Introduction
Breast cancer (BCa) remains the most common type of
malignant tumor among women, with 11.6% for incidence and 6.6% for
mortality worldwide in 2018 (1).
Clinically, breast tumors often display intra-tumoral
heterogeneity. Multiple cell subpopulations within a single tumor
may show diverse phenotypes, such as different cell surface
markers, growth rates, metastasis and therapeutic responses that
eventually drive cancer progression (2). There is currently a lack of approaches
for identifying more aggressive subpopulations in a tumor mass,
which makes it difficult to monitor cancer progression and response
to treatment. Thus, it is of urgent importance to examine novel
targets for the valuable diagnosis of BCa.
CD44 is a family of cell surface glycoproteins
ubiquitously expressed by various tumors, especially in BCa
(3). CD44 participates in cell
adhesion and migration, lymphocyte homing and activation, and
abnormal behaviors of tumor cells (4,5).
Hyaluronan (HA), the principal ligand for CD44, is a major
extracellular matrix component accumulated in stromal and tumor
cells (6). Studies have reported that
HA acts by binding to CD44 to modulate various fundamental and
pathological processes, including cell-cell adhesion, cell
proliferation, cancer migration and invasion (7–9). However,
not all CD44-expressing cells can constitutively bind HA; only
activated CD44 binds and internalizes HA. Thus, the cellular
behaviors induced by CD44-HA binding are cell-specific and strongly
depend on the activation states of CD44 (10). As previously reported, CD44
participates in three different contexts in relation to its binding
capacities with HA: Inactive, constitutively active and inducible
active (does not bind to HA unless activated by appropriate
stimuli) (11,12). Although numerous studies have
highlighted the association of CD44 expression with breast
malignancies, it remains unknown whether an activation process of
CD44 is critical for BCa development and progression (13–15).
Previous studies have revealed that CD44 activation
is a complex and dynamic process, as the ability of CD44 to engage
HA is highly regulated (16,17). In humans, the full-length CD44 gene
consists of 19 exons that have molecular masses ranging from 80–250
kDa (18). The shortest CD44 standard
isoform (CD44s) includes only constitutive exons, while the CD44
variant isoforms (CD44v) undergo alternative splicing and contain
≥1 variable exons (19). Although all
CD44 molecules share a common HA-binding domain, their affinities
for surface-bound hyaluronate are different, which suggests that
the structural heterogeneity of CD44 alternative splicing may be
responsible for modulating CD44 activation (20). Early observations using melanoma
models demonstrated that CD44s bound to HA more efficiently
compared with CD44v8-v10 (21). At
the same time, another study suggested that the splice variant of
CD44v4-v7 confers stronger HA-binding properties compared with
CD44s in rat pancreatic carcinoma cell lines (22). These different results indicated that
CD44 isoforms may have distinct roles in its binding ability with
HA. However, the effect of CD44 isoforms on CD44 activation in BCa
is poorly understood. Although there are still some controversies,
it is considered that alternative splicing determines CD44
activities. Moreover, accumulating evidence has suggested that CD44
splice isoforms contribute to cellular and functional behaviors by
mediating specific signaling pathways (23). CD44v was reported to stimulate MAPK
activation and to promote cell proliferation (24), while CD44s was suggested to trigger
the PI3K/AKT/mTOR signaling cascade and contribute to tumor cell
escape from drug-induced cell death (25). However, whether the different CD44v
isoforms share common signaling pathways to regulate the biologic
function is yet to be elucidated.
The present study aimed to investigate whether CD44
activation is critical requirement for BCa cells proliferation and
to further examine which CD44 isoform determines CD44 activities.
Firstly, HA−/low and HAhigh subpopulations
were isolated using FACS technique, and cell proliferation assays
in vitro were applied to assess whether the two subsets
exhibited different proliferative capacity. Then, a panel of small
interfering (si)RNAs targeting each CD44v exon region were employed
to identify which CD44v isoform was associated with CD44 activities
and cells proliferation. Furthermore, the current study
investigated the underlying mechanisms by studying the
proliferation-related ERK/p38 MAPK and AKT/mTOR signal pathway.
These findings identified a subset of fast-growing BCa cells
characterized by CD44v10 expression, which may serve as a promising
therapeutic target for BCa.
Materials and methods
Cell lines and culture
Human BCa cell lines (MCF-7, T-47D, BT-549 and
MDA-MB-231) were obtained from the Cell Bank of the Type Culture
Collection of the Chinese Academy of Sciences. MCF-7 cells were
cultured in MEM (Gibco; Thermo Fisher Scientific, Inc.), T-47D and
BT-549 cells were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) and MDA-MB-231 cells were cultured in
high-glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.). All cell
lines were maintained at a temperature of 37°C in humidified air
with 5% CO2. All cell culture media were supplemented
with 10% FBS (Bovogen Biologicals Pty Ltd.), 100 U/ml penicillin
and 100 mg/ml streptomycin. In addition, all cells were grown to
80% confluency for the experiments.
Isolation of HA high and
HA−/low subpopulations by FACS
Fluorescein-labeled HA (fl-HA; Merck KGaA) binding
was used to determine the CD44 activation state. In total,
~5.0×107 cells in supplemented Hank's balanced salt
solution (HBSS) were incubated on ice with a working concentration
of 40 µg/ml fl-HA for 1 h, as previously described (26). Unbounded fl-HA was removed via
centrifugation (682 × g for 5 min at room temperature) two times
with HBSS. After resuspended in the HBSS, the cells were filtered
through cell strainers and analyzed via CytoFLEX flow cytometer
(Beckman-Coulter, Inc.). Non-viable cells were excluded using
7-amino-actinomycin D dye (staining for 15 min at room
temperature). Fl-HA-bound cells were sorted using gates set to a
10% threshold on a histogram profile. Cells were sorted into
HAhigh and HA−/low subpopulations based on
differential binding to HA (10% maximum and minimum signal
selection), and collected for further in vitro analyses. In
preparation for HA binding analysis, a HA−/low
single-cell suspension (106 cells/ml) was incubated with
fl-HA or isotype control antibody for 1 h on ice. After washing
three times, the HA binding profile was subsequently analyzed using
a flow cytometer (MoFlo Astrios EQ; Beckman-Coulter, Inc.).
Cell Counting Kit (CCK-8) assay
The cell viability was measured using a CCK-8 assay
(Dojindo Molecular Technologies, Inc.) according to the
manufacturer's protocol. Briefly, cells in the logarithmic growth
phase were seeded in triplicate onto 96-well plates at a density of
3.0×103 cells per well. Cells were then incubated for 0,
1, 2, 3 and 4 days, after which the complete medium of each well
was replaced by 10 µl CCK-8 solution supplemented with 90 µl
serum-free medium and incubated at 37°C for additional 2 h. The
absorbance was then measured at 450 nm using a microplate
absorbance reader (Bio-Rad Laboratories, Inc.). In preparation for
rescue experiments, HA−/low cells were incubated (37°C)
with anti-CD44 monoclonal antibody or normal IgG (10 µg/ml;
Invitrogen) for 48 h after transfected with CD44v10 siRNA, and
measured using the CCK-8 assay.
5-Ethynyl-20- deoxyuridine (EdU)
assay
Cell proliferation was analyzed using EdU assays
with a Cell-Light EdU DNA Cell Proliferation kit (Beyotime
Institute of Biotechnology). Sorted HAhigh and
HA−/low cells were resuspended for cell density
adjustment at 1×105/ml. A total of 100 µl suspension was
seeded into each well of the 96-well plates. After incubation
overnight, 10 mM EdU was added and incubated at 37°C for another 2
h. Cells were then stained with azide (30 min, for proliferating
cells) and Hoechst 33342 (10 min, for all cells) at room
temperature. Images were captured using a fluorescence microscope
(Nikon Corporation, magnification, ×100). The percentage of
proliferating cells was calculated using ImageJ V1.50 software
(National Institutes of Health).
Plate colony formation assay
A total of 500 viable cells per well were plated
into 6-well culture plates. All plates were then incubated for 14
days at 37°C to allow colony formation. The cells were washed twice
with PBS and fixed with 4% paraformaldehyde for 30 min at room
temperature, followed by staining with 0.1% crystal violet (BaSO
Biotech Co., Ltd.) for 30 min at room temperature. After rinsing
three times, the stained colonies were imaged and the number of
colonies was counted by the naked eye. In total, three parallel
wells were set for each group.
Western blot analysis
Total proteins were extracted with RIPA lysis buffer
(Beyotime Institute of Biotechnology) supplemented with protease
and phosphatase inhibitors (Beyotime Institute of Biotechnology).
Protein concentration was quantified using a BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Proteins (~20 µg/lane) were
separated by 8% SDS-PAGE and then transferred to PVDF membranes
(Merck Millipore). The membranes were blocked with 5% skimmed milk
in 0.1% TBS-Tween-20 at room temperature for 1 h and incubated with
following primary antibodies overnight at 4°C: Pan-CD44 (dilution
1:1,000; Abcam; cat. no. ab189524), CD44v10 (dilution 1:1,000;
Sigma-Aldrich; Merck KGaA; cat. no. AB2082), phosphorylated
(p)-ERK1/2 [dilution 1:2,000; Cell Signaling Technology, Inc.
(CST); cat. no. 4370], ERK1/2 (dilution 1:1,000; CST; cat. no.
4695), p-p38 (dilution 1:1,000; CST; cat. no. 4511), p38 (dilution
1:1,000; CST; cat. no. 9212), p-AKT (dilution 1:2,000; CST; cat.
no. 4060), AKT (dilution 1:1,000; CST; cat. no. 4691), p-mTOR
(dilution 1:1,000; CST; cat. no. 5536) and mTOR (dilution 1:1,000;
CST; cat. no. 2983), GAPDH (dilution 1:1,000; Lianke Biotech Co.,
Ltd.; cat. no. Mab5465). On the following day, membranes were
incubated with horseradish peroxidase-conjugated secondary antibody
goat anti-rabbit IgG (dilution 1:5,000; Lianke Biotech Co., Ltd.;
cat. no. 70-GAR0072) or goat anti-mouse IgG (dilution 1:5,000;
Lianke Biotech Co., Ltd.; cat. no. 70-GAM0072) for 1 h at room
temperature. Subsequently, an image of the protein band was
captured using the ChemiDoc™ MP imaging system (Bio-Rad
Laboratories, Inc.) using the enhanced plus chemiluminescence
reagent (EMD Millipore; cat. no. WBKL-S0100). The band densities
were semi-quantitated using ImageJ V1.50 software (National
Institutes of Health).
siRNA for CD44 variants
The expression of CD44 variants was knocked down
using corresponding siRNAs. A series of siRNA oligonucleotides
including sicontrol, siCD44v3, siCD44v4, siCD44v5, siCD44v6,
siCD44v7, siCD44v8, siCD44v9 and siCD44v10 were designed and
synthesized by Guangzhou RiboBio Co., Ltd. The CD44v2 exon was
excluded in current study due to its considerably low expression in
BCa cells (27). The siRNA
oligonucleotides are listed in Table
SI. The MDA-MB-231 and BT-549 cells were separately seeded in
6-well plates at 3×105/well and transfected with siRNA
for 48 h at 37°C using riboFECT™ (Guangzhou RiboBio Co., Ltd.),
according to the manufacturer's protocol, and 50 nM siRNAs. Reverse
transcription-quantitative (RT-q)PCR was performed to verify the
transfection efficacy after 48 h, and cells were either used for
in vitro experiments or lysed for western blot analysis.
RT-qPCR
Total RNA was extracted from cultured cells using
RNAiso Plus (Takara Bio, Inc.), according to the manufacturer's
instructions. The RNA concentration was measured using NanoDrop
system (Thermo Fisher Scientific, Inc.). Then, 1 µg mRNA was
reverse transcribed using the PrimeScript™ RT Reagent kit with gDNA
Eraser (Takara Bio, Inc.). qPCR assays were performed with
SYBR-Green mix (Takara Bio, Inc.) according to the manufacturer's
protocol. The sequences of primers for CD44 exon-specific qPCR used
in the study are listed in Table
SII, which were those reported by Hu et al (28). The qPCR conditions included: Initial
denaturation at 95°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec annealing and extension at 60°C for
34 sec. All RT-qPCR values of each gene were normalized against
that of GAPDH. The relative gene expression was calculated using
the 2−ΔΔCq method (29).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 8.0.2 (GraphPad Software, Inc.). Data are presented the as
mean ± SD. The statistically significant of differences between the
two groups were determined with an unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Sorting of BCa cells based on HA
binding reveals subpopulations with distinct proliferative
capacities
To gain insights into the distinct functions of CD44
activities, fl-HA was used to probe CD44-HA binding abilities.
HAhigh and HA−/low subpopulations were
isolated from two less malignant BCa cell lines (MCF-7 and T-47D)
and two more malignant cells (MDA-MB-231 and BT-549) using the FACS
technique (Fig. S1). A subsequent
CCK-8 assay demonstrated that the HA−/low subpopulations
were significantly more proliferative compared with the
HAhigh subpopulations in all investigated BCa cell lines
(Fig. 1A). The basal-like cell lines
MDA-MB-231 and BT-549 were selected for the following experiments
as these are malignant triple-negative BCa cells that exhibit more
aggressive properties. Consistent with the CCK-8 results, the EdU
incorporation assay and plate colony formation assay demonstrated
that the proliferation of HA−/low subpopulations was
higher compared with that of HAhigh subpopulations in
both MDA-MB-231 and BT-549 cells (Fig. 1B
and C).
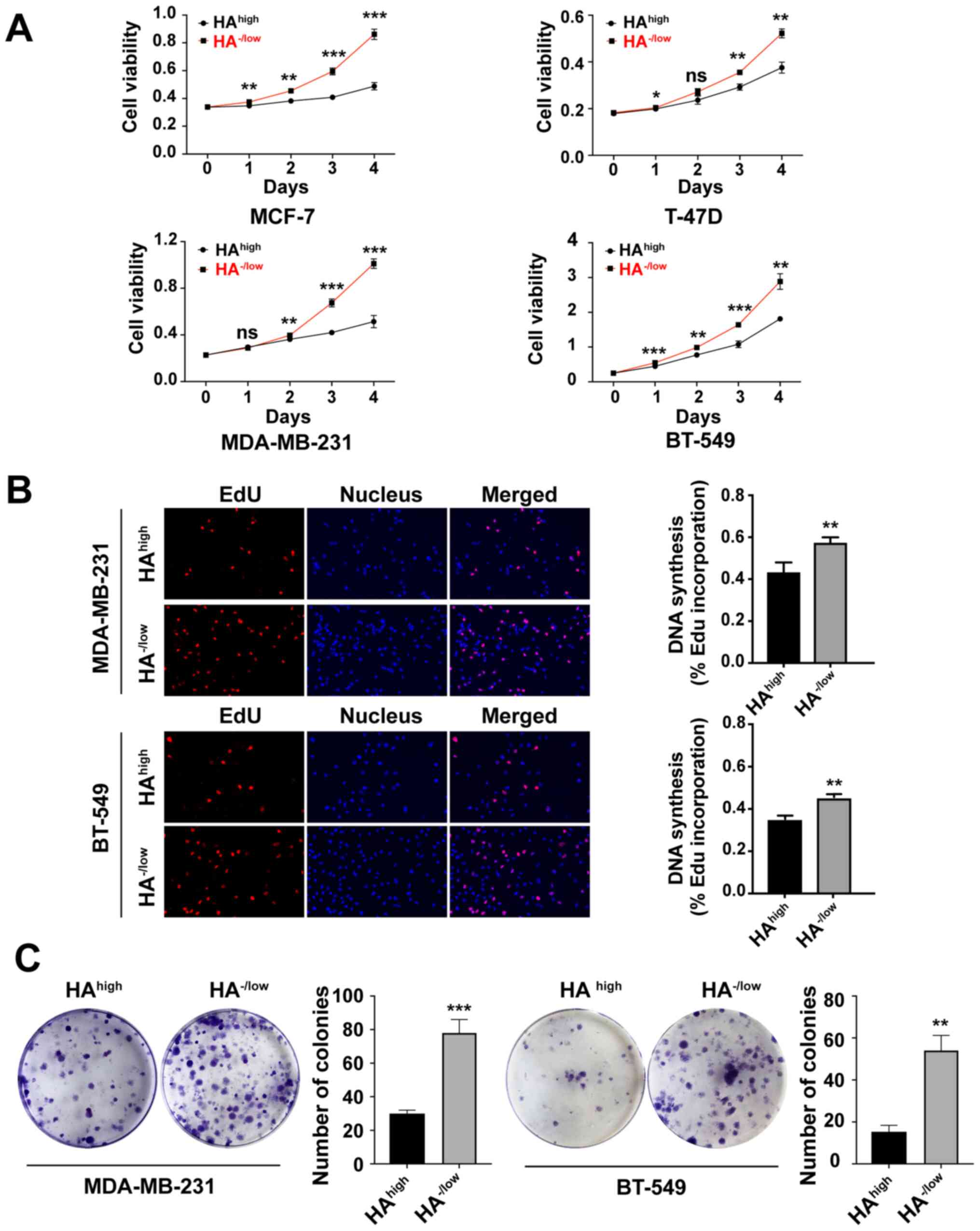 | Figure 1.HA−/low subpopulation
exhibits a significantly higher capacity of proliferation compared
with the HAhigh subset. (A) Proliferation of FACS sorted
HAhigh binding and HA−/low binding cells from
MCF-7, T-47D, MDA-MB-231 and BT-549 BCa cells was assessed using a
Cell Counting Kit-8 assay on the indicated days. (B) DNA synthesis
was detected using EdU incorporation of sorted HAhigh
and HA−/low cancer cells in MDA-MB-231 and BT-549. Cells
were fluorescently stained with EdU (red), nuclei were stained with
Hoechst 3342 (blue). Micrographs represent ≥3 experiments
(magnification, ×100). Quantitative EdU assay data are presented as
the mean ± SD of three experiments. (C) Plate colony formation
capability of HAhigh and HA−/low cells
derived from MDA-MB-231 and BT-549 cells. Cells were cultured for
14 days, and the number of colonies >10 mm2 in each
well was quantified. Unpaired Student's t-tests were used to
analyze the data. Data are presented as the mean ± SD of three
separate experiments. *P<0.05, **P<0.01, ***P<0.001 vs.
HAhigh cells. ns, no significance; EdU,
5-ethynyl-2′-deoxyuridine; HA, hyaluronan. |
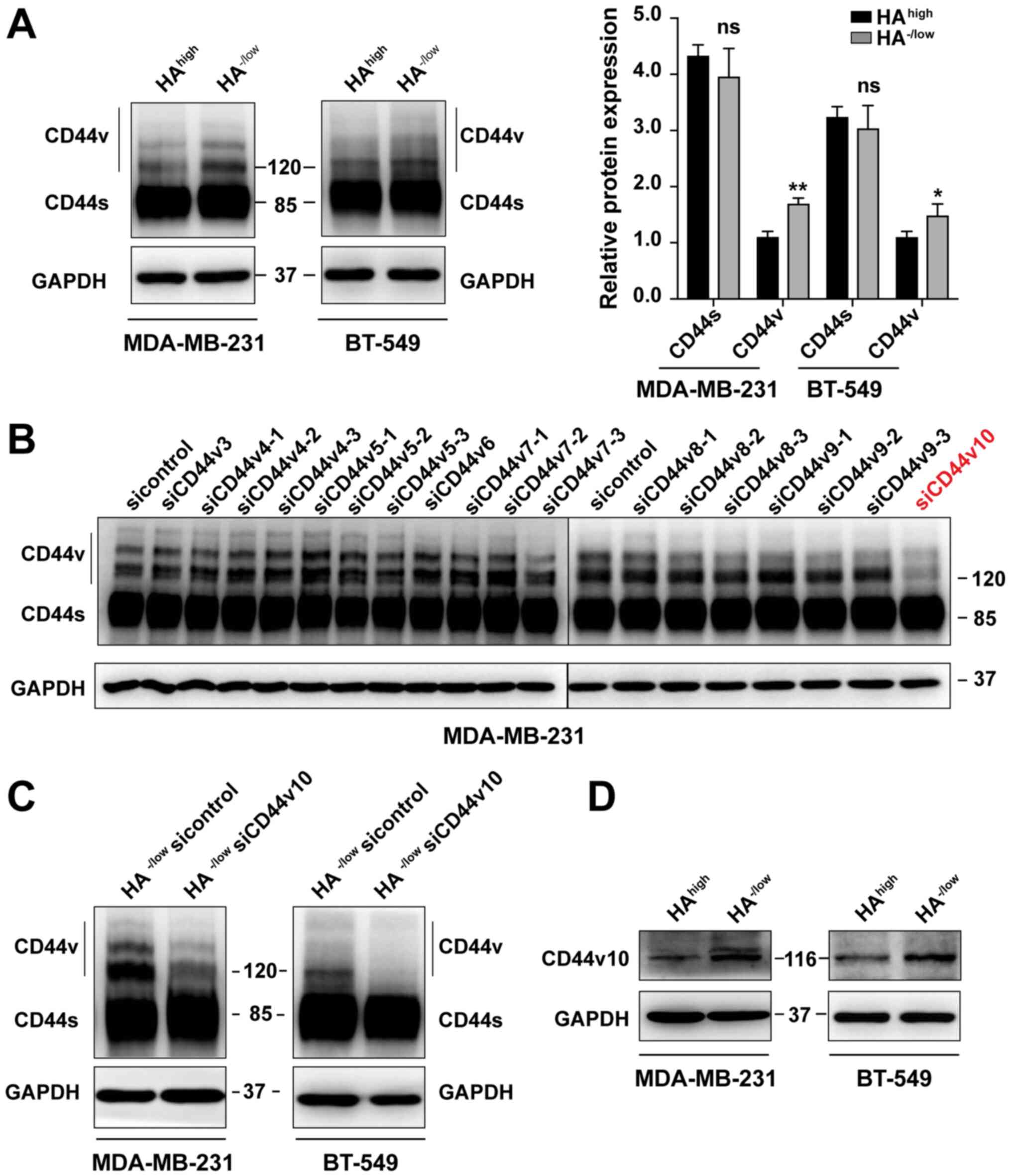
CD44v10 isoform is highly expressed in
HA−/low binding subpopulation
Based on the aforementioned finding that
HA−/low subsets grew faster than HAhigh
cells, it was next investigated whether CD44 expression patterns
were associated with CD44 activities. First, western blotting with
pan-CD44 antibody revealed that the expression levels of CD44s in
HAhigh and HA−/low subsets sorted from
MDA-MB-231 and BT-549 cells were comparable, while the CD44v in
HA−/low cells was significantly higher compared with
that in the HAhigh cells (Fig.
2A). Notably, an ~120 kDa intense band was detected in the
HA−/low subpopulations but was weaker in the
HAhigh cells (Fig.
2A).
Next, to further identify which CD44v isoform
corresponding to the 120 kDa protein determines the CD44 activation
state, a panel of siRNAs targeting each CD44v exon region was
designed and synthesized. The expression levels of each CD44v mRNA
were downregulated after siRNA transfection (Fig. S2). It was noted that the band at 120
kDa was significantly decreased in MDA-MB-231 unsorted parental
cells only after CD44v10 siRNA transfection (Fig. 2B) and was verified in
HA−/low subpopulations of both MDA-MB-231 and BT-549
cells (Fig. 2C). As confirmed
previously, the 120 kDa corresponds to a CD44v with the inclusion
of exon v10 (30). The result was
also identified at protein level by using a CD44v10-specific
antibody (Fig. 2D). Therefore, the
HA−/low subpopulation of BCa was characterized by the
preference to express the CD44v10 isoform.
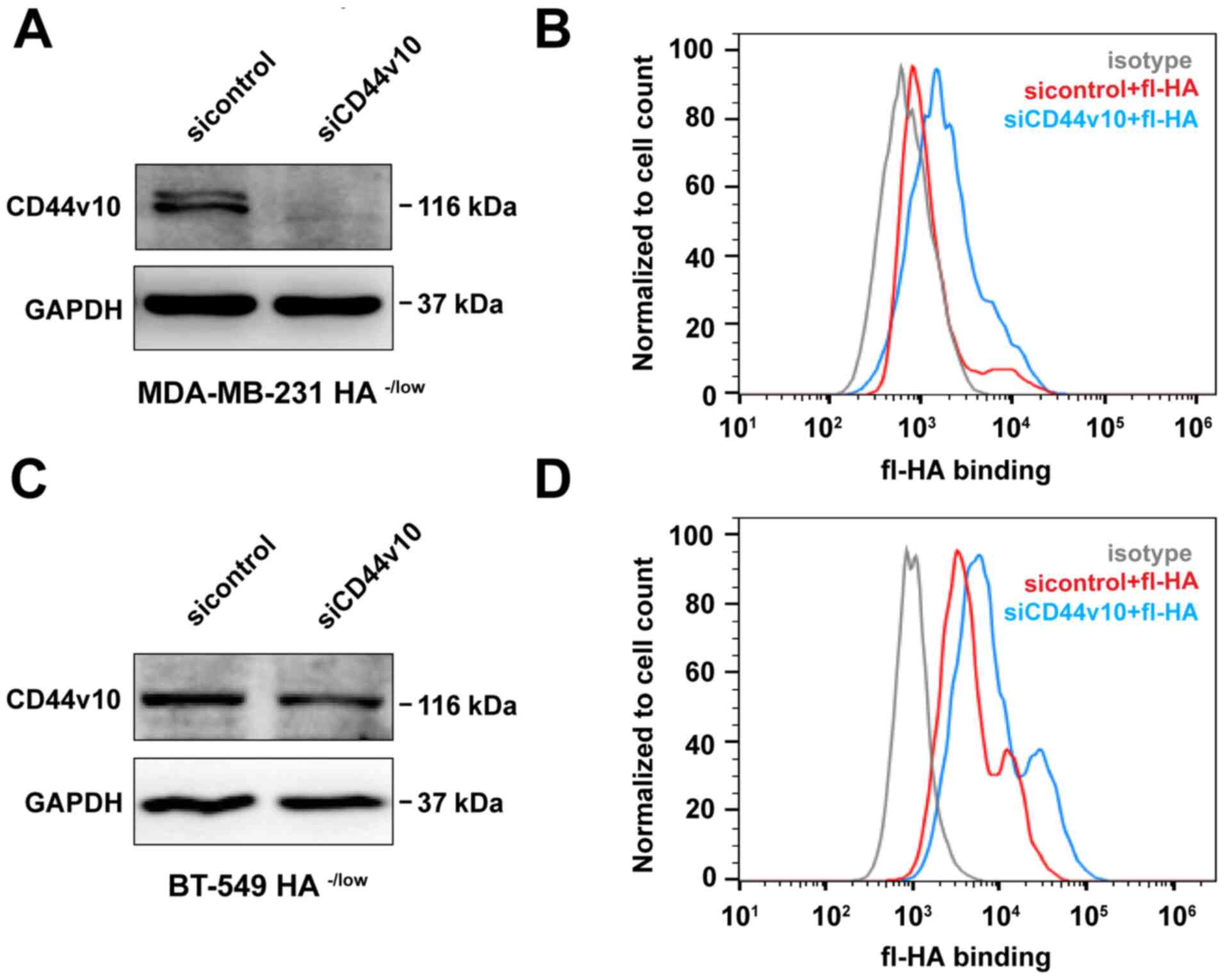
Knockdown of the CD44v10 isoform
increases the HA binding ability of HA−/low binding
subpopulation
Following the identification that CD44v10 was
preferentially expressed in HA−/low subpopulation, the
effect of the CD44v10 isoform on the capacity of HA binding was
further investigated. siRNA was used to knockdown the expression of
CD44v10 in the HA−/low binding subpopulation of
MDA-MB-231 cells. As presented in Fig.
3A, the protein expression level of CD44v10 was markedly
decreased in the HA−/low subset of MDA-MB-231 cells
after CD44v10 siRNA transfection. Subsequently, a flow cytometer
was used to evaluate the HA binding ability. As expected, HA
binding was increased as compared with the sicontrol group
following CD44v10 knockdown (Fig.
3B). Similar results were obtained in BT-549 cells, which also
showed enhanced HA binding in CD44v10-silenced HA−/low
subsets (Fig. 3C and D). Thus,
CD44v10 could give rise to inhibition of HA binding.
Knockdown of the CD44v10 isoform
inhibits the proliferation of HA−/low binding
subpopulation
Since CD44v10 was identified as the main cause
differentiating between CD44-HA high and low interactions, the
effect of the CD44v10 isoform on cell proliferation was examined.
First, a CCK-8 assay was conducted to analyze the proliferative
ability of cells after CD44v10 siRNA transfection. As indicated in
Fig. 4A, knockdown of CD44v10
significantly suppressed the proliferation of HA−/low
subsets in both MDA-MB-231 and BT-549 cells when compared with the
sicontrol group. Similarly, the EdU incorporation assay and plate
colony formation assay also confirmed the decreased proliferative
ability of HA−/low cells after CD44v10 knockdown
(Fig. 4B and C).
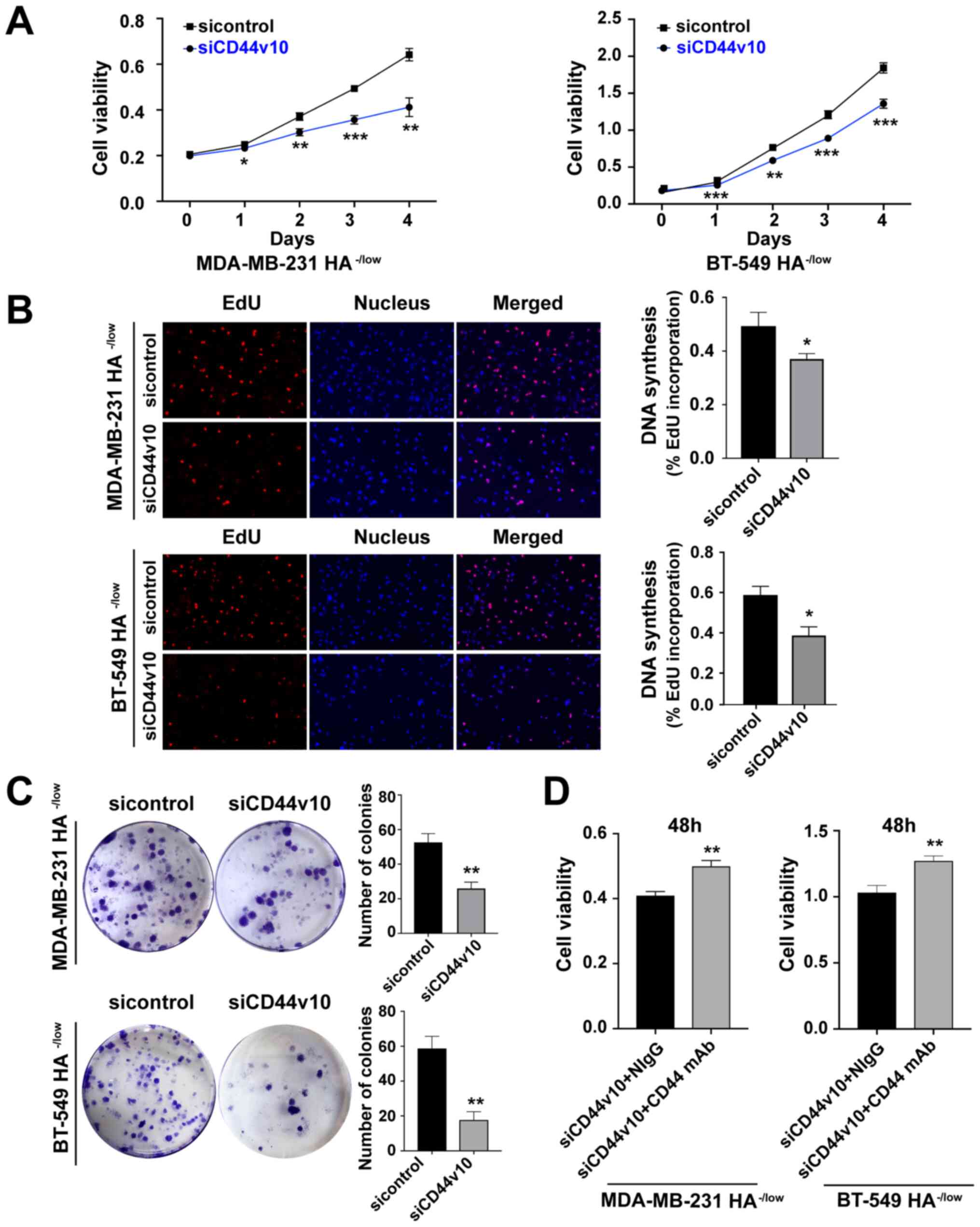 | Figure 4.Knockdown of CD44v10 isoform inhibits
the proliferation of HA−/low binding subpopulation. (A)
Cell proliferation was evaluated using a CCK-8 assay after CD44v10
knockdown. (B) Cell proliferation was also assessed using the EdU
incorporation assay (magnification, ×100). (C) Proliferative
ability of HA−/low cells transfected with CD44v10 was
detected by colony formation. *P<0.05, **P<0.01,
***P<0.001 vs. sicontrol group. (D) Viability of MDA-MB-231 and
BT-549 HA−/low cells after control siRNA or CD44v10
siRNA transfection and treatment with anti-CD44 mAb or NIgG (10
µg/ml) for 48 h was measured using a CCK-8 assay. Unpaired
Student's t-tests were used to analyze the data. Data are presented
the mean ± SD of three experiments; n=3. **P<0.01 vs. siCD44v10
+ NIgG group. NIgG, normal mouse IgG; CCK, Cell Counting Kit; HA,
hyaluronan; siRNA/si, small interfering RNA; CD44v, CD44 variant;
EdU, 5-ethynyl-2′-deoxyuridine; mAb, monoclonal antibody. |
To further verify whether the function of CD44v10
was mediated by HA binding capacity, CD44v10 was knocked down to
enhance HA binding, in which an inhibited proliferation of
HA−/low cells was observed, while the proliferative
capacity was rescued following the addition of an antibody to CD44
that blocks HA binding (Fig. 4D).
Taken together, the aforementioned results demonstrated that
knocking down CD44v10 could inhibit the proliferative and colony
formation abilities of HA−/low binding
subpopulations.
CD44v10 isoform facilitates the proliferation of BCa
cell via the ERK/p38 MAPK and AKT/mTOR signaling pathways. As the
distinct CD44 active status mediated by CD44v10 displayed different
proliferative capabilities, this study next sought to identify
whether the expression levels of cell proliferation-related
signaling molecules were altered. Previous studies have reported
that ERK/p38 MAPK and AKT/mTOR signaling pathways are involved in
the regulation of cell proliferation (31,32). Thus,
the expression levels of p-ERK, p-p38, p-AKT and p-mTOR were
examined via western blotting. As presented in Fig. 5A, p-ERK expression was upregulated in
HA−/low binding cells, while the expression of p-p38 was
slightly decreased. Similarly, both AKT and its downstream executor
mTOR displayed elevated phosphorylation in HA−/low
binding subpopulations (Fig. 5B). A
high p-ERK to p-p38 ratio is regarded as an indicator of
proliferating cells (33).
Consistently, it was observed that the ratio of p-ERK to p-p38 was
markedly increased in HA−/low binding cells (Fig. 5C). Collectively, the expression levels
of signaling molecules associated with proliferation were
significantly higher in HA−/low cells compared with in
HAhigh subsets (Fig.
5C).
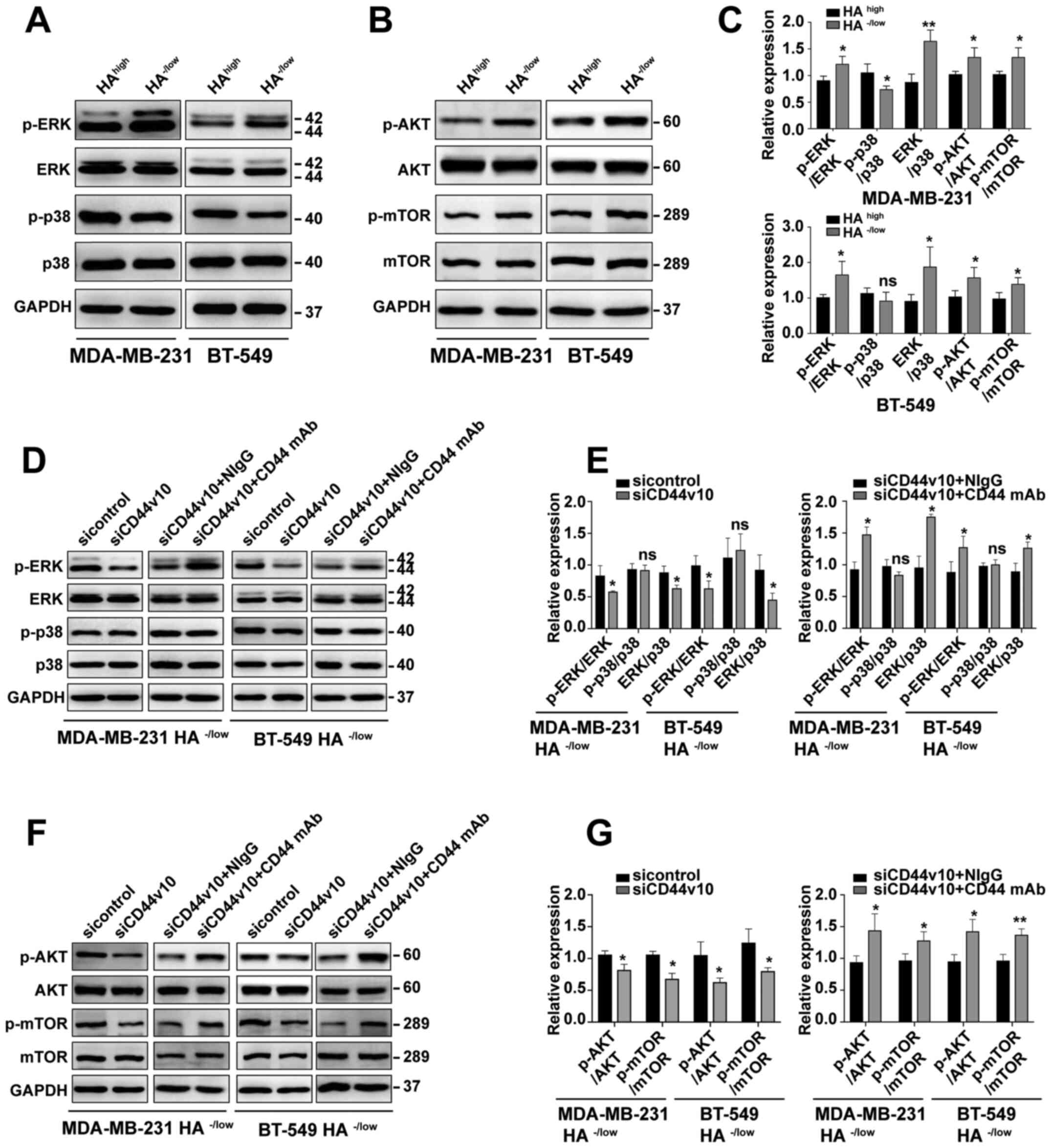 | Figure 5.CD44v10 isoform facilitates the
proliferation of BCa cells via the ERK/p38 MAPK and AKT/mTOR
signaling pathways. Expression levels of (A) ERK/p38 MAPK and (B)
AKT/mTOR signals in HAhigh and HA−/low
subpopulations were examined via western blotting. (C) Relative
protein expression of ERK/p38 MAPK and AKT/mTOR pathways in
HAhigh and HA−/low cells. *P<0.05,
**P<0.01 vs. HAhigh. (D) Western blot analysis of
ERK/p38 MAPK signals of MDA-MB-231 HA−/low and BT-549
HA−/low cells after CD44v10 knockdown. (E) Relative
protein expression of ERK/p38 MAPK in HA−/low cells
transfected with CD44v10 siRNA. (F) Western blot analysis of
AKT/mTOR signals of MDA-MB-231 HA−/low and BT-549
HA−/low cells after CD44v10 knockdown. (G) Relative
protein expression of AKT/mTOR in HA−/low cells
transfected with CD44v10 siRNA. Unpaired Student's t-tests were
used to analyze the data. *P<0.05, **P<0.01 vs. sicontrol
group or siCD44v10 + NIgG group. HA, hyaluronan; siRNA/si, small
interfering RNA; CD44v, CD44 variant; CD44s, CD44 standard; ns, no
significance; p-, phosphorylated; NIgG, normal mouse IgG; mAb,
monoclonal antibody. |
To evaluate the possible roles of ERK/p38 MAPK
pathways in CD44v10-mediated proliferation, western blotting was
conducted after knocking down CD44v10 in both MDA-MB-231 and BT-549
cells. The results demonstrated that the phosphorylation of ERK was
significantly downregulated by CD44v10 knockdown, while the
phosphorylation of p38 was not changed in HA−/low
binding cells, as compared with the sicontrol group (Fig. 5D and E). Although the expression of
p-p38 was not changed by silencing CD44v10, the ratio of p-ERK to
p-p38 was significantly decreased (Fig.
5E). To further investigate whether HA binding capacity
mediated ERK/p38 MAPK activation, rescue experiments were
performed. As presented in Fig. 5D and
E, p-ERK expression rescued following the addition of an
antibody to CD44 that blocked HA binding. Furthermore, a similar
decreasing trend was observed for the alterations in p-AKT and
p-mTOR levels induced by CD44v10 knockdown in the
HA−/low binding subsets (Fig.
5F), while the phosphorylated levels were rescued following the
addition of an antibody to CD44 that blocked HA binding (Fig. 5F and G). Taken together, these results
suggested that the CD44v10 isoform facilitated the proliferation of
BCa cells via ERK/p38 MAPK and AKT/mTOR activation.
Discussion
CD44 has long been recognized as a cell surface
receptor that mediates diverse biological functions. As previously
suggested, CD44 exists in two distinct states: It is in a resting
state on the surface of most normal cells, while it becomes active
following an inflammatory stimulus (34,35). A
feature for the ‘activated’ stage of CD44 is that it can bind to
its ligand HA (36). Usually,
tumor-derived cells often express CD44 in an active state that can
bind and internalize to HA (37).
Although CD44 expression has been reported to be closely associated
with tumor malignancies, few studies have examined whether CD44
activation is critical for cancer development (38). The present study demonstrated a
potential association between the activation status of CD44 and its
subsequent effect on BCa cell proliferation. The current results
indicated that BCa cells with inactive CD44 states had higher
proliferation, while the subsets with active CD44 grew slowly.
However, these findings are not consistent with some previous
studies, which indicated CD44 activation was likely to support
tumor growth. For example, evidence for CD44 activation giving rise
to cell proliferation was provided for smooth muscle cells,
fibroblasts, immune cells and endothelial cells (39). Moreover, a similar phenomenon was
observed in melanoma cells (40) and
mesothelioma cells (41). However,
Veiseh et al (42) reported
that a HAlow binding subpopulation sorted from BCa cells
exhibited significantly higher proliferation compared with unsorted
parental cells or the HAhigh subpopulation, which was
consistent with the present results. These studies implied that the
recognition of CD44 on HA could either positively or negatively
affect tumor growth. Such contradictory results may be ascribed to
different types of cancer or different studies investigating
various cell types. It is thus possible that CD44 activation is
essential for tumor growth derived from certain organs, such as the
skin (43) and lung (44), whereas tumors derived from other
sources have no such requirement.
The present study first sorted resting and active
CD44 expressing subpopulations from BCa cell lines according to
their binding abilities to fl-HA. Then, western blotting results
demonstrated that CD44s was highly expressed in both
HAhigh and HA−/low subsets without obvious
difference. However, the expression of CD44v was significantly
higher in HA−/low cells. This unique finding suggested
that CD44v may have an essential role in regulating CD44 activities
in BCa cells. Previous studies have revealed that elevated levels
of CD44 do not necessarily correlate with increased HA-binding,
while the binding capacities differ in various CD44 isoforms
(45,46). Some reports suggested that CD44s binds
to HA more efficiently compared with CD44v8-v10 (21,47),
whereas another study observed that CD44v4-v7 had a stronger
HA-binding capacity compared with CD44s (22). Although these inconsistencies are
still being investigated, different isoforms of CD44 subtypes or
variants coexisting within the same cell population are considered
to be responsible for such CD44-HA binding affinity
heterogeneity.
In the present study, a panel of siRNAs targeting
each CD44v exon region was designed and synthesized to identify the
CD44v isoform that may determine the CD44-HA binding difference or
CD44 activities. As expected, it was found that the differential
expression band between HAhigh and HA−/low
subpopulations was downregulated only by transfection with CD44v10
siRNA, inferring that CD44v10 was responsible for distinct HA
binding abilities between the two subsets. To further verify the
effect of CD44v10 on HA binding, CD44v10 expression was knocked
down by transfecting CD44v10 siRNA in HA−/low cells. It
was found that the HA binding ability was enhanced after knocking
down CD44v10 expression. The present findings were in line with
previous reports, which suggested that post-translational
modifications of CD44v10 induced surface rearrangements and
conformational changes in the HA binding domains, finally leading
to a loss of HA binding (48–50).
Even though the upregulation of several CD44v
isoforms has been reported to be associated with BCa progression
and prognosis (51), so far, only a
few studies have revealed the function of the CD44v10 isoform in
BCa. As previously suggested, CD44v10 is highly expressed in
various cancer types, such as Hodgkin's disease (52), multiple myeloma (53), lymphoma (54) and renal cell carcinoma (55), and is associated with an unfavorable
clinical outcome. In the present study, it was identified that
CD44v10 was highly expressed in the HA−/low binding
subset, which exhibited a stronger proliferation capability
compared with the HAhigh cells. Furthermore, knockdown
of CD44v10 markedly inhibited the proliferative capability and
colony formation ability of HA−/low binding
subpopulation. The loss- and gain-of-function experiments indicated
that the enhanced HA binding ability by CD44v10 siRNA transfection
significantly inhibited the proliferation of HA−/low
cells. At the same time, the proliferative capacity could be
rescued by the addition of an antibody to CD44 that specifically
blocks HA binding. Collectively, these data suggested that CD44v10
mediated by CD44 activities has a key role in BCa cells
proliferation.
To further understand the intracellular mechanism
via which CD44v10 enhances BCa cell proliferation, the present
study next investigated the effect of CD44v10 on cell
proliferation-related signaling molecules. CD44 isoforms can
promote BCa cell proliferation and survival by activation of two
important members of MAPK families, including ERK1/2 and p38, as
well as the AKT/mTOR signaling pathway (25,56). In
the current research, it was demonstrated that phosphorylation of
ERK1/2, AKT and mTOR was elevated in HA−/low binding
cells and was subsequently downregulated by knockdown of CD44v10.
Conversely, the expression of p-p38 was slightly lower in
HA−/low binding subpopulations, and knockdown CD44v10 in
HA−/low cells showed no effect on p38 phosphorylation.
As previously reported, although ERK was mostly involved in the
induction of proliferation, a high level of p38 activity was
considered to be a negative growth regulator that may suppress cell
proliferation by inhibiting ERK (57–59).
Accordingly, a high ERK-to-p38 ratio dictated cell proliferation,
while a low ERK-to-p38 predicted cell proliferation arrest and
dormancy (33). In the present study,
although the phosphorylation of p38 decreased or unchanged, the
ratio of ERK-to-p38 phosphorylation was significantly higher in
HA−/low subsets compared with in HAhigh
cells, and the ratio was decreased in HA−/low subsets
after silencing CD44v10. These results suggested that ERK/p38 MAPK
signals are likely to be involved in CD44v10-induced proliferation.
Additionally, the rescue experiments indicated that the
phosphorylation ERK/p38 and AKT/mTOR could be rescued following the
addition of an antibody to CD44 that blocks HA binding. Overall,
the present study demonstrated that CD44v10 regulated by CD44
activities could enhance the proliferation of BCa cells by
triggering the ERK/p38 MAPK and AKT/mTOR signaling pathway
cascades.
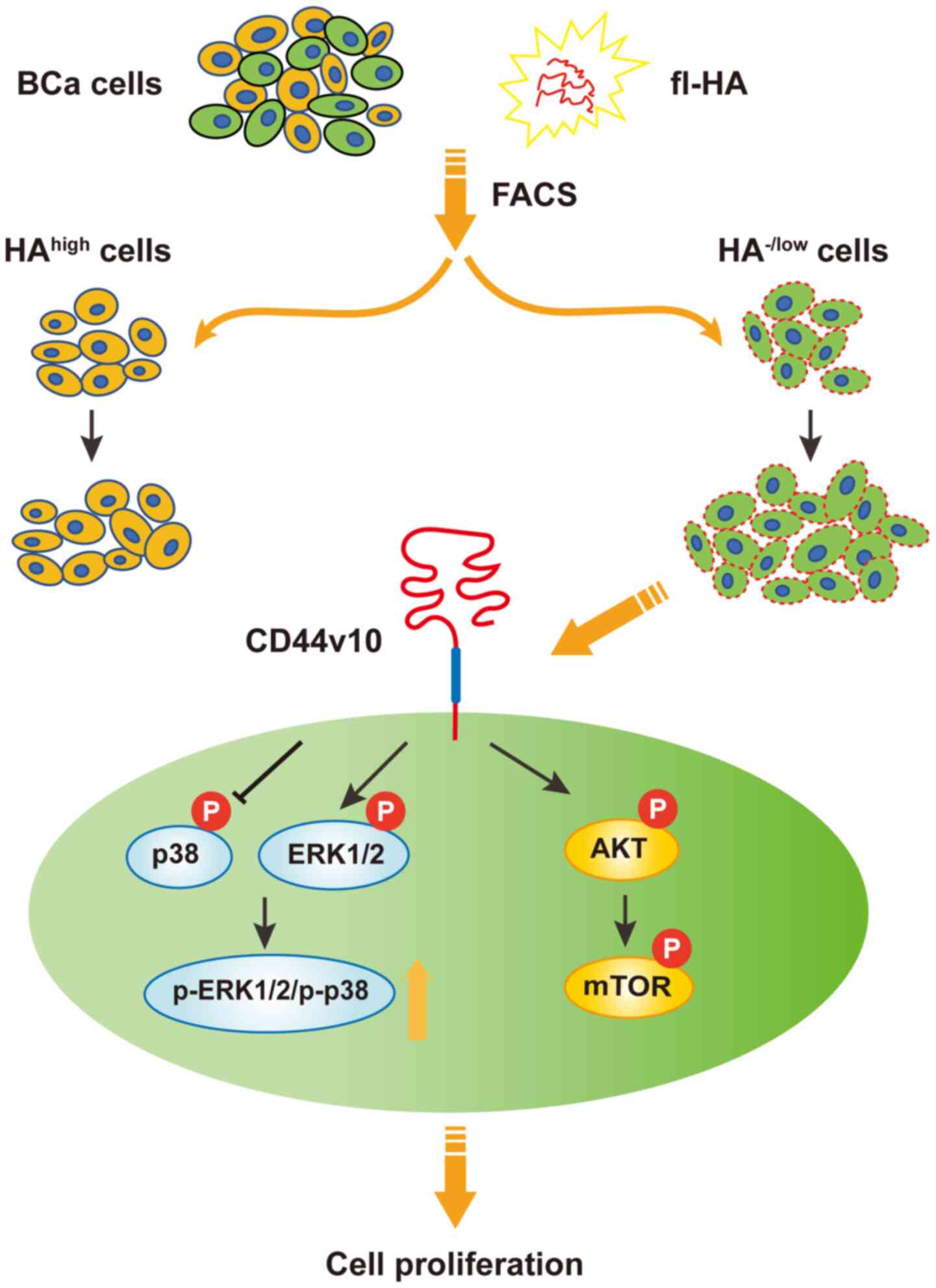
In summary, the present study demonstrated that the
biological function was distinct between inactive and active CD44
status in BCa cells, and the CD44v10 isoform was responsible for
the CD44 activities (Fig. 6). It was
suggested that CD44v10 may facilitate proliferation via the
activation of the ERK/p38 MAPK and AKT/mTOR signaling pathways.
Collectively, this study provided novel insight into the mechanisms
underlying CD44 activation and suggested that CD44v10 may be a
potential therapeutic target for BCa. However, it remains unknown
whether CD44v10 could facilitate tumor growth in vivo, thus
further studies on the clinical significance of CD44v10 are
warranted. Further work in this area is ongoing in the authors'
laboratory.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was funded by grants from the National
Natural Science Foundation of China (grant nos. 81672843, 81702852,
81872357, 81974445, 81974446 and 82073199), Shanghai Municipal
Education Commission-Gaofeng Clinical Medicine Grant Support (grant
no. 20171924), Doctor Innovation Fund of Shanghai Jiaotong
University School of Medicine (grant no. BXJ201944), Shanghai
Pujiang Program (grant no. 2019PJD037) and Shanghai ‘Rising Stars
of Medical Talent’ Youth Development Program Clinical Laboratory
Practitioners Program (grant no. 2020087).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
QG, CY and FG conceived and designed the study. QG,
YL, YH, YD and GZ performed the experiments and analyzed the data.
QG wrote the draft manuscript. CY and FG supervised the study and
revised the manuscript. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declared that they no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yeo SK and Guan JL: Breast cancer:
Multiple subtypes within a tumor? Trends Cancer. 3:753–760. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heldin P, Basu K, Kozlova I and Porsch H:
HAS2 and CD44 in breast tumorigenesis. Adv Cancer Res. 123:211–229.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Senbanjo LT and Chellaiah MA: CD44: A
multifunctional cell surface adhesion receptor is a regulator of
progression and metastasis of cancer cells. Front Cell Dev Biol.
5:182017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DeGrendele HC, Estess P and Siegelman MH:
Requirement for CD44 in activated T cell extravasation into an
inflammatory site. Science. 278:672–675. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chanmee T, Ontong P and Itano N:
Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett.
375:20–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Toole BP: Hyaluronan-CD44 interactions in
cancer: Paradoxes and possibilities. Clin Cancer Res. 15:7462–7468.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karousou E, Misra S, Ghatak S, Dobra K,
Götte M, Vigetti D, Passi A, Karamanos NK and Skandalis SS: Roles
and targeting of the HAS/hyaluronan/CD44 molecular system in
cancer. Matrix Biol. 59:3–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McDonald B and Kubes P: Interactions
between CD44 and hyaluronan in leukocyte trafficking. Front
Immunol. 6:682015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lesley J, Kincade PW and Hyman R:
Antibody-induced activation of the hyaluronan receptor function of
CD44 requires multivalent binding by antibody. Eur J Immunol.
23:1902–1909. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ogino S, Nishida N, Umemoto R, Suzuki M,
Takeda M, Terasawa H, Kitayama J, Matsumoto M, Hayasaka H, Miyasaka
M, et al: Two-state conformations in the hyaluronan-binding domain
regulate CD44 adhesiveness under flow condition. Structure.
18:649–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lesley J and Hyman R: CD44 can be
activated to function as an hyaluronic acid receptor in normal
murine T cells. Eur J Immunol. 22:2719–2723. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiraga T, Ito S and Nakamura H: Cancer
stem-like cell marker CD44 promotes bone metastases by enhancing
tumorigenicity, cell motility, and hyaluronan production. Cancer
Res. 73:4112–4122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu S, Cao M, He Y, Zhang G, Liu Y, Du Y,
Yang C and Gao F: CD44v6 targeted by miR-193b-5p in the coding
region Modulates the migration and invasion of breast cancer cells.
J Cancer. 11:260–271. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao P, Xu Y, Wei Y, Qiu Q, Chew TL, Kang
Y and Cheng C: The CD44s splice isoform is a central mediator for
invadopodia activity. J Cell Sci. 129:1355–1365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teriete P, Banerji S, Noble M, Blundell
CD, Wright AJ, Pickford AR, Lowe E, Mahoney DJ, Tammi MI, Kahmann
JD, et al: Structure of the regulatory hyaluronan binding domain in
the inflammatory leukocyte homing receptor CD44. Mol Cell.
13:483–496. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu D, Liu T, Li R and Sy MS: Mechanisms
regulating the binding activity of CD44 to hyaluronic acid. Front
Biosci. 3:d631–d636. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Louderbough JM and Schroeder JA:
Understanding the dual nature of CD44 in breast cancer progression.
Mol Cancer Res. 9:1573–1586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Naor D, Nedvetzki S, Golan I, Melnik L and
Faitelson Y: CD44 in cancer. Crit Rev Clin Lab Sci. 39:527–579.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dougherty GJ, Cooper DL, Memory JF and
Chiu RK: Ligand binding specificity of alternatively spliced CD44
isoforms. Recognition and binding of hyaluronan by CD44R1. J Biol
Chem. 269:9074–9078. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bennett KL, Modrell B, Greenfield B,
Bartolazzi A, Stamenkovic I, Peach R, Jackson DG, Spring F and
Aruffo A: Regulation of CD44 binding to hyaluronan by glycosylation
of variably spliced exons. J Cell Biol. 131:1623–1633. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sleeman J, Rudy W, Hofmann M, Moll J,
Herrlich P and Ponta H: Regulated clustering of variant CD44
proteins increases their hyaluronate binding capacity. J Cell Biol.
135:1139–1150. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Orian-Rousseau V: CD44 Acts as a signaling
platform controlling tumor progression and metastasis. Front
Immunol. 6:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen C, Zhao S, Karnad A and Freeman JW:
The biology and role of CD44 in cancer progression: Therapeutic
implications. J Hematol Oncol. 11:642018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu S and Cheng C: Akt Signaling is
sustained by a CD44 splice isoform-mediated positive feedback loop.
Cancer Res. 77:3791–3801. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang C, Cao M, Liu H, He Y, Xu J, Du Y,
Liu Y, Wang W, Cui L, Hu J, et al: The high and low molecular
weight forms of hyaluronan have distinct effects on CD44
clustering. J Biol Chem. 287:43094–43107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Brown RL, Wei Y, Zhao P, Liu S,
Liu X, Deng Y, Hu X, Zhang J, Gao XD, et al: CD44 splice isoform
switching determines breast cancer stem cell state. Genes Dev.
33:166–179. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu J, Li G, Zhang P, Zhuang X and Hu G: A
CD44v+ subpopulation of breast cancer stem-like cells
with enhanced lung metastasis capacity. Cell Death Dis.
8:e26792017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lokeshwar VB, Iida N and Bourguignon LY:
The cell adhesion molecule, GP116, is a new CD44 variant (ex14/v10)
involved in hyaluronic acid binding and endothelial cell
proliferation. J Biol Chem. 271:23853–23864. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aguirre-Ghiso JA, Estrada Y, Liu D and
Ossowski L: ERK(MAPK) activity as a determinant of tumor growth and
dormancy; regulation by p38(SAPK). Cancer Res. 63:1684–1695.
2003.PubMed/NCBI
|
|
32
|
Nan H, Han L, Ma J, Yang C, Su R and He J:
STX3 represses the stability of the tumor suppressor PTEN to
activate the PI3K-Akt-mTOR signaling and promotes the growth of
breast cancer cells. Biochim Biophys Acta Mol Basis Dis.
1864:1684–1692. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aguirre-Ghiso JA, Ossowski L and Rosenbaum
SK: Green fluorescent protein tagging of extracellular
signal-regulated kinase and p38 pathways reveals novel dynamics of
pathway activation during primary and metastatic growth. Cancer
Res. 64:7336–7345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dong Y, Poon GF, Arif AA, Lee-Sayer SS,
Dosanjh M and Johnson P: The survival of fetal and bone marrow
monocyte-derived alveolar macrophages is promoted by CD44 and its
interaction with hyaluronan. Mucosal Immunol. 11:601–614. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levesque MC and Haynes BF: Cytokine
induction of the ability of human monocyte CD44 to bind hyaluronan
is mediated primarily by TNF-alpha and is inhibited by IL-4 and
IL-13. J Immunol. 159:6184–6194. 1997.PubMed/NCBI
|
|
36
|
Siegelman MH, DeGrendele HC and Estess P:
Activation and interaction of CD44 and hyaluronan in immunological
systems. J Leukoc Biol. 66:315–321. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bachar G, Cohen K, Hod R, Feinmesser R,
Mizrachi A, Shpitzer T, Katz O and Peer D: Hyaluronan-grafted
particle clusters loaded with Mitomycin C as selective nanovectors
for primary head and neck cancers. Biomaterials. 32:4840–4848.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patrawala L, Calhoun T,
Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra
D, Zhou J, Claypool K, et al: Highly purified CD44+
prostate cancer cells from xenograft human tumors are enriched in
tumorigenic and metastatic progenitor cells. Oncogene.
25:1696–1708. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Misra S, Hascall VC, Markwald RR and
Ghatak S: Interactions between hyaluronan and its receptors (CD44,
RHAMM) regulate the activities of inflammation and cancer. Front
Immunol. 6:2012015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bartolazzi A, Peach R, Aruffo A and
Stamenkovic I: Interaction between CD44 and hyaluronate is directly
implicated in the regulation of tumor development. J Exp Med.
180:53–66. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hanagiri T, Shinohara S, Takenaka M,
Shigematsu Y, Yasuda M, Shimokawa H, Nagata Y, Nakagawa M, Uramoto
H, So T, et al: Effects of hyaluronic acid and CD44 interaction on
the proliferation and invasiveness of malignant pleural
mesothelioma. Tumour Biol. 33:2135–2141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Veiseh M, Kwon DH, Borowsky AD, Tolg C,
Leong HS, Lewis JD, Turley EA and Bissell MJ: Cellular
heterogeneity profiling by hyaluronan probes reveals an invasive
but slow-growing breast tumor subset. Proc Natl Acad Sci USA.
111:E1731–E1739. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ahrens T, Sleeman JP, Schempp CM, Howells
N, Hofmann M, Ponta H, Herrlich P and Simon JC: Soluble CD44
inhibits melanoma tumor growth by blocking cell surface CD44
binding to hyaluronic acid. Oncogene. 20:3399–3408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song JM, Im J, Nho RS, Han YH, Upadhyaya P
and Kassie F: Hyaluronan-CD44/RHAMM interaction-dependent cell
proliferation and survival in lung cancer cells. Mol Carcinog.
58:321–333. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Peach RJ, Hollenbaugh D, Stamenkovic I and
Aruffo A: Identification of hyaluronic acid binding sites in the
extracellular domain of CD44. J Cell Biol. 122:257–264. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stamenkovic I, Aruffo A, Amiot M and Seed
B: The hematopoietic and epithelial forms of CD44 are distinct
polypeptides with different adhesion potentials for
hyaluronate-bearing cells. EMBO J. 10:343–348. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
van der Voort R, Manten-Horst E, Smit L,
Ostermann E, van den Berg F and Pals ST: Binding of cell-surface
expressed CD44 to hyaluronate is dependent on splicing and cell
type. Biochem Biophys Res Commun. 214:137–144. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Weimann TK, Wagner C, Funk R, Hirche H,
Goos M and Wagner SN: Hyaluronan-independent adhesion of
CD44H+ and CD44v10+ lymphocytes to dermal microvascular
endothelial cells and keratinocytes. J Invest Dermatol.
117:949–957. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Iida N and Bourguignon LY: Coexpression of
CD44 variant (v10/ex14) and CD44S in human mammary epithelial cells
promotes tumorigenesis. J Cell Physiol. 171:152–160. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Katoh S, Zheng Z, Oritani K, Shimozato T
and Kincade PW: Glycosylation of CD44 negatively regulates its
recognition of hyaluronan. J Exp Med. 182:419–429. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bellerby R, Smith C, Kyme S, Gee J,
Günthert U, Green A, Rakha E, Barrett-Lee P and Hiscox S:
Overexpression of specific CD44 isoforms is associated with
aggressive cell features in acquired endocrine resistance. Front
Oncol. 6:1452016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Beham-Schmid C, Heider KH, Hoefler G and
Zatloukal K: Expression of CD44 splice variant v10 in Hodgkin's
disease is associated with aggressive behaviour and high risk of
relapse. J Pathol. 186:383–389. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Asosingh K, Günthert U, De Raeve H, Van
Riet I, Van Camp B and Vanderkerken K: A unique pathway in the
homing of murine multiple myeloma cells: CD44v10 mediates binding
to bone marrow endothelium. Cancer Res. 61:2862–2865.
2001.PubMed/NCBI
|
|
54
|
Megaptche AP, Erb U, Büchler MW and Zöller
M: CD44v10, osteopontin and lymphoma growth retardation by a
CD44v10-specific antibody. Immunol Cell Biol. 92:709–720. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li N, Tsuji M, Kanda K, Murakami Y,
Kanayama H and Kagawa S: Analysis of CD44 isoform v10 expression
and its prognostic value in renal cell carcinoma. BJU Int.
85:514–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ishimoto T, Nagano O, Yae T, Tamada M,
Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, et al:
CD44 variant regulates redox status in cancer cells by stabilizing
the xCT subunit of system xc(−) and thereby promotes tumor growth.
Cancer Cell. 19:387–400. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Black EJ, Clark W and Gillespie DA:
Transient deactivation of ERK signalling is sufficient for stable
entry into G0 in primary avian fibroblasts. Curr Biol.
10:1119–1122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen G, Hitomi M, Han J and Stacey DW: The
p38 pathway provides negative feedback for Ras proliferative
signaling. J Biol Chem. 275:38973–38980. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Aguirre-Ghiso JA, Liu D, Mignatti A,
Kovalski K and Ossowski L: Urokinase receptor and fibronectin
regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine
carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell.
12:863–879. 2001. View Article : Google Scholar : PubMed/NCBI
|