Introduction
Obstructive sleep apnea (OSA) is a sleep-related
breathing disorder that has increased in prevalence in recent
years. OSA is a serious public health issue associated with
multiple health problems including cerebrovascular disease,
cardiovascular disease and pulmonary disease (1–6).
Characterized by chronic intermittent hypoxia (CIH), which results
from the upper airway being recurrently obstructed during sleep,
OSA is estimated to affect 3–7% of the adult population (7,8). In
addition, OSA has been associated with increased cancer mortality
and cancer incidence in some studies (9–11).
Lung cancer is a major cause of cancer-related
deaths worldwide, accounting for 27 and 25% of cancer deaths among
men and women, respectively (12).
Three major forms of hypoxia are present in solid tumors: Chronic,
acute and intermittent (13). The
correlation between OSA-related CIH and the development of lung
cancer has been studied, and accumulating evidence indicates that
intermittent hypoxia is a key regulator of the interplay between
cancer cells and endothelial cells in tumors, and it enhances
cancer progression and metastasis (14–17).
Furthermore, previous studies have shown that hypoxia in tumors is
correlated with a greater risk of metastasis, increased
invasiveness, and resistance to systemic and radiation therapy
(18). The mechanisms underlying
these systemic and cellular responses remain unknown and warrant
further investigation.
Endothelial cell-specific molecule-1 (ESM1) is a
newly reported molecule that is expressed in the lung, regulated by
inflammatory cytokines and associated with endothelial dysfunction
(19). Recent studies have found
that ESM-1 is markedly overexpressed in several types of cancer,
including clear cell renal cell carcinoma, gastric cancer and lung
cancer, thus making ESM1 a potential endothelial cell marker and a
possible target for cancer therapy (19–22).
Furthermore, silencing of ESM1 was found to inhibit cell survival,
migration and invasion, and to modulate cell cycle progression in
hepatocellular carcinoma (23).
According to recent research, ESM1 enhances adhesion between
monocytes and endothelial cells under intermittent hypoxic
conditions (24). An investigation
of the modulation of ESM1 under hypoxia has indicated that ESM-1
expression is induced by hypoxia-inducible factor-1α (HIF-1α)
(20). HIF-1α is correlated with
apoptosis in lung cancer and is commonly overexpressed in non-small
cell lung cancer (NSCLC) (25–27).
However, the interactions among CIH, ESM1 and HIF-1α require
further study.
In the present study, we constructed a lung cancer
stem cell (LCSC) model and mouse model with tumors in situ.
The expression levels of ESM1 and HIF-1α under CIH were
investigated. We detected whether CIH exposure enhanced lung cancer
cell survival, drug resistance and stemness. Cell proliferation,
migration and cell invasion were analyzed after CIH
preconditioning. We further identified the expression of related
indicators after small interfering (si)-ESM1 plasmid transfection
or lentivirus infection. The results showed that CIH promoted ESM1
and HIF-1α expression and led to drug resistance, cell
proliferation, migration and invasion. After ESM-1 suppression,
these CIH-induced effects were reversed. Overall, our results
suggest that ESM-1 may be a potential target in OSA-associated lung
cancer progression.
Materials and methods
Cell culture
Human lung cancer cells (PC-9 and A549 cells) were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in 90% Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo
Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin in a 37°C humidified incubator with 5%
CO2.
LCSC construction
PC9 and A549 cisplatin-resistant cells were obtained
through cisplatin repeated treatment with increasing doses (1 to 10
µmol/l), and cells were cultured in serum-free stem cell culture
medium (DMEM-F12 medium; insulin, 4 U/l; B27, 1X; EGF, 20 ng/ml;
and bFGF, 20 ng/ml) to construct the spheres.
Cell CIH exposure The cells were exposed to 5
sec of 14 to 15% O2 in every 60 sec cycle for 24 or 48
h. All cells were cultured for 48 h for further experiments.
Total RNA isolation and quantitative
reverse transcription-polymerase chain reaction (RT-qPCR)
RNA was isolated with TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions and was reverse-transcribed with a miScript Reverse
Transcription kit (Qiagen, Germany). RT-qPCR was performed with a
SYBR Premium Ex Taq II kit (Takara) on an ABI PRISM 7500 Sequence
Detection System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The amplification reactions were run with 30 thermocycles of
30 sec at 95°C, 30 sec at 55°C and 30 sec at 72°C. The expression
levels were calculated using the 2−ΔΔCq method. All
reactions were performed in triplicate, and the mean value was used
to calculate expression levels after normalization to β-actin. The
primer sequences are shown in Table
I.
 | Table I.Primers for the RT-qPCR analysis. |
Table I.
Primers for the RT-qPCR analysis.
| Name | Sequences
(5′-3′) |
|---|
| CD44 | F:
GACACATATTGTTTCAATGCTTCAGC |
|
| R:
GATGCCAAGATGATCAGCCATTCTGGAAT |
| CD133 | F:
TACAACGCCAAACCACGACTGT |
|
| R:
TCTGAACCAATGGAATTCAAGACCCTTT |
| ESM1 | R:
GCCATGTCATGCTCTTTGCAG |
|
| F:
GGCGGCACCACCATGTACCCT |
| β-actin | R:
AGGGGCCGGACTCGTCATACT |
|
| F:
CTTGCTACCGCACAGTCTCA |
Protein extraction and western blot
analysis
The lung tissue and LCSCs were lysed with RIPA
buffer, and the protein concentration was determined with a BCA
protein assay kit (Thermo Fisher Scientific, Inc.). Approximately
30 µg of protein from each sample was separated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
to a polyvinylidene fluoride membrane. Membranes were blocked in 5%
skim milk in TBST and incubated with primary antibodies to one of
the following (all from Abcam) overnight at 4°C: CD44 (ab157107,
1:1,000), CD133 (ab226355, 1:1,000), OCT4 (ab18976, 1:1,000), SOX2
(ab97959, 1:1,000), E-cadherin (ab40772, 1:1,000), N-cadherin
(ab18203, 1:1,000), Snail (ab53519, 1:1,000), Vimentin (ab92547,
1:1,000), ESM1 (ab103590, 1:1,000), HIF-1α (ab51608, 1:1,000) or
β-actin (ab8226, 1:1,500). Membranes were incubated with the
corresponding secondary antibody for 1 h at room temperature and
washed in TBST. Protein signals were detected with Super ECL Plus
Detection Reagent (Merck).
Cell viability assay
The Cell Counting Kit-8 (CCK-8) assay was used to
detect cell survival rates. Transfected cells were seeded into
96-well plates at a density of 5,000 cells/well in triplicate. Cell
viability was measured with the CCK-8 system (Gibco; Thermo Fisher
Scientific, Inc.) at different cisplatin concentrations after
seeding, according to the manufacturer's instructions.
For colony formation assays, transfected cells were
seeded into 6-well plates at a density of 2,000 cells/well and
maintained in DMEM containing 10% FBS for 10 days. The colonies
were fixed with 70% ethanol for 15 min at room temperature and
stained with 2% crystal violet. The positive colonies (more than 50
cells or 3 mm2 per colony) were counted. Colonies were imaged under
a light microscope (Olympus, Tokyo, Japan) and Results were
expressed as the average number of cells in every visual field
after they were fixed and stained.
Sphere formation assay
Cells were grown in MammoCult medium (Stem Cell
Technologies) supplemented with MammoCult Proliferation Supplements
(Stem Cell Technologies) and plated in 24-well plates with
ultra-low attachment at a density of 10,000 viable cells/ml and
grown for 10 days. Spheres were counted and photographed under a
light microscope (Olympus).
Transwell migration assay
Cell migration was analyzed with Transwell chambers
(Corning Inc.) in accordance with the manufacturer's protocol.
After incubation at 37°C for 48 h, cells on the upper surfaces of
the Transwell chambers were removed with cotton swabs, and cells
located on the lower surfaces were fixed with methanol for 10 min,
then stained with 0.1% crystal violet. Stained cells were imaged
under a light microscope (Olympus) and counted in five randomly
selected fields. In invasion experiments, chamber inserts were
coated with 200 mg/ml Matrigel and dried overnight under sterile
conditions.
Mouse orthotopic assay
A total of 24 male BALB/c-nu nude mice (6 weeks old,
weight 20±2 g) were purchased from Shanghai Laboratory Animal
Center (Shanghai, China). After anesthesia, a 5-mm incision was cut
at 1.5 cm above the arcus costarum of the left anterior axillary
line in each male nude mouse, and tissues were pulled apart until
the chest wall was exposed; a PC-9 suspension with a density of
1×107 ml was prepared, and then 50 µl cell suspension
and 50 ml Matrigel were mixed and subsequently injected into the
left lung of the mice through the chest wall at a depth of 3 mm.
After the injection, the needle was held in place for 5 sec and was
then removed, and the incision was sutured. At 1 week after the
operation, magnetic resonance imaging examination was performed to
determine whether the tumor had grown. On day 7 after cell
injection, CIH exposure was initiated. The oxygen content in the
CIH exposure chamber was measured over the course of several cycles
with an oxygen sensor placed on the bottom of the chamber. Animals
were exposed to 5 sec of 14 to 15% O2 in every 60 sec
cycle. Each challenge lasted 8 h during the daytime and was
repeated on days 7, 14, 21 and 28 and 2 days after the last
challenge. si-ESM1 lentivirus was purchased from HanBio. One
hundred microliters of filter-purified lentivirus cocktail
(1×105 IU/µl) was administered by intravenous injection
weekly on days 7, 14, 21 and 28 and 2 days after the last
challenge. All mice were treated for 5 weeks and sacrificed by
cervical dislocation following anesthesia with an intraperitoneal
injection of sodium pentobarbital (60 mg/kg).
This study was performed in strict accordance with
Guide for the Care and Use of Laboratory Animals (8th edition,
2011, published by The National Academies Press, http://www.nap.edu/catalog/12910/guide-for-the-care-and-use-of-laboratory-animals-eighth).
The protocol was reviewed and approved by the Shanghai Ninth
People's Hospital Institutional Review Board (permit no.
HKDL2013001b). They were raised in a constant temperature (22±1°C)
and humidity (65±5%), with a 12-h light/dark cycle, with standard
diet and water. All surgery was performed under sodium
pentobarbital anesthesia, and all efforts were made to minimize
animal suffering.
Enzyme-linked immunosorbent assay
(ELISA)
The concentrations of ESM1 in mouse serum were
determined with an ELISA kit (Nanjing Jiancheng Bioengineering
Institute), according to standard protocols.
Immunohistochemistry
Tumor tissue samples were fixed in 10% formalin and
embedded in paraffin. Sections (5-µm) were stained with Ki67 to
evaluate proliferation at 4°C overnight. The sections were then
washed in PBS and incubated at room temperature for 20 min with
biotinylated secondary antibody (Qiagen, Germany). Sections were
examined with an Axiophot light microscope at ×200 magnification
(Zeiss) and imaged with a digital camera.
Statistical analysis
All results in this study are presented as the mean
± SD and were statistically analyzed using GraphPad Prism 9.0
(GraphPad Software, Inc.). One-way ANOVA with post hoc test was
applied among the various groups. P<0.05 was considered to
indicate statistical significance.
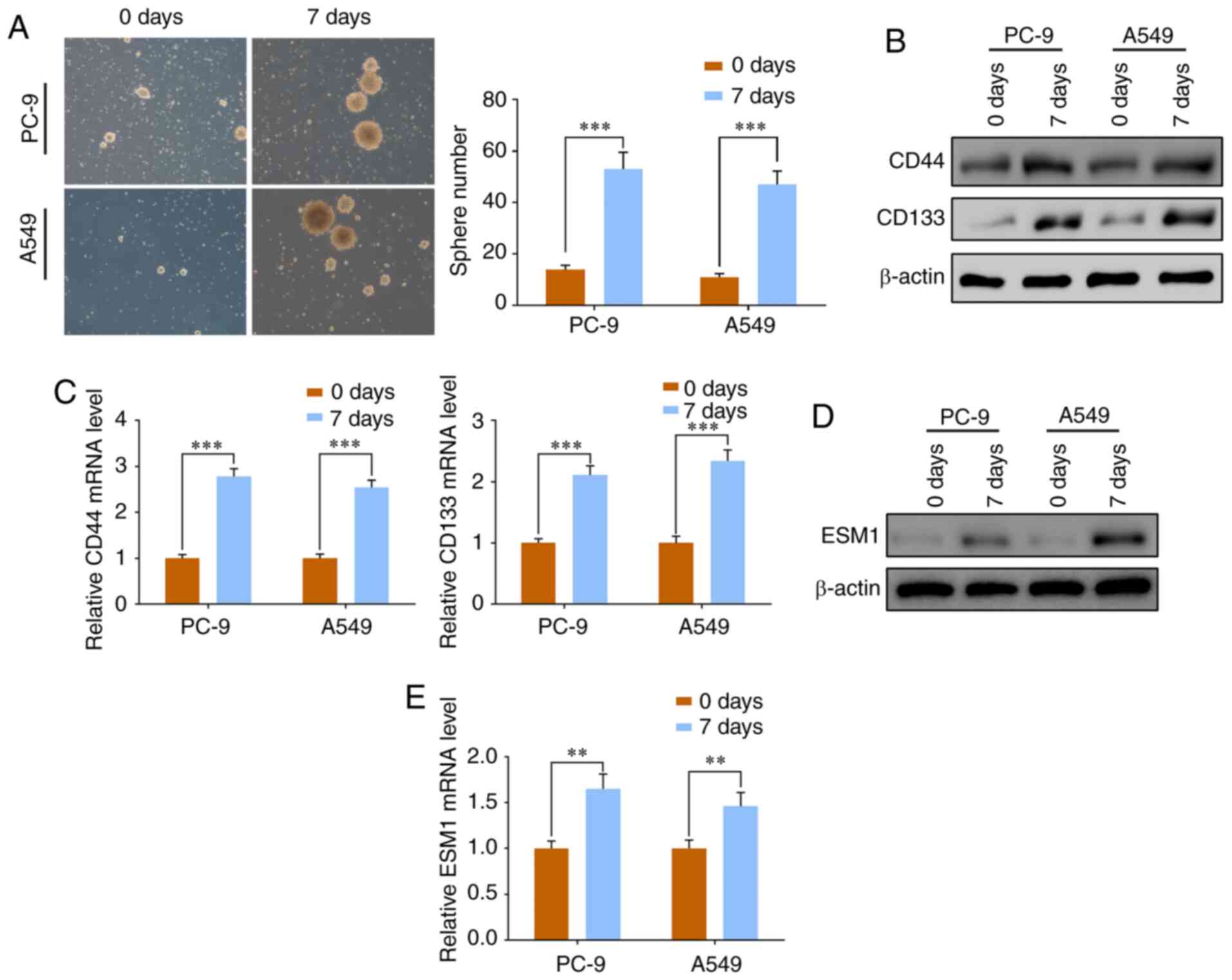
Results
ESM1 is overexpressed at both the
protein and gene levels in PC-9 and A549 cells after culture for 7
days
Repeated cisplatin treatments with increasing doses
(1 to 10 µmol/l) were applied to obtain PC-9 and A549
cisplatin-resistant cells. After 7 days in culture, the cell
proliferation led to a progressive increase in diameters and sphere
numbers, and the tumorspheres formed from NSCLC cell lines were
identified under a microscope (Fig.
1A). To further identify the cultured LCSCs, the CSC markers
CD44 and CD133 were used. Western blotting and RT-qPCR results
together showed that the cultured tumorigenic lung tumorspheres
exhibited significantly increased expression of CD133 and CD44 at
both the protein and mRNA levels (Fig.
1B and C).
Previous studies have identified that ESM1 is
markedly overexpressed in cancer cells (21,22,28).
In the present study, western blotting confirmed that the ESM1
expression increased significantly at the protein level in PC-9 and
A549 cells (Fig. 1D). RT-PCR
analysis further showed that ESM1 expression was significantly
enhanced at the mRNA level (Fig.
1E). These results indicated that the cisplatin-resistant LCSCs
obtained from PC-9 and A549 cell lines displayed stem-like features
and had enhanced expression of ESM1 at both the protein and mRNA
levels.
CIH exposure leads to drug resistance,
and promotes cell proliferation, migration and invasion in
vitro
Accumulating studies show that hypoxia in several
tumor types is associated with a greater risk of metastasis,
increased invasiveness and resistance to systemic and radiation
therapy (14–16). To investigate the cellular changes
caused by CIH, we exposed the cells to 5 sec of 14 to 15%
O2 in every 60 sec cycle for 24 or 48 h. All groups were
cultured for a total of 48 h. The viability of cisplatin-resistant
PC-9 and A549 cells was assessed with CCK-8 assays, and the results
showed greater cell viability in the 24-h CIH exposure group and
much higher cell viability in the 48-h group compared with the
control group (Fig. 2A). To further
detect the correlation between drug resistance and CIH, we added
different doses of cisplatin during the cell culture. Although the
cell viability was significantly decreased as the dose increased,
among the groups cultured with the same cisplatin dose, the groups
exposed to CIH for 24 h presented significantly higher cell
viability, and those exposed for 48 h showed the highest cell
viability (Fig. 2B).
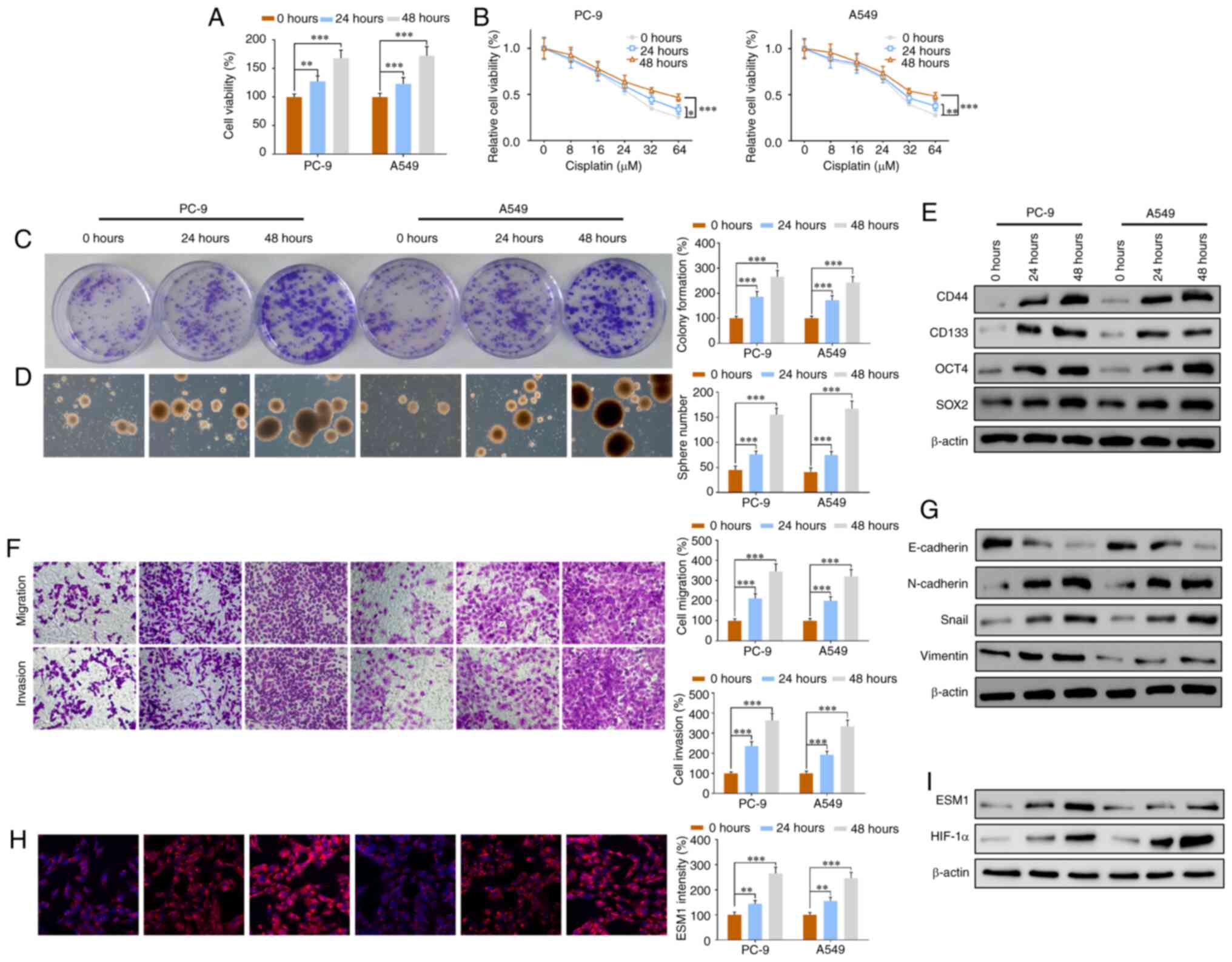 | Figure 2.CIH exposure leads to drug resistance
and promotes cell proliferation, migration and invasion in
vitro. The cells were exposed to 5 sec of 14 to 15%
O2 in every 60 sec cycle for 24 or 48 h. (A) CCK-8 assay
of cell viability in the PC-9 and A549 groups with 0, 24 or 48 h
CIH exposure. (B) Changes in cell viability with increasing doses
of cisplatin (0, 4, 1, 32 and 64 µM) in all groups. (C) Colony
formation assays of LCSCs from two cell lines before and after 24
or 48 h CIH exposure. (D) Images of PC-9 and A549 tumor spheres.
(E) Western blotting results of the CSC markers CD44, CD133, OCT4
and SOX2 in the PC-9 and A549 groups exposed to CIH for 0, 24 or 48
h. (F) Transwell assay results of cell migration and invasion in
two cell lines with or without CIH. (G) Western blotting results of
the EMT-associated proteins E-cadherin, N-cadherin, Snail and
vimentin in all groups. (H) Immunofluorescence staining results of
ESM1 and quantitative intensity in the PC-9 and A549 groups exposed
to CIH for 0, 24 or 48 h. (I) ESM1 and HIF-1α western blotting
results in all groups. Data are expressed as the means ± SEM (n=3);
*P<0.05, **P<0.01, ***P<0.001. CIH, chronic intermittent
hypoxia; LCSCs, lung cancer stem cells; ESM1, endothelial
cell-specific molecule-1; HIF-1α, hypoxia inducible factor-1α. |
Colony formation assay results showed that in the
PC-9 and A549 LCSC groups, cell proliferation increased markedly
after a 24-h CIH exposure and was further enhanced after CIH
exposure for 48 h (Fig. 2C).
Photographs of the tumorspheres provided additional confirmation of
the increasing sphere numbers (Fig.
2D). The protein expression levels of the stem cell markers
OCT-4, SOX2, CD44 and CD133 were investigated via western blotting.
OCT-4, SOX2, CD44 and CD 133 were significantly increased after
exposure to CIH for 24 h and greatly increased after exposure for
48 h, in both the PC-9 and A549 groups (Fig. 2E). Transwell migration assays
indicated that both cell migration and invasion were significantly
increased in a time-dependent manner after CIH exposure (Fig. 2F).
Epithelial-mesenchymal transition (EMT) is a key
factor in cell migration and invasion (29–31).
We analyzed the expression of EMT-associated proteins including
E-cadherin, N-cadherin, Snail and vimentin. Western blotting
results of these EMT markers showed that CIH enhanced the
expression of N-cadherin, Snail and vimentin, and inhibited the
expression of E-cadherin in a time-dependent manner (Fig. 2G). These results suggested that CIH
contributed to overexpression of EMT proteins and led to cell
migration and invasion. Immunofluorescence results showed that the
ESM1 intensity markedly increased after CIH exposure in both PC-9
and A549 LCSCs (Fig. 2H). Western
blot analysis confirmed the overexpression of ESM1 protein and
further indicated that HIF-1α expression was significantly
upregulated by CIH exposure (Fig.
2I). The results together suggested that CIH not only led to
drug resistance but also enhanced cell proliferation, migration,
invasion and EMT in vitro in a time-dependent manner.
Inhibition of ESM1 suppresses the
effects of CIH exposure in promoting drug resistance, cell
proliferation, migration and invasion in vitro
Our preceding experiments confirmed the upregulation
of ESM1 during CIH exposure. To investigate the biological role of
ESM1 in CIH-associated NSCLC progression and the underlying
cellular mechanism, we constructed si-ESM1 plasmids and transfected
them into PC-9 or A549 LCSCs to suppress the expression of ESM1.
The RT-PCR and western blotting results verified the efficiency of
si-ESM1 transfection in the PC-9 and A549 groups (Figs. 3A and S1A).
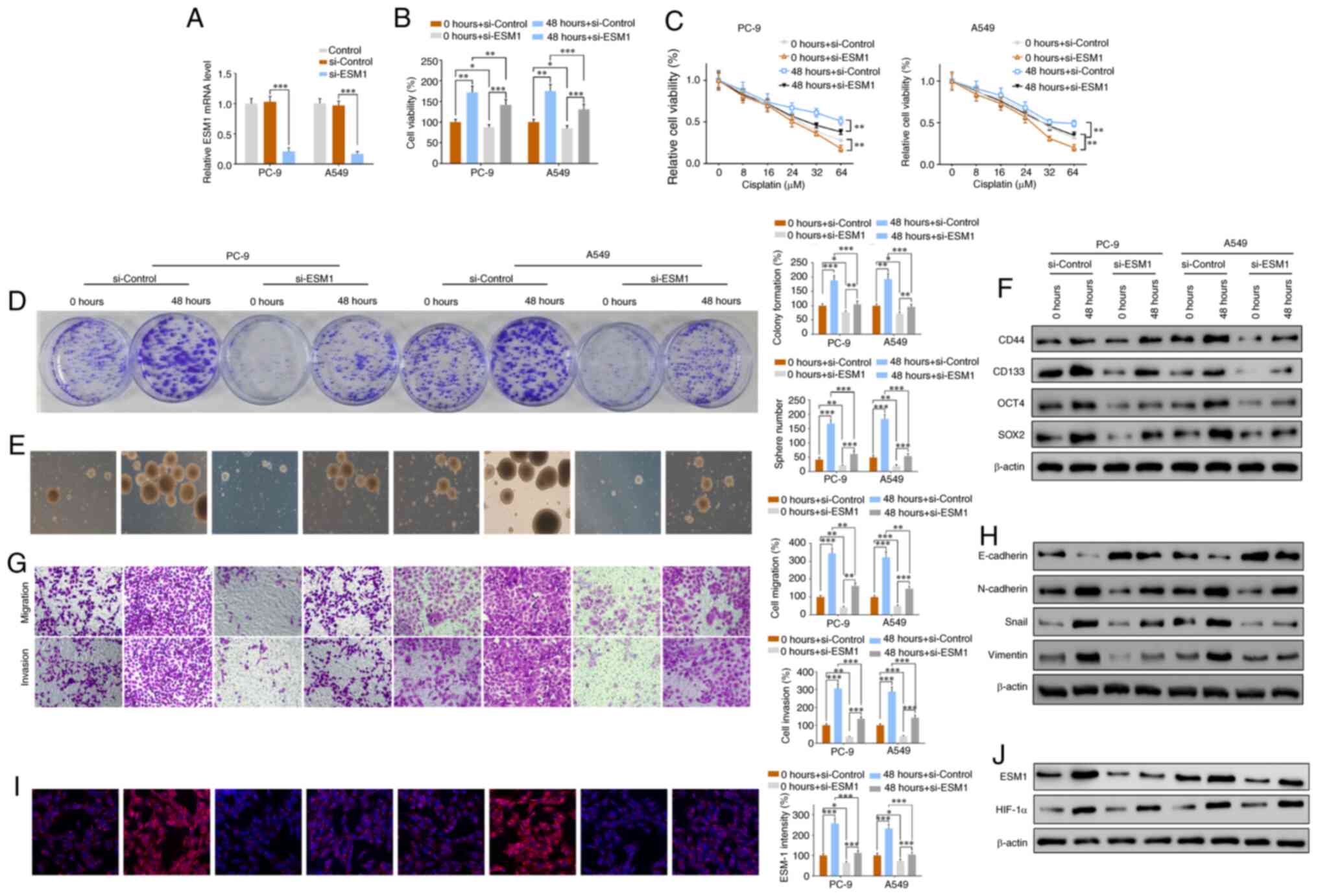 | Figure 3.si-ESM1 plasmid transfection
suppresses the effects of CIH exposure in promoting drug
resistance, cell proliferation, migration and invasion. si-ESM1
plasmids were constructed and transfected into PC-9 or A549 LCSCs
to suppress the expression of ESM1, and then exposed to a CIH
environment for 0 or 48 h. (A) RT-qPCR results verified the mRNA
level of ESM1 in the si-control and si-ESM1 groups. (B) CCK-8
assays of cell viability in the si-control and si-ESM1 groups with
0 or 48 h CIH. (C) PC-9 and A549 LCSC viability changes with
increasing doses of cisplatin (0, 4, 16, 32 and 64 µM) in the
si-control and si-ESM1 groups with 0 or 48 h CIH. (D) Colony
formation assays of LCSCs with or without si-ESM1 transfection. (E)
Images and quantitative sphere numbers. (F) Western blotting
results of the CSC markers CD44, CD133, OCT4 and SOX2 in the
si-control and si-ESM1 groups with 0 or 48 h CIH exposure. (G)
Transwell assay results of cell migration and invasion in the
si-control and si-ESM1 groups with 0 or 48 h CIH exposure. (H)
Western blotting results of the EMT-associated proteins E-cadherin,
N-cadherin, Snail and vimentin in all groups. (I)
Immunofluorescence staining results of ESM1 and quantitative
intensity in the si-control and si-ESM1 groups with 0 or 48 h CIH
exposure. (J) ESM1 and HIF-1α western blotting results in all
groups. Data are expressed as the means ± SEM (n=3); *P<0.05,
**P<0.01, ***P<0.001. CIH, chronic intermittent hypoxia;
LCSCs, lung cancer stem cells; ESM1, endothelial cell-specific
molecule-1; HIF-1α, hypoxia inducible factor-1α. |
CCK-8 assays showed that, although CIH increased the
cell viability in both the si-control and si-ESM1 groups, in the
48-h CIH exposure groups, si-ESM1 transfection, compared with
si-control transfection, inhibited the increase in cell viability
(Fig. 3B). Similar results were
obtained in drug-resistance analysis of PC-9 and A549; cell
viability decreased in a dose-dependent manner, but when the same
dose was used, si-ESM1 transfection significantly inhibited cell
viability in both the 0 and 48 h CIH groups (Figs. 3C and S1B). Cell proliferation was assessed via
colony formation assays. si-ESM1 did not prevent or eliminate the
CIH-induced effects on promoting proliferation, but compared with
the control group exposed to CIH for the same 48 h, the si-ESM1
group presented much lower colony formation (Fig. 3D).
Images and quantitative analysis of tumorspheres
showed that the sphere numbers were markedly lower in the si-ESM1
group than the control groups (Fig.
3E). To further investigate the changes in cell stemness, we
analyzed the expression of the CSC markers OCT-4, SOX2, CD44 and
CD133. In both the control and si-ESM1 groups, CIH led to
overexpression of OCT-4, SOX2, CD44 and CD 133 (Fig. 3F). However, si-ESM1 significantly
suppressed the expression of OCT-4, SOX2, CD44 and CD 133 at the
protein level, as compared with that in the si-control group, after
exposure to CIH for 48 h (Fig.
3F).
Transwell chamber assays showed that the 48-h CIH
exposure promoted cell migration and invasion in the control and
si-ESM1 groups, but the increase in the si-ESM1 groups was
significantly lower than that in the si-control groups (Fig. 3G). Western blot analysis of
EMT-associated proteins showed that CIH enhanced N-cadherin, Snail
and vimentin expression, whereas si-ESM1, compared with si-Control,
effectively inhibited the CIH-induced increase (Fig. 3H). According to immunofluorescence
assays, the ESM1 intensity increased in both the control and
si-ESM1 groups after CIH exposure, but the increase in si-ESM1 was
significantly lower than that in the control group (Fig. 3I). Western blotting results
confirmed the same ESM1 expression changes and showed that
CIH-induced overexpression of HIF-1α was inhibited by si-ESM1
transfection (Fig. 3J). These
results together suggested that si-ESM1 significantly suppresses
the CIH-mediated promotion of drug resistance, cell proliferation,
migration, invasion and EMT in PC-9 and A549 cells.
CIH promotes tumor growth,
proliferation and EMT, whereas ESM1 knockdown reverses the
CIH-induced effects in a mouse model
Mice were injected with a PC-9 cell suspension into
the left lung through the chest wall, and tumor generation was
confirmed by magnetic resonance imaging. To further investigate the
effects of CIH on histopathologic changes and tumor growth in lung
tissue in a mouse model, we infected the mice with si-ESM1
lentivirus and/or exposed them to CIH for 48 h. Gross findings
showed that CIH enhanced the tumor volumes and numbers in both the
si-control and si-ESM1 groups (Fig.
4A). However, the CIH-induced increase in the control group was
much greater than that in si-ESM1 group, thus indicating that
si-ESM1 effectively suppresses tumor growth (Fig. 4A). H&E staining, Ki-67
immunohistochemistry and TUNEL assay results together showed that
CIH promoted tumor growth and cell proliferation, whereas si-ESM1
reversed these effects (Fig.
4B-D).
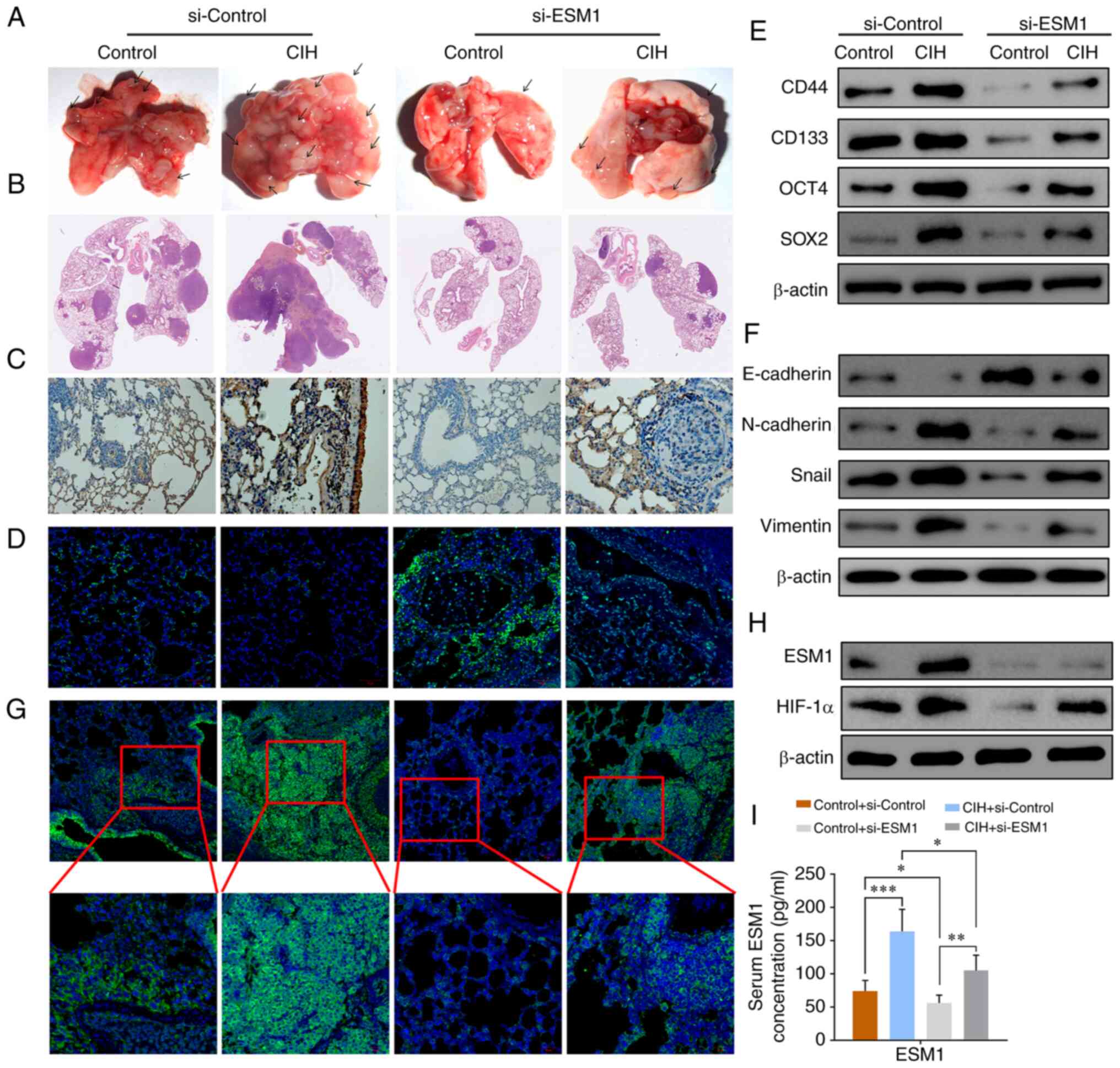 | Figure 4.CIH promotes tumor growth,
proliferation and EMT, whereas ESM1 knockout reverses the
CIH-induced effects in a mouse model. The mice were injected with a
PC-9 cell suspension into the left lung through the chest wall, and
tumor generation was confirmed by magnetic resonance imaging
examination. (A) Tumor volumes and numbers in lungs from both the
si-control and si-ESM1 groups with or without CIH. (B) H&E
staining results of lung sections from both the si-control and
si-ESM1 groups with or without CIH. (C) Immunohistochemistry
results for Ki-67, indicating cell proliferation and migration. (D)
TUNEL assays of the si-control and si-ESM1 groups. (E) Western
blotting results of the CSC markers CD44, CD133, OCT4 and SOX2 in
the si-control and si-ESM1 groups with or without CIH exposure. (F)
Western blotting results of the EMT-associated proteins E-cadherin,
N-cadherin, Snail and vimentin in all groups. (G)
Immunofluorescence assay of ESM1 intensity in all groups. (H) ESM1
and HIF-1α western blotting results in all groups. (I) ELISA
results of ESM1 in the serum from the si-control and si-ESM1 groups
with or without CIH exposure. Data are expressed as the means ± SEM
(n=6); *P<0.05, **P<0.01, ***P<0.001. CIH, chronic
intermittent hypoxia; ESM1, endothelial cell-specific molecule-1;
EMT, epithelial-mesenchymal transition; HIF-1α, hypoxia inducible
factor-1α. |
Western blot analysis showed that the CSC markers
CD44, CD133, OCT4 and SOX2 were highly overexpressed after CIH
exposure, whereas si-ESM1 lentivirus infection suppressed the
overexpression at the protein level (Fig. 4E). Overexpression of the EMT-related
proteins N-cadherin, Snail and vimentin was detected after CIH for
48 h, whereas si-ESM1 effectively inhibited the expression of these
proteins (Fig. 4F). ELISA analysis
indicated that ESM1 expression in mouse serum increased
significantly after CIH exposure, whereas si-ESM1 effectively
suppressed the CIH-enhanced ESM1 expression (Fig. 4I). Western blotting results
confirmed the function of si-ESM1 in suppressing ESM1 levels and
indicated that CIH-induced HIF-1α overexpression was also inhibited
via si-ESM1 transfection (Fig. 4H).
Similarly, the immunofluorescence results showed that the level of
ESM1 in tumor tissues was inhibited by si-ESM1 and was increased
after CIH exposure (Fig. 4G). These
results together suggested that si-ESM lentivirus transfection
effectively suppresses CIH-induced overexpression of cell
proliferation, tumor growth, EMT and HIF-1α.
Discussion
In the present study, our findings indicate a direct
correlation between chronic intermittent hypoxia (CIH) and the
enhancement of lung cancer stem cell (LCSC) NSCLC progression.
Intermittent hypoxia was previously found to enhance melanoma
metastasis to the lung in a mouse model of sleep apnea (15). In addition, accumulating evidence
shows that hypoxia is a key regulator of angiogenesis in cancer
(32). We found that CIH exposure
markedly promoted tumor growth, cell spheres, EMT-associated
proteins, and cell proliferation, migration and invasion, thereby
promoting carcinogenesis in PC-9 and A549 LCSC cell lines and in
CIH-exposed mice with lung tumors. Song and colleagues identified
that hypoxia directly induces resistance to cisplatin and
doxorubicin in NSCLC (33). We
confirmed that obstructive sleep apnea (OSA)-associated CIH
promoted drug resistance to cisplatin in vitro in a
time-dependent manner. Furthermore, our findings suggest that
endothelial cell-specific molecule-1 (ESM1) plays an important role
in CIH-mediated drug resistance, and cell proliferation, migration
and invasion in vitro, as well as in NSCLC mouse models. In
addition, si-ESM1 effectively suppressed the CIH-induced
aggravation of tumor growth, drug resistance and metastasis.
Cell proliferation and migration comprise the core
mechanism of tumor metastasis. Highly proliferative tumors are
often highly invasive (34).
Previous studies have found that hypoxia stimulates the
proliferation and migration of endothelial cells and enhances the
proliferation and tissue formation of human mesenchymal stem cells
(35,36). In this study, animals and cells were
exposed to 5 sec of 14 to 15% O2 in every 60 sec cycle
to simulate CIH exposure. CIH substantially stimulated cell
proliferation, migration and invasion, whereas si-ESM1 reversed the
effects of CIH in promoting tumor growth and metastasis. EMT plays
a key part in the progression of cell migration and invasion. We
identified that levels of N-cadherin, Snail and vimentin, which are
EMT-associated proteins, were significantly increased during CIH
exposure. Although EMT might still have occurred under CIH
conditions with si-ESM1 treatment, si-ESM1 functioned as an
EMT-suppressor and attenuated the increases in N-cadherin, Snail
and vimentin in this study. However, the specific signaling
pathways linking ESM-1 and EMT require further investigation.
According to a previous study, hypoxia can lead to
therapeutic resistance through direct effects, owing to a lack of
oxygen, which some drugs and radiation therapies require for
maximal cytotoxicity, as well as indirect effects through altered
cellular metabolism, which decreases drug cytotoxicity (37). Accumulating evidence indicates the
major contribution of hypoxia and HIF-1 to drug resistance in a
wide spectrum of neoplastic cells (38). A serious and primary problem in lung
cancer chemotherapy is the emergence of inherent and acquired drug
resistance in cancer cells (39).
In this study, PC-9 and A549 cisplatin-resistant cells were
obtained by repeated cisplatin treatment with increasing doses (1
to 10 µmol/l). Tumorsphere formation was used for enriching LCSC
cells. CIH enhanced cisplatin resistance in a CIH environment,
whereas si-ESM1 transfection effectively prevented the increase in
drug resistance. Given that OSA complications in lung cancer are
common, ESM1 might serve as a therapeutic target for inhibiting the
damage due to CIH in LCSC.
ESM1, a molecule regulated by inflammatory cytokines
and associated with endothelial dysfunction, is markedly
overexpressed in many types of cancer (19–21,23,24,28).
The expression of ESM1 is regulated by HIF-1α, which has a key role
in adaptation to hypoxic environments (40,41).
Hypoxia inducible factor-1α (HIF-1α) was highly upregulated by CIH
exposure, whereas si-ESM1 inhibited the overexpression of HIF-1α.
In some studies, HIF-1α has been reported to be an important
mediator of apoptosis and proliferation (25,42).
Accumulating evidence suggests that HIF-1α may be an upstream
regulator of ESM1; in some studies ESM1 was found to regulate
HIF-1α (43,44). Our study implies that HIF-1α and
ESM1 are both important factors in the downstream signaling
pathways involved in CIH-induced tumor progression. Targeting ESM1
inhibited CIH-induced and HIF-1α-mediated tumor growth, as well as
cell proliferation, migration and invasion. However, the specific
molecular interaction between ESM1 and HIF-1α still requires
further investigation, as does the mechanism linking CIH, ESM1 and
HIF-1α. Our research still has some limitations for many reasons.
Previous studies have shown the effect of CIH on lung cancer
(13,45,46).
Hence, our article only focused on the mechanism of CIH on LCSCs.
Whether the research results can be more widely applicable to NSCLC
still needs further research.
Collectively, our findings suggest that CIH enhances
tumorsphere formation, drug resistance, cell proliferation,
migration and invasion in LCSC progression via an ESM1 and HIF-1α
signaling pathway. ESM1-knockout sufficiently suppressed the
effects of CIH in promoting tumor growth and metastasis. Together,
our results indicate that ESM1 may serve as a new therapeutic
target in the treatment of OSA-associated NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by research grants from The
National Natural Science Foundation of China (grant no. 81700086)
and Shanghai Municipal Commission of Health and Family Planning
Foundation (no. 20134087).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
XG and LL designed the experiments. XG and JZ
performed most of the experiments with the assistance of YS and HS.
XG and YC collected and analyzed the data. YL validated the data
analysis. XG drafted the manuscript and LL revised the draft. All
authors approved the final manuscript before submission.
Ethics approval and consent to
participate
The present study was performed in strict accordance
with Guide for the Care and Use of Laboratory Animals (8th edition,
2011, published by The National Academies Press (https://www.nap.edu/catalog/12910/guide-for-the-care-and-use-of-laboratory-animals-eighth).
The protocol was reviewed and approved by the Shanghai Ninth
People's Hospital Institutional Review Board (permit no.
HKDL2013001b). All surgery was performed under sodium pentobarbital
anesthesia, and all efforts were made to minimize animal
suffering.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
OSA
|
obstructive sleep apnea
|
|
CIH
|
chronic intermittent hypoxia
|
|
NSCLC
|
non-small cell lung cancer
|
|
siRNA
|
small interfering RNA
|
|
ESM1
|
endothelial cell-specific
molecule-1
|
|
HIF-1α
|
hypoxia inducible factor-1α
|
|
CSC
|
cancer stem cell
|
|
EMT
|
epithelial-mesenchymal transition
|
|
RT-qPCR
|
quantitative reverse transcription
polymerase chain reaction
|
References
|
1
|
Aloia MS, Arnedt JT, Davis JD, Riggs RL
and Byrd D: Neuropsychological sequelae of obstructive sleep
apnea-hypopnea syndrome: A critical review. J Int Neuropsychol Soc.
10:772–785. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Golbin JM, Somers VK and Caples SM:
Obstructive sleep apnea, cardiovascular disease, and pulmonary
hypertension. Proc Am Thorac Soc. 5:200–206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olson EJ, Moore WR, Morgenthaler TI, Gay
PC and Staats BA: Obstructive sleep apnea-hypopnea syndrome. Mayo
Clinic Proceedings. Elsevier; pp. 1545–1552. 2003, View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pihtili A, Bingol Z, Kiyan E, Cuhadaroglu
C, Issever H and Gulbaran Z: Obstructive sleep apnea is common in
patients with interstitial lung disease. Sleep Breathing.
17:1281–1288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Redline S, Yenokyan G, Gottlieb DJ, Shahar
E, O'Connor GT, Resnick HE, Diener-West M, Sanders MH, Wolf PA,
Geraghty EM, et al: Obstructive sleep apnea-hypopnea and incident
stroke: The sleep heart health study. Am J Respir Crit Care Med.
182:269–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yaggi HK, Concato J, Kernan WN, Lichtman
JH, Brass LM and Mohsenin V: Obstructive sleep apnea as a risk
factor for stroke and death. N Engl J Med. 353:2034–2041. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Punjabi NM: The epidemiology of adult
obstructive sleep apnea. Proc Am Thorac Soc. 5:136–143. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Young T, Palta M, Dempsey J, Skatrud J,
Weber S and Badr S: The occurrence of sleep-disordered breathing
among middle-aged adults. N Engl J Med. 328:1230–1235. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campos-Rodriguez F, Martinez-Garcia MA,
Martinez M, Duran-Cantolla J, Peña Mde L, Masdeu MJ, Gonzalez M,
Campo FD, Gallego I, Marin JM, et al: Association between
obstructive sleep apnea and cancer incidence in a large multicenter
Spanish cohort. Am J Respir Crit Care Med. 187:99–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kendzerska T, Leung RS, Hawker G,
Tomlinson G and Gershon AS: Obstructive sleep apnea and the
prevalence and incidence of cancer. CMAJ. 186:985–992. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martínez-García MA, Campos-Rodriguez F,
Durán-Cantolla J, de la Peña M, Masdeu MJ, González M, Del Campo F,
Serra PC, Valero-Sánchez I, Ferrer MJ, et al: Obstructive sleep
apnea is associated with cancer mortality in younger patients.
Sleep Med. 15:742–748. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Y, Hou L, Yu F, Lu G, Qin S, Xie R,
Yang H, Wu T, Luo P, Chai L, et al: Incidence and physiological
mechanism of carboplatin-induced electrolyte abnormality among
patients with non-small cell lung cancer. Oncotarget.
8:18417–18423. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Song X, Wang X, Wei L, Liu X, Yuan
S and Lv L: Effect of chronic intermittent hypoxia on biological
behavior and hypoxia-associated gene expression in lung cancer
cells. J Cell Biochem. 111:554–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Almendros I, Montserrat JM, Ramirez J,
Torres M, Duran-Cantolla J, Navajas D and Farré R: Intermittent
hypoxia enhances cancer progression in a mouse model of sleep
apnoea. Eur Respir J. 39:215–217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Almendros I, Montserrat JM, Torres M,
Dalmases M, Cabañas ML, Campos-Rodríguez F, Navajas D and Farré R:
Intermittent hypoxia increases melanoma metastasis to the lung in a
mouse model of sleep apnea. Respir Physiol Neurobiol. 186:303–307.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Almendros I, Wang Y, Becker L, Lennon FE,
Zheng J, Coats BR, Schoenfelt KS, Carreras A, Hakim F, Zhang SX, et
al: Intermittent hypoxia-induced changes in tumor-associated
macrophages and tumor malignancy in a mouse model of sleep apnea.
Am J Respir Crit Care Med. 189:593–601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toffoli S and Michiels C: Intermittent
hypoxia is a key regulator of cancer cell and endothelial cell
interplay in tumours. FEBS. 275:2991–3002. 2008. View Article : Google Scholar
|
|
18
|
Verduzco D, Lloyd M, Xu L, Ibrahim-Hashim
A, Balagurunathan Y, Gatenby RA and Gillies RJ: Intermittent
hypoxia selects for genotypes and phenotypes that increase
survival, invasion, and therapy resistance. PLoS One.
10:e01209582015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lassalle P, Molet S, Janin A, Heyden JV,
Tavernier J, Fiers W, Devos R and Tonnel AB: ESM-1 is a novel human
endothelial cell-specific molecule expressed in lung and regulated
by cytokines. J Biol Chem. 271:20458–20464. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JH, Park MY, Kim CN, Kim KH, Kang HB,
Kim KD and Kim JW: Expression of endothelial cell-specific
molecule-1 regulated by hypoxia inducible factor-1α in human colon
carcinoma: Impact of ESM-1 on prognosis and its correlation with
clinicopathological features. Oncol Rep. 28:1701–1708. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu N, Zhang LH, Du H, Hu Y, Zhang GG,
Wang XH, Li JY and Ji JF: Overexpression of endothelial cell
specific molecule-1 (ESM-1) in gastric cancer. Ann Surg Oncol.
17:2628–2639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sarrazin S, Adam E, Lyon M, Depontieu F,
Motte V, Landolfi C, Lortat-Jacob H, Bechard D, Lassalle P and
Delehedde M: Endocan or endothelial cell specific molecule-1
(ESM-1): A potential novel endothelial cell marker and a new target
for cancer therapy. Biochim Biophys Acta. 1765:25–37.
2006.PubMed/NCBI
|
|
23
|
Kang YH, Ji NY, Lee CI, Lee HG, Kim JW,
Yeom YI, Kim DG, Yoon SK, Kim JW, Park PJ and Song EY: ESM-1
silencing decreased cell survival, migration, and invasion and
modulated cell cycle progression in hepatocellular carcinoma. Amino
Acids. 40:1003–1013. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun H, Zhang H, Li K, Wu H, Zhan X, Fang
F, Qin Y and Wei Y: ESM-1 promotes adhesion between monocytes and
endothelial cells under intermittent hypoxia. J Cell Physiol.
234:1512–1521. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan LF, Diao LM, Chen DJ, Liu MQ, Zhu LQ,
Li HG, Tang ZJ, Xia D, Liu X and Chen HL: Expression of HIF-1 alpha
and its relationship to apoptosis and proliferation in lung cancer.
Ai Zheng. 21:254–258. 2002.(In Chinese). PubMed/NCBI
|
|
26
|
Giatromanolaki A, Koukourakis MI, Sivridis
E, Turley H, Talks K, Pezzella F, Gatter KC and Harris AL: Relation
of hypoxia inducible factor 1 alpha and 2 alpha in operable
non-small cell lung cancer to angiogenic/molecular profile of
tumours and survival. Br J Cancer. 85:881–890. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Swinson DE, Jones JL, Cox G, Richardson D,
Harris AL and O'Byrne KJ: Hypoxia-inducible factor-1 alpha in non
small cell lung cancer: Relation to growth factor, protease and
apoptosis pathways. Int J Cancer. 111:43–50. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leroy X, Aubert S, Zini L, Franquet H,
Kervoaze G, Villers A, Delehedde M, Copin MC and Lassalle P:
Vascular endocan (ESM-1) is markedly overexpressed in clear cell
renal cell carcinoma. Histopathology. 56:180–187. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Diepenbruck M and Christofori G:
Epithelial-mesenchymal transition (EMT) and metastasis: Yes, no,
maybe? Curr Opin Cell Biol. 43:7–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guarino M: Epithelial-mesenchymal
transition and tumour invasion. Int J Biochem Cell Biol.
39:2153–2160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wong IY, Javaid S, Wong EA, Perk S, Haber
DA, Toner M and Irimia D: Collective and individual migration
following the epithelial-mesenchymal transition. Nat Mater.
13:1063–1071. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liao D and Johnson RS: Hypoxia: A key
regulator of angiogenesis in cancer. Cancer Metastasis Rev.
26:281–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song X, Liu X, Chi W, Liu Y, Wei L, Wang X
and Yu J: Hypoxia-induced resistance to cisplatin and doxorubicin
in non-small cell lung cancer is inhibited by silencing of
HIF-1alpha gene. Cancer Chemother Pharmacol. 58:776–784. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garay T, Juhász É, Molnár E, Eisenbauer M,
Czirók A, Dekan B, László V, Hoda MA, Döme B, Tímár J, et al: Cell
migration or cytokinesis and proliferation? -revisiting the ‘go or
grow’ hypothesis in cancer cells in vitro. Exp Cell Res.
319:3094–3103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grayson WL, Zhao F, Bunnell B and Ma T:
Hypoxia enhances proliferation and tissue formation of human
mesenchymal stem cells. Biochem Biophys Res Commun. 358:948–953.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meininger CJ, Schelling ME and Granger HJ:
Adenosine and hypoxia stimulate proliferation and migration of
endothelial cells. Am J Physiol. 255:H554–H562. 1988.PubMed/NCBI
|
|
37
|
Teicher BA: Hypoxia and drug resistance.
Cancer Metastasis Rev. 13:139–168. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rohwer N and Cramer T: Hypoxia-mediated
drug resistance: Novel insights on the functional interaction of
HIFs and cell death pathways. Drug Resist Updat. 14:191–201. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nishio K, Nakamura T, Koh Y, Suzuki T,
Fukumoto H and Saijo N: Drug resistance in lung cancer. Curr Opin
Oncol. 11:109–115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen C, Nettleton D, Jiang M, Kim SK and
Powell-Coffman JA: Roles of the HIF-1 hypoxia-inducible factor
during hypoxia response in Caenorhabditis elegans. J Biol Chem.
280:20580–20588. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hänze J, Eul BG, Savai R, Krick S, Goyal
P, Grimminger F, Seeger W and Rose F: RNA interference for
HIF-1alpha inhibits its downstream signalling and affects cellular
proliferation. Biochem Biophys Res Commun. 312:571–577. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jin H, Rugira T, Ko YS, Park SW, Yun SP
and Kim HJ: ESM-1 Overexpression is involved in increased
tumorigenesis of radiotherapy-resistant breast cancer cells.
Cancers (Basel). 12:13632020. View Article : Google Scholar
|
|
44
|
Zhu Y, Zhang X, Qi L, Cai Y, Yang P, Xuan
G and Jiang Y: HULC long noncoding RNA silencing suppresses
angiogenesis by regulating ESM-1 via the PI3K/Akt/mTOR signaling
pathway in human gliomas. Oncotarget. 7:14429–14440. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Campillo N, Torres M, Vilaseca A, Nonaka
PN, Gozal D, Roca-Ferrer J, Picado C, Montserrat JM, Farré R,
Navajas D and Almendros I: Role of Cyclooxygenase-2 on intermittent
hypoxia-induced lung tumor malignancy in a mouse model of sleep
apnea. Sci Rep. 7:446932017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kang HS, Kwon HY, Kim IK, Ban WH, Kim SW,
Kang HH, Yeo CD and Lee SH: Intermittent hypoxia exacerbates tumor
progression in a mouse model of lung cancer. Sci Rep. 10:18542020.
View Article : Google Scholar : PubMed/NCBI
|