Introduction
Pancreatic cancer has the worst prognosis among all
malignancies. The 5-year survival rate of patients with pancreatic
cancer is 8% despite advances in treatment (1). The majority of patients with
pancreatic cancer have distant metastasis and peritoneal
dissemination at the time of diagnosis (2), and therefore most are unable to
undergo curative surgical treatment (3). As a result, the development of
effective chemotherapy regimens is extremely important for this
population.
Tissue factor (TF), a 47-kDa transmembrane
glycoprotein, is widely known to play a role in initiating the
extrinsic blood coagulation cascade. The correlation between blood
coagulation and cancer has been extensively discussed (4–6).
Invasion of cancer cells, enhanced vascular permeability and
abnormal inflammation can trigger hyper-coagulation in tumor
tissues (7). In fact, TF
overexpression has been observed in various cancer types, including
pancreatic cancer (8–10). In addition to its role in initiating
the blood coagulation cascade, TF plays functional roles in cancer
development, for instance in the areas of tumor growth,
inflammation, and angiogenesis (11). Recently, several studies reported
that TF expression was also correlated with tumor metastasis in
several cancer types (12–15). Other studies demonstrated that the
downregulation of TF expression in cancer cells and the blockade of
TF by anti-TF monoclonal antibodies inhibited tumor metastasis
in vivo (12,16,17).
Thus, TF may be an ideal target for cancer treatments.
The use of antibody-drug conjugates (ADCs) is
currently considered to be a powerful strategy in cancer therapy.
ADCs have three components, namely a monoclonal antibody (mAb), a
potent payload, and a linker. The antigens on cancer cell membranes
are typical targets of ADCs. When these antigens are endocytosed,
the bound ADCs are internalized into the cancer cells, and the
linker is then selectively cleaved by proteases such as cathepsin B
in lysosomes. This cleavage of the linker causes the release of the
anticancer agents into the cytoplasm, thus inducing cell apoptosis.
Anti-TF ADCs have been investigated in several studies (17–23).
The efficacy of these agents was confirmed using various animal
models (including conventional subcutaneous xenograft models,
patient-derived xenograft models and mouse orthotopic models) and
in various cancer types (such as pancreatic, lung, prostate,
ovarian, bladder, and breast cancer, and cervical squamous cell
carcinoma). One of these anti-TF ADCs, tisotumab vedotin, is
currently in clinical trials, which has demonstrated clinical
responses in patients with recurrent or metastatic cervical cancer
(24,25). In the majority of cases, ADCs are
employed for the treatment of advanced cancer with metastasis.
However, few basic studies have investigated the efficacy of
anti-TF ADCs in advanced-stage cancer models such as abdominal
dissemination or distant metastasis.
In the present study, a humanized anti-TF antibody
was constructed, and its binding specificity was confirmed.
Previous studies demonstrated that the bis-alkylating conjugation
strategy was more feasible than the conventional maleimide-based
conjugation strategy for ADC construction (23,26,27).
Therefore, the bis-alkylating bis-sulfone
group-PEG12-valine-citrulline-p-amino-benzoyloxycarbonyl
(bisAlk-PEG12-vc-PABC) linker and monomethyl auristatin E (MMAE)
were used in the preparation of the humanized anti-TF ADC, and its
efficacy was evaluated in several pancreatic cancer models,
including the orthotopic model and peritoneal dissemination
model.
Materials and methods
Generation of humanized anti-TF
monoclonal antibody
Rat anti-TF mAb (clone 1084) was humanized as
previously described (28).
Briefly, the complementarity-determining regions of rat anti-TF mAb
were grafted onto a human antibody framework (IgG1). The procedure
was performed according to standard humanization protocols
(29). The heavy chain and the
kappa light chain cDNAs were cloned into a pcDNA3.3 expression
vector (Thermo Fisher Scientific, Inc.). These expression vectors
were transiently transfected into ExpiCHO cells (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Cell cultures
BxPC-3, HPAF-II, PSN-1 and Panc-1 cells were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA), while Suit-2 cells were obtained from the Japanese
Collection of Research Bioresources (JCRB, Tsukuba, Japan). To
evaluate the efficacy of anti-TF ADC in vivo, the
CRISPR-Cas9 system was used to generate BxPC-3 TF knock-out cells
(BxPC-3-TF-KO), and the CMV-GFP-T2A-Luciferase Lentivector system
(System Biosciences) was used to produce HPAF-II cells that stably
expressed firefly luciferase (HPAF-II-Luc). These cell lines were
cultured in ATCC- or JCRB-recommended medium supplemented with 10%
fetal bovine serum (FBS, Thermo Fisher Scientific, Inc.),
penicillin G (100 units/ml), streptomycin (100 µg/ml), and
amphotericin B (0.25 µg/ml; FUJIFILM Wako Pure Chemical Corp.) in a
5% CO2 atmosphere at 37°C.
Preparation of ADC
The ADC preparation protocol was previously
described (23). The
bisAlk-PEG12-vc-PABC linker was used for conjugation with MMAE to
humanized control mAb or humanized anti-TF mAb. The mAbs were
reduced by 35 mM 1,4-dithiothreitol (Sigma Aldrich; Merck KGaA),
and then reacted with the linker at 4°C overnight. After the
unreacted linker was removed, ADCs were stored at −80°C until use
in subsequent experiments.
Determination of molecular particle
size
To estimate the molecular sizes of mAbs and ADCs,
particle size analysis was performed using DelsaNano HC (Beckman
Coulter) based on photon correlation spectroscopy (PCS) and dynamic
light scattering (DLS), as previously described (30). Briefly, the particle size of each
mAb and ADC was measured by DelsaNano HC after the protein
concentration was adjusted to 1 mg/ml.
ELISA
The ELISA procedure was previously described
(22). Briefly, recombinant human
TF and mouse TF antigens were immobilized on C8 MaxiSorp plates
(Thermo Fisher Scientific, Inc.). Antibodies and ADCs were applied
at each concentration and left to react for 15 min at room
temperature (RT) (n=3). As a secondary antibody, goat anti-human
(H+L) pAb-HRP (Medical & Biological Laboratories Co., Ltd.) was
then allowed to react for 15 min at RT. The absorbance was
determined with a SpectraMax190 microplate reader (Molecular
Devices, LLC).
Flow cytometry (FCM) analysis
The FCM procedure was performed as previously
described (23). To detect human TF
antigens, goat anti-human TF polyclonal antibody (R&D Systems)
and anti-goat IgG (H+L) cross-adsorbed secondary antibody
conjugated with Alexa Fluor 647 (Thermo Fisher Scientific, Inc.)
were used. A Guava easyCyte flow cytometer (Merck Millipore) and
FlowJo analysis software (v10.6.1; Tree Star Inc.) were used to
analyze the data.
Assessment of cell cytotoxicity in
vitro
The cytotoxicity assessment procedure was previously
described (22). BxPC-3,
BxPC-3-TF-KO and HPAF-II cells were harvested on 96-well plates
(Corning) and incubated at 37°C overnight. MMAE and ADCs were
applied at each concentration, and the plates were incubated at
37°C for 72 h. MMAE was applied at the same concentrations with the
payloads conjugated with ADC. Cell Counting Kit-8 (Dojindo
Molecular Technologies) was used to evaluate cell viability
(n=3).
Animal models
A total of 125 female BALB/c nude mice (Charles
River Laboratories Japan, five-week-old, initial average weight
20.6 g) were used in the subsequent experiments. The mice were
maintained in cages under specific pathogen-free condition
(temperature; 23°C, humidity; 50%) and exposed to a 12-h light/dark
cycle. Sterilized water and standard food were offered ad
libitum. In all experiments, mice were anesthetized by
inhalation with 1–2% isoflurane (Pfizer). To prepare the
subcutaneous tumor models, 2×106 BxPC-3, BxPC-3-TF-KO or
HPAF-II cells were subcutaneously implanted into mice under deep
anesthesia. Tumor volume was calculated by the following formula:
Volume=length × (width)2 × 1/2. To prepare the
orthotopic tumor model, 1×106 HPAF-II cells were
injected into the pancreas of mice under deep anesthesia. To
prepare the peritoneal dissemination model, 1×106
HPAF-II-Luc cells were intraperitoneally injected into mice under
deep anesthesia. Tumor volume exceeding 2,000 mm3,
weight loss of 20% or more, ascites retention, and apparent
reduction in physical activity were set as humane endpoints. As a
method of euthanasia, mice were euthanized by cervical dislocation
after confirming that they were sufficiently anesthetized by
halation with isoflurane.
Antitumor effects in the subcutaneous
and orthotopic tumor models
In the subcutaneous tumor model, the treatment was
started when the tumor volume reached approximately 200
mm3. In the BxPC-3 ×enograft model, mice (n=5) underwent
three weekly treatments with intravenous DPBS, humanized anti-TF
mAb (10 mg/kg), humanized control ADC (10 mg/kg), or humanized
anti-TF ADC (2, 5 and 10 mg/kg). In the HPAF-II and BxPC-3-TF-KO
xenograft models, mice (n=5) received three weekly treatments with
DPBS or each ADC (10 mg/kg).
In the orthotopic tumor model, the treatment was
started 3 weeks after transplantation. The mice (n=8) were
administered intravenous DPBS, humanized control ADC (10 mg/kg), or
humanized anti-TF ADC (10 mg/kg) once a week for 4 weeks. After
treatment, the mice were sacrificed under deep anesthesia. The
tumors were harvested and their weights were measured.
Antitumor effects in the peritoneal
dissemination model
In the peritoneal dissemination model, the treatment
was started 2 weeks after transplantation (Day 0). In vivo
photon counting analysis was performed using the IVIS kinetic
imaging system (Caliper Life Sciences) and IVIS Living Imaging 3.0
software (Caliper Life Sciences) at Day 0, 14, and 28. At Day 0,
the mice were randomly grouped based on bioluminescence intensity
in the regions of interest (ROIs) in the abdominal area. The
average bioluminescence intensity was approximately
5.0–5.5×106 photons/sec at Day 0. The mice (n=11) were
treated weekly with intravenous DPBS, humanized control ADC (10
mg/kg) or humanized anti-TF ADC (10 mg/kg) for 4 weeks. The
bodyweight and mortality were checked twice or thrice per week. In
the survival study, the mice were sacrificed when weight loss of
20% or more, ascites retention, and apparent reduction in physical
activity were observed. In addition, the monitoring period was set
to 4 months because the survival period of this model was 1–2
months in a preliminary study.
All animal experiments were carried out with the
approval of the Committee for Animal Experimentation of the
National Cancer Center, Japan. Experiments met the ethical
standards required by law and also complied with guidelines for the
use of experimental animals in Japan.
Statistical analysis
Statistical analysis was conducted using EZR
software Ver. 1.42 (31). The error
bars in all figures represent the mean ± SD. In the subcutaneous
xenograft model, repeated-measures ANOVA was applied to evaluate
the statistical differences. ANOVA with Tukey's multiple comparison
test was used to investigate the changes in tumor weight in the
orthotopic xenograft model. Repeated-measures ANOVA was used to
compare the bioluminescence intensities in the peritoneal
dissemination model. A log-rank test of Kaplan-Meier curves for
pairwise comparisons was performed to analyze the statistical
difference in the peritoneal dissemination model. The Bonferroni's
test was applied as P-value adjustment method.
Results
Preparation of anti-TF ADC
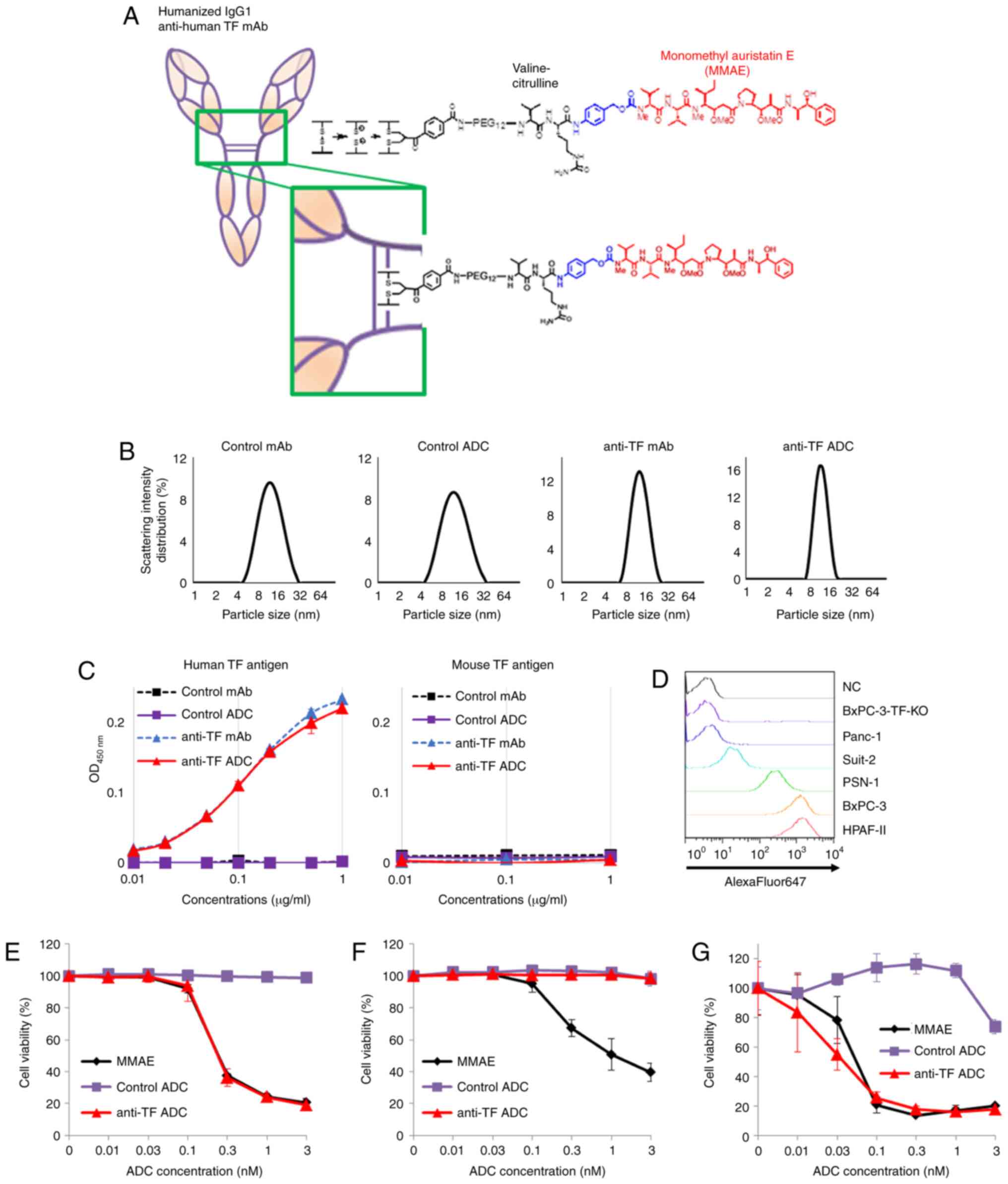
The valine-citrulline linker was adopted, as shown
in Fig. 1A. The drug antibody
ratios of control ADC and anti-TF ADC were determined to be 3.5 and
3.6, respectively. To evaluate the changes in molecular size
between mAbs and ADCs, the particle size of each mAb and ADC was
measured using a method based on PCS and DLS. The particle sizes of
control mAb, control ADC, anti-TF mAb, and anti-TF ADC were
13.0±4.7, 13.0±5.2, 13.0±3.3 and 12.0±2.4 nm, respectively
(Fig. 1B). These results indicated
that there were no significant changes in molecular size between
mAbs and ADCs. In addition, ELISA was performed to estimate the
binding affinity of each mAb and ADC to recombinant human and mouse
TF antigen (Fig. 1C). Anti-TF mAb
and anti-TF ADC showed almost equivalent affinities to recombinant
human TF antigen. However, neither reacted with recombinant mouse
TF antigen. On the other hand, control mAb and control ADC did not
recognize either recombinant human or mouse TF antigen. Before
evaluating cytotoxic activity, TF expression was determined by
performing FCM analysis using human pancreatic cancer cell lines,
including Panc-1, Suit-2, PSN-1, BxPC-3 and HPAF-II (Fig. 1D). Of these, BxPC-3 and HPAF-II
cells showed the highest TF antigen expression. To clarify whether
the cytotoxic activity of anti-TF ADC depended on cellular TF
expression, BxPC-3-TF-KO cells that did not express TF antigen on
the cell surface were prepared. These three cell lines (BxPC-3,
BxPC-3-TF-KO and HPAF-II) were used for the subsequent
experiments.
WST-8 assays were performed to investigate the
cytotoxic activity of each agent (Fig.
1E-G). The results showed that the cytotoxic effects of anti-TF
ADC and MMAE were almost the same against TF-positive cells, BxPC-3
cells and HPAF-II cells (Fig. 1E and
G). On the other hand, TF-negative cells, BxPC-3-TF-KO, were
damaged by MMAE but not by anti-TF ADC (Fig. 1F). Control ADC had no cytotoxic
activity against any of the three cell lines. In this assay, the
IC50 values of MMAE and anti-TF ADC against BxPC-3 cells
were 0.25±0.00 nM (as the ADC equivalent) and 0.25±0.02 nM,
respectively (Fig. 1E). The
IC50 value of MMAE against BxPC-3-TF-KO cells was
1.48±0.71 nM (as the ADC equivalent) (Fig. 1F). The IC50 values of
MMAE and anti-TF ADC against HPAF-II cells were 0.06±0.01 nM (as
the ADC equivalent) and 0.04±0.02 nM, respectively (Fig. 1G).
Antitumor effects in the subcutaneous
tumor models
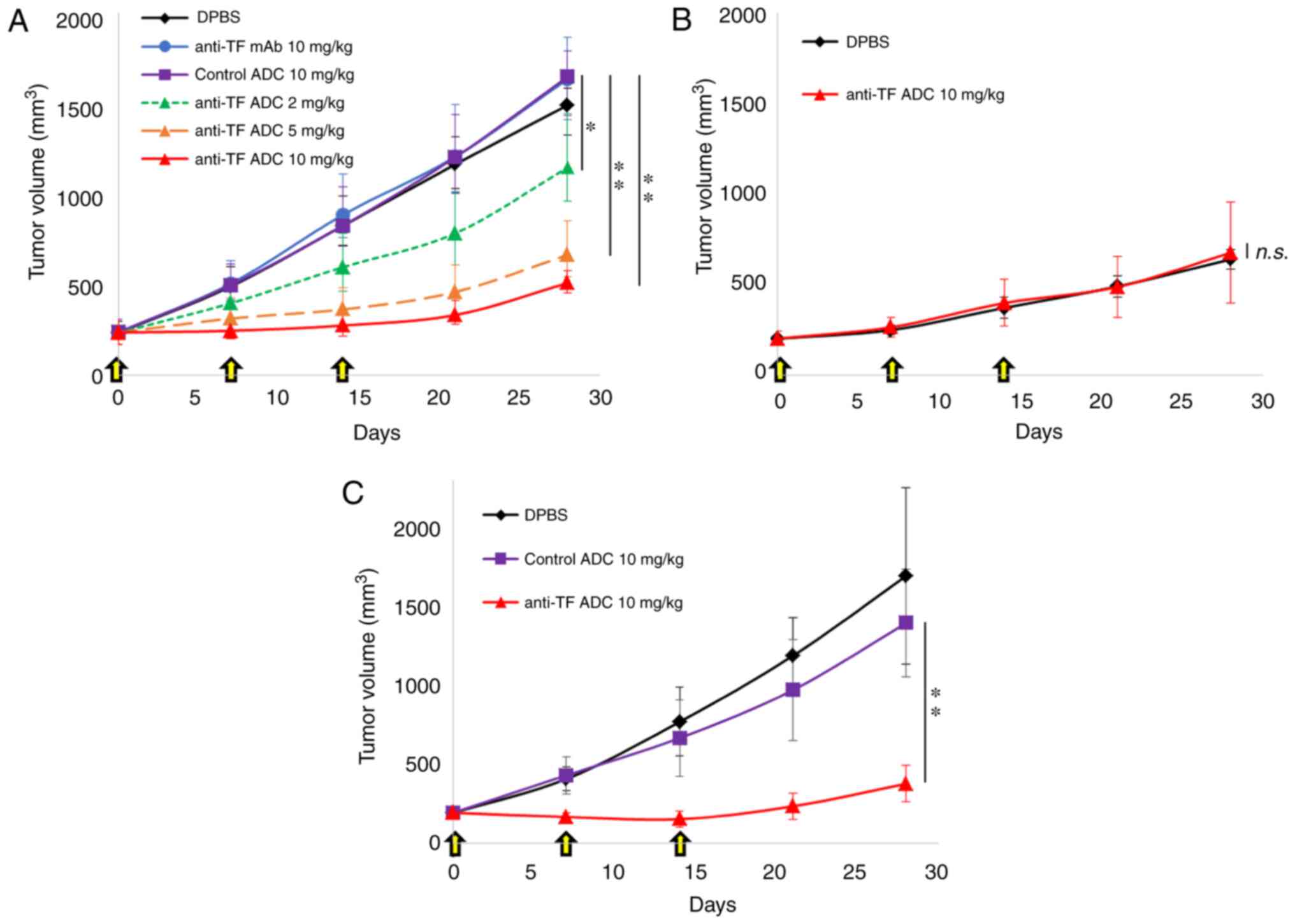
Our study next examined the antitumor effects of
anti-TF ADC in three subcutaneous xenograft models. In the BxPC-3
×enograft model, no antitumor effects were observed with anti-TF
mAb or control ADC (Fig. 2A). Our
previous reports also confirmed that the rat anti-TF mAb (IgG2b) by
itself did not show an antitumor effect in vivo (19,22).
In the present study, humanized anti-TF mAb (IgG1)
was prepared, which was expected to exert an antibody-dependent
cellular cytotoxicity effect in vivo; however, it did not
have an antitumor effect. On the other hand, anti-TF ADC showed a
significant antitumor effect (Fig.
2A). Anti-TF ADC at doses of 2, 5 and 10 mg/kg exhibited
significantly greater antitumor effects than those of control ADC
at 10 mg/kg (P<0.005, P<0.001 and P<0.001, respectively).
The antitumor effects of anti-TF ADC were clearly dose dependent.
In the BxPC-3-TF-KO xenograft model, anti-TF ADC did not inhibit
tumor growth any more than DPBS (P=0.997), indicating that the
effects of anti-TF ADC depended entirely on the expression of TF in
tumor tissues (Fig. 2B).
Furthermore, the growth speed of BxPC-3-TF-KO tumors was slower
than that of BxPC-3 tumors (Fig. 2A and
B), indicating that TF expression in BxPC-3 tumors contributed
to accelerating tumor growth in vivo. In the HPAF-II
xenograft model, control ADC did not show an antitumor effect,
whereas anti-TF ADC caused tumor shrinkage (Fig. 2C). Statistically, there was a
significant difference between treatment with control ADC at 10
mg/kg and anti-TF ADC at 10 mg/kg (P<0.001). Although tumor
growth was inhibited during treatment with anti-TF ADC (10 mg/kg)
in the BxPC-3 and HPAF-II xenograft models, tumor regrowth was
observed after the end of treatment in both models (Fig. 2A and C). There were no body weight
changes in the mice treated with anti-TF ADC in the subcutaneous
tumor models (Fig. S1A-C).
Antitumor effects in the orthotopic
tumor models
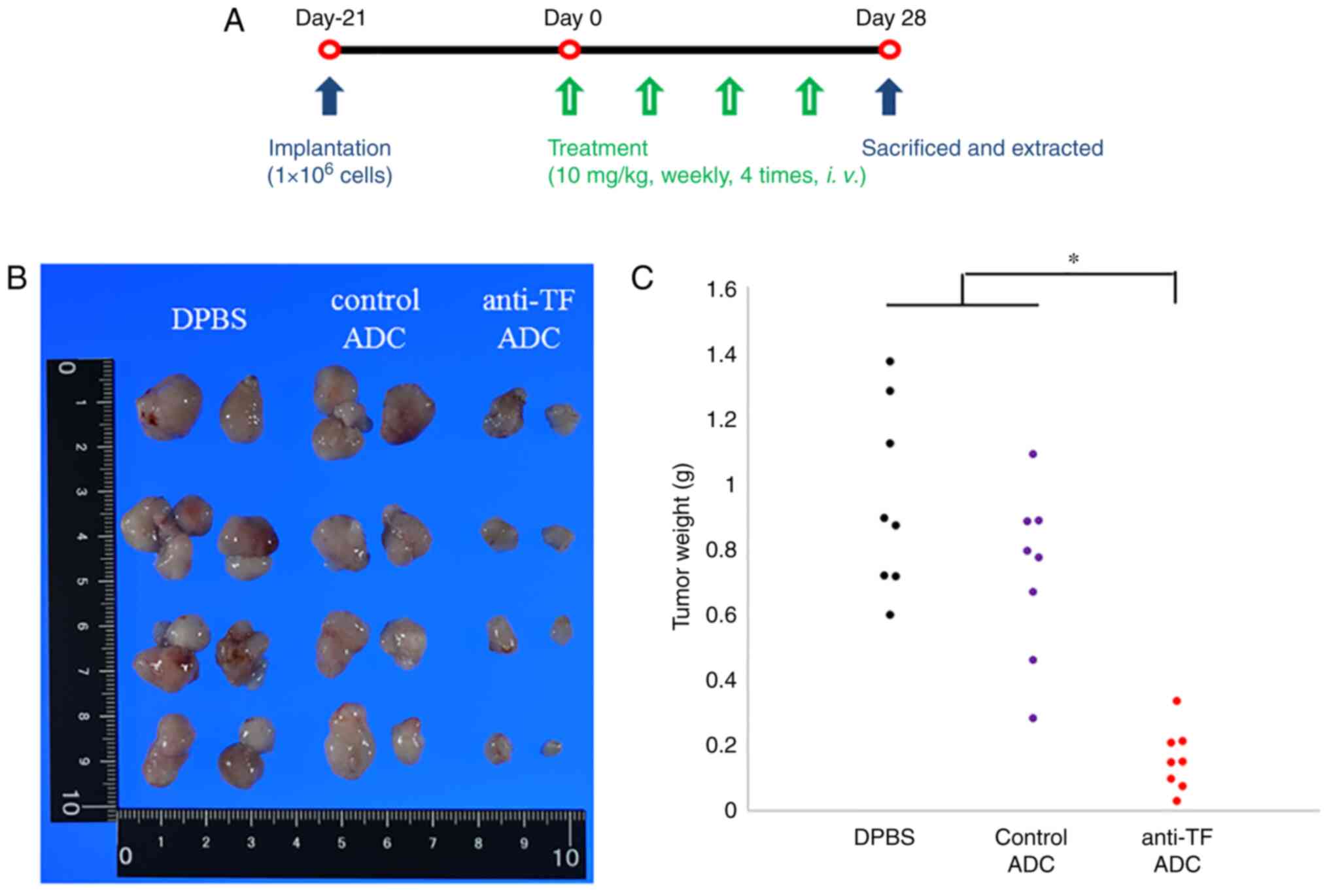
Our study examined the antitumor effects of anti-TF
ADC in an orthotopic xenograft model using the treatment schedule
shown in Fig. 3A. After treatment
with DPBS or 10/mg/kg anti-TF ADC or control ADC, each tumor was
resected (Fig. 3B). Tumor weights
are plotted in Fig. 3C. The tumors
treated with anti-TF ADC weighed significantly less than those
treated with DPBS or control ADC (P<0.001). These results
indicate that anti-TF ADC efficiently delivered the payload to the
pancreatic tumor and inhibited tumor growth in the orthotopic
pancreatic tumor model. Body weight changes of mice were not
observed in the orthotopic xenograft model (Fig. S1D).
Antitumor effects in a peritoneal
dissemination model
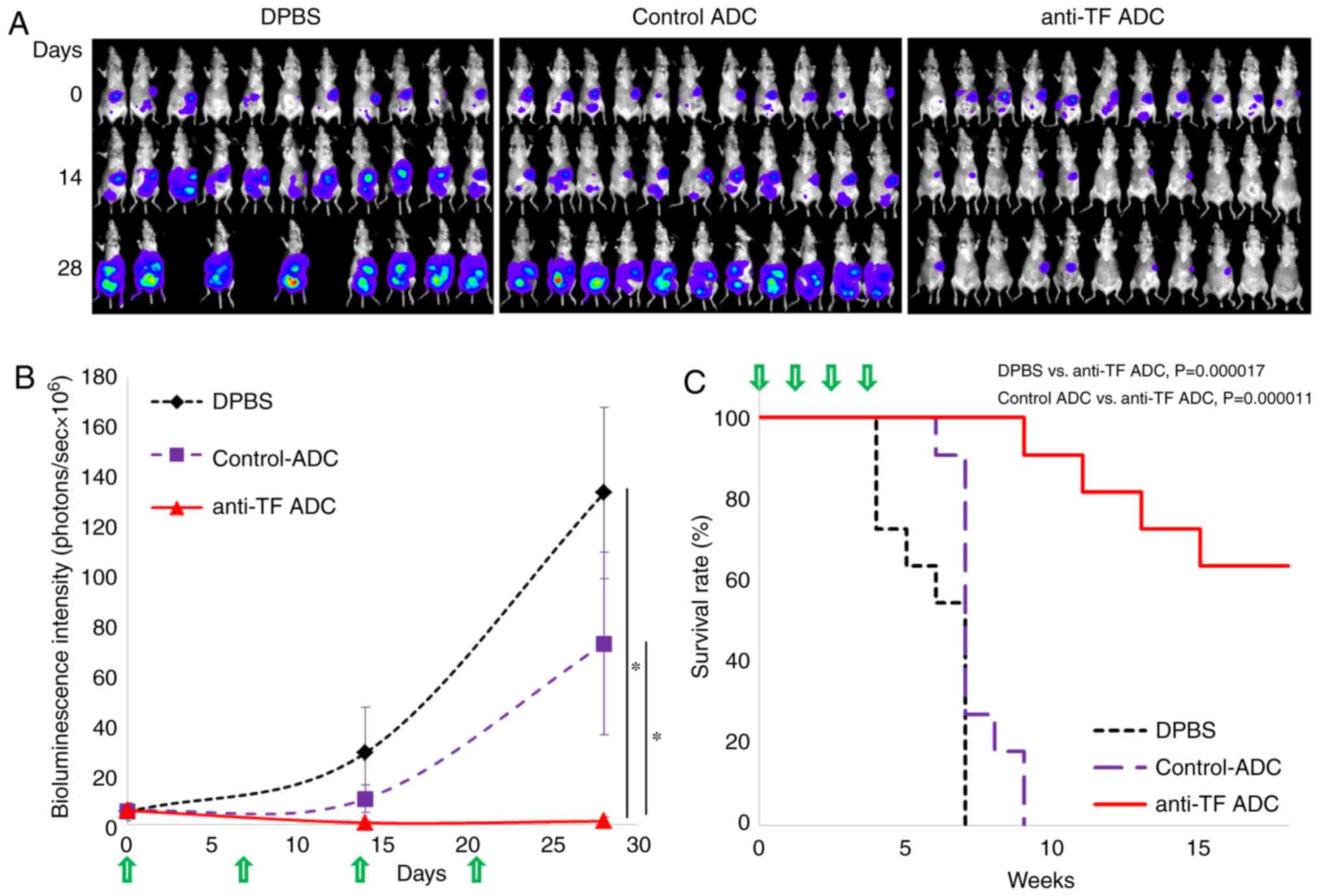
The antitumor effect of anti-TF ADC was evaluated in
a pancreatic cancer peritoneal dissemination model. One week after
implantation of HPAF-II-Luc cells into the abdominal cavity of
mice, the bioluminescence intensity of the abdominal area of the
mice was measured, and the animals were randomly grouped (Day 0).
The bioluminescence intensity of each mouse was measured at Day 14
and 28. The bioluminescence images of each mouse are shown in
Fig. 4A. In the group treated with
DPBS at Day 28, images of three mice could not be obtained due to
prior tumor-related death. Also, the bioluminescence intensity at
each time point was plotted (Fig.
4B). The mice treated with anti-TF ADC showed significantly
lower bioluminescence intensity than that exhibited by the mice
treated with DPBS or control ADC (P<0.001 both). Hence, anti-TF
ADC inhibited tumor growth in the abdominal cavity in this model.
On the other hand, widespread dissemination occurred in the mice
treated with DPBS or control ADC. It is notable that, in mice with
peritoneal dissemination, the antitumor effect of anti-TF ADC
extended the survival time compared with that caused by DPBS
(P=0.000017) and control ADC (P=0.000011) (Fig. 4C). In this experiment, the
treatments with anti-TF ADC did not affect the body weight change
of mice (Fig. S1E).
Discussion
Currently, numerous antibody-drug conjugates (ADCs)
have been investigated in clinical trials. Since tissue factor (TF)
expression has been observed in a wide range of solid tumors,
anti-TF ADC is also expected to be a promising cancer treatment.
Phase 1 and 2 clinical trials of tisotumab vedotin were conducted
in patients with advanced metastatic solid cancer (InnovaTV 201)
(24,25). In these studies, the authors
demonstrated that tisotumab vedotin had a manageable safety profile
and a preliminary antitumor effect. A different group developed
anti-TF ADCs other than tisotumab vedotin, and showed that one
anti-TF ADC did not affect blood coagulation (17,21).
Our group also established several rat anti-TF mAb clones and
compared their characteristics (19,22,32).
Our experiments indicated that mAb clone 1084 had suitable
characteristics for ADC design. Although ADC 1084 had a higher
binding affinity constant (KD) and higher dissociation
constant rate (kd) than those of other anti-TF ADCs, it
was able to penetrate more deeply into tumor clusters and exerted a
greater antitumor effect in large-sized tumors (approximately 600
mm3) in the pancreatic cancer xenograft model. Since
another report demonstrated that TF is efficient and rapidly turned
over on TF-positive tumor cells (20), it was hypothesized that the change
in kd may not affect the internalization efficiency of
anti-TF ADCs, but may instead have an effect on tumor-penetration
ability. In the present study, therefore, a humanized anti-TF mAb
was established from rat anti-TF mAb (clone 1084) and humanized
anti-TF ADC was prepared for cancer treatment.
The present study successfully produced humanized
anti-TF ADC that was able to bind to human TF antigens, but not to
mouse TF antigens. The WST-8 assay in vitro demonstrated
that anti-TF ADC showed high cytotoxic activity against two
TF-positive pancreatic cancer cell lines, namely BxPC-3 and
HPAF-II, but not against the BxPC-3-TF-KO cell line, which is
TF-negative. In subcutaneous xenograft models, anti-TF ADC also
exhibited a high antitumor effect in BxPC-3 and HPAF-II tumors, but
not in BxPC-3-TF-KO tumors. Additionally, anti-TF ADC efficiently
delivered the anticancer agent to orthotopic tumors, whose
microenvironments may be more similar to that of clinical
pancreatic cancer than to that of subcutaneous tumors, and exerted
an antitumor effect in the orthotopic model. Furthermore, anti-TF
ADC significantly extended the survival period in the peritoneal
dissemination mouse model, in which mice with no treatment showed a
shorter survival time (approximately 2 months) due to tumor
dissemination in the abdominal region. These results indicated that
anti-TF ADC has the potential to be an efficacious treatment not
only for primary tumors but also for tumors that have widely
disseminated. In future studies, other metastatic tumor models such
as a liver metastatic tumor model would be useful to evaluate the
efficacy of anti-TF ADC in advanced pancreatic cancer. Furthermore,
although a bleeding risk due to the use of tisotumab vedotin was
reported as manageable in the clinical study (24,25),
our present humanized anti-TF ADC should be carefully evaluated for
these potential adverse events and distribution in normal tissues
in the appropriate models.
As mentioned above, there have been several reports
on anti-TF ADCs. All of them, including tisotumab vedotin, adopted
the valine-citrulline linker and used microtubule inhibitors such
as MMAE and MMAF as the payload. Although MMAE has been used in a
number of ADCs, other payloads (e.g., DNA-damaging agents) have
also demonstrated in vivo efficacy (33,34).
Furthermore, multiple studies revealed that cancer cells can
develop resistance to ADCs conjugated with MMAE because MMAE can be
pumped out of the cell via transporters (e.g., P-glycoprotein)
(35–37). Therefore, other payloads are
expected to be investigated for the design of anti-TF ADCs in order
to increase their efficacy.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms. M Shimada at the
National Cancer Center, Japan for her secretarial help.
Funding
The research was supported by the National Cancer
Center Research and Development Fund (23-A-45 and 29-A-9 to YM) and
by the Japan Society for the Promotion of Science (JSPS) KAKENHI
Grant Number JP 18K14931 and 20K16021 (to RT).
Availability of data and materials
All data generated during this study are available
from the corresponding author on reasonable request.
Authors' contributions
RT mainly performed the in vitro and in
vivo experiments. TA was involved in the production and the
preparation of the humanized antibody. SM was involved in the
preparation of the linker compound. HT was involved in the
preparation of tissue factor antigen for ELISA. RT, YK, MY and YM
contributed to the study conception and design. RT and YM drafted
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were carried out with the
approval of the Committee for Animal Experimentation of the
National Cancer Center, Japan. Experiments met the ethical
standards required by law and also complied with guidelines for the
use of experimental animals in Japan.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Howlader N, Noone AM, Krapcho M, Miller D,
Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, et al:
SEER Cancer Statistics Review. 1975-2014, National Cancer
Institute; Bethesda, MD: simplehttps://seercancergov/csr/1975_2014/based on
November 2016 SEER data submission, posted to the SEER web site,
April 2017. May 11–2017
|
|
2
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar
|
|
3
|
Ferrone CR, Pieretti-Vanmarcke R, Bloom
JP, Zheng H, Szymonifka J, Wargo JA, Thayer SP, Lauwers GY,
Deshpande V, Mino-Kenudson M, et al: Pancreatic ductal
adenocarcinoma: Long-term survival does not equal cure. Surgery.
152 (3 Suppl 1):S43–S49. 2012. View Article : Google Scholar
|
|
4
|
Khorana AA and Fine RL: Pancreatic cancer
and thromboembolic disease. Lancet Oncol. 5:655–663. 2004.
View Article : Google Scholar
|
|
5
|
Rak J, Yu JL, Luyendyk J and Mackman N:
Oncogenes, trousseau syndrome, and cancer-related changes in the
coagulome of mice and humans. Cancer Res. 66:10643–10646. 2006.
View Article : Google Scholar
|
|
6
|
Stein PD, Beemath A, Meyers FA, Skaf E,
Sanchez J and Olson RE: Incidence of venous thromboembolism in
patients hospitalized with cancer. Am J Med. 119:60–68. 2006.
View Article : Google Scholar
|
|
7
|
Matsumura Y: Cancer stromal targeting
(CAST) therapy. Adv Drug Deliv Rev. 64:710–719. 2012. View Article : Google Scholar
|
|
8
|
Callander NS, Varki N and Rao LV:
Immunohistochemical identification of tissue factor in solid
tumors. Cancer. 70:1194–1201. 1992. View Article : Google Scholar
|
|
9
|
Nitori N, Ino Y, Nakanishi Y, Yamada T,
Honda K, Yanagihara K, Kosuge T, Kanai Y, Kitajima M and Hirohashi
S: Prognostic significance of tissue factor in pancreatic ductal
adenocarcinoma. Clin Cancer Res. 11:2531–2539. 2005. View Article : Google Scholar
|
|
10
|
van den Berg YW, Osanto S, Reitsma PH and
Versteeg HH: The relationship between tissue factor and cancer
progression: Insights from bench and bedside. Blood. 119:924–932.
2012. View Article : Google Scholar
|
|
11
|
Kasthuri RS, Taubman MB and Mackman N:
Role of tissue factor in cancer. J Clin Oncol. 27:4834–4838. 2009.
View Article : Google Scholar
|
|
12
|
Bourcy M, Suarez-Carmona M, Lambert J,
Francart ME, Schroeder H, Delierneux C, Skrypek N, Thompson EW,
Jerusalem G, Berx G, et al: Tissue factor induced by
epithelial-mesenchymal transition triggers a procoagulant state
that drives metastasis of circulating tumor cells. Cancer Res.
76:4270–4282. 2016. View Article : Google Scholar
|
|
13
|
Hisada Y and Mackman N: Tissue factor and
cancer: Regulation, tumor growth, and metastasis. Semin Thromb
Hemost. 45:385–395. 2019. View Article : Google Scholar
|
|
14
|
Yamashita H, Kitayama J, Ishikawa M and
Nagawa H: Tissue factor expression is a clinical indicator of
lymphatic metastasis and poor prognosis in gastric cancer with
intestinal phenotype. J Surg Oncol. 95:324–331. 2007. View Article : Google Scholar
|
|
15
|
Sawada M, Miyake S, Ohdama S, Matsubara O,
Masuda S, Yakumaru K and Yoshizawa Y: Expression of tissue factor
in non-small-cell lung cancers and its relationship to metastasis.
Br J Cancer. 79:472–477. 1999. View Article : Google Scholar
|
|
16
|
Versteeg HH, Schaffner F, Kerver M,
Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM
and Ruf W: Inhibition of tissue factor signaling suppresses tumor
growth. Blood. 111:190–199. 2008. View Article : Google Scholar
|
|
17
|
Zhang X, Li Q, Zhao H, Ma L, Meng T, Qian
J, Jin R, Shen J and Yu K: Pathological expression of tissue factor
confers promising antitumor response to a novel therapeutic
antibody SC1 in triple negative breast cancer and pancreatic
adenocarcinoma. Oncotarget. 8:59086–59102. 2017. View Article : Google Scholar
|
|
18
|
Breij EC, de Goeij BE, Verploegen S,
Schuurhuis DH, Amirkhosravi A, Francis J, Miller VB, Houtkamp M,
Bleeker WK, Satijn D and Parren PW: An antibody-drug conjugate that
targets tissue factor exhibits potent therapeutic activity against
a broad range of solid tumors. Cancer Res. 74:1214–1226. 2014.
View Article : Google Scholar
|
|
19
|
Koga Y, Manabe S, Aihara Y, Sato R,
Tsumura R, Iwafuji H, Furuya F, Fuchigami H, Fujiwara Y, Hisada Y,
et al: Antitumor effect of antitissue factor antibody-MMAE
conjugate in human pancreatic tumor xenografts. Int J Cancer.
137:1457–1466. 2015. View Article : Google Scholar
|
|
20
|
de Goeij BE, Satijn D, Freitag CM,
Wubbolts R, Bleeker WK, Khasanov A, Zhu T, Chen G, Miao D, van
Berkel PH and Parren PW: High turnover of tissue factor enables
efficient intracellular delivery of antibody-drug conjugates. Mol
Cancer Ther. 14:1130–1140. 2015. View Article : Google Scholar
|
|
21
|
Theunissen JW, Cai AG, Bhatti MM, Cooper
AB, Avery AD, Dorfman R, Guelman S, Levashova Z and Migone TS:
Treating tissue factor-positive cancers with antibody-drug
conjugates that do not affect blood clotting. Mol Cancer Ther.
17:2412–2426. 2018. View Article : Google Scholar
|
|
22
|
Tsumura R, Manabe S, Takashima H, Koga Y,
Yasunaga M and Matsumura Y: Influence of the dissociation rate
constant on the intra-tumor distribution of antibody-drug conjugate
against tissue factor. J Control Release. 284:49–56. 2018.
View Article : Google Scholar
|
|
23
|
Tsumura R, Manabe S, Takashima H, Koga Y,
Yasunaga M and Matsumura Y: Evaluation of the antitumor mechanism
of antibody-drug conjugates against tissue factor in stroma-rich
allograft models. Cancer Sci. 110:3296–3305. 2019. View Article : Google Scholar
|
|
24
|
Hong DS, Concin N, Vergote I, de Bono JS,
Slomovitz BM, Drew Y, Arkenau HT, Machiels JP, Spicer JF, Jones R,
et al: Tisotumab vedotin in previously treated recurrent or
metastatic cervical cancer. Clin Cancer Res. 26:1220–1228. 2020.
View Article : Google Scholar
|
|
25
|
de Bono JS, Concin N, Hong DS,
Thistlethwaite FC, Machiels JP, Arkenau HT, Plummer R, Jones RH,
Nielsen D, Windfeld K, et al: Tisotumab vedotin in patients with
advanced or metastatic solid tumours (InnovaTV 201): A
first-in-human, multicentre, phase 1–2 trial. Lancet Oncol.
20:383–393. 2019. View Article : Google Scholar
|
|
26
|
Badescu G, Bryant P, Bird M, Henseleit K,
Swierkosz J, Parekh V, Tommasi R, Pawlisz E, Jurlewicz K, Farys M,
et al: Bridging disulfides for stable and defined antibody drug
conjugates. Bioconjug Chem. 25:1124–1136. 2014. View Article : Google Scholar
|
|
27
|
Bryant P, Pabst M, Badescu G, Bird M,
McDowell W, Jamieson E, Swierkosz J, Jurlewicz K, Tommasi R,
Henseleit K, et al: In vitro and in vivo evaluation of cysteine
rebridged trastuzumab-MMAE antibody drug conjugates with defined
drug-to-antibody ratios. Mol Pharm. 12:1872–1879. 2015. View Article : Google Scholar
|
|
28
|
Yasunaga M, Saijou S, Hanaoka S, Anzai T,
Tsumura R and Matsumura Y: Significant antitumor effect of an
antibody against TMEM180, a new colorectal cancer-specific
molecule. Cancer Sci. 110:761–770. 2019. View Article : Google Scholar
|
|
29
|
Kuramochi T, Igawa T, Tsunoda H and
Hattori K: Humanization and simultaneous optimization of monoclonal
antibody. Methods Mol Biol. 1060:123–137. 2014. View Article : Google Scholar
|
|
30
|
Tsumura R, Sato R, Furuya F, Koga Y,
Yamamoto Y, Fujiwara Y, Yasunaga M and Matsumura Y: Feasibility
study of the Fab fragment of a monoclonal antibody against tissue
factor as a diagnostic tool. Int J Oncol. 47:2107–2114. 2015.
View Article : Google Scholar
|
|
31
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar
|
|
32
|
Saito Y, Hashimoto Y, Kuroda J, Yasunaga
M, Koga Y, Takahashi A and Matsumura Y: The inhibition of
pancreatic cancer invasion-metastasis cascade in both cellular
signal and blood coagulation cascade of tissue factor by its
neutralisation antibody. Eur J Cancer. 47:2230–2239. 2011.
View Article : Google Scholar
|
|
33
|
Jeffrey SC, Burke PJ, Lyon RP, Meyer DW,
Sussman D, Anderson M, Hunter JH, Leiske CI, Miyamoto JB, Nicholas
ND, et al: A potent anti-CD70 antibody-drug conjugate combining a
dimeric pyrrolobenzodiazepine drug with site-specific conjugation
technology. Bioconjug Chem. 24:1256–1263. 2013. View Article : Google Scholar
|
|
34
|
Ogitani Y, Aida T, Hagihara K, Yamaguchi
J, Ishii C, Harada N, Soma M, Okamoto H, Oitate M, Arakawa S, et
al: DS-8201a, a novel HER2-targeting ADC with a Novel DNA
topoisomerase I inhibitor, demonstrates a promising antitumor
efficacy with differentiation from T-DM1. Clin Cancer Res.
22:5097–5108. 2016. View Article : Google Scholar
|
|
35
|
Chen R, Hou J, Newman E, Kim Y, Donohue C,
Liu X, Thomas SH, Forman SJ and Kane SE: CD30 downregulation, MMAE
resistance, and MDR1 upregulation are all associated with
resistance to brentuximab vedotin. Mol Cancer Ther. 14:1376–1384.
2015. View Article : Google Scholar
|
|
36
|
Yu SF, Zheng B, Go M, Lau J, Spencer S,
Raab H, Soriano R, Jhunjhunwala S, Cohen R, Caruso M, et al: A
novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC)
that overcomes resistance to auristatin-based ADCs. Clin Cancer
Res. 21:3298–3306. 2015. View Article : Google Scholar
|
|
37
|
Liu-Kreyche P, Shen H, Marino AM, Iyer RA,
Humphreys WG and Lai Y: Lysosomal P-gp-MDR1 confers drug resistance
of brentuximab vedotin and its cytotoxic payload monomethyl
auristatin e in tumor cells. Front Pharmacol. 10:7492019.
View Article : Google Scholar
|