Introduction
Urothelial cancer is derived from the urothelium of
the pelvis, ureter, bladder and urethra (1). The vast majority of urothelial cancer
is bladder urothelial cancer, which accounts for ~90% of all cases
of bladder cancer (2). Bladder
cancer is more common in men than women, with ~549,393 new cases
and 199,922 mortalities worldwide, annually (3). The 5-year survival rate of patients
with bladder cancer is ~80% (2),
and it has not improved in the last three decades compared with
several other malignant tumors, such as prostate cancer, lung
cancer, kidney cancer and breast cancer (2). Investigating the genes that determine
the survival outcome of patients and attempting to block or
activate the associated pathways may help improve the therapeutic
efficacy.
The Pathology Atlas project (4) was performed based on the large
open-access transcriptome data and the clinical metadata of The
Cancer Genome Atlas (TCGA, http://www.genome.gov/Funded-Programs-Projects/Cancer-Genome-Atlas)
and the Human Protein Atlas databases (http://www.proteinatlas.org). A systems-level approach
was used to assess the transcriptome of 17 major types of cancer,
including urothelial cancer, with respect to clinical outcome, in
which several survival-associated genes were identified. However,
whether the prognostic genes serve any biological roles in cancer
development remains largely unknown.
Uncontrolled cell proliferation is one of the basic
characteristics of cancer, while cancer metastasis is considered a
major cause of cancer associated mortality (5). Previous studies have demonstrated that
the genes that regulate cancer cell proliferation or migration rate
also influence patient survival (6,7). The
present study investigated the biological function of 12 of the
most significant survival-associated genes (ABRACL, MITD1,
ZNF524, EMP1, HSPB6, CXorf38, TRIM38, ZNF182, ZNF195, SPRN,
PTPN6 and LIPT1) in urothelial cancer reported by the
Pathology Atlas project, with respect to cell proliferation and
migration. For the MIT-domain containing protein 1 (MITD1) gene,
which regulates cell migration in the present study, the downstream
molecules were assessed via RNA sequencing. Immunohistochemistry
(IHC) analysis was performed to compare the expression levels of
MITD1 between primary tumors and metastatic cancer cells, in order
to determine whether MITD1 is involved in cancer metastasis
in vivo.
Materials and methods
Searching the prognostic genes
The prognostic genes were identified as follows: The
home page of the Human Protein Atlas database (https://www.proteinatlas.org/humanproteome/pathology)
was opened and ‘urothelial cancer’ under the ‘correlation analysis
and prognostic genes’ section was selected. There are 1,092
prognostic genes in the online table. A gene is considered
prognostic by the Pathology Atlas if correlation analysis of gene
expression (FPKM) and prognosis (survival) resulted in Kaplan-Meier
plots with high significance (P<0.001) (4). Here, the P-values were calculated with
the best expression cut-offs. Based on the cut-off value of each
gene, patients were classified into two groups (high expression
group and low expression group) and the association between
survival and gene expression was assessed. The best expression
cut-off refers to the FPKM value that yields maximal difference
with regards to survival between the two groups at the lowest
log-rank P-value (4).
In addition to the P-values calculated with the best
expression cut-offs, the Pathology Atlas also provides the P-values
with the median expression cut-offs. Median expression refers to
the median FPKM value calculated based on the gene expression data
from all patients in this dataset (4). This information is attained as
follows: i) The web page of each gene in the table is opened, ii)
the median expression value under the ‘urothelial
cancer-interactive survival scatter plot & survival analysis’
section is selected and the Kaplan-Meier curve and P-value are
adjusted to provide results based on the median expression.
The candidate genes for investigation were screened
by initially selecting 29 genes with P<1×10−6 (best
expression cut-off). A total of six genes with P≥0.01 (median
expression cut-off) were excluded from the final analysis. Among
the remaining 23 genes, five genes [ZNF224 (8), CCNL1 (9), APOBEC3H (10), ANAPC4 (11) and IKBKB (12)] were further excluded as they have
previously been reported to be associated with tumor development.
Cloning primers were designed for the remaining 18 genes (Table SI). Following PCR amplification,
restriction digestion ligation and transformation, 12 genes
(ABRACL, MITD1, ZNF524, EMP1, HSPB6, CXorf38, TRIM38, ZNF182,
ZNF195, SPRN, PTPN6 and LIPT1) were successfully cloned
into the pcDNA3.1+ eukaryotic expression vector (Invitrogen; Thermo
Fisher Scientific, Inc.).
Clinical samples for IHC
A total of 10 patients with bladder cancer from the
Affiliated Hospital of Qingdao University were enrolled in the
present study between December 2016 and June 2018. The patients had
not received radiotherapy or chemotherapy prior to surgery and the
pathological type is urothelial cancer. The primary tumors,
adjacent normal tissues (the distance between the tumor tissues and
normal tissues >3 cm) and metastatic lymph nodes were surgically
removed by radical cystectomy and pelvic lymphadenectomy, and
immediately fixed in 10% formalin at room temperature for 24 h and
embedded in paraffin. The present study was approved by the Human
Ethics Committee of The Affiliated Hospital of Qingdao University
(Qingdao, China; approval no. QYFYWZLL25948). The present study was
performed in accordance with the latest version of the Declaration
of Helsinki (13). Written informed
consent was provided by all patients prior to the study start. The
detailed clinical information of each patient is listed in Table I.
 | Table I.Clinical information of patient
samples for immunohistochemistry. |
Table I.
Clinical information of patient
samples for immunohistochemistry.
| Patient number | Age, years | Sex |
Differentiation | Sample site |
|---|
| 1 | 67 | Male | Poor | Normal tissue,
Primary tumor, Lymph node metastasis |
| 2 | 77 | Female | Poor | Normal tissue,
Primary tumor, Lymph node metastasis |
| 3 | 55 | Male | Poor | Normal tissue,
Primary tumor, Lymph node metastasis |
| 4 | 60 | Male | Poor | Lymph node
metastasis |
| 5 | 64 | Male | Poor | Primary tumor,
Lymph node metastasis |
| 6 | 75 | Male | Poor | Primary tumor,
Lymph node metastasis |
| 7 | 73 | Female | Poor | Normal tissue,
Primary tumor, Lymph node metastasis |
| 8 | 70 | Male | Poor | Primary tumor,
Lymph node metastasis |
| 9 | 75 | Male | Poor | Lymph node
metastasis |
| 10 | 64 | Male | Poor | Primary tumor,
Lymph node metastasis |
Vector constructing
The gene coding sequences of ABRACL, MITD1,
ZNF524, EMP1, HSPB6, CXorf38, TRIM38, ZNF182, ZNF195, SPRN,
PTPN6 and LIPT1 were amplified via PCR. PCR
thermocycling conditions were as follows: 98°C for 1 min and 30
cycles of 98°C for 10 sec, 55°C for 15 sec, and 72°C for 30 sec.
PrimeSTAR MAX polymerase (cat. no. R045A; Takara Biomedical
Technology (Beijing), Co., Ltd.) was used and the DNA of T24 cell
was the template. The products were subsequently cloned into a
pcDNA3.1+ vector (Invitrogen; Thermo Fisher Scientific, Inc.) with
double digestion and ligation. Following transformation of the
ligation products into DH5α competent cells (Takara Biomedical
Technology, Co., Ltd.), respectively, the bacteria were cultured
using the spread plate method (14)
and 10 bacterial colonies for each gene were validated via PCR. The
PCR products of positive colonies were sent to Sanger sequencing
(BGI, Inc.) and the sequence compare was performed using Chromas
software v2.4.1 (http://technelysium.com.au/wp/chromas) for the
sequence correctness. The primer sequences used for cloning are
listed in Table SI.
DNA and small interfering (si)RNA
transfection
Transfection was performed at room temperature for 6
h. The T24 and 5637 bladder tumor cell lines (cat. no. SCSP-536 and
TCHu 1; Chinese Academy of Sciences Cell Bank) were seeded into
12-well plates at a density of 100,000 cells/well the day before
transfection. For transfection with the gene expression constructs,
1 µg of the gene expression vector (constructed by ourselves) and 2
µl p3000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) were
diluted into Opti-MEM solution (Invitrogen; Thermo Fisher
Scientific, Inc.) and mixed with equal volumes of Opti-MEM, which
contained 1.5 µl Lipofectamine® 3000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Following incubation
at room temperature for 5 min, the 12 gene mixtures were added into
the appropriate cell cultures, respectively. With regards to the
siRNA transfection of genes ABACL, CXof38, LIPT1, MITD1, ZNF524,
TRIM38 and EHBP1, 30 pmol siRNA or negative control (NC)
oligonucleotide was diluted into Opti-MEM solution and mixed with
equal volumes Opti-MEM, which contained 3 µl
Lipofectamine® 3000 reagent. Cells were collected by
trypsinization for the following experiments (15), 48 h post-transfection. The siRNAs
and NC oligonucleotide (cat. no. siN0000002-1-5) were produced by
Guangzhou RiboBio, Co., Ltd., and the siRNA targets of the assessed
genes are presented in Table
SII.
MTT assay
Cells that underwent transfection were reseeded into
96-well culture plates at a density of 2,000 cells/well, and
maintained in 100 µl RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS;
Gibco, Thermo Fisher Scientific, Inc.) at 37°C in 5%
CO2. The wells in the borders of the plates were filled
with phosphate buffered saline (PBS; Beyotime Institute of
Biotechnology) to protect against the influence of culture medium
volatilization. Cells were incubated with 10 µl MTT reagent
(Beyotime Institute of Biotechnology) at 37°C for 4 h, 72 h
post-transfection. Following the MTT incubation, the purple
formazan crystals were dissolved using 100 µl dimethyl sulfoxide
(Beyotime Biotechnology) and viability was subsequently analyzed at
a wavelength of 490 nm using a spectrophotometer (Thermo Fisher
Scientific, Inc.) (16,17). All experiments were performed in
triplicate.
Transwell assay
The Transwell assay was performed as previously
described (18). Cells that
underwent transfection were washed twice with RPMI-1640 medium
without FBS. A total of 1×105 cells were resuspended in
RPMI-1640 medium without FBS and plated in the upper chambers of
Transwell plates (cat. no. 353097; BD Biosciences), which had
transparent polyester membranes and a pore size of 8 µm. RPMI-1640
medium supplemented with 10% FBS was plated in the lower chambers.
Following incubation at 37°C for 12 h, the upper chambers were
fixed with 100% methyl alcohol at room temperature for 10 min and
subsequently stained with 0.1% crystal violet at room temperature
for 10 min, and the excess dye was washed off with water. The
unmigrated cells were removed using cotton swabs. Stained cells
were counted in five randomly selected fields using a light
microscope (magnification, ×100) (18).
Wound healing assay
T24 cells were seeded into 6-well culture plates at
a density of 1×106 cells/well the day before
transfection. Cells transfected with the negative control and
MITD1 siRNAs were further cultured in PRMI-1640 medium
supplemented with 10% FBS at 37°C for 48 h. Cells were scratched
with 200 µl pipette tips, washed once with PBS, and further
incubated in serum-free medium at 37°C cell incubator for 48 h. The
scratches were observed at 0, 24 and 48 h, respectively, using an
inverted light microscope (magnification, ×100; IX73; Olympus
Corporation). The results were quantified using ImageJ software
(version 1.50i; National Institutes of Health) and analyzed using
GraphPad Prism software (version 6; GraphPad Software, Inc.). All
experiments were performed in triplicate.
IHC staining
IHC analysis was performed as previously described
(19). Briefly, the surgically
removed tissues were fixed in 10% formalin at room temperature for
24 h and embedded in paraffin. Paraffin-embedded tissue samples
were cut into 4-µm-thick sections and heated overnight at 50°C.
Prior to immunostaining, deparaffinization and hydration were
performed in xylene and a descending ethanol series (100, 95 and
75%). Heat Induced Epitope Retrieval was performed by heating the
slides immersed in retrieval solution: 10 mM sodium citrate buffer,
pH 6.0, with 1 mM EDTA, at 125°C for 4 min in a pressure boiler.
Tissue sections were incubated with 0.3% (v/v)
H2O2 in 95% ethanol at room temperature for 5
min to inhibit endogenous peroxidase activity. Tissue sections were
washed twice with PBS. Immunohistochemical staining was performed
with Polymer HRP Detection System for Rabbit Primary Antibody (cat.
no. PV-6001; ZSGB-BIO, Inc.). All incubations were performed at
room temperature and reagents were applied at a volume of 200 µl
per slide. According to the instruction manual, blocking was not
necessary due to the special design of the primary antibody
dilution buffer. The slides were incubated with MITD1 antibody
(cat. no. HPA036162; Atlas Antibodies AB; 1:50) at room temperature
for 30 min. The slides were rinsed three times with PBS, and
subsequently incubated with peroxidase labeled polymer conjugated
to goat anti-rabbit IgG antibody working solution (cat. no.
PV-6001; ZSGB-BIO, Inc.) at room temperature for 30 min. The slides
were re-washed twice in wash buffer and developed at room
temperature for 5 min using DAB (cat. no. PV-6001; ZSGB-BIO, Inc.)
as the substrate. Counterstaining was performed using hematoxylin
solution (cat. no. C0107; Beyotime Institute of Biotechnology) for
3 min at room temperature. Cells were observed under a light
microscope (magnification ×400).
RNA sequencing
RNA was extracted from cells that underwent
transfection using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The library construction, sequencing and data analysis were
performed by Annoroad Gene Technology Corporation (http://en.annoroad.com), with an Illumina Sequencing
platform (HiSeq2500; Illumina, Inc.). Sequencing libraries were
generated using the NEBNext® Ultra™ RNA Library Prep kit
for Illumina® (cat. no. E7530L; NEB, Inc. http://www.neb.com), according to the manufacturer's
instructions. RNA integrity and concentration were assessed using
the RNA Nano 6000 Assay kit of the Bioanalyzer 2100 system (cat.
no. 5067-1511; Agilent Technologies, Inc.). The concentration of
library was measured using the Qubit® RNA Assay kit
(cat. no. Q32852; Thermo Fisher Scientific, Inc.) to preliminarily
quantify and subsequently dilute to 1 ng/µl. The accurate
concentration was measured using a StepOnePlus™ Real-Time PCR
System (Thermo Fisher Scientific, Inc.). The valid concentrations
of the libraries are greater than 10 nM. The clustering of the
index-coded samples was performed on a cBot cluster generation
system using the HiSeq PE Cluster kit v4-cBot-HS (cat. no.
PE-401-4001; Illumina, Inc.), according to the manufacturer's
instructions. Following cluster generation, the libraries were
sequenced on an Illumina platform (HiSeq2500; Illumina, Inc.) and
150-bp paired-end raw reads were generated.
RNA sequencing analysis
The Perl script was used to filter the original data
to guarantee the data quality. The steps were as follows: i) Trim
Smart-seq2 public primer sequence from the reads; ii) remove the
contaminated reads of adapters; iii) remove the low quality reads
and iv) remove the reads with N base >5% for total bases. The
reference genomes and the annotation file were downloaded from
ENSEMBL database (http://www.ensembl.org/index.html). Bowtie2 v2.2.3
(http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
was used to build the genome index, and Clean Data was subsequently
aligned to the reference genome using HISAT2 v2.1.0 (http://ccb.jhu.edu/software/hisat2/faq.shtml). Reads
Count for each gene in each sample was counted using HTSeq v0.6.0
(https://htseq.readthedocs.io), and
Fragments Per Kilobase Millon Mapped Reads (FPKM) was subsequently
calculated to estimate the expression level of genes in each
sample. DESeq2 (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html)
was used to compare differences in gene expression levels between
groups. Genes with P≤0.05 and fold change (FC)≥1.5 were classified
as differentially expressed genes (DEGs).
Enriched pathway analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analyses were performed using the Database for
Annotation, Visualization and Integration Discovery (DAVID; version
6.8; http://david.ncifcrf.gov/home.jsp). DEGs between the
control and MITD1 knockdown groups were uploaded onto DAVID.
The enriched KEGG pathways of the genes were assessed using the
‘functional annotation tool’ within DAVID. Modified fisher exact
P-value was calculated to determine the enriched extent. Gene
count=2 was used as the threshold to include the items in the
table.
Reverse transcription-quantitative
(RT-q)PCR
Transfection efficiency was demonstrated via
RT-qPCR. Total RNA were extracted from T24 and 5637 cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse transcribed at 37°C for 15 min using the PrimeScript™ RT
reagent kit with gDNA Eraser (cat. no. RR047; Takara Biomedical
Technology, Inc.). qPCR was subsequently performed using Platinum™
SYBR™ Green qPCR SuperMix-UDG (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. GAPDH was used as the
reference gene. The qPCR primers are listed in Table SIII. qPCR thermocycling conditions
were as follows: 95°C for 1 min and 40 cycles of 95°C for 15 sec,
60°C for 30 sec, and 72°C for 15 sec. All data were analyzed using
the 2−ΔΔCq method (20).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6 (GraphPad Software, Inc.). Experiments were performed at
least in triplicate, and data are presented as the mean ± standard
deviation. Unpaired Student's t-test was used to compare
differences between two groups, while one-way ANOVA followed by
Tukey's post hoc test was used to compare differences between
multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression vectors constructed for the
survival-associated genes
In order to measure the function of the prognostic
genes on cell proliferation and migration, the present study
focused on the no bias system-level analysis of the Pathology
Atlas, which is based on the genome-wide transcriptomics analysis
of TCGA project and the Human Protein Atlas project (4) (http://www.proteinatlas.org). The present study
selected the top 18 genes significantly associated with survival in
patients with urothelial cancer in the Pathology Atlas project,
which met the following criteria: i) The prognostic
P<1×10−6 (best cut-off); ii) the prognostic
P<1×10−2 (median expression cut-off) and iii) has
rarely been reported to be associated with cancer progression.
Following PCR amplification and restriction digestion, 12 genes
(ABRACL, MITD1, ZNF524, EMP1, HSPB6, CXorf38, TRIM38, ZNF182,
ZNF195, SPRN, PTPN6 and LIPT1) were successfully cloned
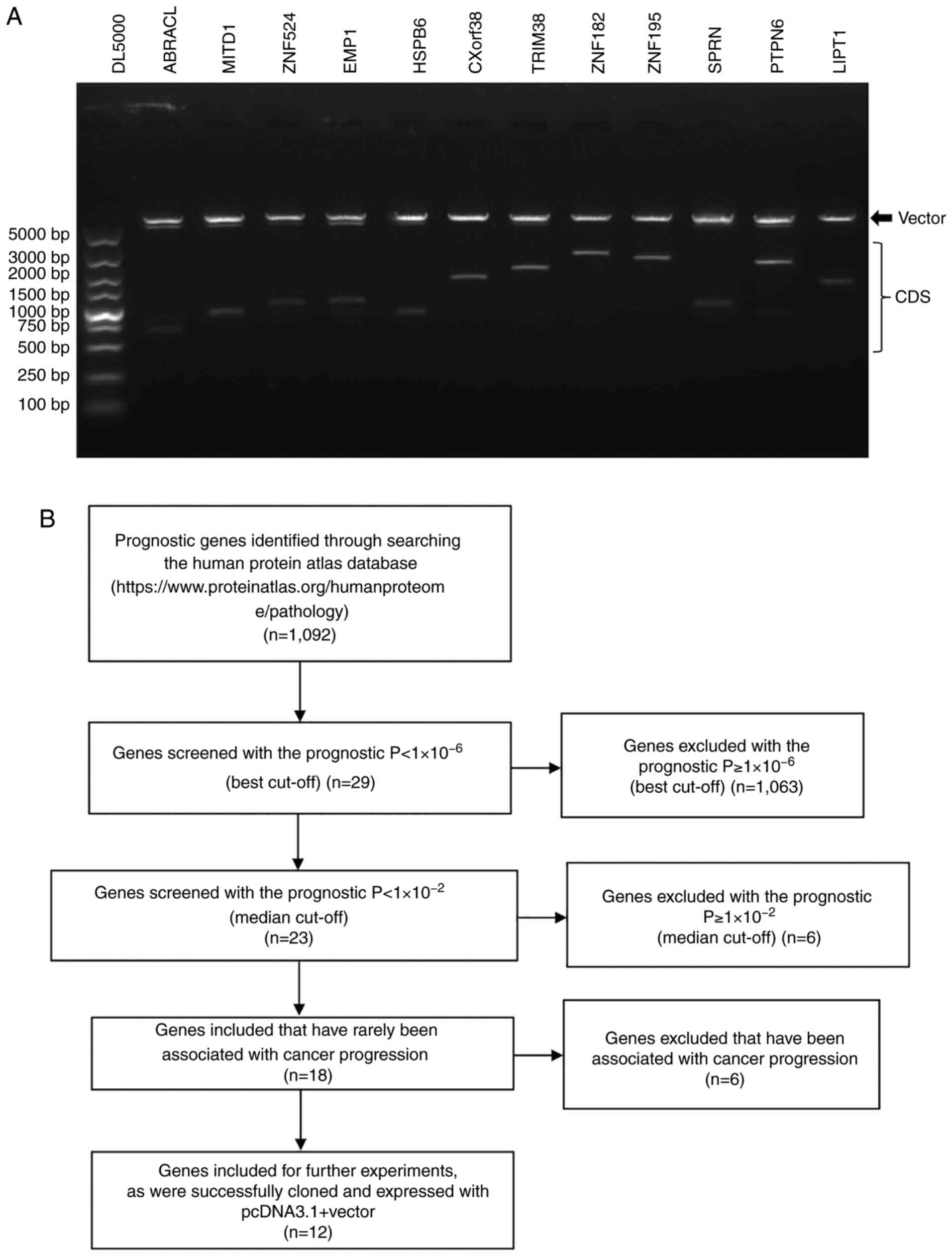
into the pcDNA3.1+ eukaryotic expression vector (Fig. 1A). The process of screening the 12
genes is presented in Fig. 1B and
detailed information of the 12 genes is presented in Table II. All 12 constructs were validated
via Sanger sequencing.
| Table II.Information on the 12 prognostic
genes. |
Table II.
Information on the 12 prognostic
genes.
| Gene | Gene
description | Prognostic
function | P-value (Best
expression cut-off) | P-value (Median
expression cut-off) |
|---|
| ABRACL | ABRA C-terminal
like | Favorable |
1.65×10−6 |
1.69×10−5 |
| CXorf38 | Chromosome X open
reading frame 38 | Favorable |
1.44×10−8 |
7.54×10−7 |
| EMP1 | Epithelial membrane
protein 1 | Unfavorable |
9.69×10−8 |
8.63×10−5 |
| HSPB6 | Heat shock protein,
alpha-crystallin-related, B6 | Unfavorable |
1.43×10−6 |
9.51×10−6 |
| LIPT1 | Lipoyltransferase
1 | Favorable |
9.60×10−8 |
9.71×10−5 |
| MITD1 | MIT, microtubule
interacting and transport, domain containing 1 | Favorable |
5.46×10−8 |
2.00×10−6 |
| PTPN6 | Protein tyrosine
phosphatase, non-receptor type 6 | Favorable |
2.64×10−8 |
2.41×10−7 |
| SPRN | Shadow of prion
protein homolog (zebrafish) | Favorable |
1.58×10−7 |
3.42×10−4 |
| TRIM38 | Tripartite motif
containing 38 | Favorable |
4.94×10−9 |
7.93×10−6 |
| ZNF182 | Zinc finger protein
182 | Favorable |
1.55×10−7 |
7.99×10−3 |
| ZNF195 | Zinc finger protein
195 | Favorable |
4.79×10−7 |
2.92×10−5 |
| ZNF524 | Zinc finger protein
524 | Favorable |
4.98×10−8 |
4.73×10−4 |
Effects of the prognostic genes on
tumor cell viability
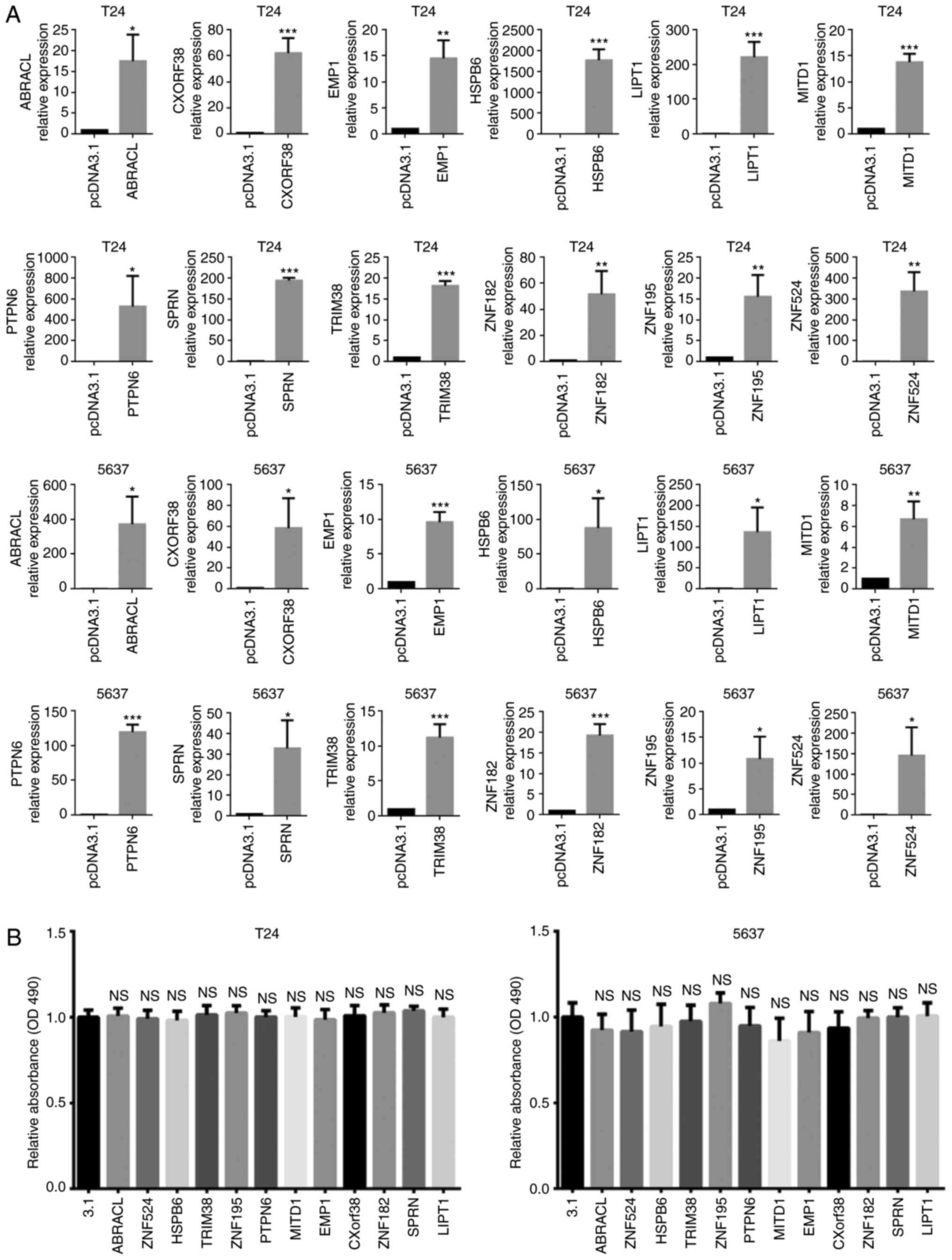
Recombinant gene transfection may induce cell death
or change cell proliferation rate (21). Thus, the present study assessed cell
viability via the MTT assay 72 h post-transfection (Fig. 2A). The results demonstrated that in
the in vitro experiments, there was no significant change in
cell viability induced by transfection of the 12 gene expression
constructors (Fig. 2B).
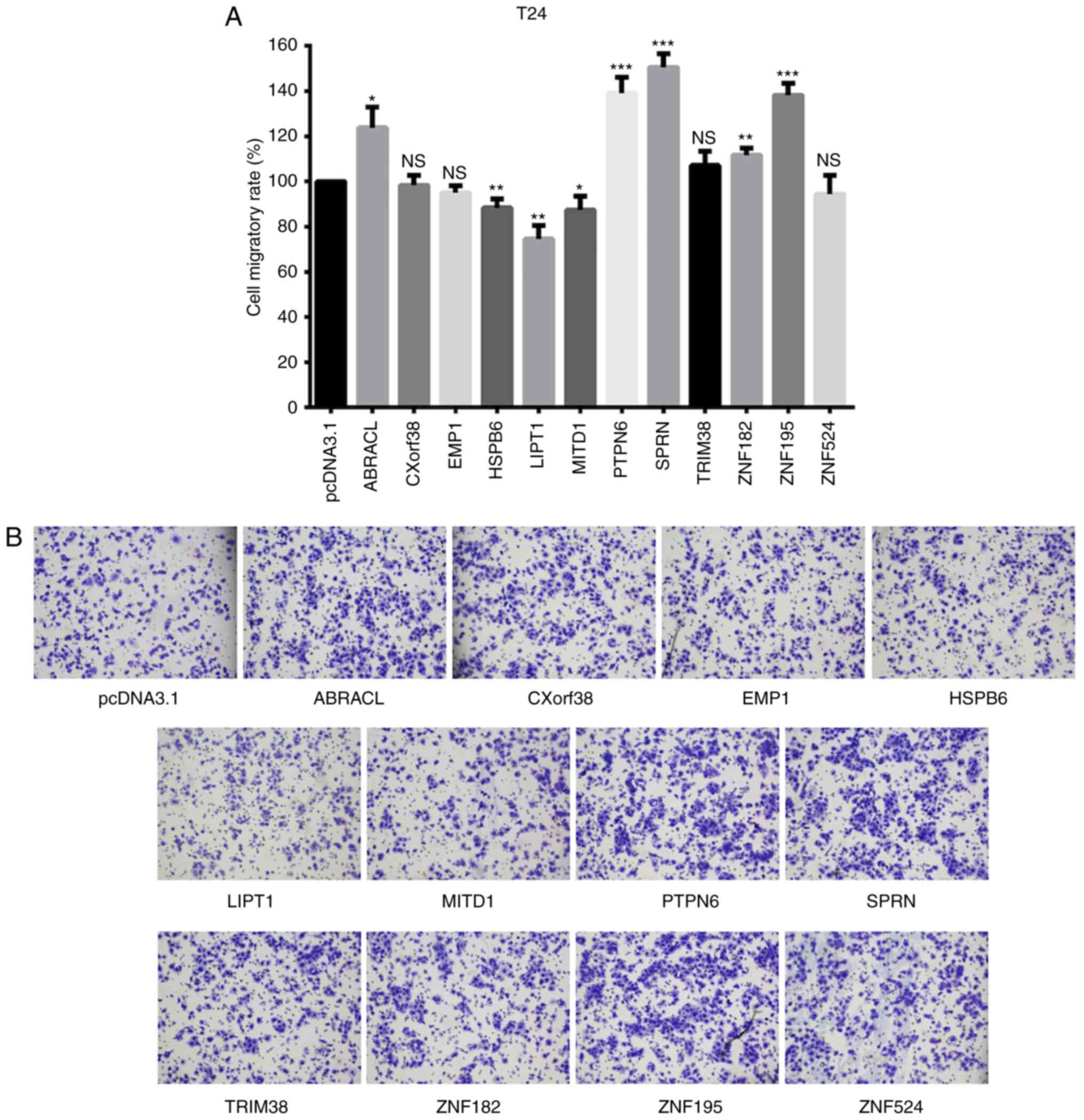
Screening migration-associated genes
among the survival-associated genes
The 12 gene expression vectors were subsequently
transfected into T24 bladder cancer cells, respectively. The
migratory ability of cells was assessed via the Transwell assay, 48
h post-transfection. The results demonstrated that the migratory
rate of cells transfected with CXorf38, EMP1, TRIM38, ZNF182
and ZNF524 did not changed compared with the mean value of
the 12 overexpression groups. However, ABRACL, PTPN6, SPRN
and ZNF195 transfection increased the cell migratory
ability. The three genes, including two favorable-survival
associated genes (MITD1 and LIPT1) and one
unfavorable-survival associated gene, HSPB6, inhibited cell
migration at a certain level (Fig.
3).
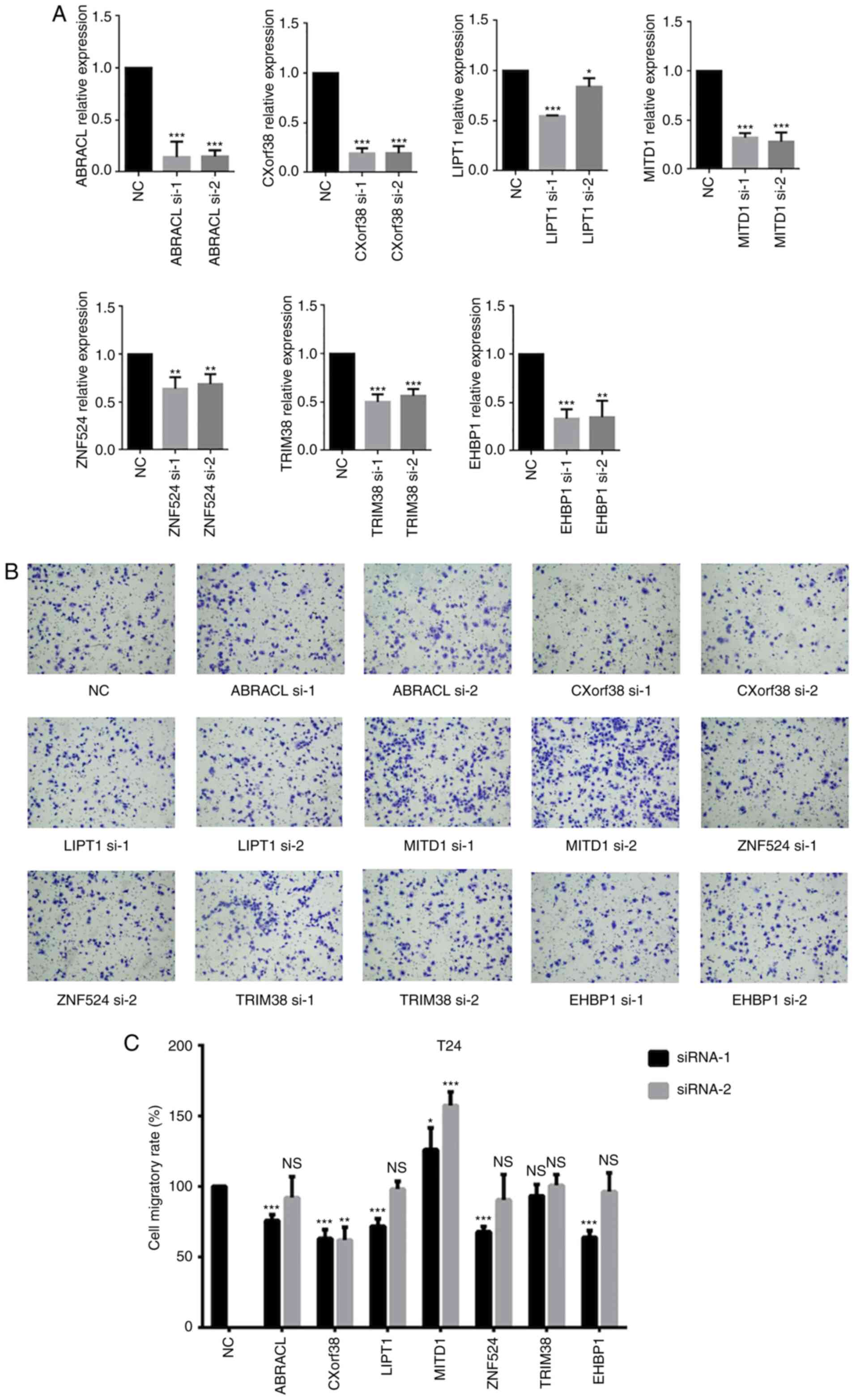
A siRNA library of 7 genes, in which each gene had
two siRNAs, was used to screen the genes regulating cell migration
with a loss-of-function strategy (Fig.
4A). MITD1 knockdown markedly increased cell migration
(Fig. 4B and C). The wound healing
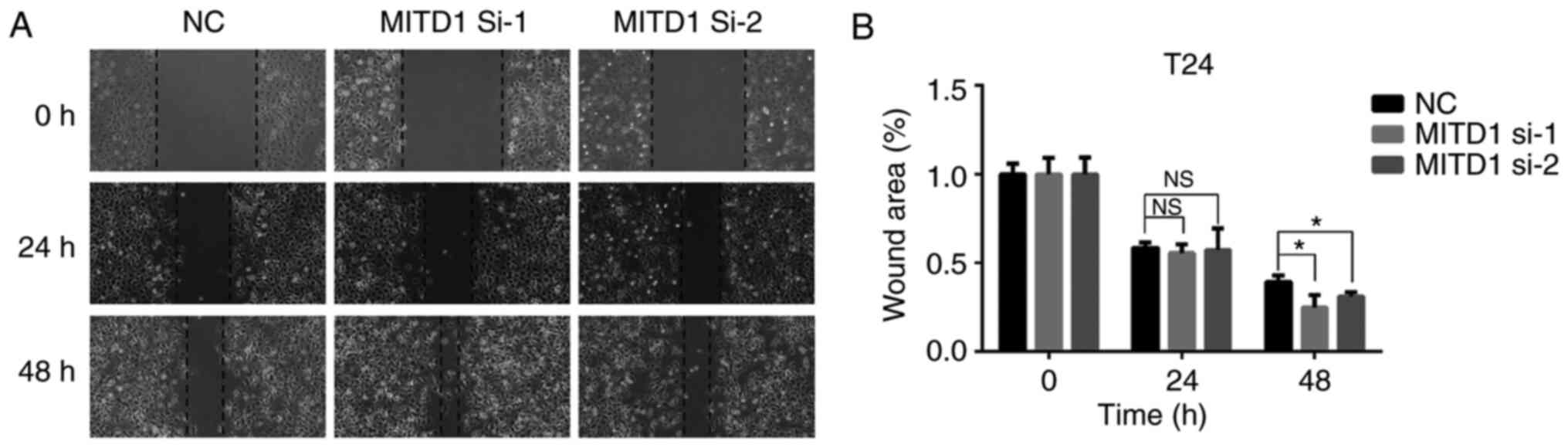
assay was performed to validate the role of MITD1 on cell migration
and the results demonstrated that MITD1 knockdown
significantly promoted cell migration (Fig. 5).
MITD1 downstream molecule
analysis
As MITD1 inhibited cell migration in
vitro, downstream genes were investigated with RNA sequencing.
T24 cells were transfected with MITD1 siRNAs or NC
oligonucleotide, while the untreated cells were cultured as the
BLANK group. Cell RNA was extracted using TRIzol®
reagent 48 h post-transfection, and transcriptome sequencing was
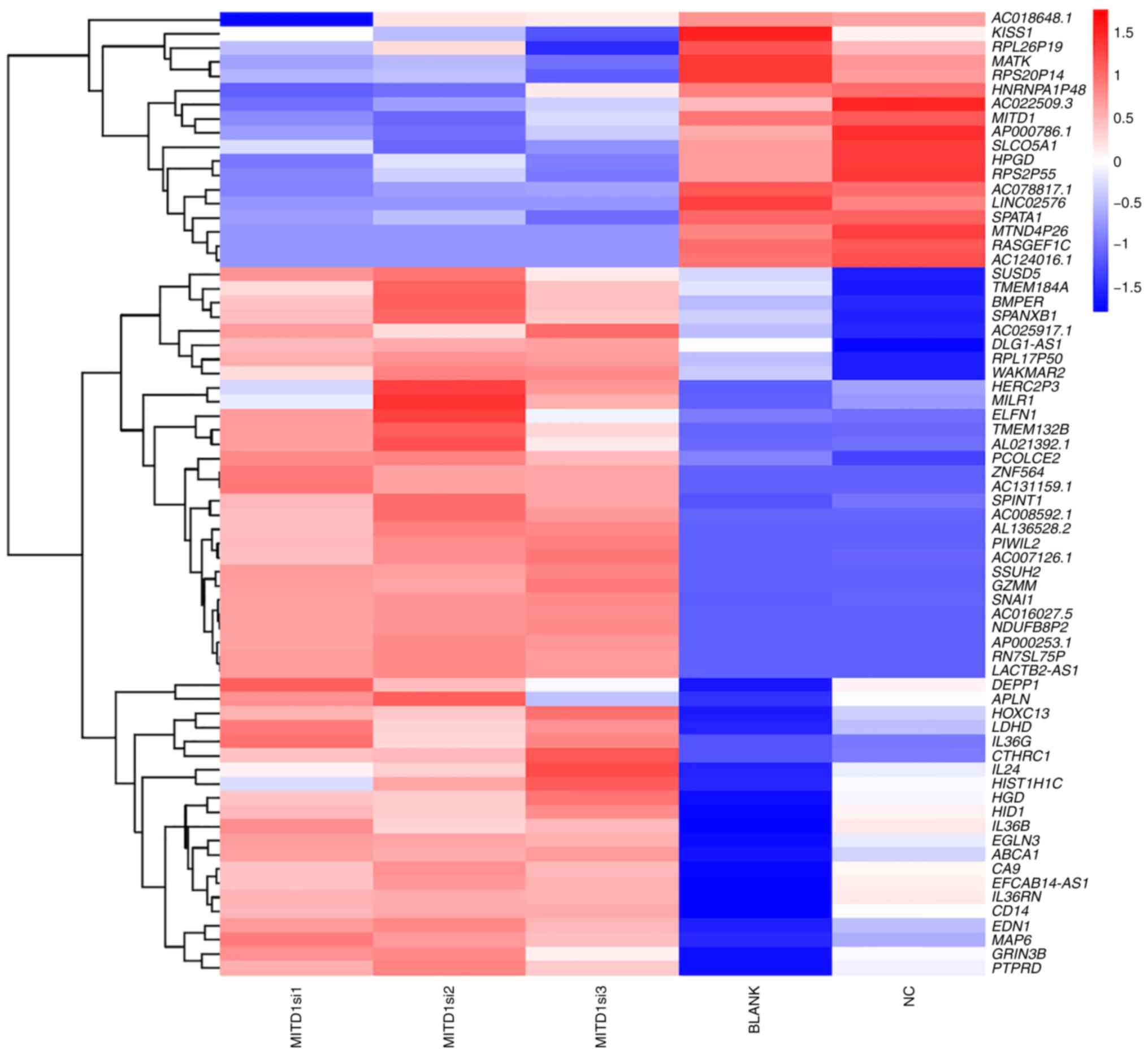
subsequently performed. The results demonstrated that cells
transfected with MITD1 siRNA exhibited dysregulation of the
migration associated genes, KISS1, SPANXB1, SPINT1, PIWIL2,
SNAI1, APLN, EDN1 and CTHRC1 compared with the NC and
BLANK groups (Fig. 6). The enriched
KEGG pathways were analyzed using DAVID Bioinformatics Resources
6.8. The results demonstrated that ‘transcriptional dysregulation
in cancer’ was significantly enriched (P=0.019) (Table SIV).
MITD1 expression is downregulated in
metastatic tumors
Lymphatic metastasis is one of the main
characteristics underlying bladder cancer progression, and a key
predictor of cancer-specific survival (22). To identify the associations between
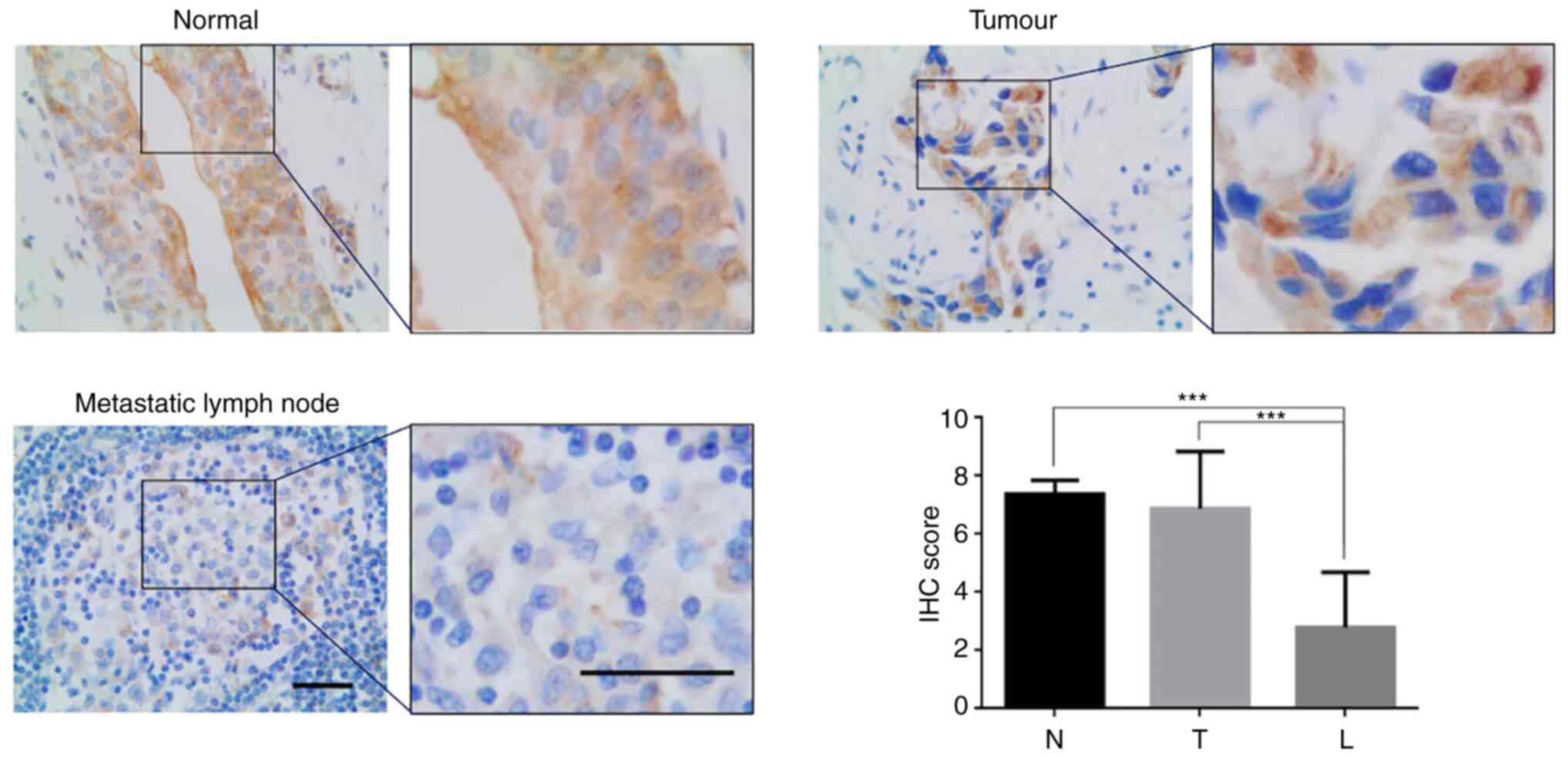
MITD1 and lymphatic metastasis, the present study assessed MITD1
protein expression in the primary tumors and paired metastasis in
lymph nodes. IHC analysis demonstrated that there was a significant
decrease in MITD1 protein expression in the metastatic tumors
compared with the primary tumors (mean IHC score, 6.875 vs. 2.800;
P<0.001; Fig. 7).
Discussion
Improving tumor prognosis remains a major challenge.
Based on the genome-wide transcriptomic data of TCGA and the
clinical metadata, the Pathology Atlas project performed a
systematical survival analysis of 17 major cancer types, and
identified the most significant survival-associated genes (4). However, the biological function and
molecular mechanisms of these genes has not yet been identified.
The present study investigated the function of prognostic genes
identified in the Pathology Atlas project with in vitro
experiments, with respect to cell proliferation and migration rate.
Among the 12 prognostic genes, a favorable gene, MITD1
exhibited a notable effect on cell migration. When exogenous
recombined MITD1 was transfected into T24 cells, the cell
migratory ability was downregulated, while knockdown of
MITD1 promoted cell migration. Using RNA sequencing, the
present study also revealed several potential downstream molecules
of MITD1 to mediate migration arrest. To validate the
function of MITD1 on cell migration in vivo, the
present study compared the MITD1 protein level in primary tumors
and lymph node metastatic tumors. The results demonstrated that
MITD1 expression decreased in metastatic tumors, which was in
accordance with the in vitro results. Taken together, these
results suggest that MITD1 may be a migration inhibitor and
a potential therapeutic target to improve bladder cancer
prognosis.
MITD1 interacts with endosomal sorting
complexes required for transport III (ESCRT-III) and may be
recruited to the midbody to participate in cytokinesis (22,23).
In the present study, both gain- and loss-of-function in
vitro experiments demonstrated that MITD1 may be a
migration inhibitor, which is consistent with the favorable role on
survival identified in the Pathology Atlas project.
To determine the molecular mechanism underlying
MITD1-induced inhibition of cell migration, RNA sequencing
was performed. The results demonstrated that several potential
downstream molecules (KISS1, SPANXB1, SPINT1, PIWIL2, SNAI1,
APLN, EDN1 and CTHRC1), which have been reported to be
associated with cell metastasis, were dysregulated following
MITD1 knockdown. For example, the EMT regulator,
SNAI1 was significantly upregulated, while the metastasis
suppressor KISS1 (24–26)
was downregulated to a certain degree.
Furthermore, although the sample size was limited,
the present study demonstrated that MITD1 expression levels were
much higher in the primary tumors compared with the metastatic
tumors. Collectively, these results suggest that tumor clones with
lower expression levels of MITD1 may be advantageous for metastasis
in vivo.
Uncontrolled cell proliferation is one of the
hallmarks of cancer, and it is widely accepted that the cell
proliferation rate may be involved in cancer progression, and
affect the prognosis (5). However,
the present study did not observe any notable effects of the
prognostic gene overexpression on cell viability. Neither serious
cell cytotoxicity or accelerated proliferation were introduced
following transfection of these genes. These results suggest that
the genes may influence tumor development independent of altering
the cell proliferation rate. Conversely, the same level of cell
viability was used in each transfection group to ensure that the
results of the migration assay were not affected by the differences
in cell cytotoxicity or proliferation.
With regards to the consistency between the in
vitro experiments and clinical survival studies, 10 of the 12
prognostic genes (ABRACL, ZNF524, EMP1, HSPB6, CXorf38, TRIM38,
ZNF182, ZNF195, SPRN and PTPN6) exhibited the unexpected
role on cell migration in the overexpression studies. In the
Pathology Atlas project, the high expression levels of ABRACL,
CXorf38, PTPN6, SPRN, TRIM38, ZNF182, ZNF195 and ZNF524
were associated with favorable survival (4). However, the results of the present
study demonstrated that overexpression of these genes did not
weaken cell migratory ability compared with overexpression of
EMP1 and HSPB6, high expression of which is
associated with poor survival outcomes (4). There were three possible reasons for
this: i) The majority of the prognostic genes identified by the
Pathology Atlas project may act as a ‘passenger’ for tumor
development, not the ‘driver’; ii) the in vitro experiments
may not completely mimic the authentic in vivo context and
iii) the prognostic genes may promote cancer progress by altering
other characteristics, such as invasion, immune escape and
angiogenesis.
One of the limitations of the present study is that
the detailed molecular mechanisms by which MITD1 regulates cell
migration and influences patient survival were not investigated.
The detailed molecular mechanisms will be investigated by in
vivo research in the future.
The present study investigated the role of the
prognostic genes identified in the Pathology Atlas project on cell
proliferation and migration, and demonstrated that the favorable
prognostic gene, MITD1 may inhibit cell migration and
metastasis, which implies a novel promising target for targeted
therapies of bladder cancer.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundations of China (grant nos. 81772713,
81472411, 81981260351 and 81972378), the Taishan Scholar Program of
Shandong Province (grant no. tsqn20161077), the Natural Science
Foundation of Shandong Province (grant no. ZR2016HQ18) and the Key
Research and Development Program of Shandong Province (grant no.
2018GSF118197).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author upon reasonable
request. The RNA-seq raw FASTQ files were deposited in the NCBI SRA
database (accession no. PRJNA658813).
Authors' contributions
YC, WJ and XY designed the present study. YC, TX and
XQ drafted the initial manuscript. YC, TX, WJ, FX and LW performed
the cell experiments. ZL, YL, KZ, XQ and DL contributed to
statistical analysis and designed the tables and figures. WJ, XY
and XQ were involved in project management and supervised the
study. WJ and XY contributed to revising the manuscript for
intellectual content and language editing. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Human Ethics
Committee of The Affiliated Hospital of Qingdao University
(Qingdao, China; approval no. QYFYWZLL25948). Written informed
consent was provided by all patients prior to the study start.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miyazaki J and Nishiyama H: Epidemiology
of urothelial carcinoma. Int J Urol. 24:730–734. 2017. View Article : Google Scholar
|
|
2
|
Berdik C: Unlocking bladder cancer.
Nature. 551:S34–S35. 2017. View
Article : Google Scholar
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar
|
|
4
|
Uhlen M, Zhang C, Lee S, Sjöstedt E,
Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et
al: A pathology atlas of the human cancer transcriptome. Science.
357:eaan25072017. View Article : Google Scholar
|
|
5
|
Fouad YA and Aanei C: Revisiting the
hallmarks of cancer. Am J Cancer Res. 7:1016–1036. 2017.
|
|
6
|
Zhang D, Jin N, Sun W, Li X, Liu B, Xie Z,
Qu J, Xu J, Yang X, Su Y, et al: Phosphoglycerate mutase 1 promotes
cancer cell migration independent of its metabolic activity.
Oncogene. 36:2900–2909. 2017. View Article : Google Scholar
|
|
7
|
Xue Y, Xiao H, Guo S, Xu B, Liao Y, Wu Y
and Zhang G: Indoleamine 2,3-dioxygenase expression regulates the
survival and proliferation of Fusobacterium nucleatum in
THP-1-derived macrophages. Cell Death Dis. 9:3552018. View Article : Google Scholar
|
|
8
|
Harada Y, Kanehira M, Fujisawa Y, Takata
R, Shuin T, Miki T, Fujioka T, Nakamura Y and Katagiri T:
Cell-permeable peptide DEPDC1-ZNF224 interferes with
transcriptional repression and oncogenicity in bladder cancer
cells. Cancer Res. 70:5829–5839. 2010. View Article : Google Scholar
|
|
9
|
Sticht C, Hofele C, Flechtenmacher C,
Bosch FX, Freier K, Lichter P and Joos S: Amplification of Cyclin
L1 is associated with lymph node metastases in head and neck
squamous cell carcinoma (HNSCC). Br J Cancer. 92:770–774. 2005.
View Article : Google Scholar
|
|
10
|
Starrett GJ, Luengas EM, McCann JL,
Ebrahimi D, Temiz NA, Love RP, Feng Y, Adolph MB, Chelico L, Law
EK, et al: The DNA cytosine deaminase APOBEC3H haplotype I likely
contributes to breast and lung cancer mutagenesis. Nat Commun.
7:129182016. View Article : Google Scholar
|
|
11
|
Eifler K, Cuijpers SAG, Willemstein E,
Raaijmakers JA, El Atmioui D, Ovaa H, Medema RH and Vertegaal ACO:
SUMO targets the APC/C to regulate transition from metaphase to
anaphase. Nat Commun. 9:11192018. View Article : Google Scholar
|
|
12
|
Carneiro-Lobo TC, Scalabrini LC, Magalhães
LDS, Cardeal LB, Rodrigues FS, Dos Santos EO, Baldwin AS, Levantini
E, Giordano RJ and Bassères DS: IKKβ targeting reduces KRAS-induced
lung cancer angiogenesis in vitro and in vivo: A potential
anti-angiogenic therapeutic target. Lung Cancer. 130:169–178. 2019.
View Article : Google Scholar
|
|
13
|
World Medical Association: World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar
|
|
14
|
Sanders ER: Aseptic laboratory techniques:
Plating methods. J Vis Exp. e30642012.
|
|
15
|
Yan S, Tang Z, Chen K, Liu Y, Yu G, Chen
Q, Dang H, Chen F, Ling J, Zhu L, et al: Long noncoding RNA MIR31HG
inhibits hepatocellular carcinoma proliferation and metastasis by
sponging microRNA-575 to modulate ST7L expression. J Exp Clin
Cancer Res. 37:2142018. View Article : Google Scholar
|
|
16
|
Liu Y, Zeng C, Bao N, Zhao J, Hu Y, Li C
and Chi S: Effect of Rab23 on the proliferation and apoptosis in
breast cancer. Oncol Rep. 34:1835–1844. 2015. View Article : Google Scholar
|
|
17
|
Huang W, Chen C, Liang Z, Qiu J, Li X, Hu
X, Xiang S, Ding X and Zhang J: AP-2α inhibits hepatocellular
carcinoma cell growth and migration. Int J Oncol. 48:1125–1134.
2016. View Article : Google Scholar
|
|
18
|
Zhou B, Wang GZ, Wen ZS, Zhou YC, Huang
YC, Chen Y and Zhou GB: Somatic mutations and splicing variants of
focal adhesion kinase in non-small cell lung cancer. J Natl Cancer
Inst. 110:2018. View Article : Google Scholar
|
|
19
|
Sun Y, Luo J, Chen Y, Cui J, Lei Y, Cui Y,
Jiang N, Jiang W, Chen L, Chen Y, et al: Combined evaluation of the
expression status of CD155 and TIGIT plays an important role in the
prognosis of LUAD (lung adenocarcinoma). Int Immunopharmacol.
80:1061982020. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Tian ZQ, Xu YZ, Zhang YF, Ma GF, He M and
Wang GY: Effects of metallothionein-3 and metallothionein-1E gene
transfection on proliferation, cell cycle, and apoptosis of
esophageal cancer cells. Genet Mol Res. 12:4595–4603. 2013.
View Article : Google Scholar
|
|
22
|
Kassouf W, Svatek RS, Shariat SF, Novara
G, Lerner SP, Fradet Y, Bastian PJ, Aprikian A, Karakiewicz PI,
Fritsche HM, et al: Critical analysis and validation of lymph node
density as prognostic variable in urothelial carcinoma of bladder.
Urol Oncol. 31:480–486. 2013. View Article : Google Scholar
|
|
23
|
Hadders MA, Agromayor M, Obita T, Perisic
O, Caballe A, Kloc M, Lamers MH, Williams RL and Martin-Serrano J:
ESCRT-III binding protein MITD1 is involved in cytokinesis and has
an unanticipated PLD fold that binds membranes. Proc Natl Acad Sci
USA. 109:17424–17429. 2012. View Article : Google Scholar
|
|
24
|
Lee JH and Welch DR: Suppression of
metastasis in human breast carcinoma MDA-MB-435 cells after
transfection with the metastasis suppressor gene, KiSS-1. Cancer
Res. 57:2384–2387. 1997.
|
|
25
|
Nash KT and Welch DR: The KISS1 metastasis
suppressor: Mechanistic insights and clinical utility. Front
Biosci. 11:647–659. 2006. View
Article : Google Scholar
|
|
26
|
Lee JH, Miele ME, Hicks DJ, Phillips KK,
Trent JM, Weissman BE and Welch DR: KiSS-1, a novel human malignant
melanoma metastasis-suppressor gene. J Natl Cancer Inst.
88:1731–1737. 1996. View Article : Google Scholar
|