Introduction
Apart from cancer cells, the tumor microenvironment
(TME) consists of heterogeneous host cells of the immune system,
the tumor vasculature and lymphatics, fibroblasts, pericytes, and
sometimes adipocytes (1).
Myeloid-derived suppressor cells (MDSCs) are crucial components of
the TME that play a pivotal role in tumor growth,
neovascularization, and metastasis (2–4). MDSCs
are a group of vastly heterogeneous immunosuppressive cells derived
from immature myeloid progenitors that have been linked to poor
patient prognosis (5). Typically,
immature myeloid cells traverse to the peripheral organs after
originating from bone marrow and rapidly mature into macrophages,
dendritic cells, or granulocytes (neutrophils, eosinophils, and
basophils) (6). That said, in the
tumor condition, multifarious factors that are present in the TME
prevent the differentiation of these immature myeloid cells and
instigate their actuation into an immunosuppressive phenotype
(7). MDSCs are usually divided into
two subpopulations: gMDSCs (granulocytic,
CD11b+Ly6G+Ly6Clow), which are
identical to neutrophils, and mMDSCs (monocytic,
CD11b+Ly6G−Ly6Chi), which are
consistent with monocytes with respect to morphology and phenotype
(8,9).
There is growing evidence that MDSCs harness various
immune and nonimmune mechanisms to promote tumor development. MDSCs
inhibit adaptive antitumor immunity by inhibiting T-cell activation
and function (T-cell receptor downregulation, T-cell cell cycle
inhibition, and immune checkpoint blockade) (9), and by driving and recruiting T
regulatory cells. Immunosuppression by MDSCs is also mediated by
the generation of reactive oxygen species (ROS) (10) and cytokine release [interleukin
(IL)-10 and tumor growth factor (TGF)-β] (11,12),
in conjunction with arginine depletion (13). They inhibit innate immunity by
polarizing macrophages toward a type 2 tumor-promoting phenotype
(M2-macrophage) (14) and by
inhibiting natural killer (NK) cell-mediated cytotoxicity (15). Likewise, MDSCs are efficient
recruiters of other immunosuppressive cells.
Although the role of MDSCs in tumor growth and
metastasis is well known, there is a significant knowledge gap for
understanding the role of MDSC-derived exosomes (MDSC exo). During
the past decade, there has been a huge surge of exosome research
and publications that are mostly focused on exosomes derived from
tumor cells and immune cells. Exosomes are 30–150 nm lipid
bi-layered extracellular bioactive vesicles of endosomal origin
that are secreted by all cells and are present in various body
fluids. Exosomes have been proposed to act as intercellular
communicators as they can transfer their cargo (proteins, lipids,
and nucleic acids) to nearby or distant recipient cells.
Previously, we observed that MDSC exo carry a significant amount of
pro-tumorigenic factors, and a large percentage of MDSC exo
injected intravenously was found to be distributed in the primary
breast tumor and metastatic sites (16). These findings warrant us to further
explore the implication of MDSC exo in immunosuppression and tumor
progression mechanisms.
In this study, we characterized the size, yield, and
contents of exosomes collected from different MDSC populations and
immature myeloid progenitor cells. We now report that, similar to
parental MDSCs, exosomes from MDSCs also play a crucial role in
inciting the immunosuppressive milieu by way of limiting the
functions of cytotoxic T cells and pro-inflammatory M1 macrophages
in the TME.
Materials and methods
Ethics statement
All experiments were performed according to the
National Institutes of Health (NIH) guidelines and regulations. The
Institutional Animal Care and Use Committee (IACUC) of Augusta
University (protocol #2014-0625) approved the experimental
procedures. All animals were kept under regular barrier conditions
at room temperature with exposure to light for 12 h and dark for 12
h. Food and water were offered ad libitum. All efforts were
made to ameliorate the suffering of animals. CO2 with a
displacement rate of 30–70% of the cage volume and delivery rate of
two liters/min into the cage followed by a secondary method was
used to euthanize animals for tissue collection.
Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was performed
using ZetaView, a second-generation instrument from Particle Metrix
for visualizing and counting individual exosome particles as
described previously (16,17). This high-performance integrated
instrument is equipped with a cell channel that is integrated into
a ‘slide-in’ cassette and a 405-nm laser. Samples were diluted
between 1:100 and 1:2,000 in PBS and injected in the sample chamber
with sterile syringes (BD Discardit II). Ten microliters of EXO
suspension was loaded into the sample chamber and all measurements
were performed at 23°C and pH 7.4. We used 11 positions with 2
cycles for the measurement mode, and a maximum pixel of 200 and
minimum of 5 for the analysis parameters. ZetaView 8.02.31 software
and Camera 0.703 µm/px were used for capturing and analyzing the
data.
Flow cytometry
For the in vivo flow cytometric analysis, the
collected fresh tissue was dispersed into single cells by filtering
through a 70-µm cell strainer, and spun at 1,200 rpm for 15 min.
For the in vitro flow cytometric analysis, cells were washed
twice with sterile PBS. The pellet was re-suspended in 1% BSA/PBS
and incubated with LEAF blocker (Stem Cell Technologies, cat.
#19867) in 100 µl volume for 15 min on ice to reduce non-specific
staining. The single cells were then labeled to detect the immune
cell populations using fluorescence conjugated antibodies for CD3
(cat. #100204), CD4 (cat. #100512), CD8 (cat. #100732), CD206 (cat.
#141708), F4/80 (cat. #123116), CD279 (cat. #135208 and 124312),
CD25 (cat. #101910), CD184 (cat. #146506), CD194 (cat. #131204),
CD69 (cat. #104506), CD62L (cat. #104432), CD11b (cat. #101208 and
101228), CD80 (cat. #1047220), CD86 (cat. #105028), Gr1 (cat.
#108406), Ly6C (cat. #128012), Ly6G (cat. #127614), and CD45 (cat.
#103108). All antibodies were mouse-specific (BioLegend), and the
samples were acquired using the Accuri C6 flow cytometer (BD
Biosciences). A minimum of 50,000 events were acquired.
Tumor model
Both 4T1 and AT3 cells expressing the luciferase
gene were orthotopically implanted in syngeneic BALB/c and C57BL/J6
mice, respectively (The Jackson Laboratory, Bar Harbor, Maine,
USA). All mice were between 5–6 weeks of age and weighed 18–20 g.
Animals were anesthetized using a mixture of xylazine (20 mg/kg)
and ketamine (100 mg/kg) administered intraperitoneally. Hair was
removed from the right half of the abdomen using hair removal
ointment, and then the abdomen was cleaned by povidone-iodine and
alcohol. A small incision was made in the middle of the abdomen,
and the skin was separated from the peritoneum using blunt forceps.
The separated skin was pulled to the right side to expose the
mammary fat pad and either 50,000 4T1 cells or 100,000 AT3 cells in
50 µl Matrigel (Corning Inc.) were injected.
Isolation of MDSCs
MDSCs were isolated from spleens and tumors of
tumor-bearing mice 3 weeks after orthotopic tumor cell
implantation. Myeloid progenitor cells were isolated from the bone
marrow of normal wild-type mice. We used anti-mouse Ly-6G, and
Ly-6C antibody-conjugated magnetic beads (BD Biosciences). The
purity of cell populations was >99%. In short, the spleen was
disrupted in PBS using the plunger of a 3 ml syringe, and cell
aggregates and debris were removed by passing the cell suspension
through a sterile 70-µm mesh nylon strainer (Fisherbrand™).
Mononuclear cells were separated by lymphocyte separation medium
(Corning®) as a white buffy coat layer. Cells were then
centrifuged at 1,500 rpm for 10 min followed by a washing step with
PBS at 1,200 rpm for 8 min. Then cells were resuspended at
1×108 cells/ml in PBS and antibodies conjugated with
magnetic beads were added followed by incubation at 4°C for 30 min.
Finally, positive cells were collected using a MACS LS column
(Miltenyi Biotec) and a MidiMACS™ magnetic stand followed by a wash
step with extra PBS. The purity of isolated MDSCs was checked by
flow cytometry using Gr1 FITC and CD11b APC antibodies (purchased
from BioLegend). Cell viability was checked with 7-AAD which was
less than 0.1–0.2% (dead cells) of the total population. MDSCs were
grown in exosome-depleted media consisting of RPMI, 2 mM
L-glutamine, 1% MEM non-essential amino acids, 1 mM sodium
pyruvate, and 10% FBS, supplemented with 100 ng/ml of GM-CSF.
Exosome isolation
Exosomes were depleted from the complete media by
ultracentrifugation for 70 min at 100,000 × g using an
ultracentrifuge (Beckman Coulter) and SW28 swinging-bucket rotor.
MDSCs (6×106) were grown in a T175 flask for 72 h under
normoxic conditions (5% CO2 and 20% oxygen) at 37°C in a
humidified incubator. The cell culture supernatant was centrifuged
at 700 × g for 15 min to remove cell debris. To isolate exosomes,
we employed a combination of two steps of the size-based method by
passing through a 0.20-µm syringe filter and centrifugation with
100k membrane tube at 3,200 × g for 30 min followed by a single
step of ultracentrifugation at 100,000 × g for 70 min [as described
in our previous publication (16)].
Protein quantification
Isolated exosomes resuspended in a minimal amount of
PBS were lysed by RIPA buffer with protease and phosphatase
inhibitor (100:1 dilution). Exosomal protein was quantified by
Bradford assay using Pierce™ BCA Protein Assay Kit (Thermo
Scientific™) and serial dilution of BSA standard (Thermo
Scientific™; Thermo Fisher Scientific, Inc.).
Protein array
Proteins were extracted from tumor cells and their
corresponding exosomes in both untreated and treated conditions to
evaluate the expression profiles of 44 factors in duplicate by
mouse cytokine antibody array (AAM-CYT-1000-8; RayBiotech, Inc.).
Protein sample (500 µg) was loaded onto the membrane according to
the manufacturer's instructions, and the chemiluminescent reaction
was detected using a LAS-3000 imaging system (Fuji Film, Japan).
All signals (expression intensity) emitted from the membrane were
normalized to the average of 6 positive control spots of the
corresponding membrane using ImageJ software version 1.53c
[National Institutes of Health (NIH)].
In vitro migration assay
A Transwell assay was performed to evaluate the
chemotaxis property of the MDSC-derived exosomes. We used 24
Transwell plates with 8-µm inserts in polyethylene terephthalate
track-etched membranes (Corning, Inc.). We collected bone marrow
cells and splenic mononuclear cells using Ficoll gradient
centrifugation, and myeloid cells from bone marrow using
CD11b+ magnetic beads from normal Balb/c mice. A total
of 1.5×106 cells/insert in serum-free media were added
into the upper compartment of the chamber. Inserts were placed in
12-well plates with DMEM containing 0.5% FBS in the presence or
absence of exosomes (20 µl containing approximately
3×108 exosomes) isolated from MDSCs. After incubating
overnight, we collected suspended immune cells (migrated) from the
media of the bottom chamber and loosely adherent immune cells on
the surface of the bottom chamber using gentle cell scraping. Then
we centrifuged and resuspended the cells in PBS and counted the
cells with a hemocytometer. Insert membranes were washed, fixed,
and stained with 0.05% crystal violet to detect the
migrated/invaded cells. The counting was made with an inverted
microscope (Nikon Eclipse E200).
In vitro scratch assay
Scratch assay was performed to detect the ability of
MDSC-derived exosomes to increase migration and invasion of tumor
cells. 4T1 luciferase positive cells were seeded in 6-well plates.
After achieving 80–90% confluency, the cells were starved overnight
with 0.5% FBS for cell cycle synchronization and a measured wound
was inflicted at the center of the culture (from top to bottom).
Then, cells were treated with 50 µl of splenic MDSC-derived
exosomes in PBS containing 7.5×108 exosomes for 48 h in
2% FBS media. Microphotographs were taken every 24 h using an
automated all-in-one microscope (BZ-X710; Keyence). The wound size
was measured using Image J software (NIH) by drawing a rectangular
region of interest to quantify the visible area of the wound.
In vivo treatment with MDSC-derived
exosomes
MDSC-derived exosomes were injected intravenously
(100 µl containing approximately 1.5×109 exosomes) into
the wild-type Balb/c and C57Bl/6 mice. The animals were treated for
a week with 3 doses (alternate days) of MDSC-derived exosomes.
After that, the animals were euthanized and organs were collected
for flow cytometric analysis.
Isolation of T cells
Both CD4+ and CD8+ cells were
isolated from normal mouse splenocytes by immune-magnetic negative
selection kit (Stemcell Technology; catalog #19852 and 19853,
respectively). In short, harvested spleens from normal mice were
disrupted in cold PBS containing 2% FBS. Clumps and debris were
removed by passing the cell suspension through a 70-µm mesh nylon
strainer. The single-cell suspension was centrifuged at 300 × g for
10 min and resuspended at 1×108 nucleated cells/ml. Rat
serum was added to the sample (50 µl/ml) followed by the addition
of isolation cocktail (50 µl/ml). After mixing, the sample mix was
incubated at room temperature for 10 min. RapidSpheres™ (75 µl/ml)
were added to the sample mix and incubated for 3 min. The tube was
placed in EASYSEP™ MAGNETS (catalog #18001; Stemcell Technologies)
for 3 min. The enriched cell suspension was collected by decanting
into a new tube. Cells were seeded in 24-well plates bound with
purified anti-mouse CD3e (5 µl/ml) in T-cell media that consists of
RPMI, 10% FBS, 1% MEAM, 2.5% HEPES, 1% penicillin-streptomycin,
0.5% β-mercaptoethanol, and purified anti-mouse CD28 (5 µl/ml).
Quantification of ROS generation by
CD8+ T-cells
ROS production from CD8+ T-cells
following MDSC-derived exosomes treatment in vitro was
estimated by labeling the CD8+ T-cells using
CM-H2DCFDA (Invitrogen™, C6827; Thermo Fisher
Scientific, Inc.). In short CD8+ T-cells were isolated
according to the above-mentioned method. After the final wash step
of the isolation procedure, cells were resuspended in 1 ml PBS.
DCFDA solution at a working concentration of 10 µM/ml was added
followed by incubation in the dark at 37°C for 30 min. The cells
were washed with an extra PBS to remove the unbound dye and
resuspended with appropriate T-cell media. A total of 100,000 cells
were seeded per well of 96 well-plate. MDSC-derived exosomes were
added in the treatment group and the same volume of PBS was added
in the control group. Hydrogen peroxide
(H2O2) was used as a positive control for ROS
production. Following 4 h of incubation, the fluorescent intensity
of each condition was measured using a Perkin Elmer Victor3 V 1420
multilabel plate reader (PerkinElmer), with excitation and emission
wavelengths of 485 and 535 nm, respectively. Each condition was
assayed in triplicate for significance.
CD8+ T-cell proliferation
assay
Following isolation, 20,000 CD8+ cells
were seeded in an anti-mouse CD3e-bound 96-well plate and treated
with 10 µl of MDSC-derived exosomes or the same volume of PBS
(control). After 48 h, 10 µl of WST-1 reagent (Alkali Scientific
Inc.) was added to each well and incubated for 4 h. The absorbance
of each well was measured at a wavelength of 450 nm by the Perkin
Elmer Victor3 V 1420 multilabel plate reader.
Statistical analysis
Quantitative data are expressed as mean ± standard
error of the mean (SEM) unless otherwise stated, and statistical
differences between more than two groups were determined by
analysis of variance (ANOVA) followed by multiple comparisons using
Tukey's multiple comparisons test. A comparison between 2 samples
was performed by the Student t-test. GraphPad Prism version 8.2.1
for Windows (GraphPad Software, Inc.) was used to perform the
statistical analysis. Differences with P-values <0.05 were
considered significant and are indicated in the figures and legends
(*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Results
Isolation and characterization of
exosomes from different MDSC populations
We isolated MDSCs from the bone marrow of normal
(non-tumor bearing) wild-type mice, and from spleen and tumors of
tumor-bearing mice by magnetic particle separation using Ly6G and
Ly6C beads. Tumors were implanted orthotopically in the mammary fat
pad and allowed to grow for 3 weeks. Following their separation, by
flow cytometry, more than 98% of the cells were estimated to be
positive for MDSC markers (CD11b+Gr+)
(Fig. 1A). Cell viability was
checked with 7-AAD and determined to be less than 0.1–0.2% of the
total population (Fig. S1). After
72 h, we isolated exosomes from culture supernatant and
characterized them by NTA. Exosomes isolated from MDSCs of normal
bone marrow (BM MDSC exo), the spleen of tumor-bearing mice (spleen
MDSC exo), and tumors (tumor MDSC exo) were similar in size and
distribution (Fig. 1B and C).
However, MDSCs from tumors released significantly more exosomes
compared to MDSCs from normal bone-marrow, which could be due to
the presence of the stressful condition, and more active and
immunosuppressive MDSCs in the primary tumor area (Fig. 1D).
Next, we tested if the protein contents of normal BM
MDSC exo differ from the spleen MDSC exo and tumor MDSC exo
isolated from tumor-bearing mice. We quantified the expression
level of cytokines in MDSC exo that are involved in tumor invasion
(Fig. 2A), myeloid cell activation
and function (Fig. 2B), and
angiogenesis (Fig. 2C) by
membrane-based protein array. All the cytokines were significantly
overexpressed in exosomes isolated from MDSCs of tumor-bearing mice
(in both spleen MDSC exo and tumor MDSC exo) compared to normal BM
MDSC exo. Interestingly, tumor MDSC exo showed a higher level of
expression than that of spleen MDSC exo indicating that
MDSC-derived exosomal cytokine contents are different based on the
microenvironment of the host tissues.
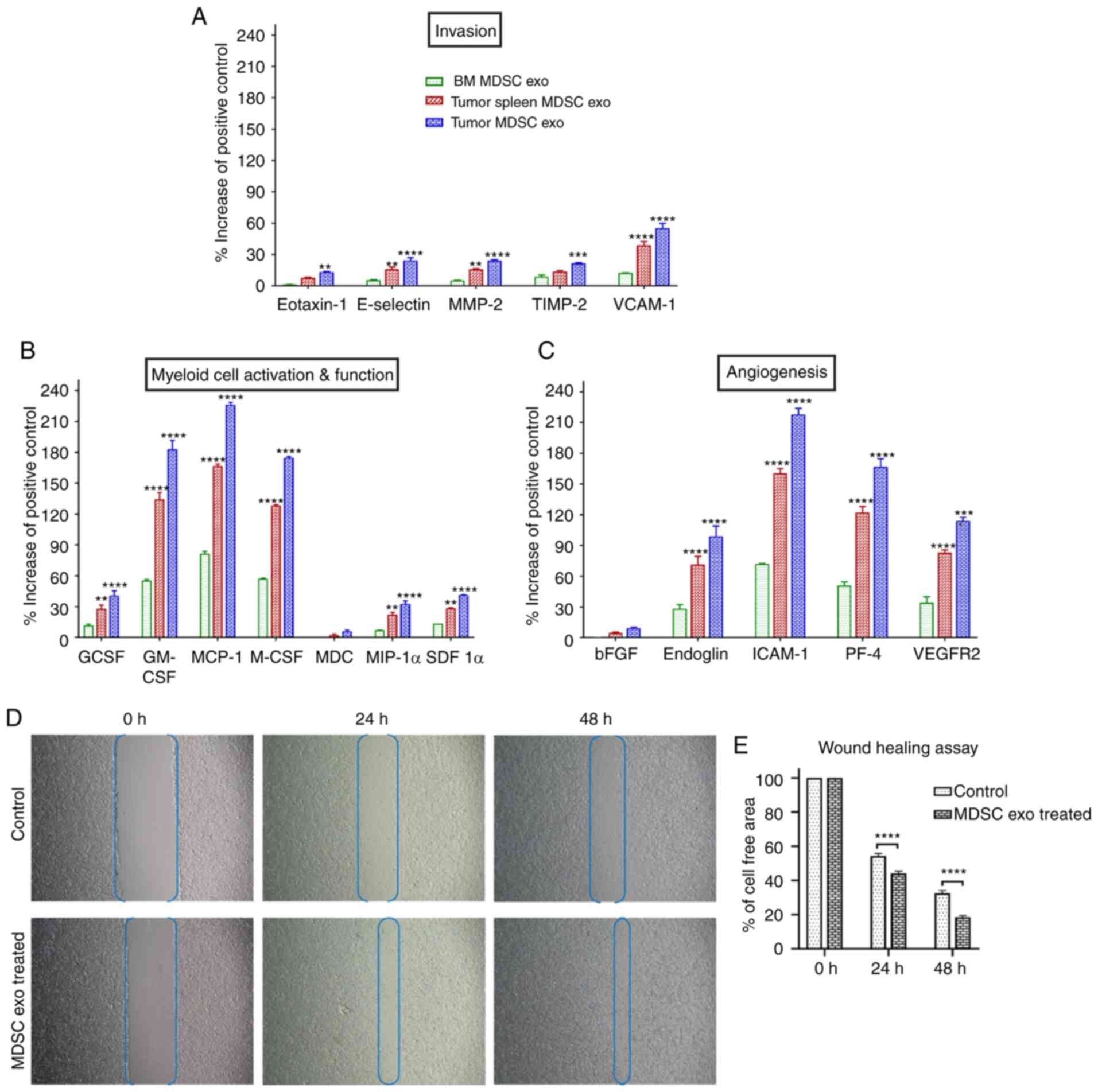 | Figure 2.The expression level of cytokines in
MDSC-derived exosomes (exo) that are involved in tumor invasion,
angiogenesis, and myeloid cell activation and function. In
vitro quantification of the level of cytokines associated with
(A) tumor invasion, (B) myeloid cell activation and function, and
(C) angiogenesis, detected in the membrane-based array in protein
samples collected from exosomes isolated from MDSCs of normal bone
marrow (BM), the spleen of tumor-bearing mice and tumors.
Quantitative data are expressed as mean ± SEM. **P<0.01,
***P<0.001, ****P<0.0001, compared to the BM MDSC exo. n=4.
(D and E) The role of MDSC-derived exosomes in tumor cell migration
was evaluated by a wound-healing assay/scratch assay, carried out
in the 4T1 murine breast cancer cell line with or without (control)
splenic MDSC-derived exosome treatment. (D) Representative
microscopic images (×4 magnification) are shown before treatment,
and 24 and 48 h after treatment. (E) Semi-quantitative analysis of
the percentage of non-covered area/cell-free area. Quantitative
data are expressed as mean ± SEM. ****P<0.0001. n=10. MDSCs,
myeloid-derived suppressor cells; MMP-2, Matrix
metalloproteinase-9; TIMP-2, tissue inhibitor of metalloproteinase
2; VCAM-1, vascular cell adhesion molecule 1; GCSF, granulocyte
colony-stimulating factor; GM-CSF, granulocyte/macrophage colony
stimulating factor; MCP-1, monocyte chemoattractant protein-1;
M-CSF, macrophage colony-stimulating factor; MDC,
macrophage-derived chemokine; MIP-1α, macrophage inflammatory
protein-1α; SDF 1α, stromal cell-derived factor 1α; bFGF, basic
fibroblast growth factor; ICAM-1, intercellular adhesion
molecule-1; PF-4, platelet factor 4; VEGFR2, vascular endothelial
growth factor receptor 2. |
MDSC-derived exosomes promote invasion
and migration of tumor cells
MDSCs were demonstrated to promote tumor invasion
and metastasis by two mechanisms: i) Increased production of
multiple matrix metalloproteinases (MMPs) for extracellular matrix
degradation and increased production of chemokines to establish a
pre-metastatic milieu (18,19), and ii) merging with tumor cells,
thus promoting the metastatic process (20,21).
We observed significantly higher expression of invasion and
migration-associated cytokines in the spleen MDSC-exo of
tumor-bearing mice compared to normal BM MDSC-exo, which led us to
further investigate the role of MDSC exo in promoting invasion and
migration of tumor cells. In vitro, the wound-healing assay
showed a significantly increased migration of 4T1 tumor cells in
the spleen of the MDSC-exo treated group compared to the untreated
control group at 24 and 48 h (Fig. 2D
and E).
MDSC exosomes promote the recruitment
of immunosuppressive cells in vitro
Tumor-specific endocrine factors systemically
stimulate the quiescent immune-compartments (bone marrow, spleen,
lymph nodes), resulting in the expansion, mobilization, and
recruitment of immunosuppressive cells. Discrete subsets of
tumor-instigated immune cells bolster tumor progression and
metastasis by governing angiogenesis, inflammation, and immune
suppression. Of the immune cells, much focus has been denoted
towards the MDSCs (22),
tumor-associated macrophages (TAMs) (23), Tie2-expressing monocytes (24), vascular endothelial
(VE)-cadherin+CD45+ vascular leukocytes, and
infiltrating mast cells and neutrophils (25,26).
We observed significant high expression levels of cytokines crucial
for immunosuppressive cell mobilization and recruitment in spleen
MDSC-exo isolated from splenic MDSCs of tumor-bearing mice compared
to normal BM MDSC exo.
To determine the chemotaxis capability of MDSC exo,
we seeded CD11b+ myeloid cells (from bone marrow) or all
bone marrow cells, or all splenic mononuclear cells (following
Ficoll separation) isolated from normal mice in the upper chamber
and with or without spleen MDSC exo in the bottom chamber of the
Transwell insert. After 24 h of incubation, the number of migrated
cells in the bottom chamber was significantly higher in the wells
treated with spleen MDSC exo compared to untreated control wells
(Fig. 3A-C). We also washed, fixed,
and stained the insert membranes (of the CD11b+
cell-incubated group) with 0.05% crystal violet to detect the
migrated/invaded cells. The number of cells that attached to the
membrane were visualized by microscopy (Fig. 3D) and later quantified by an ImageJ
cell counter. A significantly higher number of CD11b+
myeloid cells were attached to the transmembrane in the wells
treated with spleen MDSC exo compared to the untreated control
wells (Fig. 3E).
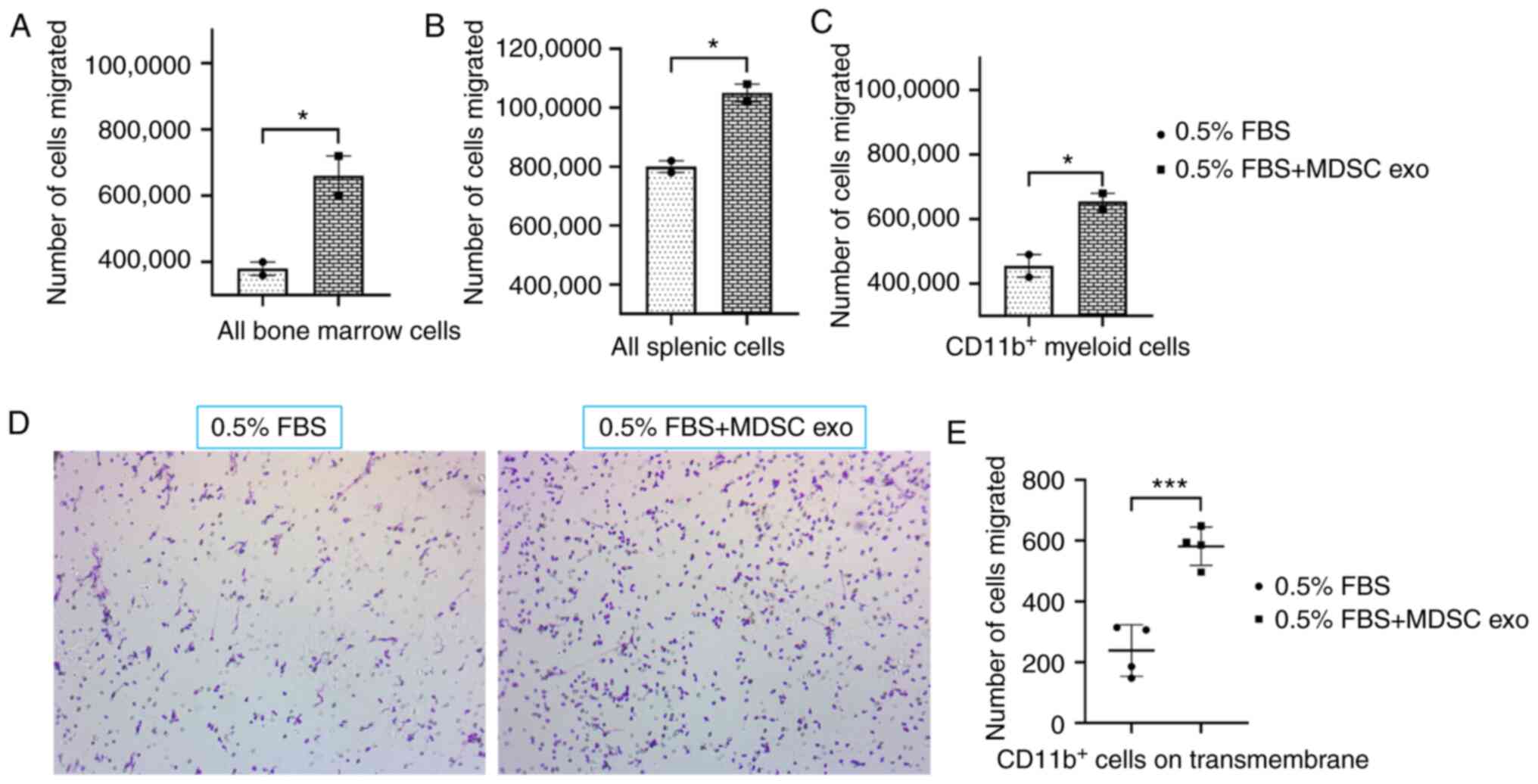 | Figure 3.Role of MDSC-derived exosomes (exo)
in immune cell migration. Isolated mouse myeloid cells, bone marrow
cells, and splenic cells were seeded on the top chamber of the
Transwell, and splenic MDSC-derived exosomes were added in the
bottom chamber with 0.5% FBS. After 24 h, migrated (A) bone marrow
cells, (B) splenic cells, and (C) myeloid cells in the bottom
chamber were counted with a hemocytometer. In addition, (D)
attached myeloid cells on the Transwell membrane were visualized
under a light microscope, and (E) quantified. Quantitative data are
expressed as mean ± SEM. *P<0.05, ***P<0.001, n=4. MDSCs,
myeloid-derived suppressor cells. |
Expression of T-cell
function-associated and immuno- modulatory cytokines in exosomes
from different MDSC populations
We further estimated the level of expression of
T-cell function-associated and immunomodulatory cytokines in the
protein contents of normal BM MDSC exo and exosomes isolated from
MDSCs (tumor and splenic) of tumor-bearing mice by protein array.
Among the immunomodulatory cytokines, the levels of IL-12, IL-13,
IL-1Ra, IL-4, C-X-C motif chemokine 5 (LIX), and tumor necrosis
factor (TNF)-α were significantly elevated in both the
tumor-MDSC-exo and spleen-MDSC-exo of tumor-bearing mice compared
to normal BM-MDSC-exo (Fig. 4A).
Among T-cell function-associated cytokines, IL-2, IL-7, L-selectin,
and thymic stromal lymphopoietin (TSLP) were significantly higher
in the exosomes derived both from the tumor and splenic MDSCs of
tumor-bearing mice compared to normal BM MDSC-exo (Fig. 4B).
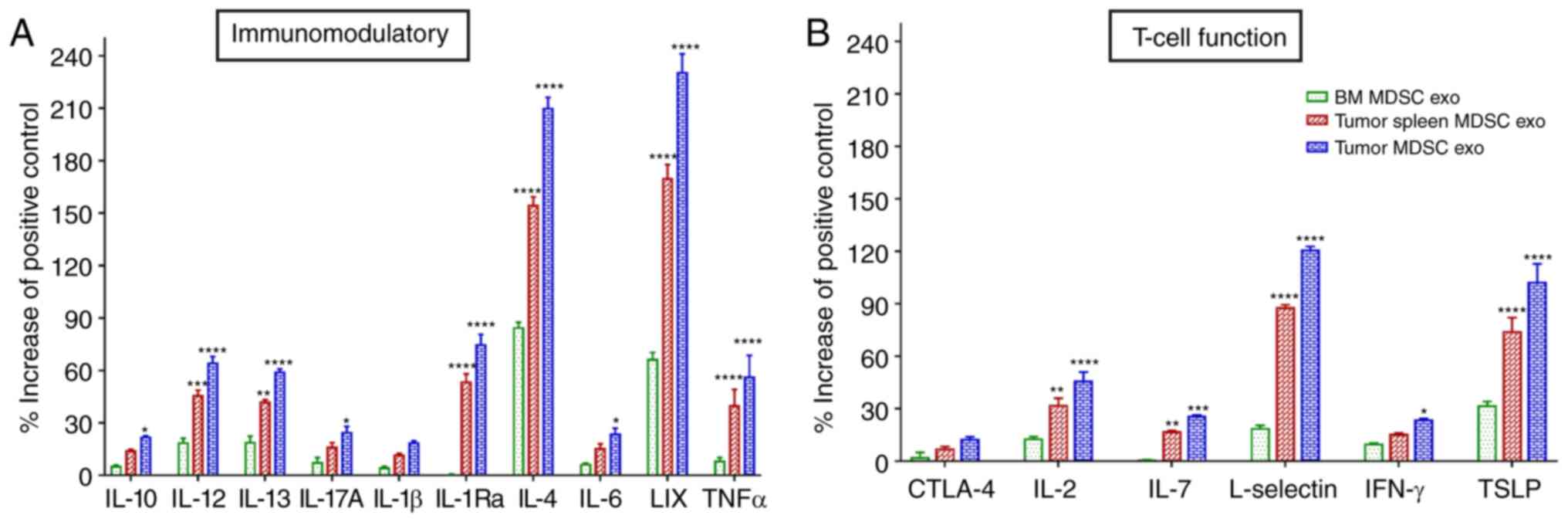 | Figure 4.Expression levels of cytokines in
MDSC-derived exosomes (exo) that are involved in T-cell function
and immunomodulation. In vitro quantification of the level
of cytokines associated with (A) immunomodulation and (B) T-cell
function, detected in the membrane-based array in protein samples
collected from the exosomes isolated from MDSCs of normal bone
marrow (BM), the spleen of tumor-bearing mice and tumors.
Quantitative data are expressed as mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001, compared with the BM
MDSC exo. n=4. MDSCs, myeloid-derived suppressor cells; IL,
interleukin; LIX, C-X-C motif chemokine 5; TNFα, tumor necrosis
factor α; CTLA-4, cytotoxic T-lymphocyte-associated protein 4;
IFN-γ, interferon-γ; TSLP, thymic stromal lymphopoietin. |
In vivo effect of MDSC-derived
exosomes on T-cells
Next, we investigated whether MDSC exo treatment
could deplete the CD8+ T-cells in mice. For this in
vivo study, we used both C57BL/6 and Balb/c normal mice. We
treated the mice with MDSC exo by intravenous (i.v.) injection
through the tail vein for a week (total of 3 doses, alternate
days). Then we euthanized the animals and harvested spleens for
flow cytometric evaluation. The CD8+ T-cell population
in splenic MDSC-exo treated animals was significantly declined
compared to the untreated control group in both animal models
(Fig. 5A and B). However, we did
not observe any significant change in the CD4+ T-cell
population.
In vivo effect of MDSC-derived
exosomes on myeloid cells
We also explored whether the MDSC exo treatment
could change the distribution of the myeloid populations in
vivo. We noticed a significant reduction in M1-macrophages
(CD11b+CD80+ and
CD11b+CD86+) and a notable increase of
M2-macrophages (CD11b+CD206+) in the spleen
of animals treated with spleen MDSC exo while we did not see a
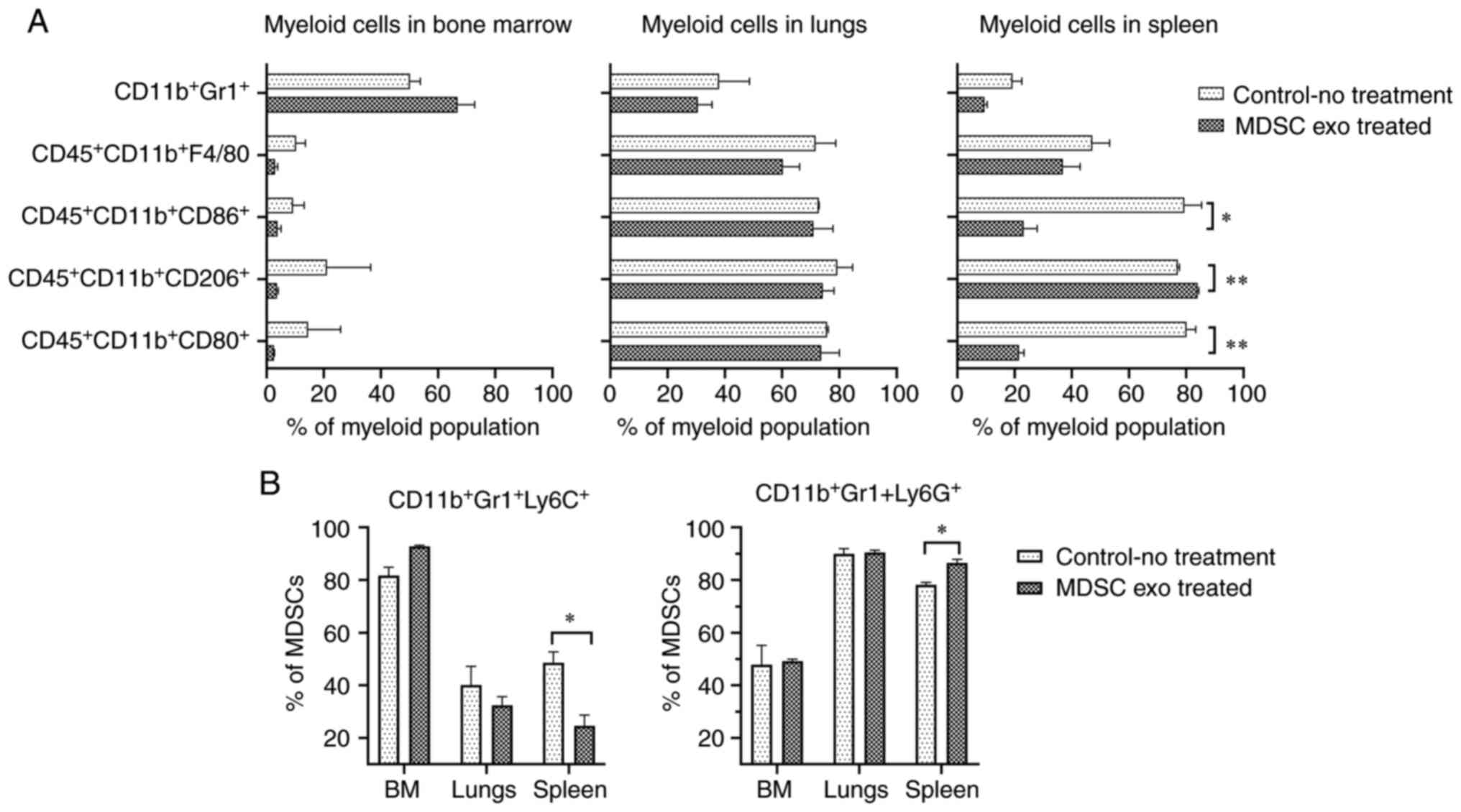
similar effect in the other organs (Fig. 6A). Representative dot-plots are
provided in Figs. S2 and S3. There was a considerable decline in
the monocytic MDSCs
(CD11b+Gr1+Ly6C+) and the
expansion of granulocytic MDSCs
(CD11b+Gr1+Ly6G+) in the spleen of
the treated animals compared to the untreated group (Fig. 6B). Representative dot-plots are
provided in Fig. S4.
In vitro effect of MDSC-derived
exosomes on T-cells
Since we demonstrated that MDSC exo express a
significantly high level of cytokines that facilitate regulatory
T-cell or Th2 cell functions and immunosuppression, we wanted to
investigate the effect of MDSC exo directly on CD4+ and
CD8+ T-cells in vitro. We isolated both types of
cells and treated them with spleen MDSC exo or with the same volume
of PBS (control). After 24 h, we collected the cells and analyzed
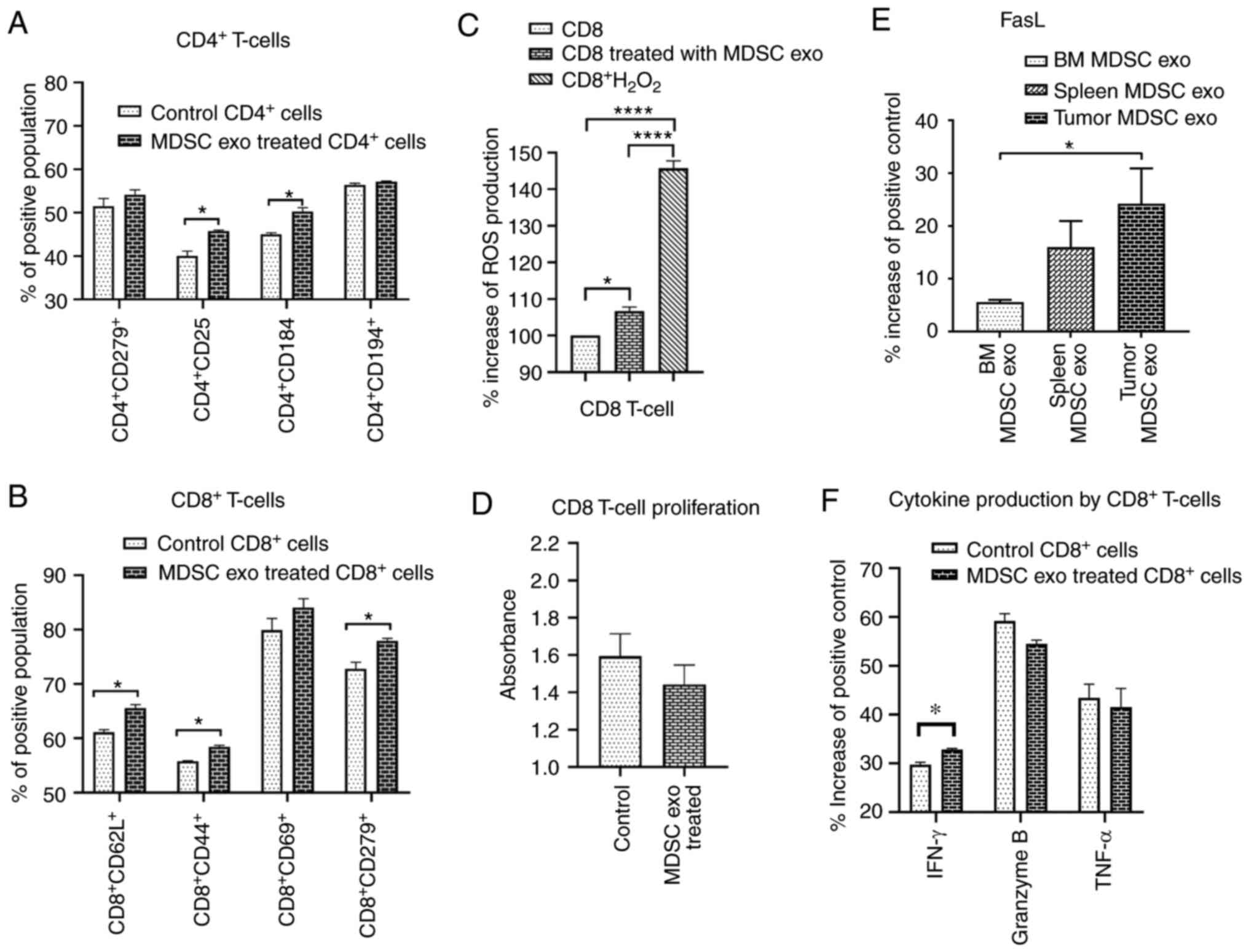
the functional marker changes by flow cytometry. Splenic MDSC
exo-treated CD4+ T-cells expressed a significantly
higher level of T-regulatory cell marker (CD25) and Th2 cell marker
(CD184) compared to the control group (Fig. 7A). There was no change in the level
of T-cell activation or exhaustion marker (CD279/PD-1). For the
CD8+ T-cells, MDSC exo-treated cells showed a
significantly higher level of T-cell activation marker (CD44),
naïve T-cell marker (CD62L), and exhaustion marker (CD279)
(Fig. 7B). Representative dot-plots
are provided in the supplementary file (Figs. S5 and S6).
In vitro effect of MDSC-derived
exosomes on CD8+ T-cell function and proliferation
MDSCs release ROS molecules as part of a primary
mechanism to suppress T-cell responses (27). Considering the previous results on
CD8+ T-cells, we further determined the level of ROS
production by CD8+ T-cells after splenic MDSC exo
treatment and also whether the treatment affects CD8+
T-cell proliferation. We labeled the CD8+ T-cells with
CM-H2DCFDA ROS probe and treated them with MDSC exo or
with PBS (control). After 4 h, MDSC exo-treated CD8+
T-cells showed a considerably higher level of ROS production
compared to the control (Fig. 7C).
Although the cell proliferation assay using WST-1 reagent showed a
decreased number of CD8+ T-cells in the MDSC exo group
compared to the control group, it was not statistically significant
(Fig. 7D).
Considering the fact that MDSC exo were able to
activate and deplete CD8+ T-cells, we determined the
level of FasL in different MDSC exo that could conceivably trigger
the apoptosis process in CD8+ T-cells. We detected
higher expression of FasL in the exosomes isolated from the MDSCs
in tumors compared to that in exosomes of MDSCs from spleen and
bone marrow (Fig. 7E). Furthermore,
we quantified the activation markers of CD8+ T-cells
with or without treatment with MDSC exo by protein array. Treatment
with MDSC exo appreciably increased the level of interferon (IFN)-γ
while no significant changes in granzyme B and TNF-α level were
observed (Fig. 7F).
Discussion
It has been perceived that functional differences
may exist in myeloid-derived suppressor cells (MDSCs) isolated from
different environments within the same host, and that MDSCs from
tumors have a stronger immunosuppressive capacity than MDSCs in the
peripheral lymphoid organs (spleen, lymph nodes) (28). We observed that exosomes were
secreted more abundantly from tumor MDSCs, presumably due to the
fact that MDSCs in the tumor microenvironment (TME) are in a more
distressing milieu (e.g., hypoxia and acidic pH). It has been
contemplated that cells may exploit exosome secretion to survive
under stressful conditions (29–31).
We also observed a higher concentration of proteins that are
crucial for tumor growth, invasion, angiogenesis, and
immunomodulation in exosomes isolated from MDSCs in tumors than
those from the spleen or bone marrow. Although Haverkamp et
al reported that MDSCs from inflammatory sites or tumor tissue
possess the immediate ability to hamper T-cell function whereas
those isolated from peripheral tissues were not suppressive without
activation of iNOS by exposure to IFN-γ (32), we observed equivalent competency in
MDSC exosomes (exo) isolated both from the tumor and spleen.
However splenic MDSC exo demonstrated that to a lesser extent. For
comparison as a control group, we used MDSCs from the bone marrow
of normal wild-type animals. Although initially we wanted to use
MDSCs from the spleen of normal/wild-type mice, it was quite
impossible to isolate enough MDSCs from multiple spleens of
wild-type animals while they were abundantly present in the spleen
of tumor-bearing animals. Since distribution of MDSCs is very low
(~1–2%) in normal spleen (8,33), we
chose to use exosomes collected from bone marrow-derived MDSC as
the control group. In spite of the fact that tumor MDSC exo had
higher immunosuppressive potential, we used splenic MDSC exo
(spleens were collected from tumor-bearing animals) for the
downstream functional assays. Part of the reason was that MDSCs in
the tumor-bearing mouse spleen were abundant for purification and
growth in culture for adequate exosome isolation. Compared to the
spleen, the number of MDSCs in the tumor was limited and we needed
to pool MDSCs from multiple tumors to obtain enough cells for
culture.
MDSCs are competent in promoting tumor growth
through remodeling the TME (21,34).
However, MDSCs are a miscellaneous population of immature myeloid
cells that is comprised of monocytic and granulocytic
subpopulations both of which have been shown to be
immunosuppressive (35). It has
been recently reported that early expansion and infiltration of
mMDSCs take place in primary tumors where they pave the way for
tumor cell dissemination by inducing epithelial to mesenchymal
transition (EMT), and higher levels of gMDSC infiltration occurs in
the metastatic site where they augment the colonization and
metastatic growth of disseminated tumor cells by reversing EMT
(36). We observed that treatment
of normal wild-type mice with MDSC exo significantly decreased the
number of monocytic (m)MDSCs and increase the number of
granulocytic (g)MDSCs in the spleen. As expected, we also detected
a decrease in M1-macrophages in the spleen.
Tumor-associated macrophages (TAMs) are part of the
heterogeneous populations of immunosuppressive myeloid cells that
produce chemokines for the activation and maintenance of
inflammatory processes in the TME (37–40).
For the most part, M1 macrophages induce T helper type 1 cell (Th1)
responses that drive cellular immunity to eliminate cancerous
cells, while M2 macrophages incite Th2 responses associated with
the anti-inflammatory and immunosuppressive TME, which promotes
tumor growth (17,41). We observed a substantial decline of
M1-macrophages and an expansion of M2 macrophages following
treatment with MDSC exo that also imply their immunosuppressive
effect in the TME.
We observed that MDSC-derived exosomes are able to
deplete CD8+ T-cells in vivo and inhibit the
proliferation of CD8+ T-cells in vitro. When
activated through their antigen-specific T-cell receptor (TCR) and
CD28 co-receptor, resting mature T lymphocytes start to proliferate
followed by the so-called activation-induced cell death (AICD),
which mechanistically is triggered by the death receptor and leads
to apoptosis. The apoptotic pathway is triggered by signals
originating from cell-surface death receptors that are activated by
several ligands such as CD95L (FasL), tumor necrosis factor (TNF),
or TNF-related apoptosis-inducing ligand (TRAIL) (42). Our protein array data demonstrated
that MDSC exo contain a high level of Fas and TNF-1α, which
indicate a potential role of these exosomes in inciting apoptotic
pathways. We noted that MDSC exo treatment increased the activation
markers of CD8+ T cells (CD69 and CD44), as well as
their exhaustion marker CD279/PD-1. CD8+ T-cells also
kill target cells by a cytokine-mediated mechanisms (e.g., by
IFN-γ, TNF-α), which are produced and secreted as long as TCR
stimulation continues. IFN-γ induces transcriptional activation of
the MHC class I antigen presentation pathway and Fas in target
cells, leading to enhanced Fas-mediated target-cell lysis (43). We noted that MDSC exo can activate
CD8+ T cells and prompt them to generate more IFN-γ.
Interestingly, ROS can control the fate of antigen-specific T cells
through reciprocal modulation of the main effector molecules FasL
and Bcl-2 (44). We detected a
significantly large amount of ROS production from the
CD8+ T-cells that were treated with MDSC exo. Therefore,
we hypothesized that MDSC exo precipitate CD8+ T-cell
apoptosis by AICD through hyper-activation or repeated stimulation,
which in turn results in increased levels of ROS production and
activation of the Fas/FasL (CD95/CD95L) pathway. According to our
data, we believe that ROS are involved in the reduction of
CD8+ T-cells and there is a possibility that ROS
inhibition such as treatment with N-acetylcysteine might prevent
this reduction. Although we did not look into the lymph node T-cell
distribution, it will be interesting to see cellular distribution
changes following MDSC exo treatment in future studies.
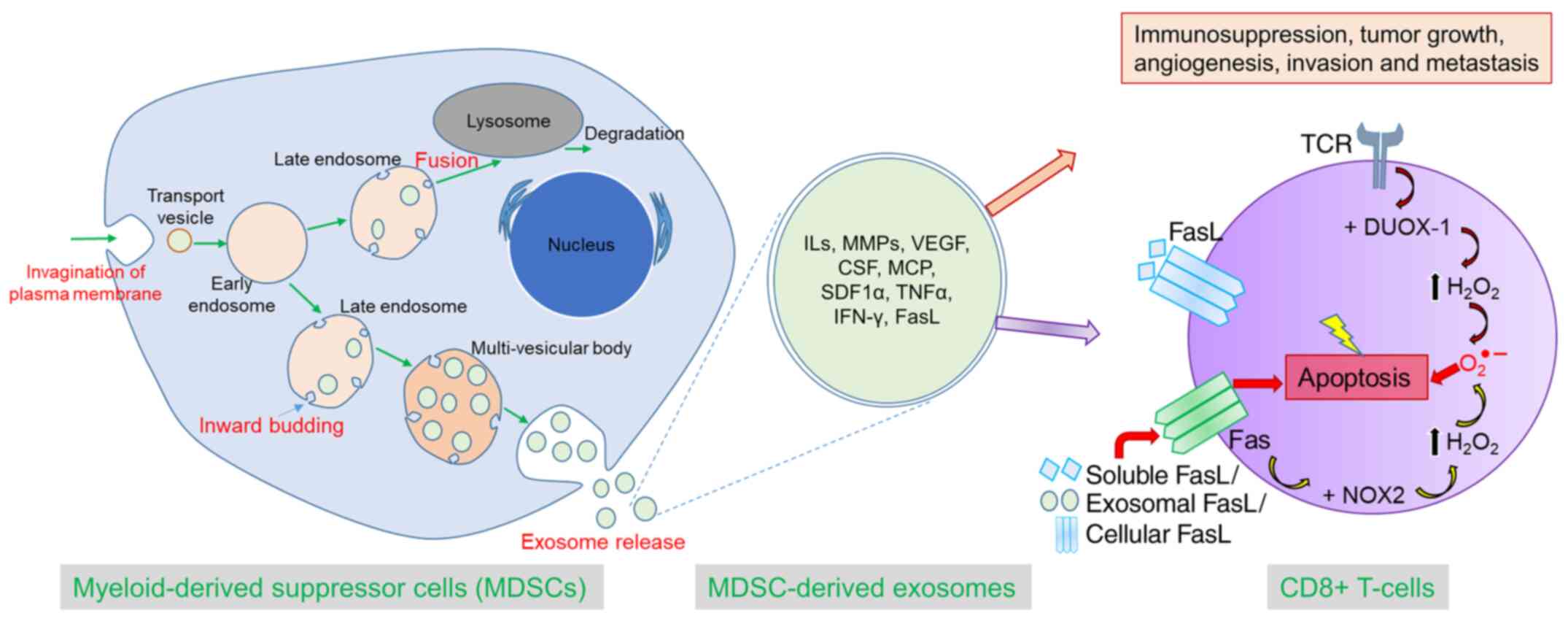
In summary, we comprehensively demonstrated that
MDSC-derived exosomes inherit pro-tumorigenic factors and
functionally resemble parental cells in immunosuppression, tumor
growth, angiogenesis, invasion, and metastasis. In addition,
MDSC-derived exosomes are capable of increasing ROS production and
inciting the Fas/FasL pathway in CD8+ T-cells, which
precipitates AICD (Fig. 8). This
novel concept would open up a new avenue of MDSC research and
MDSC-targeted therapy.
Supplementary Material
Supporting Data
Acknowledgements
The authors thank Dr Rhea-Beth Markowitz, Director,
Office of Grant Development, Georgia Cancer Center for help with
the English language editing of the manuscript.
Funding
This study was supported by the Georgia Cancer
Center Startup Fund and Intramural Grant Program at Augusta
University (Augusta, GA, USA) to ASA.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
MHR conceived the hypothesis, designed and performed
the experiments, and conducted the data collection, data analysis,
and interpretation, and wrote the manuscript. TFB conducted
acquisition of the in vitro data and edited the manuscript.
RA performed the animal experiments and treated the animals. RP
conducted the data interpretation and edited the manuscript. BRA
helped with planning the in vitro T-cell experiments. HK
guided MDSC collection and types, and helped with implantation of
breast cancers. YL provided laboratory facilities for NTA, data
interpretation, and editing of the manuscript. ASA supervised the
findings of this work, aided in interpreting the results, and
provided the funds and critical revision of the manuscript.
Ethics approval and consent to
participate
All experiments were performed according to the
National Institutes of Health (NIH) guidelines and regulations. The
Institutional Animal Care and Use Committee (IACUC) of Augusta
University (Augusta, GA, USA) (protocol #2014-0625) approved the
experimental procedures.
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest
exists.
References
|
1
|
Balkwill FR, Capasso M and Hagemann T: The
tumor microenvironment at a glance. J Cell Sci. 125:5591–5596.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dysthe M and Parihar R: Myeloid-derived
suppressor cells in the tumor microenvironment. Tumor
Microenvironment: Hematopoietic Cells-Part A. Birbrair A: Springer
International Publishing; Cham: pp. 117–140. 2020, View Article : Google Scholar
|
|
3
|
Achyut BR and Arbab AS: Myeloid derived
suppressor cells: Fuel the fire. Biochem Physiol. 3:e123.
2014.PubMed/NCBI
|
|
4
|
Arbab AS, Rashid MH, Angara K, Borin TF,
Lin PC, Jain M and Achyut BR: Major challenges and potential
microenvironment-targeted therapies in glioblastoma. Int J Mol Sci.
18:27322017. View Article : Google Scholar
|
|
5
|
Safarzadeh E, Hashemzadeh S, Duijf PHG,
Mansoori B, Khaze V, Mohammadi A, Kazemi T, Yousefi M, Asadi M,
Mohammadi H, et al: Circulating myeloid-derived suppressor cells:
An independent prognostic factor in patients with breast cancer. J
Cell Physiol. 234:3515–3525. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Trac N.T..Chung E.J.: Peptide-based
targeting of immunosuppressive cells in cancer. Bioactive
Materials. 5:92–101. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lechner MG, Liebertz DJ and Epstein AL:
Characterization of cytokine-induced myeloid-derived suppressor
cells from normal human peripheral blood mononuclear cells. J
Immunol. 185:2273–2284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bronte V, Brandau S, Chen SH, Colombo MP,
Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A,
Ostrand-Rosenberg S, et al: Recommendations for myeloid-derived
suppressor cell nomenclature and characterization standards. Nat
Commun. 7:121502016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang Z, Guo J, Weng L, Tang W, Jin S and
Ma W: Myeloid-derived suppressor cells-new and exciting players in
lung cancer. J Hematol Oncol. 13:102020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Corzo CA, Cotter MJ, Cheng P, Cheng F,
Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC and
Gabrilovich DI: Mechanism regulating reactive oxygen species in
tumor-induced myeloid-derived suppressor cells. J Immunol.
182:5693–5701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bruno A, Mortara L, Baci D, Noonan DM and
Albini A: Myeloid derived suppressor cells interactions with
natural killer cells and pro-angiogenic activities: Roles in tumor
progression. Front Immunol. 10:7712019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dai J, El Gazzar M, Li GY, Moorman JP and
Yao ZQ: Myeloid-derived suppressor cells: Paradoxical roles in
infection and immunity. J Innate Immun. 7:116–126. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Geiger R, Rieckmann JC, Wolf T, Basso C,
Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et
al: L-arginine modulates T cell metabolism and enhances survival
and anti-tumor activity. Cell. 167:829–842.e13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sinha P, Clements VK, Bunt SK, Albelda SM
and Ostrand-Rosenberg S: Cross-talk between myeloid-derived
suppressor cells and macrophages subverts tumor immunity toward a
type 2 response. J Immunol. 179:977–983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ostrand-Rosenberg S and Fenselau C:
Myeloid-derived suppressor cells: Immune-suppressive cells that
impair antitumor immunity and are sculpted by their environment. J
Immunol. 200:422–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rashid MH, Borin TF, Ara R, Angara K, Cai
J, Achyut BR, Liu Y and Arbab AS: Differential in vivo
biodistribution of 131I-labeled exosomes from diverse
cellular origins and its implication for theranostic application.
Nanomedicine. 21:1020722019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rashid MH, Borin TF, Ara R, Alptekin A,
Liu Y and Arbab AS: Generation of novel diagnostic and therapeutic
exosomes to detect and deplete protumorigenic M2 macrophages. Adv
Ther (Weinh). 3:19002092020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Du R, Lu KV, Petritsch C, Liu P, Ganss R,
Passegué E, Song H, Vandenberg S, Johnson RS, Werb Z and Bergers G:
HIF1alpha induces the recruitment of bone marrow-derived vascular
modulatory cells to regulate tumor angiogenesis and invasion.
Cancer Cell. 13:206–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hiratsuka S, Watanabe A, Aburatani H and
Maru Y: Tumour-mediated upregulation of chemoattractants and
recruitment of myeloid cells predetermines lung metastasis. Nat
Cell Biol. 8:1369–1375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pawelek JM and Chakraborty AK: Fusion of
tumour cells with bone marrow-derived cells: A unifying explanation
for metastasis. Nat Rev Cancer. 8:377–386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Umansky V, Blattner C, Gebhardt C and
Utikal J: The role of myeloid-derived suppressor cells (MDSC) in
cancer progression. Vaccines (Basel). 4:362016. View Article : Google Scholar
|
|
22
|
Yang L, Huang J, Ren X, Gorska AE, Chytil
A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC and Moses
HL: Abrogation of TGF beta signaling in mammary carcinomas recruits
Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell.
13:23–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pollard JW: Tumour-educated macrophages
promote tumour progression and metastasis. Nat Rev Cancer. 4:71–78.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Palma M, Venneri MA, Galli R, Sergi
Sergi L, Politi LS, Sampaolesi M and Naldini L: Tie2 identifies a
hematopoietic lineage of proangiogenic monocytes required for tumor
vessel formation and a mesenchymal population of pericyte
progenitors. Cancer Cell. 8:211–226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Conejo-Garcia JR, Buckanovich RJ, Benencia
F, Courreges MC, Rubin SC, Carroll RG and Coukos G: Vascular
leukocytes contribute to tumor vascularization. Blood. 105:679–681.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nozawa H, Chiu C and Hanahan D:
Infiltrating neutrophils mediate the initial angiogenic switch in a
mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA.
103:12493–12498. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ohl K and Tenbrock K: Reactive oxygen
species as regulators of MDSC-mediated immune suppression. Front
Immunol. 9:24992018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma J, Xu H and Wang S: Immunosuppressive
role of myeloid-derived suppressor cells and therapeutic targeting
in lung cancer. J Immunol Res. 2018:63196492018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
De Maio A: Extracellular heat shock
proteins, cellular export vesicles, and the stress observation
system: A form of communication during injury, infection, and cell
damage. It is never known how far a controversial finding will go!
Dedicated to Ferruccio Ritossa. Cell Stress Chaperones. 16:235–249.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kucharzewska P and Belting M: Emerging
roles of extracellular vesicles in the adaptive response of tumour
cells to microenvironmental stress. J Extracell Vesicles. 2:2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McAndrews KM and Kalluri R: Mechanisms
associated with biogenesis of exosomes in cancer. Mol Cancer.
18:522019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haverkamp JM, Crist SA, Elzey BD, Cimen C
and Ratliff TL: In vivo suppressive function of myeloid-derived
suppressor cells is limited to the inflammatory site. Eur J
Immunol. 41:749–759. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao F, Obermann S, von Wasielewski R,
Haile L, Manns MP, Korangy F and Greten TF: Increase in frequency
of myeloid-derived suppressor cells in mice with spontaneous
pancreatic carcinoma. Immunology. 128:141–149. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sevko A and Umansky V: Myeloid-derived
suppressor cells interact with tumors in terms of myelopoiesis,
tumorigenesis and immunosuppression: Thick as thieves. J Cancer.
4:3–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Veglia F, Perego M and Gabrilovich D:
Myeloid-derived suppressor cells coming of age. Nat Immunol.
19:108–119. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ouzounova M, Lee E, Piranlioglu R, El
Andaloussi A, Kolhe R, Demirci MF, Marasco D, Asm I, Chadli A,
Hassan KA, et al: Monocytic and granulocytic myeloid derived
suppressor cells differentially regulate spatiotemporal tumour
plasticity during metastatic cascade. Nat Commun. 8:149792017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gao F, Liang B, Reddy ST, Farias-Eisner R
and Su X: Role of inflammation-associated microenvironment in
tumorigenesis and metastasis. Curr Cancer Drug Targets. 14:30–45.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mou W, Xu Y, Ye Y, Chen S, Li X, Gong K,
Liu Y, Chen Y, Li X, Tian Y, et al: Expression of Sox2 in breast
cancer cells promotes the recruitment of M2 macrophages to tumor
microenvironment. Cancer Lett. 358:115–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dijkgraaf EM, Heusinkveld M, Tummers B,
Vogelpoel LT, Goedemans R, Jha V, Nortier JW, Welters MJ, Kroep JR
and van der Burg SH: Chemotherapy alters monocyte differentiation
to favor generation of cancer-supporting M2 macrophages in the
tumor microenvironment. Cancer Res. 73:2480–2492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Allavena P, Sica A, Solinas G, Porta C and
Mantovani A: The inflammatory micro-environment in tumor
progression: The role of tumor-associated macrophages. Crit Rev
Oncol Hematol. 66:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Biswas SK and Mantovani A: Macrophage
plasticity and interaction with lymphocyte subsets: Cancer as a
paradigm. Nat Immunol. 11:889–896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sikora E: Activation-induced and
damage-induced cell death in aging human T cells. Mech Ageing Dev.
151:85–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Andersen MH, Schrama D, Thor Straten P and
Becker JC: Cytotoxic T Cells. J Invest Dermatol. 126:32–41. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hildeman DA, Mitchell T, Kappler J and
Marrack P: T cell apoptosis and reactive oxygen species. J Clin
Invest. 111:575–581. 2003. View Article : Google Scholar : PubMed/NCBI
|