Introduction
Apoptosis is the main form of programmed cell death
that occurs during animal development and tissue homeostasis. It
involves several stages, including progressive nuclear and
cytoplasmic shrinkage, chromatin condensation, and the shedding of
apoptotic bodies (1). Following
apoptosis, cell bodies are cleared by phagocytes or adjacent cells
(2). At the molecular level,
apoptosis is executed by caspase proteins, which cleave cellular
substrates, leading to cell death (3). Apoptosis is an essential process that
serves key roles in morphogenesis and tissue remodeling (4), and can also affect surrounding tissues
by affecting cell division, cellular fate and the remodeling of
nearby cells (5). Therefore, it is
important to elucidate the underlying mechanism of cell
apoptosis.
Apoptosis, also known as ‘programmed cell death’,
has been identified throughout the process of mouse brain
development. It is crucial for maintaining the circuits of the
nervous system (6). Apoptosis
occurs during in utero and early postnatal life (7–9). In
the embryo, apoptosis affects different sites, including hindbrain
neural folds at embryonic day (E)8-9, the lamina terminalis at
E10.5-12.5, optic invagination at E11.5-12.5 (9), and the ventricular zone, intermediate
zone and the developing cortical plate of the cerebral cortex at
E12-18 (10,11). Due to the low rate of relative
apoptosis (0.14-0.35% of cells per day), the process is difficult
to identify histologically (10).
However, evaluation of cleaved-caspase3 levels or cell morphology
using TUNEL assays can indicate the apoptosis rate (12). Apoptosis has been recognized in most
sites of central development after birth, including the brain,
spinal cord and peripheral ganglia (13). The precise time of the apoptosis
rate peak varies, but cells mostly undergo apoptosis between
postnatal day 0 (P0) and P14 (8).
In the cortex, the apoptosis rate is highest between P4 and P8
(14). The final number of
apoptotic neurons can range between 60 and 90% (14).
In addition to tissue development, apoptosis is
associated with numerous pathologies. For example, defective
apoptosis leads to proliferative diseases, such as cancer,
excessive apoptosis has been linked to neurodegenerative disease
(15), and the dysregulation of
apoptosis is associated with stroke and traumatic brain injury
(16,17). The morbidity of neurodegenerative
diseases is increasing worldwide; however, effective treatments and
cures remain limited (18). A
series of detrimental effects emerge during the development of
neurodegenerative diseases, including mitochondrial dysfunction,
inflammation and oxidative stress, which contribute to neuronal
apoptosis, and inhibit nerve recovery and axon regeneration
(19,20).
Apoptosis can be categorized into two pathways:
Intrinsic and extrinsic. Extrinsic apoptosis is initiated by the
ligand-mediated activation of death receptors (21). Ligand binding leads to the
activation of pro-caspase-8 (21,22),
after which extrinsic apoptosis signaling converges with intrinsic
apoptosis pathways at executioner caspases (2). Bax and BCL2 antagonist/killer 1
proteins are activated during intrinsic apoptosis, leading to the
initiation of pathways that activate caspases, including mainly
caspase-3, caspase-6 and caspase-7 (23). Although the mechanisms underlying
apoptosis are well known, its regulation is poorly understood.
NF-κB is a protein complex with a REL proto-oncogene
NF-κB subunit (Rel) homology domain. The NF-κB mammalian family
contains five proteins: p65, NF-κB1/2 and Rel/RELB proto-oncogene
NF-κB subunit. NF-κB proteins are widely expressed in developing
and mature nervous systems, and NF-κB signaling has a
neuroprotective function in the latter, where it protects neurons
from glutamate-induced neurotoxicity and oxidative stress by
enhancing the expression of brain-derived neurotrophic factors and
the antiapoptotic gene Bcl-2 (24).
NF-κB also activates the expression of genes involved in cell
proliferation and protects against apoptosis (25). Although NF-κB has been implicated in
the reduction of the neuronal apoptosis rate (26), its regulation is unclear.
Rho family GTPase 3 (RND3; also known as RhoE or
Rho8) belongs to the RND subfamily of the Rho family of GTPases
(27). The RND subfamily includes
three members: RND1, RND2 and RND3. Most Rho family members
function as molecular switches that cycle between the active
GTP-bound form and the GDP-bound inactive form (28). In contrast to typical Rho family
members, RND subfamily proteins can bind GTP but are resistant to
GTPases, and are thus constitutively bound to GTP (29). RND3 was originally defined as a
repressor of Rho protein kinase 1 (28). Previous studies on RND3 have focused
mainly on its inhibitory effects on Rho kinase-mediated biological
functions, including myosin light chain phosphorylation (30), actin cytoskeleton formation and
apoptosis (31). RND3 is highly
expressed in the mouse brain where it serves an essential role in
neuronal development through its negative regulation of the Rho
signaling pathway (32).
Additionally, RND3 is associated with ependymal cell proliferation
(33); however, its function in
neuronal apoptosis remains unknown.
Genetic deletion of Rnd3 can have various effects on
multiple organs. For instance, knocking out Rnd3 can result in
aqueductal stenosis leading to hydrocephalus and heart calcium
leakage which leads to heart failure in mice (33,34).
The present study demonstrated a novel function of Rnd3 in the
regulation of brain apoptosis using an Rnd3-knockout mouse model.
The present study identified the molecular mechanism of
Rnd3-mediated apoptosis and demonstrated that Rnd3 was a regulator
of NF-κB signaling in apoptosis. Furthermore, RND3 was demonstrated
to interact with NF-κB P65 in vivo, which blocked the
anti-apoptotic action of NF-κB P65 via the downregulation of P65.
Therefore, the present study suggested the importance of
RND3-NF-κB-P65 signaling integrity in neurocyte apoptosis. The
present findings may advance the understanding of apoptosis in the
development of the nervous system, and may provide a potential
target for pharmacologic manipulations in the treatment of
neurodegenerative diseases.
Materials and methods
Rnd3−/− mouse lines
Rnd3-knockout mice were generated from a gene trap
embryonic stem (ES) cell line at Texas Institute for Genomic
Medicine (Texas, USA), provided by Dr Jiang Chang (Texas A&M
University Health Science Center, USA). The targeting vector was
inserted into an Rnd3 intron 2 as described previously
(34). A total of 16 male mice (8
Rnd3+/+ mice and 8 Rnd3− mice;
7-day-old; weight, 4-5 g) were used and raised in specified
pathogen-free conditions supplied with free access to sterilized
water and UV-irradiated food, with an environment of 22°C and 60%
humidity, 12-h light/dark cycle and noise <60 dB. Investigators
were blinded to the mouse genotype and experimental group. Animals
were identified by earmark numbers, which were unknown to the
investigators until after the experiments and analyses were
completed.
Immunostaining, immunoblotting (IB)
and immunoprecipitation (IP)
The following antibodies were used for the immune
analyses: Anti-Rho8/Rnd3 [dilution, 1:1,000 for IB, 1:200 for IP,
1:100 for immunohistochemistry (IHC) and 1:50 for
immunofluorescence (IF); cat. no. GTX81316; GeneTex, Inc.],
anti-GAPDH (dilution, 1:200 for IB; cat. no. sc-25778; Santa Cruz
Biotechnology, Inc.), anti-P65 (dilution, 1:200 for IB, 1:50 for IP
and 1:25 for IF; cat. no. sc372; Santa Cruz Biotechnology, Inc.),
anti-cleaved-caspase3 (dilution, 1:2,000 for IB and 1:100 for IF;
cat. no. 9661; Cell Signaling Technology, Inc.), anti-caspase 3
(dilution, 1:200 for IB; cat. no. sc-7148; Santa Cruz
Biotechnology, Inc.), anti-Myc-tag (dilution, 1:1,000 for IB; cat.
nos. M047-3; Medical & Biological Laboratories Co., Ltd.),
anti-phospho-(p-)inhibitor of NF-κB-α (dilution, 1:500 for IB; cat.
no. 2859; Cell Signaling Technology, Inc.), anti-IκB-α (dilution,
1:500 for IB; cat. no. CSB-RA015761A0HU; Cusabio Technology LLC),
anti-Histone-H3 (dilution, 1:1,000 for IB; cat. no. GB11102; Wuhan
Servicebio Technology Co., Ltd.), anti-β-tubulin (dilution, 1:1,000
for IB; cat. no. GB11017; Wuhan Servicebio Technology Co., Ltd.),
anti-glial fibrillary acidic protein (dilution, 1:100 for IF; cat.
no. 60190-1-lg; ProteinTech Group, Inc.), anti-neuronal nuclei
(dilution, 1:100 for IF; cat. no. 26975-1-AP; ProteinTech Group,
Inc.), HRP-goat anti mouse IgG (dilution, 1:2,000 for IB; cat. no.
GB23301; Wuhan Servicebio Technology Co., Ltd.) and HRP-goat anti
rabbit IgG (dilution, 1:2,000 for IB and 1:200 for IHC; cat. no.
GB23303; Wuhan Servicebio Technology Co., Ltd.). The following
fluorescent secondary antibodies were used: Donkey anti-goat IgG
coupled with Alexa Fluor 488 (dilution, 1:200 for IF; cat. no.
A11055; Invitrogen; Thermo Fisher Scientific, Inc.), goat
anti-mouse IgG coupled with Alexa Fluor 488 (dilution, 1:200 for
IF; cat. no. A11029; Invitrogen; Thermo Fisher Scientific, Inc.)
and goat anti-rabbit IgG coupled with Alexa Fluor 594 (dilution,
1:200 for IF; cat. no. A11037; Invitrogen; Thermo Fisher
Scientific, Inc.).
For IF and IHC analysis, previous protocols of
tissue and cell sample preparation were adopted (35). Cell samples were fixed in 4%
paraformaldehyde for 30 min, while the tissues from 7-day-old mice
brains were cut using a freezing microtome (CM1850; Leica
Microsystems GmbH), fixed in 4% paraformaldehyde for 2 weeks and
then embedded in paraffin before use. Tissue sections (5-µm-thick)
were deparaffinized at 65°C for 2 h, hydrated with 100/95/75%
ethanol for 10 min each and antigen repaired with 10 mM sodium
citrate (pH 6.0) at 100°C for 20 min. Tissue sections were
incubated with 3% H2O2 to remove endogenous
peroxidase and cell samples were permeabilized with 0.1% Triton
X-100 in PBS for 30 min at room temperature. Following treatment
with 5% BSA (cat. no. G5001; Wuhan Servicebio Technology Co., Ltd.)
for 30 min at room temperature, the samples were incubated with
primary antibody at 4°C overnight. After washing with PBS five
times, the cells were subjected to the secondary antibody and
incubated at room temperature. The nuclei were stained with DAPI
for 10 min at room temperature. IHC experiments were performed
using an avidin-biotin complex and diaminobenzidine tetrachloride
(G1210-2; Wuhan Servicebio Technology Co., Ltd.). Images were
captured under a confocal fluorescence microscope (FV1000; Olympus
Corporation).
Tissue samples from 7-day-old mouse brains were
homogenized in RIPA buffer (50 mM Tris-HCl at pH 8.0, 0.15 M NaCl,
1 mM EDTA and 0.5% NP-40) supplemented with a protease inhibitor
cocktail (04693159001; Roche Diagnostics). Following centrifugation
at 13,000 g at 4°C for 15 min, the supernatant fraction was first
subjected to IP with a weight of 5 µg for primary antibodies
against P65, RND3 or pre-immune IgG (AR1010; Wuhan Boster
Biological Technology, Ltd.) and incubated at 4°C for 4 h.
Subsequently, the samples were incubated with the beads of protein
G PLUS-agarose (Sc-2002; Santa Cruz Biotechnology, Inc.) in a low
speed oscillation at 4°C overnight. Following washing with IP
buffer (10 mM HEPES, 142.5 mM KCl, 5 mM MgCl, 1 mM EDTA and 0.5%
NP-40), the samples were subjected to western blot analysis.
Western blot analysis was performed as previously
described (36). Briefly, tissue
and cell samples were lysed in RIPA buffer (Beyotime Institute of
Biotechnology) with protease inhibitors (Roche Diagnostics). The
protein concentrations were measured using a BCA kit (Beyotime
Institute of Biotechnology). Protein samples (20 µg/lane) were
separated on 12% SDS-polyacrylamide gels and then transferred onto
0.45-mm PVDF membranes (Merck KGaA). The membranes were incubated
with anti-P65 or anti-RND3 antibodies overnight at 4°C after
blocking with 5% skim milk powder (cat. no. G5002; Wuhan Servicebio
Technology Co., Ltd.) at room temperature for 1 h. The membranes
were incubated with the indicated HRP-conjugated secondary antibody
for 1 h after washing in TBST (20 mM Tris-HCl pH 7.5, 150 mM NaCl,
0.1% Tween-20). A Super Signal Chemiluminescent Substrate system
(k-12045-D50; Advansta, Inc.) was used to detect the signals. The
amount of protein loaded in the IB analysis was verified by the
intensity of the GAPDH blots. The specificity and sensitivity of
the antibodies were validated based on the protocols used in our
previous study (37). IB
densitometry was performed using Image Lab (version 5.2; Bio-Rad
Laboratories, Inc.).
Nuclear and cytoplasmic
fractionation
Nuclear and cytoplasmic proteins were extracted
using NE-PER nuclear-cytoplasmic extraction reagents (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. When
measuring cytosolic and nuclear P65 protein expression, β-tubulin
and histone H3 were used as cytoplasmic and nuclear loading
controls, respectively.
Gene knockdown, cell culture and
transfection
PC12 cells (derived from a pheochromocytoma, an
embryonic origin from the neural crest; The Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences) and cultured
in DMEM (Hangzhou Jinuo Biomedical Technology Co., Ltd.) with 5%
FBS (Gibco; Thermo Fisher Scientific, Inc.), 10% horse serum
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Hangzhou Jinuo Biomedical Technology Co.,
Ltd.). The transient transfection of all genes studied was
performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells in each well of a 6-well plate were
transfected with 100 pmol small interfering RNA (siRNA/si) or 2 µg
plasmid for 48 h prior to experimentation. The siRNA pools
containing multiple siRNAs specific for Rnd3 and p65,
with non-targeting siRNAs used as controls, were purchased from
Thermo Fisher Scientific, Inc. Dharmacon RNAi technologies (RND3,
cat. no. L-007794-00-0005; p65, cat. no. L-040776-00-00010; and
non-targeting siRNA, cat. no. D-001206-13-20; GE Healthcare
Dharmacon, Inc.). Detailed information of siRNAs was described
previously (38) and the sequences
of siRND3, siP65 and siControl were 5′-CUACAGUGUUUGAGAAUUAUU−3′,
5′-GATTGAGGAGAAACGTAAA-3′ and 5′-CCTACGCCACCAATTTCGT-3′. The
construction of myc-RND3 and overexpression vectors was
described previously (37).
Isolation of hippocampus in mice
A cut was made along the coronal and sagittal
sutures and both sides of the parietal bone and interparietal bone
were pulled off. To expose the hippocampus, the cerebral cortex
covering it was removed. The first incision was made at the end of
the hemisphere, extending no more than ~0.7 mm deep to prevent
nicking the adult mouse hippocampus while exposing it. The second
incision was made 1.5-2 mm in front of the first one, and the
lateral ventricle was cut into. Both incisions entered the ventral
brain and met there. Both sides of the cortex that covered the
hippocampus were pulled up along the ventricle. The remainder of
the hippocampus was separated from the cortex by pulling the cortex
along the surface of the hippocampus towards the ventral part of
the hippocampus. Subsequently, the hippocampus was freed from the
surrounding tissue.
TUNEL assay
Brain tissues from 7-day-old mice were fixed with
10% neutral buffered formalin at room temperature for 2 weeks,
processed and trimmed for use in the TUNEL assay. After being
embedded in paraffin, the samples were cut into ~5-µm-thick slices.
TUNEL assays were performed following the protocol of an in situ
cell death detection kit (Roche Molecular Diagnostics). Samples
were incubated in TUNEL reagent at 37°C in a wet dark box for 60
min. Nuclei were stained with 0.5% hematoxylin at room temperature
for 5 sec, and neutral resin was used as mounting medium. Each
sample was observed in three fields of view.
Annexin-V-FITC assay of apoptotic
cells
The apoptosis rate of the PC12 cells was measured by
flow cytometry. PC12 cells (2×105/well) were seeded into
6-well plates and incubated overnight. After 48 h of transfection
with the relevant siRNA (10 µl/well) or plasmid (1 µg/well), the
cells were collected and subjected to Annexin V-FITC-propidium
iodide double staining according to the manufacturer's protocols as
previously described (39). The
cells were analyzed using a BD FACSAria flow cytometer (BD
Biosciences) with CellQuest software 5.1 (Becton-Dickinson and
Company).
Ethics statement
Human experiments were performed in accordance with
relevant approved guidelines and regulations, and were approved by
Renmin Hospital of Wuhan University's Institutional Ethics
Committee of the Faculty of Medicine [approval no. 2012LKSZ (010)
H]. The human para-glioblastoma (GBM) tissue used in the present
study was collected in a tumor resection performed at the
Department of Neurosurgery, Renmin Hospital of Wuhan University
(Wuhan, China) in December 2017, and was the temporal lobe of a
59-year-old female patient with GBM. Prior to the collection of a
normal human brain tissue next to GBM, written informed consent was
obtained from the relatives of the patient according to the
principles of the Declaration of Helsinki.
RND3 knockout mice were generated at Texas A&M
University Health Science Center. All animal experiments were
approved by the Institutional Animal Care and Use Committee of the
Texas A&M Health Science Center-Houston. According to the
Animal Research: Reporting of In Vivo Experiments checklist
(40), the animal's physical
condition was monitored daily. Chloral hydrate (350 mg/kg) was
injected intraperitoneally during anesthesia of mice. The
euthanasia of mice was conducted using 100% carbon dioxide
inhalation, and the displacement rate of carbon dioxide in the
chamber was 30%/min. The death of mice was verified by no
spontaneous breathing for 2-3 min with no eyeblink reflex. One
mouse in the RND3−/− group was excluded from the
experiment for a premature death due to malnutrition.
Statistical analysis
The data are presented as the mean ± SEM.
Statistical analysis was performed using SPSS v13.0 (SPSS, Inc.).
Significant differences between the means of two groups were
assessed by an unpaired t-test. Differences among the means of
multiple groups were assessed by one-way ANOVA and Tukey's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference and the results are representative of three
independent repeats.
Results
Rnd3 deletion suppresses apoptosis in
the mouse brain
To explore the functions of Rnd3 in the brain, the
present study first analyzed Rnd3 expression in the mouse brain
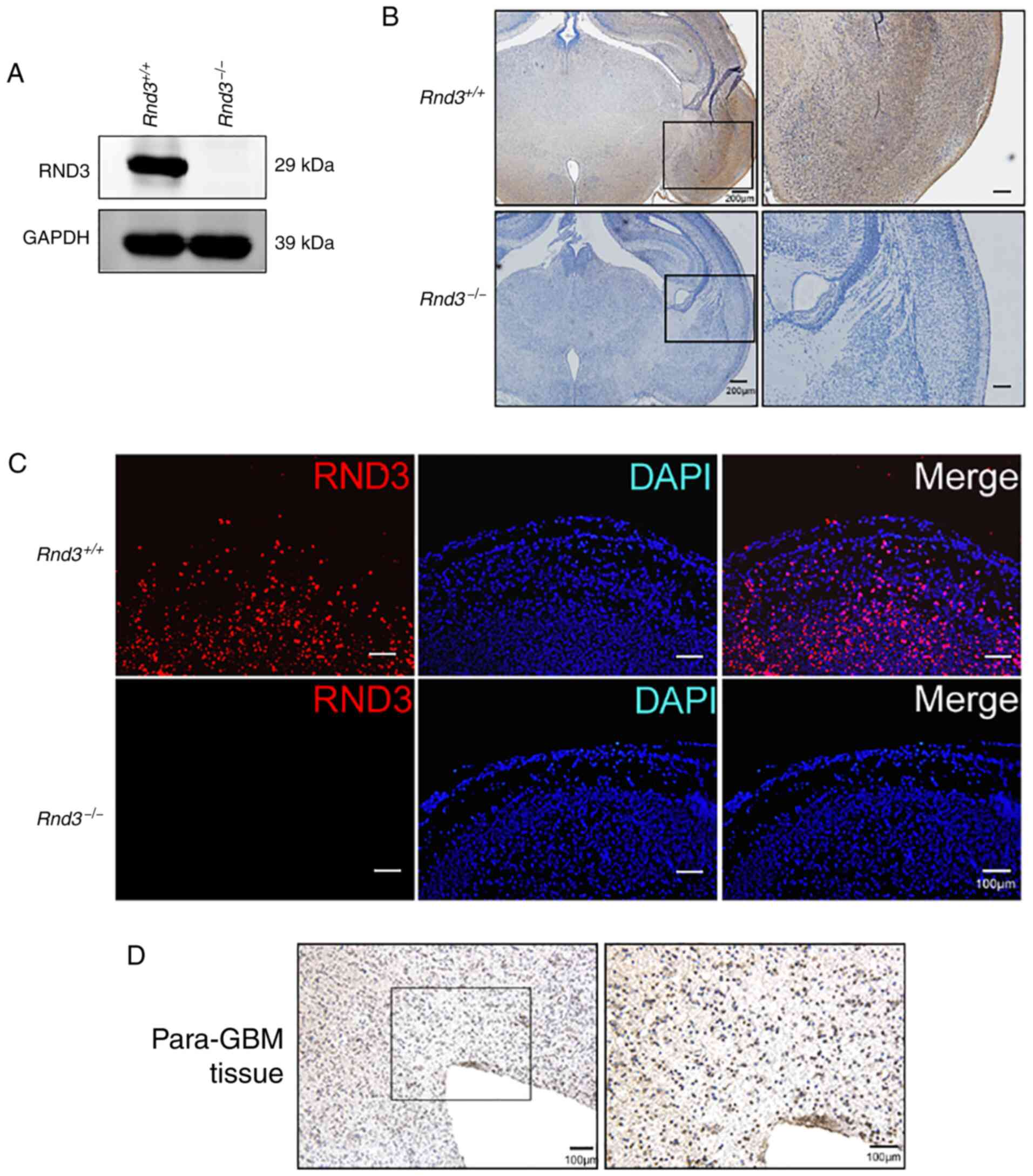
using Rnd3-knockout mice (Fig.
1A) generated from an ES cell line by gene trapping. Neither IF
nor IHC staining detected Rnd3 signals in the
Rnd3− mouse brain, while Rnd3 was mainly
expressed in the cytoplasm of the cortex in wild-type mice
(Fig. 1A-C). In the IHC staining of
the human brain adjacent to glioblastoma tissue, it was observed
that the signals of Rnd3 were not in the nucleus but in the
cytoplasm, which indicated that Rnd3 of para-GBM tissue exhibited
an expression pattern similar to that of the normal mouse brain
(Fig. 1D).
Because neuronal apoptosis is widespread in the
brain, particularly in most neurological disorders, the present
study next explored the possible roles of Rnd3 in brain cell
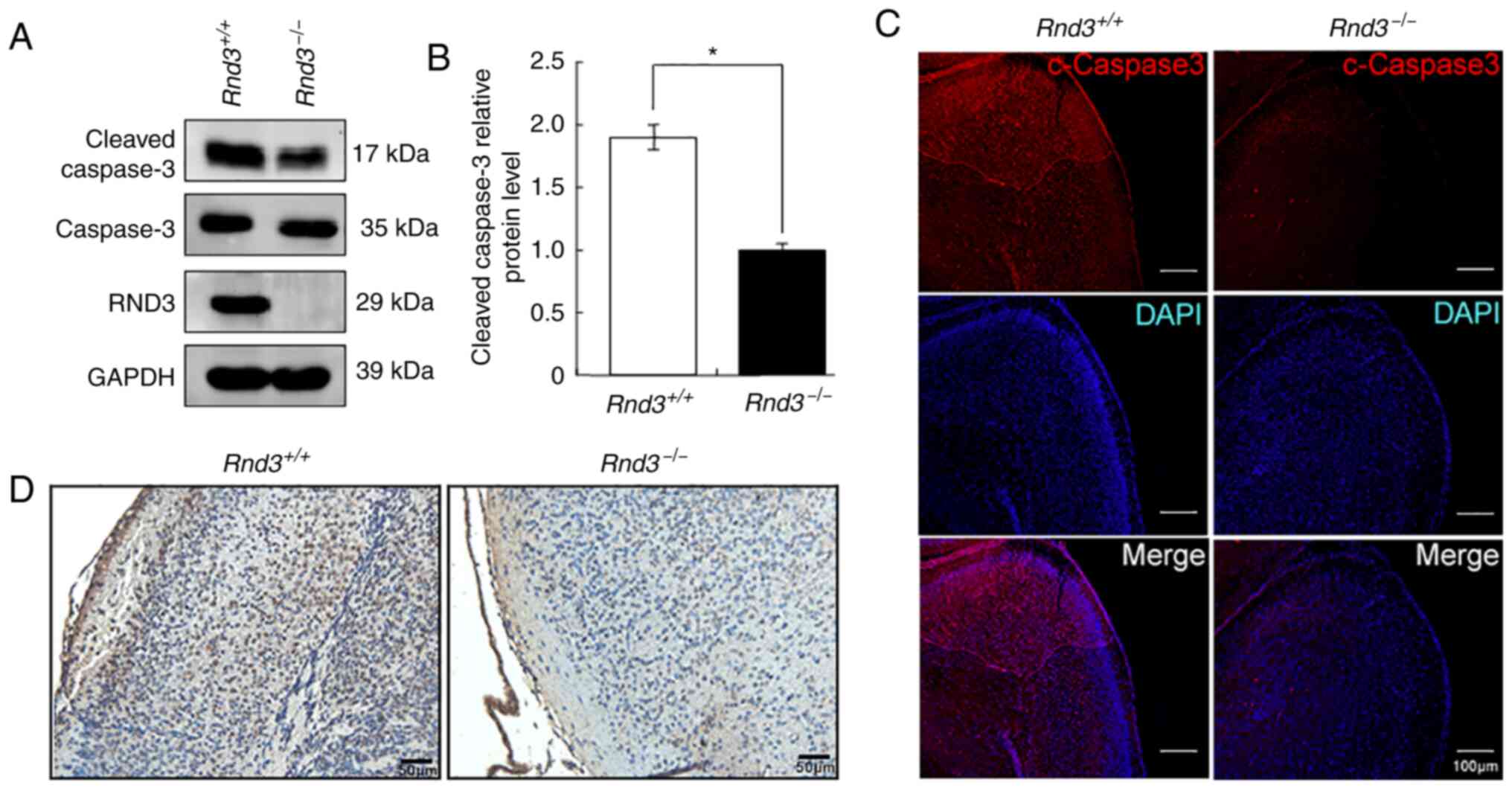
apoptosis. The western blot analysis revealed that the expression
levels of cleaved caspase-3, a key apoptotic protease, were
significantly decreased in the Rnd3−/− mouse
brains compared with in the wild-type mouse brains, in both the
cerebral cortex (P<0.05; Fig. 2A and
B) and hippocampus (P<0.05; Fig. S1A and B). The IF results also
indicated that the intensity of the cleaved caspase-3 signals was
lower in the cortex of Rnd3−/− mouse brains than
it was in the wild-type mouse brains (Fig. 2C), both in neurons (Fig. S2A) and astrocytes (Fig. S2B).
Furthermore, TUNEL assays revealed decreased
apoptotic signals in the Rnd3− mouse brains
compared with in the wild-type mouse brains (Fig. 2D). These data suggested that the
deletion of Rnd3 suppressed apoptosis in the mouse
brain.
RND3 promotes apoptosis in PC12
cells
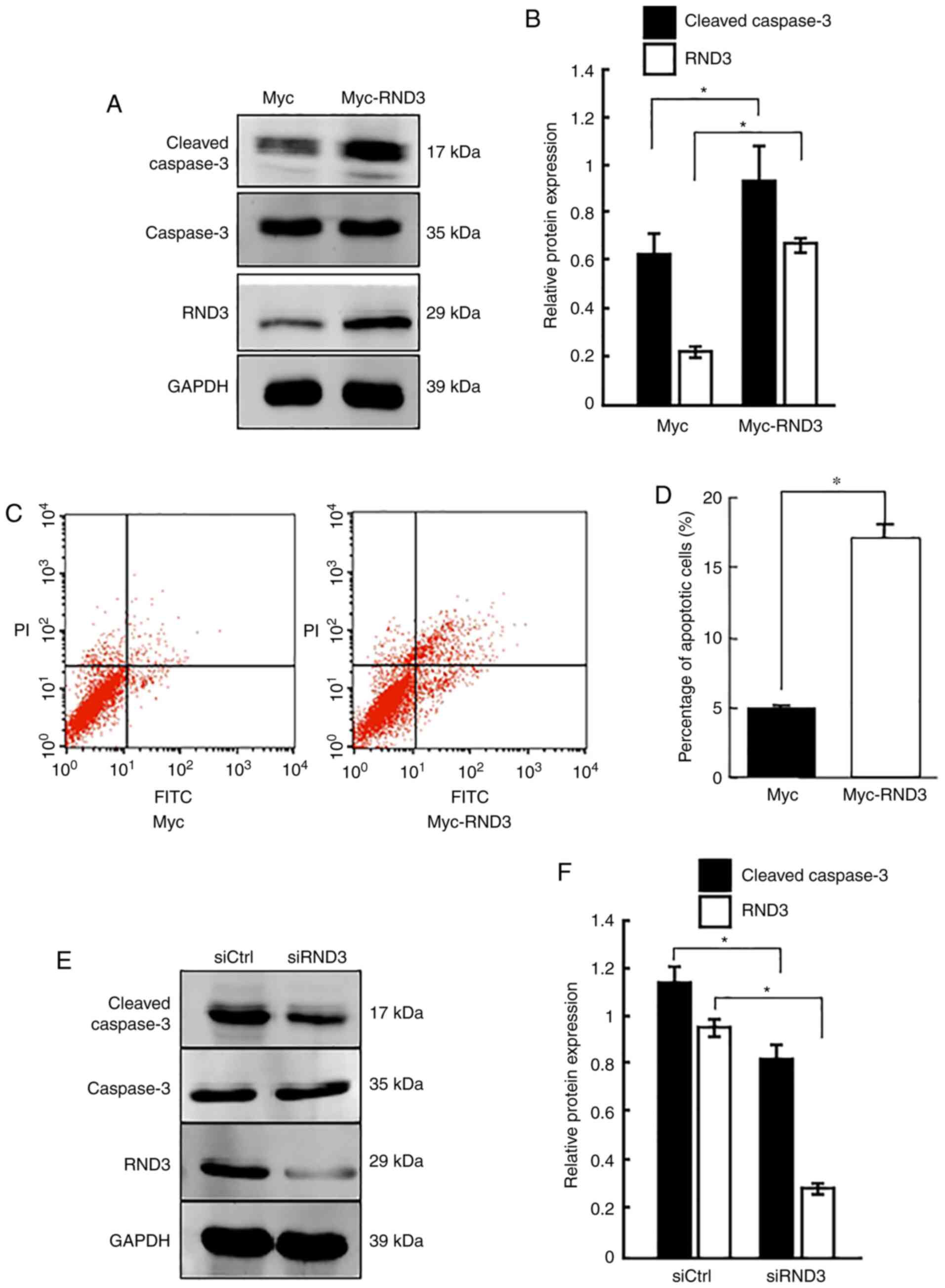
To determine the role of RND3 in the regulation of
apoptosis, the present study investigated the apoptosis rate of
PC12 cells (which have an embryonic origin from the neural crest)
using overexpression and knockdown approaches. Western blot
analysis demonstrated that cleaved caspase-3 protein expression was
significantly increased when RND3 was overexpressed (P<0.05;
Fig. 3A and B), which was
consistent with the results of flow cytometry, which revealed that
RND3 overexpression clearly increased the proportion of apoptotic
cells (P<0.05; Fig. 3C and D).
By contrast, RND3 knockdown reduced the levels of cleaved caspase-3
compared with the control (P<0.05; Fig. 3E and F). These data suggested that
RND3 could promote the apoptosis of PC12 cells.
Rnd3 regulates P65 expression and
affects the NF-κB signaling pathway
To further explore the molecular mechanisms of Rnd3
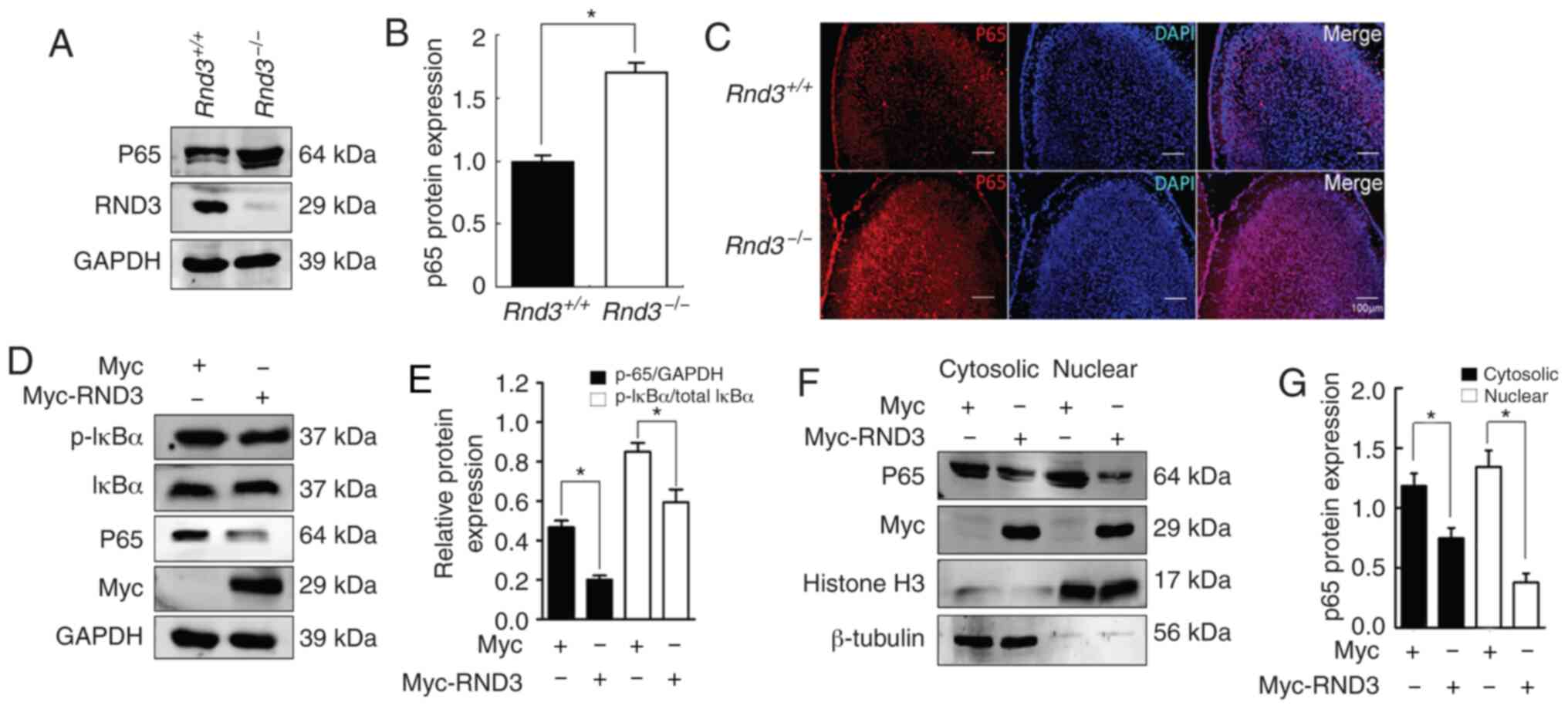
in the regulation of apoptosis, the present study analyzed NF-κB
signaling in the mouse brain. The western blot analysis results
demonstrated that NF-κB P65 was upregulated in the brains of the
Rnd3–/– mice compared with in the wild-type mice
(P<0.05; Fig. 4A and B),
indicating that Rnd3 downregulated P65 protein expression. IF
analysis revealed the upregulation of P65 protein expression in the
cortex of the Rnd3− mouse brains compared with
that in the brains of the wild-type mice (Fig. 4C). IκB-α is a protein associated
with P65, and the phosphorylation of IκB-α leads to
proteasome-mediated degradation, which results in the release of
activated P65, leading to its nuclear translocation (41,42).
To investigate the role of Rnd3 in the NF-κB signaling pathway, the
present study measured the activation of IκB-α when Rnd3 was
downregulated by Myc-Rnd3 in PC12 cells. Compared with Myc control,
p-IκB-α was downregulated (P<0.05; Fig. 4D and E). In addition, the extent of
P65 nuclear translocation in PC12 cells was examined, and compared
with those in the Myc control group, the levels of both the
cytosolic and nuclear P65 protein were decreased in the Myc-Rnd3
group (P<0.05; Fig. 4F and G).
Notably, the expression levels of P65 in the nucleus declined to a
greater extent, indicating that Rnd3 can downregulate P65
expression and decrease the nuclear entry of P65, thus suppressing
the NF-κB signaling pathway. These data suggested that Rnd3 may
regulate apoptosis via the NF-κB signaling pathway.
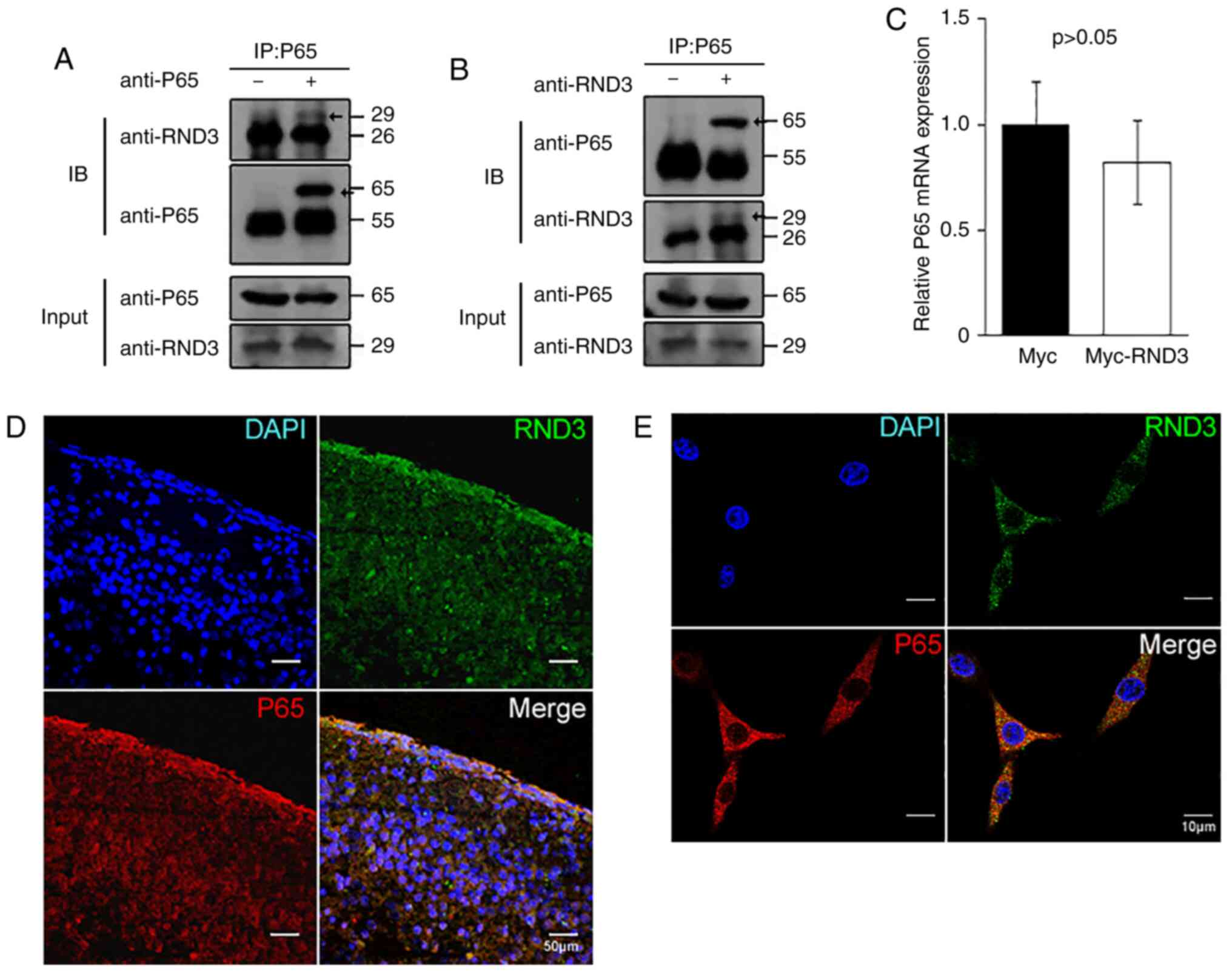
Rnd3 interacts with NF-κB P65 in
vivo
To explore the association of RND3 with P65, the
present study analyzed their interaction in both PC12 cells and the
mouse brain. Mouse brain lysates were immunoprecipitated with
anti-P65 or anti-Rnd3 antibodies, followed by IB with anti-Rnd3 or
anti-P65 antibodies. In both cases, interactive proteins were
detected (Fig. 5A and B). The IF
analysis indicated that RND3/Rnd3 colocalized with P65 in the
cytoplasm in the mouse brains (Fig.
5D). Rnd3 was also observed to colocalize with P65 in the
cytoplasm of PC12 cells using confocal microscopy, while some P65
protein was localized to the nuclei (Fig. 5E). Furthermore, the p65 mRNA level
was not altered (P>0.05) when RND3 was overexpressed (Fig. 5C). These results indicated that
RND3/Rnd3 interacts with P65 in vivo and in
vitro.
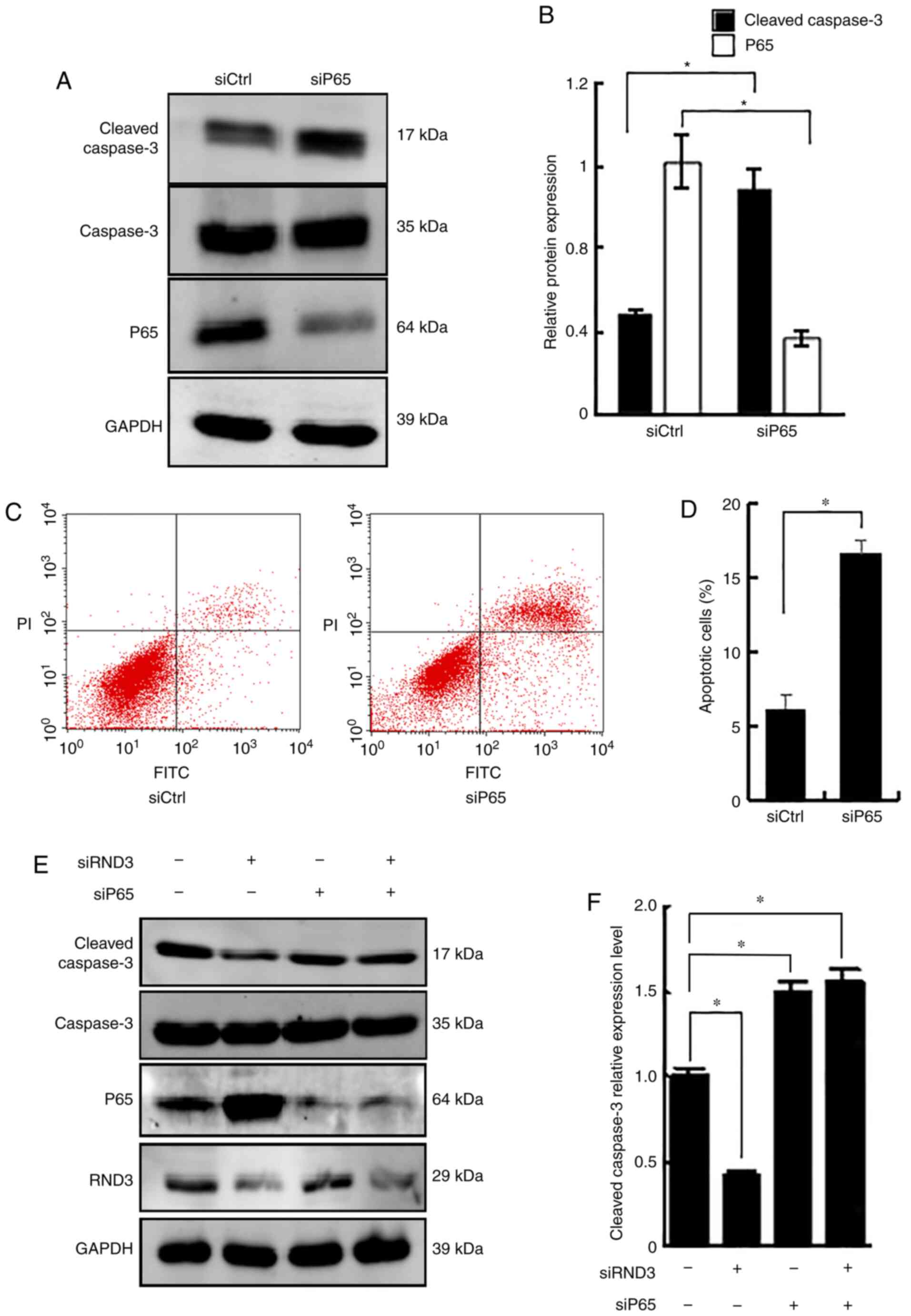
Rnd3 blocks the anti-apoptotic role of
NF-κB P65
To further analyze the role of Rnd3 and P65 in the
regulation of apoptosis, RNA interference (RNAi) was used to knock
down Rnd3, P65 or both. Knockdown of P65 upregulated cleaved
caspase-3 protein expression, compared with that in the control
RNAi group (P<0.05; Fig. 6A and
B), and increased the proportion of apoptotic cells (P<0.05;
Fig. 6C and D), demonstrating the
role of P65 in suppressing apoptosis. A notable decrease in cleaved
caspase-3 expression was detected following Rnd3 knockdown, which
was reversed by P65 knockdown (P<0.05; Fig. 6E and F). These data demonstrated
that Rnd3 blocked the anti-apoptotic action of NF-κB P65.
Discussion
The histopathology of neurodegenerative diseases
indicates that neurocytes undergo chronic progressive apoptosis
(43). Because neurocytes are
nonrenewable, efficient treatment for neurodegenerative diseases is
limited. Furthermore, the most commonly used drugs for the
treatment of Alzheimer's disease, donepezil and memantine
hydrochloride, are expensive and only slow the progression of
disease development rather than alleviating symptoms (44). Therefore, alternative treatments of
neurodegenerative diseases are required to reduce the neuronal
apoptosis rate and maintain cell numbers.
The present study identified a novel role for RND3
as a regulator of apoptosis in the central nervous system. The
function of RND3 in promoting apoptosis was demonstrated in PC12
cells using RNAi and overexpression experiments. Notably, Rnd3 was
mainly located in the cortex of wild-type mice, but the apoptosis
marker, cleaved caspase-3, seemed to decrease more obviously in the
hippocampus of Rnd3−/− mice. Although it was mainly
located in the cortex, Rnd3 expression could also be detected in
the hippocampus, and Rnd3 expression in the hippocampus of the
Rnd3−/− mice was decreased compared with that of the
Rnd3+/+ mice. This revealed a close relationship between
Rnd3 and apoptosis in the central nervous system. The present study
also demonstrated that RND3 regulated apoptosis via NF-κB P65 in
vivo. Specifically, RND3 blocked the anti-apoptotic role of
NF-κB P65 by downregulating P65, suggesting the importance of
RND3-NF-κB P65 signaling integrity in neuropathogenesis. Therefore,
RND3-NF-κB P65 represents a novel pathway in the regulation of
apoptosis, which may be utilized in the treatment of
neurodegenerative diseases. Although Pacary et al (45) reported that Rnd3 knockdown in
cortical neuron progenitors did not induce cell death at embryonic
stages, the present study mainly focused on the neurons and
neuroglial cells of 7-day-old mice, which have different activated
intracellular signaling pathways.
RND3 is a member of the GTPase family. Most previous
studies of RND3 have focused on its inhibitory effects involving
Rho kinase signaling (28).
However, evidence suggests that RND3 is involved in multiple
biological functions that are likely independent of Rho kinase
activity, including mouse neuron development (32,46),
neuronal migration (45), and
glioblastoma cell proliferation, migration and invasion (37). Additionally, our previous study
demonstrated that RND3 was involved in ependymocyte proliferation
(33). Combined with the present
findings, these studies indicated that RND3 has important roles in
the central nervous system and that in-depth studies of its
molecular mechanisms in the brain will provide novel insights into
the pathogenesis of neurodegenerative diseases.
The present study reported that RND3 decreased p65
protein expression, while the mRNA levels were not altered,
indicating that RND3 does not impact p65 mRNA transcription and may
instead regulate p65 protein expression in a post-transcriptional
manner. Although, to the best of our knowledge, no studies have
reported the regulation of p65 by RND3 in the central nervous
system, a recent study reported that RND3 regulates the
inflammatory response by interacting with p65 and p50 and
inhibiting the nuclear translocation of the p65/p50 complex,
indicating a protective role for RND3 in myocardial infarction
(47). Several previous studies
have demonstrated that RND3 is a downstream target of p53,
resulting in the inhibition of Rho associated coiled-coil
containing protein kinase 1 (ROCK1)-mediated apoptosis in certain
types of tumor (48,49). Furthermore, RND3 is associated with
the hyperactivation of ROCK1 signaling in mouse hearts, leading to
apoptotic cardiomyopathy and heart failure (50).
NF-κB is involved in multiple activities through
various extracellular signals, such as neurotransmitters,
neuropeptides, neurotrophins, cytokines and neural cell adhesion
molecules, in the nervous system (51). Furthermore, NF-κB signaling provides
neuroprotective functions through its role in the survival of
certain populations of peripheral and central neurons (52). NF-κB also protects neurons from
glutamate, oxidative stress and amyloid β-peptide (53). Notably, vascular endothelial growth
factor-mediated NF-κB activation protected PC12 cells from damage
induced by hypoxia through P65 nuclear translocation (54), while p65 siRNA and a p65-specific
inhibitor promoted apoptosis in cultured fibroblasts (55).
The aforementioned anti-apoptotic effect of P65 was
consistent with the present observations of the involvement of the
RND3-P65 signaling pathway in apoptosis regulation. However, an
opposite function of p65 in the PDCD5-mediated apoptosis in cancer
cells has been reported, in which the NF-κB signaling pathway
induced tumor cell apoptosis via P65 (56). The transfection of constructs
encoding p65 decreased the expression levels of cell proliferation
markers but not apoptosis markers in ovarian cells (57). The differences in the results of
these studies may reflect the activation of different signaling
pathways in various cell types and/or physiological conditions,
which should be clarified.
The present study demonstrated that RND3 attenuated
the NF-κB P65 signaling pathway along with cell apoptosis in the
brain. Additionally, it was demonstrated that RND3 interacted with
P65, leading to the inhibition of the NF-κB signaling pathway,
which increased cell apoptosis in the central nervous system in
vivo and in vitro. These findings may help comprehend
the regulation of the NF-κB P65 signaling pathway and potential
therapeutic uses of RND3 in the brain.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Jiang Chang, Dr
Xiangsheng Yang and Dr Dai Yuan of Texas A&M University Health
Science Center (Bryan, USA) for generating and providing RND3
knockout mice and Dr Rongjia Zhou and Dr Hanhua Cheng (College of
Life Sciences of Wuhan University, Wuhan, China) for providing the
Flag-p65 plasmid.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81502175 and
81371390) and Hubei Province Health and Family Planning Scientific
Research Project (grant no. WJ2017Q008).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BL designed the research. HD, QS, YZ, YL and FY
carried out the experimental work. SM analyzed the data and wrote
the paper. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal and human experiments were performed in
accordance with the relevant approved guidelines and regulations,
and were approved by the Ethics Committee of Wuhan University
[approval no. 2012LKSZ (010) H]. Prior to sample collection,
written informed consent was obtained from the relatives of this
patient with impaired consciousness.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
RND3
|
Rho family GTPase 3
|
|
RNAi
|
RNA interference
|
References
|
1
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fuchs Y and Steller H: Live to die another
way: Modes of programmed cell death and the signals emanating from
dying cells. Nat Rev Mol Cell Biol. 16:329–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fuchs Y and Steller H: Programmed cell
death in animal development and disease. Cell. 147:742–758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ryoo HD, Gorenc T and Steller H: Apoptotic
cells can induce compensatory cell proliferation through the JNK
and the Wingless signaling pathways. Dev Cell. 7:491–501. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamaguchi Y and Miura M: Programmed cell
death in neurodevelopment. Dev Cell. 32:478–490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Buss RR, Gould TW, Ma J, Vinsant S,
Prevette D, Winseck A, Toops KA, Hammarback JA, Smith TL and
Oppenheim RW: Neuromuscular development in the absence of
programmed cell death: Phenotypic alteration of motoneurons and
muscle. J Neurosci. 26:13413–13427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferrer I, Soriano E, del Rio JA, Alcántara
S and Auladell C: Cell death and removal in the cerebral cortex
during development. Prog Neurobiol. 39:1–43. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuan CY, Flavell RA and Rakic P:
Programmed cell death in mouse brain development. Results Probl
Cell Differ. 30:145–162. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haydar TF, Nowakowski RS, Yarowsky PJ and
Krueger BK: Role of founder cell deficit and delayed neuronogenesis
in microencephaly of the trisomy 16 mouse. J Neurosci.
20:4156–4164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yeo W and Gautier J: Early neural cell
death: Dying to become neurons. Dev Biol. 274:233–244. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de la Rosa EJ and de Pablo F: Cell death
in early neural development: Beyond the neurotrophic theory. Trends
Neurosci. 23:454–458. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oppenheim RW: Cell death during
development of the nervous system. Annu Rev Neurosci. 14:453–501.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Verney C, Takahashi T, Bhide PG,
Nowakowski RS and Caviness VS Jr: Independent controls for
neocortical neuron production and histogenetic cell death. Dev
Neurosci. 22:125–138. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Favaloro B, Allocati N, Graziano V, Di
Ilio C and De Laurenzi V: Role of apoptosis in disease. Aging
(Albany NY). 4:330–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamblin MR: Photobiomodulation for
traumatic brain injury and stroke. J Neurosci Res. 96:731–743.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sureda A, Capó X and Tejada S:
Neuroprotective effects of flavonoid compounds on neuronal death
associated to Alzheimer's disease. Curr Med Chem. 26:5124–5136.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gitler AD, Dhillon P and Shorter J:
Neurodegenerative disease: Models, mechanisms, and a new hope. Dis
Model Mech. 10:499–502. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Egawa N, Lok J, Washida K and Arai K:
Mechanisms of axonal damage and repair after central nervous system
injury. Transl Stroke Res. 8:14–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li C, Wang D, Wu W, Yang W, Ali Shah SZ,
Zhao Y, Duan Y, Wang L, Zhou X, Zhao D and Yang L: DLP1-dependent
mitochondrial fragmentation and redistribution mediate
prion-associated mitochondrial dysfunction and neuronal death.
Aging Cell. 17:e126932018. View Article : Google Scholar
|
|
21
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharma D and Kanneganti TD: Inflammatory
cell death in intestinal pathologies. Immunol Rev. 280:57–73. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bleicken S, Landeta O, Landajuela A,
Basañez G and García-Sáez AJ: Proapoptotic Bax and Bak proteins
form stable protein-permeable pores of tunable size. J Biol Chem.
288:33241–33252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu L, Zhang R, Liu K, Zhou H, Tang Y, Su
J, Yu X, Yang X, Tang M and Dong Q: Tissue kallikrein alleviates
glutamate-induced neurotoxicity by activating ERK1. J Neurosci Res.
87:3576–3590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sheikh MS and Huang Y: Death receptor
activation complexes: It takes two to activate TNF receptor 1. Cell
Cycle. 2:550–552. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bourteele S, Oesterle K, Weinzierl AO,
Paxian S, Riemann M, Schmid RM and Planz O: Alteration of NF-kappaB
activity leads to mitochondrial apoptosis after infection with
pathological prion protein. Cell Microbiol. 9:2202–2217. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fiegen D, Blumenstein L, Stege P, Vetter
IR and Ahmadian MR: Crystal structure of Rnd3/RhoE: Functional
implications. FEBS Lett. 525:100–104. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jie W, Andrade KC, Lin X, Yang X, Yue X
and Chang J: Pathophysiological functions of Rnd3/RhoE. Compr
Physiol. 6:169–186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J
and Settleman J: Identification of a novel human Rho protein with
unusual properties: GTPase deficiency and in vivo farnesylation.
Mol Cell Biol. 16:2689–2699. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Riento K, Guasch RM, Garg R, Jin B and
Ridley AJ: RhoE binds to ROCK I and inhibits downstream signaling.
Mol Cell Biol. 23:4219–4229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klein RM and Aplin AE: Rnd3 regulation of
the actin cytoskeleton promotes melanoma migration and invasive
outgrowth in three dimensions. Cancer Res. 69:2224–2233. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pacary E, Azzarelli R and Guillemot F:
Rnd3 coordinates early steps of cortical neurogenesis through
actin-dependent and -independent mechanisms. Nat Commun.
4:16352013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin X, Liu B, Yang X, Yue X, Diao L, Wang
J and Chang J: Genetic deletion of Rnd3 results in aqueductal
stenosis leading to hydrocephalus through up-regulation of Notch
signaling. Proc Natl Acad Sci USA. 110:8236–8241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang X, Wang T, Lin X, Yue X, Wang Q, Wang
G, Fu Q, Ai X, Chiang DY, Miyake CY, et al: Genetic deletion of
Rnd3/RhoE results in mouse heart calcium leakage through
upregulation of protein kinase A signaling. Circ Res. 116:e1–e10.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sheng Y, Song Y, Li Z, Wang Y, Lin H,
Cheng H and Zhou R: RAB37 interacts directly with ATG5 and promotes
autophagosome formation via regulating ATG5-12-16 complex assembly.
Cell Death Differ. 25:918–934. 2018.PubMed/NCBI
|
|
36
|
Yuan J, Zhang Y, Sheng Y, Fu X, Cheng H
and Zhou R: MYBL2 guides autophagy suppressor VDAC2 in the
developing ovary to inhibit autophagy through a complex of
VDAC2-BECN1-BCL2L1 in mammals. Autophagy. 11:1081–1098. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu B, Dong H, Lin X, Yang X, Yue X, Yang
J, Li Y, Wu L, Zhu X, Zhang S, et al: RND3 promotes Snail 1 protein
degradation and inhibits glioblastoma cell migration and invasion.
Oncotarget. 7:82411–82423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun Q, Dong H, Li Y, Yuan F, Xu Y, Mao S,
Xiong X, Chen Q and Liu B: Small GTPase RHOE/RND3, a new critical
regulator of NF-κB signalling in glioblastoma multiforme? Cell
Prolif. 52:e126652019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu B, Guo Z, Dong H, Daofeng T, Cai Q, Ji
B, Zhang S, Wu L, Wang J, Wang L, et al: LRIG1, human EGFR
inhibitor, reverses multidrug resistance through modulation of
ABCB1 and ABCG2. Brain Res. 1611:93–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG; NC3Rs Reporting Guidelines Working Group, : Animal
research: Reporting in vivo experiments: The ARRIVE guidelines. Br
J Pharmacol. 160:1577–1579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brockman JA, Scherer DC, McKinsey TA, Hall
SM, Qi X, Lee WY and Ballard DW: Coupling of a signal response
domain in I kappa B alpha to multiple pathways for NF-kappa B
activation. Mol Cell Biol. 15:2809–2818. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen ZJ, Parent L and Maniatis T:
Site-specific phosphorylation of IkappaBalpha by a novel
ubiquitination-dependent protein kinase activity. Cell. 84:853–862.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Waldmeier PC and Tatton WG: Interrupting
apoptosis in neurodegenerative disease: Potential for effective
therapy? Drug Discov Today. 9:210–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Briggs R, Kennelly SP and O'Neill D: Drug
treatments in Alzheimer's disease. Clin Med (Lond). 16:247–253.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pacary E, Heng J, Azzarelli R, Riou P,
Castro D, Lebel-Potter M, Parras C, Bell DM, Ridley AJ, Parsons M
and Guillemot F: Proneural transcription factors regulate different
steps of cortical neuron migration through Rnd-mediated inhibition
of RhoA signaling. Neuron. 69:1069–1084. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mocholí E, Ballester-Lurbe B, Arqué G,
Poch E, Peris B, Guerri C, Dierssen M, Guasch RM, Terrado J and
Pérez-Roger I: RhoE deficiency produces postnatal lethality,
profound motor deficits and neurodevelopmental delay in mice. PLoS
One. 6:e192362011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dai Y, Song J, Li W, Yang T, Yue X, Lin X,
Yang X, Luo W, Guo J, Wang X, et al: RhoE fine-tunes inflammatory
response in myocardial infarction. Circulation. 139:1185–1198.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ongusaha PP, Kim HG, Boswell SA, Ridley
AJ, Der CJ, Dotto GP, Kim YB, Aaronson SA and Lee SW: RhoE is a
pro-survival p53 target gene that inhibits ROCK I-mediated
apoptosis in response to genotoxic stress. Curr Biol. 16:2466–2472.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhu Y, Zhou J, Xia H, Chen X, Qiu M, Huang
J, Liu S, Tang Q, Lang N, Liu Z, et al: The Rho GTPase RhoE is a
p53-regulated candidate tumor suppressor in cancer cells. Int J
Oncol. 44:896–904. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yue X, Yang X, Lin X, Yang T, Yi X, Dai Y,
Guo J, Li T, Shi J, Wei L, et al: Rnd3 haploinsufficient mice are
predisposed to hemodynamic stress and develop apoptotic
cardiomyopathy with heart failure. Cell Death Dis. 5:e12842014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gutierrez H and Davies AM: Regulation of
neural process growth, elaboration and structural plasticity by
NF-κB. Trends Neurosci. 34:316–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mémet S: NF-kappaB functions in the
nervous system: From development to disease. Biochem Pharmacol.
72:1180–1195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Qin ZH, Tao LY and Chen X: Dual roles of
NF-κB in cell survival and implications of NF-kappaB inhibitors in
neuroprotective therapy. Acta Pharmacol Sin. 28:1859–1872. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mo SJ, Hong J, Chen X, Han F, Ni Y, Zheng
Y, Liu JQ, Xu L, Li Q, Yang XH, et al: VEGF-mediated NF-κB
activation protects PC12 cells from damage induced by hypoxia.
Neurosci Lett. 610:54–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen S, Jiang S, Zheng W, Tu B, Liu S,
Ruan H and Fan C: RelA/p65 inhibition prevents tendon adhesion by
modulating inflammation, cell proliferation, and apoptosis. Cell
Death Dis. 8:e27102017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Murshed F, Farhana L, Dawson MI and
Fontana JA: NF-κB p65 recruited SHP regulates PDCD5-mediated
apoptosis in cancer cells. Apoptosis. 19:506–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sirotkin AV, Dekanová P, Harrath AH,
Alwasel SH and Vašíček D: Interrelationships between sirtuin 1 and
transcription factors p53 and NF-κB (p50/p65) in the control of
ovarian cell apoptosis and proliferation. Cell Tissue Res.
358:627–632. 2014. View Article : Google Scholar : PubMed/NCBI
|