Introduction
Breast cancer is the most commonly diagnosed cancer
in women (1). Despite the
considerable progress in cancer therapy in preceding years,
chemotherapy, radiotherapy, and surgery remain the main approaches
to treating breast cancer. However, cancer cells frequently use
processes to escape cell death activated owing to chemotherapy
(2). Furthermore, resistance occurs
in endocrine therapy including tamoxifen for breast cancers,
involving the same pathways as chemotherapeutic agents (3–6). In
addition, nature-derived anticancer agents such as doxorubicin,
actinomycin D, and bleomycin confer resistance to cells. Thus, it
is important to develop strategies to oppose this obstacle
(7–9).
One main characteristic of cancer is metabolic
reprogramming and progressive dependence on glycolysis (10). This reliance on glycolysis leads to
the accumulation of impaired glycosylated and misfolded proteins in
the lumen of the endoplasmic reticulum (ER stress) and the
following unfolded protein response (UPR) (11,12).
In addition, the proliferative feature of cancer cells augments ER
stress compared to the normal cells (13).
In fact, transformed cells hijack UPR as a survival
strategy in the stressful microenvironment of tumors. Several
studies have demonstrated the importance of UPR signaling in
chemoresistance and tumor growth (14). Clinical indications have revealed
that the UPR-driven chemoresistance occurs exclusively in breast
cancers (15–18). UPR activation gives rise to
chemoresistance by modifying drug targets, increasing efflux pump
expression, drug detoxification and activating pro-survival and
anti-apoptotic gene expression (19–21).
Numerous studies have suggested that UPR signaling
molecules interact with well-known tumor suppressors and oncogenes
such as binding immunoglobulin protein (BiP; also known as GRP78)
and 14-3-3ζ. In fact, some cancer cells display constant activation
of the IRE-1α-XBP1 pathway (UPR survival pathway) by overexpression
of BiP, which is anti-apoptotic (22–25).
Anticancer drugs are an additional source of stress
(especially ER stress which promotes UPR) (26), activating the autophagy in tumor
cells as a mechanism of resistance to chemotherapy (20,27).
Autophagy is a stress-adaptive self-digestive process in which
cellular components are enveloped within autophagosomes and
degraded by lysosomal hydrolyses to eliminate misfolded proteins,
reconstruct ER homeostasis and provide cells with integral
nutrients to facilitate cancer cell survival and manage the
metabolic stresses caused by anticancer treatments (20).
BiP is the master regulator of the ER stress. In
benign breast lesions, the expression levels of BiP are low while
as it progresses into breast cancer, BiP expression greatly
increases (22,28,29).
Solid tumor cells secrete BiP to activate various cellular
processes such as tumor cell proliferation, angiogenesis,
differentiation of bone marrow-derived mesenchymal stem cells, and
the polarization of tumor-associated macrophages (30). An increased level of
membrane-resident BiP was discovered in numerous types of cancers,
including breast, ovarian, prostate, hepatocellular, gastric,
melanoma, fibroblastoma, and renal cancers (31,32).
Notably, increased expression of BiP could function in the
protection of dormant tumor cells from stress problems, including
chemotherapy (33) acting as a
chaperone for aggregation-prone misfolded proteins leading to their
degradation by autophagy. The mechanisms by which BiP upregulation
is involved in chemoresistance is not fully understood. However, it
appears that its overexpression serves an antiapoptotic role by
driving the acute requirement of protein synthesis to support
different cellular functions such as tumor proliferation,
migration, and differentiation (34–39).
Numerous studies have revealed that BiP expression is linked with
resistance of cancer cells to cytotoxic drugs, increased tumor
progression, and worse patient prognosis (40), suggesting that its knockdown confers
anticancer drug sensitization to the tumor cell (23,41–43).
In turn, BiP expression increases due to treatment with several
anticancer drugs (23,44–46).
Proteins of the 14-3-3 family play critical roles in
several regulatory mechanisms in which they function by binding to
the phospho-serine or the phospho-threonine motif in various
proteins. They act in signal transduction, apoptosis, DNA repair,
and malignant transformation (47–51).
14-3-3ζ is one of the members of this family, and increased
expression has been revealed in early stages of several cancers
including but not limited to liver cancer, head and neck squamous
cell carcinoma, skin cancer, lung cancer and breast carcinoma
(52–62).
More than 40% of advanced breast tumor tissues
exhibit a high expression of 14-3-3ζ (57). 14-3-3ζ upregulation has been
revealed to be directly associated with higher grades of tumors and
a more advanced pathologic stage (58). Positive expression of 14-3-3ζ in
early stages of cancer, such as hyperplasia, dysplasia and
carcinoma, suggest that alteration in its expression is associated
to the onset of tumorigenesis (58). Cell survival and
anchorage-independent growth are intensified in breast cancer cell
lines through 14-3-3ζ overabundance, whereas its knockdown results
in cell sensitization to stress-induced apoptosis and decreased
tumor xenograft size (54).
Apoptosis signal-regulating kinase 1 (Ask1), a
serine-threonine-protein kinase in the JNK-apoptosis pathway of UPR
(63–65), can be negatively activated by the
phosphorylation of 14-3-3ζ during excess ER stress as well as
oxidant stress which is the case in the tumor environment. This
association may highly weaken ASK1 potential to activate JNK and
apoptosis signaling (66–69).
Cisplatin, a chemotherapeutic agent, is used to
treat various malignancies, including lung, ovary and breast. There
are several studies on the effects of cisplatin on the MCF-7 cell
line (70–73). However, cisplatin exhibits limited
therapeutic efficacy. Several studies have reported that cisplatin
activates autophagy which acts as a survival factor to counteract
cisplatin-induced apoptosis (72).
Therefore, more effective therapeutic options are required.
The present study promoted the apoptotic pathway of
UPR (ASK1/JNK axis) through BiP knockdown according to previous
studies (23,74–76).
14-3-3ζ has been revealed to inactivate ASK1 and prevent apoptosis
triggers (66–69). Previous research has revealed that
14-3-3ζ dissociation from ASK1 may accelerate cell death in cancer
(66). Thus, in the present study
it was hypothesized that the concurrent knockdown of BiP and
14-3-3ζ may have a superior impact on cisplatin sensitization of
MCF-7, as a human breast cancer carcinoma cell line, compared to
knockdown of either BiP or 14-3-3ζ as previously reported.
Materials and methods
Cell culture and reagents
The MCF-7 breast cancer cell line was acquired from
the American Type Culture Collection (ATCC). The cells were
cultured in DMEM medium supplemented with 10% fetal bovine serum,
penicillin (100 U/ml), and streptomycin (100 mg/ml; all from Gibco;
Thermo Fisher Scientific, Inc.). The cell line was cultured at 37°C
in a humified incubator supplying 5% CO2. Cells were
passaged every 3-4 days and treated in the exponential growth
phase. The MCF-7 cells were tested for mycoplasma contamination by
MycoAlet Mycoplasma Detection Kit (Lonza Group, Ltd.).
Cell transfection
To silence BiP and 14-3-3ζ genes,
small interfering (si)RNAs (Table
I) against BiP and 14-3-3ζ as well as scrambled siRNA were
purchased from GE Healthcare Dharmacon, Inc. (97% purity) and used
at a final concentration of 20 nM for MCF-7 cell transfection using
Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.).
Briefly, MCF-7 cells (5×105 cells/well) were plated on
6-well plates containing an antibiotic-free medium. They were
incubated overnight at 37°C. To perform transfection, Lipofectamine
RNAiMAX and siRNA (3:1) were added in serum-free medium. The
mixture was incubated at room temperature for 20 min to allow the
siRNA-lipofectamine complex to be formed. The mixture was then
added to the cells in a proper volume of antibiotic- and serum-free
DMEM to obtain a final concentration of 20 nM for each siRNA. After
incubation for 6 h at 37°C, DMEM supplemented with serum was added
to the cells and were incubated with siRNA/Lipofectamine at 37°C
for 72 h.
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| siRNA | Sense | Antisense |
|---|
| BiP |
5′-PGGAGCGAUUGAUACUAGAdTdT-3′ |
5′-PUCUAGUAUCAAUGCGCUCCdTdT-3′ |
| 14-3-3ζ |
5′-PAAAGUUCUUGAUCCCCAUGC-3′ |
5′-PAUUGGGGAUCAAGAACUUUGC-3′ |
| Scrambled |
5′-PGAAGUAGUACCGUUGUAGUAdTdT-3′ |
5′-PUACUACAACGGUACUACUUCdTdT-3′ |
RNA purification and reverse
transcription-quantitative PCR (RT-qPCR)
To extract total cellular RNA, Ribospin II RNA
extraction kit (GeneAll Biotechnology Co., Ltd.) was used following
the manufacturers instructions. The total RNA concentration and
purity were quantified by applying the NanoDrop spectrophotometer
(ND-1000; Thermo Fisher Scientific, Inc.). Complementary DNA was
synthesized from 1 µg of total RNA employing RevertAid First
Stranded cDNA Synthesis kit (Thermo Fisher Scientific, Inc.)
according to the manufacturers protocol. RT-qPCR for RNA expression
quantification was carried out on Applied Biosystems (StepOnePlus;
Thermo Fisher Scientific, Inc.) using QuantiTect SYBR-Green (Qiagen
GmbH) with a total reaction volume of 10 µl and the initial
denaturation at 95°C for 15 min, followed by 40 cycles of PCR at
95°C for 15 sec, 63°C for 10 sec, 72°C for 20 sec. Comparative
analysis by 2(−∆∆Cq) was used to compare the expression
of target genes (77). Relative
expression software tool (REST) was applied for comparison
(78), and the statistical analysis
of relative expression results in real-time PCR. The related
primers (Ki-67, cyclin D1, Rb, p21, BiP and 14-3-3ζ) (Table II) were obtained from
Sigma-Aldrich; Merck KGaA. GAPDH served as the reference gene. All
reactions were performed in triplicate.
| Table II.Primer sequences (5→3). |
Table II.
Primer sequences (5→3).
| Primer | Forward | Reverse |
|---|
| Ki-67 |
GAAAGAGTGGCAACCTGCCTTC |
GCACCAAGTTTTACTACATCTGCC |
| Cyclin D1 |
TCTACACCGACAACTCCATCCG |
TCTGGCATTTTGGAGAGGAAGTG |
| Rb |
CAGAAGGTCTGCCAACACCAAC |
TTGAGCACACGGTCGCTGTTAC |
| p21 |
CCAGCATGACAGATTTCTACC |
AGACACACAAACTGAGACTAAGG |
| BiP/GRP78 |
TGCAGCAGGACSATCAAGTTC |
AGTTCCAGCGTCTTTGGTTG |
| 14-3-3ζ |
ACTGCTGAGCCCGTCCGTC |
TCAGCCTGCTCGGCCAGTT |
| GAPDH |
ACCTGACCTGCCGTCTAGAAAA |
TGTCGCTGTTGAAGTCAGAGGA |
Cell viability assay
Cell viability was quantified by colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(≥97% purity; Sigma-Aldrich; Merck KGaA) assay. Basically, cleavage
of tetrazolium salts by mitochondrial succinate reductase, which is
active in viable cells, leads to formazan dye formation. Cisplatin
resistance was induced via culturing the cells in solutions by
increasing the concentration of cisplatin (from 0.03 to 15 µM) for
3 months. To identify viability of MCF-7 cells in response to
cisplatin (99.9% purity; Sigma-Aldrich; Merck KGaA) exposure, the
cells were seeded at a concentration of 8×103 cells/well
in 96-well plates. They were dedicated time to attaching for 8 h
and treated with cisplatin for 48 h. Next, MTT (5 mg/ml) was added
to each well, and the cells were incubated for 2 h at 37°C. DMSO
was added to the wells to dissolve the formed formazan crystals.
The absorbance was then measured at 570 nm, using a plate reader
(Cytation Multi-Mode; BioTek Instruments, Inc.). Cell viability was
calculated as the percentage of viable treated cells against the
control group. Each treatment of cisplatin or untreated control was
performed in triplicate.
Cell lysate and immunoblotting
The MCF-7 cells were seeded into 6-well plates at
5×105 cells/well and incubated overnight at 37°C before
being subjected to transfection with BiP siRNA, 14-3-3ζ siRNA and
BiP+14-3-3ζ siRNAs for 72 h at 37°C. The cells were then treated
with cisplatin for 48 h. Cold RIPA lysis buffer [150 mM NaCl, 50 mM
Tris, 1 mol/l EDTA, Triton X-100, 0.1% sodium dodecyl sulphate, 1
mol/l phenyl methyl sulfonyl fluoride (pH 8.0): Merck KGaA] and
fresh protease inhibitor (Sigma-Aldrich; Merck KGaA) were utilized
to produce the whole cell lysate. The extracts were sonicated for
15 sec followed by the centrifugation at 8,600 × g for 20 min at
4°C to discard cell debris. Bradford assay (Bio-Rad Laboratories,
Inc.) was administered to assess the protein concentration. Then,
the total protein (20 µg) was subjected to 10% SDS-PAGE and
transferred to polyvinylidene difluoride (PVDF) membrane
(Sigma-Aldrich; Merck KGaA). After blocking the membrane with 5%
bovine serum albumin TBST [1% Tween-20 in 20 mmol/l Tris-buffered
saline (pH 7.6)] at 4°C for 1 h, the bands were probed with
monoclonal anti-BiP (1:5,000; product no. SAB5200168;
Sigma-Aldrich; Merck KGaA), anti-14-3-3ζ (1:5,000; product code
ab188368), anti-JNK (1:2,000; product code ab124956), anti-ATG5
(1:5000; product code ab238092), anti-PARP1 (1:5000; product code
ab110915), anti-cleaved PARP1 (1:2,000; product code ab110315 all
from Abcam), anti-c-Jun (1:2000; product no. 9165 Cell Signaling
Technology, Inc.) and anti-Beclin1 (1:5,000; product no.
SAB5300513; Sigma-Aldrich; Merck KGaA). GAPDH (1:2000; product code
ab125247; Abcam) served as the loading control. The horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:5,000; product code
ab6721) and rabbit anti-mouse IgG (1:5,000; product code ab6728;
both from Abcam) were used as secondary antibodies. The membrane
was incubated with secondary antibodies for 1 h at room
temperature. The bound antibodies were visualized through an
enhanced chemiluminescence reagent (Amersham Pharmacia Biotech;
Cytiva) and a Gel Logic 4000 Pro device (Bruker BioSpin
Corporation). The quantification of the band intensity was
conducted with ImageJ software (1.52c; National Institutes of
Health).
Apoptosis assay using flow
cytometry
A flow cytometric assay was conducted using an
FITC-Annexin V apoptosis detection kit (cat. no. 556547; BD
Biosciences) according to the manufacturers instructions. To
perform the apoptosis assay, the cells were seeded overnight at a
concentration of 2×105 in 24-well plates and transfected
with BiP siRNA, 14-3-3ζ siRNA and BiP+14-3-3ζ siRNAs concurrently
for a 72-h incubation followed by 2 µM cisplatin treatment for 48
h. Cells were harvested, and centrifuged at 1,500 × g for 5 min at
4°C. The cells were then washed with 1X PBS and 1X binding buffer
and were incubated with 5 µl of Annexin V for 15-20 min at room
temperature in darkness. After the incubation, the cells were
washed and stained with propidium iodide (PI) staining solution on
ice for 10 min. The cells were immediately applied to flow
cytometric analysis using FACSCanto II flow cytometer (BD
Biosciences). Distinct labeling patterns were used to determine
different cell populations; early apoptotic cells
(PI−/Annexin V+); late apoptotic cells
(PI+/Annexin V+) and viable cells
(PI−/Annexin V−). Apoptotic cells were
assessed as the percentage of early and late apoptotic cells (sum
of the numerical values of the right upper quadrant and the right
lower quadrant) of total cells. The results were analyzed using
FlowJo 7.6.1 software (FlowJo, LLC).
Terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick-end labeling (TUNEL) staining
To detect DNA fragmentation TUNEL staining (product
code ab66110; Abcam) according to the manufacturers instructions
was carried out. This assay indicates late-stage apoptosis. TdT
catalyzes the binding of red-UTP at the 3-OH terminus of fragmented
DNA in apoptotic cells. Cells were seeded at a density of
2×105 cells/well into 24-well plates. A constant final
concentration of 20 nM siRNA was added to the cells in each well,
and the cells were incubated for 72 h. The cells were then treated
with cisplatin (final concentration of 2 µM) for 48 h. The TUNEL
assay was performed according to the manufacturers instructions. In
short, the cells were fixed with 250 µl of 4% paraformaldehyde in
PBS (pH 7.4) for 15 min at 4°C. The cells were washed twice with
PBS and permeabilized through the addition of 300 µl of 70% ethanol
for 45 min on ice. Then, the cells were rinsed twice with PBS.
Subsequently, 50 µl of DNA labeling solution (10 µl of TdT reaction
buffer, 0.75 µl of TdT Enzyme, 8 µl of Br-dUTP, 32.5 µl of
ddH2O) was added to the cells and incubated at 37°C for
1 h. The cells were then washed twice with 300 µl rinse buffer and
the antibody solution (5 µl of Anti-BrdU-Red antibody, 95 µl of
rinse buffer) was prepared and added after washing the cells and
incubated for 30 min at room temperature in the dark. The 300 µl of
7-AAD/RNase solution was added at the end.
4,6-Diamidino-2-phenylindole (DAPI; product no. 4083; Cell
Signaling Technology, Inc.) at 1 µg/ml prepared in PBS was applied
to stain nucleus. The cells were incubated with DAPI for 5 min at
room temperature in the dark before mounting to the coverslips with
ProLong Gold Antifade reagent (cat. no. NC0581708; Thermo Fisher
Scientific, Inc.). Confocal microscopy (LSM 510 META; Zeiss AG) was
used to observe Red-dUTP-labeled DNA. Fluorescence intensity was
assessed in 20 randomly selected microscopic fields of view per
sample using a microscopeconfocal (magnification, ×60). ImageJ
software v.1.52c was utilized to quantify fluorescence in each
image.
Caspase-3 activity assay
Caspase-3 activity was investigated using the
Caspase-3 Activity Detection kit according to the producers
descriptions (product code ab39383; Abcam). To identify caspase-3
activity, the assay was carried out in 96-well plates at a density
of 8×103 cells/well. Cell lysates were prepared
following siRNA transfection and cisplatin treatment. Assays were
conducted in 96-well plates. Briefly, 10 µl protein of cell lysate
was incubated with 10 µl caspase-3 substrate (Ac-DEVD-pNA, 2 mM) in
80 µl of reaction buffer [10% glycerol, 1% NP-40, 137 mM NaCl and
20 mM Tris-HCL (pH 7.5)] at 37°C for 5 h. ODs were measured using
an ELISA reader (Microlisa Plus; Micro Lab Instruments) at 405 nm.
The assay and preparation of the standard curve were performed
following the manufacturers instructions. All experiments were
performed in triplicate.
SAPK/JNK kinase assay
JNK activity was performed as described by the
manufacturer (product no. 8794; Cell Signaling Technology, Inc.).
In short, the cells were seeded in 6-well plates at a concentration
of 5×105 cells/well and after transfection and treatment
with cisplatin were collected under non-denaturing conditions,
lysed on ice, and centrifuged at 8,600 × g for 10 min at 4°C. Then,
20 µl phospho-JNK rabbit monoclonal antibody (product code 4306;
Cell Signaling Technology, Inc.) linked to agarose beads was
incubated with gentle rocking an overnight at 4°C with 200 µl of
cell lysates to precipitate the JNK enzyme. Then, necessary
buffers, c-Jun substrate (2 µl) and adenosine triphosphate (200 µl)
were added, and the reaction mixture was incubated for 30 min at
30°C. The reaction was terminated by adding 25 µl of 4X SDS
(>90% purity; Sigma-Aldrich; KGaA) sample buffer, and the
samples (20 µg) were loaded onto 10% polyacrylamide gel. Proteins
were transferred to polyvinylidene difluoride (PVDF) membrane
(Sigma-Aldrich; KGaA). The membrane was then subjected to
phospho-c-Jun antibody (1:1,000, product no. 12598; Cell Signaling
Technology, Inc.) for 1 h at 4°C to determine JNK-induced
phosphorylation of c-Jun substrate at Ser63 and Ser73 residues.
Then, anti-rabbit IgG, HRP-linked antibody (1:2,000, product no.
7074) and anti-biotin, HRP-linked antibody (1:1,000; product no.
7075; both from Cell Signaling Technology, Inc.) were used to
detect biotinylated protein markers in 10 ml of Blocking Buffer
with gentle agitation for 1 h at room temperature. The membrane was
washed three times for 5 min, each with 15 ml Wash Buffer and was
incubated with 10 ml LumiGLO Substrate (product no. 7003; Cell
Signaling Technology, Inc.) with gentle agitation for 1 min at room
temperature. The membrane was then drained of excess LumiGLO
Substrate, and visualized with a Gel Logic 4000 Pro device (Bruker
BioSpin Corporation).
Autophagy assay
The autophagy assay was performed using Autophagy
Assay Kit containing the DAPI-labeled antibody against LC3-II
(Sigma-Aldrich; KGaA). Cells were seeded in 8-well chamber plates
for 24 h. The treated cells were then incubated with the autophagy
kit reagents according to the product description. In brief, 200 µl
of 1X autophagosome detection buffer solution was added to each
well after removing the medium. Cells were incubated at 37°C with
5% CO2 for 30 min. Cells were washed 3 times by gently
adding 100 µl of wash buffer to each well. The wash solution was
then removed carefully to prevent detachment of the cells. The
fluorescence (λex= 360/λem= 520 nm) intensity
was measured using confocal microscopy (LSM 510 META). The
quantification of fluorescence intensity was calculated with ImageJ
software 1.52c.
Protein-protein interaction model
If some part of two proteins functional role overlap
in the cell, it is enough for them to be considered as associated
proteins in a pathway or functional map. Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING, v. 11.0), an
online biological database to predict protein-protein interactions
(www.string-db.org) was used to design a model for
the associated proteins with Beclin1 (79).
Statistical analysis
Students t-test and one-way analysis of variance
(ANOVA) followed by post hoc Tukeys test were performed to compare
and analyze the difference of significance between groups
(P<0.05, P<0.01 and P<0.001). The mRNA and protein levels
are presented as the logarithm base 2 of the fold change in
expression among untreated and different treatments. The data were
presented as the means ± SD (standard deviation) of at least three
experiments. The statistical analyses were performed with IBM SPSS
Statistics software (version 22; IBM Corp.).
Results
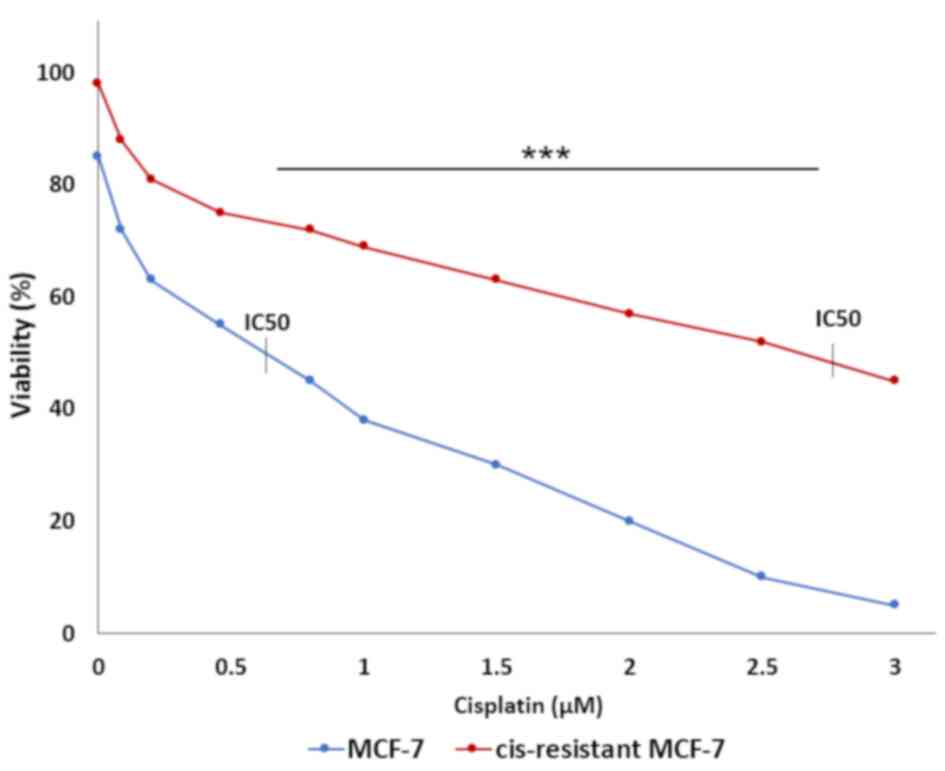
Induction of cisplatin resistance in
MCF-7 cells
Cisplatin with increasing concentration was added to
the cells twice a week after reseeding. At the end of each month,
cell viability was assessed by MTT assay. IC50 values
for MCF-7 and resistant MCF-7 cells were 0.65 and 2.8 µM
respectively (P<0.001). The IC50 value for resistant
cells was approximately four times higher than non-resistant cells
(Fig. 1). As the formation of drug
resistance to cisplatin in human breast cancer MCF-7 cells is
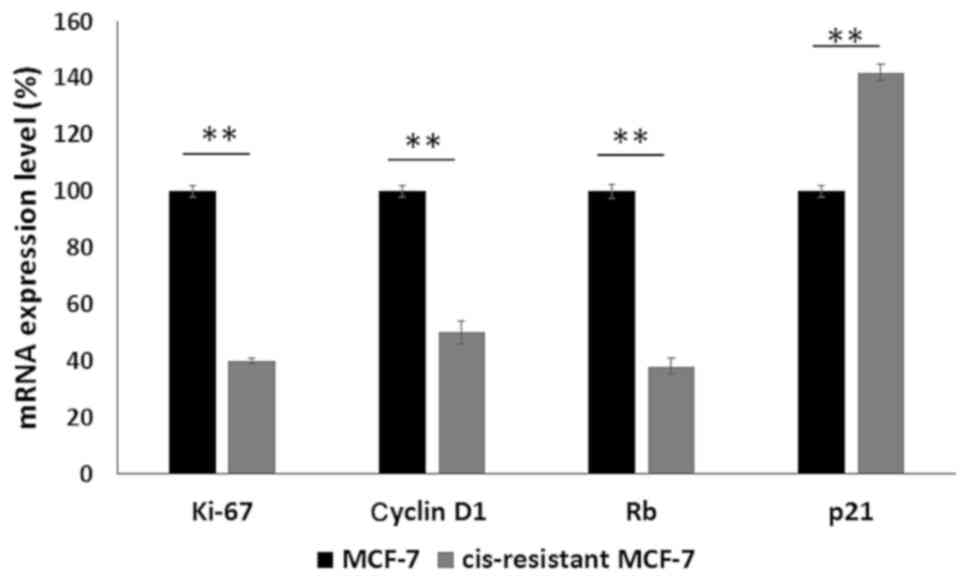
characterized by changes in the expression of proteins involved in
the control of apoptosis, the cell cycle, proliferation, and
adhesion (80–83) the expression levels of p21, cyclin
D1, Rb and Ki-67 were studied to determine whether the expression
profiles of these genes are consistent with the profile of the
resistant cells. Ki-67, cyclin D1 and Rb exhibited a reduced
expression while p21 was upregulated in cis-resistant MCF-7 cells
(Fig. 2). These experiments were
performed according to a previous study (84) to render cisplatin resistant in MCF-7
cells with which our results were consistent.
Increased apoptosis in BiP and 14-3-3ζ
co-knockdown cells compared to cells knocked down by either
gene
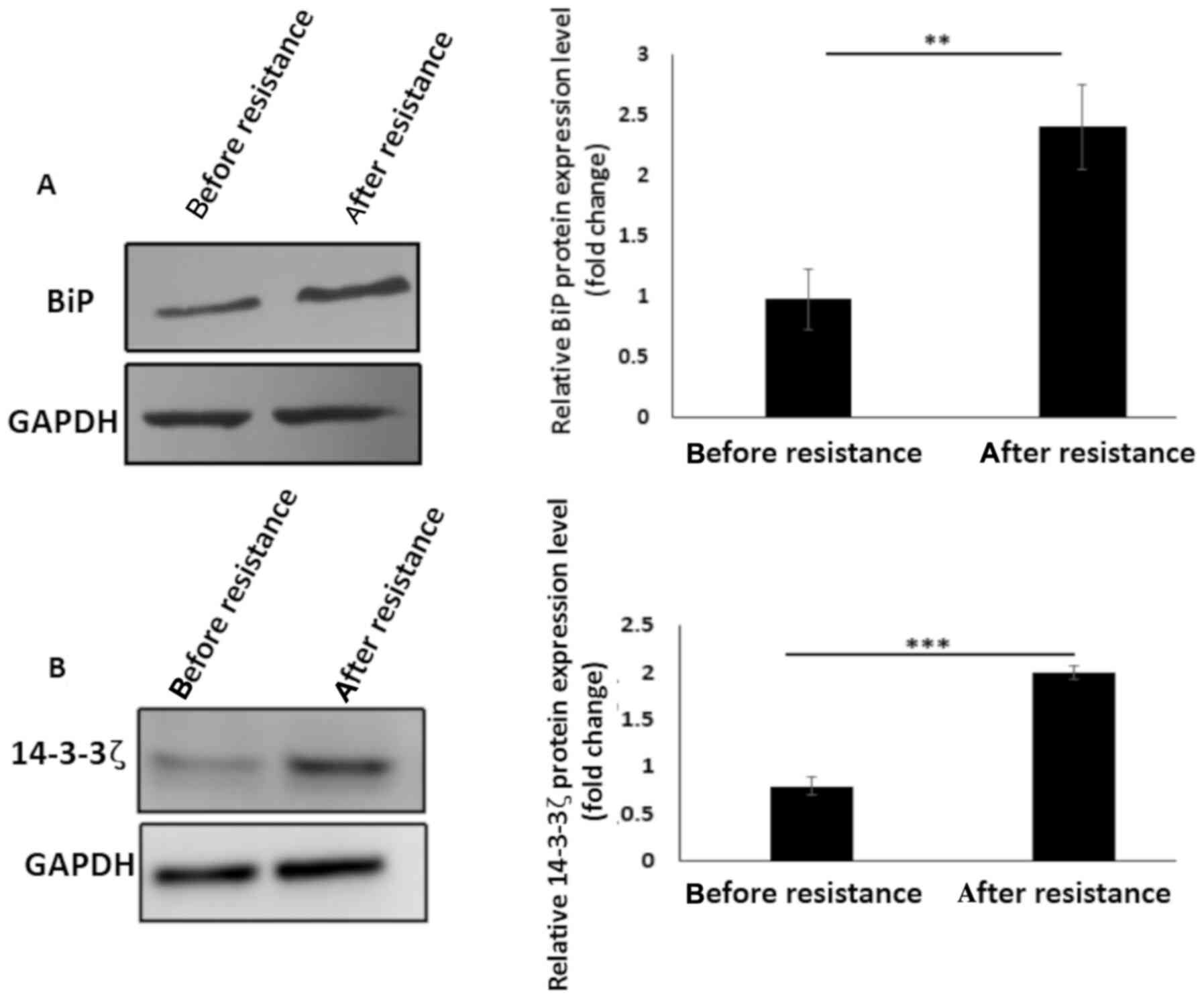
BiP and 14-3-3ζ protein levels before and after the
induction of cisplatin resistance were analyzed by performing
western blotting. An increase in BiP protein expression level
(P<0.01) was observed following cisplatin resistance (Fig. 3A) which could be associated with BiP
induction during chemotherapy (23,43–45).
In addition, 14-3-3ζ protein was overexpressed after resistance
induction (P<0.001) (Fig. 3B)
which could be as a result of the protective effects of 14-3-3ζ
against cellular stresses, here exerted through chemotherapy
exposure (60,68). Considering this evidence that
cisplatin can induce apoptosis, and that BiP or 14-3-3ζ positive
expression possibly leads to cisplatin resistance in breast cancer
cells through anti-apoptotic pathways, apoptosis induction in
cisplatin-resistant MCF-7 cells knocked down in BiP, 14-3-3ζ and
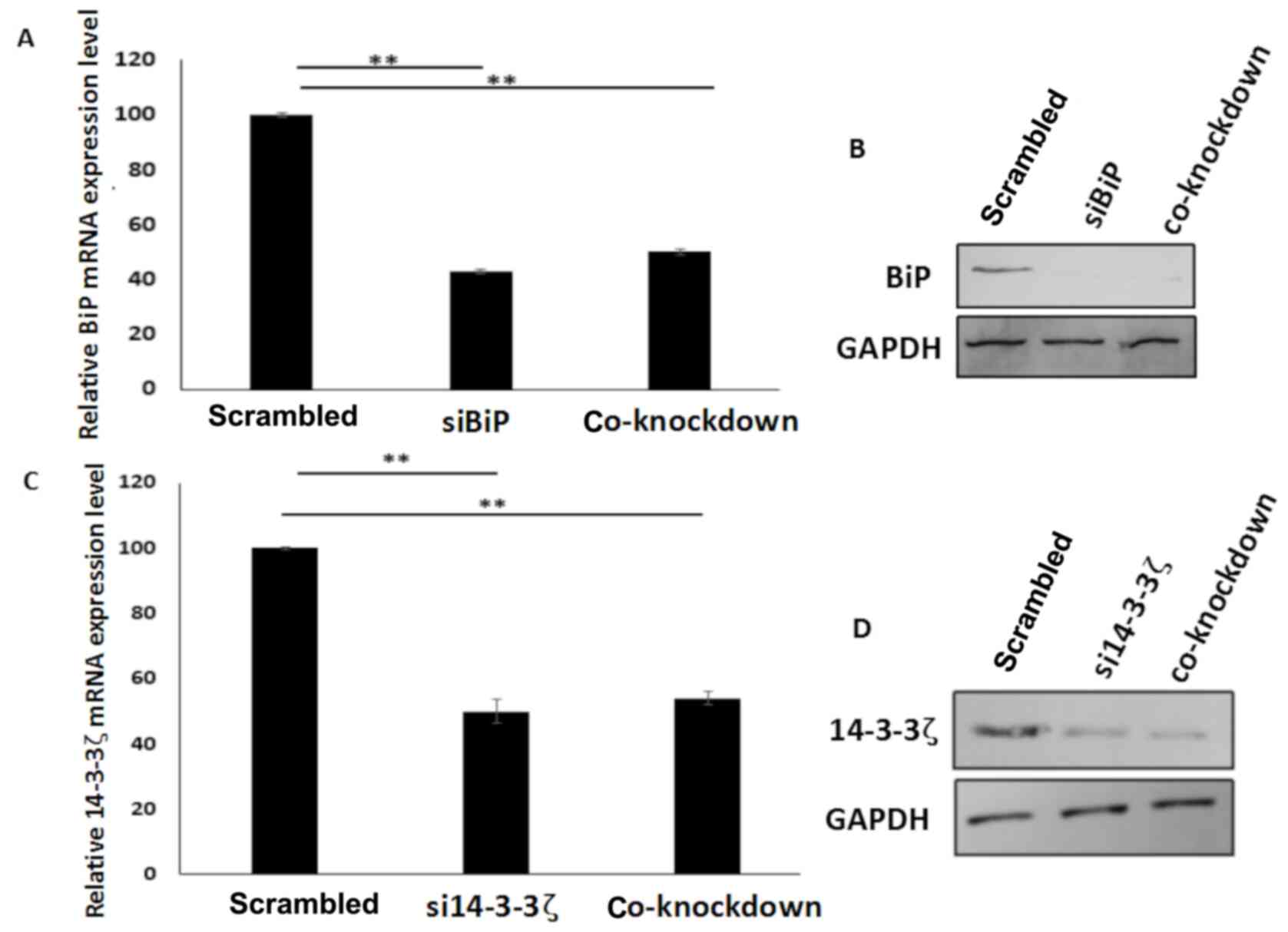
BiP+14-3-3ζ was assessed. To achieve the results, the cells were
transfected with BiP or 14-3-3ζ siRNA and co-transfected with BiP
and 14-3-3ζ siRNAs. Quantitative PCR and western blot analysis
revealed that downregulation of mRNA and protein levels of BiP and
14-3-3ζ occurred when the genes were inhibited or co-inhibited in
the cells (Fig. 4A-D). These
results revealed that there was no conflict between the two siRNAs,
when they were utilized in co-transfection of the cells.
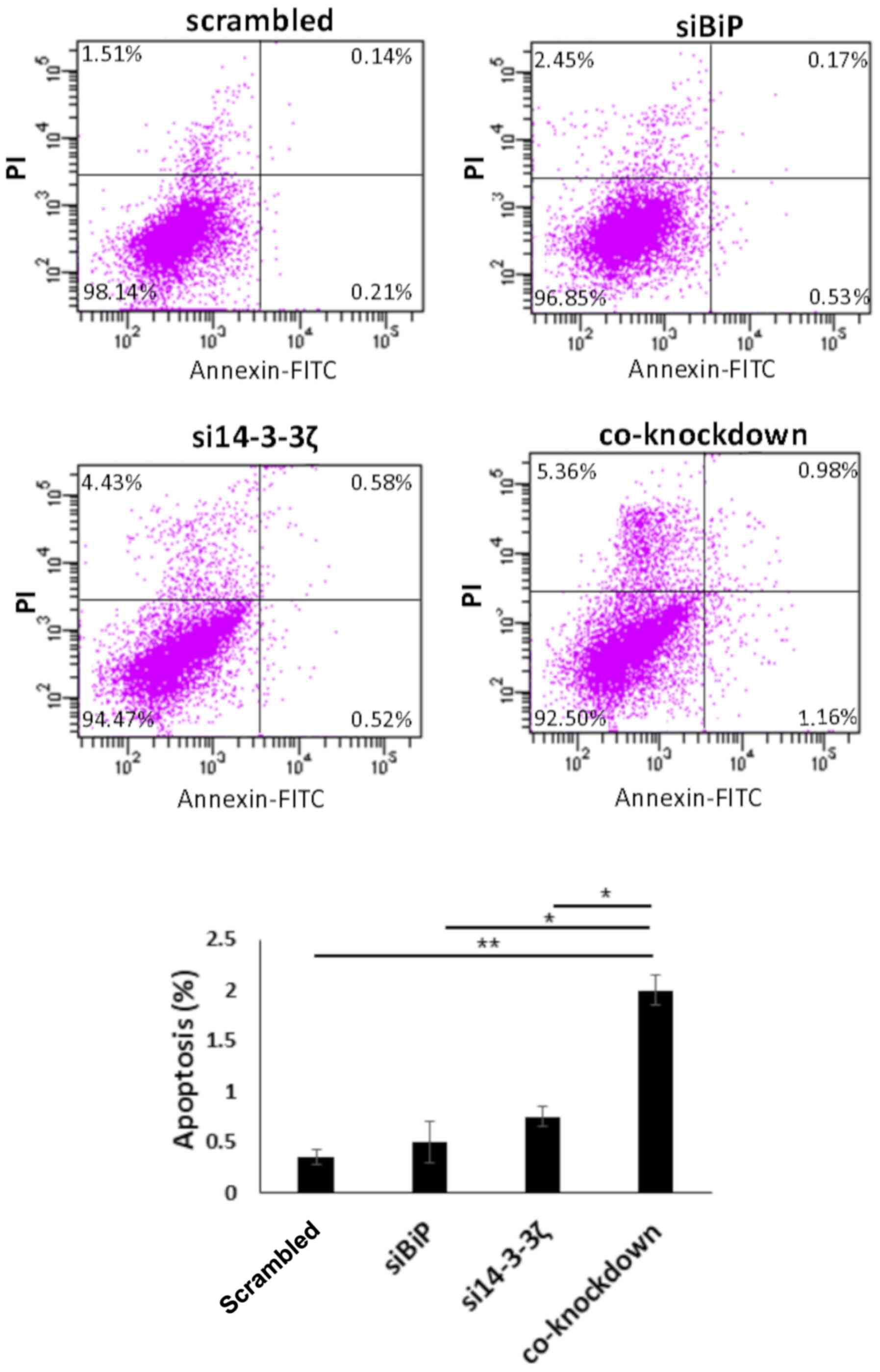
Flow cytometric results revealed that co-knockdown
with both siRNAs led to moderately increased apoptosis in the
absence of cisplatin, which was ~1.8% in the co-inhibited cells,
0.35% in BiP- and 0.75% in 14-3-3ζ-knockdown cells (normalized with
scrambled) (P<0.05) (Fig. 5). In
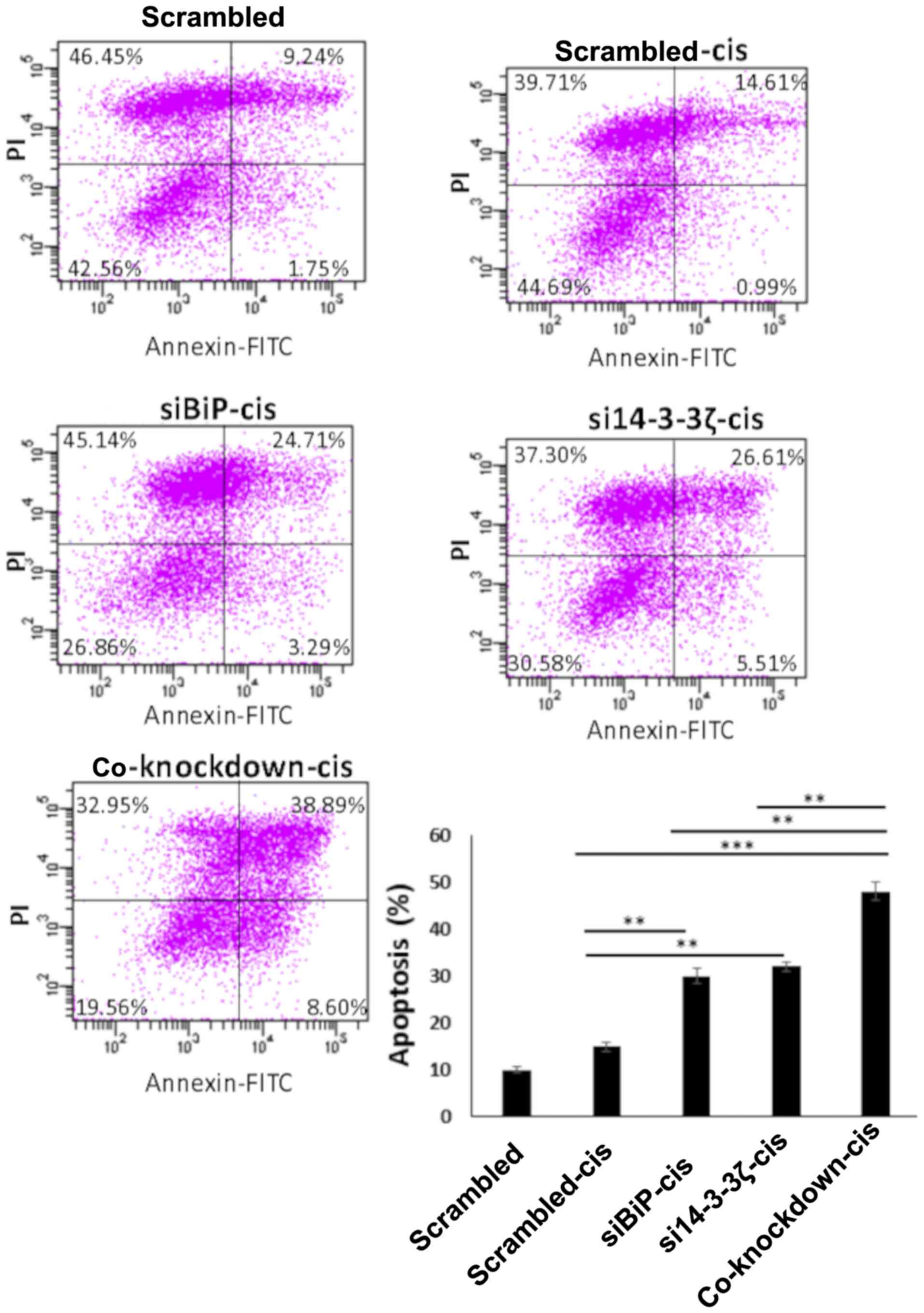
contrast, the co-inhibition of BiP and 14-3-3ζ in cisplatin-treated
MCF-7 cells significantly increased the number of apoptotic cells
compared with the cells knocked down by either BiP or 14-3-3ζ
siRNAs. This apoptosis induction was significantly higher in
cisplatin-treated cells which was ~13% in BiP, 15% in
14-3-3ζ-knockdown cells, and 32% in co-inhibited cells (normalized
with cis-scrambled) (P<0.01) (Fig.
6). The mean difference between co-knockdown and single
knockdown apoptosis in the presence of cisplatin (~18%) compared to
that in the absence of cisplatin (~1%) was statistically
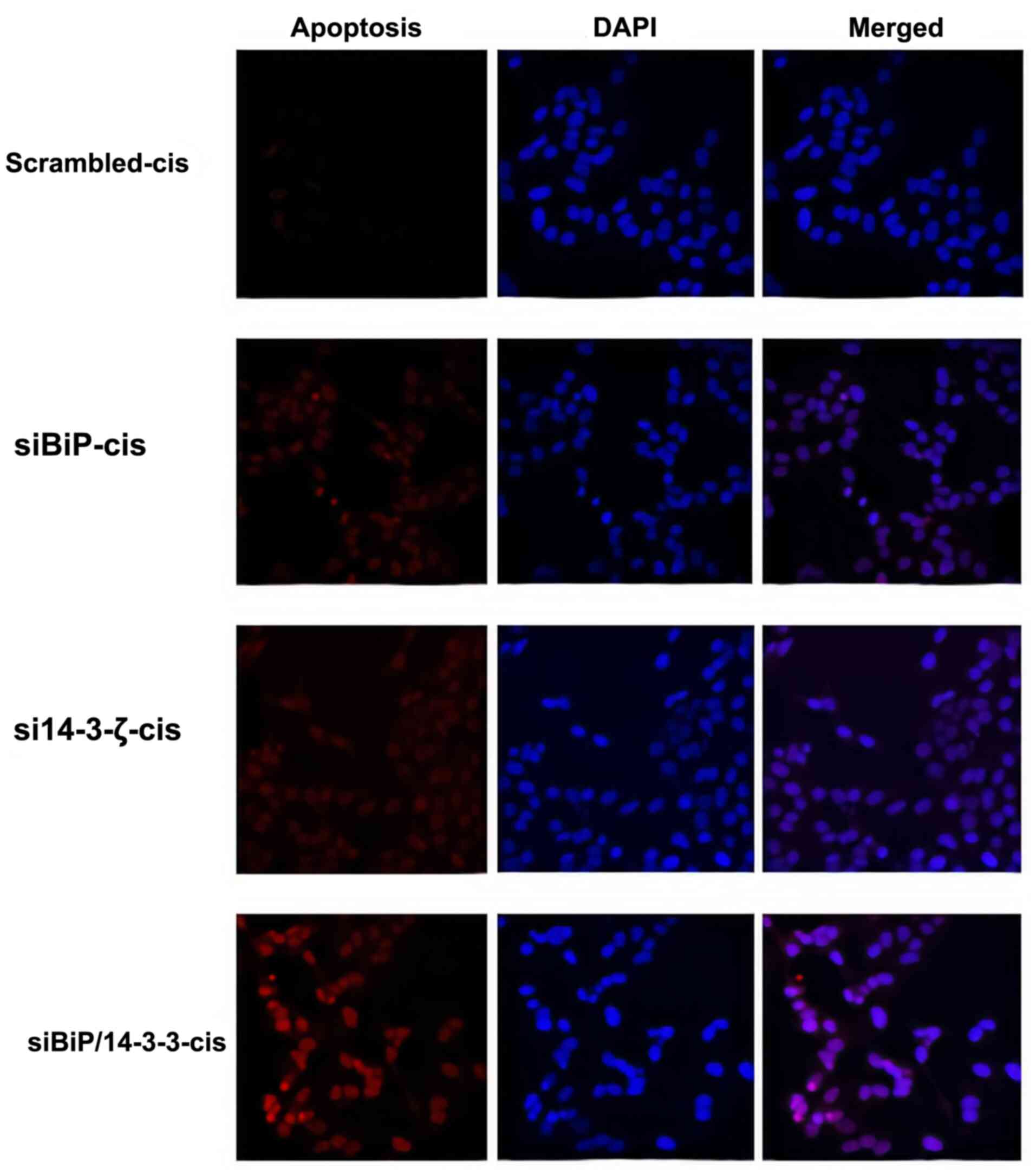
significant (P<0.001). To further explore the apoptosis rate a
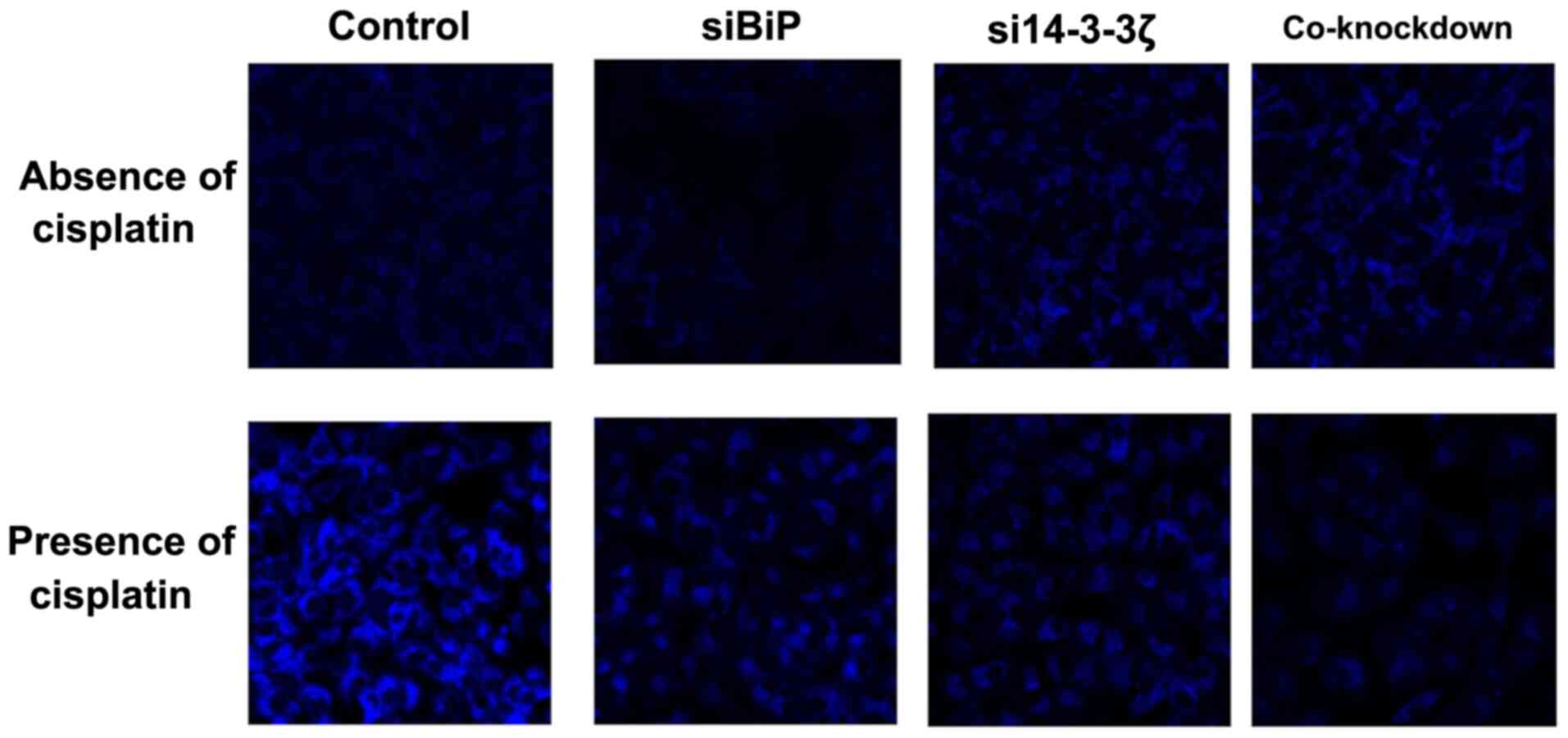
TUNEL assay was performed. The TUNEL assay demonstrated consistent
results with the flow cytometric data confirming a considerable
increase in the apoptosis of co-knockdown cells in the presence of
cisplatin (Figs. 7 and S1).
These results strongly supported the rationale that
the overexpression of 14-3-3ζ in MCF-7 cells may prevent the
intense UPR-induced apoptosis, and its downregulation along with
BiP could decrease drug resistance more potentially in comparison
to the knockdown of each protein.
Co-knockdown of BiP and 14-3-3ζ
markedly increases the protein levels of cleaved PARP1 and JNK as
well as caspase-3 and JNK activity in comparison with knockdown of
each gene alone
Apoptosis involves the mitochondrial (intrinsic) and
death receptor-mediated (extrinsic) pathways, using caspases as the
principal players of apoptosis in a series of distinct steps
(85). The present results
indicated that the overexpression of 14-3-3ζ in MCF-7 cells can
prevent intensive apoptosis induced by cisplatin in the
BiP-knockdown cells, suggesting that co-knockdown of BiP and
14-3-3ζ may increase the expression of JNK and caspase-3 activity
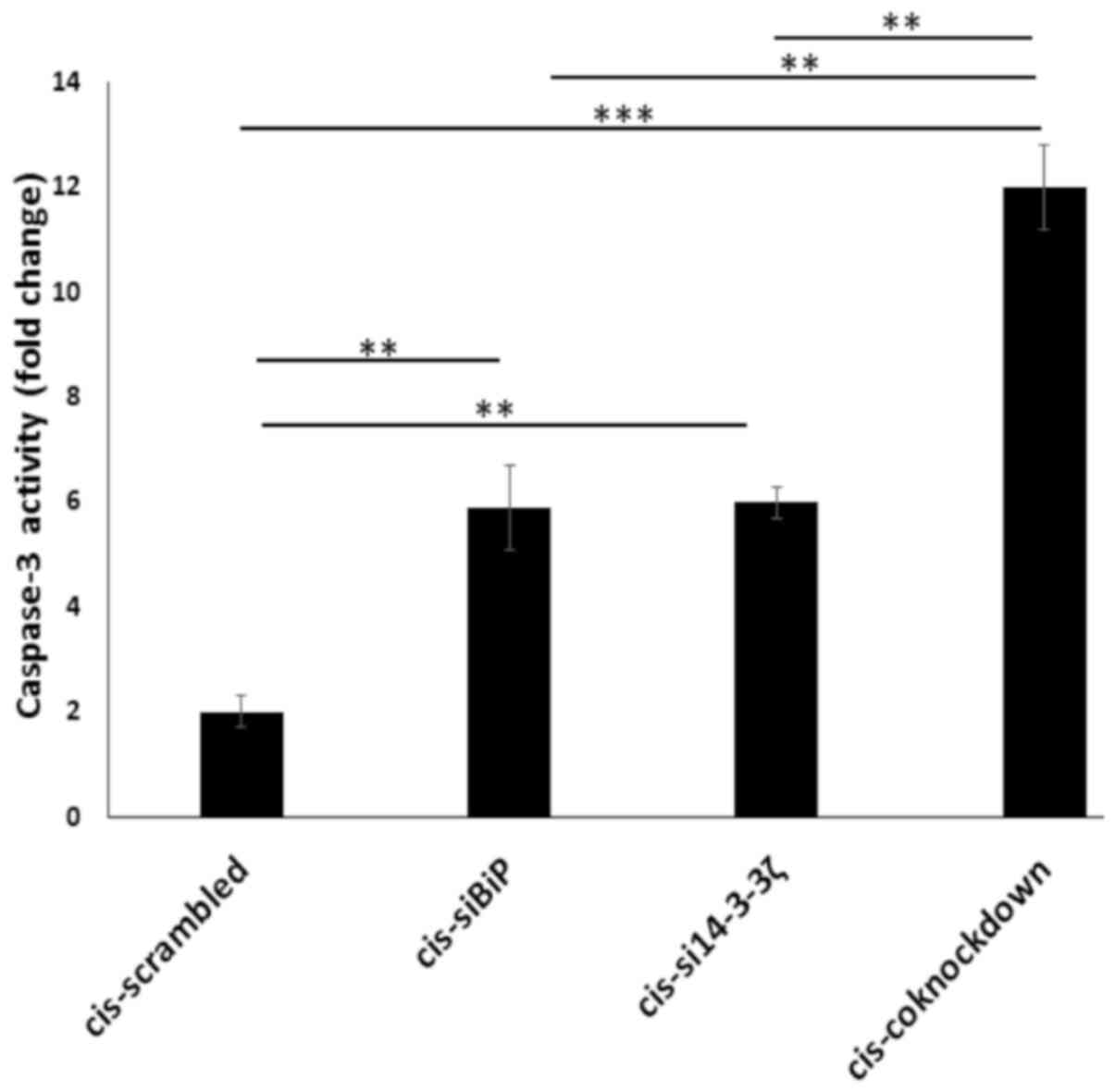
to induce apoptosis. To this end, a caspase-3 activity assay
followed by cleaved PARP1 western blotting were carried out. PARP1
is a downstream target of caspase-3 and is cleaved in the DEVD214
site leading to the formation of 24 kDa and 89 kDa fragments. Thus,
the presence of cleaved PARP1 is an indicator of caspase-3 activity
(86). The results revealed
increased caspase-3 activity (P<0.01) (Fig. 8) and cleaved PARP1 (P<0.05)
levels (Fig. 9A and C) in
co-knockdown cells as compared with knockdown ones by either gene.
Then, the JNK protein level as well as its activity in co-knockdown
and knockdown cells were examined. As revealed in Fig. 9A-C, the levels of JNK protein
(P<0.01) and phosphorylated c-Jun (P<0.01) increased to a
greater extent in co-knockdown cells compared with knockdown cells
with either BiP or 14-3-3ζ siRNA, suggesting that the reduced
protein levels of 14-3-3ζ and BiP may play a key role in the
overactivation of JNK and caspase-3 as pro-apoptotic proteins.
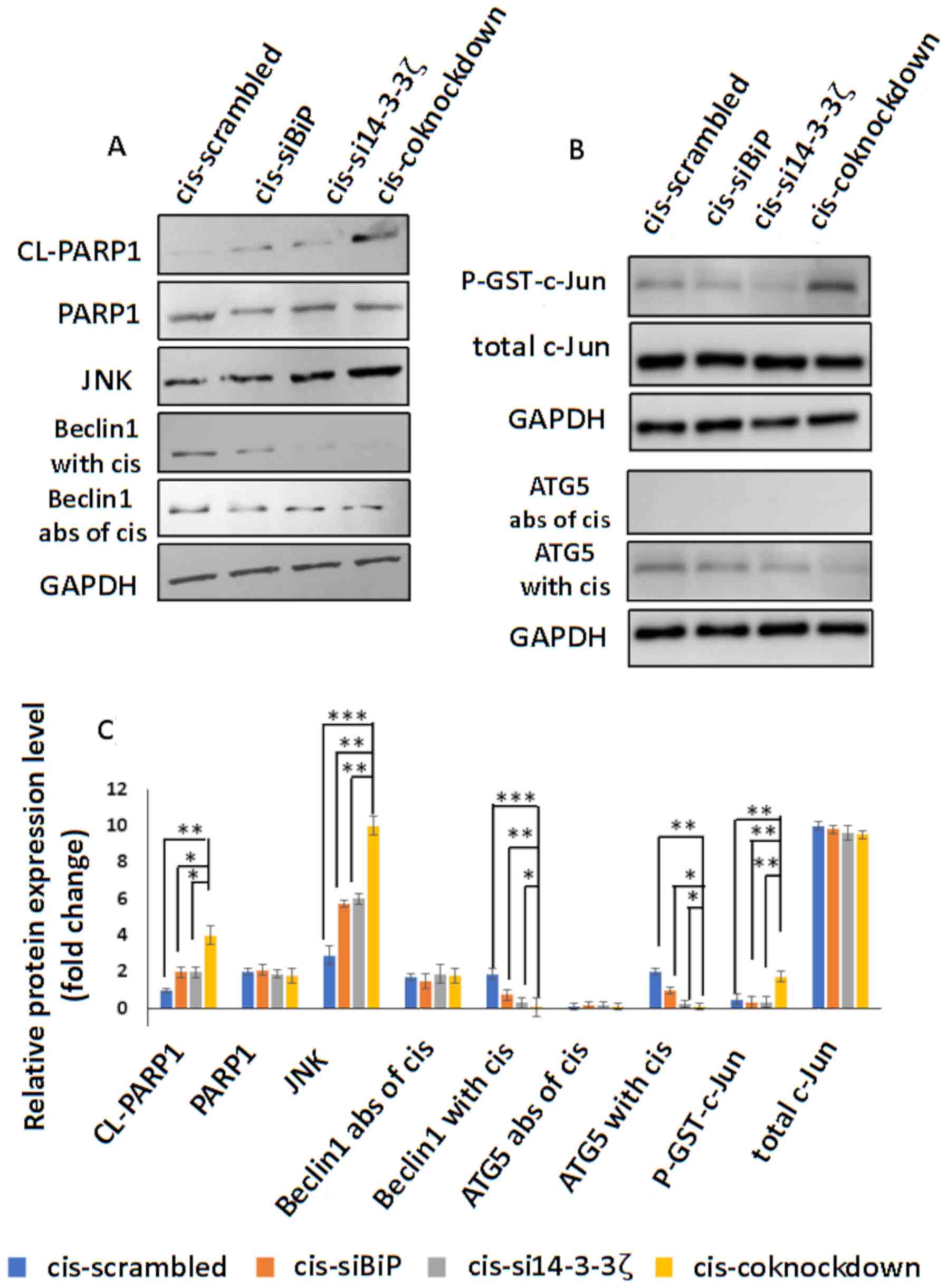 | Figure 9.(A) Western blot detection of cleaved
PARP1, PARP1, JNK, Beclin1 in the absence and presence of
cisplatin, in cisplatin-resistant MCF-7 cells transfected with
scrambled siRNA or siRNA against BiP or14-3-3ζ or BiP+14-3-3ζ. JNK
expression significantly increased in co-knockdown cells compared
to knockdown with either gene. Beclin-1 protein levels
significantly decreased in the co-knockdown cells compared to the
single-knockdown cells while the protein levels of Beclin-1
exhibited no change in the western blots. Cleaved PARP1 which is an
indicator of apoptosis promoted by caspase-3 activity decreased in
the co-knockdown cells compared to the single-knockdown cells. (B)
JNK activity (determined by p-GST-c-Jun level) and western blot of
ATG5 in the absence and presence of cisplatin. JNK activity
increased in the co-knockdown cells. ATG5 protein levels increased
in the double-knockdown cells compared to the single-knockdown
cells. (C) Analysis of data is presented in the graph using one-way
ANOVA and the significance of differences was identified using post
hoc, Tukeys test. GAPDH served as a loading control. *P<0.05,
**P<0.01 and ***P<0.001. BiP, binding immunoglobulin protein;
cis, cisplatin; si, small interfering; CL-PARP1, cleaved PARP1;
abs, absence. |
BiP and 14-3-3ζ co-knockdown
attenuates autophagy in the presence of cisplatin
Cisplatin counteracts breast cancer apoptosis by
autophagy induction. BiP and 14-3-3ζ play important roles in the
autophagy (76,88). The overexpression of BiP aids in
cancer cell survival by increasing autophagy as a mechanism of
resistance to chemotherapy (76),
whereas 14-3-3ζ overexpression reduces the rate of autophagy
(88). Thus, the effect of
BiP/14-3-3ζ double-knockdown on autophagy in absence as well as the
presence of cisplatin was investigated. A decrease in autophagy in
BiP-knockdown and an increase in 14-3-3ζ-knockdown cells in the
absence of cisplatin were observed which is based on previous
studies (76,88–90).
The co-knockdown cells exhibited a slight increase in autophagy,
while the pattern of autophagy was different in the presence of
cisplatin (Fig. 10). The present
data revealed a reduction in autophagy in all knockdown cells but
more significantly in co-knockdown cells, suggesting cisplatin can
markedly decrease the autophagosome formation in MCF-7 in BiP and
14-3-3ζ double-downregulation (P<0.001) compared to targeting
either gene (P<0.05) (ImageJ quantification) (Figs. 10 and S2). To further confirm the autophagy
results, the protein levels of ATG5 with and without cisplatin
treatment in different transfected cells was investigated. ATG5 is
essential for autophagosome formation. ATG5 knockdown or knockout
can lead to downregulation or inhibition of autophagy, suggesting
that ATG5 plays a central role in autophagy (91). The results revealed a reduction in
ATG5 protein level in co-knockdown cells compared to single gene
knockdown of BiP or 14-3-3ζ (P<0.05), while no
significant difference was observed in the absence of cisplatin
treatment (Fig. 9B and C).
Cisplatin considerably decreases
Beclin1 protein level in co-knockdown cells
JNK could regulate the crosstalk between autophagy
and apoptosis through phosphorylation of Bcl-2 (92). The stress-induced activation of JNK
(here exerted by BiP knockdown and enhanced via 14-3-3ζ
downregulation), can phosphorylate Bcl-2 leading to dissociation of
Bcl-2 and Beclin1 (92). Beclin1, a
central protein in autophagosome formation, cross-regulates
apoptosis and autophagy which can be cleaved by caspase-3 in two
cleavage sites during apoptosis. It can activate apoptosis in
apoptotic-competent cells by inactivating autophagy (92). For this reason, the effects of
apoptosis induced through the effects of BiP and 14-3-3ζ
double-knockdown on Beclin1 expression were investigated. Thus, the
expression levels of Beclin1 in the absence and presence of
cisplatin were assessed. The present results indicated a
significant decrease of Beclin1 protein in co-knockdown cells
compared to cells knocked down with either siRNA in the presence of
cisplatin (P<0.05, P<0.01) (Fig.
9A and C). This finding indicated that cisplatin may change the
cell fate from autophagy to apoptosis in knockdown cells by
reducing Beclin1. No difference in the expression level of Beclin1
was observed in the absence of cisplatin (Fig. 9A and C).
Discussion
BiP belongs to the HSP70 family which has pivotal
functions in stress during oncogenesis. In addition to contributing
to the protein folding and impeding protein aggregation, BiP
functions as a regulator of ER stress signaling. In normal and
non-stressed cells, BiP binds to three sensors in the ER membrane,
IRE-1, PERK and ATF6, rendering them inactive. Whereas under
physiological stress, following which ER function is disturbed, BiP
is dissociated from the sensors, rendering them active to send
signals to the nucleus and trigger UPR (63). This protects cells and tissues
against pathological conditions, such as arteriosclerosis,
neurotoxic stress and myocardial infarction. To highlight the
importance of BiP for survival of stressed cells such as in cancer,
one study revealed that the mice with heterozygous BiP,
which have half of the expression level of BiP than the wild-type
(WT), exhibited equivalent growth status with WT mice, while they
had markedly lower tumor development (93).
BiP has been revealed to be increased in cancer
metastasis, and for this reason its downregulation diminishes tumor
cell invasion in vitro and in vivo (93). In cancer cells, following the
accumulation of misfolded proteins and homeostasis perturbation,
BiP dissociates from IRE-1 which then dimerizes and promotes the
survival pathway of UPR, increasing the transcription of chaperones
and quality control proteins. Through BiP knockdown, ER stress
persists by virtue of BiP depletion, leading to IRE-1
oligomerization. This induces ASK1/JNK activation and signals
apoptosis (63).
14-3-3ζ overexpression occurs in an early stage of
breast cancer and contributes to the transformation of human
mammary epithelial cells and the progression of breast cancer
(47–51). There are several lines of evidence
that have revealed that 14-3-3ζ binds to ASK1 and inactivates it
(66–68). Zhang et al indicated that
ASK1 associates with 14-3-3ζ through phosphoSer697 (66). They determined that the dissociation
of 14-3-3ζ/ASK1 augments ASK1-induced apoptosis through JNK and
caspase-3 activation and that the high expression of 14-3-3ζ halts
ASK1-induced apoptosis in several cell lines.
Jiang et al indicated the cisplatin
counteracts breast cancer (MCF-7 and MDA-MB-231 cells) apoptosis by
autophagy induction (72). Since
BiP and 14-3-3ζ have roles in autophagy and JNK and Beclin1
crosstalk between apoptosis and autophagy, we examined the impact
of the double-knockdown of these two genes on cisplatin treatment
of MCF-7 cells.
Conversely, it has already been well-established
that as a result of aberrant protein folding in the ER environment,
cancer cells activate autophagy, a stress-adaptive self-degradative
process (94). In fact, cancer
cells rely on autophagy for survival more than normal cells. For
instance, PERK (one sensor of the UPR pathway)-eIF2α
phosphorylation is essential to decrease protein synthesis to
activate autophagy (95,96). It was previously reported that the
activation of autophagy and cell survival upon ER stress can be
achieved by the IRE-1α-JNK pathway, which has been suggested to act
in the interplay between cancer cell death and as a process of
chemotherapy resistance (97).
Autophagy promotes cancer cell survival during chemotherapy to deal
with metabolic stresses induced by chemotherapeutic drugs (20). In addition, in breast cancer cells,
resistance to endocrine therapy including fulvestrant and tamoxifen
is the outcome of the dynamic network between UPR activation,
apoptosis and autophagy (3–5).
JNK plays a role in the regulation between apoptosis
and autophagy through the phosphorylation of Bcl-2. Activating JNK
disrupts Bcl-2 and Beclin1 interaction. Beclin1 is the
cross-regulator of autophagy and apoptosis. Moreover, caspase-3 can
cleave Beclin1 to change the cell fate from autophagy to apoptosis
(98,99). The C-terminal fragment of Beclin1
can then move to the mitochondria and induce mitochondrial membrane
permeability and apoptosis (100).
Taking all these into consideration, we assumed that
14-3-3ζ combined with BiP may be a more effective co-target to
enhance cisplatin sensitivity compared to targeting either gene.
Cisplatin-induced resistant MCF-7 cells were generated by a
three-month cisplatin treatment in constantly increasing
concentrations. A viability MTT assay before and after the
induction of resistance was performed. The results indicated that
the IC50 of cells increased by 4-fold in the
MCF-7-resistant cells when compared to the MCF-7 cells. The
cellular adaptation was investigated by ascertaining the expression
levels of four critical genes (Ki-67, cyclin D1, Rb and p21), the
expression of which changed following cisplatin resistance
consistent with a previous study (84). The Ki-67 protein is a marker for
cell proliferation, present during all active phases of the cell
cycle (G1, S, G2 and mitosis), but is absent
in resting (quiescent) cells (G0) (80) cyclin D1 is overexpressed in breast
carcinoma (81) and has been shown
to be required for progression through the G1 phase of the cell
cycle to induce cell migration (101) and angiogenesis (102). Hyperphosphorylation of Rb promotes
proliferation and plays a role in tumor progression (82) and p21 protein is a cyclin-dependent
kinase inhibitor (CKI) that is capable of inhibiting all cyclin/CDK
complexes (83).
Furthermore, BiP and 14-3-3ζ protein expression
levels were assessed before and after resistance. The results
revealed an increase in BiP and 14-3-3ζ expression levels after
cisplatin resistance induction, which could be a result from the
fact that chemotherapy induces BiP (23,43–45)
and the fact that 14-3-3ζ plays protective roles during cellular
stresses promoted for example during chemotherapy (59,68).
This protective role of 14-3-3ζ was confirmed when it was silenced
by siRNA, revealing, it sensitized the cells to stress-induced
apoptosis (59,68). The resistant MCF-7 cells were
cultured and utilized in the next experiments. To validate the
downregulation of BiP and 14-3-3ζ, qPCR was used revealing ~60%
decrease in the mRNA levels in the single and co-transfected cells.
To support the PCR results, the reduction of the protein levels was
confirmed for BiP and 14-3-3ζ using western blot analysis.
Then, the MCF-7 cells were transfected with either
BiP or 14-3-3ζ siRNA or both and apoptosis in the absence and
presence of cisplatin was investigated. Based on the flow
cytometric and confocal microscopic results, BiP and 14-3-3ζ
double-knockdown could increase the sensitivity of resistant MCF-7
cells exposed to cisplatin treatment compared to downregulation of
either gene. Furthermore, the activities of caspase-3 and JNK were
assessed and the results revealed higher expression in the combined
BiP with 14-3-3ζ group compared to single knockdown of BiP or
14-3-3ζ and these results were confirmed with phosphorylated c-Jun
and cleaved PARP1 protein levels. These results revealed that
apoptosis may be enhanced in the double-knockdown of BiP and
14-3-3ζ compared to targeting each gene alone.
The knockdown effects of either gene in autophagy is
paradoxical, with BiP knockdown preventing autophagy formation
(76) and 14-3-3ζ downregulation
inducing autophagy (88). Thus, the
effects of cisplatin were assessed in the three types of knockdown
cells to determine whether autophagy changes after BiP/14-3-3ζ
co-knockdown. The results revealed a slight increase of autophagy
in the co-inhibited cells in the absence of cisplatin and a
significant decrease in the two single-knockdown (BiP or 14-3-3ζ)
and co-knockdown (BiP+14-3-3ζ) cells in the presence of cisplatin.
This reduction was more prominent in the latter case, supporting
the critical impact of BiP and 14-3-3ζ on autophagy in resistant
MCF-7 cells following cisplatin treatment. Collectively, this
indicated that co-knockdown of BiP and 14-3-3ζ leads to
autophagosome attenuation in the presence of cisplatin in MCF-7
cells. The autophagy-related results were confirmed by assessing
the protein levels of Beclin1 and ATG5 in the absence and presence
of cisplatin which revealed a significant decrease in their levels
in the co-knockdown compared to the knockdown cells. This was
consistent with a previous study which demonstrated that activation
of IRE-1/JNK can promote apoptosis by inactivating autophagy
(92) and BiP/14-3-3ζ
double-knockdown could enhance this pathway.
MCF-7, an HR-positive and HER2-negative cell line,
is not a perfect chemotherapy model as are triple negative cell
lines (103). The main obstacle
for these cancers is that the chemotherapeutic agents including
cisplatin increase autophagy. Autophagy is one of the chemotherapy
resistance mechanisms in breast cancer (72). Thus, certain processes involved in
chemotherapy resistance need to be tackled to overcome this
problem. The ability of a protein to perform its function depends
on its potential to interact with other proteins. Thus, a system
that can circumvent a potential chemotherapy resistance component,
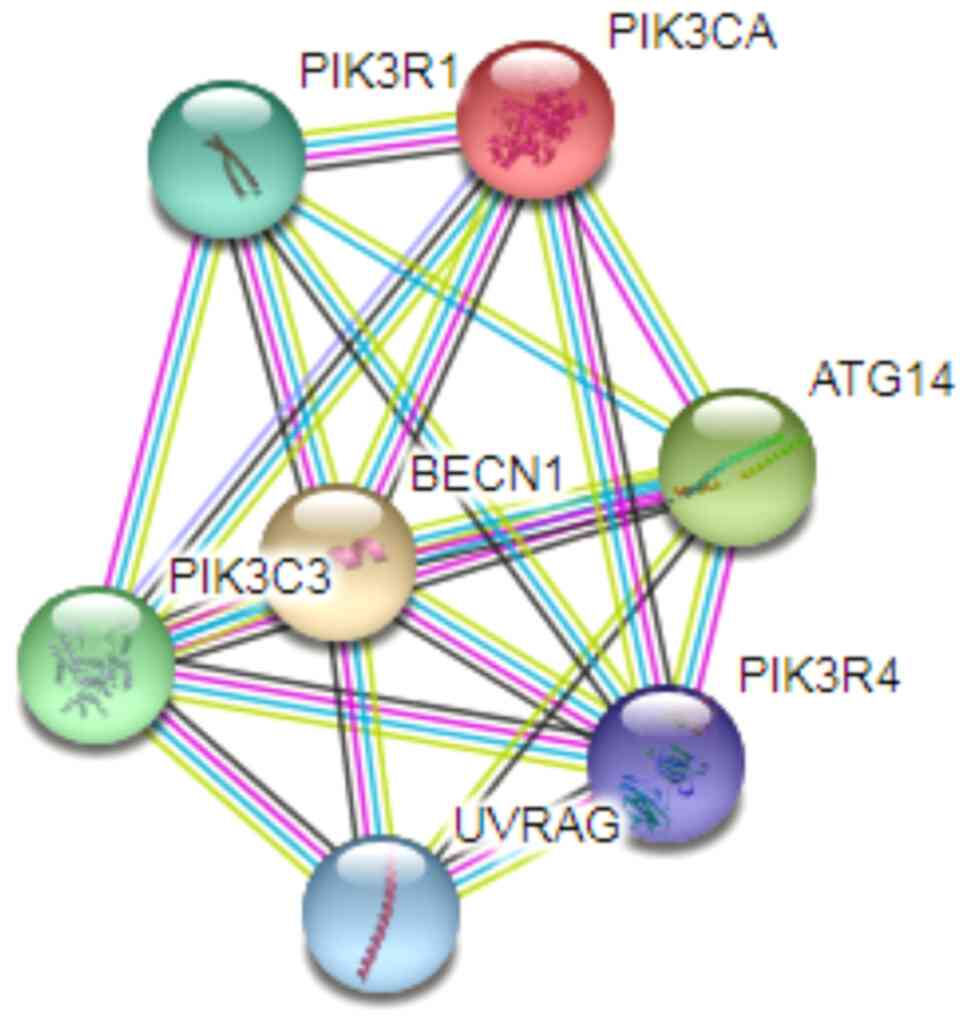
may influence other associated proteins. For example, it is well
known that PIK3CA, with which Beclin1 interacts, is mutated in some
breast cancers related to overactivation of PI3K/Akt signaling
pathways. The mutations in PIK3CA confer resistance to cisplatin in
some cancers (104). Therefore, if
Beclin1 is downregulated, it could disrupt the whole complex to
prevent the overexpressed signals in cancer. A model from the
STRING interaction network (79)
which revealed some of the proteins interacting with Beclin1 which
may be affected by Beclin1 downregulation is presented in Fig. 11.
In conclusion, cisplatin resistance occurs
relatively often in breast cancer carcinoma and the MCF-7 cell
line, and for this reason finding a method to overcome this
obstacle of chemotherapy tolerance is critical. This resistance may
occur as a consequence of autophagy induced by cisplatin. Thus,
detecting the pivotal factors involved in autophagy may give rise
to improved treatment. BiP/GRP78 and 14-3-3ζ proteins play roles in
autophagy, and their overexpression leads to chemotherapy
resistance. The dissociation of these proteins from their partners
(BiP from IRE-1 and 14-3-3ζ from ASK1) relay UPR-apoptotic signal
in a series of pathways. By targeting both genes concurrently, the
MCF-7 cells were markedly sensitized to cisplatin. The results
indicated that this sensitization was related to attenuation of
autophagy.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to acknowledge the University
of Tehran and the Research Center of CICbiomaGUNE for supporting
the project.
Funding
The authors provided the financial support for this
study. No fuding was received.
Availability of data and materials
All data generated and analyzed during this study
are included in this article.
Authors contributions
TKK designed and performed the experiments, prepared
the figures and/or tables, analyzed the data and wrote the
manuscript. SS supervised the research, designed the experiments,
analyzed the data, wrote the study and reviewed drafts of the
study. BA reviewed drafts of the manuscript and supported the
research techniques for flow cytometry and its analysis. LS
reviewed drafts of the paper and technically supported the research
for western blot experiments. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent or publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BiP
|
binding immunoglobulin protein
|
|
GRP78
|
glucose-regulated protein 78
|
|
UPR
|
unfolded protein response
|
|
ER
|
endoplasmic reticulum
|
|
siRNA
|
small interfering RNA
|
|
ROS
|
reactive oxygen species
|
|
UTP
|
uridine-5-triphosphate
|
|
dUTP
|
deoxy UTP
|
|
FITC
|
fluorescein isothiocyanate
|
|
FACS
|
fluorescence-activated cell
sorting
|
|
PI
|
propidium iodide
|
|
SDS-PAGE
|
sodium dodecyl sulfate polyacrylamide
gel electrophoresis
|
|
RIPA
|
radioimmunopercipitation
|
|
cis
|
cisplatin
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
PVDF
|
polyvinylidene difluoride
|
References
|
1
|
Torre LA, Islami F, Siegel RL, Ward EM and
Jemal A: Global cancer in women: Burden and trends. Cancer
Epidemiol Biomarkers Prev. 26:444–457. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu G, Zhou W, Pan X, Sun Y, Xu H, Shi P,
Li J, Gao L and Tian X: miR-100 reverses cisplatin resistance in
breast cancer by suppressing HAX-1. Cell Physiol Biochem.
47:2077–2087. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cook KL, Clarke PA, Parmar J, Hu R,
Schwartz-Roberts JL, Abu-Asab M, Wärri A, Baumann WT and Clarke R:
Knockdown of estrogen receptor-α induces autophagy and inhibits
antiestrogen-mediated unfolded protein response activation,
promoting ROS-induced breast cancer cell death. FASEB J.
28:3891–3905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu R, Warri A, Jin L, Zwart A, Riggins RB,
Fang HB and Clarke R: NF-κB signaling is required for XBP1
(unspliced and spliced)-mediated effects on antiestrogen
responsiveness and cell fate decisions in breast cancer. Mol Cell
Biol. 35:379–390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yeung BH, Kwan BW, He QY, Lee AS, Liu J
and Wong AS: Glucose-regulated protein 78 as a novel effector of
BRCA1 for inhibiting stress-induced apoptosis. Oncogene.
27:6782–6789. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Cook KL, Warri A, Cruz IM, Rosim
M, Riskin J, Helferich W, Doerge D, Clarke R and Hilakivi-Clarke L:
Lifetime genistein intake increases the response of mammary tumors
to tamoxifen in rats. Clin Cancer Res. 23:814–824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Christowitz C, Davis T, Isaacs A, van
Niekerk G, Hattingh S and Engelbrecht AM: Mechanisms of
doxorubicin-induced drug resistance and drug resistant tumour
growth in a murine breast tumour model. BMC Cancer. 19:7572019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma W, Teng Y, Hua H, Hou J, Luo T and
Jiang Y: Upregulation of heat shock protein 27 confers resistance
to actinomycin D-induced apoptosis in cancer cells. FEBS J.
280:4612–4624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Q, Cui K, Espin-Garcia O, Cheng D,
Qiu X, Chen Z, Moore M, Bristow RG, Xu W, Der S, et al: Resistance
to bleomycin in cancer cell lines is characterized by prolonged
doubling time, reduced DNA damage and evasion of G2/M arrest and
apoptosis. PLoS One. 8:e823632013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo B and Lee AS: The critical roles of
endoplasmic reticulum chaperones and unfolded protein response in
tumorigenesis and anticancer therapies. Oncogene. 32:805–818. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kosakowska-Cholody T, Lin J, Srideshikan
SM, Scheffer L, Tarasova NI and Acharya JK: HKH40A downregulates
GRP78-BiP expression in cancer cells. Cell Death Dis. 5:e12402014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Avril T, Vauléon E and Chevet E:
Endoplasmic reticulum stress signaling and chemotherapy resistance
in solid cancers. Oncogenesis. 6:e3732017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee E, Nichols P, Spicer D, Groshen S, Yu
MC and Lee AS: GRP78 as a novel predictor of responsiveness to
chemotherapy in breast cancer. Cancer Res. 66:7849–7853. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Andruska N, Zheng X, Yang X, Helferich WG
and Shapiro DJ: Anticipatory estrogen activation of the unfolded
protein response is linked to cell proliferation and poor survival
in estrogen receptor α-positive breast cancer. Oncogene.
34:3760–3769. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davies MPA, Barraclough DL, Stewart C,
Joyce KA, Eccles RM, Barraclough R, Rudland PS and Sibson DR:
Expression and splicing of the unfolded protein response gene XBP-1
are significantly associated with clinical outcome of
endocrine-treated breast cancer. Int J Cancer. 123:85–88. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee E, Nichols P, Groshen S, Spicer D and
Lee AS: GRP78 as potential predictor for breast cancer response to
adjuvant taxane therapy. Int J Cancer. 128:726–731. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Longley DB and Johnston PG: Molecular
mechanisms of drug resistance. J Pathol. 205:275–292. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fernandez PM, Tabbara SO, Jacobs LK,
Manning FC, Tsangaris TN, Schwartz AM, Kennedy KA and Patierno SR:
Overexpression of the glucose-regulated stress gene GRP78 in
malignant but not benign human breast lesions. Breast Cancer Res
Treat. 59:15–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pyrko P, Schönthal AH, Hofman FM, Chen TC
and Lee AS: The unfolded protein response regulator GRP78/BiP as a
novel target for increasing chemosensitivity in malignant gliomas.
Cancer Res. 67:9809–9816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee HK, Xiang C, Cazacu S, Finniss S,
Kazimirsky G, Lemke N, Lehman NL, Rempel SA, Mikkelsen T and Brodie
C: GRP78 is overexpressed in glioblastomas and regulates glioma
cell growth and apoptosis. Neuro Oncol. 10:236–243. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Epple LM, Dodd RD, Merz AL, Dechkovskaia
AM, Herring M, Winston BA, Lencioni AM, Russell RL, Madsen H, Nega
M, et al: Induction of the unfolded protein response drives
enhanced metabolism and chemoresistance in glioma cells. PLoS One.
8:e732672013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsuruo T, Naito M, Tomida A, Fujita N,
Mashima T, Sakamoto H and Haga N: Molecular targeting therapy of
cancer: Drug resistance, apoptosis and survival signal. Cancer Sci.
94:15–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Salazar M, Hernández-Tiedra S, Torres S,
Lorente M, Guzmán M and Velasco G: Detecting autophagy in response
to ER stress signals in cancer. Methods Enzymol. 489:297–317. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sokolowska I, Woods AG, Gawinowicz MA, Roy
U and Darie CC: Identification of potential tumor differentiation
factor (TDF) receptor from steroid-responsive and steroid-resistant
breast cancer cells. J Biol Chem. 287:1719–1733. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baptista MZ, Sarian LO, Vassallo J, Pinto
GA, Soares FA and de Souza GA: Prognostic significance of GRP78
expression patterns in breast cancer patients receiving adjuvant
chemotherapy. Int J Biol Markers. 26:188–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
La X, Zhang L, Li H, Li Z, Song G, Yang P
and Yang Y: Ajuba receptor mediates the internalization of
tumor-secreted GRP78 into macrophages through different endocytosis
pathways. Oncotarget. 9:15464–15479. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y, Wang JH, Zhang XL, Wang XL and
Yang L: Endoplasmic reticulum chaperone glucose-regulated protein
78 in gastric cancer: An emerging biomarker. Oncol Lett.
15:6087–6093. 2018.PubMed/NCBI
|
|
32
|
Shimizu A, Kaira K, Yasuda M, Asao T and
Ishikawa O: Clinical and pathological significance of ER stress
marker (BiP/GRP78 and PERK) expression in malignant melanoma.
Pathol Oncol Res. 23:111–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peñaranda Fajardo NM, Meijer C and Kruyt
FA: The endoplasmic reticulum stress/unfolded protein response in
gliomagenesis, tumor progression and as a therapeutic target in
glioblastoma. Biochem Pharmacol. 118:1–8. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Uramoto H, Sugio K, Oyama T, Nakata S, Ono
K, Yoshimastu T, Morita M and Yasumoto K: Expression of endoplasmic
reticulum molecular chaperone Grp78 in human lung cancer and its
clinical significance. Lung Cancer. 49:55–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scriven P, Coulson S, Haines R,
Balasubramanian S, Cross S and Wyld L: Activation and clinical
significance of the unfolded protein response in breast cancer. Br
J Cancer. 101:1692–1698. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xing X, Li Y, Liu H, Wang L and Sun L:
Glucose regulated protein 78 (GRP78) is overexpressed in colorectal
carcinoma and regulates colorectal carcinoma cell growth and
apoptosis. Acta Histochem. 113:777–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Daneshmand S, Quek ML, Lin E, Lee C, Cote
RJ, Hawes D, Cai J, Groshen S, Lieskovsky G, Skinner DG, et al:
Glucose-regulated protein GRP78 is up-regulated in prostate cancer
and correlates with recurrence and survival. Hum Pathol.
38:1547–1552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zheng HC, Takahashi H, Li XH, Hara T,
Masuda S, Guan YF and Takano Y: Overexpression of GRP78 and GRP94
are markers for aggressive behavior and poor prognosis in gastric
carcinomas. Hum Pathol. 39:1042–1049. 2007. View Article : Google Scholar
|
|
39
|
Langer R, Feith M, Siewert JR, Wester HJ
and Hoefler H: Expression and clinical significance of glucose
regulated proteins GRP78 (BiP) and GRP94 (GP96) in human
adenocarcinomas of the esophagus. BMC Cancer. 8:702008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nagelkerke A, Bussink J, Sweep FC and Span
PN: The unfolded protein response as a target for cancer therapy.
Biochim Biophys Acta. 1846:277–284. 2014.PubMed/NCBI
|
|
41
|
Baumeister P, Dong D, Fu Y and Lee AS:
Transcriptional induction of GRP78/BiP by histone deacetylase
inhibitors and resistance to histone deacetylase inhibitor-induced
apoptosis. Mol Cancer Ther. 8:1086–1094. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Roué G, Pérez-Galán P, Mozos A,
López-Guerra M, Xargay-Torrent S, Rosich L, Saborit-Villarroya I,
Normant E, Campo E and Colomer D: The Hsp90 inhibitor IPI-504
overcomes bortezomib resistance in mantle cell lymphoma in vitro
and in vivo by down-regulation of the prosurvival ER chaperone
BiP-Grp78. Blood. 117:1270–1279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Al-Rawashdeh FY, Scriven P, Cameron IC,
Vergani PV and Wyld L: Unfolded protein response activation
contributes to chemoresistance in hepatocellular carcinoma. Eur J
Gastroenterol Hepatol. 22:1099–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang L, Wang S, Wangtao, Wang Y, Wang J,
Jiang L, Li S, Hu X and Wang Q: Upregulation of GRP78 and GRP94 and
its function in chemotherapy resistance to VP-16 in human lung
cancer cell line SK-MES-1. Cancer Invest. 27:453–458. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kern J, Untergasser G, Zenzmaier C, Sarg
B, Gastl G, Gunsilius E and Steurer M: GRP-78 secreted by tumor
cells blocks the antiangiogenic activity of bortezomib. Blood.
114:3960–3967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang J, Yin Y, Hua H, Li M, Luo T, Xu L,
Wang R, Liu D, Zhang Y and Jiang Y: Blockade of GRP78 sensitizes
breast cancer cells to microtubules-interfering agents that induce
the unfolded protein response. J Cell Mol Med. 13((9B)): 3888–3897.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morrison DK: The 14-3-3 proteins:
Integrators of diverse signaling cues that impact cell fate and
cancer development. Trends Cell Biol. 19:16–23. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee JA, Park JE, Lee DH, Park SG, Myung
PK, Park BC and Cho S: G1 to S phase transition protein 1 induces
apoptosis signal-regulating kinase 1 activation by dissociating
14-3-3 from ASK1. Oncogene. 27:1297–1305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dong S, Kang S, Gu TL, Kardar S, Fu H,
Lonial S, Khoury HJ, Khuri F and Chen J: 14-3-3 integrates
prosurvival signals mediated by the AKT and MAPK pathways in
ZNF198-FGFR1-transformed hematopoietic cells. Blood. 110:360–369.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Obsilová V, Silhan J, Boura E, Teisinger J
and Obsil T: 14-3-3 proteins: A family of versatile molecular
regulators. Physiol Res. 57 (Suppl 3):S11–S21. 2008.PubMed/NCBI
|
|
51
|
Masters SC, Subramanian RR, Truong A, Yang
H, Fujii K, Zhang H and Fu H: Survival-promoting functions of
14-3-3 proteins. Biochem Soc Trans. 30:360–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ralhan R, Desouza LV, Matta A, Tripathi
SC, Ghanny S, Datta Gupta S, Bahadur S and Siu KW: Discovery and
verification of head-and-neck cancer biomarkers by differential
protein expression analysis using iTRAQ labeling, multidimensional
liquid chromatography, and tandem mass spectrometry. Mol Cell
Proteomics. 7:1162–1173. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ralhan R, Desouza LV, Matta A, Tripathi
SC, Ghanny S, Dattagupta S, Thakar A, Chauhan SS and Siu K:
WiTRAQ-multidimensional liquid chromatography and tandem mass
spectrometry-based identification of potential biomarkers of oral
epithelial dysplasia and novel networks between inflammation and
premalignancy. J Proteome Res. 8:300–309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Arora S, Matta A, Shukla NK, Deo SV and
Ralhan R: Identification of differentially expressed genes in oral
squamous cell carcinoma. Mol Carcinog. 42:97–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sharma R, Samantaray S, Shukla NK and
Ralhan R: Transcriptional gene expression profile of human
esophageal squamous cell carcinoma. Genomics. 81:481–488. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fan T, Li R, Todd NW, Qiu Q, Fang HB, Wang
H, Shen J, Zhao RY, Caraway NP, Katz RL, et al: Up-regulation of
14-3-3zeta in lung cancer and its implication as prognostic and
therapeutic target. Cancer Res. 67:7901–7906. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Neal CL, Yao J, Yang W, Zhou X, Nguyen NT,
Lu J, Danes CG, Guo H, Lan KH, Ensor J, et al: 14-3-3zeta
overexpression defines high risk for breast cancer recurrence and
promotes cancer cell survival. Cancer Res. 69:3425–3432. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang X, Cao W, Zhang L, Zhang W, Zhang X
and Lin H: Targeting 14-3-3zeta in cancer therapy. Cancer Gene
Ther. 19:153–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang X, Cao W, Zhou J, Zhang W, Zhang X,
Lin W, Fei Z, Lin H and Wang B: 14-3-3ζ positive expression is
associated with a poor prognosis in patients with glioblastoma.
Neurosurgery. 68:932–938, discussion 938. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chatterjee D, Goldman M, Braastad CD,
Darnowski J, Wyche JH, Pantazis P and Goodglick L: Reduction of
9-nitrocamptothecin-triggered apoptosis in DU-145 human prostate
cancer cells by ectopic expression of 14-3-3zeta. Int J Oncol.
25:503–509. 2004.PubMed/NCBI
|
|
61
|
Shen J, Person MD, Zhu J, Abbruzzese JL
and Li D: Protein expression profiles in pancreatic adenocarcinoma
compared with normal pancreatic tissue and tissue affected by
pancreatitis as detected by two-dimensional gel electrophoresis and
mass spectrometry. Cancer Res. 64:9018–9026. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jang JS, Cho HY, Lee YJ, Ha WS and Kim HW:
The differential proteome profile of stomach cancer: Identification
of the biomarker candidates. Oncol Res. 14:491–499. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Coelho DS and Domingos PM: Physiological
roles of regulated Ire1 dependent decay. Front Genet. 5:762014.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nishitoh H, Matsuzawa A, Tobiume K,
Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H: ASK1
is essential for endoplasmic reticulum stress-induced neuronal cell
death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang L, Chen J and Fu H: Suppression of
apoptosis signal-regulating kinase 1-induced cell death by 14-3-3
proteins. Proc Natl Acad Sci USA. 96:8511–8515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Goldman EH, Chen L and Fu H: Activation of
apoptosis signal-regulating kinase 1 by reactive oxygen species
through dephosphorylation at serine 967 and 14-3-3 dissociation. J
Biol Chem. 279:10442–10449. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Brennan GP, Jimenez-Mateos EM, McKiernan
RC, Engel T, Tzivion G and Henshall DC: Transgenic overexpression
of 14-3-3 zeta protects hippocampus against endoplasmic reticulum
stress and status epilepticus in vivo. PLoS One. 8:e544912013.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Brobey RK, Dheghani M, Foster PP, Kuro-O M
and Rosenblatt KP: Klotho regulates 14-3-3ζ monomerization and
binding to the ASK1 signaling complex in response to oxidative
stress. PLoS One. 10:e01419682015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yde CW and Issinger OG: Enhancing
cisplatin sensitivity in MCF-7 human breast cancer cells by
down-regulation of Bcl-2 and cyclin D1. Int J Oncol. 29:1397–1404.
2006.PubMed/NCBI
|
|
71
|
Lauritzen G, Jensen MB, Boedtkjer E,
Dybboe R, Aalkjaer C, Nylandsted J and Pedersen SF:
Cisplatin-induced cell death in MCF-7 breast cancer cells: Roles of
ΔNErbB2 and pH regulatory ion transporters NHE1 and NBCn1. Exp Cell
Res. 316:2538–2553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jiang Y, Ji F, Liu Y, He M, Zhang Z, Yang
J, Wang N, Zhong C, Jin Q, Ye X, et al: Cisplatin-induced autophagy
protects breast cancer cells from apoptosis by regulating
yes-associated protein. Oncol Rep. 38:3668–3676. 2017.PubMed/NCBI
|
|
73
|
Al-Taweel N, Varghese E, Florea AM and
Büsselberg D: Cisplatin (CDDP) triggers cell death of MCF-7 cells
following disruption of intracellular calcium
([Ca(2+)]i) homeostasis. J Toxicol Sci.
39:765–774. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lee AS: GRP78 induction in cancer:
Therapeutic and prognostic implications. Cancer Res. 67:3496–3499.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang M, Wey S, Zhang Y, Ye R and Lee AS:
Role of the unfolded protein response regulator GRP78/BiP in
development, cancer, and neurological disorders. Antioxid Redox
Signal. 11:2307–2316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST©) for group-wise
comparison and statistical analysis of relative expression results
in real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47(D1): D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bruno S and Darzynkiewicz Z: Cell cycle
dependent expression and stability of the nuclear protein detected
by Ki-67 antibody in HL-60 cells. Cell Prolif. 25:31–40. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
He Y, Liu Z, Qiao C, Xu M, Yu J and Li G:
Expression and significance of Wnt signaling components and their
target genes in breast carcinoma. Mol Med Rep. 9:137–143. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lukyanova NY, Rusetskya NV, Tregubova NA
and Chekhun VF: Molecular profile and cell cycle in MCF-7 cells
resistant to cisplatin and doxorubicin. Exp Oncol. 31:87–91.
2009.PubMed/NCBI
|
|
85
|
Salvesen GS and Abrams JM: Caspase
activation - stepping on the gas or releasing the brakes? Lessons
from humans and flies. Oncogene. 23:2774–2784. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Castri P, Lee YJ, Ponzio T, Maric D, Spatz
M, Bembry J and Hallenbeck J: Poly(ADP-ribose) polymerase-1 and its
cleavage products differentially modulate cellular protection
through NF-kappaB-dependent signaling. Biochim Biophys Acta.
1843:640–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lin JF, Lin YC, Tsai TF, Chen HE, Chou KY
and Hwang TIS: Cisplatin induces protective autophagy through
activation of BECN1 in human bladder cancer cells. Drug Des Devel
Ther. 2017.1517-1533.2017. View Article : Google Scholar
|
|
88
|
Pozuelo-Rubio M: Regulation of autophagic
activity by 14-3-3ζ proteins associated with class III
phosphatidylinositol-3-kinase. Cell Death Differ. 18:479–492. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lee AS: The glucose-regulated proteins:
stress induction and clinical applications. Trends Biochem Sci.
26:504–10. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hendershot LM: The ER function BiP is a
master regulator of ER function. Mt Sinai J Med. 71:289–297.
2004.PubMed/NCBI
|
|
91
|
Ye X, Zhou Xu and Zhang H: Exploring the
role of autophagy-related gene 5 (ATG5) yields important insights
into autophagy in autoimmune/autoinflammatory. Front Immunol.
9:2332018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhu Y, Zhao L, Liu L, Gao P, Tian W, Wang
X, Jin H, Xu H and Chen Q: Beclin 1 cleavage by caspase-3
inactivates autophagy and promotes apoptosis. Protein Cell.
1:468–477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lee AS, Brandhorst S, Rangel DF, Navarrete
G, Cohen P, Longo VD, Chen J, Groshen S, Morgan TE and Dubeau L:
Effects of prolonged GRP78 haploinsufficiency on organ homeostasis,
behavior, cancer and chemo-toxic resistance in aged mice. Sci Rep.
7:409192017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Suh DH, Kim MK, Kim HS, Chung HH and Song
YS: Unfolded protein response to autophagy as a promising druggable
target for anticancer therapy. Ann N Y Acad Sci. 1271:20–32. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tallóczy Z, Jiang W, Virgin HW IV, Leib
DA, Scheuner D, Kaufman RJ, Eskelinen EL and Levine B: Regulation
of starvation- and virus-induced autophagy by the eIF2α kinase
signaling pathway. Proc Natl Acad Sci USA. 99:190–195. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang L, Wang H, Ding K and Xu J: FTY720
induces autophagy related apoptosis and necroptosis in human
glioblastoma cells. Toxicol Lett. 236:432015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wirawan E, Vande Walle L, Kersse K,
Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R,
Verspurten J, Declercq W, et al: Caspase-mediated cleavage of
Beclin-1 inactivates Beclin-1-induced autophagy and enhances
apoptosis by promoting the release of proapoptotic factors from
mitochondria. Cell Death Dis. 1:e182010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Neumeister P, Pixley FJ, Xiong Y, Xie H,
Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, et al:
Cyclin D1 governs adhesion and motility of macrophages. Mol Biol
Cell. 14:2005–2015. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Holnthoner W, Pillinger M, Groger M, Wolff
K, Ashton AW, Albanese C, Neumeister P, Pestell RG and Petzelbauer
P: Fibroblast growth factor-2 induces Lef/Tcf-dependent
transcription in human endothelial cells. J Biol Chem.
277:45847–45853. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Barrios CH, Sampaio C, Vinholes J and
Caponero R: What is the role of chemotherapy in estrogen
receptor-positive, advanced breast cancer? Ann Oncol. 20:1157–1162.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Alqahtani A, Ayesh HSK and Halawani H:
PIK3CA gene mutations in solid malignancies: Association with
clinicopathological parameters and prognosis. Cancers (Basel).
12:932019. View Article : Google Scholar
|