Introduction
Cholangiocarcinoma (CCA), a bile duct carcinoma, is
a serious health concern in Northeastern Thailand (1) and its occurrence is increasing
globally (2). This type of cancer
is usually detected at the late stages, as the disease is
asymptomatic in the early stages, and effective treatments other
than resection are not available (2). In Northeastern Thailand, a suspected
etiology for CCA is a combination of the ingestion of carcinogens
and infection by Opisthorchis viverrini (Ov), a liver fluke.
The resulting tissue inflammation and DNA damage are linked to
oncogenic mutations that ultimately lead to cholangiocarcinogenesis
(1,3). Whole exome sequencing has identified
TP53, a tumor suppressor gene (‘guardian of the genome’)
(4), as the most highly mutated
gene (44.4% of cases) in Ov-associated CCA (5). In addition, TP53 is among the
most frequently mutated genes implicated in the pathogenesis and
poor prognosis of intrahepatic CCA (6). Furthermore, TP53 mutations have
been reported to reduce the efficacy and increase resistance toward
chemotherapeutic drugs (7),
diminishing the overall survival of patients with CCA (8). Similar to other types of cancer, the
overexpression of mutant p53 proteins has been detected in tissues
from patients with CCA (9), due to
the increased stability of mutant p53s compared to the wild-type
(WT) protein (10).
In response to DNA damage or oncogene activation,
p53 initiates cell cycle arrest, DNA repair, senescence and as a
last resort, apoptosis (11–13).
Mutations of tumor suppressor genes, in particular those of
TP53, allow tumor cells to suppress DNA repair and avoid
apoptosis (14). TP53
variants most often arise from missense mutations with the majority
located in 6 hotspots within the p53 DNA-binding domain: R175,
R273, R248, R249, G245 and R282 (15). These mutations compromise p53
biochemical functions, and also exert a dominant-negative effect on
WT p53 (16). In addition, some p53
variants exhibit oncogenic properties that promote a variety of
tumor malignancy behaviors (17).
Several p53 variants (e.g., A143, H175, W248 and H273) promote cell
proliferation through interaction with DNA topoisomerase 2-binding
protein 1 (TopBP1), p300 and nuclear factor (NF)-Y, leading to the
subsequent upregulation of cell cycle-related NF-Y target genes
(18). The H175 variant enhances
the transforming growth factor (TGF)-β-induced cell migration and
invasiveness of tumor cells by forming a complex with Smad and p63,
thereby inhibiting the function of p63 to oppose tumor promotion by
TGF-β (19). The variant p53 (H175
or H273)/p63 complex also enhances the ability of the Rab-coupling
protein (RCP) to promote epidermal growth factor receptor
(EGFR)/integrin receptor recycling and to induce cell motility
(20). The p53 variants (H175, H273
and K280) interact with E2F transcription factor 1 to increase the
expression of inhibitor of DNA binding 4 (ID4), which binds and
stabilizes interleukin (IL)-8 and growth-regulated oncogene-α
(GRO-α), thereby promoting the angiogenic properties of cancer
cells (21). More concerning is the
induction of resistance to chemotherapy by certain p53 variants
(H175 and H179) (22,23). The diverse ‘gain-of-function’ (GOF)
phenotypes of variant p53s require that not only the mutation
status, but also functional changes be taken into consideration
when devising appropriate cancer therapeutic strategies.
Given that mutations of TP53 are one of the
most common genetic alterations in CCA and are associated with a
poor prognosis (6,24), p53 variants in CCA may possess GOF
properties that promote the progression of CCA. The present study
investigated TP53 mutations in several CCA cell lines for
possible GOF properties. The information presented herein may have
a bearing on tumorigenesis and the treatment of cancer in patients
carrying such TP53 mutations.
Materials and methods
Cell lines and plasmids
Ov-associated human CCA cell lines, established by
Professor Banchob Sripa (Khon Kaen University, Thailand) and
Professor Stitaya Sirisinha (Mahidol University, Thailand), KKU-100
(25), KKU-M213 (26) and HuCCA-1 (27), were obtained from the Japanese
Collection Research Bioresources Cell Bank (JCRB). The human
non-small cell lung cell line, H1299, pCB6+ vector, WT p53 and p53
H175 pCB6+ plasmids were kindly provided by Professor Karen Vousden
(The Beatson Institute for Cancer Research, UK). Mycoplasma
contamination was absent in all cell lines (28). Recombinant pCB6+ based plasmids
carrying full-the length WT TP53K100 and del
TP53M213 were constructed with cDNA following the
RT-PCR amplification of RNA from KKU-100 and KKU-M213 cells. RNA
was extracted using an RNAspin Mini RNA Isolation kit (GE
Healthcare) and cDNA was amplified using the following primers:
forward, 5′-AAAATGAATTCCGCACCGTCCAGGGAGCAG-3′ and reverse,
5′-GGTGGATCCAGATCATCATATACAAG-3′. The p53-p72-Δ225-331 variant
(hereafter referred to as del p53Δ225-331) was
constructed by site-directed mutagenesis with WT p53 pCB6+ as a
template for overlapping extension-PCR (29) using the following primers: forward,
5′-CTATGAGCCGCCTGAGCAGATCCGTGGGCGTG-3′ and reverse,
5′-CACGCCCACGGATCTGCTCAGGCGGCTCATAG-3′. All PCR reactions were
performed using the MJ Mini Thermal Cycler (Bio-Rad Laboratories,
Inc.). Prior to the construction of recombinant pCB6+ vectors,
TP53 cDNA sequences were validated by insertion into the
pGEM®-T Easy Vector (Promega Corporation) followed by
transformation into the Escherichia coli strain, DH10B
(Invitrogen; Thermo Fisher Scientific, Inc.), and the sequencing of
plasmid inserts (Macrogen).
Cell culture and transfection
CCA cells were maintained in HAM's F-12 medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) and 1X antibiotic-antimycotic solution (Gibco;
Thermo Fisher Scientific, Inc.). H1299 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 44 mM NaHCO3 pH to
7.2, 10% heat-inactivated FBS (Gibco) and 1X antibiotic-antimycotic
solution (Gibco; Thermo Fisher Scientific, Inc.). All cell lines
were grown at 37°C under a humidified 5% CO2
atmosphere.
H1299 cells were transfected with 0.4 µg of
recombinant p53-pCB6+ or empty pCB6+ plasmid using
Effectene® transfection reagent (Qiagen GmbH). Following
48 h of transfection, clones were selected by incubation with 10%
FBS DMEM (Gibco; Thermo Fisher Scientific, Inc.) in the presence of
1 mg/ml G418 (Santa Cruz Biotechnology, Inc.).
TP53 gDNA status determination
Genomic DNA from HuCCA-1, KKU-100 and KKU-M213 cell
lines was extracted using the phenol-chloroform DNA extraction
method. In brief, cell pellets were first lysed using lysis buffer
[1% sodium dodecyl sulfate (SDS), 100 mM ethylenediaminetetraacetic
acid (EDTA), 1 mg/ml proteinase-K in 50 mM Tris-HCl pH 8.0]. DNA
was then extracted with a phenol/chloroform solution (25:24:1
phenol:chloroform:isoamyl alcohol), precipitated with isopropanol,
and dissolved in RNase-free water. DNA concentration was determined
with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.)
Specific TP53 regions were amplified using the primers
listed in Table I. Amplicons were
separated using 2% agarose gel electrophoresis and stained with
ethidium bromide. 18S rDNA was used as PCR control.
 | Table I.Primers used for amplification of
TP53 sequences in the study. |
Table I.
Primers used for amplification of
TP53 sequences in the study.
| Primer pair
name | Sequence
(5′→3′) | Amplicon size
(bp) |
|---|
| p53 E6-7 | Forward:
TTTGCGTGTGGAGTATTTGG | 742 |
|
| Reverse:
TCCAGTGTGATGATGGTGAG |
|
| p53 E7-I9 | Forward:
CCTCACCATCATCACACTGG | 806 |
|
| Reverse:
TGTCTTTGAGGCATCACTGC |
|
| p53 I9-E10 | Forward:
TGTTGCTTTTGATCCGTCAT | 150 |
|
| Reverse:
CCTCATTCAGCTCTCGGAAC |
|
| 18S rDNA (PCR
control) | Forward:
CCATCCAATCGGTAGTAGCG | 150 |
|
| Reverse:
GTAACCCGTTGAACCCCATT |
|
Western blot analysis
Cells were lysed with RIPA lysis buffer (1% Triton
X-100, 0.5% deoxycholic acid, 150 mM NaCl, 2 mM
Na3VO4, 50 mM NaF, 50 mM β-glycerophosphate,
1 mM dithiothreitol and protease inhibitor cocktail (Roche
Diagnostics) in 50 mM Tris-HCl pH 8.0). The protein concentration
was determined using Bradford protein dye reagent (Bio-Rad
Laboratories Inc.) and 40 µg of protein were separated by 10% SDS
polyacrylamide gel electrophoresis (SDS-PAGE) followed by transfer
onto nitrocellulose membranes. The membranes were incubated with 5%
skim milk in TBST (0.1% Tween-20 in 10 mM Tris-HCl pH 8.0), then
probed overnight at 4°C with primary antibodies (1:1,000 dilution)
against p53 (cat. no. sc-126; mouse monoclonal antibody), vimentin
(cat. no. sc-6260; mouse monoclonal antibody), GAPDH (cat. no.
sc-48166; goat polyclonal antibody) (for normalization of gel
loading; all from Santa Cruz Biotechnology, Inc.), zonula
occludens-1 (ZO-1; cat. no. 8193; rabbit monoclonal antibody), zinc
finger E-box-binding homeobox 1 (ZEB1; cat. no. 3396; rabbit
monoclonal antibody), N-cadherin (cat. no. 13116; rabbit monoclonal
antibody), E-cadherin (cat. no. 3195; rabbit monoclonal antibody),
claudin-1 (cat. no. 13255; rabbit monoclonal antibody) (all from
Cell Signaling Technology, Inc.), Cdc42 (cat. no. 05-542; mouse
monoclonal antibody), Rac (cat. no. 05-389; mouse monoclonal
antibody) and β-actin (cat. no. A1978; mouse monoclonal antibody)
(all from Merck). Membranes were then probed at room temperature
for 2 h with matched secondary antibodies [goat anti-mouse
horseradish peroxidase (HRP)-conjugated IgG (cat. no. sc-2005),
rabbit anti-goat HRP-conjugated IgG (cat. no. sc-2768; Santa Cruz
Biotechnology, Inc.) and goat anti-rabbit HRP-conjugated IgG (cat.
no. 7074; Cell Signaling Technology, Inc.)]. Immunoreactive protein
bands were visualized with the Clarity™ Western ECL kit (Bio-Rad
Laboratories, Inc.) and band intensities were quantified using a
G:BOX Chemi XT4 analysis system (Syngene).
Immunofluorescence assay
Cells were grown on slide coverslips coated with
0.01% poly-L-lysine (Sigma-Aldrich; Merck KGaA) until 80%
confluent. Samples were then treated with 4% paraformaldehyde and
2% sucrose in phosphate-buffered saline pH 7.4 (PBS) and
permeabilized with 0.25% Triton X-100, followed by treatment with
2% BSA in PBS. Cells were stained with mouse anti-p53 antibody
(1:100 dilution; cat. no. sc-126; Santa Cruz Biotechnology, Inc.)
at room temperature for 4 h, and an Alexa 488-conjugated goat
anti-mouse was used as a secondary antibody (1:200; cat. no.
A-11001; Thermo Fisher Scientific, Inc.) with 1 h incubation at
room temperature. Actin filaments and nuclei were stained at room
temperature for 30 min with Alexa 647-labeled phalloidin (1:1,000;
cat. no. A22287; Thermo Fisher Scientific, Inc.) and
4′,6′-diamidino-2-phenylindole (DAPI) (1:1,000; cat. no. P36962;
Thermo Fisher Scientific, Inc.) respectively. Coverslips were
mounted with 70% glycerol and examined under a confocal microscope
(FV10i Confocal Laser Scanning Microscope; Olympus
Corporation).
Cell growth assay
Cell growth was determined using a
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT)
colorimetric assay. In brief, cells (2×103) in
appropriate culture medium were incubated at 37°C in a 96-well
tissue culture plate. At the indicated time points (6, 24, 48 and
72 h) the culture medium was replaced with 100 µl of 0.5 mg/ml MTT
(PanReac AppliChem) in serum-free culture medium and incubated at
37°C for 4 h. The insoluble formazan dye was dissolved in 200 µl
dimethylsulfoxide and monitored at A540 nm using a
Thermo Labsystems Multiskan EX Microplate Reader (Artisan
Technology Group). Three independent experiments were performed in
quintuplet.
Colony formation assay
Cells (103) were plated onto 6-well
plates in DMEM containing 10% FBS. Following 7 days of incubation
at 37°C, cells were fixed with acetic acid/methanol (1:7 v/v) and
stained with 0.5% (w/v) crystal violet in 25% methanol. Three
independent experiments were performed each carried out in
duplicate. The number of colonies (>15 square pixels) was
counted using ImageJ software (version 1.8; NIH).
Knockdown of del p53M213 in
del p53M213-expressing H1299 cells using siRNA targeting
p53
Del p53M213-expressing H1299 cells were
transiently transfected with 30 nM siRNA specific for human
p53 mRNA (si-p53) (sense, 5′-GAAAUUUGCGUGUGGAGUAtt-3′ and
antisense, 5′-UACUCCACACGCAAAUUUCct-3′) (Ambion Inc.) or with
negative control siRNAs which has no homology to any known
mammalian gene (si-Neg) (Qiagen; cat. no. 1027280) using
Lipofectamine RNAimax Transfection Agent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Briefly, del p53M213 cells (2×105) suspended
in DMEM with 10% FBS were overlayed onto the transfection solution
containing 30 nM si-RNA in 6-well plates. Following 24 h of
transfection, the media were replaced with fresh complete media.
The invasive ability was determined at 48 h post-transfection as
described below.
Transwell migration/invasion
assay
Cells (105 in 200 µl of serum-free
medium) were plated onto a Transwell upper chamber (fitted with an
8-µm pore size polycarbonate membrane filter 6.5 mm in diameter)
(Corning, Inc.; for migration assay) or pre-coated (for invasion
assay) with 30 µg of Matrigel (Corning, Inc.). The lower chamber
was filled with 600 µl of DMEM containing 10% serum. Following a
6-h incubation at 37°C under a CO2 atmosphere,
non-migrating cells in the upper chamber were removed using a
cotton swab. The migrated/invasive cells attached to the lower
surface of the Transwell membrane were treated with 25% methanol
for 30 min, stained with a 0.5% crystal violet solution at room
temperature for 1 h and then counted (10 random fields) under a
light microscope (MEIJI VT Series; Meiji Techno Co., Ltd.; ×20
magnification).
Active Rac/Cdc42 pull-down assay
Levels of active cellular Rac/Cdc42 were determined
using the Rac/Cdc42 Activation Assay kit (Merck) following the
manufacturer's protocol with some modifications. In short, cells
(1.3×106) were grown on 100-mm culture dishes overnight,
lysed with 550 µl of ice-cold Mg2+ lysis/wash buffer
(MLB) containing a protease inhibitor cocktail. Samples were
incubated at 4°C for 10 min with 50 µl of glutathione-agarose beads
for pre-clearing. A total of 500 µl aliquots of pre-cleared cell
lysates were incubated with 15 µl of PAK1 PBD glutathione-agarose
beads for 45 min at 4°C. Bead-bound active Rac/Cdc42 was sedimented
by pulsed centrifugation (14,000 × g) for 5 sec at 4°C (Legend
Micro 17R Centrifuge; Thermo Fisher Scientific, Inc.). Prior to
agarose bead isolation, a 500 µl aliquot of pre-cleared cell lysate
was incubated for 30 min at 30°C with GTP (100 µM GTP containing 10
mM EDTA) or GDP (1 mM GDP containing 10 mM EDTA) solution as
positive and negative controls, respectively. Protein-attached
beads were resuspended in 20 µl of 2X SDS loading dye, boiled for 5
min and separated by 12% SDS-PAGE and subjected to immunoreactive
Rac/Cdc42 detection using reagents supplied with the kit.
Statistical analysis
Data were analyzed using a two-tailed Student's
t-test (comparisons between 2 groups) and one-way ANOVA with
Tukey's post hoc test (for >2 groups). Data are presented as the
means ± standard error of the mean (SEM). All statistical analysis
was carried out using SPSS version 22.0 (SPSS Inc.) and a result
was considered statistically significant when the P-value was
<0.05.
Results
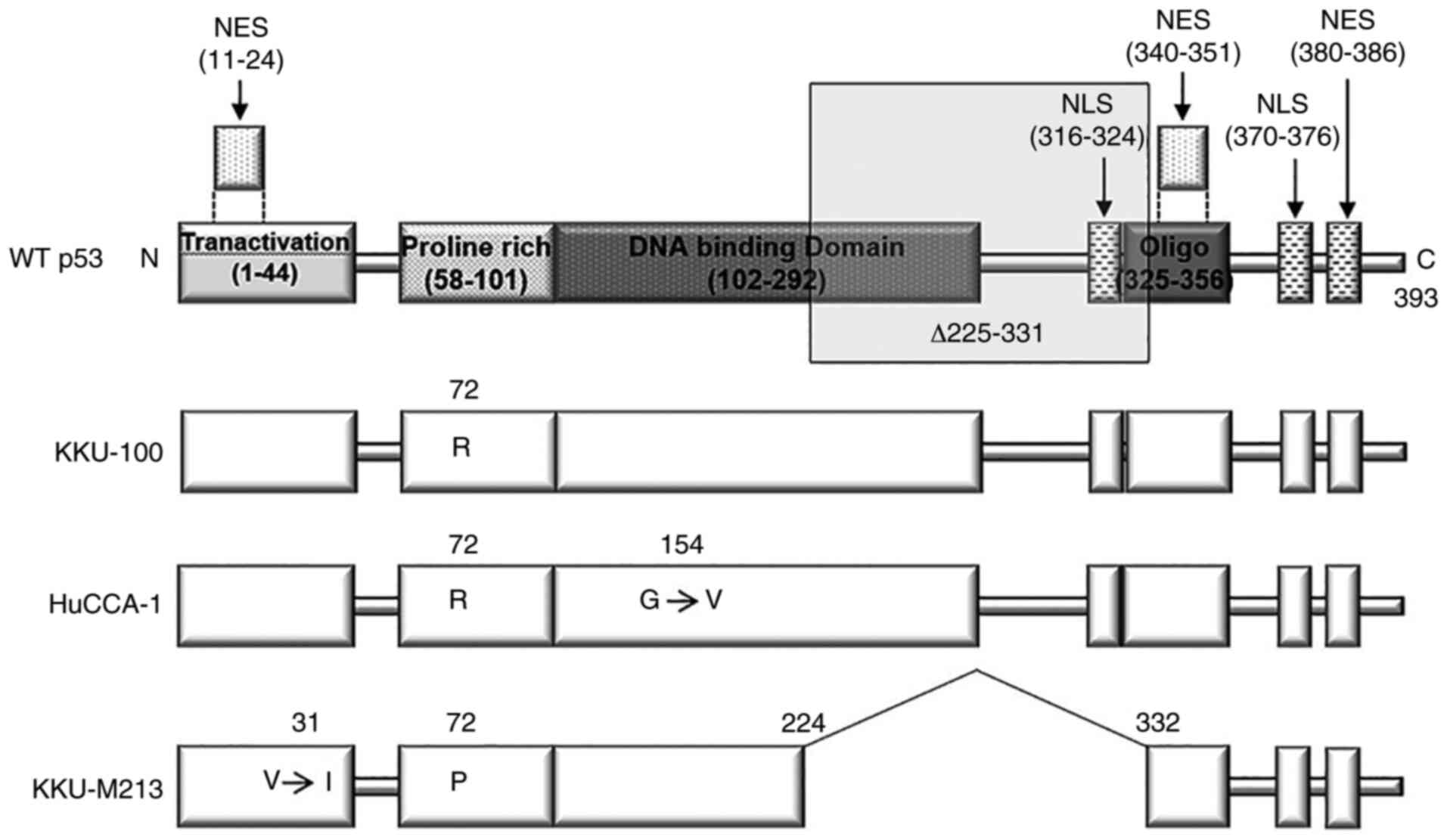
TP53 status of CCA cell lines
Mutations in TP53 are often encountered in
various types of cancer, including CCA (5,6).
TP53 sequences from 3 CCA cell lines, namely KKU-100,
KKU-M213 and HuCCA-1, were determined from their respective cDNA.
The sequencing results revealed a p53 variant R72 [p53-R72, a
common isoform (30)] in the
KKU-100 cell line (hereafter referred to as p53K100); a
second variant had a deletion of amino acids 225–331 (corresponding
to the loss of exons 7–9) together with an I31 point mutation in
the same allele (p53-p72-Δ225-331-V31I) in KKU-M213 (hereafter
referred to as del p53M213). In addition, R72 with V154
point mutations was found in cis (p53-R72-G154V) in HuCCA-1
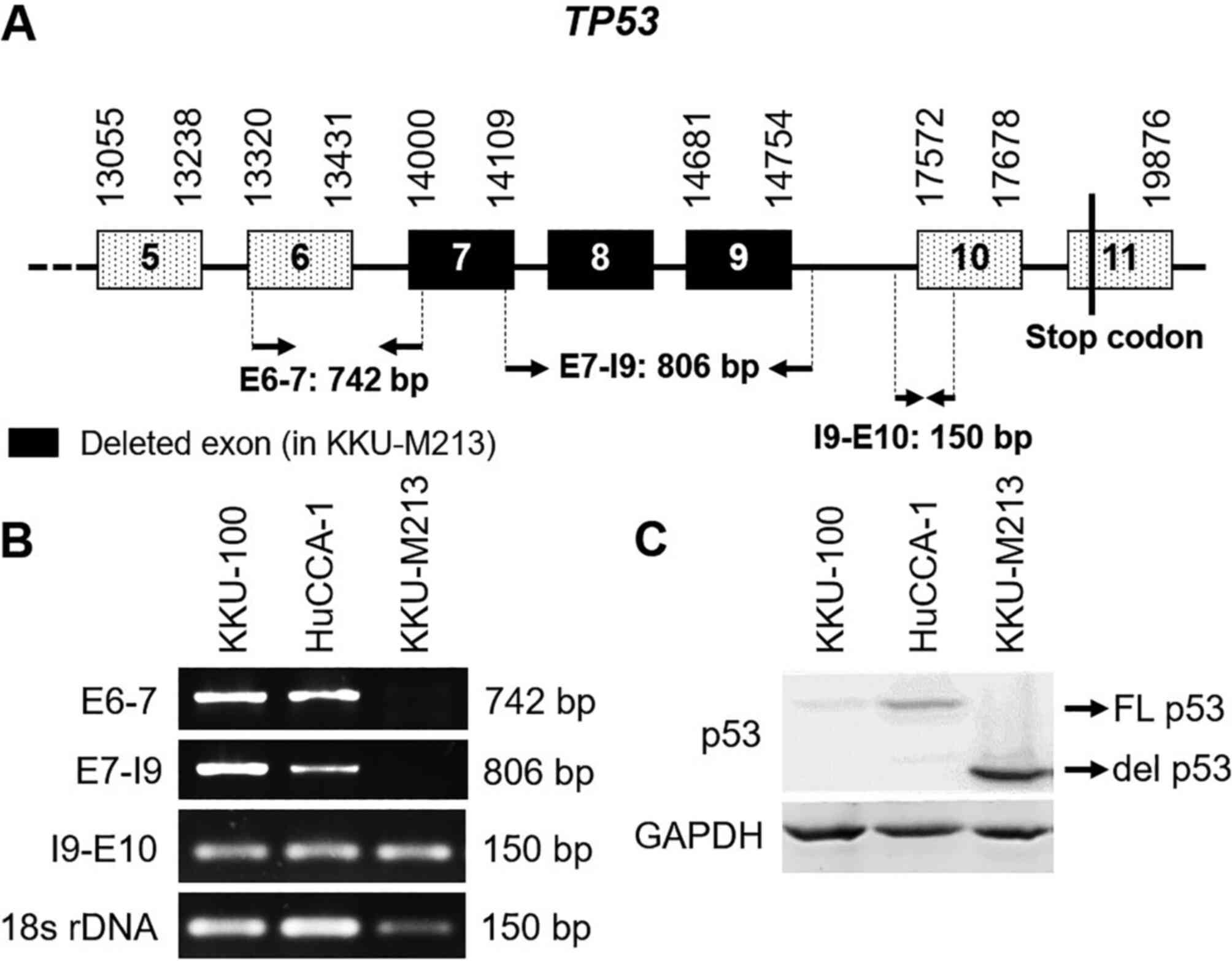
cells (Fig. 1). To determine
whether del p53M213 resulted from alternate
splicing or deletion in TP53, the gene was PCR-amplified
using 3 sets of primer pairs (Table
I) targeting exons 6 and 7 (E6-7), exon 7 and intron 9 (E7-I9),
and intron 9 and exon 10 (I9-E10). The amplification of 18S rDNA
was utilized as an internal control (Fig. 2A). In the KKU-M213 cells, a product
for I9-E10 was detected; however, amplicons for E6-7 and E7-I9 were
absent. By contrast, all 3 amplicons were obtained with the KKU-100
and HuCCA-1 cells (Fig. 2B). These
results indicate that the KKU-M213 cell line carries a single
allele of the del TP53 mutant with the normal TP53
allele absent, although the unlikely event of homozygosity of the
del TP53 alleles cannot be ruled out. The boundaries of the
deleted sequence were not determined. Western blot analysis
confirmed the presence of the smaller del p53M213
protein compared to full-length p53 present in the other 2 cell
lines (Fig. 2C). The presence of
low immunoreactive WT p53, as has been observed in KKU-100, is a
well-known phenomenon (31).
Establishment of stable p53-expressing
H1299 cell lines
To examine the functions of the del p53 variant
found in the highly invasive KKU-M213 cell line, stably transfected
p53-null H1299 cell lines expressing various p53 variants were
generated. Cell lines were established that expressed del
p53M213 variant [in cells transfected with recombinant
pCB6+ vector carrying KKU-M213 del TP53M213 cDNA
(expressing p53-Δ225-331-V31I)], del p53Δ225-331 variant
(in cells transfected with a recombinant vector carrying del
TP53Δ225-331 cDNA constructed by site-directed
mutagenesis of WT TP53 cDNA) and a common point mutant p53,
H175 variant, (in cells transfected with recombinant plasmid
carrying TP53 H175 cDNA) (Fig.
3A). The inability to generate stably transfected H1299 cell
lines expressing WT and R72 (from KKU-100) p53 may be due to their
anti-proliferative activities.
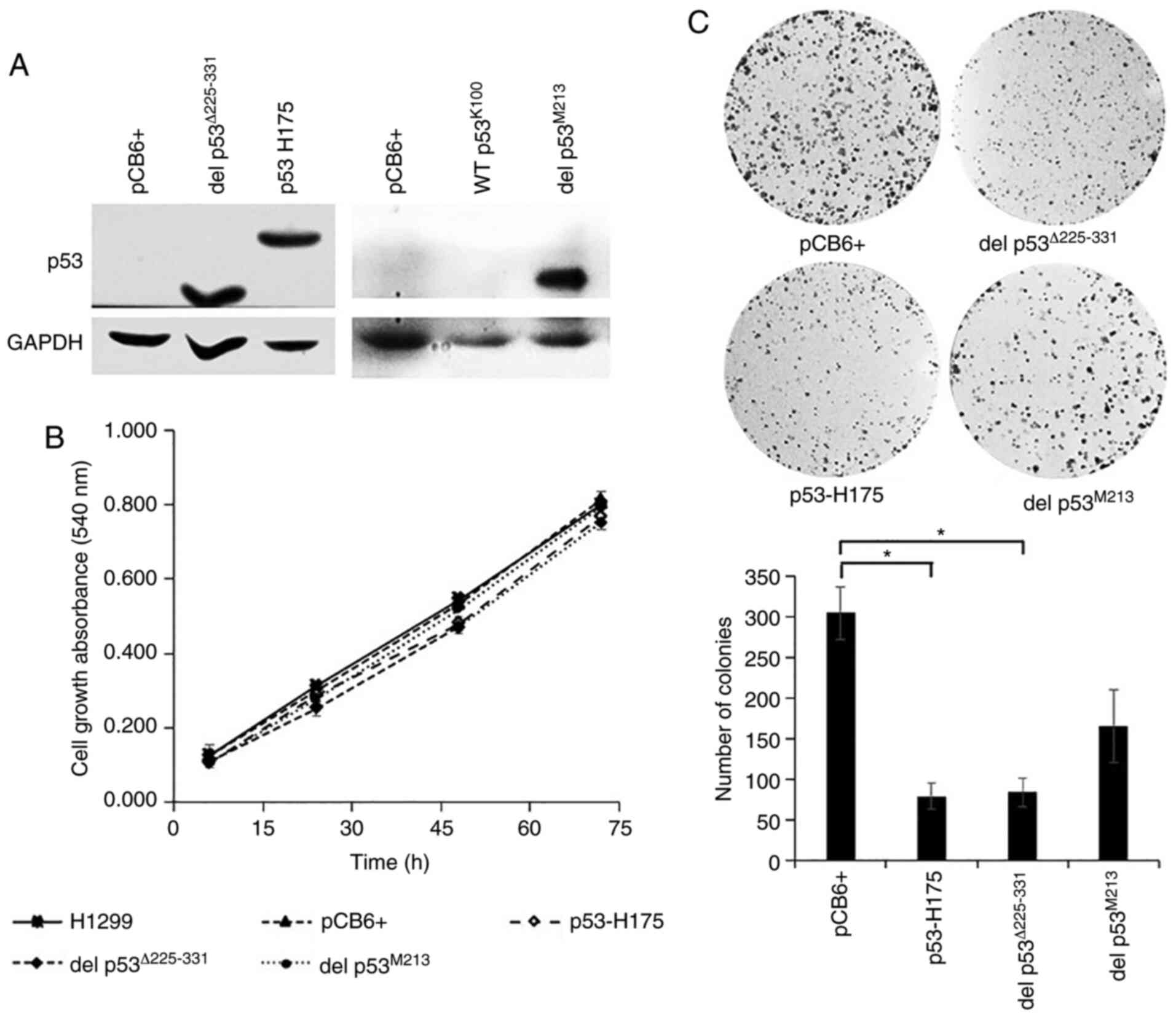 | Figure 3.Effect of p53 variants on cell growth
analyzed by MTT and colony formation assays. H1299 cells were
stably transfected with recombinant pCB6+ carrying various
TP53 cDNA using Effectene® reagent. (A) Western
blot analysis was carried out as described in the Materials and
methods section and in the legend of Fig. 2. pCB6+, del p53Δ225-331,
p53 H175, WT p53K100, and del p53M213: cells
transfected with empty vector, with vector carrying TP53
cDNA constructed by site-directed mutation of wild-type p53 to
delete amino acids 225–331, with vector carrying TP53 cDNA
expressing p53 H175 variant, with vector carrying KKU-100
TP53 cDNA expressing p53 R72 variant, and with vector
carrying KKU-M213 TP53 cDNA expressing del
p53M213 variant, respectively. (B) For cell growth
assay, cells were cultured in 10% FBS media for the indicated
periods of time. Viable cells were determined using MTT
colorimetric assay and expressed as mean ± SEM from three
independent experiments each performed in quintuplet. (C) For
colony formation assay, cells (103) were cultured in 10%
FBS media. Following 7 days of incubation, the number of colonies
was counted and compared to pCB6+ transfected cells. H1299,
untransfected H1299 cells; pCB6+, p53 H175 and del
p53M213: H1299 cells transfected with empty vector
(pCB6+), p53 H175 and del p53M213, respectively.
*P≤0.05. |
Growth inhibitory effect of del p53
variants
A common role of p53 is the inhibition of cell
growth and proliferation. Several p53 mutants exhibit the loss of
such functions, and fail to suppress cell growth and proliferation
(32). In the present study, to
determine whether del p53Δ225-331 and del
p53M213 have lost growth inhibitory activity, the growth
rate of the cells expressing these p53 variants was compared with
that of cells expressing p53 H175, a mutant that does not exhibit
growth inhibitory activity (16),
cells transfected with empty pCB6+ vector, and untransfected cells.
As shown by the results of MTT assay, for the cells that were
allowed to grow for 72 h, a similar growth rate was observed for
all 5 cell types. Cells carrying del p53Δ225-331, del
p53M213, p53 H175, empty vector and untransfected cells
exhibited a 6.8-, 7.5-, 7.1-, 6.5- and 6.3- fold increase in the
absorbance at 72 h compared to the cultures at 6 h (Fig. 3B), indicating that del
p53Δ225-331 and del p53M213 variants lacked
the growth-suppressive property.
To investigate the long-term growth-inhibitory
ability of p53 variants, colony formation assay was performed.
Surprisingly, p53 H175 and del p53Δ225-331 exhibited
some growth-suppressive effects, as shown by the decrease in the
number and size of the colonies, when compared to the cells
transfected with the empty vector. By contrast, the number of
colonies was not significantly decreased in the del
p53M213 cells (Fig. 3C).
It appeared that del p53M213 did not possess long-term
growth inhibitory activity. However, del p53Δ225-331 was
capable of limiting cell growth, suggesting that the presence of
I31 in cis with Δ225-331 in del p53M213 may contribute
to the defect in the growth-suppressive ability of this p53
variant.
Del p53M213 variant
cellular localization
The transcription factor TP53 contains 3
domains responsible for translocation into the nucleus (33). One of these regions is located
within amino acid 316–324 region (Fig.
1) and its absence (del p53M213) can compromise
nuclear import. In the present study, the localization of p53 was
monitored in H1299 cells transiently overexpressing WT
p53K100, del p53Δ225-331 and del
p53M213. The cells were fixed, permeabilized and
immunostained with anti-p53 antibody and visualized using an Alexa
488-conjugated secondary antibody. Alexa 647-labeled phalloidin was
used for the detection of actin and DAPI for nuclear localization.
Samples were viewed under a confocal microscope (×60 magnification)
with del p53Δ225-331 and del p53M213 variants
exhibiting localization to both nucleus and cytoplasm; in contrast,
WT p53K100 was only found in the nucleus (Fig. 4).
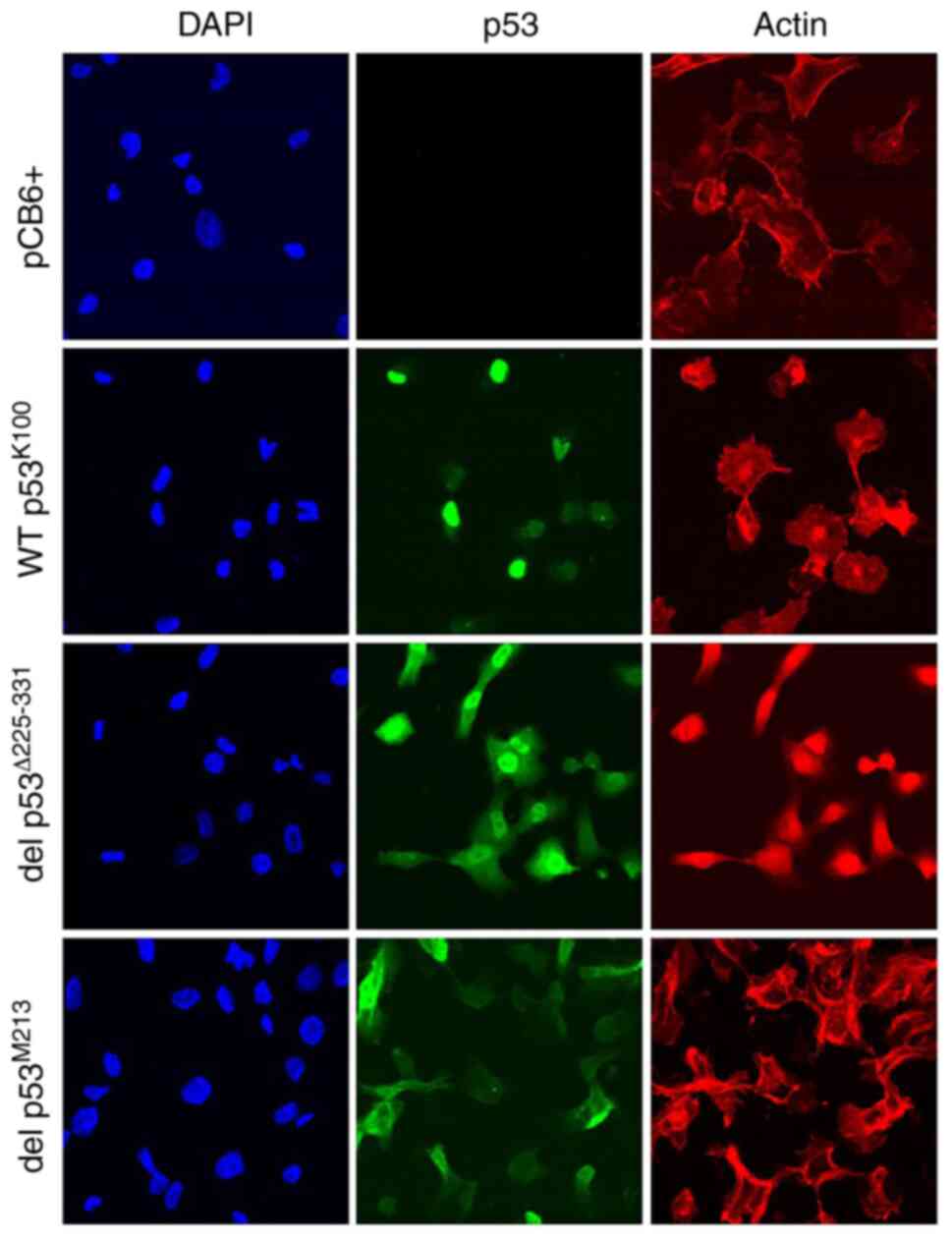 | Figure 4.Subcellular localization of WT
p53K100 and del p53M213 transiently expressed
in p53-null H1299 cells. Cells were transfected as described in the
legend to Fig. 3, fixed,
permeabilized, and treated with mouse anti-p53 antibodies followed
by Alexa 488-conjugated goat anti-mouse secondary antibodies (p53,
in green), with 4′,6-diamidino-2-phenylindole (DAPI, in blue) and
with Alexa 647-labeled phalloidin (Actin, in red), then observed
under a confocal microscope (×60 magnification). pCB6+, cells
transfected with the empty vector. |
Effects of del p53 variants on cell
migration, invasion and epithelial-mesenchymal transition
(EMT)
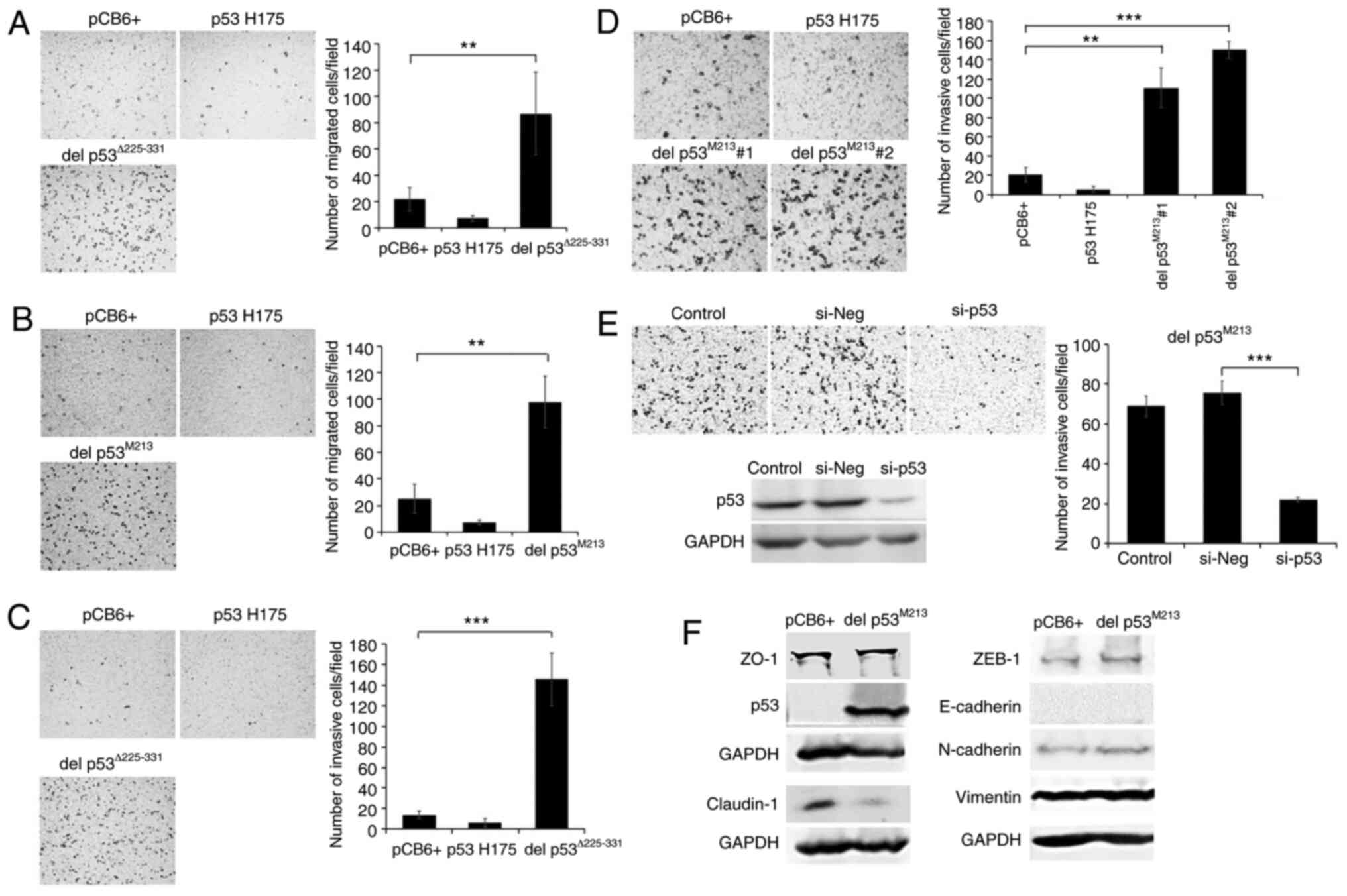
A number of p53 variants promote cancer metastasis
by inducing cancer cell migration and invasiveness (19,20,34,35).
In the present study, H1299 cells stably expressing
p53Δ225-331 and del p53M213 variants were
assayed for their migratory and invasive abilities using a
Transwell system. In addition, these cells were also examined for
their EMT status, a condition in which epithelial cells loss
cell-cell contact and acquire mesenchymal phenotypes enhancing
their migratory ability. The del p53Δ225-331- and del
p53M213-expressing H1299 cells exhibited significant
increases in migration (4–5 fold) and invasion (7–10 fold) compared
to the controls. An increase in these properties was not observed
with the p53 H175-expressing cells (Fig. 5A-D). To verify whether the
invasion-promoting effects were due to the presence of del
p53M213, this variant was knocked down using
p53-specific siRNA. The silencing of del p53M213
suppressed the invasiveness of the del p53M213 cells by
68% compared to the cells transfected with si-Neg (Fig. 5E). However, the silencing of p53
only marginally affected KKU-M213 cell invasion (data not shown),
possibly since multiple genetic alterations occurred in KKU-M213
and some of those may override the invasive effects of del
p53M213. In addition, in the transfected H1299 cells,
del p53M213 induced EMT compared to the empty
vector-transfected control cells, as demonstrated by the decrease
in the levels of the epithelial cell marker, claudin-1, a tight
junction protein involved in cell-cell adhesion (Fig. 5F). However, the levels of other
epithelial cell markers, such as E-cadherin and ZO-1, as well as
the mesenchymal cell markers, N-cadherin, vimentin and ZEB-1,
remained unaltered. It is worth noting that the H1299 cell line
exhibited an elevated EMT state, as shown by very low E-cadherin
and high vimentin levels.
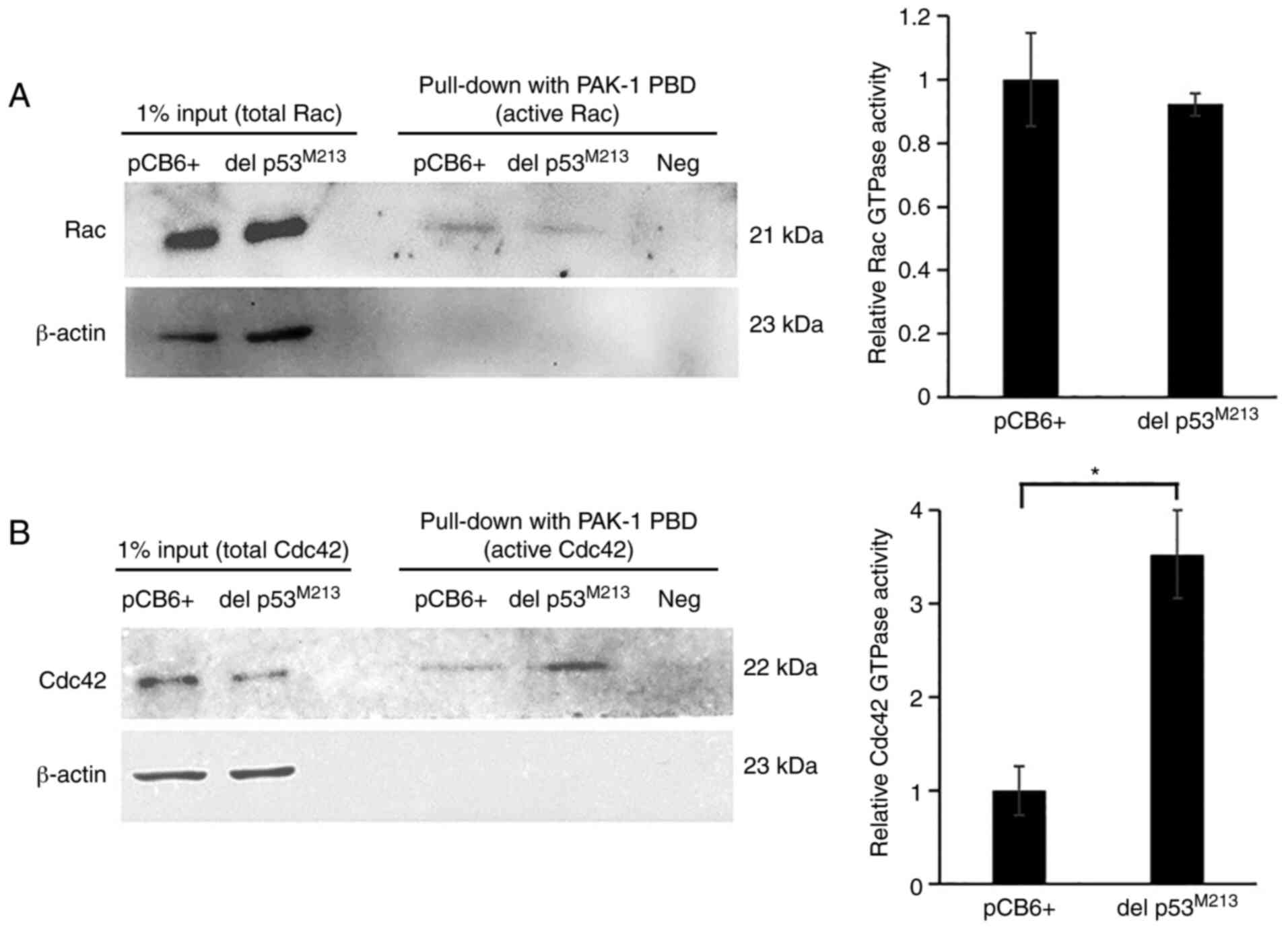
Effects of del p53 on Cdc42 and Rac
activities
In addition to EMT, Rho GTPase family members, in
particular Cdc42 and Rac, are key regulators of actin dynamics
involved in cell migration and cancer metastasis (36). Some p53 variants enhance Rac
activation (37). Therefore, the
possible role of del p53 variant in promoting H1299 cell
invasiveness, through elevating active Cdc42 and Rac levels, was
investigated in the present study. A pull-down assay was utilized
to monitor the active (GTP-bound) forms using GST-PAK fusion
protein affinity beads followed by western blot analysis of Cdc42
and Rac. In del p53M213-expressing H1299 cells, the
level of active Cdc42, but not that of active Rac, was
significantly higher than that in the empty vector-transfected
control cells (P-value, 0.048). (Fig.
6).
Discussion
Tumor suppressor TP53 is the most often gene
mutated in several human cancers (32,38)
including CCA (5,6). Variant p53s not only lose tumor
suppression functions, but often acquire oncogenic GOF properties
that promote cancer progression and are associated with a poor
prognosis (39,40). The presence of such p53 variants
poses additional problems of cell invasiveness and metastasis
(41). Among the 3 CCA cell lines
used in the present study, the KKU-M213 cells exhibits the most
highly invasive properties followed by the HuCCA-1 and KKU-100
cells, respectively (42). These
highly invasive KKU-M213 cell expressed a truncated del
p53M213 variant that lacks anti-growth activity. This
variant was localized in both cytoplasm and nucleus, presumably due
to the loss of a nuclear localizing signal (located in the missing
amino acid 316–324 region). The del p53M213 variant also
enhanced cancer cell migration and invasiveness. These effects are
possibly due to attenuation of cell-cell adhesion through
downregulation of claudin-1, and promotion of migration from Cdc42
activation. However, the oncogenic property of the del p53 was not
confirmed in animals. Together, these properties of del
p53M213 could contribute to the highly invasive nature
of the cancer.
The presence of a del p53Δ225-331 variant
has been reported in this and other CCA cell lines (31) in breast cancer patients (43,44),
as well as in a therapy-related acute myeloid leukemia (tAML)
patient (45). The del
p53Δ225-331 is encoded by a rare TP53 mutant
(45,46) and its properties are not yet well
described.
The majority of p53 variants lose the abilities to
regulate transcription of its target genes, particularly those
involved in cell cycle arrest (e.g., cyclin-dependent kinase
inhibitors) (47,48) and apoptotic pathways (e.g.,
pro-apoptosis proteins, death receptors and apoptosis execution
factors) (12,48). These loss of function mutations are
present within the p53 DNA binding domain, limiting transcriptional
activation activity due to impaired p53/DNA interactions.
Transcriptional activity, from a p53-responsive element of
p21, is absent in a del p53 variant missing amino acid
residues 225–331 within the DNA-binding domain (45). Likewise, the I31 p53 variant, found
in hematological malignancies and solid cancers (49,50),
exhibits lower transcriptional activity and lower antiproliferative
activity than WT p53 (51).
Claudin-1, a major component of tight junctions,
functions in the binding of adjacent epithelial cells and in
regulating epithelial permeability (paracellular transport)
(52). The expression of claudin-1
varies among types of cancer cells (53), with its downregulation reported in
breast (54), colon cancer
(55) and lung adenocarcinoma
(56). Low claudin-1 levels are
associated with the short-term survival of patients with several
types of cancer, including lung adenocarcinoma (56), hepatocellular carcinoma (57) and aggressive forms of colorectal
carcinoma (58). The silencing of
claudin-1 promotes cancer invasiveness and metastasis (56); by contrast, the overexpression of
claudin-1 inhibits cell dissociation and suppresses cancer
invasiveness and metastasis of lung adenocarcinoma. The
downregulation of claudin-1 found in del p53-expressing CCA cells
may result in reduced cell-cell adhesion, promote cell dissociation
and migration, and may ultimately lead to cancer cell invasion and
metastasis.
The Rho-GTPase family consists of a group of small G
proteins that include Cdc42 and Rac. These proteins are involved in
the regulation of actin cytoskeleton dynamics, a crucial process
involved in cell migration (36).
Variant p53-H175 promotes metastasis by inducing the formation of
active Rac1-GTP through the inhibition of a SUMO-specific protease
that limits Rac1 de-SUMOylation (37), leading to enhances cell migration.
In the present study, del p53M213 was found to enhance
the levels of active Cdc42-GTP, a regulator of cell migration, by
promoting filopodia formation (59). The mechanism by which del
p53M213 promotes Cdc42 activation is not yet known;
however, the del p53M213 variant may directly facilitate
Cdc42-GTP formation as it is significantly present in the cell
cytoplasm. This p53 variant may mediate, directly or indirectly,
the expression of genes encoding proteins that regulate Cdc42
activation (such as guanine nucleotide exchange factor and
GTPase-activating protein) (60). A
number of p53 variants are known to interact with other
transcription factors or nuclear proteins, allowing them to
regulate expression of genes distinct from those responsive to WT
p53 (61). A number of p53 variants
are also able to regulate receptor/integrin translocation. For
instance, p53 H175 and p53 H273 bind and inhibit p63 and enhance
the activity of the Rab-coupling protein. This is important for
recycling EGFR and integrin receptors from endosomes to the plasma
membrane to promote cell movement (39). The attachment of integrin to the
extracellular matrix proteins stimulates Cdc42-GDP/Cdc42-GTP
exchange, inducing actin polymerization and membrane protrusions
associated with cell motility (62). Thus, del p53M213 variant
may enhance cancer cell invasiveness through Cdc42 activation
together with claudin-1 downregulation; however, the actual
mechanisms require further investigation.
In conclusion, KKU-M213 CCA cells harbor a single
allele of a TP53 del mutant, del p53M213. This
variant has lost WT p53 growth inhibitory activity and acquired
gain-of-functions, such as stimulating cell migration and
invasiveness, downregulating claudin-1 and activating Cdc42. A more
complete understanding of the role of del p53M213 and
other del p53 variants in Ov-associated CCA tumorigenesis should
help in developing better therapeutic strategies tailored for
patients with tumors carrying such truncated TP53.
Acknowledgements
The authors would like to thank Professor Karen
Vousden (The Beatson Institute for Cancer Research, UK) for
providing the plasmids and the H1299 cell line, Professor Prapon
Wilairat (Mahidol University, Thailand) for the critical reading of
the manuscript, Dr Laran Jensen (Mahidol University, Thailand) for
English editing and the Center of Nanoimaging and Olympus
Bioimaging Center, Faculty of Science, Mahidol University for
providing assistance with confocal microscopy. The proofreading of
this manuscript was supported by the Editorial Office, Faculty of
Graduate Studies, Mahidol University.
Funding
The present study was supported by grants from the
Thailand Research Fund (grant no. BRG5480019 and IRG5980008) and
the Central Instrument Facility, Faculty of Science, Mahidol
University to TS, and a scholarship from the Royal Golden Jubilee
Ph.D. Program, Thailand Research Fund (scholarship no.
PHD/0255/2552) to JP.
Availability of data and materials
The data sets used and/or analyzed in the study are
available from the corresponding author on request.
Authors' contributions
JP performed all the experiments, analyzed the data,
and prepared the manuscript. TS designed the study, analyzed the
data, and help prepare the manuscript. Both authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Glossary
Abbreviations
Abbreviations:
|
CCA
|
cholangiocarcinoma
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
EDTA
|
ethylenediaminetetraacetic acid
|
|
EGFR
|
epidermal growth factor receptor
|
|
FBS
|
fetal bovine serum
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
GOF
|
gain-of-function
|
|
GRO-α
|
growth-regulated oncogene-α
|
|
HRP
|
horseradish peroxidase
|
|
ID4
|
inhibitor of DNA binding 4
|
|
IL-8
|
interleukin 8
|
|
MLB
|
Mg2+ lysis/wash buffer
|
|
MTT
|
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
|
|
NF-Y
|
nuclear factor Y
|
|
Ov
|
Opisthorchis viverrini
|
|
RCP
|
Rab-coupling protein
|
|
SDS-PAGE
|
sodium dodecyl sulfate polyacrylamide
gel electrophoresis
|
|
SEM
|
standard error of the mean
|
|
tAML
|
therapy-related acute myeloid
leukemia
|
|
TGF-β
|
transforming growth factor β1
|
|
TopBP1
|
DNA topoisomerase 2-binding protein
1
|
|
WT
|
wild-type
|
References
|
1
|
Sripa B, Kaewkes S, Sithithaworn P,
Mairiang E, Laha T, Smout M, Pairojkul C, Bhudhisawasdi V, Tesana
S, Thinkamrop B, et al: Liver fluke induces cholangiocarcinoma.
PLoS Med. 4:e2012007. View Article : Google Scholar
|
|
2
|
Khan SA, Tavolari S and Brandi G:
Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 39
(Suppl 1):S19–S31. 2019. View Article : Google Scholar
|
|
3
|
Sripa B, Brindley PJ, Mulvenna J, Laha T,
Smout MJ, Mairiang E, Bethony JM and Loukas A: The tumorigenic
liver fluke Opisthorchis viverrini-multiple pathways to
cancer. Trends Parasitol. 28:395–407. 2012. View Article : Google Scholar
|
|
4
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View Article : Google Scholar
|
|
5
|
Ong CK, Subimerb C, Pairojkul C, Wongkham
S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et
al: Exome sequencing of liver fluke-associated cholangiocarcinoma.
Nat Genet. 44:690–693. 2012. View Article : Google Scholar
|
|
6
|
Huang SB and Zheng CX: Gene alterations
and epigenetic changes in intrahepatic cholangiocarcinoma. Expert
Rev Anticancer Ther. 17:89–96. 2017. View Article : Google Scholar
|
|
7
|
Ahn DH, Javle M, Ahn CW, Jain A, Mikhail
S, Noonan AM, Ciombor K, Wu C, Shroff RT, Chen JL and Bekaii-Saab
T: Next-generation sequencing survey of biliary tract cancer
reveals the association between tumor somatic variants and
chemotherapy resistance. Cancer. 122:3657–3666. 2016. View Article : Google Scholar
|
|
8
|
Churi CR, Shroff R, Wang Y, Rashid A, Kang
HC, Weatherly J, Zuo M, Zinner R, Hong D, Meric-Bernstam F, et al:
Mutation profiling in cholangiocarcinoma: Prognostic and
therapeutic implications. PLoS One. 9:e1153832014. View Article : Google Scholar
|
|
9
|
Liu XF, Zhang H, Zhu SG, Zhou XT, Su HL,
Xu Z and Li SJ: Correlation of p53 gene mutation and expression of
P53 protein in cholangiocarcinoma. World J Gastroenterol.
12:4706–4709. 2006. View Article : Google Scholar
|
|
10
|
Vijayakumaran R, Tan KH, Miranda PJ, Haupt
S and Haupt Y: Regulation of mutant p53 protein expression. Front
Oncol. 5:2842015. View Article : Google Scholar
|
|
11
|
Yee KS and Vousden KH: Complicating the
complexity of p53. Carcinogenesis. 26:1317–1322. 2005. View Article : Google Scholar
|
|
12
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar
|
|
13
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumour suppressor p53. Nature. 458:1127–1130.
2009. View Article : Google Scholar
|
|
14
|
Hanel W and Moll UM: Links between mutant
p53 and genomic instability. J Cell Biochem. 113:433–439. 2012.
View Article : Google Scholar
|
|
15
|
Mello SS and Attardi LD: Not all p53
gain-of-function mutants are created equal. Cell Death Differ.
20:855–857. 2013. View Article : Google Scholar
|
|
16
|
Willis A, Jung EJ, Wakefield T and Chen X:
Mutant p53 exerts a dominant negative effect by preventing
wild-type p53 from binding to the promoter of its target genes.
Oncogene. 23:2330–2338. 2004. View Article : Google Scholar
|
|
17
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar
|
|
18
|
Liu K, Ling S and Lin WC: TopBP1 mediates
mutant p53 gain of function through NF-Y and p63/p73. Mol Cell
Biol. 31:4464–4481. 2011. View Article : Google Scholar
|
|
19
|
Adorno M, Cordenonsi M, Montagner M,
Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo
V, et al: A mutant-p53/Smad complex opposes p63 to empower
TGFbeta-induced metastasis. Cell. 137:87–98. 2009. View Article : Google Scholar
|
|
20
|
Muller PA, Trinidad AG, Timpson P, Morton
JP, Zanivan S, van den Berghe PV, Nixon C, Karim SA, Caswell PT,
Noll JE, et al: Mutant p53 enhances MET trafficking and signalling
to drive cell scattering and invasion. Oncogene. 32:1252–1265.
2013. View Article : Google Scholar
|
|
21
|
Fontemaggi G, Dell'Orso S, Trisciuoglio D,
Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany
E, et al: The execution of the transcriptional axis mutant p53,
E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol.
16:1086–1093. 2009. View Article : Google Scholar
|
|
22
|
Blandino G, Levine AJ and Oren M: Mutant
p53 gain of function: Differential effects of different p53 mutants
on resistance of cultured cells to chemotherapy. Oncogene.
18:477–485. 1999. View Article : Google Scholar
|
|
23
|
Do PM, Varanasi L, Fan S, Li C, Kubacka I,
Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR, et al:
Mutant p53 cooperates with ETS2 to promote etoposide resistance.
Genes Dev. 26:830–845. 2012. View Article : Google Scholar
|
|
24
|
Ross JS, Wang K, Gay L, Al-Rohil R, Rand
JV, Jones DM, Lee HJ, Sheehan CE, Otto GA, Palmer G, et al: New
routes to targeted therapy of intrahepatic cholangiocarcinomas
revealed by next-generation sequencing. Oncologist. 19:235–242.
2014. View Article : Google Scholar
|
|
25
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholangiocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar
|
|
26
|
Sripa B, Seubwai W, Vaeteewoottacharn K,
Sawanyawisuth K, Silsirivanit A, Kaewkong W, Muisuk K, Dana P,
Phoomak C, Lert-Itthiporn W, et al: Functional and genetic
characterization of three cell lines derived from a single tumor of
an Opisthorchis viverrini-associated cholangiocarcinoma
patient. Hum Cell. 33:695–708. 2020. View Article : Google Scholar
|
|
27
|
Sirisinha S, Tengchaisri T, Boonpucknavig
S, Prempracha N, Ratanarapee S and Pausawasdi A: Establishment and
characterization of a cholangiocarcinoma cell line from a Thai
patient with intrahepatic bile duct cancer. Asian Pac J Allergy
Immunol. 9:153–157. 1991.
|
|
28
|
Netto C, Thomaz-Soccol V, Sepúlveda L,
Oliveira Garcia G and Timenetsky J: Quality control of
biotechnological inputs detecting mycoplasma. Braz Arch Biol Techn.
58:239–243. 2015. View Article : Google Scholar
|
|
29
|
Hansson MD, Rzeznicka K, Rosenback M,
Hansson M and Sirijovski N: PCR-mediated deletion of plasmid DNA.
Anal Biochem. 375:373–375. 2008. View Article : Google Scholar
|
|
30
|
Matlashewski GJ, Tuck S, Pim D, Lamb P,
Schneider J and Crawford LV: Primary structure polymorphism at
amino acid residue 72 of human p53. Mol Cell Biol. 7:961–963. 1987.
View Article : Google Scholar
|
|
31
|
Phimsen S, Kuwahara K, Nakaya T, Ohta K,
Suda T, Rezano A, Kitabatake M, Vaeteewoottacharn K, Okada S, Tone
S and Sakaguchi N: Selective cell death of p53-insufficient cancer
cells is induced by knockdown of the mRNA export molecule GANP.
Apoptosis. 17:679–690. 2012. View Article : Google Scholar
|
|
32
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar
|
|
33
|
Lim YP, Lim TT, Chan YL, Song AC, Yeo BH,
Vojtesek B, Coomber D, Rajagopal G and Lane D: The p53
knowledgebase: An integrated information resource for p53 research.
Oncogene. 26:1517–1521. 2007. View Article : Google Scholar
|
|
34
|
Weissmueller S, Manchado E, Saborowski M,
Morris JP IV, Wagenblast E, Davis CA, Moon SH, Pfister NT,
Tschaharganeh DF, Kitzing T, et al: Mutant p53 drives pancreatic
cancer metastasis through cell-autonomous PDGF receptor β
signaling. Cell. 157:382–394. 2014. View Article : Google Scholar
|
|
35
|
Vogiatzi F, Brandt DT, Schneikert J, Fuchs
J, Grikscheit K, Wanzel M, Pavlakis E, Charles JP, Timofeev O, Nist
A, et al: Mutant p53 promotes tumor progression and metastasis by
the endoplasmic reticulum UDPase ENTPD5. Proc Natl Acad Sci USA.
113:E8433–E8442. 2016. View Article : Google Scholar
|
|
36
|
Schmitz AA, Govek EE, Bottner B and Van
Aelst L: Rho GTPases: Signaling, migration, and invasion. Exp Cell
Res. 261:1–12. 2000. View Article : Google Scholar
|
|
37
|
Yue X, Zhang C, Zhao Y, Liu J, Lin AW, Tan
VM, Drake JM, Liu L, Boateng MN, Li J, et al: Gain-of-function
mutant p53 activates small GTPase Rac1 through SUMOylation to
promote tumor progression. Genes Dev. 31:1641–1654. 2017.
View Article : Google Scholar
|
|
38
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar
|
|
39
|
Muller PA, Caswell PT, Doyle B, Iwanicki
MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL,
Gosselin P, et al: Mutant p53 drives invasion by promoting integrin
recycling. Cell. 139:1327–1341. 2009. View Article : Google Scholar
|
|
40
|
Ahn JH, Kim TJ, Lee JH and Choi JH: Mutant
p53 stimulates cell invasion through an interaction with Rad21 in
human ovarian cancer cells. Sci Rep. 7:90762017. View Article : Google Scholar
|
|
41
|
Mantovani F, Walerych D and Sal GD:
Targeting mutant p53 in cancer: A long road to precision therapy.
FEBS J. 284:837–850. 2017. View Article : Google Scholar
|
|
42
|
Treekitkarnmongkol W and Suthiphongchai T:
High expression of ErbB2 contributes to cholangiocarcinoma cell
invasion and proliferation through AKT/p70S6K. World J
Gastroenterol. 16:4047–4054. 2010. View Article : Google Scholar
|
|
43
|
Runnebaum IB, Nagarajan M, Bowman M, Soto
D and Sukumar S: Mutations in p53 as potential molecular markers
for human breast cancer. Proc Natl Acad Sci USA. 88:10657–10661.
1991. View Article : Google Scholar
|
|
44
|
Bertheau P, Turpin E, Rickman DS, Espie M,
de Reynies A, Feugeas JP, Plassa LF, Soliman H, Varna M, de
Roquancourt A, et al: Exquisite sensitivity of TP53 mutant and
basal breast cancers to a dose-dense epirubicin-cyclophosphamide
regimen. PLoS Med. 4:e902007. View Article : Google Scholar
|
|
45
|
Link DC, Schuettpelz LG, Shen D, Wang J,
Walter MJ, Kulkarni S, Payton JE, Ivanovich J, Goodfellow PJ, Le
Beau M, et al: Identification of a novel TP53 cancer susceptibility
mutation through whole-genome sequencing of a patient with
therapy-related AML. JAMA. 305:1568–1576. 2011. View Article : Google Scholar
|
|
46
|
Khoury MP and Bourdon JC: The isoforms of
the p53 protein. Cold Spring Harb Perspect Biol. 2:a0009272010.
View Article : Google Scholar
|
|
47
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar
|
|
48
|
Chen J: The cell-cycle arrest and
apoptotic functions of p53 in tumor initiation and progression.
Cold Spring Harb Perspect Med. 6:a0261042016. View Article : Google Scholar
|
|
49
|
Wu SJ, Lin CT, Agathangelidis A, Lin LI,
Kuo YY, Tien HF and Ghia P: Distinct molecular genetics of chronic
lymphocytic leukemia in Taiwan: Clinical and pathogenetic
implications. Haematologica. 102:1085–1090. 2017. View Article : Google Scholar
|
|
50
|
Kishimoto Y, Murakami Y, Shiraishi M,
Hayashi K and Sekiya T: Aberrations of the p53 tumor suppressor
gene in human non-small cell carcinomas of the lung. Cancer Res.
52:4799–4804. 1992.
|
|
51
|
Yamada H, Shinmura K, Okudela K, Goto M,
Suzuki M, Kuriki K, Tsuneyoshi T and Sugimura H: Identification and
characterization of a novel germ line p53 mutation in familial
gastric cancer in the Japanese population. Carcinogenesis.
28:2013–2018. 2007. View Article : Google Scholar
|
|
52
|
Lal-Nag M and Morin PJ: The claudins.
Genome Biol. 10:2352009. View Article : Google Scholar
|
|
53
|
Ding L, Lu Z, Lu Q and Chen YH: The
claudin family of proteins in human malignancy: A clinical
perspective. Cancer Manag Res. 5:367–375. 2013.
|
|
54
|
Kramer F, White K, Kubbies M, Swisshelm K
and Weber BH: Genomic organization of claudin-1 and its assessment
in hereditary and sporadic breast cancer. Hum Genet. 107:249–256.
2000. View Article : Google Scholar
|
|
55
|
Resnick MB, Konkin T, Routhier J, Sabo E
and Pricolo VE: Claudin-1 is a strong prognostic indicator in stage
II colonic cancer: A tissue microarray study. Mod Pathol.
18:511–518. 2005. View Article : Google Scholar
|
|
56
|
Chao YC, Pan SH, Yang SC, Yu SL, Che TF,
Lin CW, Tsai MS, Chang GC, Wu CH, Wu YY, et al: Claudin-1 is a
metastasis suppressor and correlates with clinical outcome in lung
adenocarcinoma. Am J Respir Crit Care Med. 179:123–133. 2009.
View Article : Google Scholar
|
|
57
|
Higashi Y, Suzuki S, Sakaguchi T, Nakamura
T, Baba S, Reinecker HC, Nakamura S and Konno H: Loss of claudin-1
expression correlates with malignancy of hepatocellular carcinoma.
J Surg Res. 139:68–76. 2007. View Article : Google Scholar
|
|
58
|
Suren D, Yildirim M, Kaya V, Alikanoglu
AS, Bulbuller N, Yildiz M and Sezer C: Loss of tight junction
proteins (Claudin 1, 4, and 7) correlates with aggressive behavior
in colorectal carcinoma. Med Sci Monit. 20:1255–1262. 2014.
View Article : Google Scholar
|
|
59
|
Hall A: Rho GTPases and the actin
cytoskeleton. Science. 279:509–514. 1998. View Article : Google Scholar
|
|
60
|
Schmidt A and Hall A: Guanine nucleotide
exchange factors for Rho GTPases: Turning on the switch. Genes Dev.
16:1587–1609. 2002. View Article : Google Scholar
|
|
61
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar
|
|
62
|
DeMali KA and Burridge K: Coupling
membrane protrusion and cell adhesion. J Cell Sci. 116:2389–2397.
2003. View Article : Google Scholar
|