Introduction
Despite the recent developments in novel
therapeutics and improvements in early detection, the 5-year
survival rates for lung cancer (LC) and pancreatic cancer (PC)
patients remain at 19 and 9%, respectively. To date, these two
diseases are the first and fourth most common cancer-related deaths
in the US (1). These data establish
the magnitude of the clinical problem, underlying the need for
identifying novel therapeutic approaches.
Dysregulated cell cycle progression and rapid cell
proliferation is a common attribute of cancer cells (2,3). In normal
cells, the sequential progression through the stages of the cell
cycle is regulated by the differential expression and activation of
proteins, with cyclins and cyclin-dependent kinases (CDKs) playing
a central role (4). The activity of
the cyclin/CDK complexes prevents the premature entry of cells into
the cell cycle and ensure its appropriate progression. In cancer
cells, these highly conserved pathways are believed to enable the
viability of cancer cells in the face of deregulated cellular
proliferation (5) and consequently
have been identified as therapeutic targets for cancer treatment
(4). For example, numerous clinical
trials have taken place using molecules that inhibit the activity
of CDKs and block cell cycle progression. Recently, three such
molecules (ribociclib, palbociclib and abemaciclib) were approved
for the treatment of hormone receptor-positive, epidermal growth
factor receptor 2 (EGFR2)-negative metastatic breast cancer
(6–12).
G1/S transition of the cell cycle is
associated with the activity of CDK4/6. In LC, CDK4 has an elevated
expression, which negatively correlates to prognosis (13), while CDK6 has been reported to be
either upregulated or downregulated in different subtypes of LC
(14). Early research on halting
G1/S cell cycle transition using CDK4 inhibition has
been promising, with induced apoptotic activity and tumor growth
inhibition (15,16). Unfortunately, clinical trials have
indicated that CDK4/6 monotherapy is unlikely to exert sufficient
therapeutic outcomes, even in cases with CDK4/6 activation
(17).
Similar to LC, PC displays a broad spectrum of
genetic alterations that drive the cancer phenotype and supports
uncontrolled proliferation. For example, activating KRAS,
observed in >90% of cases (18,19),
drives deregulated proliferation and activates survival signals,
but can also elicit replication stress, induction of reactive
oxygen species, and promote oncogene-induced senescence (20,21). Rapid
proliferation and deregulated cell cycle progression are also
observed in PC, which is associated with the activation of the CDKs
(22). CDKs are upregulated in PC,
and their expression is also negatively correlated with
chemoresistance and prognosis (23).
Efforts to regulate the cell cycle in PC through CDK inhibitors,
focusing primarily on halting the G1/S transition have
taken place (22). Still, PC exhibits
intrinsic resistance to CDK4/6 inhibition mediated through a
KRAS-dependent response and blocking of the Rb tumor suppressor
activity (23,24), which can also lead to increased
metastatic potential (25).
Thus, although inducing a G1/S transition
halt can be a promising approach for cancer treatment, combination
therapies targeting complementary modulators of the cell cycle are
required to overcome these limitations in LC and PC (26). During cell cycle progression,
transitioning from G2 to M phase is a crucial step for
the successful mitosis process. CDK1 is a critical protein for that
transition and the completion of cell cycle progression (8,27,28). Furthermore, CDK1 expression is
increased in LC and PC samples, with a negative correlation to
survival rates (29–31), also classifying this protein as a
predictive biomarker and potential target for treatment in these
two cancers (29,32). Targeting two cell cycle transitions,
at the G1/S and G2/M represents a highly
significant strategy for LC and PC therapy.
miR-based therapeutics have emerged as promising
mRNA regulators for cancer treatment, capable to control multiple
cell functions (33). In particular,
the miR combination of miR-143 and miR-506 inhibits the cell cycle
progression in two phase transitions, through downregulation of
CDK1, CDK4 and CDK6, and causes strong apoptotic activity, as we
recently reported (34,35). Here, we report on the combinatorial
treatment effect of these two miRs downregulating CDK1, CDK4 and
CDK6, and inhibiting cell growth in LC and PC cells. Our analysis
reveals the efficacy and activity of the combinatorial miR-143/506
treatment with broader implications in LC and PC.
Materials and methods
Materials
Cell culture reagents were obtained from Gibco™
(Thermo Fisher Scientific, Inc.) and VWR International, LLC.
miR-143-3p and miR-506-3p mimics were purchased from ABM. Negative
control scramble-siRNA was purchased from Ambion. Opti-MEM and
Lipofectamine 2000 reagent were obtained from Thermo Fisher
Scientific, Inc., and the Quick-RNA miniprep kit was obtained from
Zymo Research. Rabbit polyclonal anti-human CDK1 (dilution:
1:10,000; product #ab140847), rabbit monoclonal anti-human CDK4
(dilution: 1:2,000; product #ab108357), rabbit polyclonal
anti-human β-tubulin (dilution: 1:500; product #ab6046), and goat
anti-rabbit IgG H&L (HRP) secondary (dilution: 1:10,000;
product #ab97051) antibodies were purchased from Abcam. Other
chemicals and kits were purchased from Thermo Fisher Scientific,
Inc., VWR, or Sigma-Aldrich/Merck KGaA. H69-AR, Calu3, H358, and
H1975 LC cell lines, normal fibroblast cell line HFL-1, MIA-Paca-2,
and Panc-1 PC cell lines were obtained from and cultured according
to ATCC. Cells were accordingly cultured with DMEM, F12K media, or
RPMI-1640 media supplemented with 10% fetal bovine serum (FBS) and
1% penicillin/streptomycin, at 37°C with 5% CO2.
Cell culture and transfection
We transfected the respective cells with
miR-143(−3p), and miR-506(−3p) mimics or scrambled siRNA, using
Lipofectamine 2000, following the manufacturer's protocol and as
previously described (34,35). Briefly, H358 and H1975 LC cells were
cultured in RPMI medium. MIA-Paca-2 and Panc-1 PC cells were
cultured in DMEM supplemented with 10% fetal serum and 1%
penicillin/streptomycin. Although these cells lines were not
authenticated in our laboratory they were characterized by cell
morphology and growth rate, and cultured in our laboratory less
than six months after being received. For different experiments,
the cells were seeded overnight in T25 cm2
flask/6well/96-well plates, accordingly, and we transfected them
with the respective miR mimics or scramble at 100 nM using
Lipofectamine 2000, for 6 h, and the media were subsequently
replaced with fresh media for incubation of up to 48 h.
Quantitative real-time qPCR (qPCR)
analysis
We isolated total RNA using the Quick-RNA Miniprep
kit and used Verso cDNA Synthesis Kit to develop complementary DNA
to performed quantitative RT-qPCR using PowerUP SYBR-Green Master
Mix (Applied Biosystems/Thermo Fisher Scientific, Inc.), as
previously described (34,35). In Table
SI, we present the primer sequences used for the detection of
CDK1, CDK4, CDK6, and CYPA. We normalized all of the
results to the untreated cells and calculated differential gene
expression using the -ΔΔCq method (36). Scrambled siRNA with Lipofectamine were
used as a negative control. All P-values are in comparison to
untreated (control) samples unless stated otherwise.
Western blot analysis
We performed western blot (WB) analysis on protein
extracts from H358, H1975, MIA-Paca-2, and Panc-1 cells, following
previously established protocols (34,35).
Briefly, protein extracts from the cells transfected with miR-143
and/or miR-506 for 24 and/or 48 h were aliquoted. We used 10–12%
(w/v) polyacrylamide gel electrophoresis to separate proteins
according to their size and transferred them to PVFD membranes,
followed by incubation with the respective antibodies to detect
CDK1, CDK4 or β-tubulin. After incubation at room temperature with
the secondary antibody, we identified the protein bands using
chemiluminescent substrate, visualized under a Chemidoc imaging
system (Bio-Rad Laboratories, Inc.). We performed the histogram
analysis using BioRad Image Lab V-6.0 software (Bio-Rad
Laboratories, Inc.).
Cell cycle assay
We performed the cell cycle analysis using a flow
cytometric technique, as previously described (34,35).
Briefly, we stained cells with propidium iodide (PI) (MP
Biomedicals, LLC) and analyzed the cells using a BD FACSCalibur
Flow Cytometer, with Cellquest Pro software (BD Biosciences). We
measured 10,000 events of the gated population. We identified the
percentage of cell distributions in the various cell cycle phases
using ModFit LT 5.0 (Verity Software House).
Apoptosis assay
We harvested H358, H1975, and HFL-1 cells 24 or 48 h
after transfection with miR-143 and miR-506, at 100 nM, as
described above, and stained them with FITC-Annexin V and PI, as
previously described (34,35). Subsequently, we analyzed the samples
using a BD FACSCalibur Flow Cytometer to determine apoptotic
behavior due to treatment. We measured 10,000 events of the gated
population. Untreated cells and cells treated with scrambled siRNA
(at 200 nM) with Lipofectamine were used as negative controls.
Cell proliferation
Twenty-four hours after transfection, the cells were
seeded in 96-well plates at a density of 2,000 cells per well. Cell
proliferation was determined after 48 h, using the reduction of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye
(MTT), according to the manufacture's protocol (Millipore
Sigma).
Statistical analysis and database
sources
We performed one way analysis of variance (ANOVA)
followed by post hoc Tukey's test to determine the significance of
differences among groups, unless otherwise specified. We present
the mean values ± standard errors; P-values <0.05 were
considered statistically significant. We used two database sources
to acquire gene expression relevant information in human patients.
Data from the Pan-Cancer Analysis, utilizing the ENCORI Platform
(http://starbase.sysu.edu.cn/panCancer.php) were used
to determine gene expression in human cancer and normal tissues
(37,38). Data from Protein Atlas were used to
determine survival probability vs. gene expression in patients
(http://www.proteinatlas.org; v19.proteinatlas.org) (39,40).
Results
CDK1 and CDK4 are upregulated while
miR-143 and miR-506 are downregulated in LC cell lines compared to
normal human fetal lung fibroblasts
We quantified the relative gene expression levels of
CDK1 and CDK4, using qPCR, in a panel of LC cell
lines and compared them to a normal human fetal lung fibroblast
cell line, HFL-1. As shown in Fig. 1A and
B, both CDK1 and CDK4 expression were significantly higher in
all of the tested LC cell lines (P<0.05), compared to HFL-1,
except in the case of CDK1 and CDK4 expression in A549 cells and
CDK1 expression in H358 cells, which did not achieve significant
difference compared to the normal cell line. Specifically, the
highest upregulation of CDK1 was in the H69-AR cell line (4.1-fold
increase vs. HFL-1; P<0.001), while A549 cells had the lowest
levels among the different LC cell lines (1.8-fold increase). In
contrast, H358 had the highest levels of CDK4 (4.8-fold increase
vs. HFL-1; P<0.05), while A549 also had the lowest levels of
CDK4 among the tested cancer cell lines, without achieving
significant difference to HFL-1.
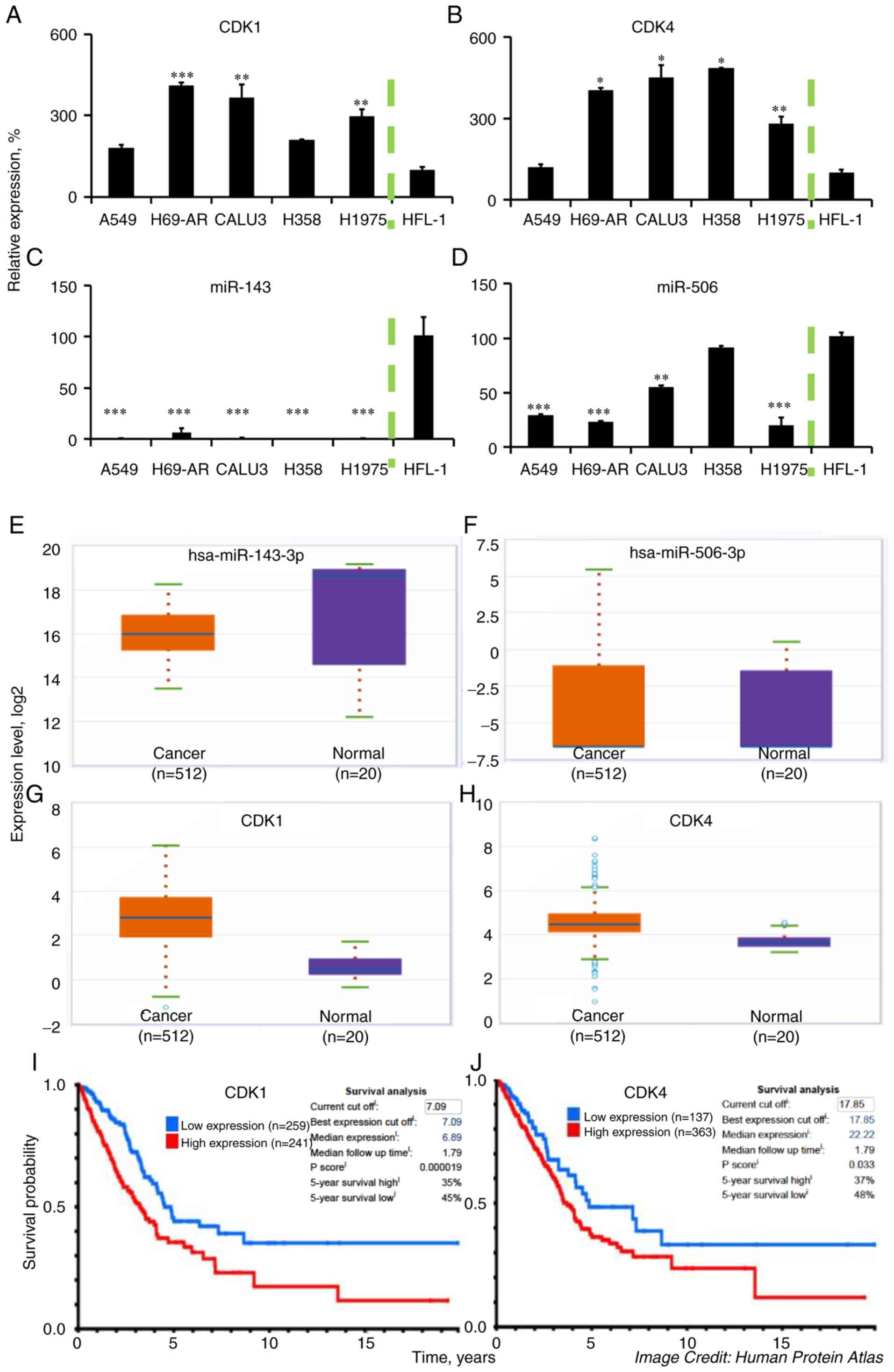 | Figure 1.CDK1, CDK4, miR-143, and miR-506
expression in LC cells and LC tissue samples. Higher CDK1 and CDK4
expression levels are associated with poor prognosis in patients
with LC adenocarcinomas. We detected the (A) CDK1, (B) CDK4, (C)
miR-143, and (D) miR-506, levels in a panel of LC cell lines and
compared them to the normal human fetal lung fibroblasts HFL-1,
through qPCR. U6 RNA and GAPDH were used as reference genes, where
applicable. (E-H) We assessed the relative expression of the same
genes in tumor and normal tissues using the ENCORI Pan-Cancer
Analysis Platform. (I and J) Kaplan-Meier plots were generated from
patient survival using data from the Proteinatlas database
(v19.proteinatlas.org/humancell). ***P<0.001;
**P<0.01; *P<0.05 compared to HFL-1 cell line. CDK,
cyclin-dependent kinase; LC, lung cancer. |
Similarly, we quantified the expression of
miR-143-3p and miR-506-3p in the LC cell lines, using qPCR, and
compared their expression to the normal cell line HFL-1 (Fig. 1C and D). Our analysis indicated a
strong and consistent downregulation of miR-143 in the LC cell
lines (all P<0.001) compared to HFL-1, which aligns with
previous reports (41). In contrast,
miR-506 had a variable expression among the different LC cell
lines, being significantly lower in A549 (71% downregulation;
P<0.001), H69-AR (77% downregulation, P<0.001), H1975 (81%
downregulation; P<0.001) and Calu-3 (45% downregulation,
P<0.01) compared to the HFL-1 cell line. H358 had approximately
similar levels of the gene to HFL-1. Processing data from the
Pan-Cancer Analysis, utilizing the ENCORI Platform, we determined
the relative expression of CDK1, CDK4, miR-143, and miR-506 genes
in lung adenocarcinomas (LUAD) vs. normal tissues (37,38).
Briefly, to acquire the relative expression between the LUAD and
normal tissue, we used the Starbase website (http://starbase.sysu.edu.cn/panCancer.php) and looked
for the relative gene expression by selecting the mRNA or miRNA
expression. miR-143-3p was found to be relatively lower in the
cancer samples (median: 15.4) compared to the normal samples
(median: 18.6; Fig. 1E), while
miR-506-3p had low expression but similar levels between cancer
(median: −6.6) and normal samples (median: −6.6; Fig. 1F). The expression of CDK1 was higher
in the cancer samples (median: 2.7) compared to the normal samples
(median: 0.4; Fig. 1G), as well as
CDK4 was elevated in the cancer samples (median: 4.4) compared to
the normal samples (median: 3.6; Fig.
1H). These data align with our presented analysis of the
expression of these genes. Moreover, using the Protein atlas
database, we identified a negative correlation between the protein
expression for CDK1 and CDK4 vs. median survival in patients with
lung adenocarcinomas (Fig. 1I and
J).
Combinatorial treatment with miR-143
and miR-506 downregulates the CDK1, CDK4, and CDK6 mRNA and protein
expression in cancer cell lines
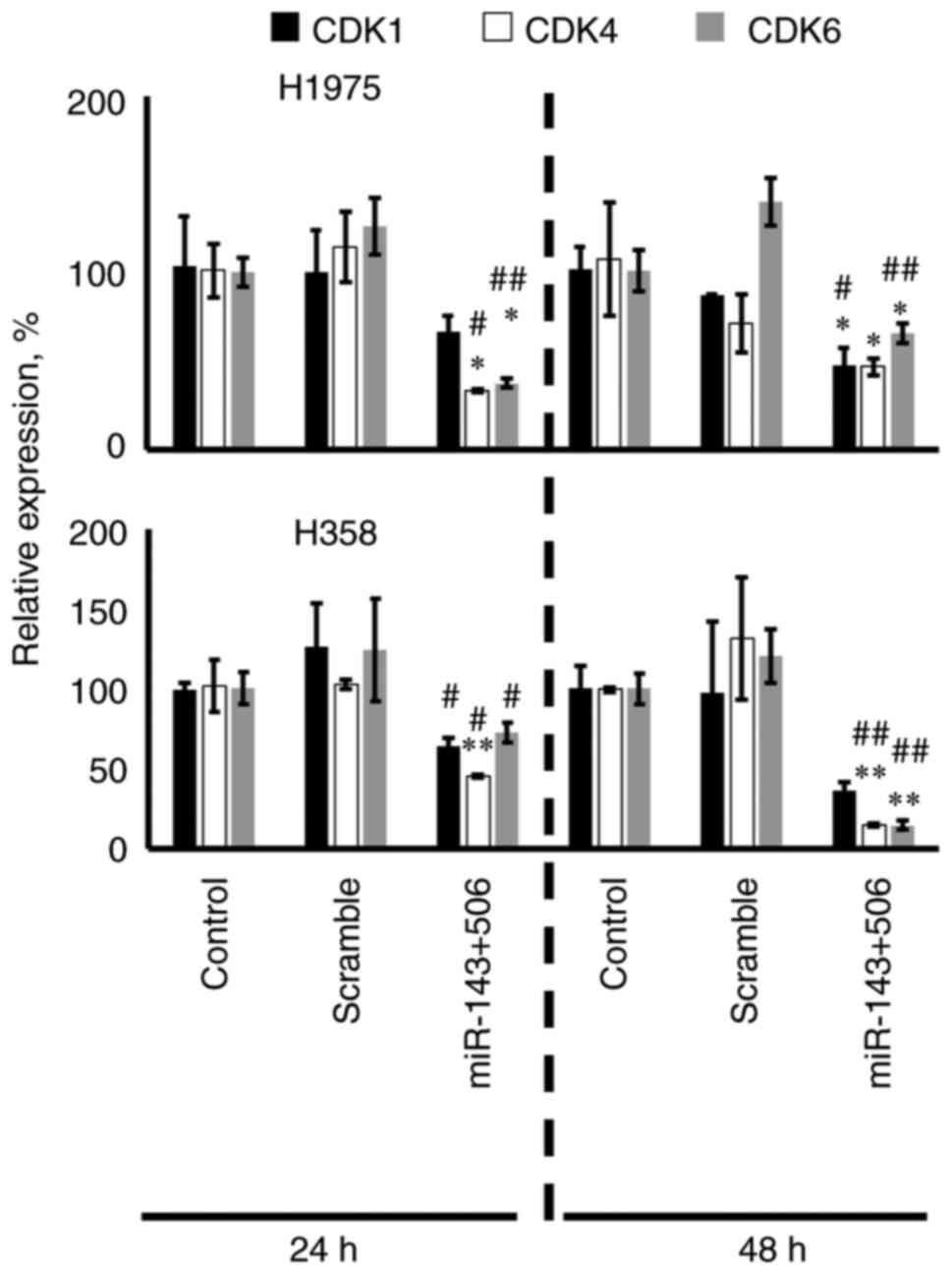
We transfected the H358 and H1975 cells with the
combinatorial treatment of miR-143/506 at 100 nM using
Lipofectamine, and analyzed the CDK1, CDK4, and CDK6
gene expression, using qPCR at 24 and 48 h post-transfection. The
chosen two cell lines carry driver mutations that can complement
and correlate with our previously published work on A549 cells
(34,35). The treatment downregulated these three
genes compared to the untreated or scramble-treated (negative
control) groups in both of the cell lines (Fig. 2). In H1975 cells, the combinatorial
treatment downregulated the CDK1, CDK4 and CDK6 genes
by 38% (no significance), 68% (P<0.05) and 63% (P<0.05) at 24
h, respectively, and by 54% (P<0.05), 61% (P<0.05) and 35%
(P<0.05) at 48 h, respectively, compared to the untreated
control. In H358 cells, the combinatorial treatment downregulated
the CDK1, CDK4 and CDK6 genes by 36% (no
significance), 54% (P<0.01) and 27% (no significance) at 24 h,
respectively, and by 73% (no significance), 85% (P<0.01), 85%
(P<0.01) at 48 h, respectively, compared to the untreated
control. Interestingly, in H358 cells, CDK1 was only significantly
downregulated when compared to the scramble control at 24 h.
Fig. 2 also shows detailed analysis
on comparison against the scramble controls.
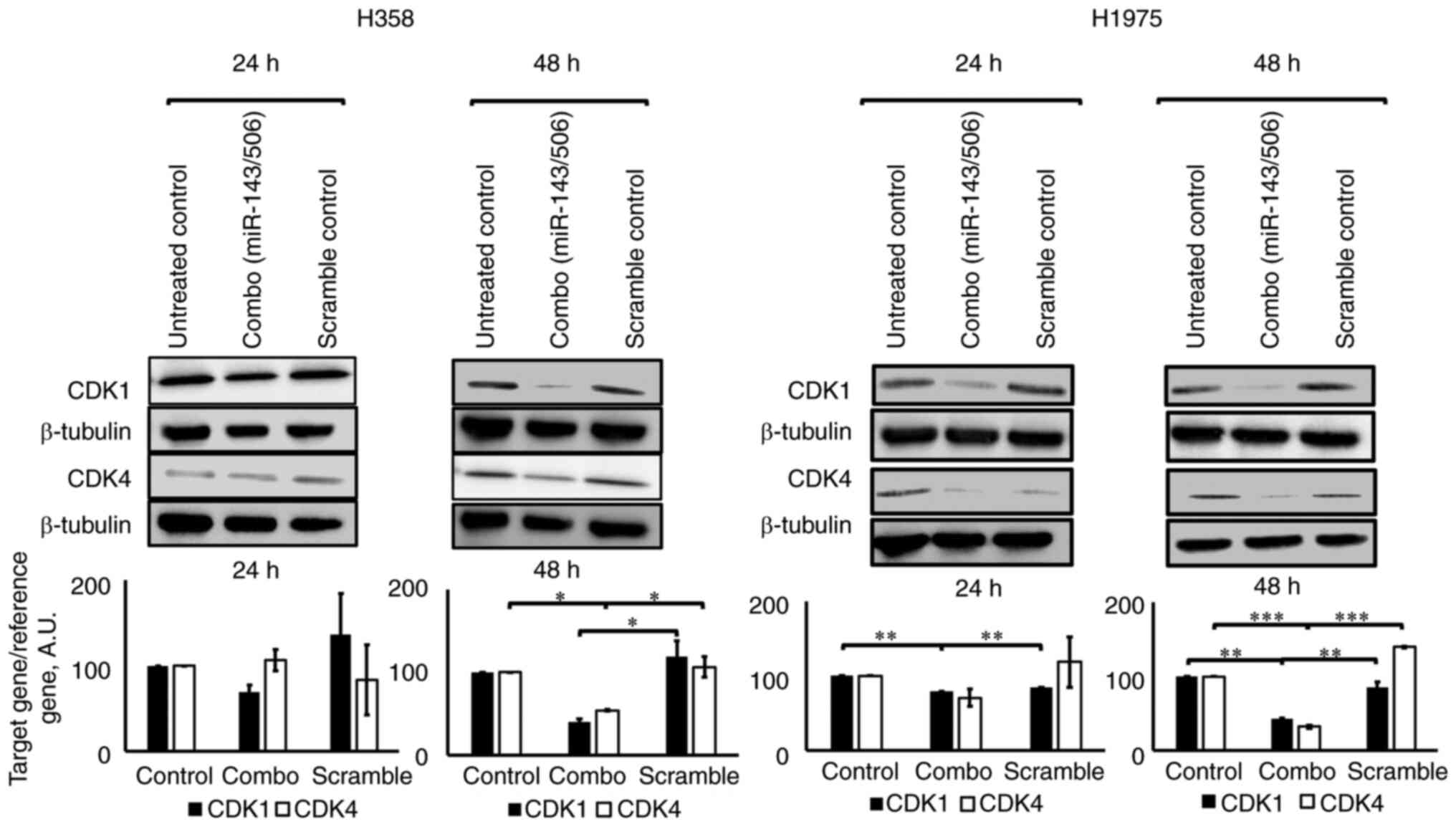
WB analysis confirmed the downregulation of CDK1 and
CDK4 by miR-143/506 transfection. Briefly, the combinatorial
treatment significantly reduced the expression levels of both CDK1
and CDK4 proteins after 48 h in both H358 and H1975 cell lines
(Fig. 3). Compared to the untreated
control at 48 h, we observed a 60% (no significance) and 46%
(P<0.05) downregulation of CDK1 and CDK4 genes, respectively, in
H358 cells, and a 58% (P<0.01) and 68% (P<0.01)
downregulation of CDK1 and CDK4, respectively, in H1975 cells.
Compared to scramble treatment at 48 h, we observed a 66%
(P<0.05) and 49% (P<0.05) downregulation of CDK1 and CDK4
genes, respectively, in H358 cells, and a 51% (P<0.01) and 77%
(P<0.001) downregulation of CDK1 and CDK4, respectively, in
H1975 cells. Of note, treatment with scrambled siRNA at equimolar
concentrations did not significantly alter the expression of the
studied genes.
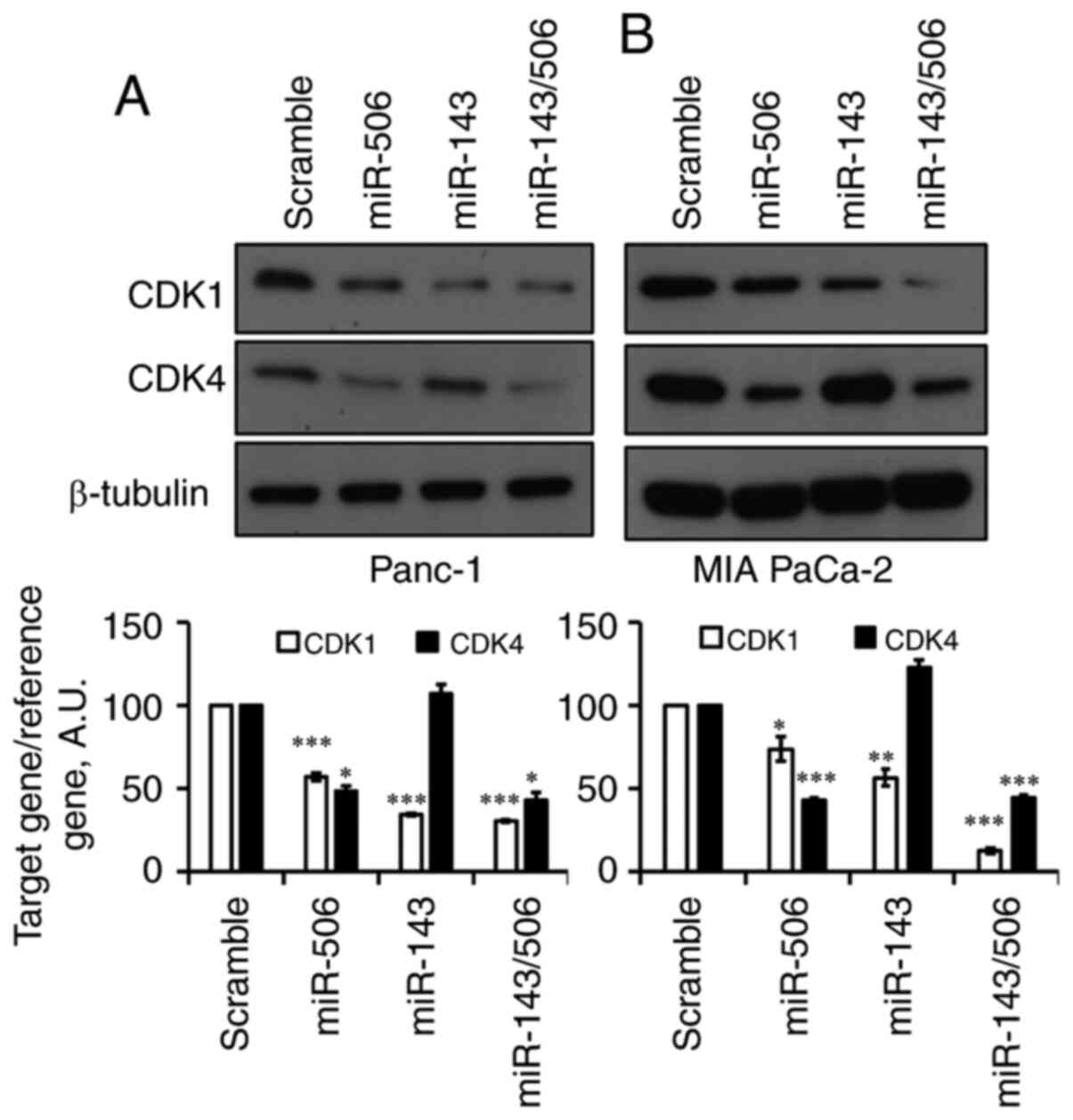
We next evaluated the effect of the individual miRs,
as well as their combinatorial treatment in human PC cells
(Fig. 4A and B). Forty-eight hours
post-transfection with 100 nM miR-143, CDK1 levels were
significantly reduced by 66% (P<0.001) in Panc-1 and 44%
(P<0.01) in MIA-PaCa-2 cells. miR-143 did not affect CDK4 levels
in both of the pancreatic cell lines. Following treatment with
equimolar miR-506, CDK4 levels were significantly reduced by 52%
(P<0.05) in Panc-1 cells and 57% (P<0.001) in MIA-PaCa-2
cells. Interestingly, miR-506 reduced CDK1 levels by 43%
(P<0.001) in Panc-1 cells and 23% (P<0.05) in MIA-PaCa-2
cells. The combination of miR-143 and miR-506 reduced CDK1 levels
by 70 and 88% (P<0.001 for both) in Panc-1 and MIA-PaCa-2 cells,
respectively. Moreover, the combinatorial miR treatment reduced
CDK4 levels by 58% (P<0.05) and 56% (P<0.001) in Panc-1 and
MIA-PaCa-2 cells, respectively.
miR-143 and miR-506 combinatorial
treatment increased the G0/G1 and
G2 cell populations
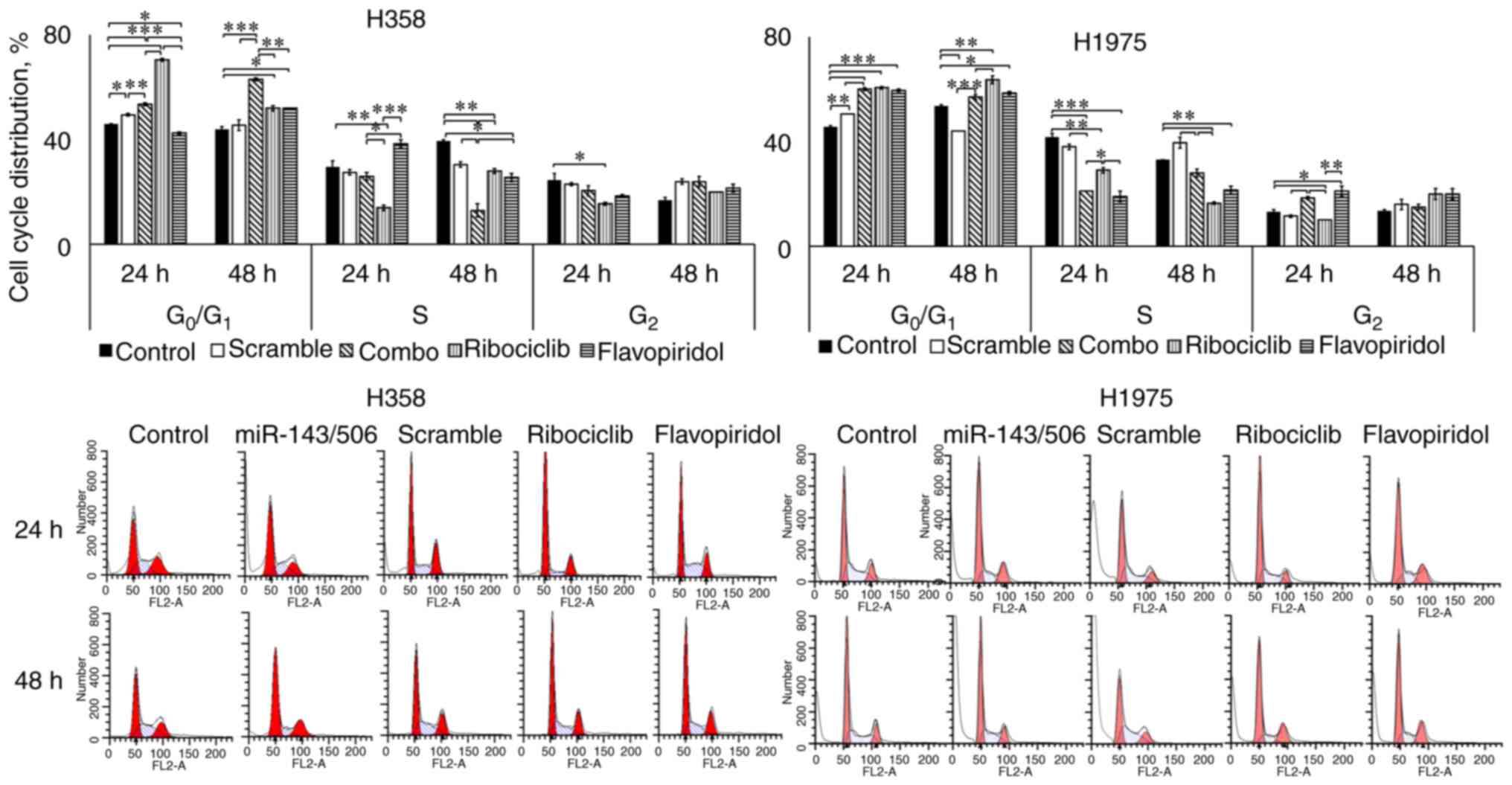
We evaluated the cell cycle regulatory effect of
miR-143/506 combination using flow cytometry in H1975 and H358 cell
lines. We transfected the cells, as described above, and performed
the PI staining method to analyze the cell cycle phase of the
cells. For comparison, we utilized two cell cycle inhibitors,
ribociclib at 3 µM (42,43) and flavopiridol at 100 nM (44,45).
Ribociclib is an FDA-approved, clinically used CDK4/6 inhibitor,
and flavopiridol is a clinically tested CDK1, CDK2, CDK4, and CDK7
inhibitor (43,44,46).
Our analysis on H358 cells indicated that the
combinatorial treatment significantly increased the
G0/G1 cell population (53.5 and 63%; all
values in parenthesis indicate the percent of the specific
population to the total) compared to the untreated (46 and 44%;
P<0.001) and scramble (49.5 and 45.5%; P<0.001) control
groups, at both 24 and 48 h, respectively. Interestingly,
ribociclib more robustly increased the G0/G1
cell population (70.5%; P<0.001) at 24 h compared to the miR
treatment. In contrast, ribociclib had significantly lower
G0/G1 cell population (52%; P<0.01) at 48
h compared to the miR treatment, which indicates a potentially
prolonged inhibition of the cell cycle progression due to the miR
treatment compared to ribociclib. Flavopiridol decreased the
G0/G1 phase population (42.5%; P<0.05) at
24 h, while it increased the G0/G1 population
(52%; P<0.05) at 48 h, compared to untreated control. However,
in both time points, the combinatorial treatment more potently
increased the G0/G1 populations compared to
flavopiridol (P<0.01). The combinatorial treatment did not alter
the cell population at the S phase (26%), compared to the control
(29.5%; no significance) at 24 h, but decreased the S phase
population (13%) compared to the control (39.5%; P<0.01) at 48
h. Ribociclib treatment decreased the cell population in the S
phase (14%; P<0.01), compared to the untreated control at 24 h,
while the decrease in the S phase population was less potent (28%;
P<0.01) at 48 h. In contrast, flavopiridol modestly increased
the S phase population (38%) compared to the untreated (no
significance) at 24 h, and lowered the S phase population (25.5%;
P<0.05) compared to the untreated control group at 48 h. No
significant changes were observed for any treatment for both time
points at the G2 cell populations, with the exception of
ribociclib reducing the G2 population (15.5%) compared
to the untreated control (24.5%, P<0.05) at 24 h (Fig. 5).
Our analysis on H1975 indicated that the
combinatorial miR treatment increased the
G0/G1 phase cell population (60 and 57%)
compared to the untreated control (46 and 54%) at 24 (P<0.001)
and 48 h (no significance), respectively. In comparison to the
scramble control though, a significant increase in the
G0/G1 was observed for the combinatorial miR
treatment for both time points (P<0.001). Unlike H358 cells, we
did not observe any significant increase in the
G0/G1 phase population due to ribociclib
treatment (61%; no significance) compared to the combinatorial
treatment at 24 h, though this was higher (64%; P<0.05) at 48 h.
In both time points, ribociclib increased the
G0/G1 cell population compared to untreated
control (P<0.01). Flavopiridol did not produce a significant
difference compared to the combinatorial miR treatment at the
G0/G1 phase cell populations (60 and 59% at
24 and 48 h, respectively). However, it was higher compared to the
untreated controls (P<0.05), at both 24 and 48 h. The
combinatorial treatment reduced the S phase population at 24 h
(21%) compared to the untreated (42%; P<0.001) and scramble
controls (38%; P<0.01). In contrast, the combinatorial treatment
did not alter the S phase population (28%) compared to the
untreated control (33%; no significance) at 48 h, but lowered the S
phase population compared to the scramble control (40%; P<0.01).
The combinatorial miR treatment more potently reduced the S phase
population compared to ribocliclib (29%; P<0.05) at 24 h, but
ribociclib more potently reduced the S phase population (17%)
compared to the combinatorial miR treatment (28%; P<0.01) at 48
h. For both time points, flavopiridol did not demonstrate
significant differences compared to the combinatorial treatment.
Both CDK-inhibitors decreased the S phase populations significantly
compared to the respective untreated controls for both time points.
Finally, at 24 h, the combinatorial miR treatment increased the
G2 phase population (18.5%) compared to the untreated
control (13%; P<0.05), scramble control (12%; P<0.05), which
was similar to flavopiridol (21%; no significance compared to
combinatorial treatment) and significantly higher compared to
ribociclib (10%; P<0.05 compared to combinatorial miR treatment;
Fig. 5). At 48 h, no significant
differences were observed among any of the groups. Detailed data on
the cell cycle cell distributions are presented in Table I.
 | Table I.Detailed data of cell cycle
distribution depending on cell cycle phases for H358 and H1975
cells following treatment with miR-143/506, ribociclib or
flavopiridol. |
Table I.
Detailed data of cell cycle
distribution depending on cell cycle phases for H358 and H1975
cells following treatment with miR-143/506, ribociclib or
flavopiridol.
|
| H358 cell line |
|---|
|
|
|
|---|
|
|
G0/G1 | S | G2 |
|---|
| Avg ± SEM | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Control | 46±0 | 44±1 | 29.5±2.5 | 39.5±0.5 | 24.5±2.5 | 17±1 |
| Scramble | 49.5±0.5 | 45.5±2 | 27.5±1 | 30.5±1 | 23±0.5 | 24±1 |
| Combo | 53.5±0.5 | 63±0.5 | 26±1.5 | 13±2.5 | 20.5±2 | 24±2 |
| Ribociclib | 70.5±0.5 | 52±1 | 14±1 | 28±1 | 15.5±0.5 | 20±0 |
| Flavopiridol | 42.5±0.5 | 52±0 | 38.5±1.5 | 25.5±1.5 | 18.5±0.5 | 21.5±1.5 |
|
|
G0/G1 | S | G2 |
| Tukey's test vs.
control group | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Scramble | <0.05 | >0.05 | >0.05 | <0.05 | >0.05 | >0.05 |
| Combo | <0.001 | <0.001 | >0.05 | <0.001 | >0.05 | >0.05 |
| Ribociclib | <0.001 | <0.05 | <0.01 | <0.05 | <0.05 | >0.05 |
| Flavopiridol | <0.05 | <0.05 | >0.05 | <0.05 | >0.05 | >0.05 |
|
|
| H1975 cell
line |
|
|
|
|
|
G0/G1 | S |
G2 |
|
| Avg ± SEM | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Control | 45.5±0.5 | 53.5±0.5 | 41.5±1.5 | 33±0 | 13±1 | 13.5±0.5 |
| Scramble | 50.5±0 | 44±0 | 38±1 | 39.5±2 | 11.5±0.5 | 16±2 |
| Combo | 60±0.5 | 57±1 | 21±0 | 28±1.5 | 18.5±0.5 | 15±1 |
| Ribociclib | 60.5±0.5 | 63.5±1.5 | 29±1 | 16.5±0.5 | 10±0 | 20±2 |
| Flavopiridol | 59.5±0.5 | 58.5±0.5 | 19±2 | 21.5±1.5 | 21±2 | 20±2 |
|
|
G0/G1 | S | G2 |
| Tukey's test vs.
control group | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h |
| Scramble | <0.01 | <0.01 | >0.05 | >0.05 | >0.05 | >0.05 |
| Combo | <0.001 | >0.05 | <0.001 | >0.05 | >0.05 | >0.05 |
| Ribociclib | <0.001 | <0.01 | <0.01 | <0.01 | >0.05 | >0.05 |
| Flavopiridol | <0.001 | <0.05 | <0.001 | <0.01 | <0.05 | >0.05 |
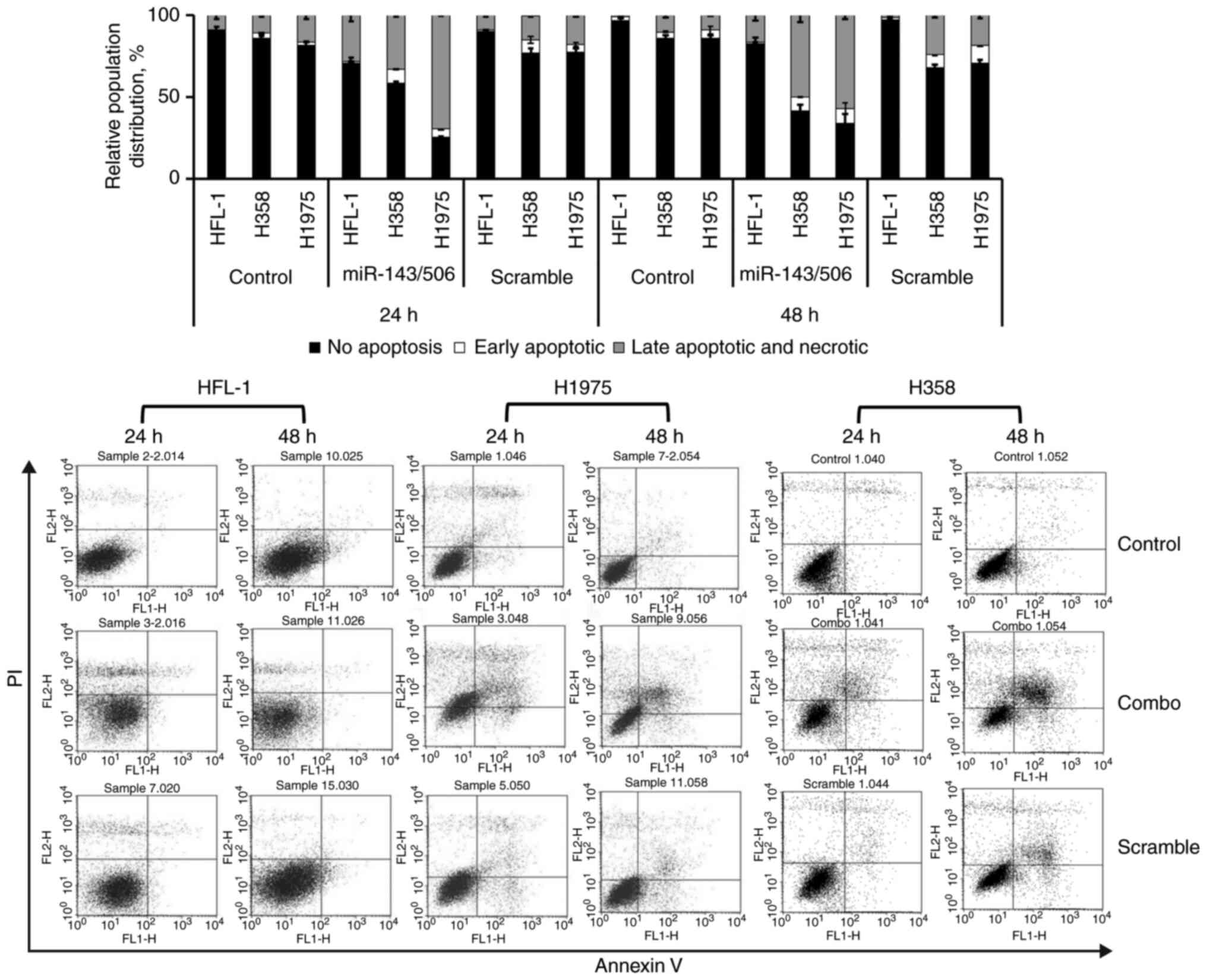
The miR-143/506 combination induces
apoptotic activity in LC cells, but has a lesser effect on normal
lung fibroblasts, and reduces PC cell growth
We investigated the capacity of the combinatorial
miR-143 and miR-506 treatment to induce apoptosis in H358 and H1975
LC cell lines, as well as in the normal human fetal lung fibroblast
HFL-1 cells, using flow cytometry and Annexin V/PI (Fig. 6). Our analysis indicated that the
combinatorial miR-143/506 treatment strongly induced apoptosis in
both LC cell lines. In contrast, the apoptotic activity in the
normal cell line was more modest and peaked at 24 h. Detailed
information for all cell populations is documented in Table SII, as here we focus on the late
apoptotic and necrotic populations.
Briefly, in the H358 cells and at 24 h, the
combinatorial miR treatment increased (33%) the late apoptotic and
necrotic cell populations compared to the untreated (11%,
P<0.001) and the scramble controls (14.63%, P<0.01). At 48 h,
the miR-143/506 treatment induced more potent apoptotic activity,
where the majority of the cells were in the late apoptotic and
necrotic populations (50%) compared to the untreated control (10%;
P<0.01) and the scramble control (24%; P<0.05).
Comparably, in the H1975 cells and at 24 h, our
analysis indicated that the combinatorial treatment induced an
increase in the late apoptotic and necrotic populations (69%)
compared to the untreated control (17%; P<0.001) and scramble
control (18%; P<0.001). At 48 h, the observed apoptotic activity
of the combinatorial treatment was maintained, as the combinatorial
treatment significantly increased the late apoptosis and necrotic
cell populations (57%) compared to the untreated control (9%;
P<0.001) and scramble treated control (18%; P<0.01).
In the normal HFL-1 cell line and at 24 h, we
detected an increase in late apoptotic and necrotic populations due
to the combinatorial treatment (28%) compared to the untreated
control (9%; P<0.05) and compared to the scramble control (9%;
P<0.05). At 48 h, we observed an increase in late apoptotic and
necrotic populations due to the combinatorial miR treatment (16%)
compared to the untreated control (1%; P<0.05) and to scramble
treatment (1%; P<0.05).
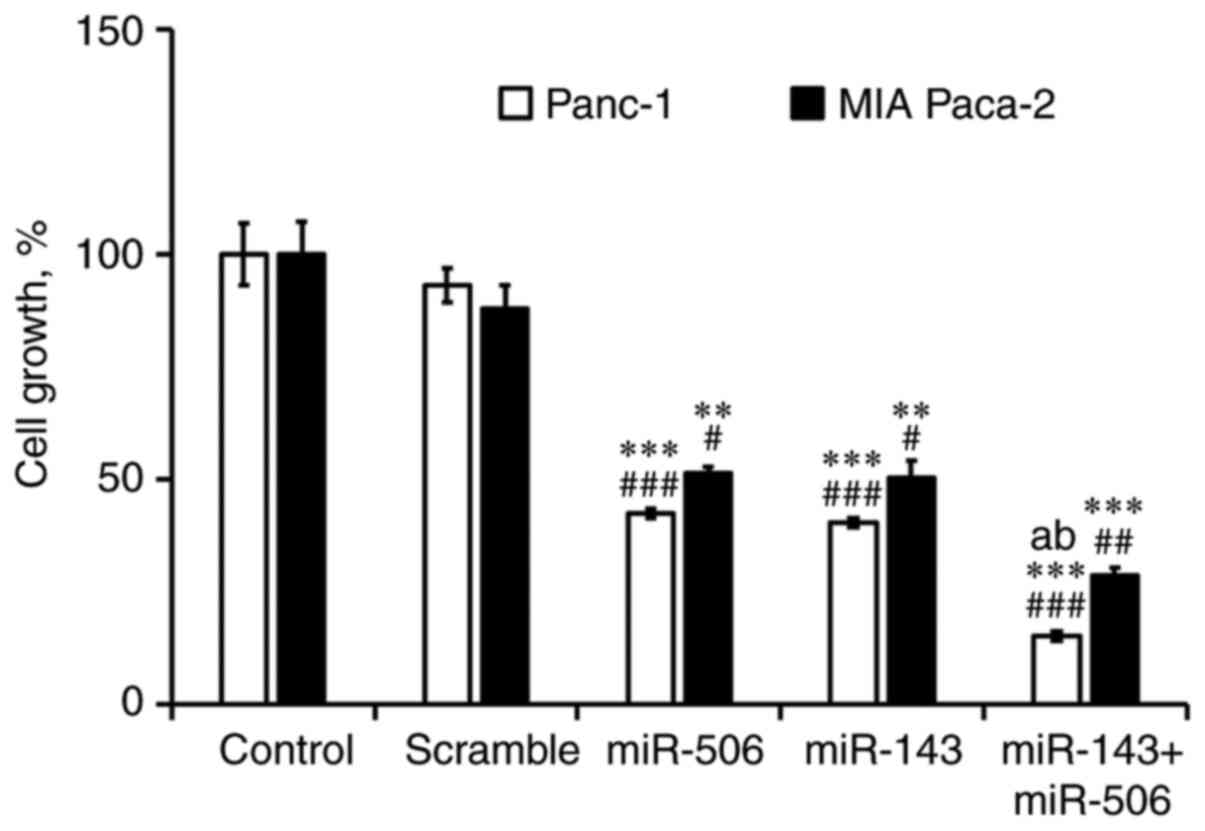
Finally, we performed a cytotoxicity analysis of
miR-143 and miR-506 treatment, individually and in combination in
Panc-1 and MIA-PaCa-2 cells, for 48 h (Fig. 7). We observed a significant
(P<0.001) reduction of viable Panc-1 (85%) and MIA-PaCa-2 (72%)
cells, due to the combinatorial miR treatment. The cytotoxic effect
of the combinatorial miR treatment was also significantly stronger
(P<0.001) when compared to the individual miR-143 or miR-506 in
Panc-1 cells. Briefly, we observed a significant reduction in
viable cells in both PC cell lines due to treatment with the
individual miRs compared to the untreated (P<0.01) and scramble
(P<0.05) controls. At 48 h post-transfection, miR-143 reduced by
60 and 50% the viable cell population in Panc-1 and MIA-PaCa-2
cells, respectively, whereas miR-506 reduced by 58 and 49% the cell
population in Panc-1 and MIA-PaCa-2 cell lines, respectively (all
values had at least P<0.05).
Discussion
MicroRNAs (miRNAs/miRs) are critical regulators of
mRNA expression, and various diseases, including cancer, have been
associated with miR dysregulation (47,48). We
recently reported on two miRs, miR-143 and miR-506, previously
recognized with essential functions in cancer and other diseases
(34,35,49,50).
miR-143 has base-pair complementarity with the 3′-UTR region of
cyclin-dependent kinase 1 (CDK1), and miR-506 has base-pair
complementarity with the 3′-UTR regions of CDK4 and CDK6 (35,51,52). The
data presented here support the notion that the combinatorial miR
treatment has a potent cell cycle inhibiting capacity in two cell
cycle phase transitions, through the downregulation of CDKs. This
represents a promising therapeutic option for cancers in which
CDK4/6 monotherapy is unlikely to exert sufficient therapeutic
outcomes, such as in lung cancer (LC) and pancreatic cancer
(PC).
Initially, we determined the expression of miR-143
and miR-506 in LC cells. Expression of the two miRs were found to
be lower in LC cell lines compared to a normal human fetal lung
fibroblast cell line (HFL-1). Although not all cancer cell lines
showed a significant miR-506 expression difference compared to
normal cell lines, our data align with previously reported analyses
showing a dysregulation of miR-143/506 in LC cells vs. normal cells
(41,53). Interestingly, CDK1 and
CDK4 gene expression levels were significantly higher in all
of the LC cell lines tested compared to HLF-1, with the exception
of A549 for CDK1 and CDK4 and H358 for CDK1.
Of interest, H358 showed increased CDK4 expression, without a
significant miR-506 downregulation. This indicates potentially that
even though miR-506 can regulate CDK4 expression, it is not its
sole regulator. Furthermore, as the miR-506 expression levels in
normal or cancer cells were found to be very low (as presented by
the tissue samples and Pan-Cancer analysis), exogenous
overexpression of miR-506 could potentially have more potent effect
than its basal expression changes on CDK4 expression. Briefly, we
used data from the Pan-Cancer Analysis to determine the
differential expression of the above genes in tumor tissues from
patients with lung adenocarcinomas compared to normal tissues.
Consistent with our findings, miR-143-3p was lower in cancer vs.
normal samples. Interestingly, miR-506-3p was expressed relatively
low in tumor and normal tissue samples, without a difference
between the two sample groups. Finally, both CDK1 and CDK4 were
higher in cancer samples compared to normal samples, and more
importantly, protein expression for CDK1 and CDK4 vs. survival in
patients with LC adenocarcinomas indicated a negative correlation
between CDK1 and CDK4 expression and survival. We recently reported
that miR-143 and miR-506 have CDK1 and CDK4 mRNAs as their
predicted targets, respectively (34,35). These
data align with previously reported work on these genes (13,32). Here,
our analysis of the miR-143/506 combination on H358, and H1975 LC
cell lines, confirmed our previous findings on CDK1, CDK4 and CDK6
downregulation (34,35). The downregulation of these genes took
place at both the post-transcriptional and translational level. We
selected the two cell lines for analysis, as they carry driver
mutations that complement or correlate with our previously
published work on A549 cells, as we further analyze below. This
analysis correlates also with the reported activity of miR-143 or
miR-506 alone (35,51,54).
Interestingly, although the combinatorial miR treatment
downregulated CDK1 in H358 cells, the downregulation achieved
statistical significance only for the 24 h time point, compared to
the scramble control. An important feature of our study is that the
combinatorial treatment inhibited the cell cycle progression in the
H358 and H1975 cells, comparably to clinically used cell cycle
inhibitors. Specifically, we used as positive controls two cell
cycle inhibitors, ribociclib and flavopiridol, which the former is
currently clinically used for breast cancer treatment, and the
latter has been evaluated in clinical trials. Flavopiridol inhibits
the activity of the CDK1/2/4/7, and causes cell cycle halt at the
G1/S and G2/M transitions (55,56) and
ribociclib (LEE011) is a selective CDK4/6 inhibitor, halting cell
cycle progression at the G1/S transition (3,12).
Flow-cytometric analysis indicated a significant increase in the
G0/G1 cell populations due to the
combinatorial miR treatment in both cell lines at either 24 or 48 h
post-transfection. The combinatorial miR treatment's increase of
the G0/G1 population was comparable to, and,
in some cases, higher than either ribociclib or flavopiridol. In
contrast, the combinatorial miR treatment significantly decreased
the cell population in the S phase compared to the untreated and
scramble controls, comparably to the two cell cycle inhibitors,
albeit at different time points in some cases. These results
suggest that the miR-143/506 treatment potentiates an inhibition of
the G1/S transition progression, and is comparable to
the activity of the G1/S cell cycle inhibitors.
The combinatorial miR treatment increased the
G2 cell population in both cell lines compared to the
untreated cells, though the increase was significant only in the
H1975 when compared to the untreated and scramble group. The
increase in the G2 cell population due to miR treatment
was similar to the effect of flavopiridol in the same cell
population for both cell lines, which is considered a
G2/M inhibitor. In contrast, ribociclib, which does not
inhibit G2/M transition, did not increase the
G2 population. This indicates that the miR-143/506
treatment can induce a two phase transition inhibition, i.e., at
both G1/S and a G2/M transitions, in LC
cells. Interestingly, the G2/M inhibition activity for
both miR-143/506 combination and flavopiridol was more potent in
the H1975 cells, while modest in H358. This potentially could be
attributed to a potential different molecular mechanism, as the two
cell lines carry different driver mutations. There is a correlation
between the activities of the combinatorial miR treatment and
flavopiridol, the G1/S and G2/M inhibitor.
Though both treatments induced a significant increase of the
G2 cell populations in H1975, this was not observed in
H358. In fact, this observation also correlates with the inability
of the combinatorial miR treatment to significantly downregulate
the CDK1 expression in the H358 cells. These results reinforce the
notion that the miR-treatment correlates with a G1/S and
G2/M inhibitor, albeit the mutational background of each
cell line needs to be taken into consideration. Briefly, H358 cells
carry a mutated KRAS and have an inactive (deleted)
TP53 gene, while H1975 have a wt-KRAS, and mutated
CDKN2A, PIK3CA, TP53 and EGFR genes (57–59). Our
work compares and expands to our previously presented data on A549
cells, which carry wt-p53, mutated KRAS and mutated
CDKN2A. Our results merit future analyses based on the
different driver mutations, as they will assist in identifying the
mechanistic background on the activity of the combinatorial miR
treatment.
Subsequently, we evaluated the apoptotic activity of
the combinatorial miR treatment in LC cell lines and compared them
to HFL-1 normal human fetal lung fibroblasts. We observed a
significant apoptotic activity in the LC cells, due to the
combinatorial miR treatment, at 24 and 48 h post-transfection.
Interestingly, the combinatorial miR treatment had a modest
apoptotic effect in the HFL-1 cell line. Briefly, the combinatorial
miR treatment induced apoptotic activity in HFL-1 cells that was
less potent when compared to the H358 and H1975 cells (P<0.05
for both cell lines) at 24 h, and was practically normalized at 48
h. In contrast, the apoptotic activity of the miR treatment
remained potent in the H1975 cell at both 24 and 48 h, while in
H358 cells, the miR treatment induced a modest apoptotic activity
at 24 h, but became more robust and comparable to H1975 cells at 48
h. This behavior difference between the two LC cell lines
correlates to the observed activity of the miR treatment on the
cell cycle analysis, where the strongest decrease in the S
population and strongest increase of the G2 population
was observed at 24 h for the H1975 and 48 h for the H358. The
modest apoptotic activity of the combinatorial treatment in the
HFL-1 cells could potentially be justified by the relative higher
expression of these miRs in this normal cell line compared to the
cancer cells, and can potentially indicate a preferential activity
against tumor cells vs. normal cells. This could potentially
translate to decreased side effects (i.e., toxicity to normal
cells) if the combination miR therapy is clinically translated.
Having established the capacity of the two miRs to
downregulate the CDKs in LC cells and induce apoptosis, we
determined whether the effects were cancer-type specific. Though
significant differences exist between PC and LC cells, mutations in
similar genes have been detected in the both cancer types (60,61). For
example, both Panc-1 and MIA-PacCa-2 cell lines carry mutated or
deleted TP53, CDKN2A, and KRAS genes, partially
similar to either H358 or H1975. Thus, the genetic background
similarities prompted us to expand to the PC cells and use Panc-1
and MIA-PaCa-2 cell lines as proof-of-concept. We confirmed our
findings in these two PC cell lines that the two miRs downregulated
CDK1 and CDK4 expression and induced cytotoxicity (62). Similar to the LC cells, the
miR-143/506 treatment strongly downregulated both CDK1 and CDK4
protein expression. Furthermore, the individual miR treatment
specifically downregulated their respective target, i.e., miR-143
downregulated CDK1 and miR-506 downregulated mostly CDK4. This led
to a significant reduction in PC cell growth, with the combination
treatment being substantially more potent that the individual miR
treatments. These results are in agreement with our previous
analysis of the individual miRs on the A549 LC cells (35), and indicate that the two miRs have an
activity that spans outside a specific cancer type.
Our data presented here, as well as in our previous
studies, reaffirm that the combinatorial miR treatment have a
potent cell cycle inhibiting capacity in two phase transitions,
through the downregulation of CDKs, comparable to respective CDK
inhibitors. This represents a promising therapeutic option for
cancers that CDK4/6 monotherapy is unlikely to exert sufficient
therapeutic outcomes. The combinatorial miR treatment acts in both
LC and PC, which presents a non-cancer-type specific therapeutic
benefit for potential cancer treatment.
In conclusion, the combinatorial miR-143 and miR-506
therapy, through the downregulation of CDK1, CDK4 and CDK6,
strongly inhibits the cell cycle progression to a comparable degree
as the clinically used CDK-inhibitor ribociclib and the clinically
evaluated CDK-inhibitor flavopiridol. In addition, the miR-143/506
combinatorial treatment selectively induces apoptotic activity in
the LC cells, with comparably diminished effect on normal
fibroblast cells. Finally, this combination of miR treatment is
effective in PC cells, suggesting a broader application for the
miR-143/506 combinatorial treatment. Overall, these data support
the notion that the miR-143/506 combinatorial treatment is a
promising approach for regulating the cell cycle progression that
merits further evaluation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the College of Pharmacy,
University of Louisiana Monroe start-up funding, and the National
Institutes of Health (NIH) through the National Institute of
General Medical Science Grants 5 P20 GM103424-15, 3 P20
GM103424-15S1.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
AKMNH collected, analyzed and interpreted the data
regarding the LC cell lines, and was a major contributor in writing
the manuscript. GGM collected, analyzed and interpreted the data
regarding the PC cell lines, and was a contributor in writing the
manuscript. GM analyzed and interpreted the data, and was a major
contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors have no competing interests to
report.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collins K, Jacks T and Pavletich NP: The
cell cycle and cancer. Proc Natl Acad Sci USA. 94:2776–2778. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dominguez-Brauer C, Thu KL, Mason JM,
Blaser H, Bray MR and Mak TW: Targeting Mitosis in cancer: Emerging
strategies. Mol Cell. 60:524–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Otto T and Sicinski P: Cell cycle proteins
as promising targets in cancer therapy. Nat Rev Cancer. 17:93–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sanchez-Martinez C, Gelbert LM, Lallena MJ
and de Dios A: Cyclin dependent kinase (CDK) inhibitors as
anticancer drugs. Bioorg Med Chem Lett. 25:3420–3435. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shah A, Bloomquist E, Tang S, Fu W, Bi Y,
Liu Q, Yu J, Zhao P, Palmby TR, Goldberg KB, et al: FDA approval:
Ribociclib for the treatment of postmenopausal women with hormone
receptor-positive, HER2-negative advanced or metastatic breast
cancer. Clin Cancer Res. 24:2999–3004. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wedam S, Fashoyin-Aje L, Bloomquist E,
Tang S, Sridhara R, Goldberg KB, Theoret MR, Amiri-Kordestani L,
Pazdur R and Beaver JA: FDA approval summary: Palbociclib for male
patients with metastatic breast cancer. Clin Cancer Res.
26:1208–1212. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
FDA expands approved use of ibrance
(Palbociclib) for HR+, HER2-metastatic breast cancer. Oncol Times.
38:222016. View Article : Google Scholar
|
|
11
|
Martin JM and Goldstein LJ: Profile of
abemaciclib and its potential in the treatment of breast cancer.
Onco Targets Ther. 11:5253–5259. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin D, Tran N, Thomas N and Tran DD:
Combining CDK4/6 inhibitors ribociclib and palbociclib with
cytotoxic agents does not enhance cytotoxicity. PLoS One.
14:e02235552019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu A, Wu B, Guo J, Luo W, Wu D, Yang H,
Zhen Y, Yu X, Wang H, Zhou Y, et al: Elevated expression of CDK4 in
lung cancer. J Transl Med. 9:382011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gong W, Wang L, Zheng Z, Chen W, Du P and
Zhao H: Cyclin-dependent kinase 6 (CDK6) is a candidate diagnostic
biomarker for early non-small cell lung cancer. Transl Cancer Res.
9:95–103. 2019. View Article : Google Scholar
|
|
15
|
Thangavel C, Boopathi E, Liu Y, McNair C,
Haber A, Perepelyuk M, Bhardwaj A, Addya S, Ertel A, Shoyele S, et
al: Therapeutic challenge with a CDK 4/6 Inhibitor Induces an
RB-Dependent SMAC-Mediated apoptotic response in non-small cell
lung cancer. Clin Cancer Res. 24:1402–1414. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patnaik A, Rosen LS, Tolaney SM, Tolcher
AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW,
Hilton JF, et al: Efficacy and safety of abemaciclib, an inhibitor
of CDK4 and CDK6, for patients with breast cancer, non-small cell
lung cancer, and other solid tumors. Cancer Discov. 6:740–753.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pacheco J and Schenk E: CDK4/6 inhibition
alone and in combination for non-small cell lung cancer.
Oncotarget. 10:618–619. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Whole genomes redefine the mutational landscape of pancreatic
cancer. Nature. 518:495–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Witkiewicz AK, McMillan EA, Balaji U, Baek
G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et
al: Whole-exome sequencing of pancreatic cancer defines genetic
diversity and therapeutic targets. Nat Commun. 6:67442015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Garcia-Reyes B, Kretz AL, Ruff JP, von
Karstedt S, Hillenbrand A, Knippschild U, Henne-Bruns D and Lemke
J: The emerging role of cyclin-dependent kinases (CDKs) in
pancreatic ductal adenocarcinoma. Int J Mol Sci. 19:32192018.
View Article : Google Scholar
|
|
23
|
Knudsen ES, Kumarasamy V, Ruiz A, Sivinski
J, Chung S, Grant A, Vail P, Chauhan SS, Jie T, Riall TS and
Witkiewicz AK: Cell cycle plasticity driven by MTOR signaling:
Integral resistance to CDK4/6 inhibition in patient-derived models
of pancreatic cancer. Oncogene. 38:3355–3370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Franco J, Balaji U, Freinkman E,
Witkiewicz AK and Knudsen ES: Metabolic reprogramming of pancreatic
cancer mediated by CDK4/6 inhibition elicits unique
vulnerabilities. Cell Rep. 14:979–990. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu F and Korc M: Cdk4/6 inhibition
induces epithelial-mesenchymal transition and enhances invasiveness
in pancreatic cancer cells. Mol Cancer Ther. 11:2138–2148. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Franco J, Witkiewicz AK and Knudsen ES:
CDK4/6 inhibitors have potent activity in combination with pathway
selective therapeutic agents in models of pancreatic cancer.
Oncotarget. 5:6512–6525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Santamaria D, Barriere C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Malumbres M, Sotillo R, Santamaria D,
Galán J, Cerezo A, Ortega S, Dubus P and Barbacid M: Mammalian
cells cycle without the D-type cyclin-dependent kinases Cdk4 and
Cdk6. Cell. 118:493–504. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piao J, Zhu L, Sun J, Li N, Dong B, Yang Y
and Chen L: High expression of CDK1 and BUB1 predicts poor
prognosis of pancreatic ductal adenocarcinoma. Gene. 701:15–22.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong S, Huang F, Zhang H and Chen Q:
Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues
predicts poor survival in pancreatic ductal adenocarcinoma. Biosci
Rep. 39:BSR201823062019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi YX, Zhu T, Zou T, Zhuo W, Chen YX,
Huang MS, Zheng W, Wang CJ, Li X, Mao XY, et al: Prognostic and
predictive values of CDK1 and MAD2L1 in lung adenocarcinoma.
Oncotarget. 7:85235–85243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li M, He F, Zhang Z, Xiang Z and Hu D:
CDK1 serves as a potential prognostic biomarker and target for lung
cancer. J Int Med Res. 48:3000605198975082020.PubMed/NCBI
|
|
33
|
O'Brien J, Hayder H, Zayed Y and Peng C:
Overview of MicroRNA biogenesis, mechanisms of actions, and
circulation. Front Endocrinol (Lausanne). 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hossian AK, Muthumula CM, Sajib MS, Tullar
PE, Stelly AM, Briski KP, Mikelis CM and Matthaiolampakis G:
Analysis of combinatorial miRNA treatments to regulate cell cycle
and angiogenesis. J Vis Ex. e594602019.
|
|
35
|
Hossian AKMN, Sajib MS, Tullar PE, Mikelis
CM and Mattheolabakis G: Multipronged activity of combinatorial
miR-143 and miR-506 inhibits lung cancer cell cycle progression and
angiogenesis in vitro. Sci Rep. 8:104952018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42:D92–D97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang JH, Li JH, Shao P, Zhou H, Chen YQ
and Qu LH: StarBase: A database for exploring microRNA-mRNA
interaction maps from Argonaute CLIP-Seq and degradome-Seq data.
Nucleic Acids Res. 39:D202–D209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thul PJ and Lindskog C: The human protein
atlas: A spatial map of the human proteome. Protein Sci.
27:233–244. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Uhlen M, Fagerberg L, Hallstrom BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C,
Sjöstedt E, Asplund A, et al: Proteomics. Tissue-based map of the
human proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang N, Su Y and Xu L: Targeting PKCε by
miR-143 regulates cell apoptosis in lung cancer. FEBS Lett.
587:3661–3667. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
van Caloen G, Schmitz S, El Baroudi M,
Caignet X, Pyr Dit Ruys S, Roger PP, Vertommen D and Machiels JP:
Preclinical activity of ribociclib in squamous cell carcinoma of
the head and neck. Mol Cancer Ther. 19:777–789. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wong CH, Ma BB, Hui CW, Lo KW, Hui EP and
Chan AT: Preclinical evaluation of ribociclib and its synergistic
effect in combination with alpelisib in non-keratinizing
nasopharyngeal carcinoma. Sci Rep. 8:80102018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kelland LR: Flavopiridol, the first
cyclin-dependent kinase inhibitor to enter the clinic: Current
status. Expert Opin Investig Drugs. 9:2903–2911. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saisomboon S, Kariya R, Vaeteewoottacharn
K, Wongkham S, Sawanyawisuth K and Okada S: Antitumor effects of
flavopiridol, a cyclin-dependent kinase inhibitor, on human
cholangiocarcinoma in vitro and in an in vivo xenograft model.
Heliyon. 5:e016752019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Senderowicz AM and Sausville EA:
Preclinical and clinical development of cyclin-dependent kinase
modulators. J Natl Cancer Inst. 92:376–387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hossian AKMN, Mackenzie GG and
Mattheolabakis G: miRNAs in gastrointestinal diseases: Can we
effectively deliver RNA-based therapeutics orally? Nanomedicine
(Lond). 14:2873–2889. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Labatut AE and Mattheolabakis G: Non-viral
based miR delivery and recent developments. Eur J Pharm Biopharm.
128:82–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao W, Zhao SP and Zhao YH:
MicroRNA-143/-145 in cardiovascular diseases. Biomed Res Int.
2015:5317402015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lovendorf MB, Zibert JR, Gyldenlove M,
Ropke MA and Skov L: MicroRNA-223 and miR-143 are important
systemic biomarkers for disease activity in psoriasis. J Dermatol
Sci. 75:133–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Anton L, DeVine A, Sierra LJ, Brown AG and
Elovitz MA: miR-143 and miR-145 disrupt the cervical epithelial
barrier through dysregulation of cell adhesion, apoptosis and
proliferation. Sci Rep. 7:30202017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu G, Sun Y, Ji P, Li X, Cogdell D, Yang
D, Parker Kerrigan BC, Shmulevich I, Chen K, Sood AK, et al:
MiR-506 suppresses proliferation and induces senescence by directly
targeting the CDK4/6-FOXM1 axis in ovarian cancer. J Pathol.
233:308–318. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yin M, Ren X, Zhang X, Luo Y, Wang G,
Huang K, Feng S, Bao X, Huang K, He X, et al: Selective killing of
lung cancer cells by miRNA-506 molecule through inhibiting NF-κB
p65 to evoke reactive oxygen species generation and p53 activation.
Oncogene. 34:691–703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li J, Wu H, Li W, Yin L, Guo S, Xu X,
Ouyang Y, Zhao Z, Liu S, Tian Y, et al: Downregulated miR-506
expression facilitates pancreatic cancer progression and
chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene.
35:5501–5514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Senderowicz AM: Flavopiridol: The first
cyclin-dependent kinase inhibitor in human clinical trials. Invest
New Drugs. 17:313–320. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Deep A, Marwaha RK, Marwaha MG, Jyoti,
Nandal R and Sharma AK: Flavopiridol as cyclin dependent kinase
(CDK) inhibitor: A review. New J Chem. 42:18500–18507. 2018.
View Article : Google Scholar
|
|
57
|
Sunaga N, Shames DS, Girard L, Peyton M,
Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, et al:
Knockdown of oncogenic KRAS in non-small cell lung cancers
suppresses tumor growth and sensitizes tumor cells to targeted
therapy. Mol Cancer Ther. 10:336–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Berger AH, Brooks AN, Wu X, Shrestha Y,
Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M,
Hogstrom L, et al: High-throughput phenotyping of lung cancer
somatic mutations. Cancer Cell. 30:214–228. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hung MS, Chen IC, Lin PY, Lung JH, Li YC,
Lin YC, Yang CT and Tsai YH: Epidermal growth factor receptor
mutation enhances expression of vascular endothelial growth factor
in lung cancer. Oncol Lett. 12:4598–4604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Korc M: Driver mutations: A roadmap for
getting close and personal in pancreatic cancer. Cancer Biol Ther.
10:588–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Timar J and Kashofer K: Molecular
epidemiology and diagnostics of KRAS mutations in human cancer.
Cancer Metastasis Rev. 39:1029–1038. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gradiz R, Silva HC, Carvalho L, Botelho MF
and Mota-Pinto A: MIA PaCa-2 and PANC-1-pancreas ductal
adenocarcinoma cell lines with neuroendocrine differentiation and
somatostatin receptors. Sci Rep. 6:216482016. View Article : Google Scholar : PubMed/NCBI
|