Introduction
Breast cancer, a primary cause of female mortality
(1–3),
is characterized by a high mortality rate owing to the invasive and
metastatic potential of cancer cells (4). In 2018 worldwide, the incidence of
breast cancer was 24.2%, and the invasive cancer rate was 11.6% in
woman cancer patents (2).
Furthermore, in Korea in 2017, the incidence of breast cancer was
20.3%, and the invasive cancer rate was 15.6% in woman cancer
patents (3). One of the primary
therapeutic approaches against breast cancer metastasis involves
the development of effective anti-invasive agents (5,6). Cancer
cell invasion, induced by extracellular matrix (ECM) degradation,
initiates the metastatic process; the cancer cells find their way
through adjacent tissues, invade blood vessels, and move along the
vessel walls to migrate to other organs (7,8). ECM
degradation is caused by various extracellular proteases, of which
matrix metalloproteinases (MMPs) play a crucial role in breast
cancer (7,8).
Bruton's tyrosine kinase (BTK) is a member of the
Tec family of cytoplasmic tyrosine kinases in the B-cell receptor
signaling pathway and a driving force for CLL and other B-cell
malignancies (9–12). BTK, a multidomain protein, can
interact with and activate other molecules, including phospholipase
C (PLC) γ2 (13–15). PLCγ2, a member of
phosphoinositide-specific PLCs, enhances the activation of protein
kinase C (PKC) by catalyzing the degradation of
phosphatidylinositol-4,5-bisphosphate (PIP2) into
diacylglycerol (DAG) and inositol-3,4,5-trisphosphate
(IP3). IP3 induces the elevation of
intracellular calcium levels (16).
Activated PKC, in turn, induces the activation of mitogen-activated
protein kinase (MAPK) signaling, which promotes cell proliferation,
viability, apoptosis, and metastasis (17,18).
Previous research on BTK in breast cancer cells has mainly focused
on its role in the regulation of cell viability (19,20). The
effects of BTK on the invasion and metastasis of breast cancer
cells and its signaling mechanism remain unclear.
Matrix metalloproteinases (MMPs) are a family of
zinc-dependent endopeptidases that can be divided into six
subclasses: Collagenases, stromelysins, gelatinases, matrilysins,
membrane-associated MMPs, and other MMPs (21). MMP-9 has been identified as a crucial
MMP involved in cancer cell invasion, and it is directly linked to
the induction of cancer cell metastasis and poor prognosis in
cancer patients (22,23). MMP-9 expression is induced by various
stimuli, including cytokines, growth factors, and
12-O-tetradecanoylphorbol-13-acetate (TPA) (24–27). In
particular, TPA significantly stimulates MMP synthesis and
secretion by activating PKC (28–30).
TPA-induced MMP-9 expression is triggered by the activation of
transcription factors, including nuclear factor-κB (NF-κB) and
activator protein-1 (AP-1) (31,32). NF-κB
and AP-1 play pivotal roles in TPA-induced MMP-9 expression in
breast cancer cell metastasis and are regulated by MAPKs (33–35). The
signaling pathways mediated by MAPKs activate IκB kinase (IKK),
extracellular signal-regulated kinase (ERK), c-Jun N-terminal
kinase (JNK), or p38 MAPK, depending on the cell type (24,36–38).
Findings of those studies highlight the potential importance of
suppressing MMP-9 expression or its upstream regulatory pathways
for the treatment of metastasis in breast cancer.
In this study, we investigated the regulatory
effects of BTK inhibitors on PKC-mediated MMP-9 expression and
invasion in MCF-7 cells. Our data demonstrated that BTK inhibitors
suppressed TPA-induced MMP-9 expression by blocking MAPK/NF-κB/AP-1
signal transduction via the PLCγ2/PKC pathway. Thus, the data
confirmed that BTK inhibitors suppress MCF-7 cell metastasis by
regulating MMP-9 expression.
Materials and methods
Cell line and culture
MCF-7 human breast cancer cells were purchased from
American Type Culture Collection (cat. no. HTB-22). MCF-7 cells
were cultured in high-glucose Dulbecco's modified Eagle's medium
(DMEM; WelGENE Inc.) supplemented with 10% fetal bovine serum (Life
Technologies; cat. no. 160000-044) and 1% antibiotics (anti-anti).
Cultures were maintained in a humidified incubator with 5%
CO2 at 37°C.
Reagents
The BTK inhibitor, ibrutinib was purchased from
Wuhan NCE Biomedical Co., Ltd., while CNX-774 was purchased from
Selleck Chemicals. TPA (P1585) and dimethyl sulfoxide (DMSO; cat.
no. 472301) were obtained from Sigma-Aldrich; Merck KGaA. Matrigel
was obtained from Corning Inc. (cat. no. 356234). Before cell
treatment, each reagent was dissolved in DMSO.
Measurement of cell viability
The viability of MCF-7 cells was assessed as
previously described (39). Cell
viability was analyzed using the EZ-Cytox reagent Cell Viability
Assay Kit (DoGen) following the manufacturer's instructions. MCF-7
cells (3×104) well were seeded onto 96-well plates,
treated with the indicated concentrations of BTK inhibitors, and
incubated for 24 h at 37°C in a humidified atmosphere containing 5%
CO2. After 24 h, EZ-cytox reagent solution (10 µl) was
added to each well of the 96-well plate, and the cells were further
incubated for 4 h at 37°C. The absorbance was measured at 450 nm
using a Sunrise™ ELISA reader (Tecan). The optical density of the
control was considered 100%.
Western blot analysis
MCF-7 cells (7×105) were pretreated with
10 µM of either of the two BTK inhibitors for 1 h and then
incubated with TPA for 24 h at 37°C. Total protein was extracted
using RIPA buffer (Thermo Fisher Scientific) containing protease
and phosphatase inhibitors (Calbiochem), as previously described
(39). The lysates were then
centrifuged at 16,000 × g for 10 min at 4°C, and the protein
concentrations in the lysates were assessed using a BioSpec-nano
device (Shimadzu). Equal quantities of protein (10 µg) were
resolved using 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to Hybond™ polyvinylidene fluoride
membranes (GE Healthcare Life Sciences) using a western blot
apparatus. Each membrane was blocked for 2 h with 5% bovine serum
albumin (BSA) or 5% skim milk in TTBS with Tween-20, and the blots
were incubated overnight at 4°C with primary antibodies (1:2,500
dilution); anti-β-actin antibodies were obtained from
Sigma-Aldrich; Merck KGaA. Primary antibodies (1:2,500 dilution)
against the following antigens were used: p38 (cat. no. 9212), JNK
(cat. no. 9252), ERK (cat. no. 9102), IKKα (cat. no. 2682) and IKKβ
(cat. no. 2678), phosphorylated forms of p38 (cat. no. 9211), JNK
(cat. no. 9251), and ERK (cat. no. 9101); c-Jun (cat. no. 9261);
inhibitory subunit of NF-κBα (IκBα; cat. no. 2859); IKKαβ (cat. no.
2697); and PLCγ2 (cat. no. 3874). These were obtained from Cell
Signaling Technology (Beverly). Polyclonal antibodies (1:2,500
dilution) against p50 (SC-7178), p65 (SC-372), MMP-9 (SC-12759),
IκBα (SC-371), PLC γ2 (SC-5283) and proliferating cell nuclear
antigen (PCNA) were obtained from Santa Cruz Biotechnology, Inc.
Antibodies (1:2,500 dilution) against PKCα (ab32376), PKCβ
(ab32026), and PKCδ (ab182126) were purchased from Abcam. The blots
were washed in TBS with 0.2% Tween-20 (TBST) and secondary
horseradish peroxidase-conjugated goat anti-mouse (cat. no.
sc-2005; Santa Cruz Biotechnology, Inc.) or anti-rabbit (cat. no.
sc-2004; Santa Cruz Biotechnology, Inc.) antibody (1:2,500
dilution) at 4°C for 1 h. The protein bands were detected by HRP
Substrate Luminol Reagent (EMD Millipore Corporation), and protein
expression levels were determined using a Mini HD6 image analyzer
(UVItec). The blot was re-probed with an anti-β-actin antibody to
confirm equal loading.
Gelatin zymography analysis
MMP-9 activity was measured by gelatin zymography,
as previously described previously (39). MCF-7 cells (7×105) were
pretreated with either of the two BTK inhibitors (10 µM) in
serum-free medium for 1 h and then incubated with TPA (20 µM) for
24 h at 37°C. The conditioned medium was collected after 24 h of
stimulation, cleared by centrifugation at 1,500 × g for 4 min at
4°C, mixed with non-reducing sample buffer, and electrophoresed on
a polyacrylamide gel containing 0.1% (w/v) gelatin. Following
electrophoresis, the gel was washed at room temperature for 30 min
with 2.5% Triton X-100, followed by incubation at 37°C for 16 h in
5 mM CaCl2, 0.02% Brij-35, and 50 mM Tris-HCl (pH 7.5).
Finally, the gels were stained with 0.25% (w/v) Coomassie Brilliant
Blue in 40% (v/v) methanol and 7% (v/v) acetic acid. Proteolysis
was detected as a white zone in a dark blue field using a digital
imaging system (Cell Biosciences).
RNA isolation and quantitative
polymerase chain reaction (PCR)
This method was performed as previously described
(39). Total RNA was isolated from
MCF-7 cells using TRIzol reagent (Invitrogen) according to the
manufacturer's instructions. cDNA (1 µg) was synthesized using a
High Capacity cDNA synthesis kit from PrimeScript™ RT Reagent Kit
(Takara). mRNA levels were determined by quantitative PCR analysis
using the StepOnePlus™ Real-time PCR System and SYBR-Green PCR
Master Mix (Applied Biosystems). The PCR cycling conditions were:
Initial denaturation at 95°C for 5 min, followed by 40 cycles of
95°C for 30 sec and 60°C for 30 sec. The primers used were: MMP-9
(NM 004994), 5-CCTGGAGACCTGAGAACCAATCT-3 (forward),
5-CCACCCGAGTGTAACCATAGC-3 (reverse); and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH; NM 002046), 5-ATGGAAATCCCATCACCATCTT-3
(forward), and 5-CGCCCCACTTGATTTTGG-3 (reverse). Gene expression
data were normalized to the expression levels of the housekeeping
gene GAPDH. Relative quantitation was performed using the
comparative ∆∆Ct method according to the manufacturer's
instructions (40).
Preparation of nuclear extracts
This method was performed as previously described
(39). MCF-7 cells (2×106)
were treated with either BTK inhibitor and then incubated with TPA
for 3 h at 37°C. The cells were then washed twice with ice-cold
PBS, resuspended in 1.5 ml ice-cold PBS (pH 7.5), and centrifuged
at 1,500 × g for 4 min at 4°C. Cytoplasmic and nuclear fractions
were prepared using the NE-PER Nuclear and Cytoplasmic Extraction
Kit (Thermo Fisher Scientific).
Membrane fractionation
MCF-7 cells (5×107) were treated with 10
µM of either BTK inhibitor for 1 h and then incubated with TPA for
1 h at 37°C. The cells were immediately washed twice, scraped in
1.5 ml of ice-cold PBS (pH 7.5), and pelleted at 300 × g for 3 min
at 4°C. After incubation on ice for 30 min, the cells were lysed in
homogenization buffer (20 mM Tris-HCl, 5 mM dithiothreitol, 2 mM
EDTA, 5 mM EGTA, and protease and phosphatase inhibitors; pH 7.5)
with brief sonication (5 times for 10 sec, each at 10% amplitude).
The resulting cell lysate was centrifuged at 16,000 × g for 15 min
at 4°C to separate the soluble (cytosolic) and pellet (membrane)
fractions. The pellet was resuspended in solubilization buffer and
incubated on ice for 30 min. The suspension was centrifuged at
16,000 × g for 15 min at 4°C. The supernatant was then collected as
the membrane fraction.
Dual luciferase reporter assay
MCF-7 cells (1×105) were seeded onto
24-well plates. Cells were transfected with NF-κB or AP-1 reporter
and Renilla luciferase thymidine kinase reporter vector were
co-transfected using Lipofectamine 2000 (Invitrogen) at 70–80%
confluency. The transfected cells were pretreated with either BTK
inhibitor at the indicated concentration for 1 h and then treated
with 100 nM TPA at 37°C. Whole-cell lysates were prepared, and
luciferase activity was measured using a Dual-Luciferase Reporter
Assay System (Promega) and Lumat LB 9507 luminometer (EG&G
Berthold). Firefly luciferase activity was normalized against
Renilla luciferase activity.
Matrigel invasion and migration
assays
This method was performed as previously described
(39). The invasion assay was carried
out in 24-well chambers (8-µm pore size) coated with 20 µl Matrigel
(diluted in DMEM). The Matrigel coating was rehydrated in 0.5 ml
DMEM for 30 min immediately before the experiments. The migration
assay was performed using an insert (8-µm pore size) in 24-well
chambers without Matrigel coating. Cells (3×105) were
added to the upper chamber, and TPA, alone or with 10 µM of either
BTK inhibitor was added to the bottom well. Additionally, cells
(2×105) transfected with BTK small-interfering RNA
(siRNA) were added to the upper chamber, and TPA was added to the
bottom well. The chambers were incubated for 24 h at 37°C in an
atmosphere containing 5% CO2. After 24-h incubation, the
cells on the upper surface of the chamber were removed with cotton
swabs, and cells that had migrated were fixed and stained with 0.2%
crystal violet for 30 min at room temperature and rinsed with water
to remove any unbound dye. The membranes were dried, and invading
cells were counted in five random areas of the membrane under a
light microscope.
Statistical analysis
Data are presented as the mean ± SEM from three in
experiments performed in triplicate. Statistical analysis was
performed using ANOVA with Scheffe post hoc test (SAS software,
version 9.3; SAS Institute Inc.). Differences between groups were
considered statistically significant at P<0.05.
Results
Inhibition of BTK expression
attenuates TPA-induced MMP-9 expression and secretion in MCF-7
cells
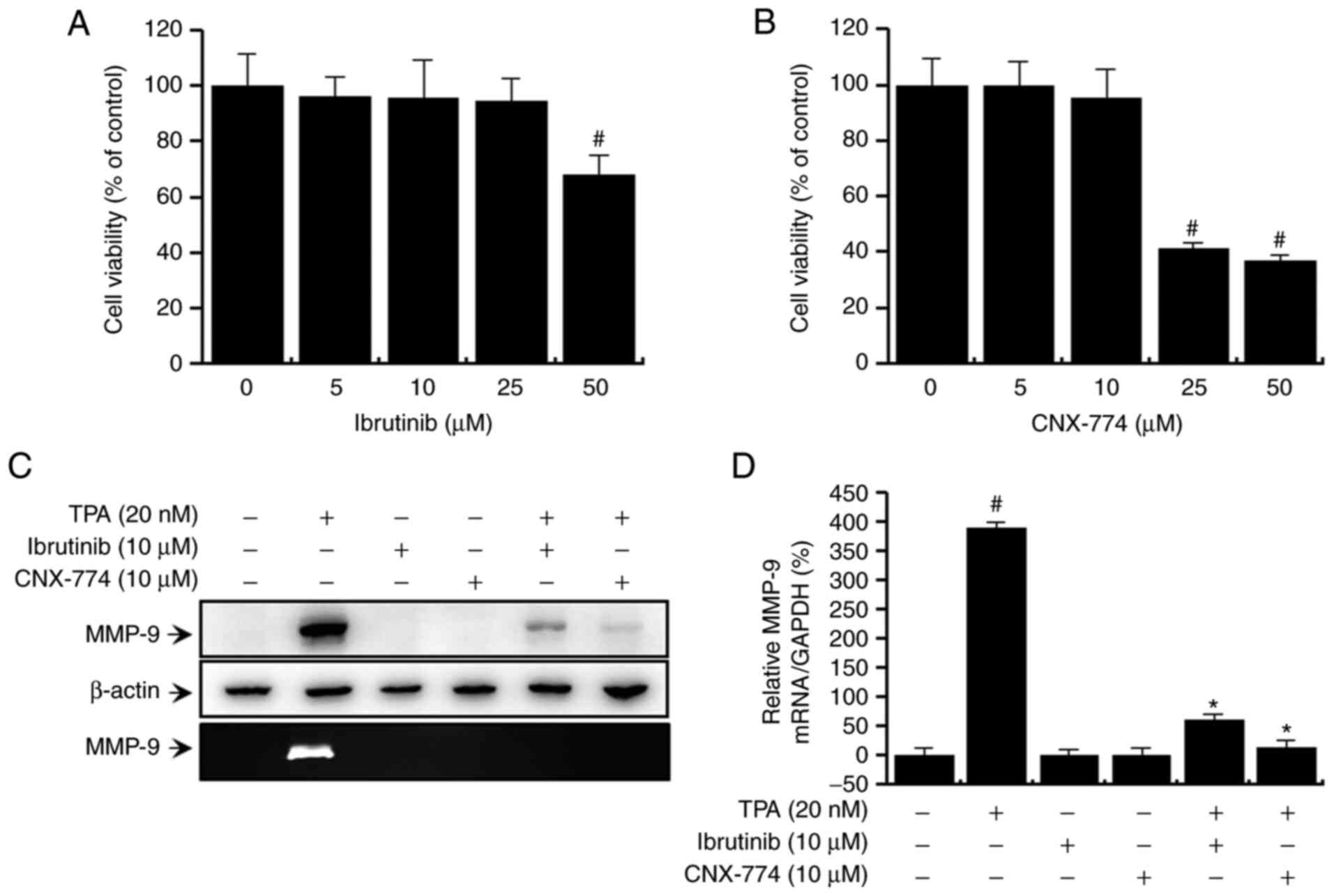
First, we evaluated the potential cytotoxic effects
of the BTK inhibitors, ibrutinib and CNX-774, on MCF-7 cells. The
inhibitors did not affect cell viability (Fig. 1A and B) or morphology at the
experimental concentrations used (≤50 µM). Therefore, we chose the
optimal non-toxic concentration (10 µM) of the BTK inhibitors for
subsequent experiments. Next, to validate the effects of BTK on
TPA-induced MMP-9 expression, we performed quantitative PCR
analysis, western blot analysis, and zymography. Western blot
analysis revealed that treatment with ibrutinib and CNX-774
suppressed the TPA-induced upregulation of MMP-9 expression
(Fig. 1C, upper panel). Quantitative
PCR also confirmed that a TPA-induced increase in MMP-9 mRNA
expression in MCF-7 cells was suppressed after treatment with
ibrutinib and CNX-774 (Fig. 1D). The
degree of TPA-induced exocytosis of MMP-9 was assessed by
zymography, which revealed that treatment with ibrutinib and
CNX-774 significantly reduced TPA-induced MMP-9 secretion (Fig. 1C, lower panel). Overall, these
findings show that the inhibition of BTK expression suppresses
TPA-induced MMP-9 mRNA and protein expression while simultaneously
suppressing MMP-9 enzymatic activation.
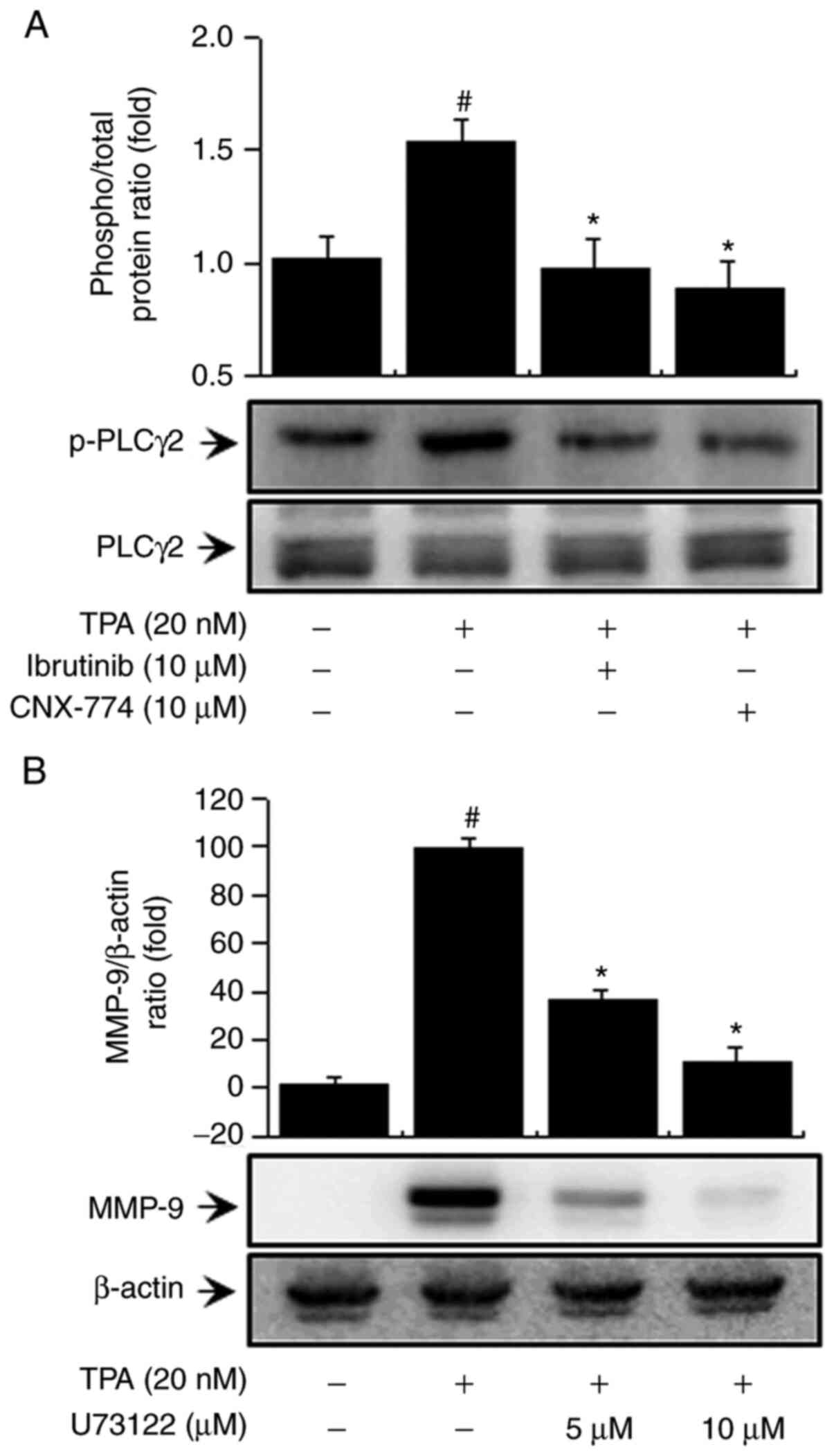
Inhibition of BTK expression regulates
PLC signaling in MCF-7 cells
BTK reportedly regulates the activation of PLCγ2
(13,15). We examined the influence of BTK on PLC
signaling regulation. First, to determine the effects of BTK
inhibitors on PLCγ2 activation in MCF-7 cells, we verified PLCγ2
phosphorylation and confirmed that treatment with ibrutinib and
CNX-774 suppressed PLCγ2 phosphorylation (Fig. 2A). In addition, to validate the
regulatory effects of PLC on MMP-9 expression, we treated the cells
with U-73122, a PLC inhibitor, and confirmed that it suppressed
MMP-9 expression (Fig. 2B). This
result suggests that PLCγ2 regulates MMP-9 expression by inhibiting
BTK expression in MCF-7 cells.
Inhibition of BTK expression regulates
the TPA-induced PKC/MAPK/IKK signaling pathway in MCF-7 cells
In previous studies, we have demonstrated that PKC
is involved in TPA-induced MMP-9 expression via MAPK and IKK
signaling (31,32). In the current study, we aimed to
validate the effects of BTK on the PKC, MAPK, and IKK-mediated
signaling pathways. First, we assessed PKC phosphorylation to
determine whether the inhibition of BTK expression influences PKC
activation in MCF-7 cells. As shown in Fig. 3A, treatment with ibrutinib and CNX-774
reduced TPA-induced translocation of PKCβ to the membrane. We also
assessed the phosphorylation of p38, ERK, and JNK after treatment
with ibrutinib and CNX-774 to determine the effects of BTK
inhibitors on TPA-induced MAPK activation. Ibrutinib and CNX-774
treatment reduced p-p38, p-ERK, and p-JNK expression (Fig. 3B). In addition, we examined the
effects of ibrutinib and CNX-774 on the activation of the IKK
protein, a signal transducer downstream of the TPA-induced PKC
signaling pathway, but upstream of the NF-κB signal transduction
cascade. We also confirmed that ibrutinib and CNX-774 treatment
inhibited TPA-induced phosphorylation of IKKαβ and that TPA induced
phosphorylation and degradation of IκBα However, IκBα
phosphorylation was suppressed in cells treated with the BTK
inhibitors, thereby preventing its degradation (Fig. 3C). The results indicate that the
inhibition of BTK expression regulates PKCβ activation and MAPK/IKK
signal transduction, and, in turn, regulates TPA-induced MMP-9
expression in MCF-7 cells.
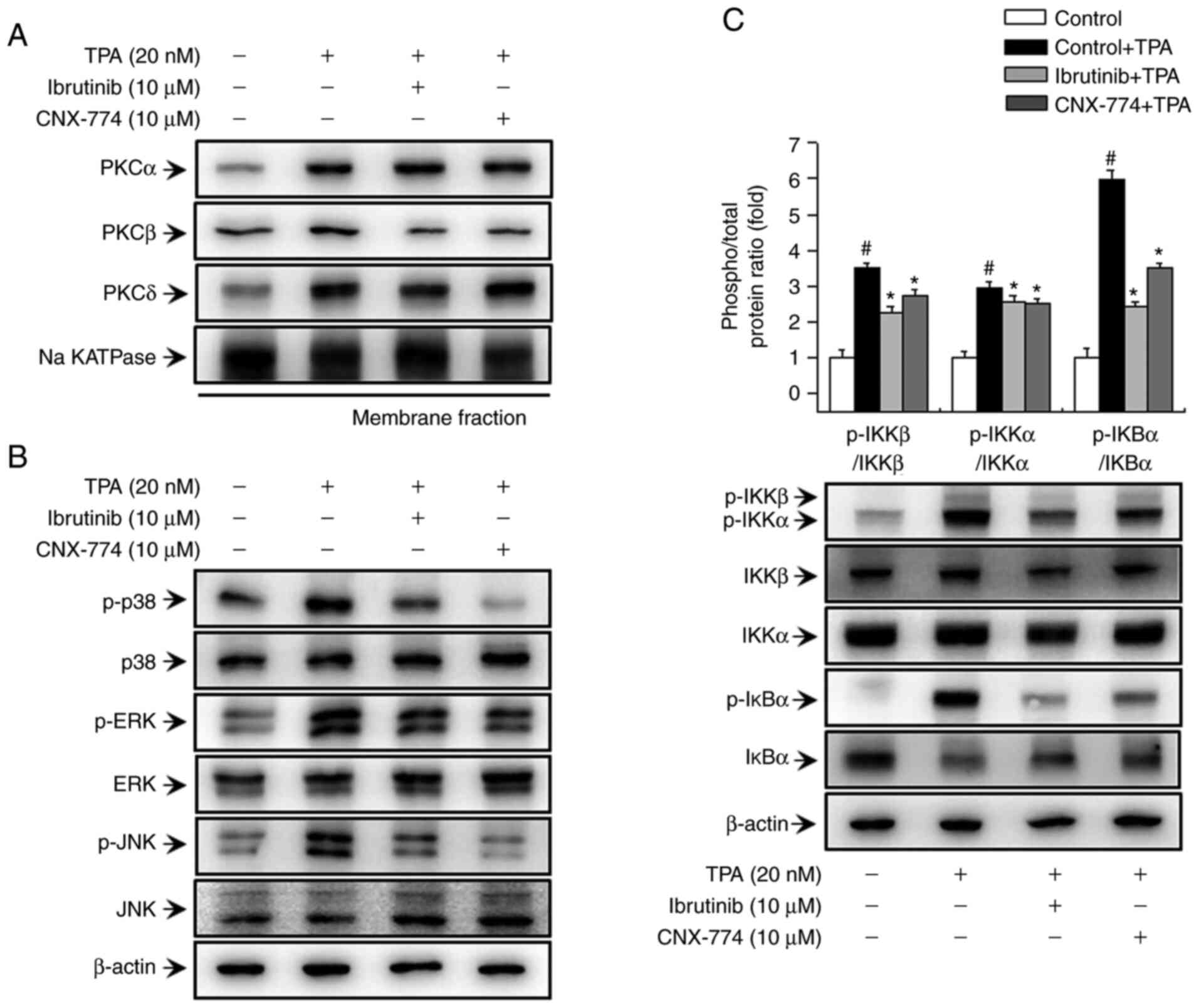 | Figure 3.BTK inhibitors decrease TPA-induced
activation of the PKC, MAPK, and IKK signaling pathways in MCF-7
cells. The cells were pretreated with BTK inhibitors for 1 h and
then stimulated with TPA. The cell lysates were analyzed by western
blotting of the PKCα, PKC δ, and PKCβ levels in the cytosolic and
membrane fractions. The blot was reprobed with an antibody against
Na k-ATPase to confirm equal loading (A). Cells (1×106)
were pretreated with BTK inhibitors and then stimulated with TPA
for 15 min. Cell lysates were assessed by western blotting using
antibodies against p38, extracellular signal-regulated kinase
(ERK), c-Jun N-terminal kinase (JNK), and their phosphorylated
forms (B), and the levels of p-IκBα, IκBα, p-IKKαβ, IKKα, and IKKβ
were determined (C). The blot was reprobed with an anti-β-actin
antibody to confirm equal loading. Data are the mean ± SEM of three
independent experiments. #P<0.01 vs. untreated
control; *P<0.01 vs. TPA. The blot was reprobed with an
anti-β-actin antibody to confirm equal loading. |
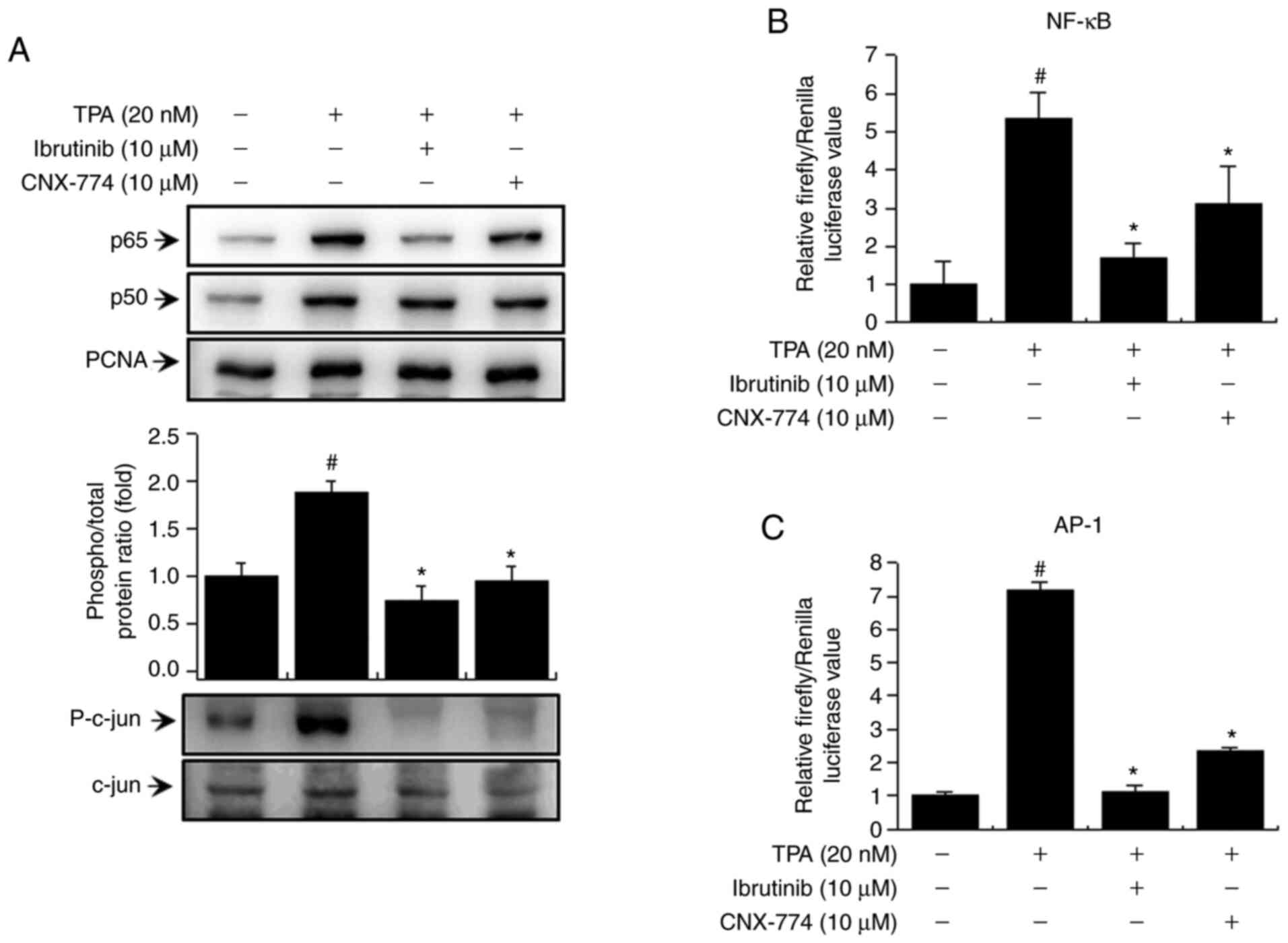
Inhibition of BTK expression reduces
TPA-induced activation of NF-κB and AP-1 in MCF-7 cells
We examined the activation of NF-κB and AP-1 to
verify the signaling mechanism downstream of PKC/MAPK and IKK. We
confirmed that the BTK inhibitors reduced the levels of p65 and
p50, the subunits of NF-κB, and p-c-Jun (Fig. 4A). In addition, to validate NF-κB and
AP-1 activation, we examined the effects of ibrutinib and CNX-774
on promoter binding using the luciferase reporter assay. We
observed an increase in TPA-induced activation of NF-κB and AP-1 in
MCF-7 cells and suppression of TPA-induced NF-κB and AP-1
activation after treatment with the BTK inhibitors (Fig. 4B and C). These findings suggest that
the inhibition of BTK expression regulates TPA-induced MMP-9
expression in MCF-7 cells by suppressing the activation of NF-κB
and AP-1 via various signaling mechanisms.
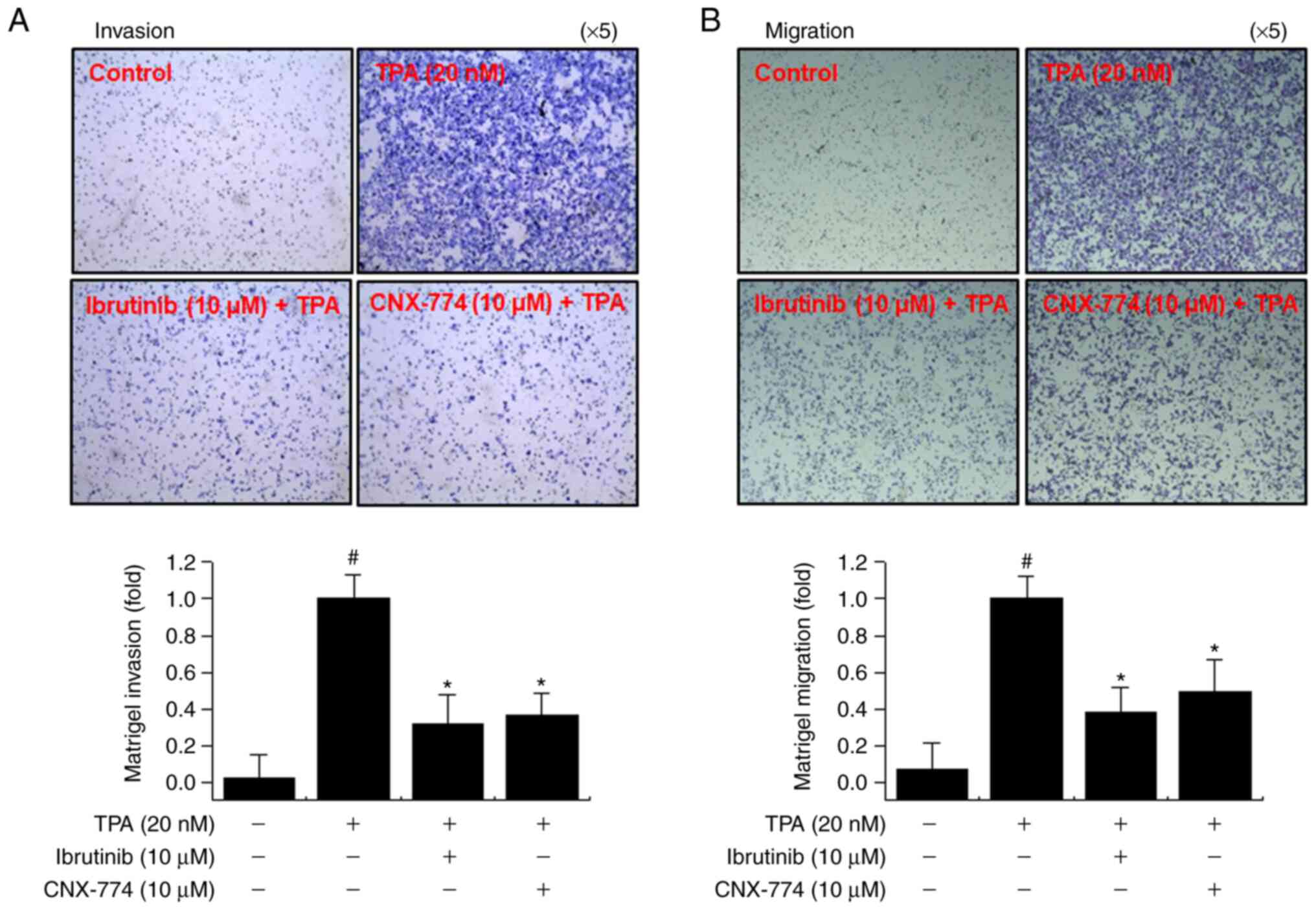
Inhibition of BTK expression
suppresses TPA-mediated invasion and migration of MCF-7 cells
The upregulation of MMP-9 expression contributes to
the metastasis of cancer cells, including breast cancer (21,41). We
performed Matrigel invasion and cell migration assays to determine
whether the inhibition of BTK expression suppresses MCF-7 cell
invasion and migration in vitro. We observed a marked
decrease in the invasion and migration of MCF-7 cells following
treatment with ibrutinib and CNX-774 (Fig. 5). These results suggest that the
inhibition of BTK expression suppresses cancer cell metastasis.
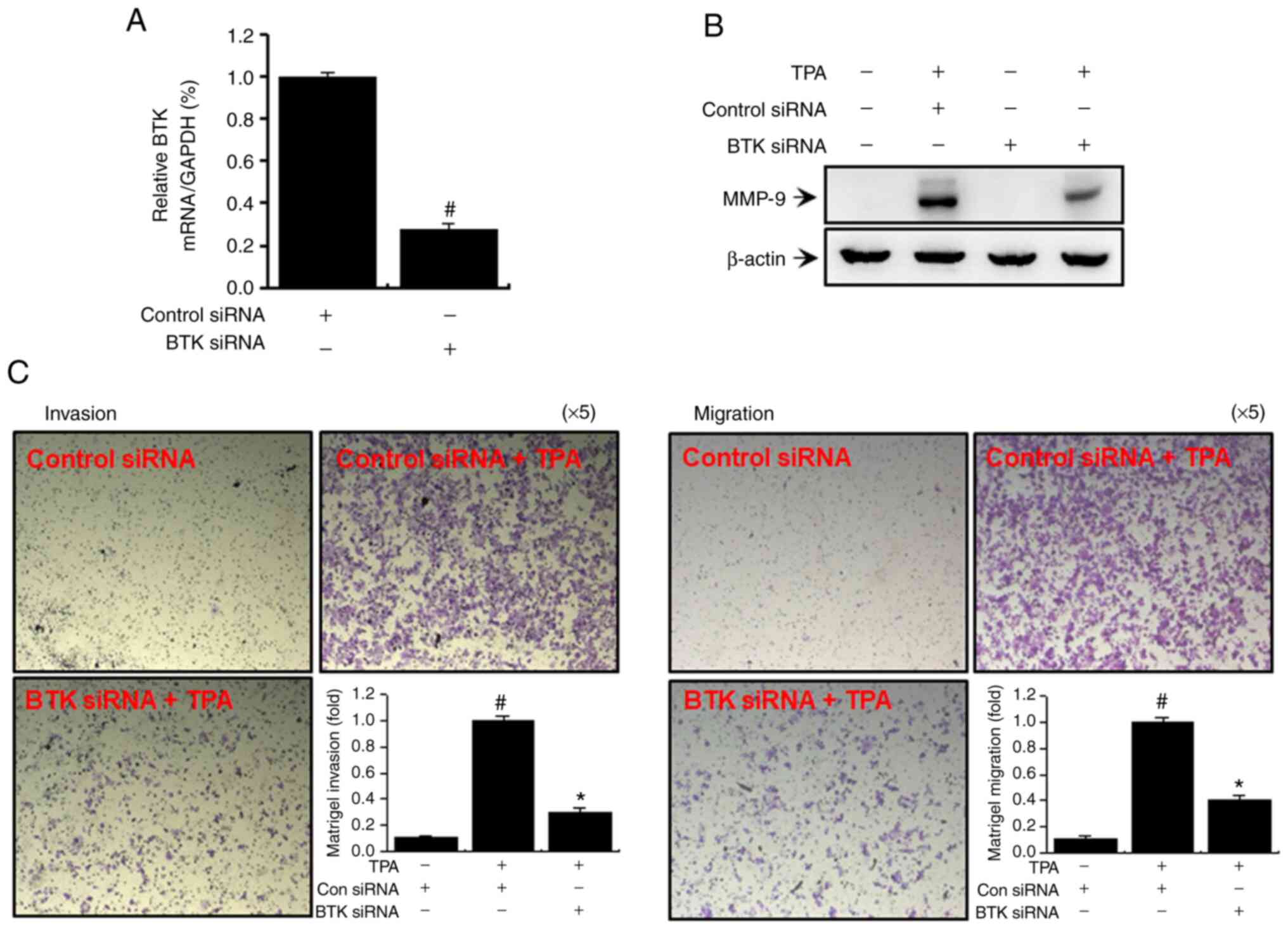
Downregulation of BTK expression
suppresses TPA-induced MMP-9 expression along with MCF-7 breast
cancer cell invasion and metastasis
We regulated BTK expression in MCF-7 cells by gene
silencing using siRNA to further substantiate our findings based on
inhibitor-induced suppression of BTK expression. As shown in
Fig. 6A, we transfected MCF-7 cells
with BTK and control siRNAs and performed quantitative PCR after 24
h to validate the knockdown of BTK. A significant decrease in
TPA-induced MMP-9 expression was observed when BTK was silenced
(Fig. 6B). Transfection with BTK
siRNA significantly reduced TPA-induced cellular invasion and
metastasis (Fig. 6C).
Discussion
Breast cancer-related mortality commonly results
from the metastasis of breast cancer cells to the bones, lung,
liver, brain, and kidney (4).
Metastasis is considered a defining characteristic of breast cancer
and primary cause of patient mortality. The initial phase of
invasion and metastasis of cancer cells involves the degradation of
ECM, which functions as a biochemical and mechanical barrier
(7,42). ECM degradation requires the expression
and activation of MMPs, which play a major role in breast cancer
progression (41,42). Among the MMPs, MMP-9 is a crucial
protein involved in tumor progression and, metastasis, including
breast cancer (26,43). MMP-9 expression activates various
intracellular signaling pathways in breast cancer cells via
inflammatory cytokines, hormones, growth factors, and TPA (41,43).
In this study, we aimed to establish the role of BTK
in TPA-induced MMP-9 expression as well as in the invasion and
migration of MCF-7 cells. A previous study has reported that BTK
translocates to the plasma membrane and, is phosphorylated by Src
family kinases, and, in turn, phosphorylates and activates PLCγ2
(13). Activated PLCγ2 catalyzes PIP2
hydrolysis to generate IP3 and DAG. DAG then promotes
Ca2+ discharge from IP3 intracellular storage. DAG and
Ca2+, in turn, activate PKCb, which then induces
activation of the Ras/RAF/MEK/ERK signaling cascade that promotes
cell growth and proliferation (36,44–46).
Previous studies have focused more on the role of BTK in B-cell
leukemia and lymphomas (47,48), which provides the basis for
kinase-targeted approaches to treat malignant tumors. However, the
role of BTK in metastatic cancer, including breast cancer, remains
unclear. Therefore, we examined whether suppressing BTK2 expression
regulates PLCγ2/PKC-mediated MMP-9 expression and invasion or
migration in MCF-7 cells. Our results confirmed that the inhibition
of BTK expression suppresses TPA-induced MMP-9 expression,
invasion, and migration and that PLCγ2 is involved in the
regulation of TPA-induced MMP-9 expression.
Another major objective of this study was to
investigate the anti-invasive activity of the downregulation of BTK
expression in the regulation of PKC-activated MMP-9 expression in
MCF-7 cells. MCF-7 cells express various types of PKCs that play an
important role in cell metastasis (18,49).
Therefore, this study was mainly performed on MCF-7 cells. In
addition, it was confirmed that the inhibition of BTK expression
inhibited MMP-2 and −9 expression and invasion or migration in
MDA-MB-231 cells (Fig. S1). However,
our results may be limited to the inhibitory effects of metastasis
in ER+ MCF-7 breast cancer cells. The ability of TPA to activate
PKC is possible due to the similarity of TPA to DAG, a natural
activator of classical PKC isoforms. Activated PKC enhances the
invasion of breast cancer cells by promoting MMP-9 expression
(50). TPA binds to the C1 domain of
PKC isoforms to activate them (51,52).
TPA-mediated PKC activation induces PKC isozymes to translocate to
the cell membranes, thereby leading to differential gene
expression, proliferation, apoptosis, differentiation, and
malignant regulation in cancer cells (51,53). The
activation of PKC isozymes is achieved by the binding of DAG and
Ca2+ (18). Various
signaling molecules are situated downstream of the PKC isozymes,
including Ras/Raf/MAPKs, phosphoinositide 3 kinase (PI3K)/Akt, and
transcription factors (NF-κB, AP-1, and STAT-3) (18,32). In
previous studies, we have shown that TPA-mediated activation of
PKCα, PKCb, and PKCδ mediates the expression and secretion of MMP-9
(31,54). The present study demonstrated that the
inhibition of BTK expression reduced TPA-mediated activation of PKC
isozymes in MCF-7 cells.
We evaluated the DNA binding of transcription
factors (NF-κB and AP-1) downstream of MAPKs and IKK and determined
the TPA-induced PKC-mediated downstream signaling cascade with
respect to MMP-9 expression in MCF-7 cells. MAPKs (ERK, p38, and
JNK), as upstream regulators of NF-κB, have been reported to induce
MMP-9 expression and activation (55). The activation of MAPK in MCF-7 cells
has been confirmed through phosphorylation (56). NF-κB and AP-1 are activated by the
IKK, MAPKs, or PI3K/Akt depending on the cell type (37,57–60).
Activation of NF-κB and AP-1 is important in MMP-9 regulation
because the NF-κB and AP-1 binding sites in the MMP-9 promoter are
involved in this activation (29,58). We
confirmed that the inhibition of BTK expression suppressed NF-κB
and AP-1 activation, while it suppressed MAPK and IKK activation at
the upstream level.
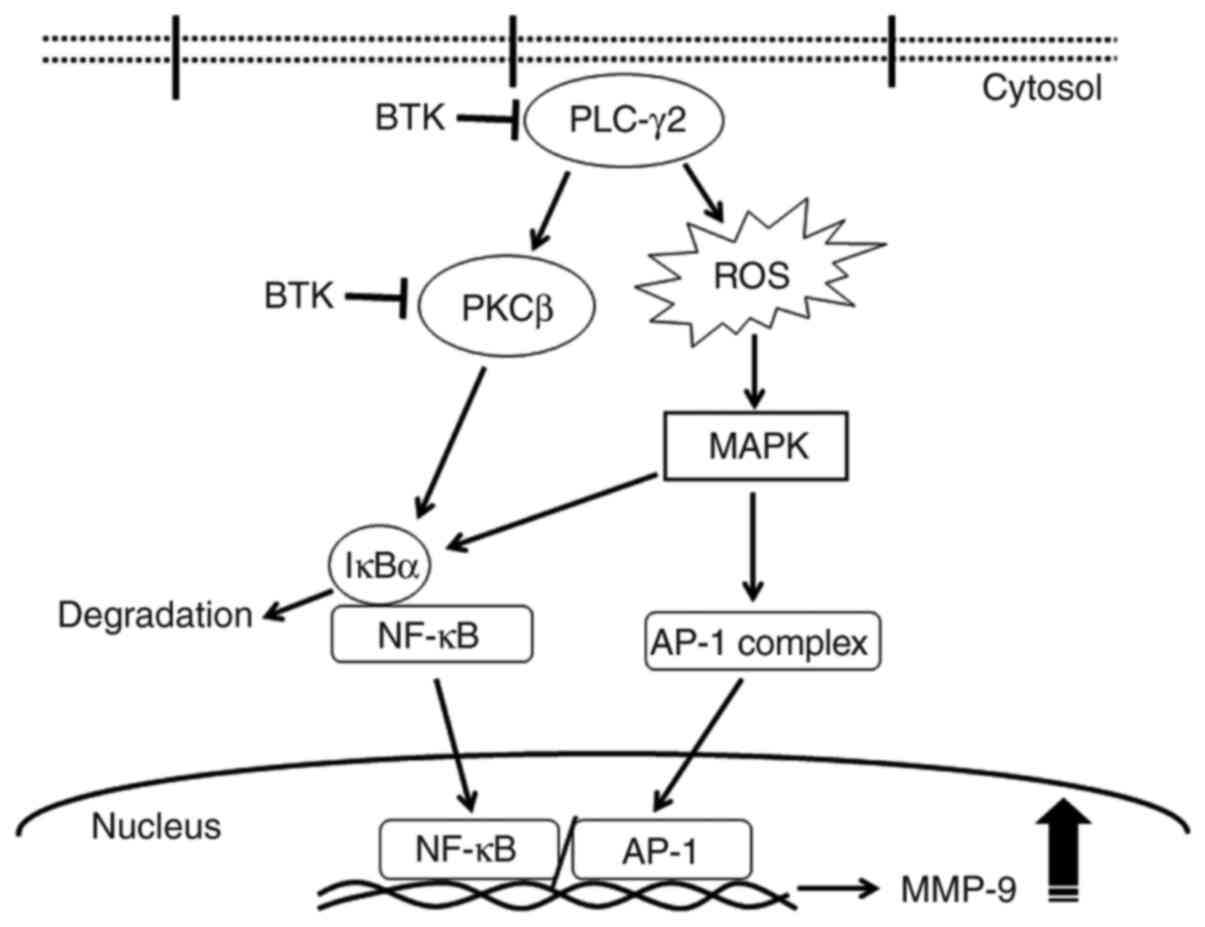
In conclusion, the inhibition of BTK expression
reduced TPA-induced MMP-9 expression and metastasis by blocking
NF-κB and AP-1 activation via the PLCγ/PKC/MAPK and IKK signaling
pathways (Fig. 7). To the best of our
knowledge, the present study is the first to validate that BTK
mediates the metastasis of MCF-7 cells by regulating the PLCγ2/PKC
signaling pathways, consequently suppressing MMP-9 expression.
Therefore, we suggest that regulating BTK expression may serve as a
therapeutic strategy to inhibit metastasis of MCF-7 breast cancer
cells.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Editage (www.editage.co.kr) for English language editing.
Funding
This work was supported by a National Research
Foundation of Korea (NRF) grant funded by the Korean Government
(NRF-2015R1D1A1A01057605, 2017R1A5A2015061), Republic of Korea. The
funding bodies had no role in study design, data collection and
analysis, decision to publish, or preparation of the
manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YRL, JP, and HJY designed the study. JMK, EMN, and
HKS performed the experiments. SYK and SHJ analyzed the western
blot and RT-PCR data. JSK and BHP provided additional experimental
comments and analyzed the data. YRL drafted the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang SY, Kim YS, Kim Z, Kim HY, Kim HJ,
Park S, Bae SY, Yoon KH, Lee SB, Lee SK, et al: Breast cancer
statistics in Korea in 2017: Data from a breast cancer registry. J
Breast Cancer. 23:115–128. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Redig AJ and McAllister SS: Breast cancer
as a systemic disease: A view of metastasis. J Intern Med.
274:113–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leber MF and Efferth T: Molecular
principles of cancer invasion and metastasis (review). Int J Oncol.
34:881–895. 2009.PubMed/NCBI
|
|
7
|
Jiang WG, Sanders AJ, Katoh M, Ungefroren
H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P,
et al: Tissue invasion and metastasis: Molecular, biological and
clinical perspectives. Semin Cancer Biol. 35 (Suppl 1):S244–S275.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Zijl F, Krupitza G and Mikulits W:
Initial steps of metastasis: Cell invasion and endothelial
transmigration. Mutat Res. 728:23–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Treon SP, Tripsas CK, Meid K, Warren D,
Varma G, Green R, Argyropoulos KV, Yang G, Cao Y, Xu L, et al:
Ibrutinib in previously treated Waldenström's macroglobulinemia. N
Engl J Med. 372:1430–1440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cao Y, Yang G, Hunter ZR, Liu X, Xu L,
Chen J, Tsakmaklis N, Hatjiharissi E, Kanan S, Davids MS, et al:
The BCL2 antagonist ABT-199 triggers apoptosis, and augments
ibrutinib and idelalisib mediated cytotoxicity in CXCR4 Wild-type
and CXCR4 WHIM mutated Waldenstrom macroglobulinaemia cells. Br J
Haematol. 170:134–138. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pal Singh S, Dammeijer F and Hendriks RW:
Role of Bruton's tyrosine kinase in B cells and malignancies. Mol
Cancer. 17:572018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HO: Development of BTK inhibitors for
the treatment of B-cell malignancies. Arch Pharm Res. 42:171–181.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mohamed AJ, Yu L, Bäckesjö CM, Vargas L,
Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF
and Smith CI: Bruton's tyrosine kinase (Btk): Function, regulation,
and transformation with special emphasis on the PH domain. Immunol
Rev. 228:58–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lampson BL and Brown JR: Are BTK and PLCG2
mutations necessary and sufficient for ibrutinib resistance in
chronic lymphocytic leukemia? Expert Rev Hematol. 11:185–194. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Halcomb KE, Contreras CM, Hinman RM,
Coursey TG, Wright HL and Satterthwaite AB: Btk and phospholipase C
gamma 2 can function independently during B cell development. Eur J
Immunol. 37:1033–1042. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilde JI and Watson SP: Regulation of
phospholipase C gamma isoforms in haematopoietic cells: Why one,
not the other? Cell Signal. 13:691–701. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koivunen J, Aaltonen V and Peltonen J:
Protein kinase C (PKC) family in cancer progression. Cancer Lett.
235:1–10. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Isakov N: Protein kinase C (PKC) isoforms
in cancer, tumor promotion and tumor suppression. Semin Cancer
Biol. 48:36–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grabinski N and Ewald F: Ibrutinib
(ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and
cell viability of ErbB2-positive breast cancer cells. Invest New
Drugs. 32:1096–1104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Kinoshita T, Sukbuntherng J, Chang
BY and Elias L: Ibrutinib Inhibits ERBB receptor tyrosine kinases
and HER2-amplified breast cancer cell growth. Mol Cancer Ther.
15:2835–2844. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stetler-Stevenson WG, Hewitt R and
Corcoran M: Matrix metalloproteinases and tumor invasion: From
correlation and causality to the clinic. Semin Cancer Biol.
7:147–154. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Itoh Y and Nagase H: Matrix
metalloproteinases in cancer. Essays Biochem. 38:21–36. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brinckerhoff CE and Matrisian LM: Matrix
metalloproteinases: A tail of a frog that became a prince. Nat Rev
Mol Cell Biol. 3:207–214. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weng CJ, Chau CF, Hsieh YS, Yang SF and
Yen GC: Lucidenic acid inhibits PMA-induced invasion of human
hepatoma cells through inactivating MAPK/ERK signal transduction
pathway and reducing binding activities of NF-kappaB and AP-1.
Carcinogenesis. 29:147–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vandooren J, Van den Steen PE and
Opdenakker G: Biochemistry and molecular biology of gelatinase B or
matrix metalloproteinase-9 (MMP-9): The next decade. Crit Rev
Biochem Mol Biol. 48:222–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scorilas A, Karameris A, Arnogiannaki N,
Ardavanis A, Bassilopoulos P, Trangas T and Talieri M:
Overexpression of matrix-metalloproteinase-9 in human breast
cancer: A potential favourable indicator in node-negative patients.
Br J Cancer. 84:1488–1496. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roomi MW, Kalinovsky T, Rath M and
Niedzwiecki A: Modulation of MMP-2 and MMP-9 secretion by
cytokines, inducers and inhibitors in human glioblastoma T-98G
cells. Oncol Rep. 37:1907–1913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Newton AC: Regulation of protein kinase C.
Curr Opin Cell Biol. 9:161–167. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin CW, Hou WC, Shen SC, Juan SH, Ko CH,
Wang LM and Chen YC: Quercetin inhibition of tumor invasion via
suppressing PKC delta/ERK/AP-1-dependent matrix metalloproteinase-9
activation in breast carcinoma cells. Carcinogenesis. 29:1807–1815.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee SO, Jeong YJ, Kim M, Kim CH and Lee
IS: Suppression of PMA-induced tumor cell invasion by capillarisin
via the inhibition of NF-kappaB-dependent MMP-9 expression. Biochem
Biophys Res Commun. 366:1019–1024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Noh EM, Park YJ, Kim JM, Kim MS, Kim HR,
Song HK, Hong OY, So HS, Yang SH, Kim JS, et al: Fisetin regulates
TPA-induced breast cell invasion by suppressing matrix
metalloproteinase-9 activation via the PKC/ROS/MAPK pathways. Eur J
Pharmacol. 764:79–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim JM, Noh EM, Kwon KB, Kim JS, You YO,
Hwang JK, Hwang BM, Kim BS, Lee SH, Lee SJ, et al: Curcumin
suppresses the TPA-induced invasion through inhibition of
PKCα-dependent MMP-expression in MCF-7 human breast cancer cells.
Phytomedicine. 19:1085–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim HR, Kim JM, Kim MS, Hwang JK, Park YJ,
Yang SH, Kim HJ, Ryu DG, Lee DS, Oh H, et al: Saussurea lappa
extract suppresses TPA-induced cell invasion via inhibition of
NF-κB-dependent MMP-9 expression in MCF-7 breast cancer cells. BMC
Complement Altern Med. 14:1702014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park SY, Kim YH, Kim Y and Lee SJ:
Frondoside A has an anti-invasive effect by inhibiting TPA-induced
MMP-9 activation via NF-κB and AP-1 signaling in human breast
cancer cells. Int J Oncol. 41:933–940. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park SH, Kim JH, Lee DH, Kang JW, Song HH,
Oh SR and Yoon DY: Luteolin 8-C-β-fucopyranoside inhibits invasion
and suppresses TPA-induced MMP-9 and IL-8 via ERK/AP-1 and
ERK/NF-κB signaling in MCF-7 breast cancer cells. Biochimie.
95:2082–2090. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Roskoski R Jr: ERK1/2 MAP kinases:
Structure, function, and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y and
Hu LL: ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med.
19:1997–2007. 2020.PubMed/NCBI
|
|
38
|
Lee SO, Jeong YJ, Im HG, Kim CH, Chang YC
and Lee IS: Silibinin suppresses PMA-induced MMP-9 expression by
blocking the AP-1 activation via MAPK signaling pathways in MCF-7
human breast carcinoma cells. Biochem Biophys Res Commun.
354:165–171. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim JM, Noh EM, Song HK, You YO, Jung SH,
Kim JS, Kwon KB, Lee YR and Youn HJ: Silencing of casein kinase 2
inhibits PKC-induced cell invasion by targeting MMP-9 in MCF-7
cells. Mol Med Rep. 17:8397–8402. 2018.PubMed/NCBI
|
|
40
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Duffy MJ, Maguire TM, Hill A, McDermott E
and O'Higgins N: Metalloproteinases: Role in breast carcinogenesis,
invasion and metastasis. Breast Cancer Res. 2:252–257. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Woessner JF Jr: Matrix metalloproteinases
and their inhibitors in connective tissue remodeling. FASEB J.
5:2145–2154. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yousef EM, Tahir MR, St-Pierre Y and
Gaboury LA: MMP-9 expression varies according to molecular subtypes
of breast cancer. BMC Cancer. 14:6092014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Roskoski R Jr: RAF
protein-serine/threonine kinases: Structure and regulation. Biochem
Biophys Res Commun. 399:313–317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roskoski R Jr: MEK1/2 dual-specificity
protein kinases: Structure and regulation. Biochem Biophys Res
Commun. 417:5–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Roskoski R Jr: A historical overview of
protein kinases and their targeted small molecule inhibitors.
Pharmacol Res. 100:1–23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hendriks RW, Yuvaraj S and Kil LP:
Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev
Cancer. 14:219–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Buggy JJ and Elias L: Bruton tyrosine
kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol.
31:119–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kawakami T, Kawakami Y and Kitaura J:
Protein kinase C beta (PKC beta): Normal functions and diseases. J
Biochem. 132:677–682. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang J, Anastasiadis PZ, Liu Y, Thompson
EA and Fields AP: Protein kinase C (PKC) betaII induces cell
invasion through a Ras/Mek-, PKC iota/Rac 1-dependent signaling
pathway. J Biol Chem. 279:22118–22123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Barry OP and Kazanietz MG: Protein kinase
C isozymes, novel phorbol ester receptors and cancer chemotherapy.
Curr Pharm Des. 7:1725–1744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kazanietz MG: Novel ‘nonkinase’ phorbol
ester receptors: The C1 domain connection. Mol Pharmacol.
61:759–767. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Urtreger AJ, Kazanietz MG and Bal de Kier
Joffé ED: Contribution of individual PKC isoforms to breast cancer
progression. IUBMB Life. 64:18–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chakraborti S, Mandal M, Das S, Mandal A
and Chakraborti T: Regulation of matrix metalloproteinases: An
overview. Mol Cell Biochem. 253:269–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qi M and Elion EA: MAP kinase pathways. J
Cell Sci. 118:3569–3572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yao J, Xiong S, Klos K, Nguyen N, Grijalva
R, Li P and Yu D: Multiple signaling pathways involved in
activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1
in human breast cancer cells. Oncogene. 20:8066–8074. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Whitmarsh AJ: Regulation of gene
transcription by mitogen-activated protein kinase signaling
pathways. Biochim Biophys Acta. 1773:1285–1298. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jiang N, Dai Q, Su X, Fu J, Feng X and
Peng J: Role of PI3K/AKT pathway in cancer: The framework of
malignant behavior. Mol Biol Rep. 47:4587–4629. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Karin M: The regulation of AP-1 activity
by mitogen-activated protein kinases. J Biol Chem. 270:16483–16486.
1995. View Article : Google Scholar : PubMed/NCBI
|