Introduction
Non-small cell lung cancer (NSCLC), which accounts
for 85% of all lung cancer cases worldwide, is the most common and
aggressive malignant type of lung cancer (1,2).
Epidermal growth factor receptor (EGFR)-activating mutations (exon
19 deletion or exon 21 L858R point mutation) were found to be
correlated with a 70% response rate to tyrosine kinase inhibitor
(TKI) treatment (3–5). However, nearly all patients inevitably
develop an acquired resistance to TKIs after a median
progression-free survival (PFS) period of 9 to 14 months, despite a
dramatic initial response (6). The
main cause of acquired resistance is due to the EGFR T790M
mutation, tyrosine-protein kinase Met (c-MET) amplification and
epithelial-mesenchymal transition (EMT) (7). A previous study demonstrated that
transforming growth factor (TGF)-β could induce TKI resistance by
activating the signal transducer and activator of transcription 3
(STAT3) signalling pathway and EMT. Intriguingly, restraining the
activation of interleukin (IL)-6/Janus kinase (JAK)/STAT3
signalling and EMT could overcome the cellular resistance to TKIs
(8). Therefore, inhibition of the
IL-6/JAK/STAT3 signalling pathway would potentially be an effective
strategy for overcoming TKI resistance in the desensitised
cells.
Zoledronic acid (ZA) is a bisphosphonate compound
that alters bone formation and breakdown in the body, which can
slow down bone loss and effectively reduce the incidence of
skeletal-related events (9). ZA is
widely used to treat abnormally high blood calcium (hypercalcemia)
and bone pain caused by multiple myeloma. ZA is not a
chemotherapeutic drug; however, preclinical studies have suggested
that ZA might have antitumour activity in lung cancer cells, such
as inhibiting cancer cell growth, angiogenesis, invasion, and
metastasis (10,11). Our preclinical study demonstrated
that ZA combined with gefitinib could synergistically inhibit the
proliferation of lung cancer cells by suppressing STAT3 activity,
suggesting that ZA may overcome TKI resistance (12). However, the underlying mechanism for
this effect remains unclear. Clinical research has shown that
bisphosphonates may exert antitumour effects and delay the
progression of NSCLC (13). Our
study, and another retrospective clinical study, demonstrated that
ZA prolonged the overall survival (OS) of bone metastatic NSCLC
patients receiving TKI as first-line therapy (13,14).
In this present study, we found that ZA had the
potential to reverse the TKI (i.e. gefitinib) resistance of lung
cancer cells. The mechanisms involved may be attributed to the
inhibition of IL-6/JAK/STA3 signalling and reversal of EMT. In
addition, combination therapy significantly reduced the tumour size
as compared to treatment with single treatment in vivo. The
synergistic action of ZA might be due to its capability to reverse
EMT and inhibition of STAT3 phosphorylation. The present work
highlights the potential for combinatory treatment of patients with
advanced NSCLC with gefitinib and ZA, although more work is clearly
needed in the future to verify this.
Materials and methods
Cell culture and reagents
The human NSCLC cell line H1975 (EGFR L858R/T790M),
HCC827 (exon19del E746-A750), and gefitinib-resistant HCC827 cells
(HCC827GR) were a kind gift by Professor Wang Yongshen from West
China Hospital (China). The H1975, HCC827, and HCC827GR cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% foetal bovine serum (FBS) (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin-streptomycin, at 37°C in
5% CO2 and 90% humidity. Gefitinib (ZD1839), ZA, Cell
Counting Kit-8 (CCK-8), Annexin V-FITC, propidium iodide (PI) and
Hoechst were purchased from Selleck Company. The IL-6 High
Sensitivity Human ELISA Kit (ab46042) was purchased from Abcam
Company.
Cytotoxicity assay
Cells were seeded in a 96-well plate at the density
of 4×103 cells per well. The cells were then treated
with various concentrations of gefitinib and ZA for 48 h.
Thereafter, cell viability was measured using the CCK-8 assay
according to the manufacturer's instructions.
Detection of cell apoptosis
H1975 cells (1×106) were treated with
gefitinib (4 µM) and/or ZA (10 µM) for 48 h, and then stained with
Annexin V-FITC. Cell apoptosis was analysed with a Becton Dickinson
FACS Calibur flow cytometer, and the data were analysed using
FlowJo software V10.6.2 (FlowJo LLC).
Enzyme-linked immunosorbent assay
H1975 cells (0.8×106) were treated with
various concentrations of gefitinib (4 µM) and/or ZA (10 µM) for 48
h. Then, the cell culture medium was centrifuged at 1,000 × g for
10 min to remove debris. The supernatant was collected and the
level of IL-6 was assayed using the IL-6 High Sensitivity Human
ELISA kit according to the manufacturer's instructions.
Western blot assay
H1975 or HCC827GR cells (0.8×106) were
seeded in a 60-mm plate and allowed to adhere overnight. The cells
were then treated with gefitinib, or ZA, or gefitinib + ZA, or
vehicle for 48 h. Cells were lysed by RIPA lysis buffer, and 30 µg
protein was resolved by 10% sodium dodecyl sulphate-polyacrylamide
gel, following which the protein bands were transferred to
nitrocellulose membranes. The membrane was blocked with 5% skim
milk and probed with primary antibodies with gentle shaking at 4°C
overnight. The membranes were then washed three times with TBST,
and then incubated with the appropriated secondary antibodies for 2
h. Antibody-bound proteins were detected by ECL Select Western
Blotting Detection Reagent (GE Healthcare). The following primary
antibodies were used: anti-JAK1 (ab133666, Abcam),
anti-phosphorylated (p)-JAK (ab138005, Abcam), anti-STAT3
(ab119352, Abcam), anti-p-STAT3 (ab76315, Abcam), anti-E-cadherin
(ab76055, Abcam), anti-vimentin (ab8978, Abcam), and anti-SNAIL
(ab180714, Abcam). ImageJ software v1.43 (National Institutes of
Health, Bethesda, MD, USA) was used to quantify the density and
size of the blots.
Animal experiments
A total of 24 female Nu/Nu mice (15-16 g, 4 weeks
old) were used for the experiment. All animals were kept in
standard housing conditions with a temperature of 21-23°C, 50-70%
relative humidity and 12-h light/dark cycle, and access to food and
water ad libitum. H1975 cells (1×107 in Hank's
balanced salt solution) were subcutaneously implanted into the left
thigh of 6-week-old female Nu/Nu mice (Chongqing Medical
University, China). Once the average volume of the tumour had
reached 200 mm3, the mice were randomly divided into the
following 4 groups (6 mice in each group): Those given 1 mg/ml ZA
only (100 µg/kg every 2 days for 2 weeks), those given 250 mg/l
gefitinib only (50 mg/kg/day for 2 weeks), those administered both
ZA and gefitinib combined (at the same dosages as for their single
use, for 2 weeks), and those administered PBS only. All treatments
were administered by intraperitoneal injection. The tumours were
measured every 4 days, and the tumour volume was calculated
according to the following formula: Tumour Volume (mm3)
= a × b2/2. Tumour size did not exceed 10% of body
weight (approximately 1.5 cm diameter for each mouse). The maximum
diameter of the tumours was 1.45 cm and the maximum tumour volume
observed in this study was 1206 mm3. Symptoms such as
pain, weight loss, loss of appetite, or weakness were set as humane
endpoints for the present study. Mice were euthanized with an
intraperitoneal injection of pentobarbital sodium (200 mg/kg) at
the end of the experiment, and then tumours were harvested and
weighed. All animal protocols were approved by the Ethics Committee
of Chongqing Medical University.
Immunohistochemistry
The H1975 ×enograft tumours were collected 14 days
after treatment and fixed in 10% formalin. Immunohistochemistry was
used to detect the expression of target proteins in the tumour
tissues, and PBS was used as a negative control (12,13).
The protein expression in the cytoplasm was analyzed by ImageJ
software v1.43 (National Institutes of Health) and was expressed as
mean intensity optical density (IOD).
Kaplan-Meier plotter
The Kaplan-Meier plotter is an open-access analysis
tool, (KM plotter, http://kmplot.com/analysis) which has survival
information for 1,926 patients with lung cancer, with a mean
follow-up of 33 months (15,16).
In this study, we used the KM plotter database to analyse the
prognostic value of the mRNA expression levels of IL-6, JAK, and
STAT3 in lung cancer.
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analyses were carried out using the
unpaired, 2-tailed Student t-test. Differences between groups were
evaluated using one-way analysis of variance. Multiple comparisons
between the groups were performed using the Tukey post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Zoledronic acid re-sensitises
EGFR-TKI-resistant cells in vitro
ZA inhibited the proliferation of NSCLC cell lines
in a dose-dependent manner (Fig.
1A). To investigate whether zoledronic acid can re-sensitise
EGFR-TKI-resistant cells, the H1975 and HCC827GR cell lines were
used. The human gefitinib-insensitive NSCLC cell line H1975,
harbours an EGFR mutation T790M at exon 20. The results showed that
H1975 cells were insensitive to ZA as compared to HCC827 and
HCC827GR cells (Fig. 1A). Treatment
with ZA significantly decreased the viability of the HCC827GR and
H1975 cells and re-sensitised them to gefitinib in a dose-dependent
manner (Fig. 1B and C). To
elucidate the potential mechanism by which ZA reinforces the
cytotoxicity of gefitinib, H1975 cells were treated with 4 µM
gefitinib alone, 10 µM ZA alone, or both drugs combined for 48 h.
Treatment-induced apoptosis was detected using flow cytometry. As
shown in Fig. 1D and E,
combinatorial treatment with gefitinib and ZA significantly
enhanced apoptosis of the H1975 cells as compared to treatments
alone (P<0.01). Taken together, these results demonstrate that
gefitinib and ZA treatment combined could overcome the resistance
of lung cancer cells to TKIs.
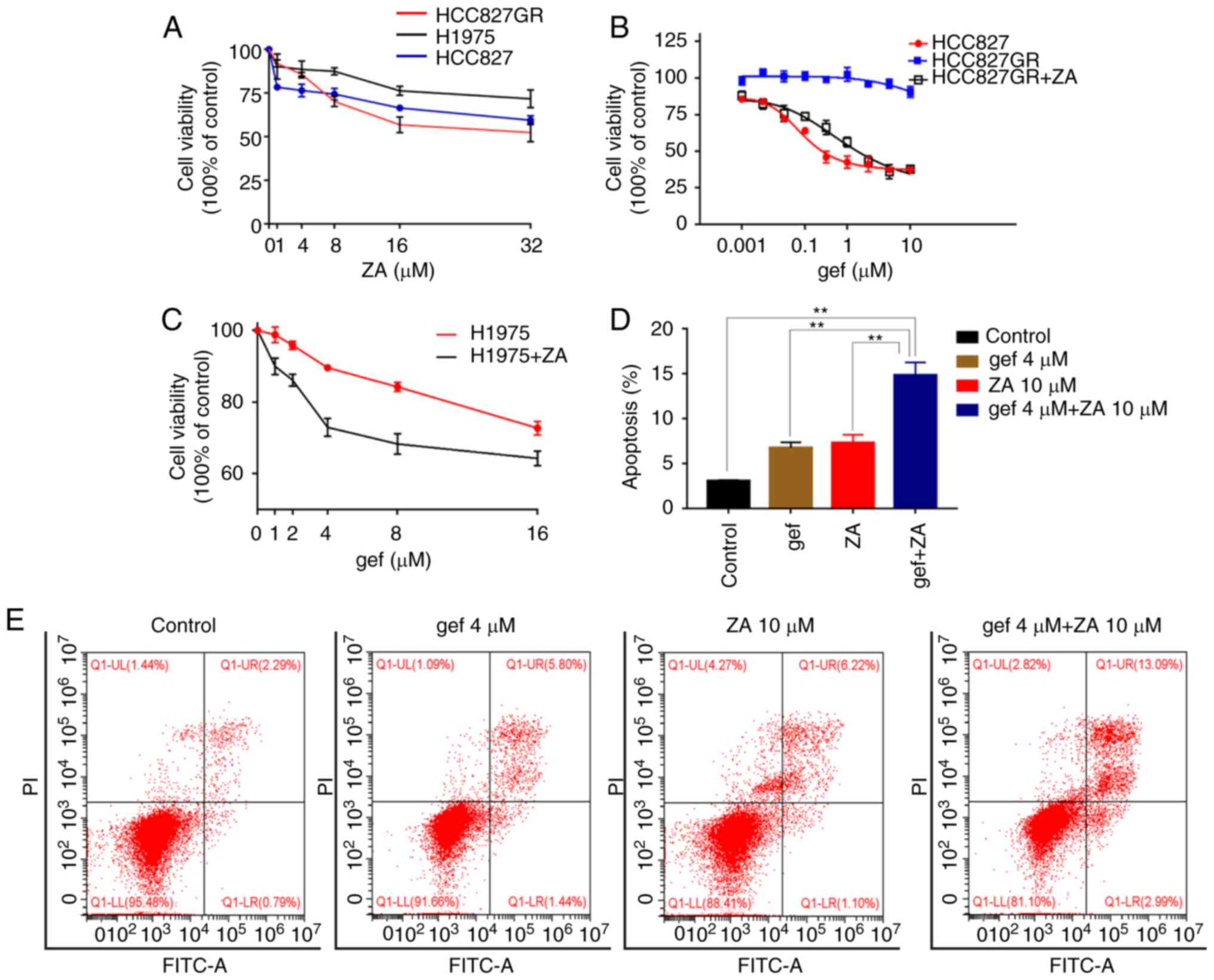 | Figure 1.ZA increases the sensitivity of
gefitinib-resistant cells to gefitinib. (A) ZA inhibited the
proliferation of NSCLC tumour cells. Cell viability of H1975,
HCC827 and HCC8278GR cells treated with the indicated doses of ZA
(0, 1, 4, 8, 16 and 32 µM) for 72 h were quantified by CCK-8 assay.
(B) Cell viability was analyzed in HCC827 and HCC827GR cells
treated with various concentrations of gefitinib alone or in
combination with ZA (4 µM) for 48 h. (C) The cell viability was
assayed for H1975 cells following treatment with gefitinib (0, 1,
4, 8, 16 µM), ZA (10 µM) or combined treatment for 48 h. (D and E)
H1975 cells were treated with gefitinib (4 µM) (gef 4) or ZA (10
µM) (ZA10) or combined for 48 h, and analyzed for apoptosis using
flow cytometry (n=3). **P<0.01. gef, gefitinib; ZA, zoledronic
acid; NSCLC, non-small cell lung cancer. |
Zoledronic acid decreases the
IL-6-induced activation of STAT3
IL-6 has been shown to positively impact the
proliferation of lung cancer cells and to decrease the sensitivity
of erlotinib or gefitinib in killing cancer cells (17). To clarify the molecular mechanisms
by which ZA overcomes acquired TKI resistance, we further
investigated the effect of ZA upon IL-6 stimulation. Firstly, we
used an enzyme-linked immunosorbent assay to quantify the level of
IL-6 in H1975 cells after treatment with 10 µM ZA alone, gefitinib
(at 0, 1, 2, 4, 8, and 16 µM) alone, or with both drugs combined.
Our results demonstrated that the combinatory treatment with ZA and
gefitinib affected cellular IL-6 levels as compared to gefitinib
monotherapy (Fig. 2A). Since STAT3
is a key component in IL-6 signalling, we performed an
immunofluorescence assay to ascertain whether IL-6 induces
activation of STAT3. The results clearly showed that IL-6 treatment
significantly increased the levels of phosphorylated STAT3
(p-STAT3) whereas STAT3 control remained unchanged (Fig. 2D-F). Further analysis using western
blot analysis confirmed an IL-6 (10 ng/ml)-dependent induction of
p-STAT3 (Fig. 2B and C).
Nevertheless, combined treatment of the cells with IL-6 and 10 µM
ZA significantly decreased STAT3 phosphorylation (Fig. 2C and F), implying that ZA could
block IL-6-induced p-STAT3 in H1975 cells.
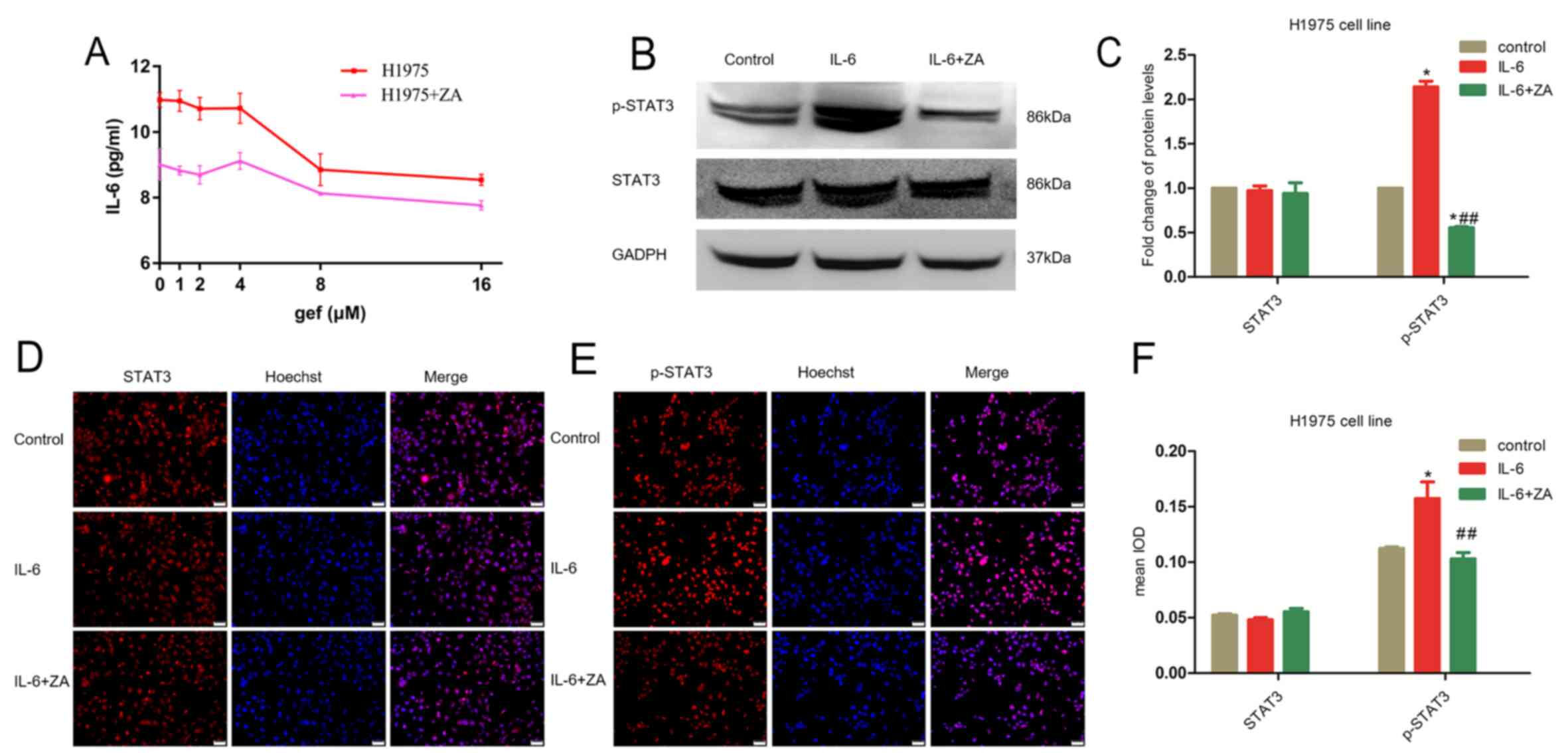 | Figure 2.ZA decreases the IL-6 levels in H1975
cells. (A) The levels of IL-6 in H1975 cells, treated with various
concentrations of gefitinib alone (0, 1, 2, 4, 8 and 16 µM) or in
combination with ZA (4 µM) for 24 h, were determined by ELISA. (B)
Phosphorylated (p-)STAT3 was determined as well as total STAT3
after induction by IL-6 and in combination with ZA. (C)
Quantification of blots in B. *P<0.05, compared with the control
(n=3 independent biological replicates); ##P<0.01
compared with IL-6 treatment alone. (D and E) Immunofluorescence
staining showed that IL-6 induced high levels of STAT3, which was
reversed by ZA treatment. Scale bar, 30 µm. (F) Quantification of
the immunofluorescence in D and E (n=3 independent biological
replicates). *P<0.05, compared with the control;
##P<0.01 compared with IL-6 treatment alone. gef,
gefitinib; ZA, zoledronic acid; IL-6, interleukin 6; STAT3,
activator of transcription 3. |
Zoledronic acid decreases
IL-6/JAK/STAT3 signalling in H1975 cells
To better understand the mechanism by which ZA
re-sensitises the H1975 cells to gefitinib, we further tested the
effect of ZA on cell survival signalling pathways downstream of
IL-6, such as STAT3. We analysed p-STAT3 in H1975 and HCC827GR
treated with gefitinib with and without ZA, using western blot
analysis. Intriguingly, ZA alone was able to decrease
IL-6-dependent p-STAT3 in both cell lines (Fig. 3A-D). Moreover, ZA treatment in
combination with gefitinib further significantly decreased p-STAT3
in H1975 and HCC827GR cells, respectively (Fig. 3A-D). Taken together, the results
demonstrated that the IL-6/JAK/STAT3 signalling pathway may
contribute to TKI resistance in lung cancer cell and that
inhibition of this pathway could overcome resistance.
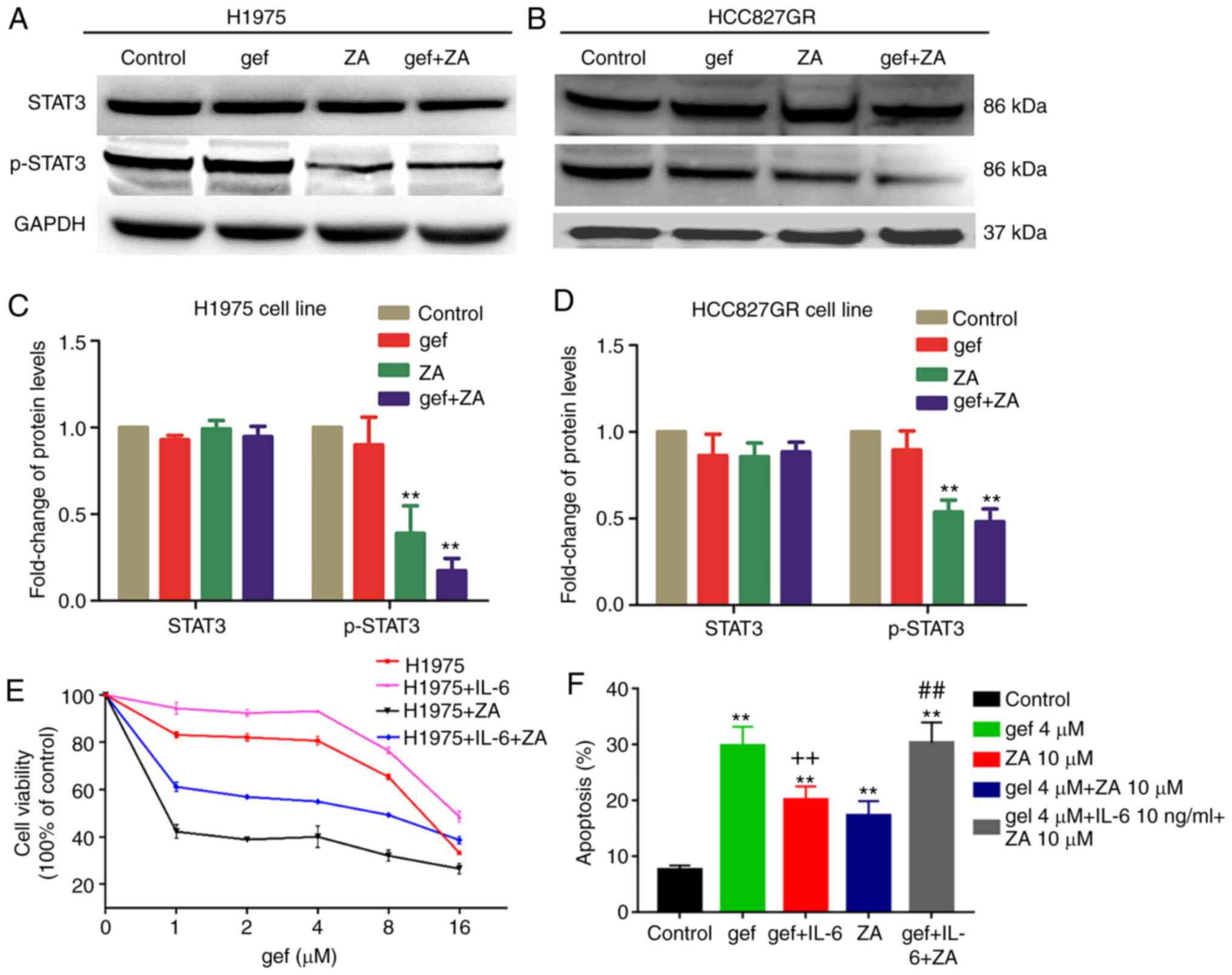 | Figure 3.(A) Western-blot analysis of STAT3
and phosphorylated (p-)STAT3 levels in H1975 cells following
gefitinib (4 µM) (gef) or ZA (10 µM) or combined treatment (gef+ZA)
for 48 h. (B) Western blot analysis of STAT3, p-STAT3 in HCC827GR
cells following gefitinib (1 µM) (gef), ZA (4 µM) or combined
treatment (gef+ZA) for 48 h. (C and D) Quantification of blots in A
and B. **P<0.01, compared with the control (n=3 independent
biological replicates). Inhibition of IL-6 signaling is essential
for ZA to re-sensitise H1975 cells to gefitinib. (E) IL-6 induced
gefitinib-resistance in H1975 cells, and ZA reversed this process.
(F) H1975 cells were pretreated with 10 µM ZA for 48 h, and then
treated with 4 µM gefitinib (gef), 10 µM ZA, 4 µM gefitinib+ IL-6
(10 ng/ml) (gef+IL-6), 4 µM gefitinib+ IL-6 (10 ng/ml) +10 µM ZA
(gef+IL-6+ZA) for 48 h, and analyzed for apoptosis using flow
cytometry (n=3). **P<0.01, compared with that without the
addition of IL-6; ++P<0.01, compared with the
gefitinib group; ##P<0.01, compared with the gef+IL-6
group. ZA, zoledronic acid; IL-6, interleukin 6; STAT3, activator
of transcription 3. |
Zoledronic acid overcomes the TKI
resistance induced by IL-6 in H1975 cells
To elucidate the role of IL-6 in TKI resistance, we
treated H1975 cells with various concentrations of gefitinib (0, 1,
2, 4, 8, and 16 µM) and IL-6 (10 ng/ml) or ZA (10 µM). The results
demonstrated that the IL-6-dependent resistance to gefitinib in
H1975 cells could be abrogated by treatment with ZA (Fig. 3E). To further characterize the role
of ZA, H1975 cells were pre-treated with ZA for 48 h and then
ZA-pre-treated cells were treated with 10 µM ZA, or 4 µM gefitinib,
or 4 µM gefitinib + IL-6 (10 ng/ml), or 4 µM gefitinib + IL-6 (10
ng/ml) + 10 µM ZA. The results showed that the pretreatment of the
cells with 10 µM ZA potently enhanced gefitinib-induced cellular
apoptosis by 22% compared with gefitinib-induced apoptosis in
Fig. 1D (Fig. 1D, 3F). The addition of IL-6 in ZA-pre-treated
cells (gef+IL-6 group) made the cells resistant to gefitinib
treatment (gef group), however, ZA (gef+IL-6+ZA group) could
reverse this phenomenon (Fig. 3F).
Thus, combining ZA with IL-6 and gefitinib significantly restored
the IL-6-induced TKI resistance.
Zoledronic acid reverses
epithelial-mesenchymal transition in H1975 and HCC827GR cells
Since EMT accounts for 5% of the acquired TKI
resistance, it was important to examine whether ZA could reverse
EMT in H1975 cells. Immunofluorescence analysis confirmed that ZA
was able to inhibit EMT as shown by increased expression of
E-cadherin and decreased expression of SNAIL and vimentin in the
H1975 cells (Fig. 4A and B).
Western blot analysis further showed that ZA significantly
suppressed the expression of vimentin and Snail, and increased the
expression of E-cadherin in the H1975 and HCC827GR cell lines
(Fig. 4C-F). Thus, these data
clearly demonstrated that ZA was able to inhibit the EMT
process.
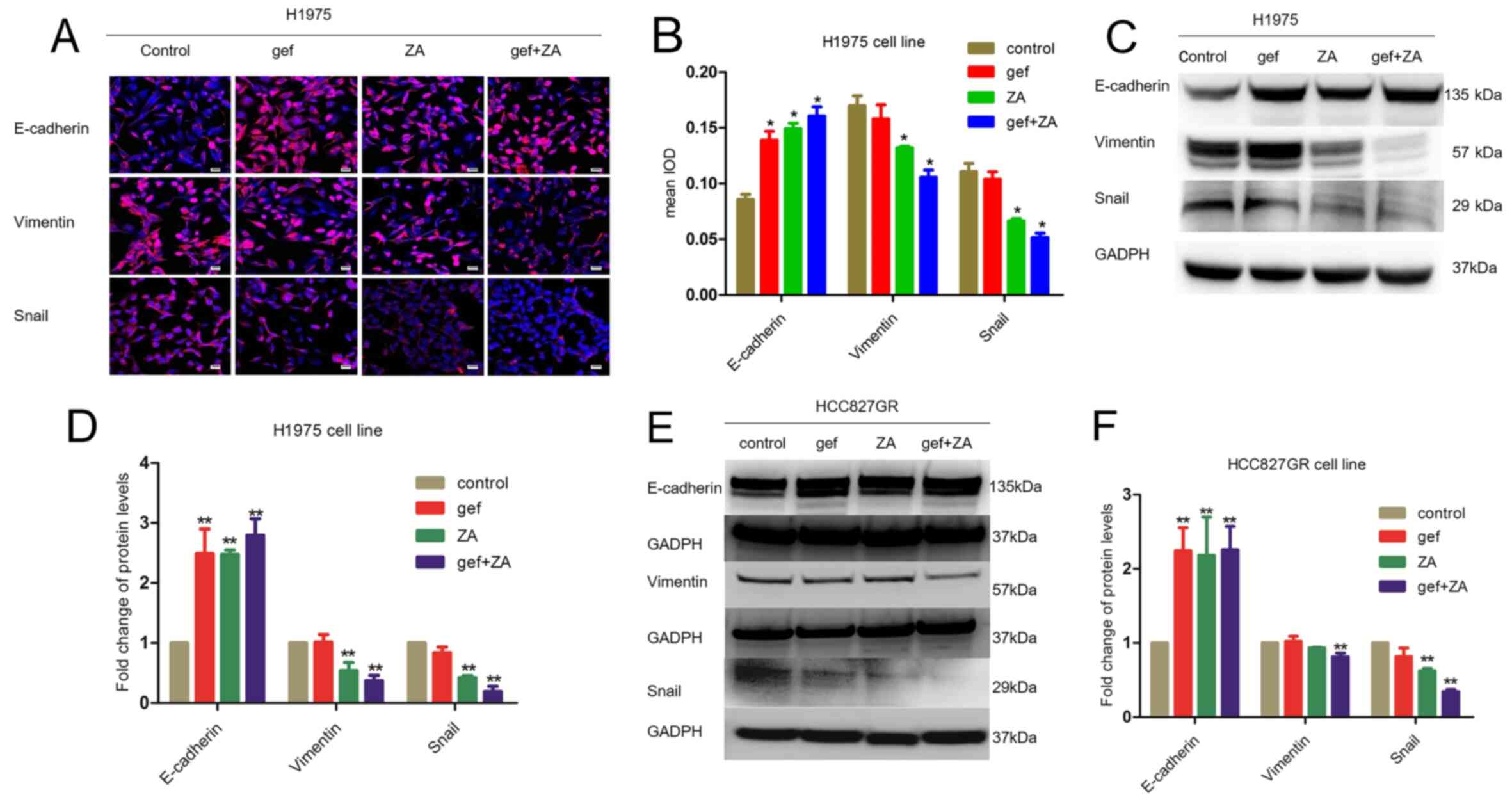 | Figure 4.ZA inhibits EMT in H1975 and HCC827GR
cells. (A) Immunofluorescence analysis of EMT marker expression
levels in response to gefitinib (4 µM) (gef), ZA (10 µM), and
gefitinib (4 µM) + ZA (10 µM) (gef+ZA). Scale bar, 30 µm. (B)
Quantification of the immunofluorescence shown in A (n=3
independent biological replicates). *P<0.05, compared with the
control. (C and E) Western blot analysis of EMT marker expression
levels in H1975 and HCC837GR cells in response to gefitinib (gef)
(4 µM for H1975 cells, 1 µM for HCC827GR cells), ZA (10 µM for
H1975 cells, 4 µM for HCC827GR cells), and gef+ZA. (D and F)
Quantification of the blots in C and E (n=3 independent biological
replicates). **P<0.01, compared with the control. ZA, zoledronic
acid; EMT, epithelial-mesenchymal transition. |
The synergistic effect of zoledronic
acid and gefitinib in a lung cancer xenograft model correlates with
the inhibition of the IL-6/JAK/STAT3 signalling pathway and EMT
reversal
To investigate why the combination of ZA and
gefitinib was more effective than the single agents in inhibiting
cell proliferation, we performed xenograft studies using H1975
cells. Treatment with ZA or gefitinib alone showed a slight
inhibition of tumour growth. In contrast, the combinatory treatment
with ZA and gefitinib significantly inhibited the growth of H1975
×enograft tumours (P<0.05) (Fig.
5A). In this study, we also investigated the systemic tolerance
of the mice towards the therapies through measurement of body
weight. The treatments did not cause any adverse effects in terms
of decreased body weight, suggesting that co-treatment with
gefitinib and ZA was well tolerated (Fig. 5B).
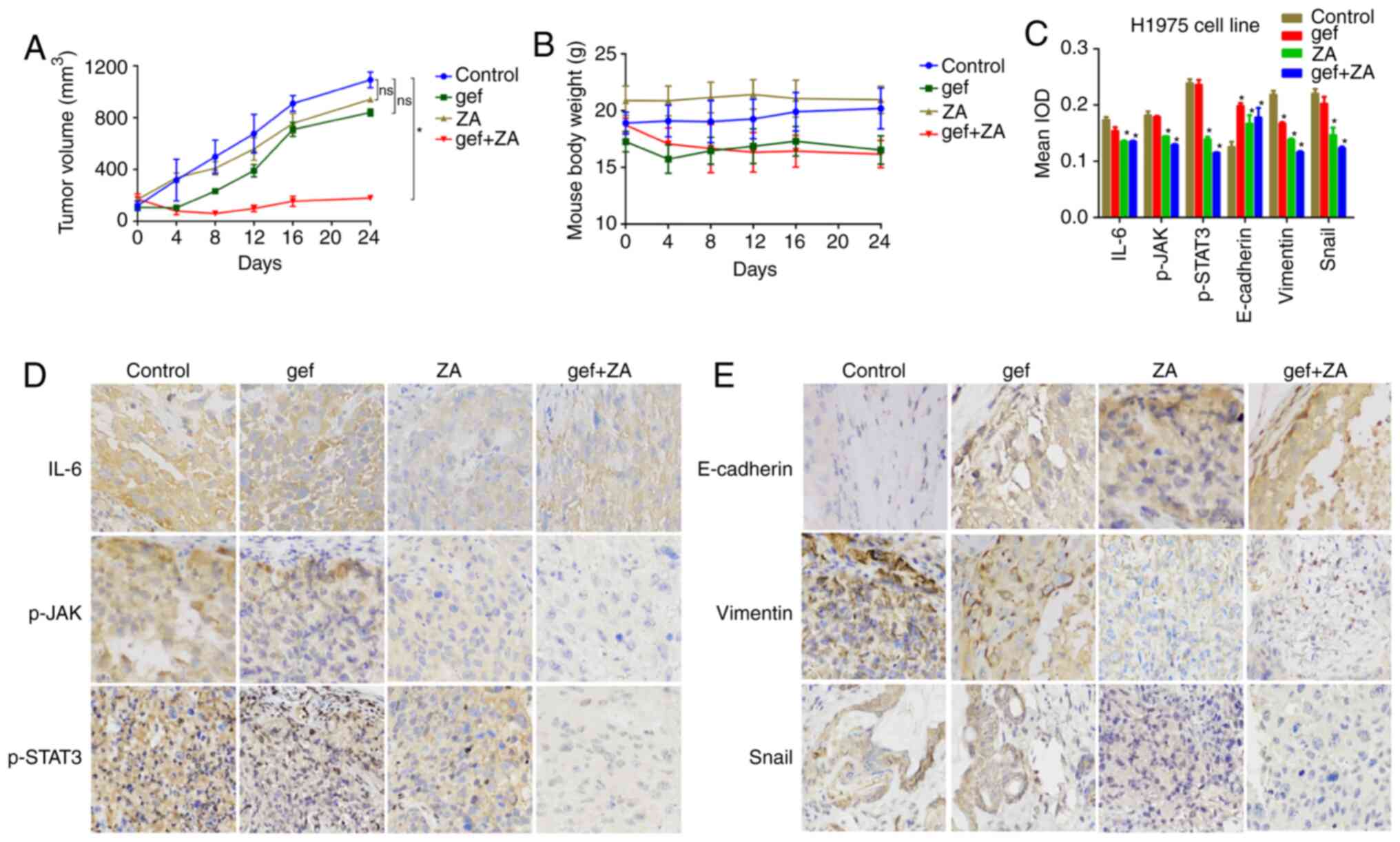 | Figure 5.Effects of gefitinib and ZA on H1975
cell-derived xenograft in nude mice. (A) ZA enhanced tumour growth
(mm3) inhibition in response to gefitinib treatment in
an H1975 ×enograft model. (B) Changes in mouse body weight during
the treatment period. (C) Statistical histograms of protein
expression in the cytoplasm, respectively (n=4). ns, no statistical
significance, *P<0.05, compared with the control. (D and E)
Representative IHC images for the indicated proteins in the
xenograft tumours, magnification ×200. gef, gefitinib; ZA,
zoledronic acid; JAK, Janus kinase; IL-6, interleukin 6; STAT3,
activator of transcription 3; p-, phosphorylated. |
Next, we investigated the markers of EMT
(E-cadherin, vimentin, and SNAIL) in tumour tissues derived from
the mice in the different treatment groups. The results showed that
ZA significantly increased the expression of E-cadherin and
significantly decreased vimentin and SNAIL compared with control
group suggesting a decrease in EMT progression (Fig. 5C and E).
Immunohistochemistry analysis showed that gefitinib
treatment alone did not decrease levels of IL-6 compared to the
control group. IL-6 levels were also significantly decreased in the
ZA treatment alone as well as in the combined treatment group,
consistent with the in vitro findings. Furthermore, ZA
treatment alone significantly reduced p-JAK and p-STAT3 in the
H1975 ×enograft tumours, whereas gefitinib treatment alone slightly
decreased the p-JAK level. The combined treatments decreased the
levels of p-JAK and p-STAT3 even further (Fig. 5C and D).
Increased IL-6/JAK/STAT3 signalling is
associated with poor survival in patients with NSCLC
We further investigated whether the IL-6/JAK/STAT3
signalling pathway could be associated with cancer survival. To
this end, we analysed the correlation between the mRNA expression
of IL-6, JAK, and STAT3 and the survival of patients with lung
adenocarcinoma, using a KM plotter database. In our present study,
all the IL-6/JAK/STAT3 KM survival data was determined using data
from http://www.kmplot.com. Fig. 6 shows the survival curves for all
patients with lung adenocarcinoma. For the IL-6 data, the valid
Affymetrix ID was 205207_at IL-6. Although the mRNA expression of
IL-6 was not significantly related to post-progression survival
(PPS) of the patients (HR=1.08, 95% CI: 0.68-1.72, P=0.76; Fig. 6A), it was significantly related to
poor overall survival (OS) in these patients (HR=1.68, 95% CI:
1.33-2.13, P=1.2e-05; Fig. 6B). We
next explored the relationship between JAK expression and survival
prognosis (valid Affymetrix ID: 201648_at JAK). The mRNA expression
of JAK was significantly correlated with favourable PPS and OS in
patients with adenocarcinoma (HR=0.5, 95% CI: 0.31-0.8, P=0.0035;
and HR=0.43, 95% CI: 0.33-0.55, P=3.3e-12, respectively; Fig. 6C and D). As shown in Fig. 6E and F, the mRNA expression of STAT3
was significantly correlated with worse PPS and OS in these
patients (HR=1.6, 95% CI: 1-2.57, P=0.048; and HR=1.33, 95% CI:
1.05-1.69, P=0.017, respectively).
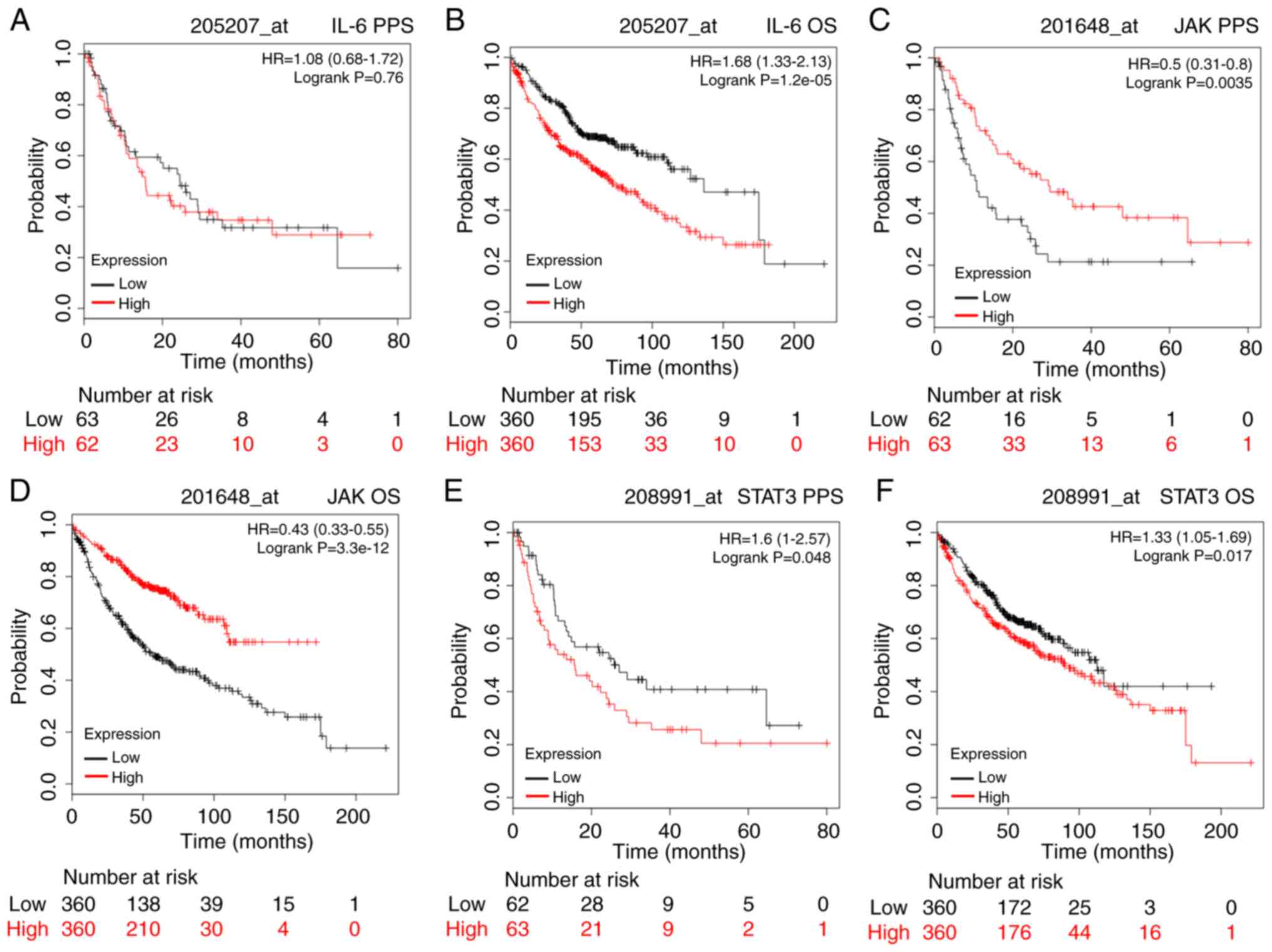 | Figure 6.The prognostic value of IL-6, JAK,
and STAT3 expression according to the database of Kaplan-Meier
plotter. Notes: The desired Affymetrix ID is valid: 205207_at
(IL-6), 201648_at (JAK), 208991_at (STAT3). Survival curves are
plotted for adenocarcinoma (A, C and E: n=125; B, D and F: n=720).
Probability: Post-progression survival (PPS) (A, C and E), overall
survival (OS) (B, D and F). JAK, Janus kinase; IL-6, interleukin 6;
STAT3, activator of transcription 3. |
Discussion
With the development of new drugs, the treatment
options for lung cancer patients have increased, yet the 5-year
survival rate remains at only 15% (1). Although epidermal growth factor
receptor (EGFR)-tyrosine kinase inhibitor (TKI) treatment can
acquire a 70% response rate, drug resistance will eventually
develop as a result of EGFR secondary mutation,
epithelial-mesenchymal transition (EMT), or IL-6 overexpression
(17–19). Therefore, the search for novel
compounds to overcome the acquired resistance of cancer cells to
EGFR-TKI is extremely urgent.
Our previous clinical study demonstrated that
bisphosphonates could sensitise patients with EGFR
mutation-positive non-small cell lung cancer (NSCLC) to gefitinib
and significantly prolong their survival (12). Another retrospective clinical study
also showed that bisphosphonates could improve the progression-free
survival (PFS) time of patients with advanced EGFR
mutation-positive NSCLC (14).
However, the molecular mechanism behind the synergistic effect of
zoledronic acid (ZA) and gefitinib is not clear. It is therefore
important to further characterise how ZA is able to overcome
acquired TKI resistance. In this study, we found that ZA may
potentially achieve this in gefitinib-resistant lung cancer cells
by reversal of EMT and suppression of STAT3 activation via
inhibition of the IL-6/JAK/STAT3 signalling pathway.
Interleukin (IL)-6, a 26-kDa molecular-weight
protein containing 185 amino acids, plays a key role in many
biological and pathobiological events (20–22).
Many recent studies have suggested that IL-6 may play a critical
role in EMT and chemoresistance in cancers (23). Database analysis verified that IL-6
expression was significantly correlated with poor OS in patients
with lung adenocarcinoma. IL-6 activation confers the acquisition
of TKI resistance in lung cancer cells. However, inhibition of IL-6
signalling has been shown to reverse EGFR TKI resistance in human
lung cancer cells both in vitro and in vivo (24). In this study, we verified that ZA
could decrease the levels of IL-6 in vivo as well as in
vitro. It has been reported that ZA could induce apoptosis in
bone marrow stromal cells (BMSCs) and significantly inhibit the
constitutive production of IL-6 by BMSCs (25). IL-6 secretion was significantly
reduced after ZA treatment in hormone-independent prostate cancer
cell lines and bone marrow-derived mesenchymal stem cells (26,27).
However, the exact mechanism of how ZA regulates IL-6 in lung
cancer cells is still unclear. A previous study demonstrated that
sorafenib, sunitinib, and pazopanib could strongly (10-fold) induce
the secretion of IL-6 in 786-O cells, even at a low concentration
of these inducers (28). Other
studies have demonstrated that high circulating levels of IL-6 in
patients with locally advanced or metastatic lung cancer, and high
levels of serum IL-6 are correlated with short survival and weight
loss (29). In our present study,
gefitinib treatment did not induce secretion of IL-6 in lung cancer
cells. A previous study demonstrated that there was a strong
correlation between mutant EGFRs and p-STAT3 (30). EGFR-TKI treatment could upregulate
the IL-6-induced phosphorylation of STAT3 in an autocrine manner
(8,31). In this present study, IL-6 alone
upregulated the expression of p-STAT3. Moreover, ZA significantly
decreased IL-6-induced activation of STAT3 in the H1975 cells.
Furthermore, adding IL-6 to ZA-pretreated TKI-resistant cells
abolished the ZA-dependent effect and restored the TKI-resistance
phenotype. Using the KM plotter database, we found that high mRNA
expression of IL-6 was significantly correlated with poor OS in
patients with adenocarcinoma, and high mRNA expression of STAT3 was
significantly correlated with worse PFS and OS. Surprisingly, high
mRNA expression of JAK was significantly related to favourable PFS
and OS in these patients. Further research with a large sample size
is needed to clarify the prognostic value of JAK mRNA expression in
patients with lung cancer. Taken together, these results suggest
that ZA effectively can overcome TKI resistance in lung cancer
cells by inhibiting the IL-6/JAK/STAT3 signalling pathway.
Apart from the inhibitory effect on the
IL-6/JAK/STAT3 signalling pathway, the inhibition of EMT may also
play a critical role in the reversal of acquired TKI resistance in
the cells. EMT is a crucial biological event which is involved in
cellular transformation, tumourigenesis, and metastasis in many
cancers (23). Previous studies
have shown that EGFR-TKI resistance is associated with EMT
(32–38). In this present study, IL-6 treatment
resulted in an increased expression of SNAIL and vimentin and
decreased expression of E-cadherin in H1975 cells. ZA was able to
reverse the IL-6-induced EMT process and restore the sensitivity of
H1975 cells to gefitinib. Another study demonstrated that STAT3
activation occurred in the IL-6-mediated induction of EMT, and
metformin reversal of EMT in the TKI-resistant lung cancer cell
lines and IL-6-stimulated PC-9 cells (23). The reversal of EMT may be a
promising strategy for improving patient outcomes in advanced
cancer. Taken together, our findings indicate that the combination
of gefitinib and ZA could be a novel therapeutic strategy for
treating advanced NSCLC.
In conclusion, we showed here that activation of
IL-6/JAK/STAT3 signalling may be linked to the acquisition of TKI
resistance and the EMT phenotype in EGFR secondary mutated NSCLC.
Treatment with ZA was able to reverse the acquired TKI resistance
in vitro and in vivo through inhibition of the
IL-6/JAK/STAT3 signalling pathway and thereby reversal of the EMT
process. On the basis of these findings, a clinical trial has been
registered in the Chinese Clinical Trial Registry, with the aim to
further evaluate whether the combination of ZA with EGFR-TKIs could
elicit a synergistic effect in patients with advanced EGFR
mutation-positive NSCLC.
Acknowledgements
We acknowledge the help of Markus Dagnell to revise
the manuscript.
Funding
This work was supported by the National Natural
Science Foundation of China (81660503), the Natural Science
Foundation of Hubei Province (2016CFB394) and the Natural Science
Foundation of Enshi (E20170003).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XY, YG, QL, LW and HL made an equal contribution to
this work. XY, YG and QL participated in the cell experiments and
drafted the manuscript. LW and HL participated in the animal
experiments. CH, WB and YD participated in the design of the study
and performed the statistical analysis. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the Ethics
Committee of Chongqing Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al: Gefitinib or chemotherapy for non-small-cell lung cancer
with mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al: Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): An open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gazdar AF: Activating and resistance
mutations of EGFR in non-small-cell lung cancer: Role in clinical
response to EGFR tyrosine kinase inhibitors. Oncogene. 28 (Suppl
1):S24–S31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F,
Ha SJ, Soo RA, Christensen JG, Lee JH and Cho BC: Activation of
IL-6R/JAK1/STAT3 signaling induces de novo resistance to
irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neville-Webbe HL and Coleman RE:
Bisphosphonates and RANK ligand inhibitors for the treatment and
prevention of metastatic bone disease. Eur J Cancer. 46:1211–1222.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Di Salvatore M, Orlandi A, Bagalà C,
Quirino M, Cassano A, Astone A and Barone C: Anti-tumour and
anti-angiogenetic effects of zoledronic acid on human
non-small-cell lung cancer cell line. Cell Prolif. 44:139–146.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morgan G and Lipton A: Antitumour effects
and anticancer applications of bisphosphonates. Semin Oncol. 37
(Suppl 2):S30–S40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng C, Liu X, Li X, Guo F, Huang C, Qin Q
and Wang Y: Zoledronic acid increases the antitumour effect of
gefitinib treatment for non-small cell lung cancer with EGFR
mutations. Oncol Rep. 35:3460–3470. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang CY, Wang L, Feng CJ, Yu P, Cai XH,
Yao WX, Xu Y, Liu XK, Zhu WJ, Wang Y, et al: Bisphosphonates
enhance EGFR-TKIs efficacy in advanced NSCLC patients with EGFR
activating mutation: A retrospective study. Oncotarget.
7:664802016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang G, Cheng R, Zhang Z, Jiang T, Ren S,
Ma Z, Zhao S, Zhou C and Zhang J: Bisphosphonates enhance
antitumour effect of EGFR-TKIs in patients with advanced EGFR
mutant NSCLC and bone metastases. Sci Rep. 7:429792017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen L, Huang C, Yang X, Zhang Q and Chen
F: Prognostic roles of mRNA expression of peroxiredoxins in lung
cancer. Onco Targets Ther. 11:8381–8388. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su KY, Chen HY, Li KC, Kuo ML, Yang JC,
Chan WK, Ho BC, Chang GC, Shih JY, Yu SL and Yang PC: Pretreatment
epidermal growth factor receptor (EGFR) T790M mutation predicts
shorter EGFR tyrosine kinase inhibitor response duration in
patients with non-small-cell lung cancer. J Clin Oncol. 30:433–440.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shih JY, Gow CH and Yang PC: EGFR mutation
conferring primary resistance to gefitinib in non-small-cell lung
cancer. N Engl J Med. 353:207–208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kosaka T, Yatabe Y, Endoh H, Yoshida K,
Hida T, Tsuboi M, Tada H, Kuwano H and Mitsudomi T: Analysis of
epidermal growth factor receptor gene mutation in patients with
non-small cell lung cancer and acquired resistance to gefitinib.
Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cohen S, Bruchim I, Graiver D, Evron Z,
Oron-Karni V, Pasmanik-Chor M, Eitan R, Bernheim J, Levavi H,
Fishman A and Flescher E: Platinum-resistance in ovarian cancer
cells is mediated by IL-6 secretion via the increased expression of
its target cIAP-2. J Mol Med (Berl). 91:357–368. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nilsson MB, Langley RR and Fidler IJ:
Interleukin-6, secreted by human ovarian carcinoma cells, is a
potent proangiogenic cytokine. Cancer Res. 65:10794–10800. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Middleton K, Jones J, Lwin Z and Coward
JI: Interleukin-6: An angiogenic target in solid tumours. Crit Rev
Oncol Hematol. 89:129–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bharti R, Dey G and Mandal M: Cancer
development, chemoresistance, epithelial to mesenchymal transition
and stem cells: A snapshot of IL-6 mediated involvement. Cancer
Lett. 375:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Derenne S, Amiot M, Barillé S, Collette M,
Robillard N, Berthaud P, Harousseau JL and Bataille R: Zoledronate
is a potent inhibitor of myeloma cell growth and secretion of IL-6
and MMP-1 by the tumoural environment. J Bone Miner Res.
14:2048–2056. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gallo M, De Luca A, Lamura L and Normanno
N: Zoledronic acid blocks the interaction between mesenchymal stem
cells and breast cancer cells: Implications for adjuvant therapy of
breast cancer. Ann Oncol. 23:597–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Asbagh LA, Uzunoglu S and Cal C:
Zoledronic acid effects interleukin-6 expression in
hormone-independent prostate cancer cell lines. Int Braz J Urol.
34:355–363; discussion 364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishibashi K, Haber T, Breuksch I, Gebhard
S, Sugino T, Kubo H, Hata J, Koguchi T, Yabe M, Kataoka M, et al:
Overriding TKI resistance of renal cell carcinoma by combination
therapy with IL-6 receptor blockade. Oncotarget. 8:55230–55245.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scott HR, McMillan DC, Crilly A, McArdle
CS and Milroy R: The relationship between weight loss and
interleukin 6 in non-small-cell lung cancer. Br J Cancer.
73:1560–1562. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B and Bromberg
JF: Mutations in the EGFR kinase domain mediate STAT3 activation
via IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Che Q, Liu BY, Wang FY, He YY, Lu W, Liao
Y, Gu W and Wan XP: Interleukin 6 promotes endometrial cancer
growth through an autocrine feedback loop involving ERK-NF-κB
signaling pathway. Biochem Biophys Res Commun. 446:167–172. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumour specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uramoto H, Iwata T, Onitsuka T, Shimokawa
H, Hanagiri T and Oyama T: Epithelial-mesenchymal transition in
EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res.
30:2513–2517. 2010.PubMed/NCBI
|
|
34
|
Byers LA, Diao L, Wang J, Saintigny P,
Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al: An
epithelial-mesenchymal transition gene signature predicts
resistance to EGFR and PI3K inhibitors and identifies Axl as a
therapeutic target for overcoming EGFR inhibitor resistance. Clin
Cancer Res. 19:279–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Davis FM, Stewart TA, Thompson EW and
Monteith GR: Targeting EMT in cancer: Opportunities for
pharmacological intervention. Trends Pharmacol Sci. 35:479–488.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marcucci F, Stassi G and De Maria R:
Epithelial-mesenchymal transition: A new target in anticancer drug
discovery. Nat Rev Drug Discov. 15:311–325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chung JH, Rho JK, Xu X, Lee JS, Yoon HI,
Lee CT, Choi YJ, Kim HR, Kim CH and Lee JC: Clinical and molecular
evidences of epithelial to mesenchymal transition in acquired
resistance to EGFR-TKIs. Lung Cancer. 73:176–182. 2011. View Article : Google Scholar : PubMed/NCBI
|














