Mitotic cells stained with
Hoechst 33342 were identified morphologically, and the mean number
of mitotic cells in 10 random visual fields was used as the mitotic
index. For imaging of HeLa-Fucci and Vero-Fucci cells,
3×104 cells were grown in 35-mm dishes in DMEM with 10%
FBS. Following overnight incubation at 37°C and 5% CO2,
cells were synchronized at the G1/S phase by the double
thymidine block (2 mM) method (32); thymidine was added into the culture
medium at a final concentration of 2 mM and incubated for 14 h. The
medium was removed, and cells were washed with PBS. Fresh cell
culture medium was added, the cells were incubated for 9 h and 2 mM
thymidine blocking solution was added for the second time and
incubated for an additional 14 h. The medium was removed and washed
with PBS, and fresh culture medium was added. Next, the cells were
exposed to 10 µM CCL113 and visualized by time-lapse imaging using
a computer-assisted fluorescence microscope (FV10i; Olympus
Corporation). Images were recorded every 30 min. Two filter cubes
were chosen for fluorescence imaging: mKusabira-orange (excitation
wavelength 548 nm; emission wavelength 559 nm) to observe Fucci
orange, and Azami-Green (excitation wavelength 493 nm; emission
wavelength 505 nm) to observe Fucci green. Fluoview version 3.1
software (Olympus Corporation) was used for acquiring and analyzing
images.
Cell cycle analysis
HeLa, Vero, HepG2, and TIG-1-20 cells were treated
with CycleTEST™ PLUS DNA Reagent kit (Becton, Dickinson and
Company) according to the manufacturer's protocol and analyzed
using a BD Accuri™ C6 Flow Cytometer (Becton-Dickinson and Company)
equipped with the FACScan fluorescence 2 (FL2) detector. The data
were analyzed using FlowJo 7.6.5 software (FlowJo, LLC).
Western blotting
Cells were collected and washed twice with PBS, and
then resuspended in M-PER® Mammalian Protein Extraction
reagent (Thermo Fisher Scientific, Inc.) with a cocktail of
protease inhibitor (Sigma-Aldrich; Merck KGaA) and mixed gently for
10 min. The cell lysates were centrifuged at 14,000 × g for 10 min
at 4°C. The Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Inc.)
was used to determine the protein concentration. Equal amounts (50
µg) of extracted protein were resolved on 10% SDS-PAGE (ATTO
Corporation) and transferred onto PVDF membranes (Trans-Blot Turbo™
Transfer Pack; Bio-Rad Laboratories, Inc.). The membranes were
blocked with 5% skim milk for 1 h at room temperature and then
probed at 4°C overnight with the following mouse monoclonal
antibodies: Anti-p-histone H3 (Ser10) (1:1,1000; cat. no. 9706S;
Cell Signaling Technology, Inc.), anti-histone H3 (1:1,1000; cat.
no. 14269S; Cell Signaling Technology, Inc.), anti-cyclin B1
(1:2,000; cat. no. 4135S; Cell Signaling Technology, Inc.),
anti-phosphorylated (p)-CDK1 (Tyr15) (1:1,1000; cat. no. 9111S;
Cell Signaling Technology, Inc.), anti-p-CDK1 (Thr14) (1:1,000;
cat. no. 2543S; Cell Signaling Technology, Inc.), anti-p-CDK1
(Thr161) (1:1,000; cat. no. 9114S; Cell Signaling Technology,
Inc.), anti-CDK1 (1:1,000; cat. no. 9116S; Cell Signaling
Technology, Inc.), anti-cyclin D1 (1:1,000; cat. no. 2922S; Cell
Signaling Technology, Inc.), anti-CDK4 (1:1,000; cat. no. 2906;
Cell Signaling Technology, Inc.), anti-cyclin E1 (1:1,000; cat. no.
4129T; Cell Signaling Technology, Inc.), anti-Cdc25B (1:1,000; cat.
no. 9525S; Cell Signaling Technology, Inc.), anti-Cdc25C (1:1,000;
cat. no. 9522; Cell Signaling Technology, Inc.), anti-p-Cdc25C
(Thr48) (1:1,000; cat. no. 9527T; Cell Signaling Technology, Inc.),
anti-Plk1 (1:500; cat. no. 4535S; Cell Signaling Technology, Inc.),
anti-p-Plk1 (Thr210) (1:1,000; cat. no. 5472S; Cell Signaling
Technology, Inc.), anti-CDK2 (1:200; cat. no. sc-6248; Santa Cruz
Biotechnology, Inc.), anti-p-CDK2 (Thr160) (1:1,000; cat. no.
2561S; Cell Signaling Technology, Inc.), anti-ATM (1:1,000; cat.
no. 92356S; Cell Signaling Technology, Inc.) anti-Chk2 (1:1,000;
cat. no. 3440T; Cell Signaling Technology, Inc.), anti-p-Chk2
(Thr68) (1:1,000; cat. no. 2661S; Cell Signaling Technology, Inc.),
anti-p-p53 (Ser15) (1:1,000; cat. no. 9286; Cell Signaling
Technology, Inc.), anti-p21 (1:2,000; cat. no. 2946S; Cell
Signaling Technology, Inc.), anti-poly (ADP-ribose) polymerase
(PARP) (1:1,000; cat. no. 9542S; Cell Signaling Technology, Inc.),
anti-β-actin (1:1,000; cat. no. 3700S; Cell Signaling Technology,
Inc.), anti-cyclin A (1:1,000; cat. no. sc-239; Santa Cruz
Biotechnology, Inc.), anti-p53 (1:1,000; cat. no. sc-126; Santa
Cruz Biotechnology, Inc.) and p-ATM (Ser1981) (Cell Signaling
Technology, Inc. #4526S, dilution: 1:1,000). After washing three
times with TBS, the blots were incubated with an HRP-linked
anti-mouse IgG (1:1,000; cat. no. 7076S; Cell Signaling Technology,
Inc.) for 1 h at room temperature. Clarity Western ECL substrate
(Bio-Rad Laboratories, Inc.) was used to visualize protein signals.
The bands were analyzed using ImageJ v1.46r software (National
Institutes of Health).
Docking studies
The geometrical details of the αβ-tubulin protein
were obtained from the Research Collaboratory for Structural
Bioinformatics Protein Data Bank (https://www.rcsb.org) with the accession code 5syf
(33). The structure was cleared of
all solvent molecules and compounds before proceeding with the
analysis. Docking analysis was performed separately for the α- and
β-tubulin subunits. The 3D chemical structures of taxol and CCL113
were designed using Avogadro software (version 1.1.1) (https://avogadro.cc) (34) and optimized in Gaussian 16 (Gaussian
Inc.) at the B3LYP/6-31G (d, p) theoretical level (35,36).
AutoDock 4.2 software (http://autodock.scripps.edu) (37) was used for molecular docking
simulations. Structure files for proteins and ligands were made in
the PDBQT format using default charges. Furthermore, non-polar
hydrogen atoms were conjoined using AutoDockTools (version 1.5.6)
(http://autodock.scripps.edu). A grid box
with the dimensions 76 Å × 76 Å × 76 Å XYZ was centered on the
tubulin structure. A precalculated value for binding affinity for
each ligand was prepared using AutoGrid 4.2 (http://autodock.scripps.edu), and rigid docking was
accomplished using the Lamarckian genetic algorithm parameter
(37). All other parameters in the
AutoDock 4.2 software were considered in their default settings.
The best pose for docking was selected based on the lowest values
of binding energies obtained. The free binding energies were
computed using the in-built linear regression model in AutoDock
4.2. The root-mean-square deviation (RMSD) values for ligands (with
the dock input file) and the hydrogen bond occupancy were
calculated using PyMOL (https://pymol.org/2/) (38).
Statistical analysis
All values were expressed as the mean ± SD.
Statistical analyses were performed using data obtained from three
independent experiments. Data were analyzed using the Mann-Whitney
U test with the software Statcel4 version 4 (OMS). One-way ANOVA
followed by Bonferroni's correction were used for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of CCL113 on cell
viabilities
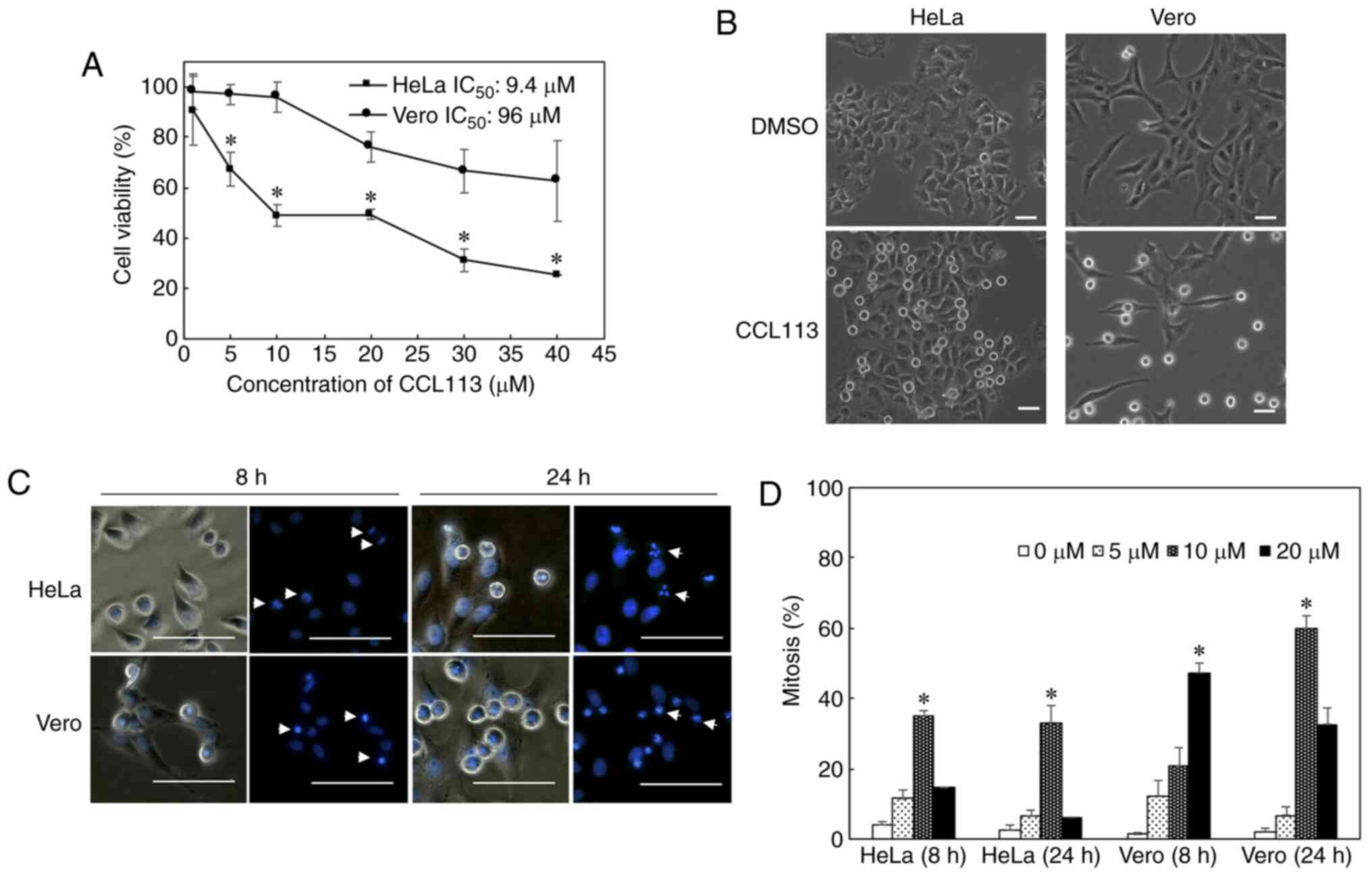
The anti-proliferative effects of the novel
sulfonamide CCL113 was observed in HeLa and Vero cells during the
anticancer drug screening of the Chiba Chemical Library. For the
screening, cervical cancer-derived HeLa cells were chosen as a
typical cancer cell line, in which wild-type p53 is expressed
(39). Monkey kidney-derived Vero
cells were chosen as a noncancerous cell line (40). CCL113 was one of the four compounds
with an IC50 that was 10-fold lower for HeLa than for
Vero cells. Further screening using HepG2 and TIG-1-20 showed only
CCL113 exhibited cancer-cell specific cytotoxicities (data not
shown). The cytotoxicity of CCL113 was assessed in HeLa and Vero
cells using an ATP assay. The IC50 of CCL113 for HeLa
and Vero cells was 9.4 and 96 µM, respectively. The results showed
that the cell viability of HeLa, but not Vero cells, was
significantly decreased following 24 h treatment with 5 and 10 µM
CCL113 (Fig. 2A). Significant
differences in cytotoxicity were also observed between HeLa and
Vero cells at 20–40 µM, although CCL113 caused Vero cells
cytotoxicity at concentrations of >10 µM. Furthermore, the
effects of 5–20 µM CCL113 on cell cycle progression in both cell
lines were investigated. Both HeLa and Vero cells showed a marked
increase in the number of round cells after 8 h of 10 µM CCL113
treatment (Fig. 2B). Hoechst 33342
staining of cell nuclei indicated that by 8 h, the chromosomes
condensed and congregated to the equatorial plane, in a manner
typical of cells in M phase in both HeLa and Vero cells. By 24 h,
abnormal nuclei were observed in mitotic HeLa cells (Fig. 2C). These observations suggested that
CCL113 might induce cell cycle arrest in the M phase. Both cells
showed an increased mitotic index after treatment with CCL113, and
the effect was more evident in Vero cells (Fig. 2D).
Effects of CCL113 on cell cycle
progression
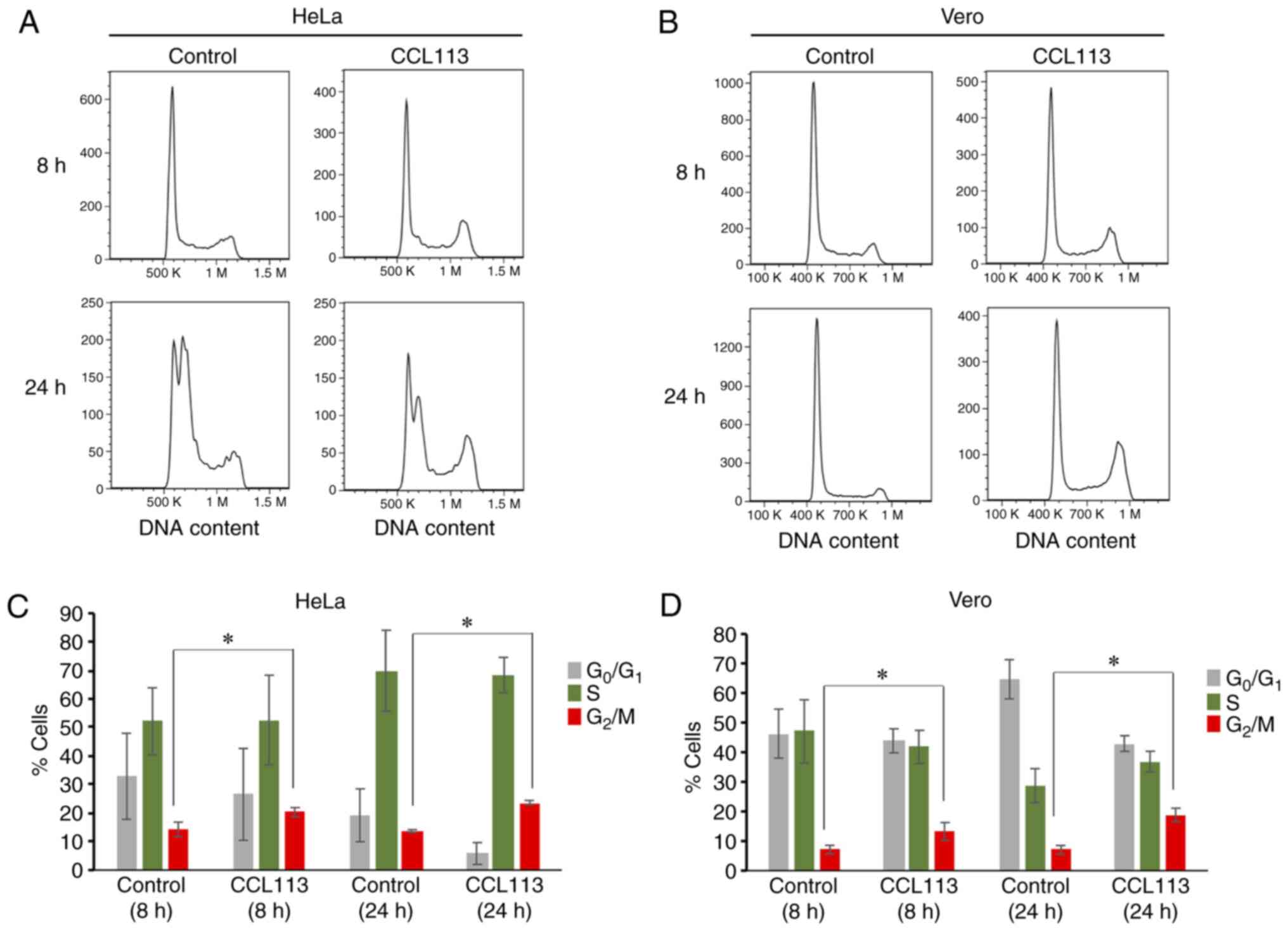
To further verify CCL113-induced cell cycle arrest,
CCL113-treated (10 µM; 8 and 24 h) HeLa and Vero cells were
analyzed by flow cytometry (Fig. 3A and
B). Compared with control groups, a significantly higher
proportion of CCL113-treated cells was observed in the
G2/M phase (Fig. 3C and
D). The results were consistent with the morphological
observations.
Effects of CCL113 on the expression of
cell cycle regulators
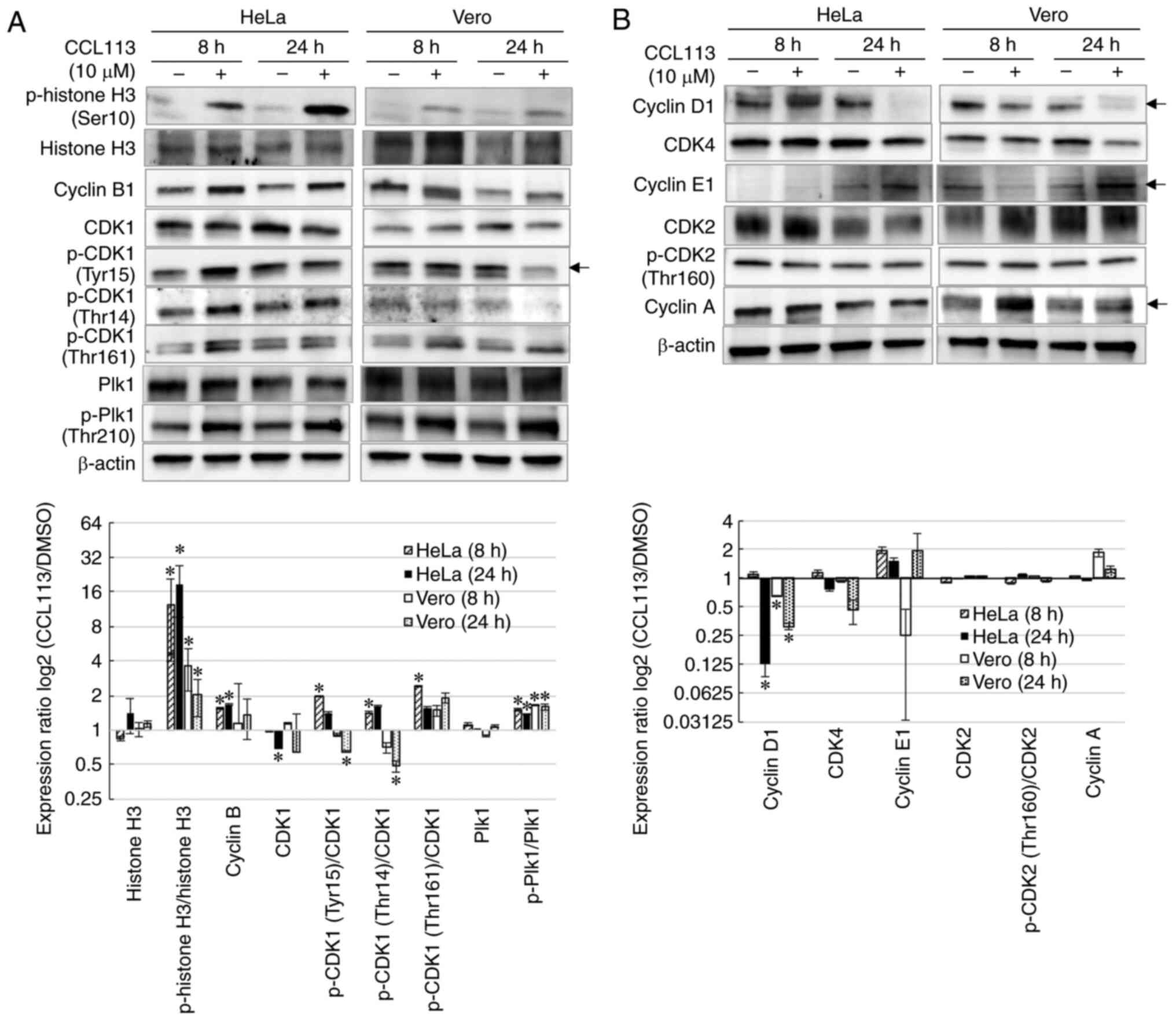
To elucidate the molecular basis of CCL113-induced M
phase arrest in HeLa and Vero cells, the levels of cell cycle
regulatory proteins were assessed using western blotting. Total
cellular protein was isolated from cells treated with 10 µM CCL113
for 8 and 24 h. Since phosphorylation at Ser10 of histone H3 is
significantly related to mitotic chromosome condensation (41–44),
phosphorylation levels of histone H3 (Ser10) were first examined to
confirm mitotic arrest. The results showed that the phosphorylation
levels of histone H3 (Ser10) were significantly upregulated in
CCL113-treated cells, suggesting that CCL113 induced M phase arrest
by facilitating phosphorylation at Ser10 of histone H3 (Fig. 4A).
Activation of the CDK1-cyclin B1 complex initiates
entry into mitosis and facilitates mitotic progression (13,14).
CDK1 activation, in turn, requires dephosphorylation at Tyr15 and
Thr14 residues and phosphorylation at Thr161 (16). To investigate the activity of
CDK1-cyclin B1, the levels of cyclin B1 and p-CDK1 in
CCL113-treated HeLa and Vero cells were measured. As shown in
Fig. 4A, the levels of cyclin B1 in
CCL113-treated HeLa cells was significantly higher compared with
DMSO-treated cells. This could be explained by the increase in the
number of cells in M phase. As depicted in Fig. 4A, the results showed that in HeLa
cells, the relative phosphorylation levels at Tyr15 and Thr14
[p-CDK1(Tyr15)/CDK1 and p-CDK1(Thr14)/CDK1] were significantly
elevated at 8 h, indicating an increase in CDK1 inhibitory
phosphorylation. Additionally, in Vero cells, the relative
phosphorylation levels at Tyr15 and Thr14 of CDK were significantly
lower at 24 h, indicating a relative decrease in the CDK1
inhibitory phosphorylation. CCL113-mediated inactivation of CDK1 by
inhibitory phosphorylation at Tyr15 and Thr14 in HeLa cells might
inhibit mitotic entry and progression, leading to an increase in
the number of cells in the G2/M phase. By contrast, the
elevated expression of cyclin B1 and decrease in CDK1
phosphorylation at Tyr15 and Thr14 in Vero cells (CCL113 treatment,
24 h) could explain the increase in cells in M phase with activated
CDK1-cyclin B1 complexes.
The Thr210 residue of Plk1 protein is the primary
phosphorylation site responsible for its activation during mitosis;
moreover, activated P1k1 regulates the CDK1-cyclin B1 complex
during mitosis (32,45). Following treatment with CCL113,
significantly increased Thr210 phosphorylation of Plk1 was observed
in both HeLa and Vero cells, consistent with the increased number
of cells in the G2/M phase (Fig. 4A). Therefore, CCL113 treatment might
inhibit the activation of CDK1 indirectly by upregulating Plk1.
Subsequently, the present study examined whether
CCL113 affected the expression of regulatory proteins during other
phases of the cell cycle, including key regulators of G1
progression (cyclin D1 and CDK4), G1/S transition
(cyclin E1 and p-CDK2) and S phase progression (cyclin A and
p-CDK2). As shown in Fig. 4B,
CCL113 had little effect on the expression of CDK4 and cyclin E1,
key regulators of G1 and G1/S transition, at
8 h. However, CCL113 significantly reduced the protein levels of
cyclin D1 after 24 h of treatment, suggesting the decrease in the
number of cells in the G1 phase. Aside from a temporal
upregulation of cyclin A in Vero cells after 8 h, CCL113 seemed to
have a negligible direct effect on the expression levels of cyclin
A and p-CDK2 (Thr160) in both HeLa and Vero cells, suggesting
CCL113 had little effect on G1/S transition. These
results suggested that although CCL113 treatment led to a slight
modulation in the regulatory proteins during other phases of the
cell cycle, its main impact was observed during the M phase in
treated cells.
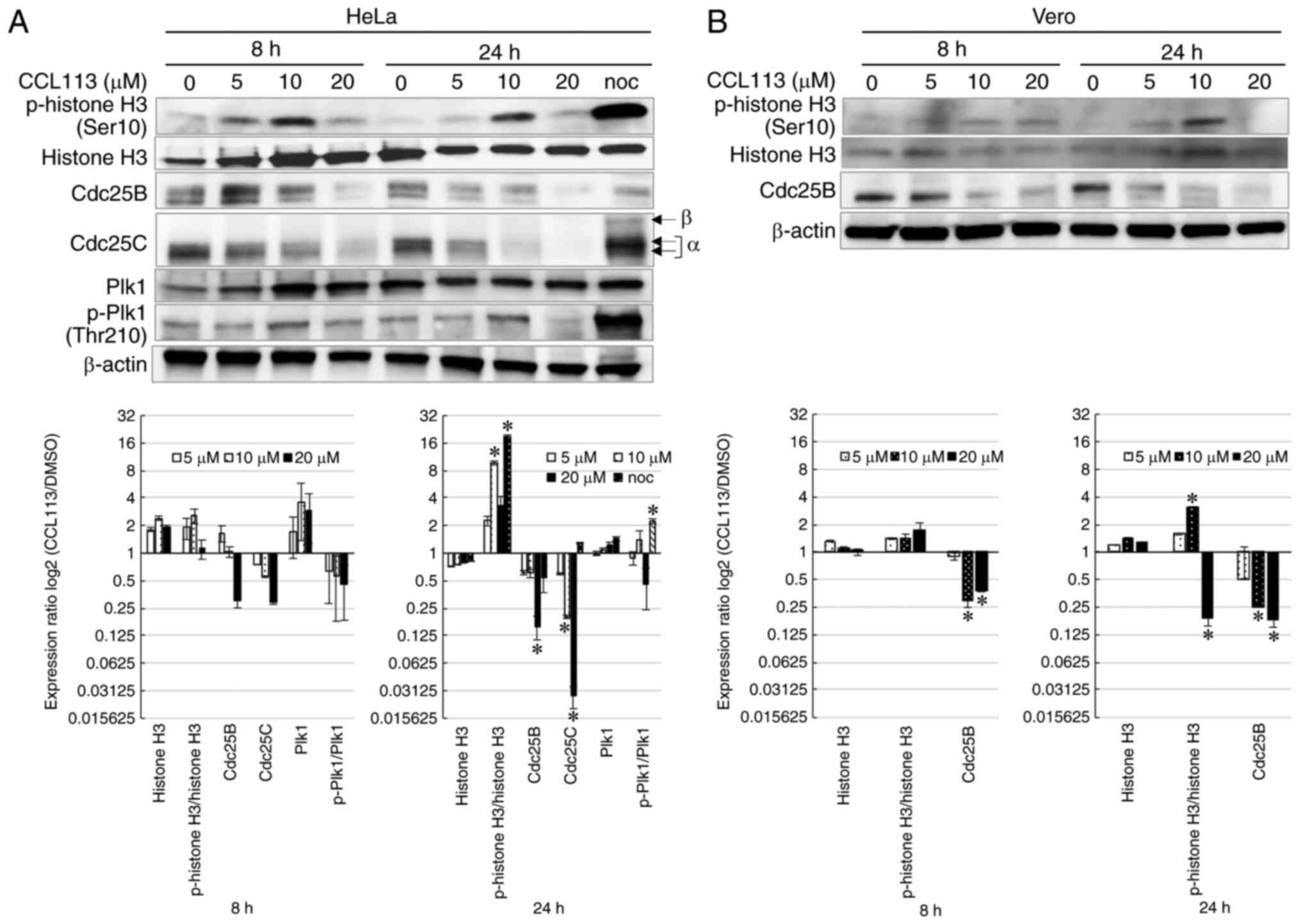
Dose-dependent effects of CCL113 on
the cell cycle
Cdc25 phosphatases activated by Plk1 are responsible
for the dephosphorylation and activation of CDK1 (46). Cdc25 phosphatases include three
different isoforms: Cdc25A, B and C (46). To examine the effects of CCL113 on
the expression levels of Cdc25 and cell cycle progression, cells
were treated with various concentrations (0, 5, 10 and 20 µM) of
CCL113 and the levels of Cdc25 phosphatases were examined by
western blotting. Nocodazole, an inducer of M phase arrest, served
as a positive control.
In HeLa cells, the expression of p-histone H3
(Ser10) increased as the concentration of CCL113 increased from 5
to 10 µM but decreased at 20 mM (Fig.
5A), suggesting that in these cells, CCL113 might inhibit
mitotic entry at 20 mM. The levels of Cdc25B and total Cdc25C
protein decreased in HeLa cells in a time- and dose-dependent
manner in response to CCL113 treatment (Fig. 5A). RT-qPCR analysis did not detect
any apparent changes in Cdc25B/C mRNA levels in HeLa cells after
treatment with 5 to 20 µM CCL113 for 8 h (data not shown). The
levels of phosphorylated Plk1 (p-Plk1, Thr210) and p-histone H3
(Ser10) showed a similar response after treatment with various
concentrations of CCL113 (Fig. 5A),
indicating that 5 to 10 µM CCL113 might inhibit mitotic progression
but not mitotic entry. In addition, a reduced amount of actin in
HeLa cells was observed following exposure to 20 µM CCL113,
suggesting that the cytotoxicity of 20 µM CCL113 might cause
nonspecific protein degradation.
As shown in Fig. 5B,
Vero cells showed a similar expression pattern of p-histone H3
(Ser10) and Cdc25B protein in HeLa cells in response to treatment
with various concentrations of CCL113.
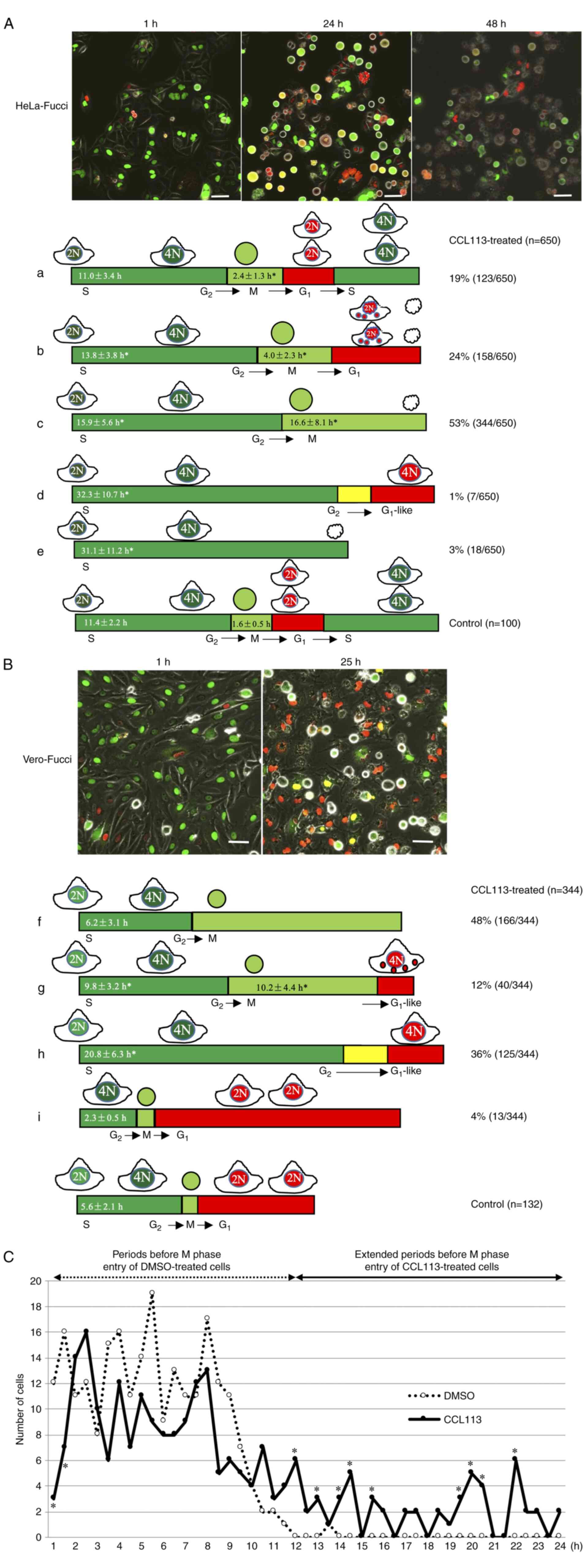
Visualization of CCL113-induced cell
cycle alterations in HeLa-Fucci and Vero-Fucci cells
The Fucci system, based on fluorescent
ubiquitination-based cell cycle indicator (FUCCI), allows
visualization of cell cycle progression in real time at the
single-cell level, and the dynamic data can aid in understanding
how individual cells respond to a drug (47). CCL113-induced cell cycle alterations
in synchronized HeLa-Fucci and Vero-Fucci cells were monitored.
FUCCI can be used to determine the distribution of a cell
population through different stages of the cell cycle and
distinguish G1 and S/G2/M phases of the cell
cycle by labeling G1 nuclei in red and S/G2/M
nuclei in green. During the G1/S transition
(red-to-green conversion), red and green fluorescence overlap,
resulting in yellow nuclei.
The results showed that most HeLa-Fucci and
Vero-Fucci cells were in the S phase at the beginning of imaging
(Fig. 6A and B) and showed
differential progression following exposure to CCL113. The serially
collected images of 650 HeLa-Fucci and 344 Vero-Fucci cells were
traced and analyzed. In synchronized HeLa-Fucci cells, 19% of cells
required more time than control cells to progress through the M
phase (mean, 2.4 vs. 1.6 h for control cells) and passed through
the G1 phase to S/G2 phase as shown in
Fig. 6A-a. A total of 24% of cells
underwent abnormal cell division and subsequent cell death at the
G1 phase (Fig. 6A-b).
These cells required more time to progress through the M phase
(mean, 4.0 h) than the control cells. As shown in Fig. 6A-c, 53% of cells entered the M phase
and exhibited long-term mitotic cell rounding (mean, 16.6 h) before
eventual cell death. The nuclei of very few HeLa-Fucci cells (1%)
changed color from green to G1-like red without
undergoing mitosis (Fig. 6A-d),
while a few cells (3%) were arrested at the S/G2 phase
and subsequently exhibited cell death (Fig. 6A-e). Therefore, CCL113 treatment
resulted in a longer S/G2 and M phase in HeLa cells
compared with control cells.
In contrast to HeLa-Fucci cells, Vero-Fucci cells
exhibited different responses to CCL113. Nearly half the cells
(48%) progressed through the S/G2 phase at approximately
the same time (mean, 6.2 h) as synchronized control cells but were
arrested in the M phase (Fig.
6B-f). A total of 12% of treated cells required more time to
progress through the S/G2 phase (mean, 9.8 h) and
underwent prolonged mitosis (mean, 10.2 h) to the
G1-like phase without cytokinesis (Fig. 6B-g). Furthermore, 36% of cells
progressed slowly through the S/G2 phase (mean, 20.8 h)
and entered the G1-like phase without undergoing mitosis
(Fig. 6B-h). Prior to transitioning
to the G1-like phase, the nuclei of these cells stained
yellow for a brief period, similar to G1/S transition
(red-to-green conversion), suggesting that the G1-like
phase did not correspond to the beginning of G1 phase,
at which the red color is weak.
As shown in Fig.
6B-i, the 4% of CCL113-treated Vero-Fucci cells that required a
shorter time to progress through the S/G2 phase than the
control cells (mean, 2.3 vs. 5.6 h for control cells) underwent
normal division. A small fraction of the Vero cell population may
have experienced inefficient thymidine block, which reflected the
cell population in the G2 phase and suggested that
CCL113 may not be effective during the G2 phase. To
confirm and validate the cell cycle phase regulated by CCL113,
CCL113-treated Vero-Fucci cells were further analyzed. As shown in
Fig. 6C, CCL113 treatment
significantly reduced the number of cells 1 to 1.5 h before M phase
entry. Treament with CCL113 increased the number of cells with
extended periods before M phase entry, suggesting that the
G2/M checkpoint hindered the cells from entering the M
phase.
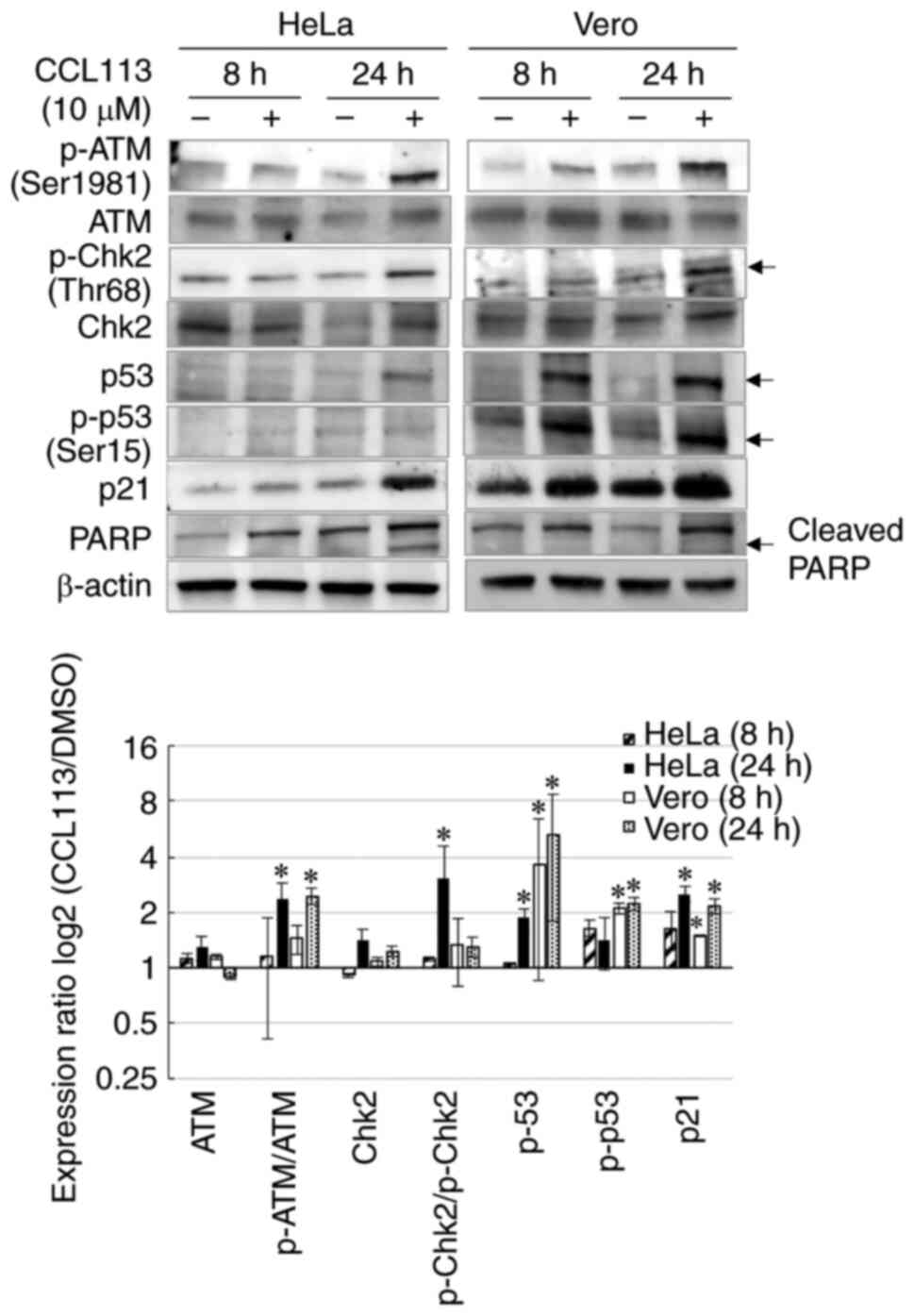
Effects of CCL113 on DNA damage
response and apoptosis
Cells that fail to progress through mitosis
accumulate de novo DNA damage foci and experience mitotic
failure (23,48). To confirm whether CCL113 activated
the DNA damage response, its central regulators, such as ATM and
other proteins including Chk2, p53, and p21, were examined in cells
treated with CCL113. The results showed that exposure to CCL113 (10
µM) increased the levels of p-ATM (Ser1981), p-Chk2 (Thr68), p53
(Ser15), and p21 in HeLa cells (Fig.
7). Increase in p-p53 at Ser15 reduces the interaction with E3
ubiquitin-protein ligase MDM2 (49), which might explain the increase in
p53 levels in CCL113-treated Vero cells (Fig. 7). To examine the effects of CCL113
on apoptosis, the present study analyzed the expression of PARP
fragment, which is an indicator of apoptosis (10). Cleaved PARP fragment (89 kDa) was
observed in CCL113-treated HeLa cells at 24 h (Fig. 7), indicating the induction of
apoptosis; however, the cleaved PARP band could not be detected in
CCL113-treated Vero cells at 24 h, suggesting that CCL113-induced
apoptosis might not be frequent in normal cells.
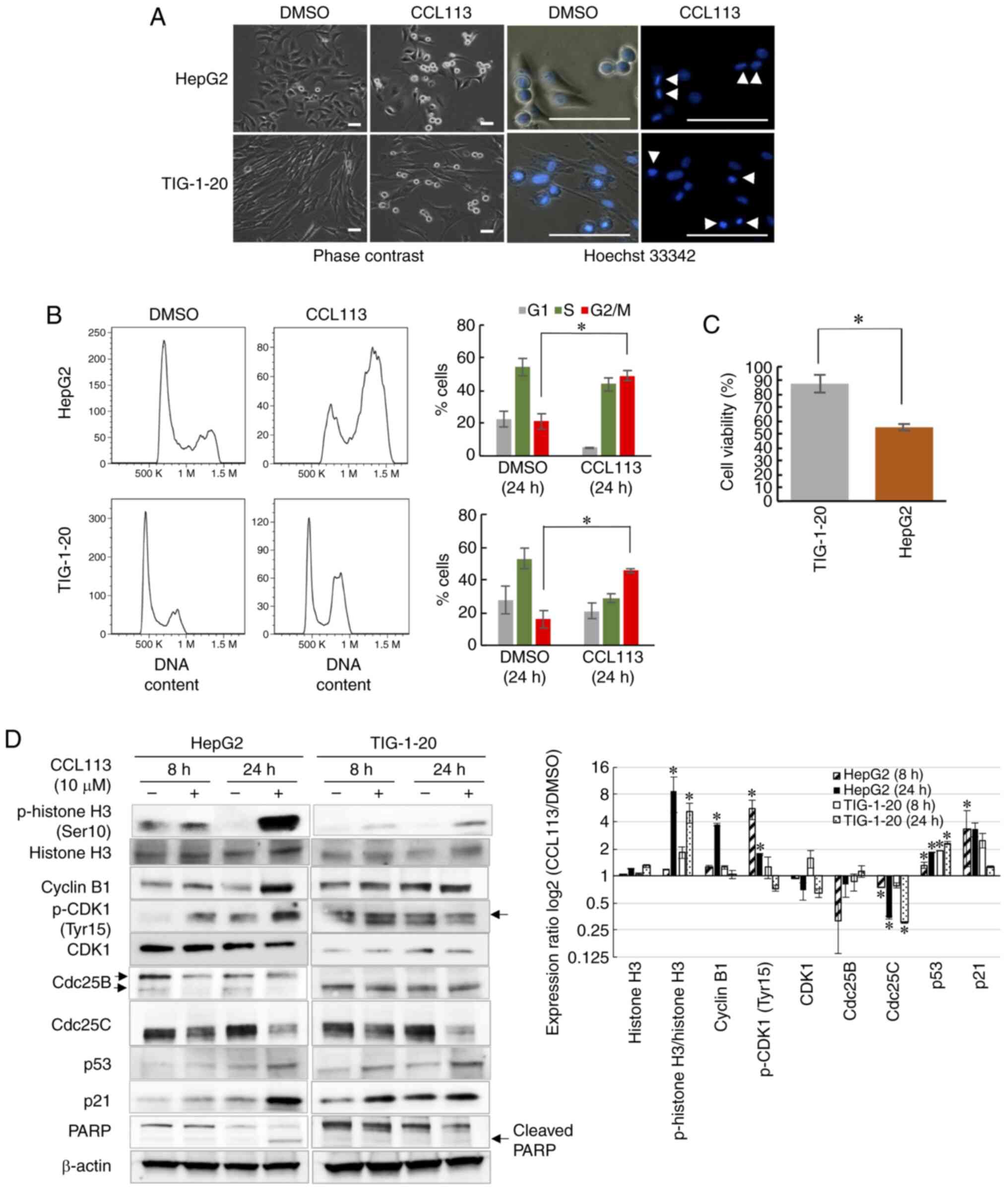
Effects of CCL113 on cell cycle
progression in HepG2 cells and normal human fibroblasts
To investigate the effects of CCL113 on other cancer
cells and normal cells, HepG2, a human liver cancer cell line, and
TIG-1-20, normal human fibroblasts, were employed. Similar to HeLa
cells, HepG2 cells are known to express wild-type p53 (39). Consistent with the results observed
in HeLa and Vero cells, both HepG2 and TIG-1-20 cells showed a
marked increase in the number of round cells after 24 h of exposure
to 10 µM CCL113. Hoechst 33342 staining of cell nuclei confirmed
that CCL113 treatment arrested HepG2 cells in metaphase, whereas
TIG-1-20 cells were arrested in prometaphase (Fig. 8A). Flow cytometry analyses showed
that the number of both HepG2 and TIG-1-20 cells significantly
increased in the G2/M phase following CCL113 treatment
(Fig. 8B). Treatment with CCL113
significantly decreased the cell viability of HepG2 compared with
TIG-1-20 cells, after 24 h of treatment (Fig. 8C).
As shown in Fig. 8D,
the expression of cell cycle regulators in HepG2 cells in response
to CCL113 treatment was comparable to the response observed in HeLa
cells; the levels of cyclin B1 and phosphorylation of CDK1 at Tyr15
significantly increased, while the levels of Cdc25B and Cdc25C
protein were decreased. Similarly, CCL113-treated TIG-1-20 cells
showed responses comparable to those of Vero cells. Furthermore,
CCL113 treatment led to a significant increase in the levels of p53
and p21 in HepG2 and TIG-1-20 cells. Meanwhile, the cleaved PARP
fragment was observed only in CCL113-treated HepG2 cells and not in
TIG-1-20 cells at 24 h (Fig. 8D),
suggesting that apoptosis was induced only in HepG2 cells and not
in normal cells, consistent with the results observed in HeLa and
Vero cells.
Docking studies of CCL113 with a- and
β-tubulin
Since microtubule-targeting agents (MTAs), which are
commonly used anticancer drugs, often function by blocking cell
entry into mitosis (50,51) the possibility of CCL113 acting as an
MTA was investigated. Molecular docking studies between CCL113 and
tubulins were performed using AutoDock 4.2. CCL113 exhibited the
highest binding affinity at the taxol-binding site of β-tubulin
(Table I). The free binding energy
of CCL113 at the taxol-binding site of β-tubulin (−4.24) was
favorable to facilitate the interaction, unlike that of a-tubulin
and taxol (−3.41 and −4.81, respectively). The RMSD value of CCL113
with b-tubulin was lower compared with taxol with β-tubulin.
Furthermore, CCL113 exhibited a higher number of hydrogen bond
interactions with the taxol-binding sites of β-tubulin than taxol,
suggesting that CCL113 could bind to the taxol-binding site of
β-tubulin.
 | Table I.Docking scores of CCL113 with the
taxol-binding sites of α- and β-tubulin. |
Table I.
Docking scores of CCL113 with the
taxol-binding sites of α- and β-tubulin.
| Compounds
(Tubulins) | CCL113
(α-tubulin) | CCL113
(β-tubulin) | Taxol
(β-tubulin) |
|---|
| Free binding energy
(kcal/mol) | −3.41 | −4.24 | −4.81 |
| RMSD | 1.52 | 0.80 | 2.18 |
| Hydrogen bond
(H-bond) | 3 | 3 | 2 |
Discussion
In HeLa cells, >1,000 proteins are reported to
exhibit enhanced phosphorylation during mitosis, and most of them
are expected to be CDK1 substrates (52). The CDK1-cyclin B1 complex plays a
crucial role in mitotic entry and progression (13,14),
and its activity is regulated by CDK1-mediated phosphorylation at
Tyr15 and Thr14 residues, while Cdc25 phosphatases are responsible
for the dephosphorylation of these sites (16,17).
The present study identified a novel sulfonamide, CCL113, that
resulted in cell cycle arrest in the M phase and induced apoptosis
in cancer cells, but not in normal cells. The effects of CCL113 on
cell cycle regulatory proteins were similar in cancer and
noncancerous cells. However, the consequence of these effects was
cell death in HeLa cells, but not in Vero cells. Although these
cell lines are from different origins and future studies should
test CCL113 in additional cell lines, these differential effects
mediated by CCL113 were possibly achieved by inhibition of Cdc25
protein via induction of the DNA damage response, suppression of
CDK1-cyclin B activity, a prolonged G2 phase at the
G2/M checkpoint and cell cycle arrest in the M
phase.
Activation of the CDK1-cyclin B complex is regulated
by several feedback loops involving Myt1, Wee1, Cdc25A/B/C, Plk1
and the Bora-Aurora-Plk1 complex (15). CDK1 is rendered inactive by the
phosphorylation of Tyr15 and Thr14 by Myt1 and Wee1 kinases
(15). Although relatively low
CDK1-cyclin B activity is required to enter mitosis and some of
these feedback loops may even be redundant, full activation of
multiple feedback loops is essential for successful completion of a
mitotic cycle (14,15). For instance, the present results
showed that CCL113-induced reduction of Cdc25 prolonged the
G2 phase at the G2/M checkpoint but did not
block mitotic entry, while lower levels of Cdc25 phosphatases were
sufficient to block the completion of mitosis.
Cdc25 phosphatases are critical regulators of the
cell cycle and are required to control CDK dephosphorylation and
activation (15). The expression
and activity of Cdc25 are regulated by numerous mechanisms, such as
cell cycle-controlled phosphorylation-dephosphorylation,
transcription, post-translational regulation and protein
degradation (15). In the present
study, since the mRNA levels of Cdc25 were not affected by CCL113
treatment, the inhibition of Cdc25 was most likely due to protein
degradation rather than transcriptional regulation. Since CCL113
activates the DNA damage response, which in turn promotes Cdc25
turnover, inhibition of Cdc25 could also be a result of indirect
regulation by CCL113. Furthermore, the possible interaction of
CCL113 at the taxol-binding site of b-tubulin might interfere with
microtubule dynamics causing prolonged mitosis, which can lead to
DNA damage and p53 induction (53).
The terminal cell cycle exit in the G2
phase is dependent on the induction of p53 and p21, resulting in
the nuclear retention of cyclin B1 and the activation of
APC/Ccdh1 (19–21). In the present study, significantly
elevated levels of p53 and p21 were detected in noncancerous cells
8 h after CCL113 treatment. Meanwhile, FUCCI analyses revealed that
CCL113-treated Vero cells could transition to the
G1-like phase from prolonged G2 and M phase
with the degradation of Geminin (Fucci red), the S/G2/M
marker, which is a target of APC/Ccdh1. These results
suggested that noncancerous cells could exit the cell cycle in
response to CCL113 treatment and may explain why apoptosis was not
observed in Vero and TIG-1-20 cells. In cancer cell lines with
compromised p21 function, its induction is delayed, and cells are
unable to terminally exit the cell cycle (2). In CCL113-treated HeLa and HepG2 cells,
a significant increase in p53 and p21 levels was detected at 24 h.
FUCCI analyses revealed that very few CCL113-treated HeLa cells
could exit the cell cycle, and most of the cells underwent mitotic
arrest followed by apoptosis.
DNA damage response arrests the cell cycle to
initiate DNA repair and prevent the proliferation of potentially
unstable cells by promoting cell cycle exit (21). However, CCL113-treated cancer cells
with compromised p21 function are unable to exit the cell cycle and
undergo apoptosis (21). The
docking study showed that CCL113 might act similarly to taxol,
which disrupts mitosis. Although the mechanisms by which anticancer
drugs that disrupt the cell cycle and then induce apoptosis are
poorly understood, the present study showed that mitotic stress
induced by these drugs might trigger DNA damage response.
In summary, the novel sulfonamide CCL113 was
identified as a candidate anticancer drug by screening a chemical
compound library. Although the widespread use of CCL113 may be
restricted due to its cytotoxicity, this compound may serve as a
lead compound for development of a more potent drug. The present
results suggested that CCL113 induces apoptosis in cancer cells and
cell cycle exit in noncancerous cells. These effects of CCL113 are
presumably mediated by upregulation of the DNA damage response.
Results of the docking analysis suggested the possible interaction
of CCL113 with the taxol-binding site of β-tubulin, which can
facilitate mitotic arrest and DNA damage and induce p53
induction.
Supplementary Material
Supporting Data
Acknowledgements
We thank Dr Takayoshi Arai (Molecular Chirality
Research Center, Department of Chemistry, Graduate School of
Science, Chiba University) for the generous donation of all the
compounds from the Chiba Chemical Library.
Funding
This work was supported by JSPS KAKENHI (grant no.
JP26430155).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RY, YO, KS and HS designed the study. RY and XM
performed most of the experiments. RY and YO screened the chemical
library, which was provided by AS, KY, SH, YT and AN. RY, ZT, SG,
WC, NNW, QL, MV, KS, NI and SN analyzed the data. RY and HS wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cicenas J and Valius M: The CDK inhibitors
in cancer research and therapy. J Cancer Res Clin Oncol.
137:1409–1418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Diaz-Moralli S, Tarrado-Castellarnau M,
Miranda A and Cascante M: Targeting cell cycle regulation in cancer
therapy. Pharmacol Ther. 138:255–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sedlacek HH: Mechanisms of action of
flavopiridol. Crit Rev Oncol Hematol. 38:139–170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khalil HS, Mitev V, Vlaykova T, Cavicchi L
and Zhelev N: Discovery and development of seliciclib. How systems
biology approaches can lead to better drug performance. J
Biotechnol. 202:40–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tong WG, Chen R, Plunkett W, Siegel D,
Sinha R, Harvey RD, Badros AZ, Popplewell L, Coutre S, Fox JA, et
al: Phase I and pharmacologic study of SNS-032, a potent and
selective Cdk2, 7, and 9 inhibitor, in patients with advanced
chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol.
28:3015–3022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Strebhardt K and Ullrich A: Targeting
polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 6:321–330.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murugan RN, Park JE, Kim EH, Shin SY,
Cheong C, Lee KS and Bang JK: Plk1-Targeted small molecule
inhibitors: Molecular basis for their potency and specificity. Mol
Cells. 32:209–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kollareddy M, Zheleva D, Dzubak P,
Brahmkshatriya PS, Lepsik M and Hajduch M: Aurora kinase
inhibitors: Progress towards the clinic. Invest New Drugs.
30:2411–2432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boutros R, Lobjois V and Ducommun B: CDC25
phosphatases in cancer cells: Key players? Good targets? Nat Rev
Cancer. 7:495–507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lavecchia A, Di Giovanni C and Novellino
E: CDC25 phosphatase inhibitors: An update. Mini Rev Med Chem.
12:62–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nigg EA and Nigg EA: Mitotic kinases as
regulators of cell division and its checkpoints. Nat Rev Mol Cell
Biol. 2:21–32. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lindqvist A, van Zon W, Karlsson C and
Wolthuis RM: Cyclin B1-cdk1 activation continues after centrosome
separation to control mitotic progression. PLoS Biol. 5:e1232007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lindqvist A, Rodríguez-Bravo V and Medema
RH: The decision to enter mitosis: Feedback and redundancy in the
mitotic entry network. J Cell Biol. 185:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dunphy WG: The decision to enter mitosis.
Trends Cell Biol. 4:202–207. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Porter LA and Donoghue DJ: Cyclin B1 and
CDK1: Nuclear localization and upstream regulators. Prog Cell Cycle
Res. 5:335–347. 2003.PubMed/NCBI
|
|
18
|
Harper JW and Elledge SJ: The DNA damage
response: Ten years after. Mol Cell. 28:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krenning L, Feringa FM, Shaltiel IA, van
den Berg J and Medema RH: Transient activation of p53 in G2 phase
is sufficient to induce senescence. Mol Cell. 55:59–72. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baus F, Baus F, Gire V, Fisher D, Piette J
and Dulić V: Permanent cell cycle exit in G2 phase after DNA damage
in normal human fibroblasts. EMBO J. 22:3992–4002. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Müllers E, Cascales HS, Jaiswal H, Saurin
AT and Lindqvist A: Nuclear translocation of Cyclin B1 marks the
restriction point for terminal cell cycle exit in G2 phase. Cell
Cycle. 13:2733–2743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lossaint G, Besnard E, Fisher D, Piette J
and Dulić V: Chk1 is dispensable for G2 arrest in response to
sustained DNA damage when the ATM/p53/p21 pathway is functional.
Oncogene. 30:4261–4274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hayashi MT and Karlseder J: DNA damage
associated with mitosis and cytokinesis failure. Oncogene.
32:4593–4601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dumontet C and Jordan MA:
Microtubule-binding agents: A dynamic field of cancer therapeutics.
Nat Rev Drug Discov. 9:790–803. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perez EA: Microtubule inhibitors:
Differentiating tubulin-inhibiting agents based on mechanisms of
action, clinical activity, and resistance. Mol Cancer Ther.
8:2086–2095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Risinger AL, Giles FJ and Mooberry SL:
Microtubule dynamics as a target in oncology. Cancer Treat Rev.
35:255–261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhalla KN: Microtubule-Targeted anticancer
agents and apoptosis. Oncogene. 22:9075–9086. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yi R, Saito K, Isegawa N and Shirasawa H:
Alteration of cell cycle progression by Sindbis virus infection.
Biochem Biophys Res Commun. 462:426–432. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshida K, Morikawa T, Yokozuka N, Harada
S and Nishida A: Stereoselective synthesis of chiral
hydrocarbazoles via the catalytic diels-alder reaction of
siloxyvinylindole and cyclic Z-olefin. Tetrahedron Lett.
55:6907–6910. 2014. View Article : Google Scholar
|
|
31
|
Yuan Z, Lourenco SD, Sage EK, Kolluri KK,
Lowdell MW and Janes SM: Cryopreservation of human mesenchymal
stromal cells expressing TRAIL for human anti-cancer therapy.
Cytotherapy. 18:860–869. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jang YJ, Ma S, Terada Y and Erikson RL:
Phosphorylation of threonine 210 and the role of serine 137 in the
regulation of mammalian polo-like kinase. J Biol Chem.
277:44115–44120. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kellogg EH, Hejab NM, Howes S, Northcote
P, Miller JH, Díaz JF, Downing KH and Nogales E: Insights into the
distinct mechanisms of action of taxane and non-taxane microtubule
stabilizers from Cryo-EM structures. J Mol Biol. 429:633–646. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hanwell MD, Curtis DE, Lonie DC,
Vandermeersch T, Zurek E and Hutchison GR: Avogadro: An advanced
semantic chemical editor, visualization, and analysis platform. J
Cheminform. 4:172012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Becke AD: Density-functional
thermochemistry. III. The role of exact exchange. J Chem Phys.
98:5648–5652. 1998. View Article : Google Scholar
|
|
36
|
Lee C, Yang W and Parr RG: Development of
the colle-salvetti correlation-energy formula into a functional of
the electron density. Phys Rev B Condens Matter. 37:785–789. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morris GM, Huey R, Lindstrom W, Sanner MF,
Belew RK, Goodsell DS and Olson AJ: AutoDock4 and autodocktools4:
Automated docking with selective receptor flexibility. J Comp Chem.
30:2785–2791. 2009. View Article : Google Scholar
|
|
38
|
Baker NA, Sept D, Joseph S, Holst MJ and
McCammon JA: Electrostatics of nanosystems: Application to
microtubules and the ribosome. Proc Natl Acad Sci USA.
98:10037–10041. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vollmer CM, Ribas A, Butterfield LH,
Dissette VB, Andrews KJ, Eilber FC, Montejo LD, Chen AY, Hu B,
Glaspy JA, et al: P53 selective and nonselective replication of an
E1B-deleted adenovirus in hepatocellular carcinoma. Cancer Res.
59:4369–4374. 1999.PubMed/NCBI
|
|
40
|
Yasumura Y and Kawakita Y: A line of cells
derived from African green monkey kidney. Nippon Rinsho.
21:1209–1210. 1963.
|
|
41
|
Castedo M, Perfettini JL, Roumier T,
Andreau K, Medema R and Kroemer G: Cell death by mitotic
catastrophe: A molecular definition. Oncogene. 23:2825–2837. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vitale I, Galluzzi L, Castedo M and
Kroemer G: Mitotic catastrophe: A mechanism for avoiding genomic
instability. Nat Rev Mol Cell Biol. 12:385–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wei Y, Mizzen CA, Cook RG, Gorovsky MA and
Allis CD: Phosphorylation of histone H3 at serine 10 is correlated
with chromosome condensation during mitosis and meiosis in
tetrahymena. Proc Natl Acad Sci USA. 95:7480–7484. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Crosio C, Fimia GM, Loury R, Kimura M,
Okano Y, Zhou H, Sen S, Allis CD and Sassone-Corsi P: Mitotic
phosphorylation of histone H3: Spatio-Temporal regulation by
mammalian aurora kinases. Mol Cell Biol. 22:874–885. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van Vugt MA and Medema RH: Getting in and
out of mitosis with polo-like kinase-1. Oncogene. 24:2844–2859.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Donzelli M and Draetta GF: Regulating
mammalian checkpoints through Cdc25 inactivation. EMBO Rep.
4:671–677. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sakaue-Sawano A, Kobayashi T, Ohtawa K and
Miyawaki A: Drug-Induced cell cycle modulation leading to
cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC
Cell Biol. 12:22011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dalton WB, Yu B and Yang VW: P53
suppresses structural chromosome instability after mitotic arrest
in human cells. Oncogene. 29:1929–1940. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shieh SY, Shieh SY, Ikeda M and Prives C:
DNA damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen W, Yi R, Vahed M, Ohno Y, Tian Z, Guo
S, Ma X, Win NN, Li Q, Tsubosaka A, et al: Novel chiral chalcone
analogs that induce M phase arrest and apoptosis in HeLa cells. Med
Chem. 9:74–82. 2019.
|
|
51
|
Colin DJ, Hain KO, Allan LA and Clarke PR:
Cellular responses to a prolonged delay in mitosis are determined
by a DNA damage response controlled by Bcl-2 family proteins. Open
Biol. 5:1401562014. View Article : Google Scholar
|
|
52
|
Dephoure N, Dephoure N, Zhou C, Villén J,
Beausoleil SA, Bakalarski CE, Elledge SJ and Gygi SP: A
quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci
USA. 105:10762–10767. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Orth JD, Loewer A, Lahav G and Mitchison
TJ: Prolonged mitotic arrest triggers partial activation of
apoptosis, resulting in DNA damage and p53 induction. Mol Biol
Cell. 23:567–576. 2012. View Article : Google Scholar : PubMed/NCBI
|