Introduction
Glioblastoma (GBM) is the most aggressive primary
intracranial malignancy of the central nervous system, and is often
lethal (1–3). Currently, gliomas are classified into
four grades (I–IV) by the World Health Organization according to
their prognostic histopathological characteristics, of which, grade
IV GBM is the most lethal (4).
Despite significant advances in GBM research and treatment
strategies including surgery, adjuvant postoperative radiotherapy
and chemotherapy, the 5-year survival rate of glioma patients
remains <5% after diagnosis (5).
Therefore, novel treatment regimens, such as the development of
novel drugs that target proliferating tumor cells, are required to
prolong the survival of patients with brain tumors, particularly
those with GBM.
Reactive oxygen species (ROS) are a group of
short-lived, highly reactive, oxygen-containing byproducts of
aerobic metabolism, including superoxide, hydroxyl radical, and
hydroxides (6,7). Cellular ROS are primarily produced by
mitochondria, NADPH oxidase, peroxisomes and in the endoplasmic
reticulum (8–10). Emerging data have demonstrated that
ROS serve as a double-edged sword in cellular processes (11). Low levels of ROS are required for
cellular survival and proliferation (12,13).
However, excessive ROS results in oxidative stress, resulting in
DNA damage, apoptosis, and necrosis (14,15).
DNA damage, induced by chemical and physical factors, is capable of
initiating a series of processes, including cell cycle arrest, DNA
repair, regulation of cell checkpoints and initiation of apoptosis
(16). Increasing cellular ROS
production, to induce DNA damage and cell death is a well-known
effective anticancer strategy.
Parthenolide (PTL), a sesquiterpene lactone isolated
from the shoots of Tanacetum parthenium (feverfew), has
attracted increased attention owing to its antitumor activities in
various human cancer cell lines (17). PTL induces cytotoxicity in a wide
variety of solid tumors, including colorectal cancer, melanoma,
pancreatic cancer, breast cancer, prostate cancer and GBM (18–27),
but does not exert a notable effect on normal tissues (28). Although PTL possesses potency in
vitro, lack of stability under both acidic and basic conditions
as well as in media containing 0.5% serum and its poor solubility
are the major limitations for preventing its pharmacological
applications (29–31). Moreover, the detailed mechanism of
how PTL suppresses GBM growth still has not been fully elucidated.
In particular, the effect on DNA damage and cell death induced by
ROS has not been reported yet in GBM.
The aim of the present study was to synthesize PTL
derivatives by improving its solubility for potential future
medical use. DMAPT-D6, a derivative of dimethylaminoparthenolide
(DMAPT), was shown to suppress the growth of GBM cells by inducing
DNA damage initiated by excessive ROS accumulation. Moreover,
mechanism analysis demonstrated that the DNA damage induced by
DMAPT-D6 further induced cell cycle arrest at the S-phase and cell
death receptor-mediated extrinsic apoptosis, suggesting that
DMAPT-D6 is a promising anticancer agent, which increases ROS
levels, resulting in the death of cancer cells.
Materials and methods
Reagents and antibodies
The derivatives of PTL were dissolved in DMSO
(Thermo Fisher Scientific, Inc.) to obtain a stock solution of 40
mM, followed by further dilution with culture medium at various
concentrations. Cells were treated with the compounds at the
designated concentrations for 48 h, and DMSO was used as the
vehicle. Penicillin-streptomycin, MTT and propidium iodide (PI)
were purchased from Sigma-Aldrich; Merck KGaA. DMEM was purchased
from HyClone (Cytiva) and FBS was purchased from Gibco (Thermo
Fisher Scientific, Inc.). Z-DEVD-FMK was purchased from Topscience,
and Z-IETD-FMK and N-acetylcysteine (NAC) were purchased from
MedChemExpress. All the primary antibodies used in the present
study were purchased from Cell Signaling Technology, Inc.: Anti-P27
(cat. no. 3686S, 1:1,000), anti-CDK1 (cat. no. 9116S, 1:1,000),
anti-CDK2 (cat. no. 2546S, 1:1,000), anti-cyclin B1 (cat. no.
4138S, 1:1,000), anti-cyclin E1 (cat. no. 20808S, 1:1,000),
anti-β-actin (cat. no. 4970S, 1:1,000), anti-nuclear factor-like 2
(NRF2; cat. no. 12721S, 1:1,000), anti-λH2AX (cat. no. 9718S,
1:1,000), anti-p53-binding protein 1 (53BP1; cat. no. 88439S,
1:1,000), anti-DNA Ligase IV (cat. no. 14649S, 1:1,000), anti-DR3
(cat. no. 45901S, 1:1,000), anti-DR5 (cat. no. 3696S, 1:1,000),
anti-FADD (cat. no. 2782S, 1:1,000), anti-TRADD (cat. no. 364S,
1:1,000), anti-cleaved caspase-8 (cat. no. 9496S, 1:1,000),
anti-PARP (cat. no. 9532S, 1:1,000), anti-caspase-3 (cat. no.
9662S, 1:1,000) and anti-cleaved caspase-3 (cat. no. 9664S,
1:1,000). The secondary antibodies used for western blotting were
IRDye® 680RD donkey anti-mouse IgG secondary antibody
(cat. no. 926-68072, 1:15,000) and IRDye® 800CW donkey
anti-rabbit IgG secondary antibody (cat. no. 926-32213, 1:15,000),
and were purchased from LI-COR Biosciences. The secondary
antibodies: anti-rabbit IgG (H + L) ReadyProbes™ secondary
antibody, Alexa Fluor 488 (cat. no. R37118, 1:2,000), used for
immunofluorescence, were purchased from Thermo Fisher Scientific,
Inc.
Cell lines and culture
Human glioblastoma cell lines U87 and LN229 were
obtained from Cobioer, and both cell lines were free of mycoplasma
and were authenticated using STR detection. The origin of
glioblastoma cell line U87 (ATCC® HTB-14™) used in the
present study was unknown as this cell line was originally obtained
from American Type Culture Collection. Cells were cultured in
high-glucose DMEM supplemented with 10% FBS, 1%
penicillin-streptomycin at 37°C in a humidified incubator with 5%
CO2.
Cell viability assay
U87 and LN229 cells were plated in 96-well plates
(3×103 cells/well) and incubated overnight at 37°C.
After treatment with various concentrations of test compounds for
designed times, MTT solution was added (20 µl/well), and the plates
were incubated for an additional 4 h. Next, the supernatant medium
was removed and the crystals were solubilized in DMSO (200
µl/well). A microplate reader (Bio-Tek Instruments, Inc.) was used
to detect the absorbance value at 570 nm. All experiments were
performed in triplicate, independently.
Colony formation assay
A total of 1×103 cells/well were seeded
in 6-well plates, respectively, and allowed to culture overnight
prior to treatment. After exposure to the designated concentrations
of DMAPT-D6 for 48 h, the cells were cultured with fresh DMEM for
another 15 days. Following fixation at 4°C with 4% paraformaldehyde
solution for 30 min, the cells were stained with 1% crystal violet
at room temperature for 30 min to visualize colonies. The number of
colonies with ≥50 cells was counted under a scanner.
Western blotting
The harvested glioma cells were lysed in RIPA lysis
buffer (Beyotime Institute of Biotechnology) supplemented with
Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher
Scientific, Inc.) at 4°C for 30 min. After quantification of total
protein concentration using a BCA Protein assay kit (Beyotime
Institute of Biotechnology), protein samples (30 µg/lane) were
loaded on 8, 10 and 12% SDS gels, resolved by electrophoresis and
transferred to PVDF membranes (Millipore). Subsequently, the
membranes were blocked with 5% BSA in TBST for 2 h at room
temperature, and then incubated with the indicated primary
antibodies overnight at 4°C. Following incubation with the
corresponding IRDye 800CW donkey anti-mouse IgG (H + L) or IRDye
680LT donkey anti-rabbit IgG (H + L) secondary antibody,
immunoreactivity was visualized using an odyssey two-color infrared
fluorescence imaging system (LI-COR Biosciences). β-actin was used
as the loading control.
Flow cytometry analysis
Glioma cells in the logarithmic growth phase were
harvested and transferred into 6-well plates at an estimated
confluence of 30% per well. After pretreatment with NAC (5 mM) for
2 h, the cells were exposed to the different concentrations of
DMAPT-D6 for 48 h. Additionally, other cells were treated with or
without DMAPT-D6, Z-VAD-FMK, Z-IETD-FMK, DMAPT-D6 and Z-VAD-FMK or
DMAPT-D6 and Z-IETD-FMK treatment. Then, all treated cells were
collected and analyzed using flow cytometry analysis. For cell
cycle analysis, the harvested cells were fixed with 70% ethanol at
4°C for 24 h, and then washed three times with PBS. Subsequently,
cells were incubated with PBS containing 50 mg/ml PI (BD
Biosciences) and 100 mg/ml RNase (Sigma-Aldrich; Merck KGaA) at
37°C for 30 min. Finally, the stained cells were analyzed using a
BD Accuri™ C6 flow cytometry (BD Biosciences), and the results were
statistically compared and automatically visualized using FlowJo
7.6 (FlowJo LLC). For cell apoptosis analysis, cells were harvested
and stained with an Annexin V-FITC/PI apoptosis assay kit according
to the manufacturer's protocol (BD Biosciences). The
fluorescence-positive cells were analyzed using a flow cytometer
within 1 h to assess the proportion of apoptotic cells, and the
apoptotic rate was visualized using FlowJo.
Immunofluorescence
Glioma cells were grown on cover slips in 24-well
plates prior to treatment with DMAPT-D6 for 48 h. After washing
three times with PBS, cells were fixed in 4% paraformaldehyde at
4°C for 30 min, permeabilized using 0.1% Triton X-100 for 10 min,
and blocked in Immunol staining blocking buffer (Beyotime Institute
of Biotechnology) for 30 min at 37°C. Cells were then incubated
overnight at 4°C with anti-γH2AX (1:250) antibody. Subsequently,
the stained cells were washed three times with PBS and incubated
with Alexa Fluor-conjugated secondary antibodies (1:2,000) for 1 h
at room temperature. For EdU cell proliferation staining, U87 and
LN229 cells were stained using BeyoClick™ EdU Cell Proliferation
kit with Alexa Fluor 594 according to the manufacturer's protocol
(cat. no. C0078S; Beyotime Institute of Biotechnology). Then, 1
mg/ml DAPI was used to label nuclei for 30 min. Images were
captured using a fluorescence microscope (Olympus IX53/DP80;
Olympus Corp.).
Determination of ROS formation
ROS formation was detected using a ROS assay kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Briefly, following pretreatment with or
without NAC (5 mM) diluted in serum-free DMEM for 2 h, glioma cells
were further treated with DMAPT-D6 for 48 h before staining with
DCFH-DA (10 µM) for 30 min. Subsequently, cell suspensions were
centrifuged and washed three times with serum-free DMEM, and then
visualized using a fluorescence microscope (Olympus IX53/DP80;
Olympus Corp.).
Statistical analysis
GraphPad Prism version 7.0 (GraphPad Software, Inc.)
was used for the statistical analysis. Data are presented as the
mean ± standard deviation, and the ANOVA method was used to
compare differences between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
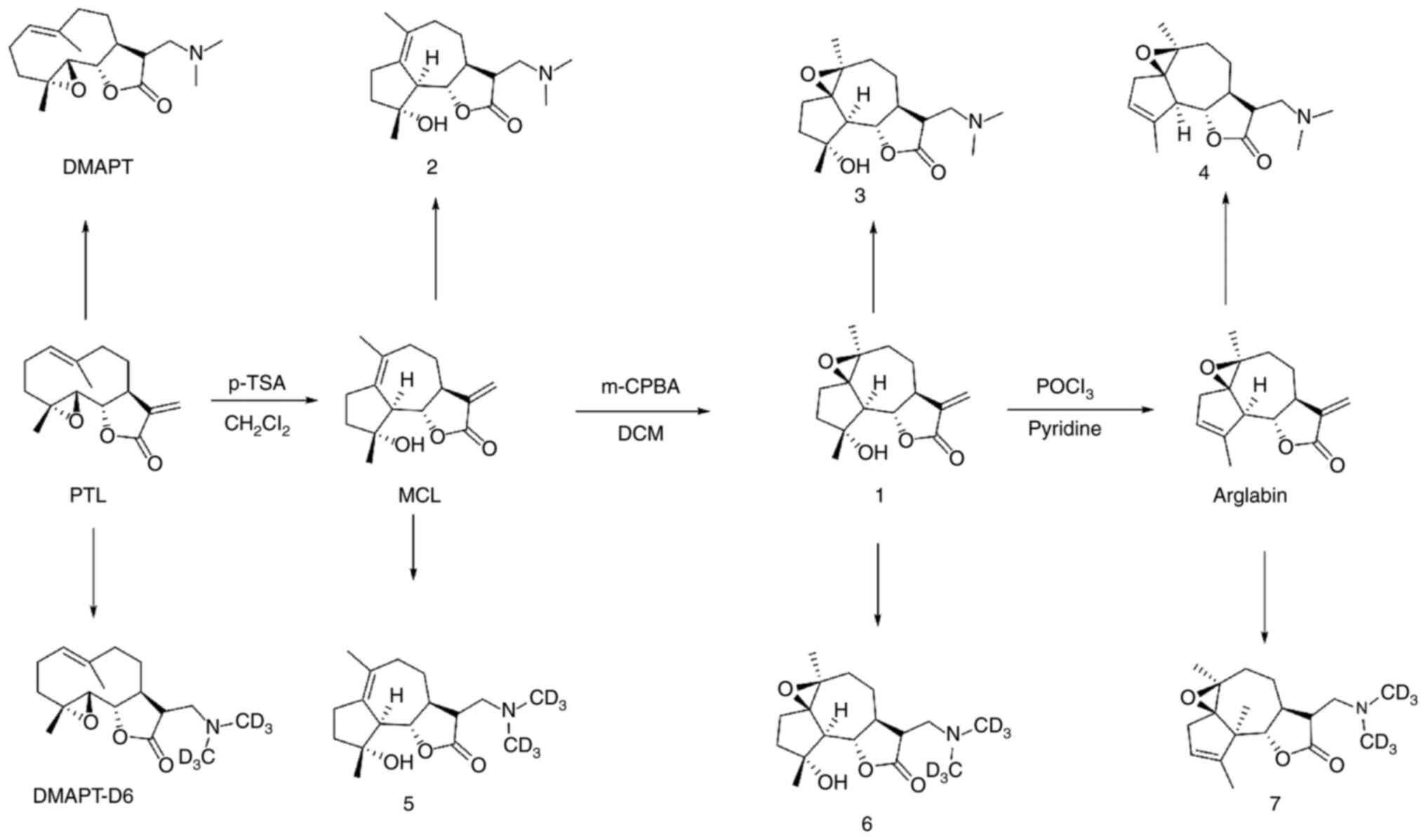
Synthesis of PTL derivatives
The syntheses of the PTL derivatives was initiated
using readily available PTL (Fig.
1). PTL was treated with p-toluenesulfonic acid (p-TSA)
to obtain micheliolide (MCL) and then subjected to epoxidation with
m-CPBA to obtain compound 1. Then, this compound was
subjected to elimination with POCl3/pyridine to obtain
arglabin. PTL, MCL, compound 1 and arglabin were treated
with dimethylamine in tetrahydrofuran under base conditions to
generate DMAPT, compound 2, compound 3, and compound
4, respectively. Similarly, DMAPT-D6, compound 5,
compound 6 and compound 7 were obtained by treating
PTL, MCL, compound 1 and arglabin with dimethyl-d6-amine under the
same conditions. All compounds were characterized by 1H
NMR and 13C NMR (Figs.
S1–22) and the detailed
synthesis process of the PTL derivatives is shown in Appendix S1.
Cytotoxic effect of DMAPT-D6 on GBM
cells
Given the anticancer effects of PTL, whether the
newly synthesized PTL-related derivatives could inhibit
proliferation and growth of GBM cells was assessed. The effects of
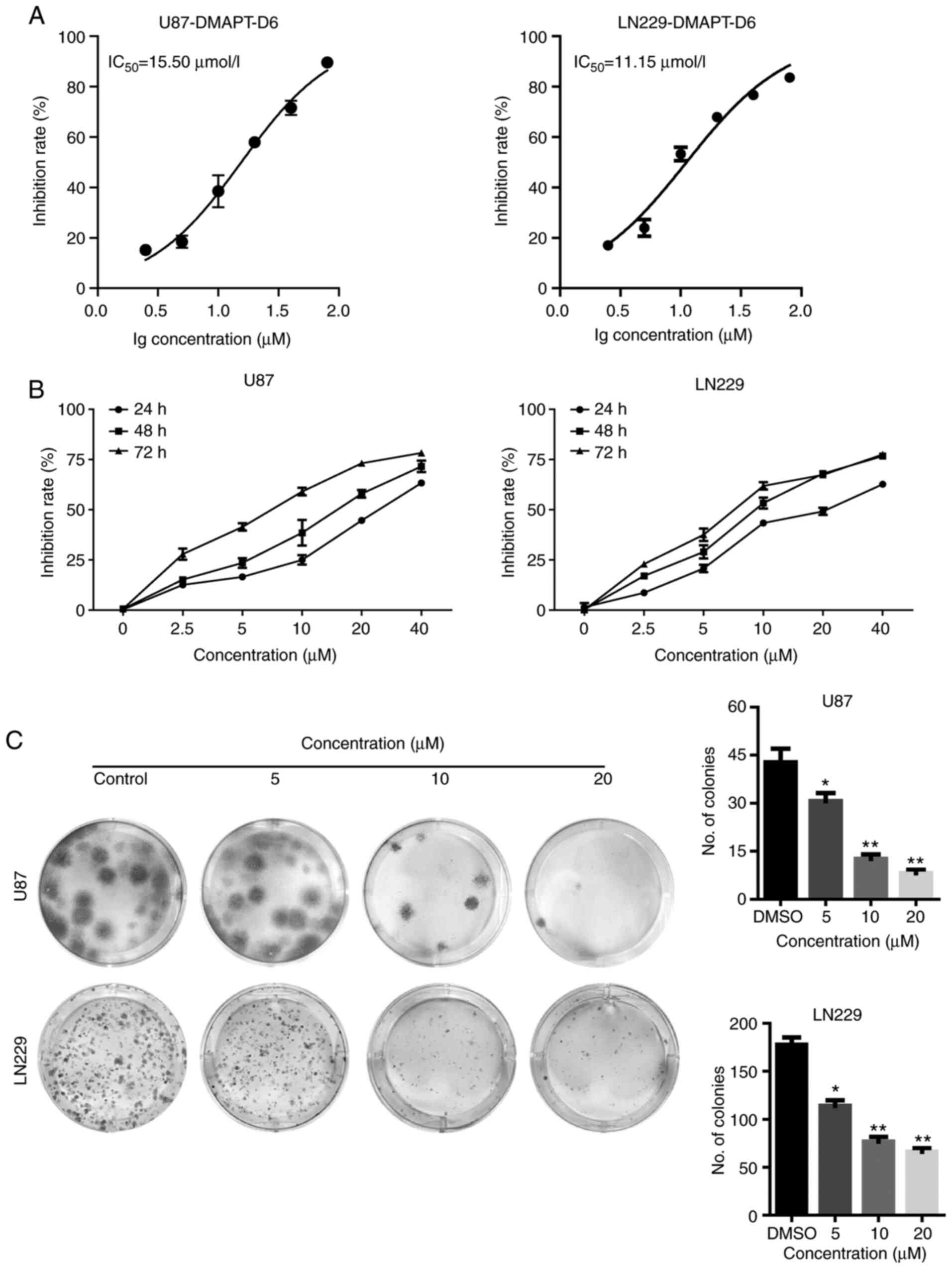
PTL-related derivatives on cell viability were determined (Table SI) and the IC50 values
of DMAPT-D6 were 15.5 and 11.15 µM in the U87 and LN229 cells,
respectively (Fig. 2A). To further
examine the inhibitory effects of DMAPT-D6, GBM cells were treated
with 2.5, 5, 10, 20 and 40 µM for 24, 48 and 72 h and the
relationship between cell growth and concentration, as well as
period of treatment were assessed. The cell growth curve showed
that DMAPT-D6 reduced the proliferative ability of both U87 and
LN229 cells in a dose- and time-dependent manner, highlighting the
dose- and time-dependent cytotoxicity of this compound on GBM cells
(Fig. 2B). A colony formation assay
was performed to further confirm the growth-inhibitory effects of
DMAPT-D6 on GBM cells. The results indicated that cells exposed to
DMAPT-D6 exhibited a significantly reduced cell growth in a
dose-dependent manner, as evidenced by the smaller colonies and
decreased colony numbers compared with the control group (Fig. 2C). Consistently, in comparison with
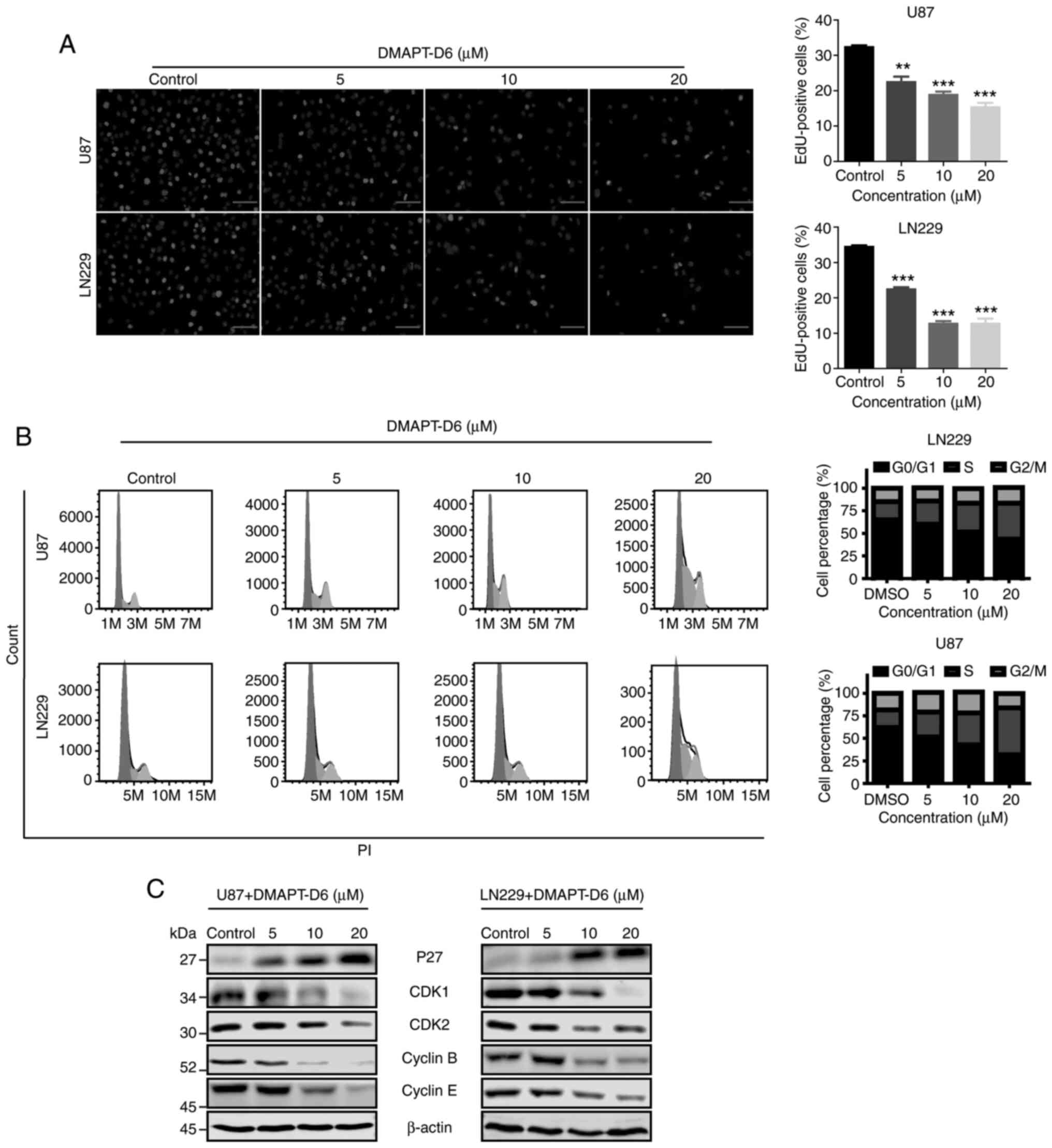
the untreated group, the number of EdU-positive cells was
significantly decreased in a dose-dependent manner following
exposure to DMAPT-D6, demonstrating its ability to suppress
proliferation in U87 and LN229 cells (Fig. 3A).
DMAPT-D6 arrests cell cycle
progression by arresting GBM cells at the S-phase
To determine the mechanism underlying cytotoxicity
of the DMAPT-D6 in GBM cells, cell cycle analysis was performed on
both U87 and LN229 cells treated with or without the inhibitor.
Flow cytometry indicated that 5 µM DMAPT-D6 resulted in cell cycle
arrest at the S-phase, whereas 20 µM DMAPT-D6 significantly induced
arrest at S-phase by reducing the proportion of cells in the G0/G1
phase (Fig. 3B). Since the effect
of DMAPT-D6 on cell cycle arrest in U87 and LN229 cells was
determined, its effect on the expression levels of S-phase-related
proteins was further examined to confirm the regulatory role in
cell cycle distribution. As shown in Fig. 3C, immunoblotting showed that
DMAPT-D6 considerably decreased the levels of cyclin B, cyclin E,
cyclin-dependent kinase 1 (CDK1) and CDK2, whereas p27 expression
was increased following exposure to DMAPT-D6 in a dose-dependent
manner in U87 and LN229 cell lines. Taken together, these data
suggest that DMAPT-D6 may suppress cell proliferation by inducing
S-phase cell cycle arrest in GBM cells.
DMAPT-D6 induces DNA damage through
excessive generation of ROS in GBM cells
To gain additional insight into the mode of
DMAPT-D6-induced inhibition of proliferation, intracellular ROS
generation was evaluated by fluorescence microcopy after incubation
with the specific ROS-detecting fluorescent dye, DCFH-DA in U87 and
LN229 cells. As indicated in Fig.
4A, ROS levels were significantly induced in response to
different concentrations of DMAPT-D6 when compared with the
control. Interestingly, the expression of the oxidative stress
responsive gene, NRF2, was significantly upregulated after
treatment with DMAPT-D6 in a dose-dependent manner, suggesting
excessive ROS accumulation within cells and the initiation of the
ROS stress response (Fig. 4C). It
has been reported that accumulation of intracellular ROS could
induce DNA damage and influence the DNA damage response caused by
genotoxic therapy, particularly in the context of double-strand
breaks (32). To evaluate whether
DMAPT-D6 could initiate DNA damage through accumulation of
excessive intracellular ROS, γH2AX, the phosphorylated of histone
H2AX and a marker of DNA double-strand breaks, was detected in both
U87 and LN229 cell lines using immunofluorescence and
immunoblotting. As illustrated in Fig.
4B, the results showed that the green signal representing γH2AX
significantly accumulated in the nuclei in a dose-dependent manner
compared with the control group. Consistent with this,
immunoblotting indicated that γH2AX was significantly upregulated
in the presence of DMAPT-D6 in U87 and LN229 cells (Fig. 4C), suggesting that DNA double-strand
breaks were generated in GBM cells in response to the treatment.
Furthermore, expression of p53, 53BP1 and DNA ligase IV (LIG IV),
which are both proteins involved in the DNA repair process, were
significantly decreased in a dose-dependent manner, demonstrating
DNA repair suppression in response to DMAPT-D6 (Fig. 4C).
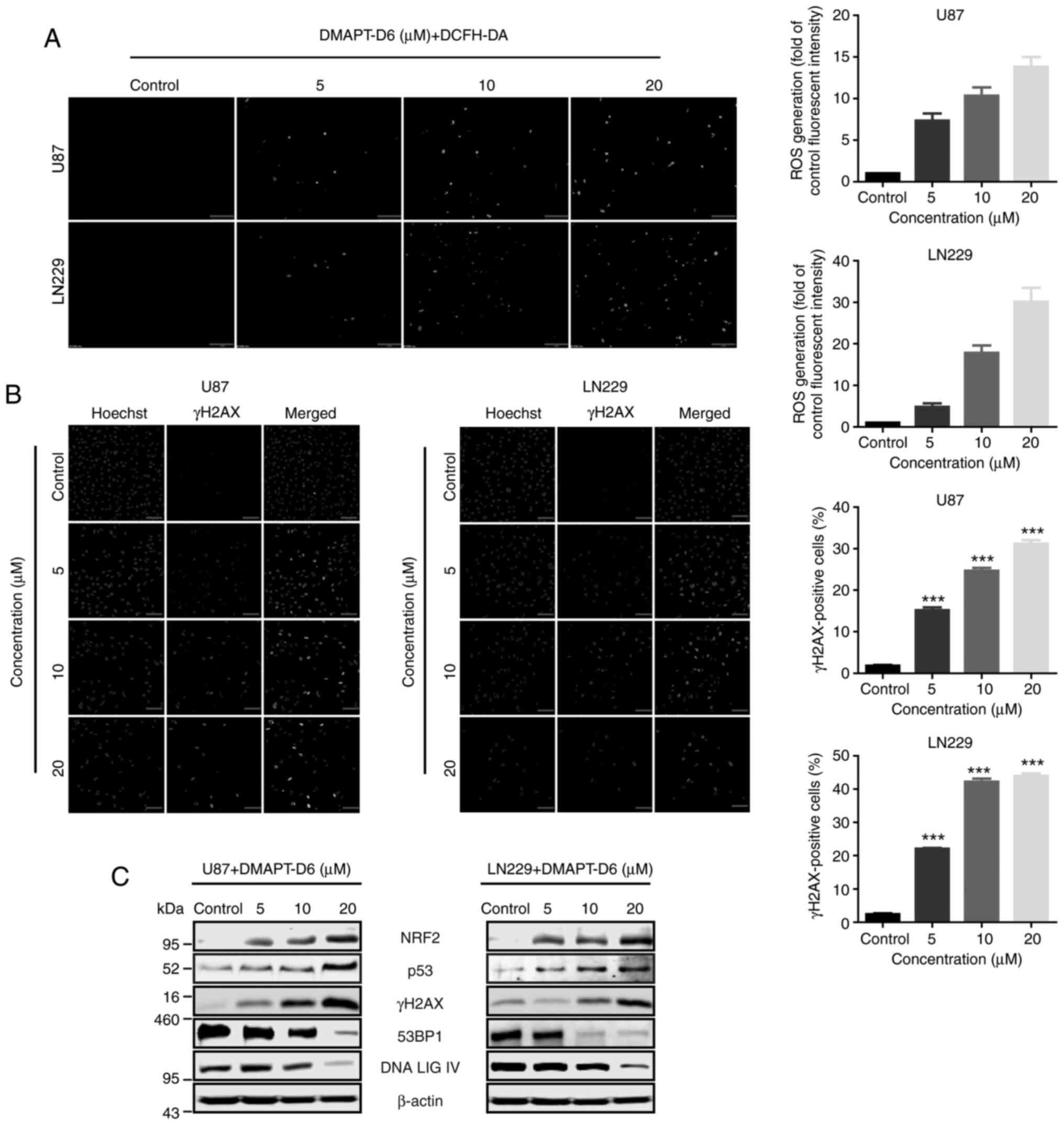 | Figure 4.DMAPT-D6 induces accumulation of ROS
and DNA damage, and impairs DNA repair. (A) GBM U87 and LN229 cells
were treated with the indicated concentrations of DMAPT-D6 for 48
h, and then were exposed to DCFH-DA for another 30 min.
Immunofluorescence analysis was performed to detect the green
signal, indicative of ROS generation. Cells with a green signal
were quantified. Scale bar, 100 µm. (B) γH2AX signal, the histone
H2AX phosphorylation representing double-strand breaks, was
detected using immunofluorescence, to assess the effects of
DMAPT-D6 on DNA damage. γH2AX-positive cells were quantified. Scale
bar, 100 µm. (C) Following treatment with different concentrations
of DMAPT-D6, cells were lysed, and expression of γH2AX, oxidative
stress response and DNA repair-related proteins, such as Nrf2, p53,
53BP1 and DNA ligase IV were examined by western blotting. β-actin
was used as the loading control. Data are presented as the mean ±
standard deviation of three independent experiments. ***P<0.001
vs. control. DMAPT, dimethylaminoparthenolide; GBM, glioblastoma;
ROS, reactive oxygen species; LIG IV, ligase IV; NRF2, nuclear
factor-like 2. |
Induction of ROS and DNA damage by
DMAPT-D6 contributes to death receptor-mediated external-apoptosis
in GBM cells
A disproportional increase in ROS and severe DNA
damage can induce the intrinsic and extrinsic apoptotic pathways,
which are mediated by mitochondria and cell death receptor
signaling, respectively (33–37).
Given the effects of DMAPT-D6 on the induction of ROS and
subsequent DNA damage, whether apoptosis was initiated in response
to DMAPT-D6 was assessed. Of note, the PI staining assay showed
that the percentage of PI-positive cells was significantly
increased following treatment with DMAPT-D6 in U87 and LN229 cells
in a dose-dependent manner (Fig.
5A), indicating the induction of apoptosis. To further confirm
the effects of DMAPT-D6 on apoptosis, an Annexin V-FITC/PI assay
was performed using flow cytometry after U87 and LN229 cells were
treated with the DMAPT-D6 (at 5, 10 and 20 µM) for 48 h. As shown
in Fig. 5B, at a low dose of 5 µM,
DMAPT-D6 induced 7.28 and 10.7% late-phase apoptosis (both
Annexin-V and PI positive-cells) in U87 and LN229 cell lines,
respectively. At a dose of 20 µM, the percentage of apoptosis rose
to 73.2 and 52.7%, respectively. Subsequently, the expression of
extrinsic apoptotic signaling-related proteins was assessed to
determine whether the cell death receptor was involved in the
apoptosis in response to DMAPT-D6 treatment. Consistently, cell
death receptor signaling pathway-related proteins such as death
receptor (DR)3, DR5, Fas-associated death domain (FADD) and tumor
necrosis factor receptor-associated death domain (TRADD) were
significantly upregulated in a dose-dependent manner after exposure
to DMAPT-D6 (Fig. 5C).
Subsequently, procaspase 8 was cleaved and the active enzyme form,
caspase 8, was produced due to the increase in FADD and TRADD.
Then, the downstream procaspase 3 and PARP were cleaved and
activated in both U87 and LN229 cells after treatment with
DMAPT-D6, suggesting that it primarily promotes extrinsic apoptosis
by inducing death receptor-mediated apoptotic signaling (Fig. 5C).
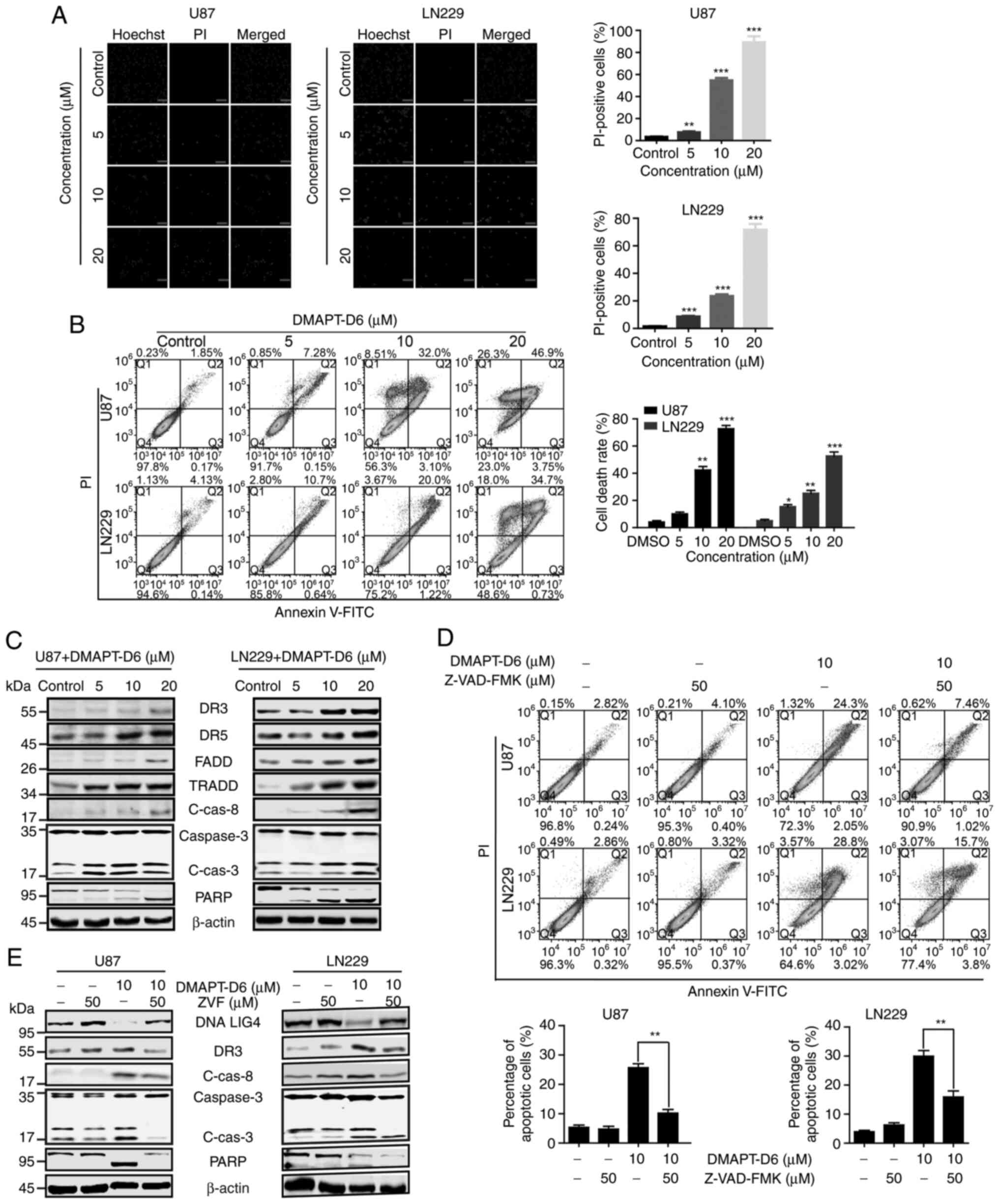 | Figure 5.DMAPT-D6 promotes caspase-dependent
death-receptor-mediated extrinsic apoptosis in both GBM U87 and
LN229 cell lines. (A) PI-staining was performed to examine whether
late apoptosis was induced after treatment with DMAPT-D6 in U87 and
LN229 cells. Subsequently, cells were analyzed using
immunofluorescence. (B) U87 and LN229 cells were treated with the
indicated concentrations of DMAPT-D6 for 48 h. Subsequently, cells
were harvested and stained with Annexin-V/PI. The Q4
(Annexin-V−/PI−), Q3
(Annexin-V+/PI−) and Q2
(Annexin-V+/PI+) quadrants represent the
populations of normal, early apoptotic and late apoptotic cells,
respectively. (C) Expression of death receptor-mediated extrinsic
apoptosis-related proteins were detected using western blotting.
(D) U87 and LN229 cells were treated with DMAPT-D6 (10 µM) either
alone or combination with the apoptosis inhibitor Z-VAD-FMK (50 µM)
for 48 h, and cells were stained with Annexin-V/PI to examine the
recovery of apoptosis. (E) Expression of death receptor-mediated
apoptosis-related proteins was examined using western blotting
after treatment with DMAPT-D6 (10 µM) either alone or in
combination with apoptosis inhibitor Z-VAD-FMK (ZVF) (50 µM).
*P<0.05, **P<0.01, ***P<0.001 vs. control. DMAPT,
dimethylaminoparthenolide; DR death receptor; FADD, Fas-associated
death domain; TRADD, tumor necrosis factor receptor-associated
death domain; PARP, poly(ADP-ribose) polymerase; C-Cas8, cleaved
caspase-8; C-Cas3, cleaved caspase-3. |
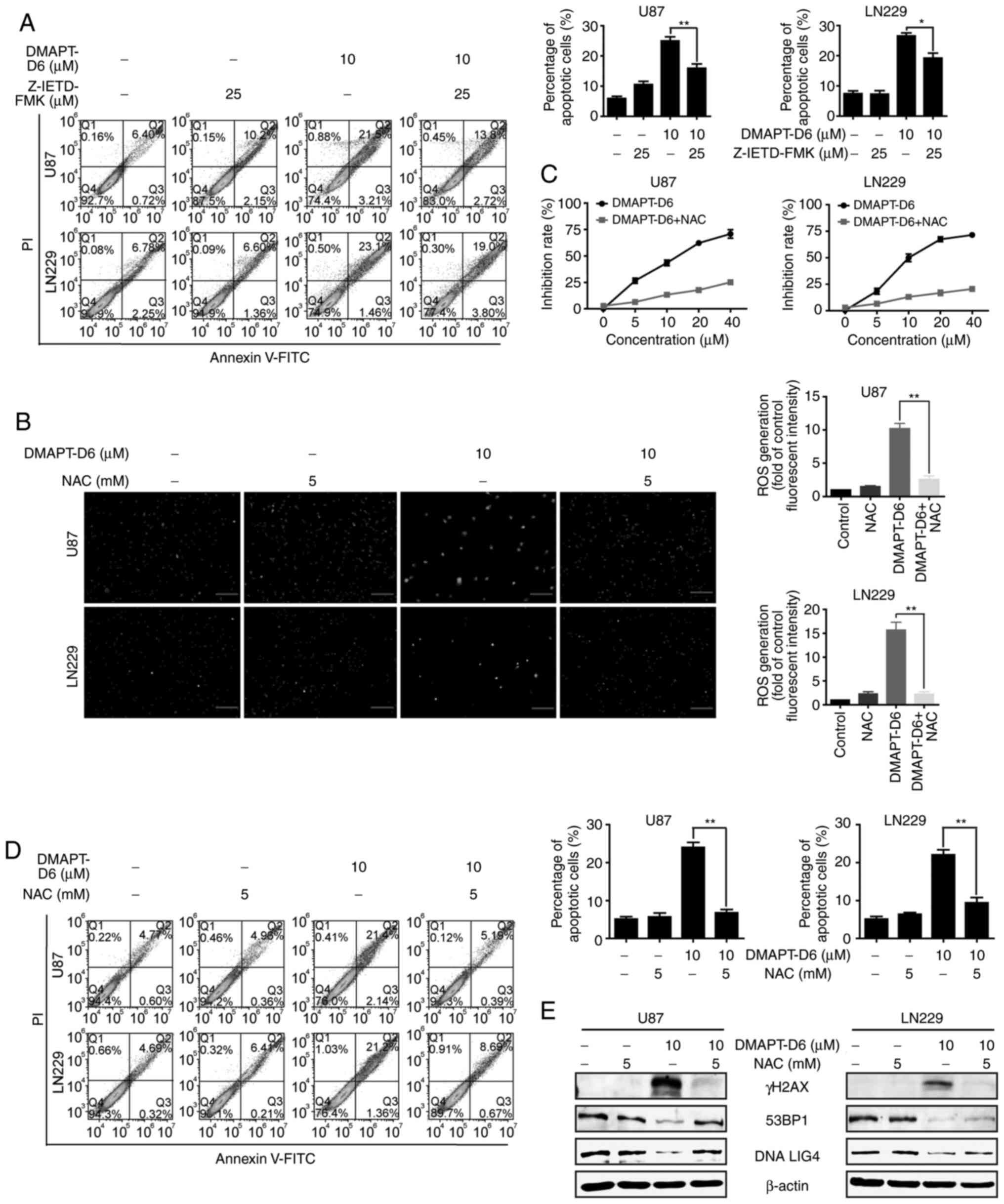
To further confirm that DMAPT-D6 induced cell death
receptor mediated-apoptosis, a pan-caspase inhibitor, Z-VAD-FMK,
was used to detect the restorative effect on apoptosis caused by
DMAPT-D6. As shown in Fig. 5D,
Z-VAD-FMK significantly rescued cell apoptosis induced by DMAPT-D6.
Interestingly, Z-VAD-FMK also partially restored the protein levels
of DNA repair protein DNA Lig IV, DR3, cleaved caspases-8 and −3,
and PARP when treated with both reagents together (Fig. 5E). Furthermore, a specific inhibitor
of caspase 8, Z-IETD-FMK, was used to examine the rescuing effect
on death-receptor-mediated apoptosis induced by DMAPT-D6. The
results showed that Z-IETD-FMK could partially reverse
DMAPT-D6-induced apoptosis in both U87 and LN229 cell lines
(Fig. 6A), demonstrating that
caspase-dependent cell death was the primary cause of GBM cell
death induced by DMAPT-D6.
A ROS scavenger, NAC, decreases the
cytotoxicity of DMAPT-D6 in GBM cells
To further demonstrate whether the anticancer
ability of DMAPT-D6 on GBM was actually initiated by generating
excessive ROS, the effects of DMAPT-D6 on U87 and LN229 cells after
treatment with an ROS scavenger, NAC, was determined. As expected,
pretreating the cells with NAC significantly reduced the number of
DCFH-DA-positive cells (Fig. 6B).
NAC pretreatment improved the DMAPT-D6-mediated decrease in cell
viability (Fig. 6C). Additionally,
the effect of NAC on apoptosis induced by DMAPT-D6 was assessed.
Flow cytometry showed that NAC could dramatically reduce the
DMAPT-D6-mediated apoptotic cells (Fig.
6D). Subsequently, the essential effects of ROS on
DMAPT-D6-induced DNA damage responses was examined after treatment
with NAC. The results indicated that pretreating the cells with NAC
significantly decreased the production of ROS and γH2AX, and
upregulated the protein expression level involved in DNA damage
response, such as 53BP1 and DNA LIG IV, suggesting causative
involvement of ROS in DMAPT-D6-induced DNA damage (Fig. 6E). Collectively, these results
showed that DMAPT-D6 induced apoptosis in GBM cells by resulting in
the generation of excessive ROS, which acts as a trigger of
downstream apoptotic pathways, such as DNA damage and
caspase-dependent apoptosis.
Discussion
Although notable advances have been made in the
treatment of glioblastoma (GBM), due to a high prevalence of
development of resistance to radiotherapy and chemotherapy, patient
prognosis remains poor (38,39).
Thus, there is an urgent need to develop novel agents that can
inhibit the proliferation and growth of GBM, and reverse
acquisition of multidrug resistance in GBM cells. It has been
reported that parthenolide (PTL) exhibits excellent anticancer,
anti-inflammatory and other beneficial properties without cytotoxic
effects on normal cells (17,40).
Nevertheless, PTL has some disadvantages that prevent it from being
developed as a clinical drug. For example, PTL is unstable under
physiological conditions, and has poor water solubility (29–31).
Modifications of PTL have been reported to improve its activity,
water solubility, stability and bioavailability. DMAPT, a prodrug
formed by addition of dimethylamine to PTL, can improve its
solubility and bioavailability (31,41,42).
Interestingly, deuteration of existing drugs can further exhibit
improved pharmacokinetic or toxicological properties, due to the
stronger deuterium-carbon bond resulting in modified metabolism
(31,42,43).
Therefore, development of this approach and evaluation of the
effects of deuterated derivatives on cancer cells and its potential
anticancer mechanisms are being intensively studied. In the present
study, 12 PTL derivatives, including 4 deuterated compounds were
synthesized and exhibited some extent of anti-proliferative
activity against GBM cells. Among all of the synthesized compounds,
DMAPT-D6 exhibited potent activity against both U87 and LN229 cell
lines, prompting further exploration of its anti-proliferative
effects, and the underlying molecular mechanism.
Despite ROS promoting the progression of cancer to
some extent, when excessive accumulation of ROS reaches a certain
threshold, this causes DNA damage, apoptosis and necrosis (12–15).
In the present study, the levels of intracellular ROS in both U87
and LN229 cells were measured after exposure to DMAPT-D6, and found
that DMAPT-D6 significantly upregulated ROS levels in a
dose-dependent manner. Therefore, these results suggested that the
cells were under high oxidative stress. Recently, Carlisi et
al (44) showed PTL
downregulated Nrf2 expression in spheroids such as MDA-MB-232 and
BT20, and a lesser effect on Nrf2 expression was exerted by DMAPT.
Reduction of Nrf2 results in the transcriptional decrease of
oxidative stress response genes, suggesting that ROS were not
removed immediately and instead accumulated within cells (44). In contrast, the present study showed
that DMAPT-D6 significantly upregulated the expression of the Nrf2
transcriptional activator. Thus, the results of the present study
conflict with the previous study regarding changes in expression
levels of Nrf2. Hypothetically, although Nrf2 was significantly
upregulated in the present study, resulting in an increase in
expression of genes to counteract the increased oxidative stress,
this was insufficient in removing the excess ROS due to the potency
of DMAPT-D6 on induction of oxidative stress compared with PTL.
Moreover, PTL may reduce Nrf2 levels through enhanced
ubiquitination and degradation of the Nrf2 protein by Keap1, a
factor that mediates ubiquitination and the consequent proteasomal
degradation of Nrf2 (44–46). However, the exact mechanism by which
the drug increased ROS levels in U87 and LN229 cells requires
further study.
It has been reported that γH2AX, (phosphorylated
histone H2AX) is a marker of DNA double-strand breaks that
accumulates in the nucleus (47).
Consistent with this, γH2AX expression was enhanced in the nucleus
in a dose-dependent manner in the present study, indicating that
DNA damage was significantly induced by upregulation of
intracellular ROS. Since DNA damage signaling is a major pathway
for induction of cell cycle arrest, it was hypothesized that cell
cycle arrest would be induced following treatment with DMAPT-D6 in
response to DNA damage (48,49).
The results showed that the U87 and LN229 cells were arrested at
S-phase induced by DMAPT-D6 treatment, and that S-phase-related
proteins were involved in this progression.
Apoptosis, which is initiated by two major pathways,
the death-receptor-mediated extrinsic pathway and the
mitochondrial-induced intrinsic pathway (50–52),
is abnormally regulated in various cancer types, and regarded as a
major defense mechanism against tumorigenesis (53,54).
Numerous anticancer agents exhibit their inhibitory effects by
promoting apoptosis (50–52). Production of caspase-8 induced by
the extrinsic pathway can eventually activate caspase 3 which is
able to cleave the downstream cellular substrates such as PARP,
resulting in progression of apoptosis (55,56).
Evidence has shown that certain pharmaceutical compounds cause
apoptosis through DNA damage via induction of intracellular ROS
(57–59). In agreement with these previous
studies, the results of the present study showed that DMAPT-D6
significantly upregulated death receptor signaling pathway-related
proteins, such as DR3, DR5, FADD, TRADD and active forms of
caspases-3, caspases-8 and PARP, suggesting that
death-receptor-mediated extrinsic apoptosis was induced in response
to DMAPT-D6 treatment. Based on these data, it was hypothesized
that DMAPT-D6 exerted its anticancer effects on GBM cells by
inducing extrinsic apoptosis following treatment, and this was due
to intracellular accumulation of ROS and the subsequent induction
of DNA damage.
In summary, a series of PTL derivatives were
synthesized using pharmaceutical methods, and a novel inhibitor
with inhibitory effects on GBM cells was identified and termed
DMAPT-D6. In DMAPT-D6, hydrogen was substituted for its isotope
deuterium in the dimethylamino group. Interestingly, DMAPT-D6
promoted cell cycle arrest at S-phase and cell
death-receptor-mediated extrinsic apoptosis pathway due to DNA
damage caused by intracellular accumulation of ROS which was
induced following treatment with DMAPT-D6. Therefore, the study
provides evidence for the use of DMAPT-D6 as an anti-GBM therapy.
Future studies should assess the efficacy and value of DMAPT-D6
in vivo.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was funded by the Basic Research and
Frontier Program of Chongqing Science and Technology Bureau
(cstc2018jcyjAX0219, cstc2020jcyj-msxmX0733 and
cstc2020jcyj-msxmX0595), Science and Technology Research Program of
Chongqing Municipal Education Commission (KJZD-K20200130,
KJQN201801304, KJQN201801308 and KJQN201901331) and Scientific
Research Foundation of the Chongqing University of Arts and
Sciences (2017RBX11, 2017RBX09, Y2020XY14, 2017ZBX05 and
2017ZBX07).
Availability of data and materials
Compounds synthesized in this present study are
available from the authors. The datasets used and/or analyzed
during the current study are available from the corresponding
author on reasonable request.
Authors' contributions
DLY, YL and ZGX conceived and designed the
experiments. YJZ, LJH, HXQ and YL conducted the experiments. JHH,
YL, DLY and ZGX analyzed and interpreted the data. DYT, ZZC and JHH
contributed analysis tools, planned the experiments and
participated in the study design and revision process. DLY and YL
wrote the paper. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stupp R, Mason WP, Van Den Bent MJ, Fisher
B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U,
Curschmann J, et al: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. New Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wen PY and Kesari S: Malignant gliomas in
adults. New Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrom QT, Gittleman H, Xu J, Kromer C,
Wolinsky Y, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2009–2013. Neuro Oncol.
18:v1–v75. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang B, Wu ZS and Wu Q: CMIP promotes
proliferation and metastasis in human glioma. Biomed Res Int.
2017:53401602017.PubMed/NCBI
|
|
6
|
Commoner B, Townsend J and Pake GE: Free
radicals in biological materials. Nature. 174:689–691. 1954.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sies H: Oxidative stress: Oxidants and
antioxidants. Exp Physiol. 82:291–295. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry JJ, Shin DS, Getzoff ED and Tainer
JA: The structural biochemistry of the superoxide dismutases.
Biochim Biophys Acta. 1804:245–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meitzler JL, Antony S, Wu Y, Juhasz A, Liu
H, Jiang G, Lu J, Roy K and Doroshow JH: NADPH oxidases: A
perspective on reactive oxygen species production in tumor biology.
Antioxid Redox Sign. 20:2873–2889. 2014. View Article : Google Scholar
|
|
10
|
Fransen M, Nordgren M, Wang B and
Apanasets O: Role of peroxisomes in ROS/RNS-metabolism:
Implications for human disease. Biochim Biophys Acta.
1822:1363–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martin KR and Barrett JC: Reactive oxygen
species as double-edged swords in cellular processes: Low-Dose cell
signaling versus high-dose toxicity. Hum Exp Toxicol. 21:71–75.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: A breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schumacker PT: Reactive oxygen species in
cancer: A dance with the devil. Cancer Cell. 27:156–157. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tasdogan A, Kumar S, Allies G, Bausinger
J, Beckel F, Hofemeister H, Mulaw M, Madan V, Scharffetter-Kochanek
K, Feuring-Buske M, et al: DNA damage-induced HSPC malfunction
depends on ROS accumulation downstream of IFN-1 signaling and bid
mobilization. Cell Stem Cell. 19:752–767. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodriguez-Rocha H, Garcia-Garcia A,
Panayiotidis MI and Franco R: DNA damage and autophagy. Mut Res.
711:158–166. 2011. View Article : Google Scholar
|
|
17
|
Knight DW: Feverfew: Chemistry and
biological activity. Nat Prod Rep. 12:271–276. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun J, Zhang C, Bao YL, Wu Y, Chen ZL, Yu
CL, Huang YX, Sun Y, Zheng LH, Wang X and Li YX:
Parthenolide-Induced apoptosis, autophagy and suppression of
proliferation in HepG2 cells. Asian Pac J Cancer Prev.
15:4897–4902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carlisi D, D'Anneo A, Martinez R, Emanuele
S, Buttitta G, Di Fiore R, Vento R, Tesoriere G and Lauricella M:
The oxygen radicals involved in the toxicity induced by
parthenolide in MDA-MB-231 cells. Oncol Rep. 32:167–172. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Fatlawi AA, Al-Fatlawi AA, Irshad M,
Rahisuddin and Ahmad A: Effect of parthenolide on growth and
apoptosis regulatory genes of human cancer cell lines. Pharm Biol.
53:104–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu C, Wang W, Jia Y, Liu X, Tong Z and Li
B: Inhibition of AMPK/autophagy potentiates parthenolide-induced
apoptosis in human breast cancer cells. J Cell Biochem.
115:1458–1466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu HJ, Jung JY, Jeong JH, Cho SD and Lee
JS: Induction of apoptosis by parthenolide in human oral cancer
cell lines and tumor xenografts. Oral Oncol. 51:602–609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang S, Ong CN and Shen HM: Critical
roles of intracellular thiols and calcium in parthenolide-induced
apoptosis in human colorectal cancer cells. Cancer Lett.
208:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
D'Anneo A, Carlisi D, Lauricella M,
Emanuele S, Di Fiore R, Vento R and Tesoriere G: Parthenolide
induces caspase-independent and AIF-mediated cell death in human
osteosarcoma and melanoma cells. J Cell Physiol. 228:952–967. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu JW, Cai MX, Xin Y, Wu QS, Ma J, Yang
P, Xie HY and Huang DS: Parthenolide induces proliferation
inhibition and apoptosis of pancreatic cancer cells in vitro. J Exp
Clin Canc Res. 29:1082010. View Article : Google Scholar
|
|
26
|
Sun Y, St Clair DK, Xu Y, Crooks PA and St
Clair WH: A NADPH oxidase–dependent redox signaling pathway
mediates the selective radiosensitization effect of parthenolide in
prostate cancer cells. Cancer Res. 70:2880–2890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Anderson KN and Bejcek BE: Parthenolide
induces apoptosis in glioblastomas without affecting NF-kappaB. J
Pharmacol Sci. 106:318–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guzman ML, Rossi RM, Karnischky L, Li X,
Peterson DR, Howard DS and Jordan CT: The sesquiterpene lactone
parthenolide induces apoptosis of human acute myelogenous leukemia
stem and progenitor cells. Blood. 105:4163–4169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nasim S and Crooks PA: Antileukemic
activity of aminoparthenolide analogs. Bioorg Med Chem Lett.
18:3870–3873. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Q, Lu Y, Ding Y, Zhai J, Ji Q, Ma W,
Yang M, Fan H, Long J, Tong Z, et al: Guaianolide sesquiterpene
lactones, a source to discover agents that selectively inhibit
acute myelogenous leukemia stem and progenitor cells. J Med Chem.
55:8757–8769. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Long J, Ding YH, Wang PP, Zhang Q and Chen
Y: Protection-Group-Free semisyntheses of parthenolide and its
cyclopropyl analogue. J Org Chem. 78:10512–10518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Srinivas US, Tan BW, Vellayappan BA and
Jeyasekharan AD: ROS and the DNA damage response in cancer. Redox
Biol. 25:1010842019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Czarny P, Pawlowska E, Bialkowska-Warzecha
J, Kaarniranta K and Blasiak J: Autophagy in DNA damage response.
Int J Mol Sci. 16:2641–2662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ichijo H, Nishida E, Irie K, ten Dijke P,
Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K and Gotoh
Y: Induction of apoptosis by ASK1, a mammalian MAPKKK that
activates SAPK/JNK and p38 signaling pathways. Science. 275:90–94.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moon DO, Kim MO, Choi YH, Hyun JW, Chang
WY and Kim GY: Butein induces G(2)/M phase arrest and apoptosis in
human hepatoma cancer cells through ROS generation. Cancer Lett.
288:204–213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pelicano H, Feng L, Zhou Y, Carew JS,
Hileman EO, Plunkett W, Keating MJ and Huang P: Inhibition of
mitochondrial respiration a novel strategy to enhance drug-induced
apoptosis in human leukemia cells by a reactive oxygen
species-mediated mechanism. J Biol Chem. 278:37832–37839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gorrini C, Harris IS and Mak TW:
Modulation of oxidative stress as an anticancer strategy. Nat Rev
Drug Discov. 12:931–947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hou J, Deng Q, Zhou J, Zou J, Zhang Y, Tan
P, Zhang W and Cui H: CSN6 controls the proliferation and
metastasis of glioblastoma by CHIP-mediated degradation of EGFR.
Oncogene. 23:1134–1144. 2017. View Article : Google Scholar
|
|
39
|
Li Q, Lu XH, de Wang C, Cai L, Lu JL, Wu
JS, Zhuge QC, Zheng WM and Su ZP: Antiproliferative and
apoptosis-inducing activity of schisandrin B against human glioma
cells. Cancer Cell Int. 15:122015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ghantous A, Sinjab A, Herceg Z and
Darwiche N: Parthenolide: From plant shoots to cancer roots. Drug
Discov Today. 18:894–905. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang Z, Kuang B, Kang N, Ding Y, Ge W,
Lian L, Gao Y, Wei Y, Chen Y and Zhang Q: Synthesis and anti-acute
myeloid leukemia activity of C-14 modified parthenolide
derivatives. Eur J Med Chem. 127:296–304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Neelakantan S, Nasim S, Guzman ML, Jordan
CT and Crooks PA: Aminoparthenolides as novel anti-leukemic agents:
Discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg Med Chem
Lett. 19:4346–4349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Timmins GS: Deuterated drugs: Where are we
now? Expert Opin Ther Pat. 24:1067–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Carlisi D, Buttitta G, Di Fiore R, Scerri
C, Drago-Ferrante R, Vento R and Tesoriere G: Parthenolide and
DMAPT exert cytotoxic effects on breast cancer stem-like cells by
inducing oxidative stress, mitochondrial dysfunction and necrosis.
Cell Death Dis. 7:e21942016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cullinan SB, Gordan JD, Jin J, Harper JW
and Diehl JA: The Keap1-BTB protein is an adaptor that bridges Nrf2
to a cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1
ligase. Mol Cell Biol. 24:8477–8486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ren D, Villeneuve NF, Jiang T, Wu T, Lau
A, Toppin HA and Zhang DD: Brusatol enhances the efficacy of
chemotherapy by inhibiting the Nrf2-mediated defense mechanism.
Proc Natl Acad Sci USA. 108:1433–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bonner WM, Redon CE, Dickey JS, Nakamura
AJ, Sedelnikova OA, Solier S and Pommier Y: GammaH2AX and cancer.
Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Luo YR, Zhou ST, Yang L, Liu YP, Jiang SY,
Dawuli Y, Hou YX, Zhou TX and Yang ZB: Porcine epidemic diarrhoea
virus induces cell-cycle arrest through the DNA damage-signalling
pathway. J Vet Res. 64:25–32. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feng Y, Huang H, Gao J and Liu S:
Salinomycin induces cell cycle arrest of glioma growth through
ROS-mediated DNA damage and AKT inactivation. ACTA Medica Mediterr.
36:249–253. 2020.
|
|
50
|
Huang C, Lu CK, Tu MC, Chang JH, Chen YJ,
Tu YH and Huang HC: Polyphenol-Rich avicennia marinaleaf extracts
induce apoptosis in human breast and liver cancer cells and in a
nude mouse xenograft model. Oncotarget. 7:35874–35893. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hilliard T, Miklossy G, Chock C, Yue P,
Williams P and Turkson J: 15α-methoxypuupehenol induces antitumor
effects in vitro and in vivo against human
glioblastoma and breast cancer models. Mol Cancer Ther. 16:601–613.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nayak VL, Nagesh N, Ravikumar A, Bagul C,
Vishnuvardhan MV, Srinivasulu V and Kama A: 2-Aryl benzimidazole
conjugate induced apoptosis in human breast cancer MCF-7 cells
through caspase independent pathway. Apoptosis. 22:118–134. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xiang M, Su H, Shu G, Wan D, He F, Loaec
M, Ding Y, Li J, Dovat S, Yang G and Song C: Amplexicaule A exerts
anti-tumor effects by inducing apoptosis in human breast cancer.
Oncotarget. 7:18521–18530. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gao L, Wang Y, Xu Z, Li X, Wu J, Liu S,
Chu P, Sun Z, Sun B, Lin Y, et al: SZC017, a novel oleanolic acid
derivative, induces apoptosis and autophagy in human breast cancer
cells. Apoptosis. 20:1636–1650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hamzeloo-Moghadam M, Aghaei M, Fallahian
F, Jafari SM, Dolati M, Abdolmohammadi MH, Hajiahmadi S and
Esmaeili S: Britannin, a sesquiterpene lactone, inhibits
proliferation and induces apoptosis through the mitochondrial
signaling pathway in human breast cancer cells. Tumour Biol.
36:1191–1198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cheng YC, Hueng DY, Huang HY, Chen JY and
Chen Y: Magnolol and honokiol exert a synergistic anti-tumor effect
through autophagy and apoptosis in human glioblastomas. Oncotarget.
7:29116–29130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hang W, Yin ZX, Liu G, Zeng Q, Shen XF,
Sun QH, Li DD, Jian YP, Zhang YH, Wang YS, et al: Piperlongumine
and p53-reactivator APR-246 selectively induce cell death in HNSCC
by targeting GSTP1. Oncogene. 37:3384–3398. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang Y, Zhang Y, Wang L and Lee S:
Levistolide A induces apoptosis via ROS-mediated ER stress pathway
in colon cancer cells. Cell Physiol Biochem. 42:929–938. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Colin D, Limagne E, Ragot K, Lizard G,
Ghiringhelli F, Solary É, Chauffert B, Latruffe N and Delmas D: The
role of reactive oxygen species and subsequent DNA-damage response
in the emergence of resistance towards resveratrol in colon cancer
models. Cell Death Dis. 5:e15332014. View Article : Google Scholar : PubMed/NCBI
|