Introduction
Metabolic changes have been recognized as one of the
hallmarks of cancer (1). These
changes, which can be genetically determined by specific oncogenic
alterations and be impacted by tumor microenvironmental conditions,
serve multiple adaptive roles that are incompletely understood.
Among them are the growing tumors' high anabolic demands, and the
defense from pathological conditions created by this uncontrolled
growth.
Pancreatic cancer is often hypoxic with a poor 8%
survival rate at 5 years (2), and is
immunologically privileged (3).
Mutated KRAS is the most commonly found oncogenic
event in pancreatic ductal adenocarcinoma (PDAC) and is responsible
for the initiation of tumor metabolic reprograming (4,5). Similarly
to other Ras-driven cancers, the metabolic needs of PDAC have been
shown to depend on scavenging of extracellular nutrient sources
(6). These nutrients, such as
proteins, nucleotides and lipids enter cells through
macropinocytosis, a well-described KRAS-dependent mechanism of
membrane budding and subsequent cargo vesicle trafficking (6–8). Fatty
acid (FA) synthesis in PDAC may be attenuated and intracellular
pool of FAs derived predominantly from exogenous sources, such as
serum lysophospholipids (9–12). Accordingly, cholesterol uptake in PDAC
has been shown to be indispensable for sustaining proliferative
capacity of PDAC. Silencing of low-density lipoprotein receptor
(LDLR) that translocates cholesterol-rich LDLs sensitizes PDAC to
chemotherapy (13). Glucose and
glutamine metabolism are also regulated by oncogenic Kras, which
can change the source of acetyl-CoA production that is being used
for fatty acid synthesis (14).
Part of the metabolic rewiring involves the
increased storage of neutral lipids inside lipid droplets (LDs).
The core of LDs contains esterified fatty acids and cholesterol
species and is separated from the hydrophilic cytosol by a
phospholipid monolayer. On the periphery, associated proteins
control the access of enzymes in a regulated manner and determine
the dynamics of LD turnover (15).
HILPDA is a small, evolutionarily young protein that
was originally identified through its induction by oxygen- or
glucose deprivation (16). It
localizes to LDs and the endoplasmic reticulum and promotes lipid
storage in a large number of cell types tested, including cancer
cells, hepatocytes, and macrophages (17–20). Whole
body ablation of Hilpda in mice results in a thermoregulatory
defect in fasted mice, suggesting a systemic role in fuel
utilization (21). Various cell
type-specific genetic models have identified defects in lipid
turnover by Hilpda loss, however, the precise molecular target of
Hilpda's action remained elusive (18,19,22,23).
Recently, it was demonstrated that Hilpda can promote LD formation
by binding and inhibiting Adipose Triglyceride Lipase
(ATGL/PNPLA2), which is the first and rate-limiting enzyme in
triglyceride hydrolysis (24,25). Furthermore, we identified
HILPDA-dependent inhibition of ATGL during states of high lipid
turnover such as fatty acid supplementation or starvation-induced
LD remodeling (26). This molecular
mechanism controlling triglyceride accumulation was important for
colon and lung model tumor growth (24,26).
The aim of the present study was to determine
whether Hilpda-dependent regulation of lipid metabolism plays a
role in the in vivo growth of model murine pancreatic tumors
and which biochemical perturbations are caused by Hilpda
deletion.
Materials and methods
Cell culture and treatments
KPC cells were originally established from the
Tuveson LSL-Kras G12D/+; LSL-Trp53R172H/+; Pdx-1-Cre model
(27) and were grown in DMEM + 10%
FBS (Gibco) in a humidified incubator at 37°C. ATGListatin (20 µM),
drug vehicle control DMSO, docosahexanoic acid (DHA) (60.7 µM) (all
from Sigma-Aldrich; Merck KGaA) were used as indicated for various
experiments. Western blots were repeated twice, biochemical assays
were performed in four independent biological replicates. All
treatments were performed at 37°C.
Molecular cloning and
transfections
HILPDA KO cell lines were generated using a double
nickase strategy. Two gRNAs targeting Hilpda: A
(5′-TCTAACAAAGATGGAAAGCA-3′) and B (5′-GGAGTCTCTGGGAGGCTTAC-3′)
were individually cloned in pX462-Cas9n backbone
[pSpCas9n(BB)-2A-Puro V2.0, Addgene: 62987] using BbsI
restriction site. Constructs targeting Hilpda were
sequence-verified and used to create Hilpda KO cell lines. Cells
were transfected with 2 µg DNA using Lipofectamine 2000 (Thermo
Fisher Scientific). Single clones were selected by antibiotic
resistance for 3 days and further expansion for 2 weeks, screened
by westerns blotting and 7 successful KO clones were combined to
generate the KO pool. The pIRES-neo-Hilpda-myc-flag expression
vector has been described previously (26). Cells were transfected with
pIRES-neo-Hilpda-myc-flag or empty vector pIRES-neo (Origene) and
underwent G418 (Sigma-Aldrich; Merck KGaA) selection at 2 mg/ml for
2 weeks before being screened for transgene expression.
Western blot analysis
KPC cells were lysed in RIPA buffer (150 mM NaCl, 1%
NP 40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0)
supplemented with 100X Halt protease inhibitor cocktail (Thermo
Scientific Fisher), 100X phosphatase inhibitor cocktail (Cell
Signaling Technology) and 1 mM PMSF (Thermo Fisher Scientific).
Lysates were cleared by centrifugation for 5 min at 12,000 × g and
at 4°C. Protein concentrations were measured with a bicinchoninic
acid (BCA) protein kit (Thermo Scientific Fisher). Then, 20–30 µg
of whole protein lysates were resolved in an 11% acrylamide gel and
transferred onto PVDF membrane. For immunodetection, primary
antibodies used were: Custom-made rabbit anti-Hilpda 1:50 (21), rabbit a-Perilipin2 (1:1,000; Origene,
cat. no. TA321279), mouse a-myc (1:1,000; Cell Signaling
Technology, cat. no. 2276S), and rabbit a-ATGL (1:1,000; Cell
Signaling Technology, cat. no. 2138S), and mouse a-tubulin
(1:1,000; Invitrogen; Thermo Fisher Scientific, cat. no.
PIMA516308). Primary antibodies were detected using Licor goat
anti-mouse (1:2,000, Licor, cat. no. 926-68070) or goat anti-rabbit
(1:2,000; Licor, cat. no. 926-32211) secondary antibodies, and
visualized using a Licor Odyssey CLx near infrared imager.
Fluorescence microscopy
Cells were grown on glass coverslips, treated as
required and fixed with 4% paraformaldehyde. Lipid droplets were
stained with 0.1 µg/ml Nile Red (Santa Cruz Biotechnology) for 20
min at room temperature. Nuclei were counterstained with 10 µg/ml
Hoechst-33342, and samples were mounted with Slowfade Diamond
antifade mounting media (Life Technologies, cat. no. S36968). The
slides were imaged on a Zeiss Axioskope widefield microscope at the
OSUCCC microscopy and imaging facility. Lipid droplet images were
visualized with ImageJ.
Triglyceride quantification
Cells were grown on 10 cm culture dishes in regular
DMEM media, and fatty acid was loaded (using DHA), or starved of
FBS for 24 h. Where indicated, ATGListatin was added at the
beginning of treatment. Triglycerides were measured using the
colorimentric Triglyceride Quantification Assay Kit (Abcam; cat.
no. ab65336) as per the manufacturer's recommendation (sensitivity
>2 µM).
In vivo xenograft growth
All animal experiments were approved by the Ohio
State University's Institutional Animal Care and Use Committee.
Five hundred thousand cells in PBS were injected subcutaneously on
the back of non-anesthetized 7- to 9-week-old female nu/nu mice
(18–21 g) obtained from the OSUCCC Target Validation Shared
Resource (n=11/group). Groups of 3–4 animals were housed in
autoclaved cages, were fed ad libitum, and maintained on a
12-h light/dark cycle. Room temperature was maintained at 22°C and
humidity at 30%. Cages were randomly assigned to experimental
groups. Tumor growth was measured using calipers. Tumor volume was
calculated using the formula: S × S × W × 0.52. Animals were
euthanized by CO2 asphyxiation followed by cervical
dislocation according to the approved protocol. Maximal tumor
dimensions at the time of sacrifice were 12 and 8 mm in the WT and
KO groups, respectively.
Statistical analysis
Using SPSS v25, data were screened for normality and
homogeneity of variance using the Shapiro-Wilk and Levene tests,
respectively. When normality and equal variance was met, a
Student's t-test was used. When normality and equal variance was
not met, a non-parametric Mann-Whitney U test was used. Data were
considered to be statistically significant if P<0.05.
Kaplan-Meier curves were compared by the Xena Browser using the
log-rank test (www.xenabrowser.net).
Results
Microenvironmental stresses regulate
Hilpda levels in KPC cells
We and others have shown that conditions that
increase lipid flux can induce Hilpda protein (17,24,26). To
determine whether similar mechanisms exist in murine pancreatic
tumor cells, we exposed KPC cells to regular normoxic conditions in
DMEM media, to oxygen deprivation (1% O2) or to
exogenous fatty acid (docosahexaenoic acid) for 24 h and examined
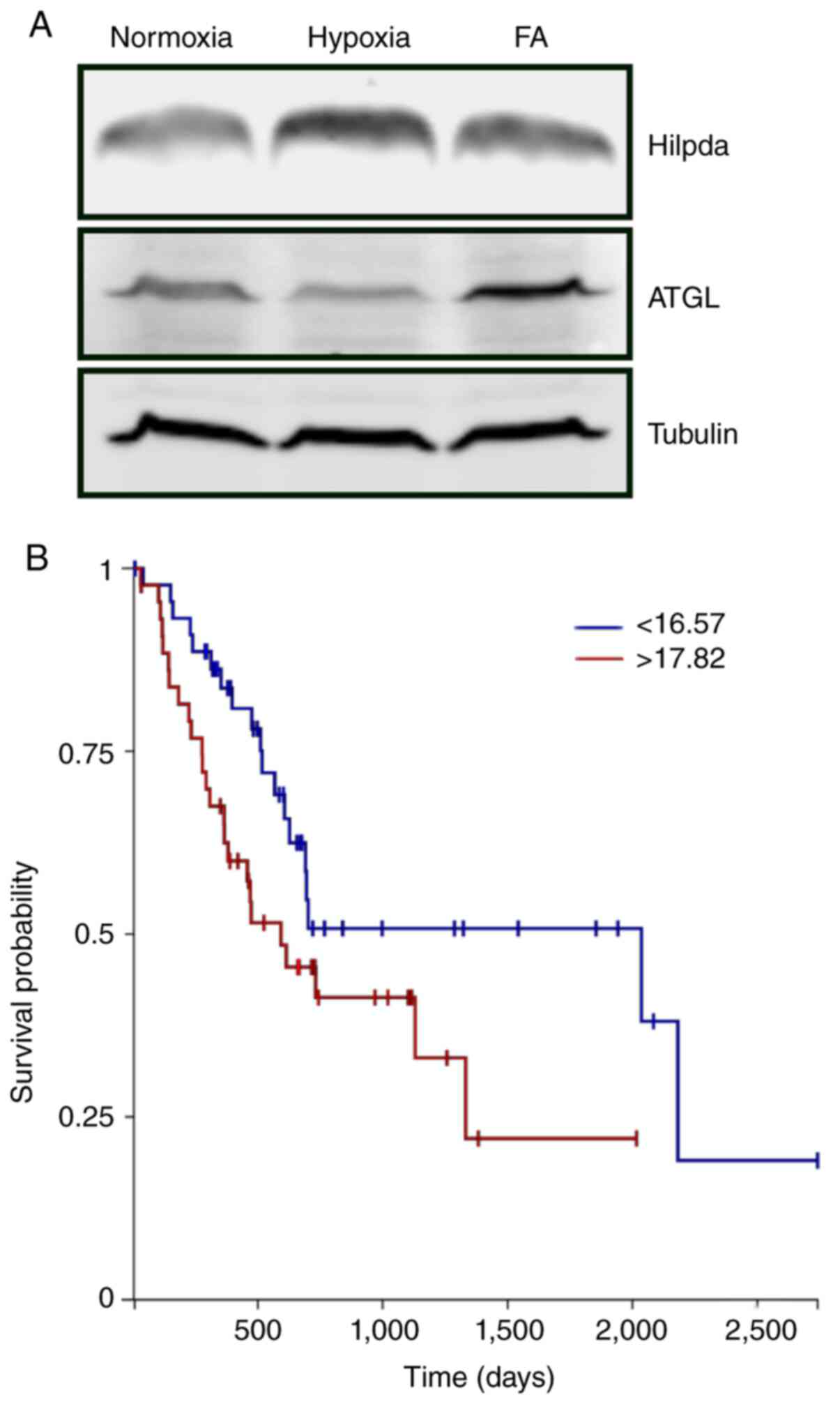
Hilpda protein expression by western blot analysis (Fig. 1A). As has been reported in other tumor
types, both hypoxia and fatty acid loading increased Hilpda levels
in KPC cells. Expression of ATGL was detectable under all
conditions but was not stress-responsive. To evaluate the possible
impact of HILPDA expression in the clinical behavior of human
pancreatic cancers we assessed the TCGA PDAC dataset using the Xena
functional genomics explorer (xenabrowser.net). PDAC tumors with the highest
quartile HILPDA expression had a significantly shorter overall
survival than those with the lowest expression (Fig. 1B), suggesting that HILPDA may be
associated with more aggressive cancers.
Hilpda promotes LD abundance
We genetically manipulated Hilpda in KPC cells and
asked whether it is necessary or sufficient for LD growth in
vitro under different growth conditions. The impact of Hilpda
on the ability of cells to form lipid droplets appears to be
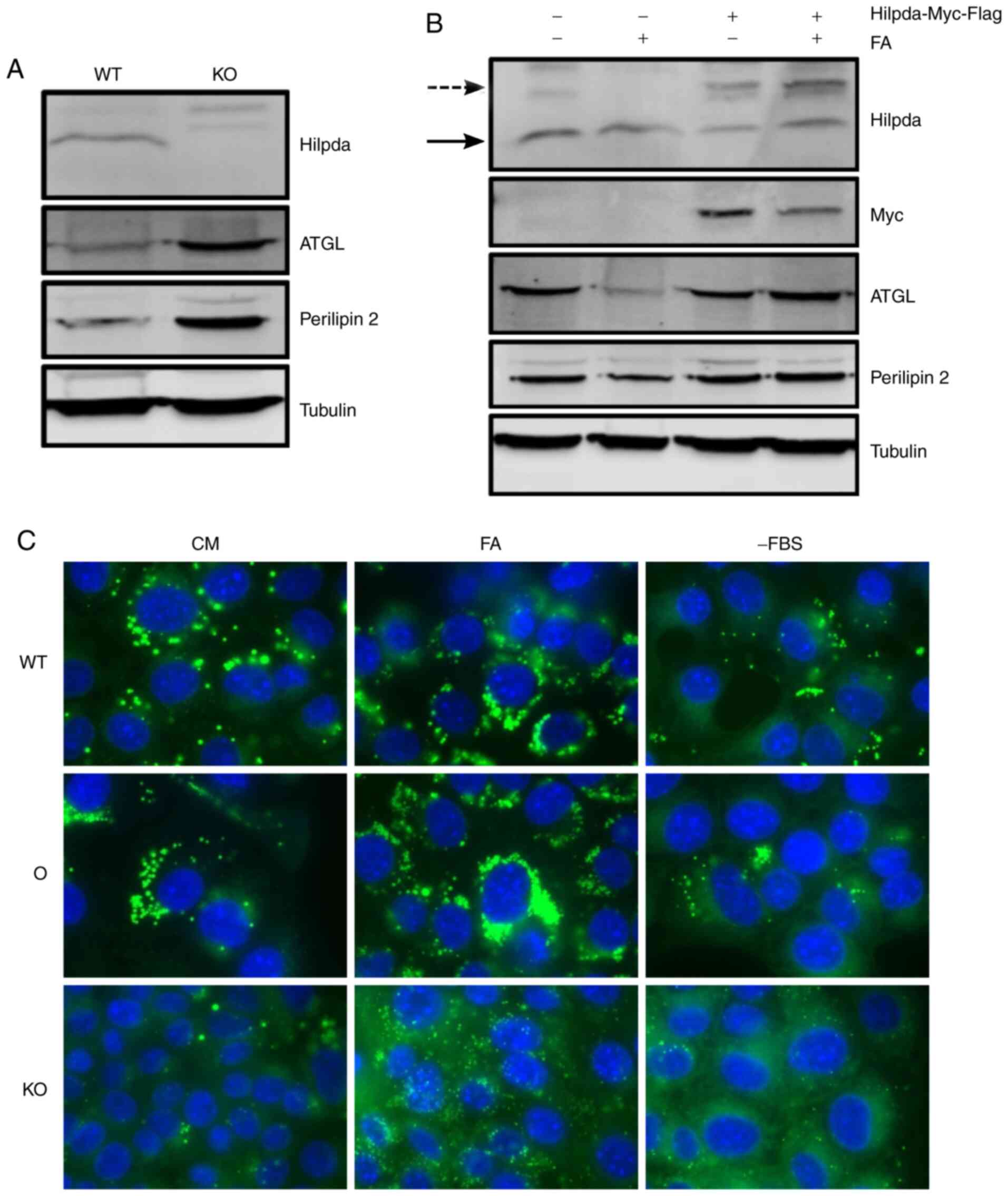
cell-type specific. First, Hilpda KO cells were generated by
CRISPR-Cas9 gene editing and single clones were screened for
successful gene deletion (data not shown). A pool of 7 KO clones
was established and loss of Hilpda protein expression was confirmed
(Fig. 2A). In parallel, a KPC cell
line stably overexpressing myc-Flag tagged Hilpda driven by the CMV
promoter was generated (Fig. 2B).
Next, we determined whether the engineered cells have perturbations
in LD dynamics. Cells were incubated in different nutritional
states that are known to increase lipid flux: Exogenous fatty acid
supplementation and lipid deprivation through serum removal. After
24 h, cells were fixed with 4% paraformaldehyde and lipid droplets
were visualized by fluorescence microscopy after staining with the
neutral lipid dye Nile Red. Hilpda overexpression led to an
increase in LD abundance compared to empty vector cells, under all
conditions. Conversely, under all environmental conditions, the
Hilpda KO cells had smaller and fewer lipid droplets,
suggesting that Hilpda positively regulates lipid droplet
abundance.
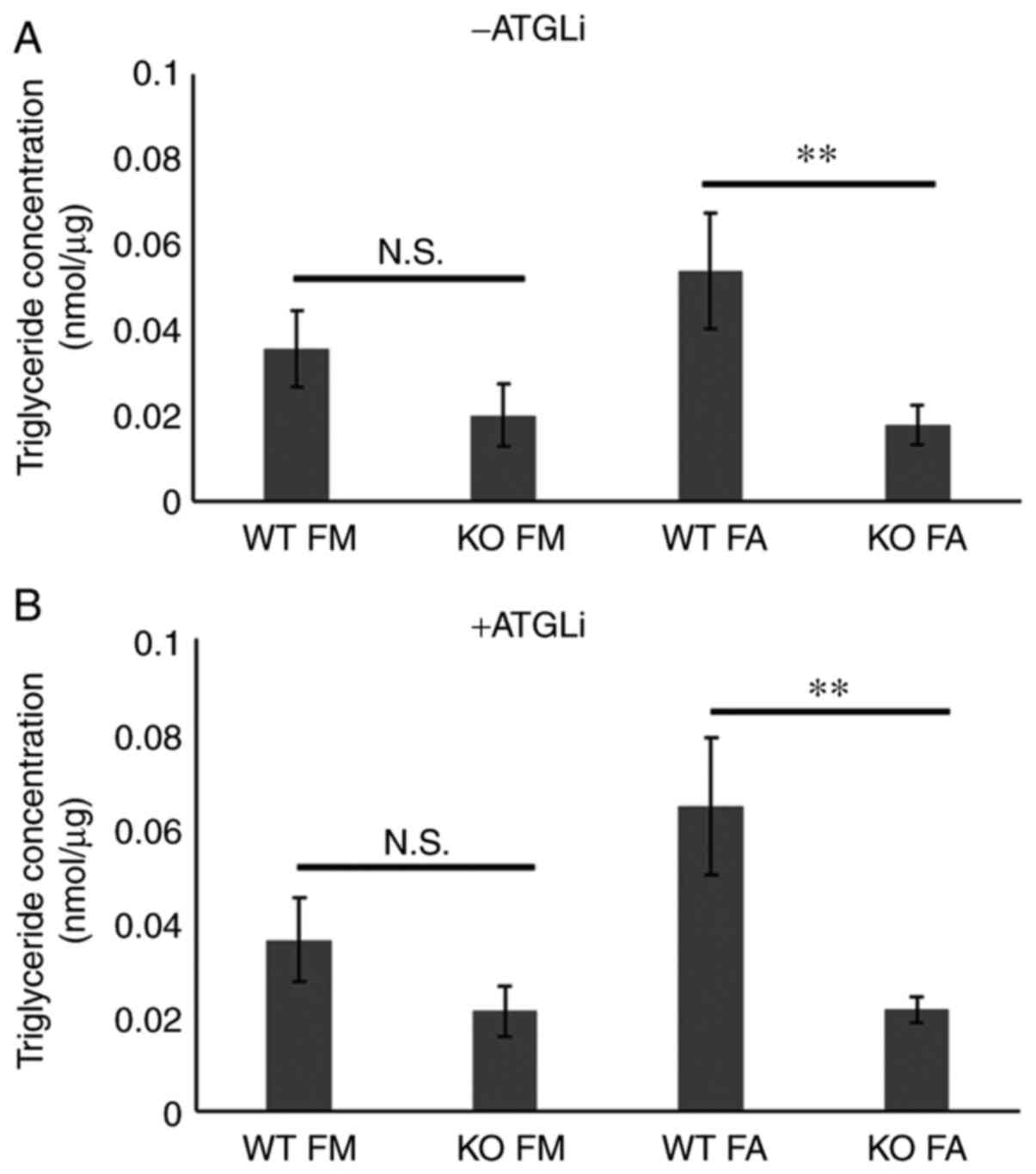
Hilpda promotes triglyceride storage in KPC cells,
independently of ATGL inhibition. Qualitative and quantitative
differences in the constitution of LD's neutral lipid core have
been identified (28). To ascertain
whether the differences in LD abundance caused by Hilpda loss in
KPC results from deregulated triglyceride metabolism, we
biochemically quantified triglyceride levels under basal- and fatty
acid-loaded conditions (Fig. 3A). In
accordance with the LD staining results, the Hilpda KO cells were
significantly impaired in their maximum triglyceride storage
capacity. In basal conditions there was a trend towards lower
triglycerides in the KOs but did not reach statistical
significance.
We and others have shown that in certain tissue
contexts Hilpda promotes triglyceride storage by inhibiting the
rate-limiting lipase ATGL/PNPLA2 (24–26). In
order to establish if Hilpda functions as a molecular ATGL
inhibitor in murine pancreatic tumors, we pharmacologically
inhibited ATGL in Hilpda WT and KO cells with the small molecule
inhibitor ATGListatin (ATGLi), and quantified triglycerides
(Fig. 3B). Notably, the chemical
inhibition of ATGL was not able to correct the defect in the KOs
and to restore their triglyceride content to the level of the
Hilpda WT cells. This finding suggests that, in KPC cells,
decreased lipid storage following Hilpda ablation is not caused by
elevated ATGL activity and enhanced lipolysis.
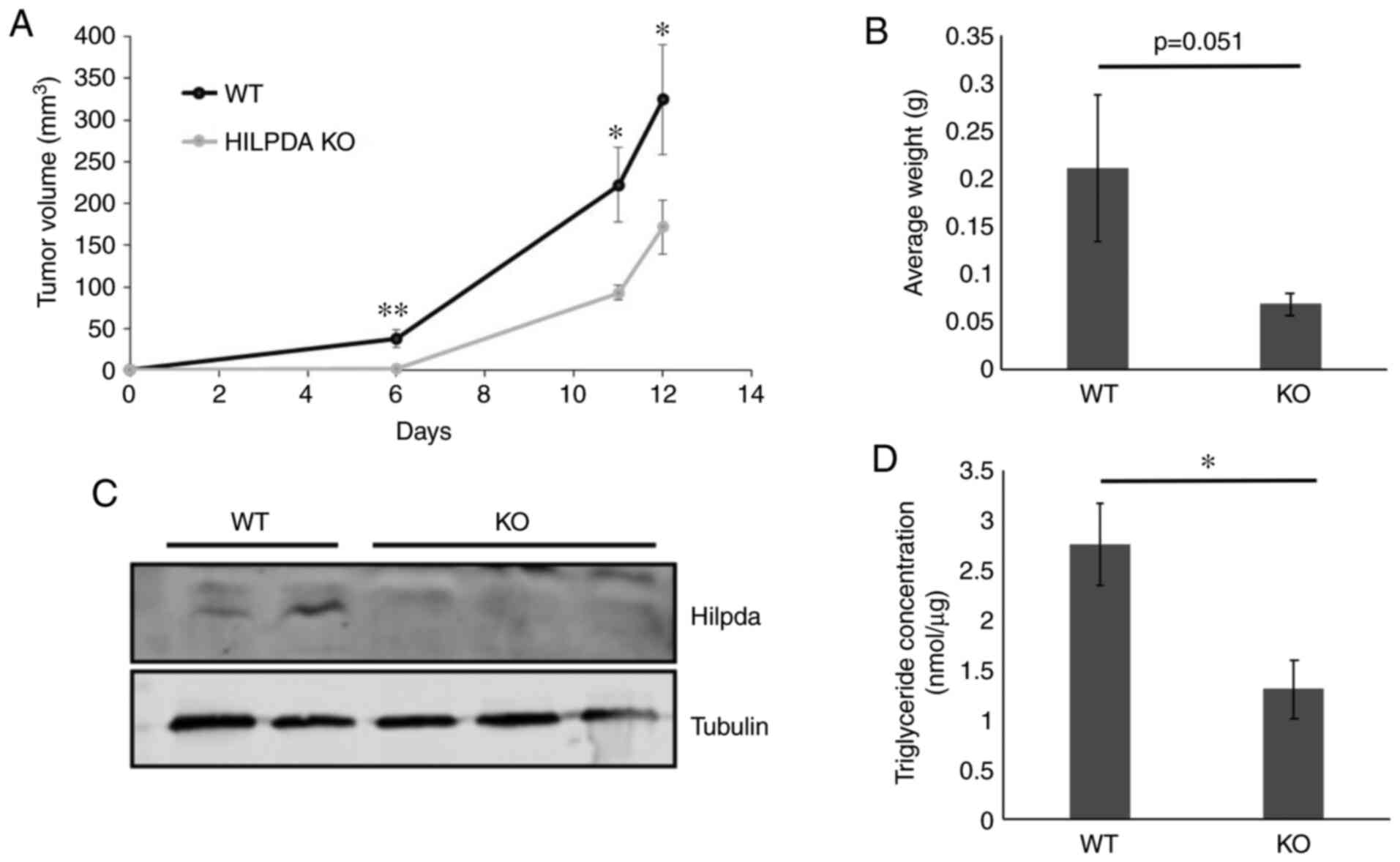
Hilpda promotes model tumor growth. Loss of
Hilpda-dependent ATGL regulation has been shown to be growth
inhibitory in model tumors (24,26). Owing
to the in vitro findings of ATGL-independent Hilpda
functions in KPC we examined whether Hilpda exerts tumor-promoting
properties in pancreatic cancer xenografts. WT and KO cells were
injected subcutaneously into the backs of nude mice (11 mice per
genotype) and tumor sizes were measured with calipers. The results
showed that loss of Hilpda significantly decreased the growth rate
of KPC tumors, suggesting that Hilpda can positively regulate tumor
growth, independently of lipolytic control (Fig. 4A and B). At the completion of the
xenograft growth, we excised the tumors and confirmed their Hilpda
genotype by western blot analysis (Fig.
4C) and measured their triglyceride content (P<0.05)
(Fig. 4D). The WT tumors contained
two times more triglycerides than the KOs, indicating that tumor
microenvironmental conditions, such as hypoxia, stimulate
Hilpda-dependent lipid storage.
Discussion
Rewiring of lipid flux pathways is a common feature
of malignancies and has important biological and clinical
implications. In the context of Ras-driven cancers, inhibition of
Fatty Acid Synthase (FASN) impaired growth, suggesting an active
pathway of de novo fatty acid synthesis (29,30). Other
reports have shown an increase in exogenous lipid uptake, storage,
and utilization, as mechanisms that support cell growth and
malignant progression (9,31). Although the source of lipids may
depend on many genetic and experimental factors, hypoxia and
nutrient availability in the tumor microenvironment can shift the
balance towards storage of esterified lipids (32), in part through the upregulation of
LD-associated proteins.
A key question surrounding the pro-tumorigenic
effects of LDs is how they can protect from cell death or promote
proliferation. Several biological mechanisms that mitigate, through
LD dynamics, nutrient fluctuations in the tumor microenvironment
have been identified. These include protection from oxidative
stress during reoxygenation after hypoxia (33), from membrane disruption and ER stress
(34,35), protection of mitochondrial integrity
and function during starvation (36–38), and
sequestration of death-inducing fatty acid metabolites (39,40).
In particular, HILPDA expression is regulated by
both hypoxia and fatty acid supplementation (41). In turn, that determines the
biochemical composition of LD content and promotes tumor growth
in vivo. In the present study, we confirmed that Hilpda
expression is induced by hypoxia in murine pancreatic cells, as has
been shown for other anatomical sites (17,24,26). In
agreement with previous studies conducted by us and others on other
model systems, HILPDA ablation significantly decreased triglyceride
content and retarded KPC xenograft tumor growth (24,26,42).
Previously, we have shown that uncontrolled ATGL activity is
responsible for triglyceride loss after Hilpda ablation in MEFs and
colorectal cancer models (26);
however, our data suggest that in pancreatic cancer Hilpda's major
biological mechanism does not involve inhibition of ATGL-initiated
lipolysis. This explanation is based on the inability of a specific
ATGL inhibitor to restore LD abundance in the HILPDA-deficient
cells. Interestingly, a recent preprint provides evidence for a
novel function of Hilpda as a positive regulator of triglyceride
synthesis, via the stimulation of DGAT1 activity (43). Based on this, it may be speculated
that, in murine pancreatic cancers, Hilpda is involved in the
growth of LDs rather than their shrinkage. The precise mechanism
for Hilpda-dependent lipid deposition may depend on the balance of
fatty acid uptake, triglyceride biosynthesis and hydrolysis in
different cell types and the presence of interacting partners
and/or of signals that direct Hilpda's localization in specific
subcellular compartments or LD subpopulations.
Acknowledgements
We would like to thank Erich Auer for technical
assistance during the early stages of the study.
Funding
This study was in part supported by NCI awards
CA191653 (I.P.) and CA197713 (A.J.G.). The OSUCCC shared resources
are supported by Cancer Center Support Grant CA016058. NIH had no
role in the study design, data generation, the writing of this
report or the decision to submit it for publication.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JJG and MK contributed to data acquisition and
analysis. AJG contributed to study design and funding. NCD
substantially contributed to the study design. IP contributed to
study design, data analysis, funding, manuscript preparation. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ohio
State University's Institutional Animal Care and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koong AC, Mehta VK, Le QT, Fisher GA,
Terris DJ, Brown JM, Bastidas AJ and Vierra M: Pancreatic tumors
show high levels of hypoxia. Int J Radiat Oncol Biol Phys.
48:919–922. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stromnes IM, Hulbert A, Pierce RH,
Greenberg PD and Hingorani SR: T-cell localization, activation, and
clonal expansion in human pancreatic ductal adenocarcinoma. Cancer
Immunol Res. 5:978–991. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biankin AV, Waddell N, Kassahn KS, Gingras
MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J,
et al: Pancreatic cancer genomes reveal aberrations in axon
guidance pathway genes. Nature. 491:399–405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kamphorst JJ, Nofal M, Commisso C, Hackett
SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA,
Bar-Sagi D, et al: Human pancreatic cancer tumors are nutrient poor
and tumor cells actively scavenge extracellular protein. Cancer
Res. 75:544–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Palm W, Park Y, Wright K, Pavlova NN,
Tuveson DA and Thompson CB: The utilization of extracellular
proteins as nutrients is suppressed by mTORC1. Cell. 162:259–270.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamphorst JJ, Cross JR, Fan J, de
Stanchina E, Mathew R, White EP, Thompson CB and Rabinowitz JD:
Hypoxic and Ras-transformed cells support growth by scavenging
unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci
USA. 110:8882–8887. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma X, Zhao X, Ouyang H, Sun F, Zhang H,
Zhou C and Shen H: The metabolic features of normal pancreas and
pancreatic adenocarcinoma: Preliminary result of in vivo proton
magnetic resonance spectroscopy at 3.0 T. J Comput Assist Tomogr.
35:539–544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yabushita S, Fukamachi K, Tanaka H, Fukuda
T, Sumida K, Deguchi Y, Mikata K, Nishioka K, Kawamura S, Uwagawa
S, et al: Metabolomic and transcriptomic profiling of human K-ras
oncogene transgenic rats with pancreatic ductal adenocarcinomas.
Carcinogenesis. 34:1251–1259. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang G, He P, Tan H, Budhu A, Gaedcke J,
Ghadimi BM, Ried T, Yfantis HG, Lee DH, Maitra A, et al:
Integration of metabolomics and transcriptomics revealed a fatty
acid network exerting growth inhibitory effects in human pancreatic
cancer. Clinical Cancer Res. 19:4983–4993. 2013. View Article : Google Scholar
|
|
13
|
Guillaumond F, Bidaut G, Ouaissi M,
Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet
O, et al: Cholesterol uptake disruption, in association with
chemotherapy, is a promising combined metabolic therapy for
pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 112:2473–2478.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gaglio D, Metallo CM, Gameiro PA, Hiller
K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G and
Chiaradonna F: Oncogenic K-Ras decouples glucose and glutamine
metabolism to support cancer cell growth. Mol Syst Biol. 7:5232011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodman JM: The gregarious lipid droplet.
J Biol Chem. 283:28005–28009. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Denko N, Schindler C, Koong A, Laderoute
K, Green C and Giaccia A: Epigenetic regulation of gene expression
in cervical cancer cells by the tumor microenvironment. Clin Cancer
Res. 6:480–487. 2000.PubMed/NCBI
|
|
17
|
Gimm T, Wiese M, Teschemacher B, Deggerich
A, Schodel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K,
Wiesener MS, et al: Hypoxia-inducible protein 2 is a novel lipid
droplet protein and a specific target gene of hypoxia-inducible
factor-1. FASEB J. 24:4443–4458. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mattijssen F, Georgiadi A, Andasarie T,
Szalowska E, Zota A, Krones-Herzig A, Heier C, Ratman D, De
Bosscher K, Qi L, et al: Hypoxia-inducible lipid droplet-associated
(HILPDA) is a novel peroxisome proliferator-activated receptor
(PPAR) target involved in hepatic triglyceride secretion. J Biol
Chem. 289:19279–19293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
DiStefano MT, Danai LV, Roth Flach RJ,
Chawla A, Pedersen DJ, Guilherme A and Czech MP: The lipid droplet
protein hypoxia-inducible gene 2 promotes hepatic triglyceride
deposition by inhibiting lipolysis. J Biol Chem. 290:15175–15184.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maier A, Wu H, Cordasic N, Oefner P,
Dietel B, Thiele C, Weidemann A, Eckardt KU and Warnecke C:
Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid
accumulation in macrophages and contributes to atherosclerosis in
apolipoprotein E-deficient mice. FASEB J. 31:4971–4984. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
VandeKopple MJ, Wu J, Baer LA, Bal NC,
Maurya SK, Kalyanasundaram A, Periasamy M, Stanford KI, Giaccia AJ,
Denko NC and Papandreou I: Stress-responsive HILPDA is necessary
for thermoregulation during fasting. J Endocrinol. 235:27–38. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DiStefano MT, Roth Flach RJ, Senol-Cosar
O, Danai LV, Virbasius JV, Nicoloro SM, Straubhaar J, Dagdeviren S,
Wabitsch M, Gupta OT, et al: Adipocyte-specific Hypoxia-inducible
gene 2 promotes fat deposition and diet-induced insulin resistance.
Mol Metab. 5:1149–1161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dijk W, Mattijssen F, de la Rosa Rodriguez
M, Loza Valdes A, Loft A, Mandrup S, Kalkhoven E, Qi L, Borst JW
and Kersten S: Hypoxia-inducible lipid droplet-associated is not a
direct physiological regulator of lipolysis in adipose tissue.
Endocrinology. 158:1231–1251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Saarinen AM, Hitosugi T, Wang Z,
Wang L, Ho TH and Liu J: Inhibition of intracellular lipolysis
promotes human cancer cell adaptation to hypoxia. Elife.
6:e311322017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Padmanabha Das KM, Wechselberger L,
Liziczai M, De la Rosa Rodriguez M, Grabner GF, Heier C, Viertlmayr
R, Radler C, Lichtenegger J, Zimmermann R, et al: Hypoxia-inducible
lipid droplet-associated protein inhibits adipose triglyceride
lipase. J Lipid Res. 59:531–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
VandeKopple MJ, Wu J, Auer EN, Giaccia AJ,
Denko NC and Papandreou I: HILPDA regulates lipid metabolism, lipid
droplet abundance, and response to microenvironmental stress in
solid tumors. Mol Cancer Res. 17:2089–2101. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hingorani SR, Wang L, Multani AS, Combs C,
Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA:
Trp53R172H and KrasG12D cooperate to promote chromosomal
instability and widely metastatic pancreatic ductal adenocarcinoma
in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khor VK, Ahrends R, Lin Y, Shen WJ, Adams
CM, Roseman AN, Cortez Y, Teruel MN, Azhar S and Kraemer FB: The
proteome of cholesteryl-ester-enriched versus
triacylglycerol-enriched lipid droplets. PLoS One. 9:e1050472014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bian Y, Yu Y, Wang S and Li L:
Up-regulation of fatty acid synthase induced by EGFR/ERK activation
promotes tumor growth in pancreatic cancer. Biochem Biophys Res
Commun. 463:612–617. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh A, Ruiz C, Bhalla K, Haley JA, Li
QK, Acquaah-Mensah G, Montal E, Sudini KR, Skoulidis F, Wistuba II,
et al: De novo lipogenesis represents a therapeutic target in
mutant Kras non-small cell lung cancer. FASEB J.
32:fj2018002042018. View Article : Google Scholar
|
|
31
|
Qiao S, Koh SB, Vivekanandan V, Salunke D,
Patra KC, Zaganjor E, Ross K, Mizukami Y, Jeanfavre S, Chen A, et
al: REDD1 loss reprograms lipid metabolism to drive progression of
RAS mutant tumors. Genes Dev. 34:751–766. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Downes DP, Daurio NA, McLaren DG,
Carrington P, Previs SF and Williams KB: Impact of extracellular
fatty acids and oxygen tension on lipid synthesis and assembly in
pancreatic cancer cells. ACS Chem Biol. 15:1892–1900. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bensaad K, Favaro E, Lewis CA, Peck B,
Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al:
Fatty acid uptake and lipid storage induced by HIF-1α contribute to
cell growth and survival after hypoxia-reoxygenation. Cell Rep.
9:349–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qiu B, Ackerman D, Sanchez DJ, Li B,
Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl JA, Keith B
and Simon MC: HIF2α-dependent lipid storage promotes endoplasmic
reticulum homeostasis in clear-cell renal cell carcinoma. Cancer
Discov. 5:652–667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ackerman D, Tumanov S, Qiu B,
Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC and Kamphorst
JJ: Triglycerides promote lipid homeostasis during hypoxic stress
by balancing fatty acid saturation. Cell Rep. 24:2596–2605.e5.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rambold AS, Cohen S and
Lippincott-Schwartz J: Fatty acid trafficking in starved cells:
Regulation by lipid droplet lipolysis, autophagy, and mitochondrial
fusion dynamics. Dev Cell. 32:678–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nguyen TB, Louie SM, Daniele JR, Tran Q,
Dillin A, Zoncu R, Nomura DK and Olzmann JA: DGAT1-dependent lipid
droplet biogenesis protects mitochondrial function during
starvation-induced autophagy. Dev Cell. 42:9–21.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herms A, Bosch M, Reddy BJ, Schieber NL,
Fajardo A, Ruperéz C, Fernández-Vidal A, Ferguson C, Rentero C,
Tebar F, et al: AMPK activation promotes lipid droplet dispersion
on detyrosinated microtubules to increase mitochondrial fatty acid
oxidation. Nat Commun. 6:71762015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Listenberger LL, Han X, Lewis SE, Cases S,
Farese RV Jr, Ory DS and Schaffer JE: Triglyceride accumulation
protects against fatty acid-induced lipotoxicity. Proc Natl Acad
Sci USA. 100:3077–3082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Senkal CE, Salama MF, Snider AJ, Allopenna
JJ, Rana NA, Koller A, Hannun YA and Obeid LM: Ceramide is
metabolized to acylceramide and stored in lipid droplets. Cell
Metab. 25:686–697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de la Rosa Rodriguez MA and Kersten S:
Regulation of lipid droplet homeostasis by hypoxia inducible lipid
droplet associated HILPDA. Biochim Biophys Acta Mol Cell Biol
Lipids. 1865:1587382020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim SH, Wang D, Park YY, Katoh H, Margalit
O, Sheffer M, Wu H, Holla VR, Lee JS and DuBois RN: HIG2 promotes
colorectal cancer progression via hypoxia-dependent and independent
pathways. Cancer Lett. 341:159–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
de la Rosa Rodriguez MA, Gemmink A, van
Weeghel M, Aoun ML, Singh R, Borst JW and Kersten S:
Hypoxia-inducible lipid droplet-associated interacts with DGAT1 to
promote lipid storage in hepatocytes. bioRxiv. Feb 27–2020.(Epub
ahead of print). doi: org/10.1101/2020.02.26.966374.
|