Introduction
The BCR-ABL1 fusion gene, derived from the
t(9;22) translocation, is the most common cytogenetic abnormality
found in adult B-acute lymphoblastic leukemia (B-ALL), accounting
for approximately 25% of all B-ALL (1–3).
BCR-ABL1 rearrangement results in a fusion protein with
constitutively active tyrosine kinase activity that fosters B-cell
progenitor malignant transformation (4). Bcr-Abl kinase activates a series of
intracellular signaling pathways, resulting in increased cell
survival and limited growth factor dependence (5–8).
Numerous studies have suggested that additional genomic alterations
are the hallmark of BCR-ABL1+ B-ALL (9,10).
These genomic alterations could be created by two classes of
enzymes required for the diversity of immunoglobulins in B-cell
development: the Rag proteins, encoded by recombination-activating
genes (RAG1 and RAG2), which introduce DNA
rearrangement of the variable (V), diversity (D), and joining (J)
segments of immunoglobulin (Ig) genes in pro-B and pre-B cells, and
activation-induced cytidine deaminase (Aid) that initiate somatic
hypermutation (SHM) and class-switch recombination (CSR) on Ig
genes in activated B cells (11).
Mistargeting of Rag mediates the deletion of IKZF1, leading
to the loss of Ikaros function, which is highly relevant for the
leukemogenesis of BCR-ABL1+ B-ALL (12). Studies in mice and humans have
suggested that the aberrant expression of Aid induced by Bcr-Abl
kinase activity enhances genetic instability by augmenting the
frequency of amplifications, deletions, and aberrant somatic
hypermutation, which reportedly contribute to genomic instability
and malignant transformation of BCR-ABL1+ B-ALL
(13–16). Although the development of tyrosine
kinase inhibitors (TKIs) has revolutionized therapy, resistance to
TKIs leads to short-lasting responses for TKIs in patients with
relapsed ALL (17,18). Consequently, new molecular
mechanisms for controlling Bcr-Abl kinase need to be developed to
cure BCR-ABL1+ B-ALL.
During the B-cell lineage commitment at the pro-B
stage, the successful rearrangement of immunoglobulin heavy chain
variable (V), diversity (D), and joining (J) gene segments
generates the μ heavy chain (μHC) proteins. The μHC pairs with
non-polymorphic surrogate light-chain components, λ5 and VpreB, to
form the pre-B cell receptor (pre-BCR). The pre-BCR is an
autonomously active receptor whose signals drive pre-B cell
proliferation and differentiation into immature B cells (19,20).
The initial step of pre-BCR signaling is the activation of the
spleen tyrosine kinase (Syk), which has a crucial role in the
activation of downstream pathways involved in the proliferation and
differentiation of pre-B cells. Syk stimulates pre-B cell
proliferation by activating PI3K and its downstream mediator Akt,
which promotes phosphorylation and nuclear exclusion forkhead box
protein O (Foxo) transcription factors, namely Foxo1, Foxo3a, and
Foxo4. Thus, the expression levels of cell cycle arrest- and
apoptosis induction-related, downstream target genes of Foxos, such
as BIM, TRAIL, CDKN1B, and GADD45α, are decreased
(21,22). As explained below, the proliferation
process is also inhibited by extensive feedback of pre-BCR
signaling components. Syk mediates differentiation by activating
the adaptor protein Blnk, which could activate phosphatase and
tensin homolog (Pten) or SH2-containing inositol phosphatase (Ship)
to decrease Akt activity, resulting in decreased phosphorylation
and increased stabilization of Foxo proteins. In addition, Blnk
recruits Bruton's tyrosine kinase (Btk), phospholipase C-γ2
(Plcγ2), and growth factor receptor-bound protein 2 (Grb2) to
initiate calcium-dependent signaling pathways, which is the key
event in the signaling pathway of the development and maturation of
B cells (23). Finally, the
proliferation phase is terminated, and the differentiation process
begins (24,25). Therefore, pre-BCR signaling
activates seemingly opposing cellular programs, such as
proliferation and differentiation. A large body of evidence has
demonstrated that Bcr-Abl functions as a constitutively active
kinase to initiate the downstream survival signaling in
BCR-ABL1+ B-ALL blasts by mimicking pre-BCR
pathways (26). Bcr-Abl activates
the PI3K/Akt pathway to induce Foxo phosphorylation, leading to the
nuclear exclusion of Foxos to stimulate abnormal cell growth
(27–30). Although there are trillions of
potential target cells, each containing hundreds of susceptible
oncogenes, cancer occurs less than once in a lifetime. There are a
variety of innate tumor inhibition mechanisms in mammalian cells.
Once the proliferation is aberrant, these mechanisms will trigger
apoptosis or senescence to prevent uncontrolled cell division
(31). The differentiation
signaling components Blnk and Btk have a synergistic effect in
pre-B cell tumor suppression, because the incidence of pre-B cell
leukemia in Blnk/Btk double-defect mice is significantly enhanced
than that in Blnk single-deficient mice (32–34).
However, the functional roles of differentiation signaling
components have not been previously characterized in
BCR-ABL1+ B-ALL.
The aim of the present study was to explore the
tumor suppressive mechanism of differentiation signaling molecules
in BCR-ABL1-transformed pro-B cells. The functional roles
and the participation of differentiation signaling components Blnk
and Foxo1 in BCR-ABL1-transformed pro-B cells were
identified, which may provide a rationale for future
BCR-ABL1+ B-ALL therapy.
Materials and methods
Cell lines
The Rag1 mutant mouse pro-B (D345) cell line was
established by infection of bone marrow cells from BCL-2
transgenic mice with the v-Abl retrovirus, which was was kindly
provided by Dr David Schatz (Yale University, New Haven, USA) and
stored in our laboratory (35). The
D345 cells were cultured in RPMI-1640 medium containing 10% fetal
bovine serum (FBS), non-essential amino acids and 1%
penicillin-streptomycin (all from Hyclone; Cytiva) and
β-mercaptoethanol (50 µM) at 37°C with 5% CO2. The 293T
cells were obtained from the American Type Culture Collection
(ATCC) and cultured in DMEM (Hyclone; Cytiva) supplemented with 10%
FBS, non-essential amino acids and penicillin-streptomycin (1%) at
37°C with 5% CO2.
Retroviral production and
transduction
MSCV vector co-expressing human BCR-ABL1
(p210) and hCD4 (MSCV-BCR-ABL1-IRES-hCD4) and MSCV vector
expressing hCD4 have been previously described (36). 293T cells were transfected with MSCV
vectors and PKAT2 package vector using X-tremeGENE HP DNA
transfection reagent (Roche Diagnostics) at 37°C. After 48 h of
transfection, viral supernatants were collected, filtered, and
stored at −80°C.
BCR-ABL1-transformed pro-B cells and the
control cells were obtained by transduction of D345 cells with
pMSCV-BCR-ABL1-hCD4 or pMSCV-EV-hCD4 viral supernatants with
1 µg/ml polybrene; then the six-well dishes were spun at 1,000 × g
for 1.5 h at room temperature. The medium was changed after 4 h and
incubated at 37°C. After 72 h, the cells were incubated with
APC-conjugated anti-human CD4 antibody (cat. no. 561840; BD
Biosciences) at a 1:10 dilution for 20 min at 4°C, and sorted by BD
FACSAria II (BD Biosciences). Flow cytometric data were analyzed
with Kaluza Analysis Software 2.1 (Beckman Coulter, Inc.).
Cell growth and viability
A Cell Counting Kit-8 (CCK-8) assay was used to
detect cell growth according to the manufacturer's instructions.
The cells were suspended with FBS-deficient RPMI-1640 medium
supplemented with non-essential amino acids,
penicillin-streptomycin, and β-mercaptoethanol (50 µM). Six
replicates of 2×104 cells were seeded in a 96-well plate
with 100 µl/well. Cells were cultured for 24, 48 and 72 h, then
incubated with 10 µl CCK-8 solution (product no. FXP132-500;
Beijing 4A Biotech Co., Ltd.) for 2 h. The absorbance at 450 nm was
measured by a microplate reader.
The CCK-8 assay was also used to measure cell
viability. Cells (2×105) were seeded in a 96-well plate
with 100 µl/well in six replicates. Cells were treated with vehicle
(PBS) or imatinib (0, 3, 6 and 9 µM) for 24 h. Subsequently, the
cells were incubated with 10 µl CCK-8 solution for 2 h. The
absorbance values at 450 nm were measured by a microplate
reader.
Generation of Aid-, Blnk- and
Foxo1-knockout cell lines
The pL-CRISPR.EFS.PAC was a gift from Dr Junjie
Zhang of the University of Southern California (Los Angeles, USA).
The AID, BLNK and FOXO1 single guide RNA (sgRNA) were
designed at the CRISPR design website from Zhang laboratory
(http://crispr.mit.edu/) and sequenced (Sunny
Biotech Co., Ltd.), sgRNA that does not target the genome was used
as a control, which was acquired from the Mouse GeCKOv2 Library
(37,38). The gRNA sequences are listed in
Table SI. The 293T cells were then
independently transfected with the pCas9-AID,
pCas9-BLNK and pCas9-FOXO1, pCas9-non-targeting
guides along with ΔR9 and pVSVG helper plasmids using the
X-tremeGENE HP DNA transfection reagent (Roche Diagnostics) at
37°C. The viral supernatants were collected 48 h after
transfection.
BCR-ABL1-transformed pro-B cells were each infected
with control, Aid-, Blnk-, or Foxo1-knockout viral supernatants in
the presence of 1 µg/ml polybrene. Then the six-well dishes were
spun at 1,000 × g for 1.5 h at room temperature. The medium was
changed after 4 h and incubated at 37°C. After 72 h, cells were
selected with puromycin (0.6 µg/ml) to enrich the transduced
cells.
Apoptosis and BrdU proliferation
assays
Cells (1×106) were treated with imatinib
(0, 3, 6 and 9 µM), and after 24 h, the cells were collected and
washed twice with cold PBS, and incubated for 20 min at room
temperature in 1:20 diluted PE-conjugated Annexin V and
anti-7-aminoactinomycin D (7-AAD) (cat. no. 559763; BD
Biosciences), according to the manufacturer's instructions. Then,
the cells were analyzed by flow cytometer (Beckman Coulter, Inc.).
The Aid KO and Aid WT BCR-ABL1-transformed pro-B cells were
starved in FBS-free medium for 24 h, and measured for apoptosis by
flow cytometry. Flow cytometric data were analyzed with Kaluza
Analysis Software 2.1 (Beckman Coulter, Inc.).
Proliferation assays were performed using the BrdU
kit (cat. no. 552598; BD Biosciences) according to the
manufacturer's instructions. Briefly, a population of
1×106 cells were cultured in 6-well plates and labeled
with BrdU labeling reagent for 45 min before harvesting. The cells
were centrifuged at 300 × g for 5 min at 4°C, then fixed and
permeabilized with Cytofix/Cytoperm buffer and Cytoperm
permeabilization buffer plus. Next, cells were treated with DNase
for 1 h at 37°C and further incubated with APC-conjugated anti-BrdU
for 20 min at room temperature. BrdU incorporation was measured
using a flow cytometer (Beckman Coulter, Inc.). Flow cytometric
data were analyzed with Kaluza Analysis Software 2.1 (Beckman
Coulter, Inc.).
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
The total RNA of cell pellets was isolated by TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Before cDNA synthesis, 1 µg of total
RNA was digested with 1 U/µl RNA-free recombination DNAse I. The
cDNA was then transcribed with the PrimeScript™ RT reagent Kit
(TaKaRa Bio, Inc.) according to the manufacturer's instructions.
Quantitative PCR experiments were performed in triplicate with
SYBR-Green dye (TaKaRa Bio, Inc.) on an Mx3000P qPCR system
(Agilent Technologies, Inc.). All data were analyzed using the
2-ΔΔCq (relative quantification) method (39). The following thermocycling
conditions were used for the qPCR: Initial denaturation at 95°C for
10 min; 40 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C for
30 sec; and a final step at 95°C for 1 min, 55°C for 30 sec and
95°C for 30 sec. Gene expression was normalized to GAPDH.
The primer sequences used for quantitative PCR are listed in
Table SII. The gene expression
levels of B-cell differentiation components were normalized to the
GAPDH level in the corresponding cell line and represented
as a heatmap using GraphPad Prism 8 (GraphPad Software, Inc.).
Western blotting
The cell pellets were lysed in RIPA buffer (150 mM
NaCl, 50 mM Tris pH 7.4, 1% Triton X-100, 0.5% NaDoc, 10% glycerol,
and 2.5% sodium deoxycholate) with protease and phosphatase
inhibitors. Protein concentration was determined using a BCA kit
(cat. no. P0010; Beyotime Institute of Biotechnology) and
Multiskan™ FC Microplate Reader (Thermo Fisher Scientific Inc.),
and analyzed using Skanit software version 3.1 (Thermo Fisher
Scientific Inc.). A total of 60 µg of proteins were loaded onto 12%
sodium dodecyl sulfide-polyacrylamide gel electrophoresis
(SDS-PAGE) for electrophoresis and transferred to polyvinylidene
difluoride (PVDF) membranes (Immobilon-P; cat. no. IPVH00010; 0.45
µm; EMD Millipore). Membranes were blocked in 5% milk solution for
2 h at room temperature, and then incubated overnight at 4°C with
indicated primary monoclonal antibodies with their respective
dilutions (Table SIII). The
membranes were then incubated with goat anti-rabbit IgG (H+L)-HRP
(1:5,000; cat. no. 31466; Thermo Fisher Scientific Inc.), goat
anti-rat IgG (H+L)-HRP (1:5,000; cat. no. 31470; Thermo Fisher
Scientific Inc.) and goat anti-mouse IgG (H+L)-HRP (1:5,000; cat.
no. 31431; Thermo Fisher Scientific Inc.) for 1 h at room
temperature, and blots were visualized with Western Lighting Pro
ECL (Fusion FX5; Vilber).
Immunofluorescence
Cells (1×104) were affixed to frosted X
slides and fixed with 4% paraformaldehyde solution for 15 min at
room temperature, followed by permeabilization with 0.2% Triton
X-100 for 10 min at room temperature. The slides were blocked with
5% BSA and 10% horse serum in PBS at room temperature for 1 h and
were incubated with anti-γH2AX (1:200; product no. 9718; Cell
Signaling Technology, Inc.) at 4°C overnight. After three rinses
with PBS, the cells were incubated with anti-rabbit IgG (H+L) Alexa
Fluor® 555 Conjugate (1:500; product no. 4413; Cell
Signaling Technology, Inc.) for 1 h. Then, the cells were washed
twice and stained with 1 µg/ml DAPI for 10 min at room temperature.
Images were acquired with a confocal microscope (Leica TCS SP8 STED
3X; Leica Microsystems, Inc.) at a magnification of ×40 and
processed with ImageJ v.1.8.0 software (National Institutes of
Health).
Mutation analysis of the ABL1
gene
Genomic DNA was extracted from 5×106
cells using the DNA Extraction Kit (cat. no. D824A; TaKaRa Bio,
Inc.) according to the manufacturer's instructions. The ABL1
kinase portion of the BCR-ABL1 gene was amplified with two
rounds of PCR. In the first round, the BCR-ABL1 fragments
were amplified using BCR- (exon 13) and ABL1- (exon
9) specific primers to prevent co-amplification of normal
ABL1. Primers for the second round of amplification focused
on the ABL1 kinase domain (exons 3-8). The genomic fragments
were amplified using high fidelity DNA polymerase KOD-Plus-Neo
(code no. KOD-401; Toyobo Life Science). PCR products were purified
with the Gel Extraction Kit (SKU no. D2500-01; Omega Bio-Tek, Inc.)
and cloned into the pMD® 18-T vector (cat. no. 6011;
TaKaRa Bio, Inc.). Clones were sequenced commercially (Sunny
Biotech Co., Ltd.). PCR primers used for amplification are listed
in Table SIV.
Statistical analysis
Unpaired t-test was performed for comparison of two
groups and one-way ANOVA followed by Bonferroni's post hoc tests
were performed for multiple comparisons using the SPSS 18.0
statistical software package (SPSS, Inc.). Results were presented
as the mean ± SEM. P<0.05 was considered to indicate a
statistically significant difference.
Results
Bcr-Abl kinase activity promotes
proliferation of pro-B cells via the PI3K/Akt pathway
To demonstrate that Bcr-Abl kinase was sufficient to
induce pro-B cell transformation, the retroviral vector
pMSCV-BCR-ABL1-IRES-hCD4 and empty vector
pMSCV–IRES-hCD4 were individually introduced into the D345 cell
line. The D345 cell line harbors the Rag1 catalytic mutant (D708A),
which retains the native ability to interact with Rag2 and bind to
DNA but lacks catalytic activity, thus failing to induce the
rearrangement of immunoglobulin genes, ultimately leading to
arrested B-cell development at the pro-B stage (35). After flow cytometric sorting, over
90% of infected cells were positive for hCD4 expression (Fig. S1A and B). A large amount of Bcr-Abl
expression was detected in pro-B cells infected with retroviral
vectors carrying BCR-ABL1 (Fig.
1A). Faster growth of BCR-ABL1-transformed pro-B cells
than the control cells in the absence of nutrition for 72 h was
observed (Fig. 1B). After treatment
with the tyrosine kinase inhibitor, imatinib, at the indicated
concentrations for 24 h, the cell viability of
BCR-ABL1-transformed pro-B cells significantly decreased
compared to that of the control cells (Fig. 1C). The percentage of apoptotic cells
was quantified by 7-AAD and Annexin V staining, and
BCR-ABL1-transformed pro-B cells exhibited higher apoptotic
cell populations than the control cells (Fig. 1D). Moreover, phosphorylated Crkl
(p-Crkl) is the main target of Bcr-Abl tyrosine kinase and serves
as the connector for downstream effector molecules (40). The p-Crkl levels as indicators of
Bcr-Abl kinase activity were therefore determined, and it was
revealed that p-Crkl was upregulated following BCR-ABL1
overexpression, while its levels were significantly reduced after
imatinib treatment (Fig. 1E). The
data revealed that Bcr-Abl kinase activity was required to promote
pro-B cell growth in vitro.
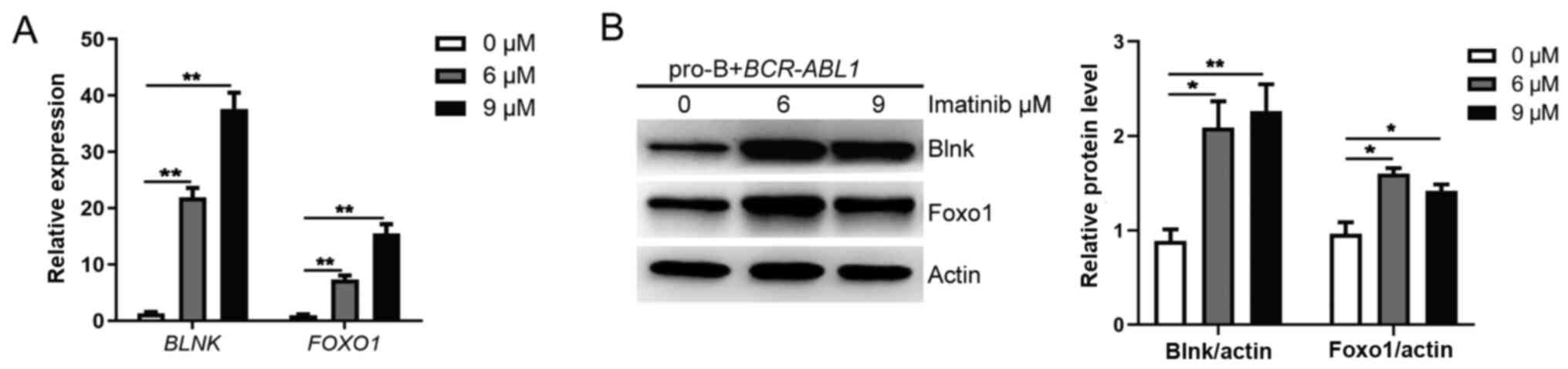
 | Figure 1.Enhanced pro-B cell survival is
mediated by BCR-ABL1 overexpression. The pro-B cells were
stably transfected with BCR-ABL1 or EV control. (A) Bcr-Abl
expression was detected by western blotting. The protein levels
were quantified by the ImageJ program and normalized to actin. (B)
Cells were seeded and grown in a 96-well plate with an
FBS-deficient culture medium; the absorbance of the CCK-8 solution
was determined at 450 nm. (C) Cells were treated with imatinib (0,
3, 6 and 9 µM) for 24 h, and the surviving cells were analyzed by
CCK-8 assay. (D) Cells were treated with imatinib (0, 3, 6 and 9
µM) for 24 h and harvested to analyze apoptosis by flow cytometry
after Annexin V and 7-AAD staining. The cell population of
apoptosis is indicated (left); the histogram reveals the
percentages of apoptotic cells (right). (E)
BCR-ABL1-transformed pro-B cells were treated or not with
imatinib (6 µM) for 24 h. Western blotting analysis for
phospho-specific antibodies against Foxo1 (S256), Akt (S473), Crkl
(Y207), blots were next stripped and reprobed with antibodies
against total-Foxo1, total-Akt, total Crkl. Phosphorylated protein
levels were quantified by the ImageJ program and normalized to
total form antibodies. The total protein levels were quantified by
the ImageJ program and normalized to actin. (F) The mRNA levels of
BIM, TRAIL, CDKN1B, and GADD45α were measured by
RT-qPCR in BCR-ABL1-transformed pro-B cells treated with
imatinib (0, 6 and 9 µM) for 24 h. GAPDH was used for normalization
of cDNA amounts. Data are representative of at least three
independent experiments. Error bars represent the mean ± SEM.
*P<0.05, **P<0.01 and ***P<0.001. EV, empty vector; CCK-8,
Cell Counting Kit-8; Foxo1, forkhead box protein O1; RT-qPCR,
reverse transcription-quantitative PCR; p-phosphorylated; ns, not
significant. |
Considering the possibility that Bcr-Abl kinase
enhances cell survival by activating the PI3K/Akt pathway to induce
Foxo1 phosphorylation in BCR-ABL1-transformed pro-B cells
(29), western blotting was
performed to examine the phosphorylation of Akt and Foxo1 in
BCR-ABL1-transformed pro-B cells. The data indicated that
the levels of p-Akt and p-Foxo1 were increased in
BCR-ABL1-transformed pro-B cells compared with that in
control cells but were significantly reduced in the presence of 6
µM imatinib (Fig. 1E). Furthermore,
the expression of Foxo1 target genes BIM, TRAIL, CDKN1B, and
GADD45α, which are critical apoptosis and cell-cycle
mediators (30), were upregulated
after imatinib treatment in BCR-ABL1-transformed pro-B cells
(Fig. 1F). Collectively, these
results are consistent with previous studies revealing that Bcr-Abl
kinase stimulates the PI3K/Akt pathway to inactivate Foxo1, leading
to aberrant proliferation of pro-B cells (8,41,42).
Bcr-Abl kinase activity upregulates
Aid expression causing genomic instability in pro-B cells
Previous studies have reported that aberrant Aid
expression depends on Bcr-Abl kinase activity in
BCR-ABL1+ B-ALL (14,43).
Consistent with these findings, the mRNA and protein expression
levels of AID gene were induced in BCR-ABL1-transformed
pro-B cells, while imatinib treatment suppressed Aid expression
(Fig. 2A and B). When Aid was
deleted using CRISPR/Cas9 gene-editing, the protein level of Aid
was significantly reduced in Aid-knockout (KO)
BCR-ABL1-transformed pro-B cells compared to
BCR-ABL1-transformed pro-B cells infected with non-targeting
control viral supernatants (hereinafter named WT cells) (Fig. S2). Cell growth and apoptosis assays
were used to determine the role of Aid in the cell survival of
BCR-ABL1-transformed pro-B cells. By using the CCK-8 assay,
significant inhibition of cell growth was revealed in Aid KO
BCR-ABL1-transformed pro-B cells in the absence of nutrition
for 72 h (Fig. 2C). The proportion
of apoptotic cells was significantly increased in Aid KO cells
compared to WT cells (Fig. 2D).
These results revealed that Aid can enhance
BCR-ABL1-transformed pro-B cell survival.
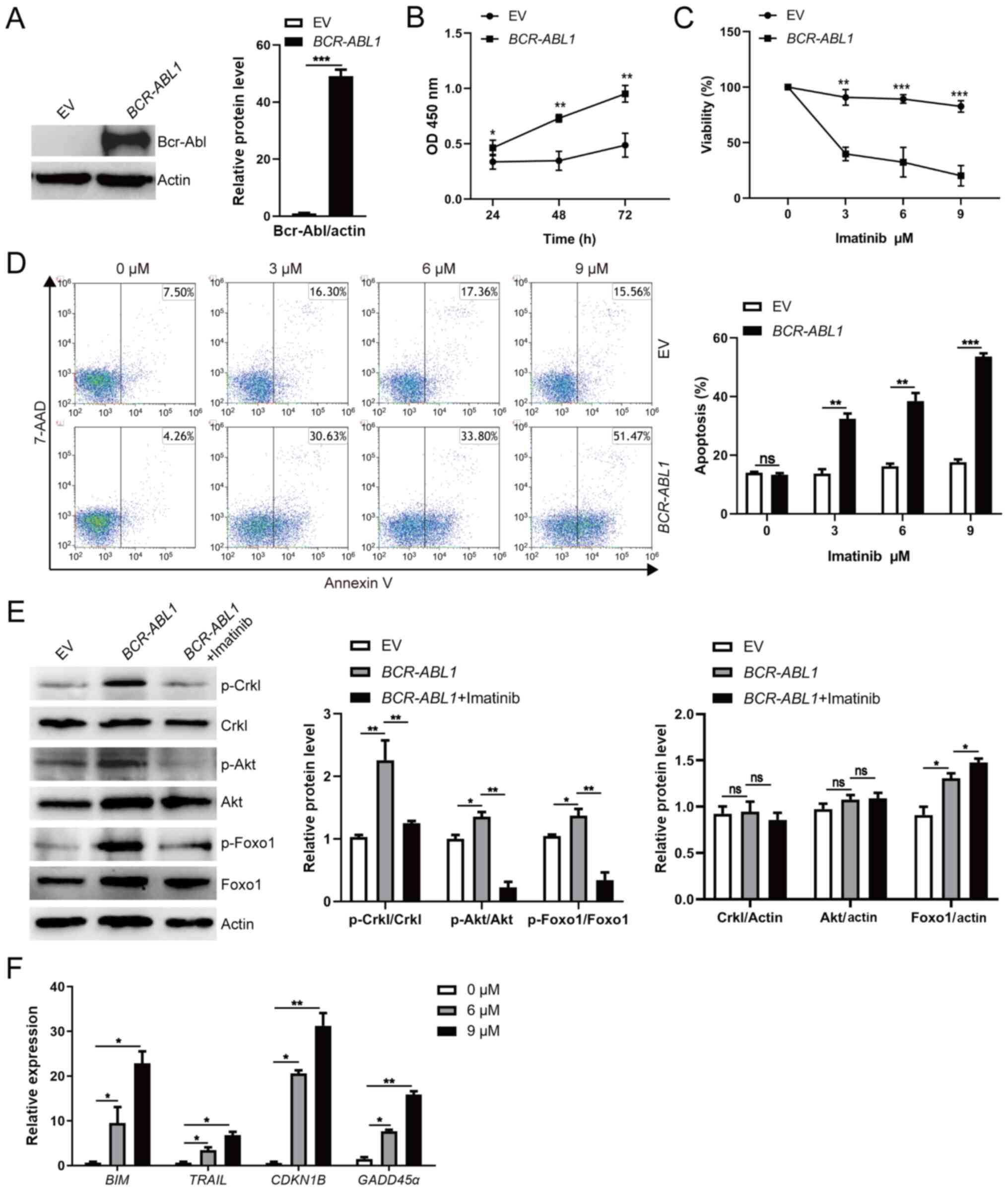
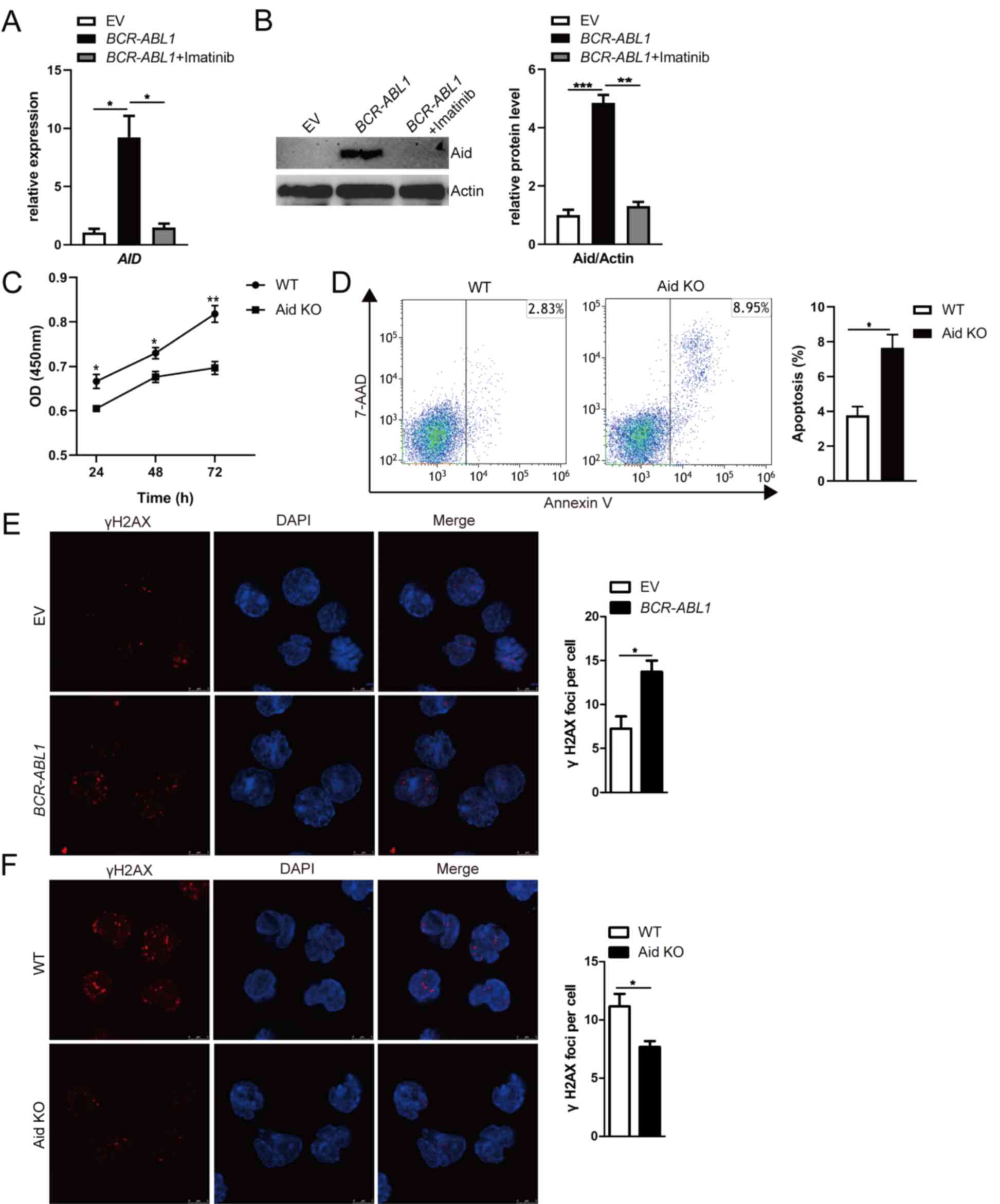 | Figure 2.Aid induces genomic instability
dependent on Bcr-Abl kinase activity in pro-B cells. (A and B)
AID mRNA and Aid protein expression were detected by (A)
RT-qPCR and (B) western blotting; BCR-ABL1-transformed pro-B
cells were treated with or without imatinib (6 µM) for 24 h.
GAPDH was used to normalize cDNA amounts, and actin was
selected as the loading control for western blotting; the protein
levels were quantified by the ImageJ program and normalized to
actin. (C) Cells were cultured in a 96-well plate with
FBS-deficient culture medium, and the absorbance of the CCK-8
solution was determined at 450 nm. (D) Cells were starved for 24 h,
and cell apoptosis was detected by flow cytometry. The cell
population of apoptosis is indicated (left); the histogram reveals
the percentages of apoptotic cells (right). (E and F)
Immunofluorescence of indicated cells stained with γH2AX antibodies
(red) and counterstained with DAPI (blue). Bars, 5 µm. The
representative images of γH2AX foci of these cells are presented
(left), and the histogram revealed the number of γH2AX foci/cell
(right). Data are representative of three independent experiments.
Error bars represent the mean ± SEM. *P<0.05, **P<0.01 and
***P<0.001. Aid, activation-induced cytidine deaminase; CKK-8,
Cell Counting Kit-8; RT-qPCR, reverse transcription-quantitative
PCR; EV, empty vector; WT, wild-type; KO, knockout. |
The Rag mutant cell line was utilized to directly
examine the contribution of dysregulated Aid on genomic
instability. The γH2AX foci, a reliable marker of DNA double-strand
breaks (DSB), was assessed through immunofluorescence to
characterize the genomic instability (44). As anticipated, increased γH2AX foci
per cell were observed in BCR-ABL1-transformed pro-B cells
compared with control cells (Fig.
2E). However, the γH2AX foci per cell were less numerous in
Aid-KO BCR-ABL1-transformed pro-B cells than WT cells
(Fig. 2F), suggesting that Bcr-Abl
kinase activity-mediated aberrant Aid expression contributed to
genomic instability in BCR-ABL1-transformed pro-B cells. It
was concluded that Bcr-Abl kinase activity initiated malignant
transformation characterized by pro-B cell proliferation and
genomic instability.
B-cell differentiation components Blnk
and Foxo1 suppress the survival of BCR-ABL1-transformed pro-B
cells
To investigate B-cell differentiation function of
components in BCR-ABL1-transformed pro-B cells, the
expression of eight key components associated with the pre-BCR
differentiation pathway was analyzed. The results revealed that the
mRNA and protein expression levels of BLNK and FOXO1 were
upregulated in BCR-ABL1-transformed pro-B cells compared to
control cells (Figs. 1E and
3A and B). To ascertain the exact
role of Blnk and Foxo1 in BCR-ABL1-transformed pro-B cells,
the Blnk- or Foxo1-KO BCR-ABL1-transformed pro-B cell lines
were generated using CRISPR/Cas9-based gene-editing technology. The
Blnk KO3 cells and the Foxo1 KO3 cells exhibited the greatest
knockout efficiency (Fig. 3C-3F).
After being treated with imatinib, the depletion of Blnk or Foxo1
in BCR-ABL1-transformed pro-B cells revealed the higher
levels of viable cells and lower levels of apoptotic cells,
indicating that either Blnk or Foxo1 deletion improved the
anti-apoptosis ability (Fig. 4A and
B). In addition, the cell growth ability of Blnk-KO3 or
Foxo1-KO3 BCR-ABL1-transformed pro-B cells was assessed by
the BrdU incorporation assay and the results revealed that
depletion of Blnk or Foxo1 induced a significant increase in the S
phase and a reduction in the G0/G1 phase (Fig. 4C). Collectively, these observations
indicated the inhibitory effects of Blnk and Foxo1 on cell growth
in BCR-ABL1-transformed pro-B cells.
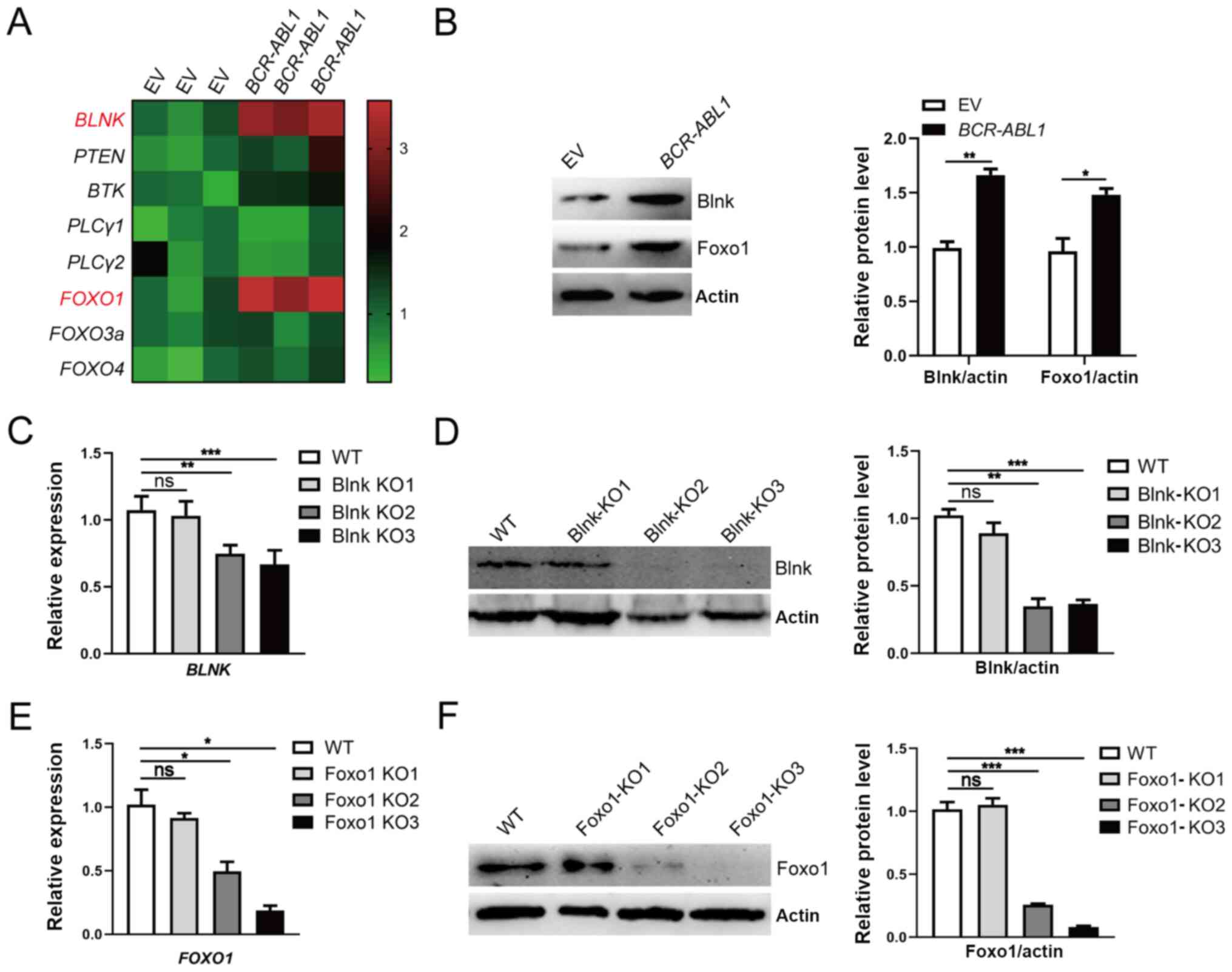 | Figure 3.Upregulation of Blnk and Foxo1
responding to oncogenic stress in BCR-ABL1-transformed pro-B
cells. (A) Heatmap of the transcription levels of B-cell
differentiation components. The gene expression levels were
measured by RT-qPCR, and the relative expression of each gene was
normalized to the GAPDH level and presented as a heatmap.
(B) Western blotting analysis for Blnk and Foxo1 expression. The
protein levels were quantified by the ImageJ program and normalized
to actin. (C and D) Blnk-knockout BCR-ABL1-transformed pro-B
cells was generated by CRISPR/Cas9-mediated deletion using three
guide RNAs (Blnk KO1-KO3). The depleting efficiency was confirmed
by (C) RT-qPCR and (D) western blotting. Blnk KO3 exhibited the
best efficiency for Blnk knockout. GAPDH was used to
normalize cDNA, and the protein levels were quantified by the
ImageJ program and normalized to actin. (E and F) Foxo1-knockout
BCR-ABL1-transformed pro-B cells were generated by
CRISPR/Cas9-mediated deletion using three guide RNAs (Foxo1
KO1-KO3). The depleting efficiency was confirmed by (E) RT-qPCR and
(F) western blotting. Foxo1-KO3 cells exhibited the best depletion
efficiency. GAPDH was used to normalize cDNA, and the
protein levels were quantified by the ImageJ program and normalized
to actin. Data are representative of three independent experiments.
Error bars represent the mean ± SEM. *P<0.05, **P<0.01 and
***P<0.001. Blnk, B-cell linker; Foxo1, forkhead box protein O1;
RT-qPCR, reverse transcription-quantitative PCR; KO, knockout; EV,
empty vector; WT, wild-type; ns, not significant. |
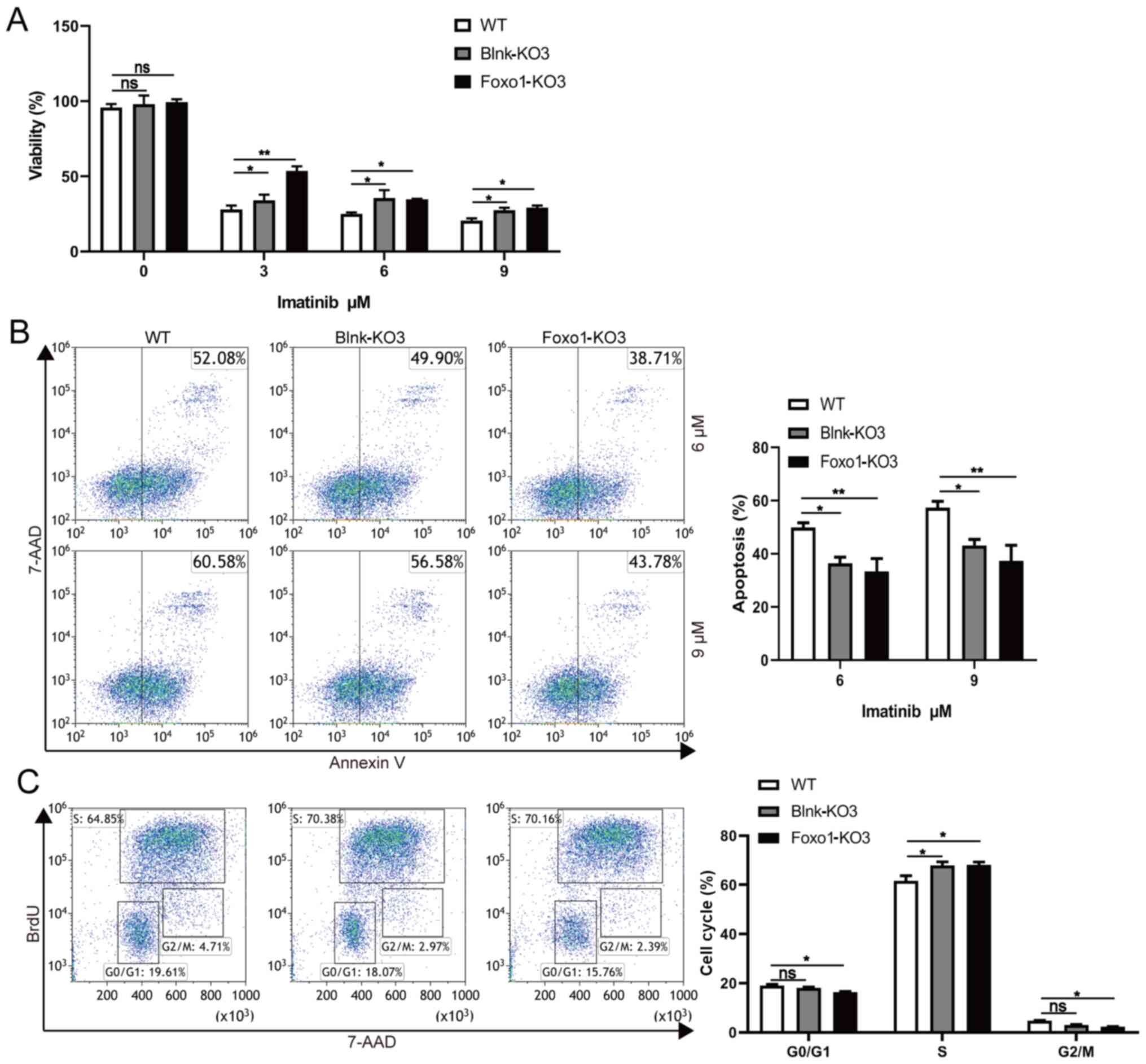 | Figure 4.Blnk or Foxo1 deletion promotes the
cell growth in BCR-ABL1-transformed pro-B cells. (A) WT,
Blnk-KO3 and Foxo1-KO3 BCR-ABL1-transformed pro-B cells were
treated with imatinib (0, 3, 6 and 9 µM) for 24 h, respectively.
After treatment, cell viability was detected by CCK-8 assay. (B)
Flow cytometric analysis of apoptosis cells in WT, Blnk-KO3 and
Foxo1-KO3 BCR-ABL1-transformed pro-B cells treated with
imatinib (6 and 9 µM) for 24 h. The cell population of apoptosis is
indicated (left); the histogram reveals the percentages of
apoptotic cells (right). (C) Flow cytometric analysis of the cell
cycle of WT, Blnk-KO3 and Foxo1-KO3 BCR-ABL1-transformed
pro-B cells. The cell population of G0/G1, S, G2/M phase is
indicated (left); the histogram reveals the percentages of each
phase (right). Data are representative of at least three
independent experiments. Error bars represent the mean ± SEM.
*P<0.05, **P<0.01. Blnk, B-cell linker; Foxo1, forkhead box
protein O1; CCK-8, Cell Counting Kit-8; KO, knockout; WT,
wild-type; ns, not significant. |
Blnk and Foxo1 reduce the genomic
instability of BCR-ABL1-transformed pro-B cells
The effects of Blnk or Foxo1 deletion on genomic
instability were then further determined in
BCR-ABL1-transformed pro-B cells. Increased levels of γH2AX
were detected in Blnk- or Foxo1-deficient
BCR-ABL1-transformed pro-B cells (Fig. 5A). Furthermore, higher AID
transcript levels were accompanied by a corresponding change of
protein in Blnk- or Foxo1-deficient BCR-ABL1-transformed
pro-B cells (Fig. 5B-E). To analyze
the somatic mutations of non-Ig genes dependent on Aid, genomic DNA
was purified from WT, Blnk-KO3, and Foxo1-KO3
BCR-ABL1-transformed pro-B cells. An 852-bp segment located
in the ABL1 kinase domain (exons 3-8), a known target of
Aid, was amplified, sequenced, and analyzed for point mutations
(Fig. S3A-C). The data revealed
that the ABL1 region exhibited higher mutation frequencies
in Blnk-KO3 cells (6.607×10−5 mutations per bp) and
Foxo1-KO3 cells (8.533×10−5 mutations per bp) than that
in WT counterparts (2.171×10−5 mutations per bp)
(Fig. 5F; Table SV). Thus, the results indicated
that Blnk or Foxo1 negatively regulated the genomic instability in
BCR-ABL1-transformed pro-B cells.
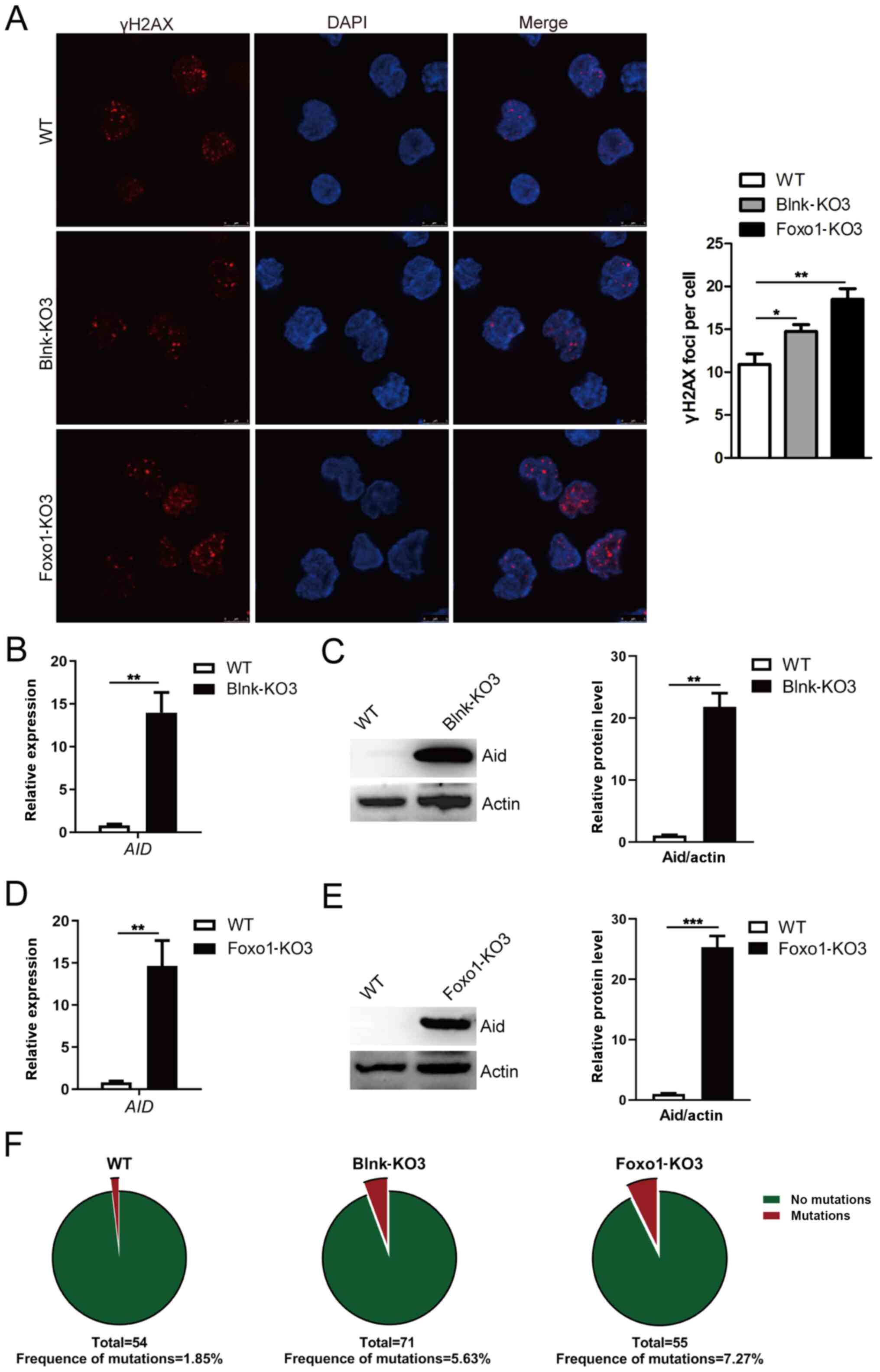 | Figure 5.Blnk or Foxo1 deletion leads to
increased genomic instability in BCR-ABL1-transformed pro-B
cells. (A) Immunofluorescence of WT, Blnk-KO3 and Foxo1-KO3
BCR-ABL1-transformed pro-B cells stained with γH2AX
antibodies (red) and counterstained with DAPI (blue). Bars, 5 µm.
The representative images of γH2AX foci of these cells are
presented (left), and the histogram reveals the number of γH2AX
foci/cell (right). The (B) mRNA and (C) protein levels of
AID gene in WT and Blnk-KO3 BCR-ABL1-transformed
pro-B cells. The (D) mRNA and (E) protein levels of AID in
WT and Foxo1-KO3 BCR-ABL1-transformed pro-B cells. GAPDH was
used for normalization of cDNA; the protein levels were normalized
to actin. (F) WT, Blnk-KO3 and Foxo1-KO3
BCR-ABL1-transformed pro-B cells were subjected to
amplification and sequence analysis for ABL1 kinase domain
mutation; the frequencies of mutation are presented at the bottom
for each sample. Data are representative of at least three
independent experiments. Error bars represent the mean ± SEM.
*P<0.05, **P<0.01 and ***P<0.001. Blnk, B-cell linker;
Foxo1, forkhead box protein O1; Aid, activation-induced cytidine
deaminase; WT, wild-type; KO, knockout. |
Blnk and Foxo1 participate in
regulation of Bcr-Abl kinase in BCR-ABL1-transformed pro-B
cells
As Blnk and Foxo1 decreased cell growth ability and
genomic instability in BCR-ABL1-transformed pro-B cells, it
was hypothesized that Blnk and Foxo1 regulated Bcr-Abl kinase
during the pro-B cell stage. p-Crkl in Blnk-KO3 and Foxo1-KO3
BCR-ABL1-transformed pro-B cells were detected. Notably,
following Blnk or Foxo1 deletion, the level of p-Crkl was
significantly increased (Fig. 6A,
lanes 1, 4, and 7). Notably, imatinib treatment attenuated p-Crkl
in WT, Blnk-KO3, and Foxo1-KO3 BCR-ABL1-transformed pro-B
cells (Fig. 6A, lanes 2, 3, 5, 6, 8
and 9), indicating that either Blnk- or Foxo1-deficient
BCR-ABL1-transformed pro-B cells still responded to imatinib
treatment. Thus, based on these results, it was argued that Blnk
and Foxo1 served as tumor suppressors by inhibiting the Bcr-Abl
kinase activity, hindering tumor initiation and progression in
BCR-ABL1+ B-ALL.
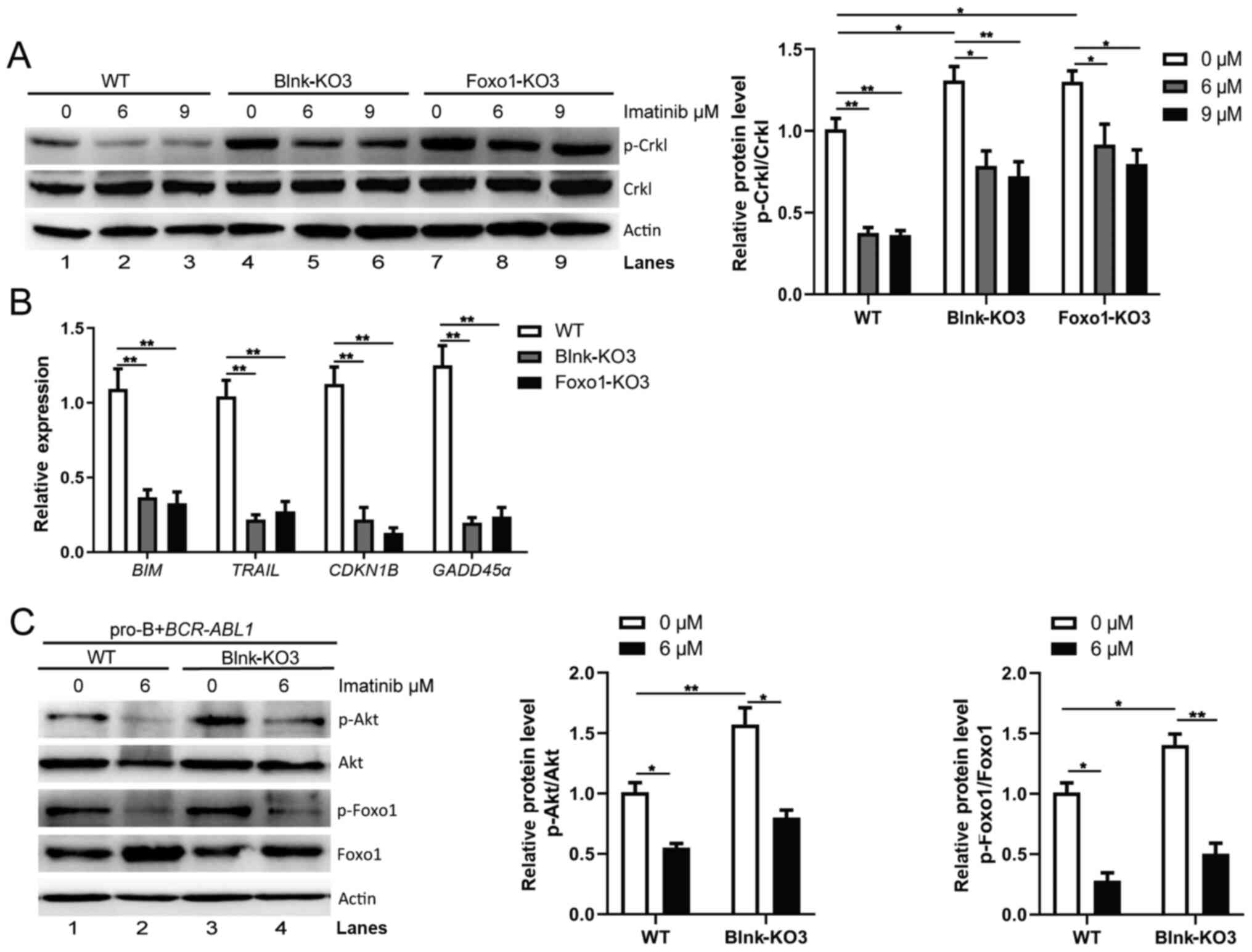 | Figure 6.Blnk and Foxo1 limit the activity of
Bcr-Abl kinase in BCR-ABL1-transformed pro-B cells. (A)
Western blotting analysis of p-Crkl was performed in WT, Blnk-KO3
and Foxo1-KO3 BCR-ABL1-transformed pro-B cells treated with
imatinib (0, 6 and 9 µM) for 24 h, respectively. The p-Crkl levels
were quantified by the ImageJ program and normalized to total Crkl.
Actin was used as a loading control. (B) The transcription of
BIM, TRAIL, CDKN1B, and GADD45α in WT, Blnk-KO3 and
Foxo1-KO3 BCR-ABL1-transformed pro-B cells. Gene mRNA fold
expression values were normalized to the GAPDH. (C) Effects
of deletion of Blnk on p-Foxo1 and p-Akt were measured with
phospho-specific antibodies in WT and Blnk-KO3
BCR-ABL1-transformed pro-B cells treated with imatinib (0
and 6 µM). Blots were next stripped and reprobed with antibodies
against total-Foxo1, total-Akt. Phosphorylated protein levels were
quantified by the ImageJ program and normalized to total form
antibodies. Actin served as a loading control. Data are
representative of three independent experiments. Error bars
represent the mean ± SEM. *P<0.05 and **P<0.01. Blnk, B-cell
linker; Foxo1, forkhead box protein O1; WT, wild-type; KO,
knockout; p-phosphorylated. |
To elucidate the interaction of Blnk and Foxo1 in
BCR-ABL1-transformed pro-B cells, gene expression changes of
Foxo1 target genes were assayed following Blnk or Foxo1 knockout.
The results revealed that the expression levels of Foxo1 target
genes BIM, TRAIL, CDKN1B and GADD45α were lower in
Blnk- or Foxo1-deficient BCR-ABL1-transformed cells than in
the WT cells (Fig. 6B). During
B-cell development, Blnk inhibits Akt activity and activates Foxo
proteins to initiate differentiation (22). Consistent with these findings, the
present results revealed higher levels of p-Foxo1 and p-Akt
(Fig. 6C, lanes 1 and 3) in
Blnk-KO3 BCR-ABL1-transformed pro-B cells compared to the WT
counterparts, and imatinib caused a decrease in p-Foxo1 and p-Akt
levels (Fig. 6C, lanes 2 and 4),
suggesting that Blnk restrains Akt activity-mediated reduction of
Foxo1 phosphorylation in BCR-ABL1-transformed pro-B cells.
The present findings indicated that, in BCR-ABL1-transformed
pro-B cells, Blnk reduced Foxo1 phosphorylation and maintained
Foxo1 activity via PI3K/Akt pathway inactivation, which ultimately
was involved in regulation of the Bcr-Abl kinase.
Although overactive oncogenes induce apoptosis or
senescence, the oncogenes could also suppress these ‘commits
suicide’ processes to allow aberrant proliferation (45). It is worth mentioning that the mRNA
and protein expression levels of BLNK and FOXO1 were
significantly increased in the BCR-ABL1-transformed pro-B
cells treated with imatinib (Figs.
1E and 7A and B). These results
indicated the inhibitory effect of Bcr-Abl kinase on the expression
of Blnk and Foxo1 in BCR-ABL1-transformed pro-B cells.
Therefore, it was inferred that pro-B cells have an intrinsic
antitumor ability, mediated by Blnk and Foxo1 upregulation in
response to oncogenic stress. In contrast, Bcr-Abl kinase inhibited
the expression of Blnk and Foxo1, thus affecting their tumor
suppressor function and fueling malignant transformation of pro-B
cells, marking the first step toward tumorigenesis.
Discussion
BCR-ABL1+ B-ALL defines the most
common and prognostically unfavorable subtype of ALL. Although
recent studies indicated that TKIs combined with conventional
chemotherapy could improve clinical outcomes, the overall prognosis
of BCR-ABL1+ B-ALL patients remains poor due to
high rates of resistance to therapy (46,47).
Therefore, novel, more effective therapy is required to improve the
outcome of BCR-ABL1+ B-ALL. In the present study,
BCR-ABL1-transformed pro-B cells characterized by cell
growth advantage and genomic instability caused by Bcr-Abl kinase
activity were established. It was demonstrated that the expression
of Blnk and Foxo1 were enhanced in response to oncogenic stress of
BCR-ABL1. Blnk and Foxo1 reduced Bcr-Abl kinase activity,
thus weakening the aberrant proliferation and preventing genomic
instability in BCR-ABL1-transformed pro-B cells. The present
findings established the antitumor role of Blnk and Foxo1 in
Bcr-Abl kinase regulation in BCR-ABL1-transformed pro-B
cells.
BCR-ABL1+ B-ALL initiates with
great efficiency in B-cell progenitors, including pro-B cells, but
not in more mature pre-B cells (48). The D345 pro-B cell line was utilized
to simulate the initiation stage of BCR-ABL1+
B-ALL. Consistent with previous studies, the present results
demonstrated that Bcr-Abl kinase activity promoted cell
proliferation by activating the PI3K/Akt signaling pathway to
facilitate the phosphorylation of Foxo1, which was reversed by
imatinib treatment (49,50). In the experimental setting, the Rag1
mutant (D708A) of the pro-B cell line lacked catalytic activity,
which excludes the role of Rag recombinase and focuses on Aid
contribution to genomic instability. Aid induced global genetic
instability by hypermutation on the tumor suppressor and DNA repair
genes, leading to poor clinical outcomes of patients (51–53).
Previous studies have revealed that Bcr-Abl kinase activity
upregulates Aid through transcriptional inhibition of ID2 or
the absence of miR155 (14,43). Similar to this data, the present
results verified that Bcr-Abl kinase induced aberrant Aid
expression to mediate genomic instability in
BCR-ABL1-transformed pro-B cells, while the mechanism of
Bcr-Abl signaling pathway-regulation of Aid expression in
BCR-ABL1-transformed pro-B cells warrants further study.
The present data established a tight association
between Bcr-Abl and the B-cell differentiation components Blnk and
Foxo1 in BCR-ABL1-transformed pro-B cells. Aberrant
activation of oncogenic signaling are necessary to produce
pre-malignant tumors. Concurrently, oncogenic signaling triggers
cell apoptosis or senescence through the Arf/p53/p21, the p16/pRb
and the DDR pathways (54–57). Previously published results have
highlighted the mechanism that BCR-ABL1+ B-ALL
initiation in the B-cell lineage is developmentally regulated; the
oncogene-induced stress leads to higher expression of the tumor
suppressor gene ARF in pre-B cells rather than pro-B cells,
and Arf induces apoptosis and suppresses the occurrence of ALL at
pre-B cell stage (48). However,
the studies left important gaps regarding whether there are tumor
suppressors that regulate B-ALL initiation at the pro-B cell stage.
Studying the specific process in response to oncogenic stress at
the pro-B cell stage, in the present study it was determined that
the expression levels of the B-cell differentiation components Blnk
and Foxo1 were increased after BCR-ABL1 oncogene expression.
In fact, Blnk and Foxo1 are considered initiators of the
differentiation program of the pre-BCR signaling pathway and
critical tumor suppressors (20).
One interpretation for the enhanced expression of Blnk and Foxo1 in
BCR-ABL1-transformed pro-B cells is that Blnk and Foxo1
antagonize the function of Bcr-Abl kinase activity. Consistent with
this argument, Blnk- or Foxo1-deficient BCR-ABL1-transformed
pro-B cells revealed enhanced Bcr-Abl kinase activity, accompanied
by marked cell survival and increased genomic instability mediated
by enhanced Aid expression. It has been reported that the aberrant
expression of Aid initiating SHM on Ig and non-Ig genes is driven
by carcinogenic Bcr-Abl kinase and is related to the poor prognosis
of BCR-ABL1+ leukemia (43) Continuous upregulation of
BCR-ABL1 not only induces the survival advantage of leukemic
cells, but also provides a target for Aid (58,59).
Numerous studies have suggested that Aid promotes the mutations of
BCR-ABL1 to cause imatinib-resistance and leads to the rapid
progression of CML-blast crisis progression (13,60,61).
We question whether Aid may lead to imatinib-resistance in
BCR-ABL1+ B-ALL. Sequencing of the
BCR-ABL1 gene in Blnk-KO3 cells and Foxo1-KO3 cells revealed
increased kinase domain mutation frequencies while it failed to
cause drug resistance, which may be explained by leukemic cell
dependency shift from Bcr-Abl signaling toward the SRC kinase
signaling (62,63). Furthermore, a previous study by Plas
and Thompson revealed that, Foxo1 is a major downstream effect
factor of the PI3K/Akt pathway. The ultilization of Akt inhibitor
LY294002 confirmed that the protein levels of Foxo1 were regulated
by proteasome-dependent Akt activation (64). Akt-mediated Foxo1 phosphorylation
promoting the transport of phosphorylated Foxo1 from the nucleus to
the cytoplasm, and then phosphorylated Foxo1 are rapidly degraded
(4,5,65).
This is a key step for cell proliferation. However, Blnk can change
the fate of cells from proliferation to differentiation (32,66).
In previous studies, the relationship of Blnk/PI3K-Akt/Foxo1 was
clarified by Blnk ectopic expression; Blnk counteracted the
function of the PI3K/Akt pathway and enabled Foxo1 to activate
target genes in the nucleus (23,25).
According to the present data, the expression levels of Foxo1
target genes were lower in Blnk-deficient
BCR-ABL1-transformed cells than in WT cells, and the levels
of p-Foxo1 and p-Akt were increased in Blnk-KO3
BCR-ABL1-transformed pro-B cells, indicating that Blnk
maintained Foxo1 activity by inhibiting the PI3K/Akt pathway in
BCR-ABL1-transformed pro-B cells. We will further explore
the specific interaction of Blnk/PI3K-Akt/Foxo1 in subsequent
studies. Based on these observations, it was concluded that Blnk
and Foxo1 exerted an antitumor role by inhibiting Bcr-Abl kinase in
BCR-ABL1-transformed pro-B cells. However, oncogenes may
bypass and/or inhibit these mechanisms, further fueling tumor cell
survival (45,67). The present results indicated that
Bcr-Abl kinase activity inhibition results in stronger upregulation
of the tumor suppressor genes Blnk and Foxo1, demonstrating that
Bcr-Abl kinase reversely inhibited Blnk and Foxo1 expression in
pro-B cells. The present data indicated a network of interaction
between tumor suppressors and Bcr-Abl, which requires further study
on the more detailed regulatory mechanism.
In conclusion, the present study provided evidence
of the pivotal function of Blnk and Foxo1 for Bcr-Abl kinase
regulation in BCR-ABL1-transformed pro-B cells. The results
provided novel insights into the roles of Blnk and Foxo1 tumor
suppressors in BCR-ABL1-transformed pro-B cells, which will
aid in developing new therapeutic strategies for
BCR-ABL1+ B-ALL.
Supplementary Material
Supporting Data
Acknowledgments
The authors would like to thank Dr David G. Schatz
(Yale University, New Haven, USA) for providing D345 pro-B and BD
pre-B v-abl cell lines, and we thank Dr Junjie Zhang (University of
Southern California, Los Angeles, USA) for providing
pL-CRISPR.EFS.PAC plasmids. The authors also thank the Core
Facilities Sharing Platform of Xi'an Jiaotong University for
providing the confocal microscope (Leica TCS SP8 STED 3X; Leica
Microsystems, Inc.).
Funding
This work was supported by grants (grant nos.
81670157 and 81801581) from the National Natural Scientific
Foundation of China (to YJ and YD) and by a grant (grant no.
2016JZ030) from the Natural Scientific Foundation of Shaanxi (to
YJ).
Availability of data and materials
The datasets analyzed during the present study are
available from the corresponding author upon reasonable
request.
Author's contributions
PZ and YJ conceived and designed the experiments.
PZ and YW performed the majority of experiments and wrote the
manuscript. MQ and DL performed the retroviral production and
transfection. WOO was involved in manuscript review and DNA
extraction. MY and ZL performed the CCK-8 assay. CL and YM revised
the work critically for important intellectual content. YD and YJ
were major contributors in funding acquisition and assisted in the
data analysis. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
B-ALL
|
B-acute lymphoblastic leukemia
|
|
BCR-ABL1+ B-ALL
|
BCR-ABL1-positive B-cell acute
lymphoblastic leukemia
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
pre-BCR
|
pre-B cell receptor
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
EV
|
empty vector
|
|
CCK-8
|
Cell Counting Kit-8
|
|
p-
|
phosphorylated
|
|
Aid
|
activation-induced cytidine
deaminase
|
|
Blnk
|
B-cell linker
|
|
Foxo1
|
forkhead box protein O1
|
|
WT
|
wild-type
|
|
KO
|
knockout
|
|
ns
|
not significant
|
References
|
1
|
El Fakih R, Jabbour E, Ravandi F,
Hassanein M, Anjum F, Ahmed S and Kantarjian H: Current paradigms
in the management of Philadelphia chromosome positive acute
lymphoblastic leukemia in adults. Am J Hematol. 93:286–295. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee HJ, Thompson JE, Wang ES and Wetzler
M: Philadelphia chromosome-positive acute lymphoblastic leukemia:
current treatment and future perspectives. Cancer. 117:1583–1594.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vinhas R, Lourenço A, Santos S, Lemos M,
Ribeiro P, de Sousa AB, Baptista PV and Fernandes AR: A novel
BCR-ABL1 mutation in a patient with Philadelphia
chromosome-positive B-cell acute lymphoblastic leukemia.
OncoTargets Ther. 11:8589–8598. 2018. View Article : Google Scholar
|
|
4
|
Köhrer S, Havranek O, Seyfried F, Hurtz C,
Coffey GP, Kim E, Ten HE, Jäger U, Vanura K and O'Brien S: Pre-BCR
signaling in precursor B-cell acute lymphoblastic leukemia
regulates PI3K/AKT, FOXO1, and MYC, and can be targeted by SYK
inhibition. Leukemia. 30:1246–1254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neshat MS, Raitano AB, Wang HG, Reed JC
and Sawyers CL: The survival function of the Bcr-Abl oncogene is
mediated by Bad-dependent and -independent pathways: Roles for
phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 20:1179–1186.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roumiantsev S, Aos IED, Varticovski L,
Ilaria RL and Etten RAV: The Src homology 2 domain of Bcr/Abl is
required for efficient induction of chronic myeloid leukemia–like
disease in mice but not for lymphoid leukemogenesis or activation
of phosphatidylinositol 3-kinase. Blood. 97:4–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheng Z, Ma L, Sun JE, Zhu LJ and Green
MR: BCR-ABL suppresses autophagy through ATF5-mediated regulation
of mTOR transcription. Blood. 118:2840–2848. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and Mccubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iacobucci I, Lonetti A, Messa F, Ferrari
A, Cilloni D, Soverini S, Paoloni F, Arruga F, Ottaviani E,
Chiaretti S, et al: Different isoforms of the B-cell mutator
activation-induced cytidine deaminase are aberrantly expressed in
BCR-ABL1-positive acute lymphoblastic leukemia patients. Leukemia.
24:66–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsai AG, Lu H, Raghavan SC, Muschen M,
Hsieh CL and Lieber MR: Human chromosomal translocations at CpG
sites and a theoretical basis for their lineage and stage
specificity. Cell. 135:1130–1142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swaminathan S, Klemm L, Park E,
Papaemmanuil E, Ford A, Kweon SM, Trageser D, Hasselfeld B, Henke
N, Mooster J, et al: Mechanisms of clonal evolution in childhood
acute lymphoblastic leukemia. Nat Immunol. 16:766–774. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dong Y, Liu F, Wu C, Li S, Zhao X, Zhang
P, Jiao J, Yu X, Ji Y and Zhang M: Illegitimate RAG-mediated
recombination events are involved in IKZF1 Δ3-6 deletion in
BCR-ABL1 lymphoblastic leukaemia. Clin Exp Immunol. 185:320–331.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gruber TA, Mi SC, Sposto R and Müschen M:
Activation-induced cytidine deaminase accelerates clonal evolution
in BCR-ABL1-driven B cell lineage acute lymphoblastic leukemia.
Cancer Res. 70:7411–7420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Klemm L, Duy C, Iacobucci I, Kuchen S,
Levetzow GV, Feldhahn N, Henke N, Li Z, Hoffmann TK and Kim YM: The
B cell mutator AID promotes B lymphoid blast crisis and drug
resistance in chronic myeloid leukemia. Cancer Cell. 16:232–245.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Messina M, Chiaretti S, Iacobucci I,
Tavolaro S, Lonetti A, Santangelo S, Elia L, Papayannidis C,
Paoloni F, Vitale A, et al: AICDA expression in BCR/ABL1-positive
acute lymphoblastic leukaemia is associated with a peculiar gene
expression profile. Br J Haematol. 152:727–732. 2015. View Article : Google Scholar
|
|
16
|
Robbiani DF, Bunting S, Feldhahn N,
Bothmer A, Camps J, Deroubaix S, Mcbride KM, Klein IA, Stone G,
Eisenreich TR, et al: AID produces DNA double-strand breaks in
non-Ig genes and mature B cell lymphomas with reciprocal chromosome
translocations. Mol Cell. 36:631–641. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fielding AK: Treatment of Philadelphia
chromosome-positive acute lymphoblastic leukemia in adults: a
broader range of options, improved outcomes, and more therapeutic
dilemmas. Am Soc Clin Oncol Educ Book. 35:e352–9. 2015. View Article : Google Scholar
|
|
18
|
Xing H, Yang X, Liu T, Lin J, Chen X and
Gong Y: The study of resistant mechanisms and reversal in an
imatinib resistant Ph+ acute lymphoblastic leukemia cell line. Leuk
Res. 36:509–513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Donahue AC and Fruman DA: Proliferation
and survival of activated B cells requires sustained antigen
receptor engagement and phosphoinositide 3-kinase activation. J
Immunol. 170:5851–5860. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Übelhart R, Werner M and Jumaa H: Assembly
and function of the precursor B-cell receptor. Curr Top Microbiol
Immunol. 393:3–25. 2016.PubMed/NCBI
|
|
21
|
Burgering B: A brief introduction to
FOXOlogy. Oncogene. 27:2258–2262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Herzog S, Reth M and Jumaa H: Regulation
of B-cell proliferation and differentiation by pre-B-cell receptor
signalling. Nat Rev Immunol. 9:195–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koretzky GA, Abtahian F and Silverman MA:
SLP76 and SLP65: Complex regulation of signalling in lymphocytes
and beyond. Nat Rev Immunol. 6:67–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garg M, Wahid M and Khan F: Regulation of
peripheral and central immunity: Understanding the role of Src
homology 2 domain-containing tyrosine phosphatases, SHP-1 &
SHP-2. Immunobiology. 225:1518472020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ochiai K, Maienschein-Cline M, Mandal M,
Triggs JR, Bertolino E, Sciammas R, Dinner AR, Clark MR and Singh
H: A self-reinforcing regulatory network triggered by limiting IL-7
activates pre-BCR signaling and differentiation. Nat Immunol.
13:300–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feldhahn N, Klein F, Mooster JL, Hadweh P,
Sprangers M, Wartenberg M, Bekhite MM, Hofmann WK, Herzog S, Jumaa
H, et al: Mimicry of a constitutively active pre–B cell receptor in
acute lymphoblastic leukemia cells. J Exp Med. 201:1837–1852. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hickey FB, England K and Cotter TG:
Bcr-Abl regulates osteopontin transcription via Ras, PI-3K, aPKC,
Raf-1, and MEK. J Leukoc Biol. 78:289–300. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim E, Koehrer S, Wang Z, O'Brien S,
Wierda WG, Thomas DA, Estrov Z, Kantarjian HM, Lannutti B and Davis
RE: The PI3K delta inhibitor idelalisib interferes with pre-B cell
receptor signaling in acute lymphoblastic leukemia (ALL): a new
therapeutic concept. Blood. 122:26322013. View Article : Google Scholar
|
|
29
|
Pellicano F, Scott MT, Helgason GV,
Hopcroft LE, Allan EK, Aspinall-O'Dea M, Copland M, Pierce A,
Huntly BJ, Whetton AD, et al: The antiproliferative activity of
kinase inhibitors in chronic myeloid leukemia cells is mediated by
FOXO transcription factors. Stem Cells. 32:2324–2337. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Szydlowski M, Kiliszek P, Sewastianik T,
Jablonska E, Bialopiotrowicz E, Gorniak P, Polak A, Markowicz S,
Nowak E, Grygorowicz MA, et al: FOXO1 activation is an effector of
SYK and AKT inhibition in tonic BCR signal-dependent diffuse large
B-cell lymphomas. Blood. 127:739–748. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lowe SW, Cepero E and Evan G: Intrinsic
tumour suppression. Nature. 432:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Flemming A, Brummer T, Reth M and Jumaa H:
The adaptor protein SLP-65 acts as a tumor suppressor that limits
pre-B cell expansion. Nat Immunol. 4:38–43. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hendriks RW and Kersseboom R: Involvement
of SLP-65 and Btk in tumor suppression and malignant transformation
of pre-B cells. Semin Immunol. 18:67–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kersseboom R, Middendorp S, Dingjan GM,
Dahlenborg K, Reth M, Jumaa H and Hendriks RW: Bruton's tyrosine
kinase cooperates with the B cell linker protein SLP-65 as a tumor
suppressor in Pre-B cells. J Exp Med. 198:91–98. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ji Y, Resch W, Corbett E, Yamane A,
Casellas R and Schatz DG: The in vivo pattern of binding of RAG1
and RAG2 to antigen receptor loci. Cell. 141:419–431. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen
Y, Sullivan C, Cerny J, Hutchinson L, Higgins A, et al: The Blk
pathway functions as a tumor suppressor in chronic myeloid leukemia
stem cells. Nat Genet. 44:861–871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Joung J, Konermann S, Gootenberg JS,
Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE and Zhang F:
Genome-scale CRISPR-Cas9 knockout and transcriptional activation
screening. Nat Protoc. 12:828–863. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et
al: Genome-scale CRISPR-Cas9 knockout screening in human cells.
Science. 343:84–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
de Jong R, ten Hoeve J, Heisterkamp N and
Groffen J: ten HJ, Heisterkamp N and Groffen J: Tyrosine 207 in
CRKL is the BCR/ABL phosphorylation site. Oncogene. 14:507–513.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang R, Liu H, Chen Y, He Y, Kang Q, Tu
S, He Y, Zhou X, Wang L, Yang J, et al: EPS8 regulates
proliferation, apoptosis and chemosensitivity in BCR-ABL positive
cells via the BCR-ABL/PI3K/AKT/mTOR pathway. Oncol Rep. 39:119–128.
2018.PubMed/NCBI
|
|
42
|
Zhou Q, Chen Y, Chen X, Zhao W, Zhong Y,
Wang R, Jin M, Qiu Y and Kong D: In Vitro Antileukemia Activity of
ZSTK474 on K562 and Multidrug Resistant K562/A02 Cells. Int J Biol
Sci. 12:631–638. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Giebel B: Activation-induced cytidine
deaminase acts as a mutator in BCR-ABL1-transformed acute
lymphoblastic leukemia cells. J Exp Med. 204:1157–1166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Valdiglesias V, Giunta S, Fenech M, Neri M
and Bonassi S: γH2AX as a marker of DNA double strand breaks and
genomic instability in human population studies. Mutat Res.
753:24–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Larsson LG: Oncogene- and tumor suppressor
gene-mediated suppression of cellular senescence. Semin Cancer
Biol. 21:367–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bassan R, Rohatiner AZ, Lerede T, Di BE,
Rambaldi A, Pogliani E, Rossi G, Fabris P, Morandi S, Casula P, et
al: Role of early anthracycline dose-intensity according to
expression of Philadelphia chromosome/BCR-ABL rearrangements in
B-precursor adult acute lymphoblastic leukemia. Hematol J Off J Eur
Haematol Assoc. 1:226–234. 2000.
|
|
47
|
Daenen S: Imatinib in Philadelphia
chromosome positive acute lymphoblastic leukemia (ALL). Blood.
102:97–100. 2003.
|
|
48
|
Signer RA, Montecino-Rodriguez E, Witte ON
and Dorshkind K: Immature B-cell progenitors survive oncogenic
stress and efficiently initiate Ph+ B-acute lymphoblastic leukemia.
Blood. 116:2522–2530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gishizky ML: Molecular mechanisms of
Bcr-Abl-induced oncogenesis. Cytokines Mol Ther. 2:251–261.
1996.PubMed/NCBI
|
|
50
|
Skorski T, Kanakaraj P,
Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM,
Perussia B and Calabretta B: Phosphatidylinositol-3 kinase activity
is regulated by BCR/ABL and is required for the growth of
Philadelphia chromosome-positive cells. Blood. 86:726–736. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Greaves MF and Wiemels J: Origins of
chromosome translocations in childhood leukaemia. Nat Rev Cancer.
3:639–649. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lieber MR, Yu K and Raghavan SC: Roles of
nonhomologous DNA end joining, V(D)J recombination, and class
switch recombination in chromosomal translocations. DNA Repair
(Amst). 5:1234–1245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mullighan CG, Miller CB, Radtke I,
Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui
CH, et al: BCR-ABL1 lymphoblastic leukaemia is characterized by the
deletion of Ikaros. Nature. 453:110–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Collado M and Serrano M: Senescence in
tumours: Evidence from mice and humans. Nat Rev Cancer. 10:51–57.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
d'Adda di Fagagna F: Living on a break:
cellular senescence as a DNA-damage response. Nat Rev Cancer.
8:512–522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Haigis KM and Sweet-Cordero A: New
insights into oncogenic stress. Nat Genet. 43:177–178. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sharma N, Magistroni V, Piazza R, Citterio
S, Mezzatesta C, Khandelwal P, Pirola A and Gambacorti-Passerini C:
BCR/ABL1 and BCR are under the transcriptional control of the MYC
oncogene. Mol Cancer. 14:1322015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Clapper E, Wang S, Raninga PV, Di TG and
Tonissen KF: Cross-talk between Bcr-abl and the thioredoxin system
in chronic myeloid leukaemia: implications for CML treatment.
Antioxidants. 9:92020. View Article : Google Scholar
|
|
60
|
Melo JV and Barnes DJ: Chronic myeloid
leukaemia as a model of disease evolution in human cancer. Nat Rev
Cancer. 7:441–453. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
McCarron SL, Maher K, Kelly J, Ryan MF and
Langabeer SE: Rapid evolution to blast crisis associated with a
Q252H ABL1 kinase domain mutation in e19a2 BCR-ABL1 chronic myeloid
leukaemia. Case Rep Hematol. 2013:4907402013.PubMed/NCBI
|
|
62
|
Popp C, Dean W, Feng S, Cokus SJ, Andrews
S, Pellegrini M, Jacobsen SE and Reik W: Genome-wide erasure of DNA
methylation in mouse primordial germ cells is affected by AID
deficiency. Nature. 463:1101–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Blake RA, Broome MA, Liu X, Wu J, Gishizky
M, Sun L and Courtneidge SA: SU6656, a selective src family kinase
inhibitor, used to probe growth factor signaling. Mol Cell Biol.
20:9018–9027. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Plas DR and Thompson CB: Akt activation
promotes degradation of tuberin and FOXO3a via the proteasome. J
Biol Chem. 278:12361–12366. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Coffer PJ and Burgering BM: Forkhead-box
transcription factors and their role in the immune system. Nat Rev
Immunol. 4:889–899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Meixlsperger S, Köhler F, Wossning T,
Reppel M, Müschen M and Jumaa H: Conventional light chains inhibit
the autonomous signaling capacity of the B cell receptor. Immunity.
26:323–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schmitt CA: Cellular senescence and cancer
treatment. Biochim Biophys Acta. 1775:5–20. 2007.PubMed/NCBI
|