Introduction
Lung cancer is one of most common types of cancer
worldwide in terms of both incidence and mortality. In 2018,
approximately 2.1 million new cases of lung cancer were reported
and 1.8 million deaths were predicted (1,2). In China
in particular, recent statistics provided by the National Central
Cancer Registry of China (NCCRC) indicated that lung cancer had the
highest incidence rate in males as well as in the whole population.
Lung cancer was also the top contributor to cancer-related deaths
in China (3,4).
Over 80% of lung cancers are non-small cell lung
cancer (NSCLC), which is treated primarily by chemotherapy,
immunotherapy, surgery, targeted therapy, and radiation therapy
(5–7).
Some significant advances have been made in NSCLC treatment;
however, the survival rate remains poor due to the large number of
late-stage diagnoses (8). The call
for precision medicine in cancer therapy has also raised an urgent
need for the discovery of new drug targets for NSCLC (9).
One promising approach for identifying new
therapeutics is epigenetic editing, which is now undergoing
extensive studies in cancer biology. Epigenetic editing mediates
gene expression by changing the patterns of chromatin accessibility
(10). Epigenetic changes are an
important feature of NSCLC, making this a valuable approach for
lung cancer therapy (11,12). One family of epigenetic regulators is
represented by the bromodomain and extra-terminal domain (BET)
family proteins. These proteins bind to acetylated histones or
transcription factors and serve as scaffolds for the recruitment of
mediator complexes to promote protein transcription (13). Members of the BET family, such as
BRD2, BRD3, BRD4, and BRDT, are all structured with two highly
conserved N-terminal bromodomains, an extra-terminal domain, and a
divergent C-terminal recruitment domain (14,15).
Numerous studies have confirmed that BET proteins are highly
expressed in benign tumors and in malignancies, such as lung,
prostate, breast, colon, intestine, pancreas, liver, and brain
tumors. The roles and mechanisms of BRD4 have been extensively
studied (16–18); whereas studies on BRD2 and BRD3 are
extremely limited, especially in lung cancer. Moreover, the
correlations of BRD2, BRD3, and BRD4 expression and the progression
of lung cancer remain elusive. Nevertheless, inhibition of BET
protein activity may represent a promising therapeutic strategy for
treating NSCLC.
The first potent and selective BET family protein
inhibitor (‘BET inhibitor’, henceforth) was JQ1, a
thienotriazolodiazepine. Since its discovery, numerous other BET
inhibitors, such as I-BET151 and OTX015, have been used in clinical
trials and some have exhibited clear therapeutic effects (19). However, BET inhibitors have not
exhibited the anticipated therapeutic efficacy in solid tumors,
such as lung cancer, for reasons that remain unknown. One important
strategy for enhancing the anticancer effect of BET inhibitors
could therefore be to combine these inhibitors with other
drugs.
In small-cell lung cancer, both in vitro and
in vivo experiments have revealed a strong synergistic
apoptosis-promoting effect when cells are co-treated with the BCL2
inhibitor venetoclax (ABT-199) and the BET inhibitor ABBV-075
(20). In NSCLC, combined treatments
using the BET inhibitor JQ1 and the DDR2 inhibitor dasatinib
inhibited tumor growth both in vivo and in vitro;
however, the mechanism remains unclear (21). Therefore, identifying the underlying
molecular mechanism could aid in the development of more effective
combination therapies for NSCLC.
NSCLC is currently treated with chemotherapy as the
standard treatment for late-stage (III and IV) patients with NSCLC.
Doublet chemotherapy, such as a combination of platinum (cisplatin
or carboplatin) with taxanes (paclitaxel, docetaxel or
vinorelbine), is a common first-line therapy. Paclitaxel promotes
the assembly of microtubules and stabilizes the microtubules, and
paclitaxel-induced apoptosis in lung cancer has been confirmed in
numerous studies (22–24). The anticancer effect of cisplatin
largely relies on the formation of platinum-DNA adducts by
interactions between platinum and the nucleotide bases (25). Some patients benefit from doublet
chemotherapy; however, others undergo no benefits or experience
severe adverse drug reactions (ADRs) (5,26).
In the present study, the association between the
expression of BET and patient survival was analyzed according to
online databases. Henceforth, BRD2, BRD3, and BRD4 are indicated as
‘BET’ and their genes or mRNAs are designated as ‘BET.’ The
growth-inhibitory effects of BET silencing were evaluated using
siRNAs and BET inhibitors, such as JQ1 and I-BET151. The effects of
BET inhibitors on each BET member at both the mRNA and protein
levels were also revealed. Lastly, the growth-inhibitory effect of
a combined therapy of a BET inhibitor and a chemotherapy drug on
NSCLC cells was assessed. The effects of combined therapy on cell
autophagy and apoptosis were also explored. The present findings
provide new strategies for enhancing the anticancer effects of BET
inhibitors.
Materials and methods
UALCAN analysis
UALCAN is a comprehensive online tool for analyzing
cancer transcriptome data (27). We
utilized this tool to explore the correlation between BET
expression and two types of NSCLC, lung squamous cell carcinoma
(LUSC) and lung adenocarcinoma (LUAD).
Cell lines and culture conditions
All cell lines [human bronchial epithelial (HBE),
A549, H157, H1299] were purchased from the American Type Culture
Collection (ATCC). Cells were maintained in RPMI-1640 medium
(Hyclone; Cytiva) supplemented with 5% fetal bovine serum (Gibco
BRL; Thermo Fisher Scientific, Inc.) in a 5% humidified incubator
at 37°C. All cell lines were authenticated via short tandem repeat
(STR) profiling of 9 genomic loci with the Powerplex 16 HS system
(Promega Corporation).
Antibodies and reagents
Antibodies were purchased as follows: Anti-BRD4
(E2A7X) (product no. 13440), anti- BRD2 (D89B4) (product no. 5848),
anti-PARP (product no. 9542), and anti-caspase-3 (product no. 9665)
were purchased from Cell Signaling Technology, Inc.. Anti-BRD3
(2088C3a; product code ab50818) was purchased from Abcam,
anti-LC3I/II (cat. no. NB100-2220) was purchased from Novus
Biologicals, Ltd., anti-GAPDH (cat. no. AP0063) was purchased from
Bioworld Technology, Inc.. All antibodies were utilized at a
1:1,000 dilution except for GAPDH (1:10,000). JQ1 (cat. no.
HY-13030) was purchased from Haoyuan Chemexpress Co., Ltd. I-BET151
(cat. no. SML0666) and chloroquine (CQ) (product no. C6628) were
purchased from Sigma-Aldrich; Merck KGaA. Reagents were dissolved
in DMSO at 20 mmol/l, stored at −20°C, and diluted just before use.
Paclitaxel (PTX) (cat. no. HY-B0015) was purchased from Taiji
industry (Group) Co., Ltd., cisplatin (cat. no. HY-173940) was
purchased from MedChemexpress Co., Ltd., and diluted to 1 mg/ml,
and stored at 4°C.
Sulforhodamine B (SRB) assays
Cells were seeded into 96-well plates at 3,000
cells/well, and treated with either 2, 5, 10 ng/ml of paclitaxel or
1, 2, 4 µl/ml of cisplatin along with respective controls on the
second day of cell culture. After 3 days, cells were subjected to
SRB staining assay as previously described (28). The IC50 and the combination
index (CI) were determined using Compusyn software (29). CI <1 was defined as a synergistic
effect.
siRNAs and transfection
All siRNAs were obtained from Shanghai GenePharma
Co., Ltd.. Cells were seeded in 6 well plates and transfected with
either 2 µg of targeting siRNAs or scrambled control to obtain a
final concentration of 100 nmol/l using Lipofectamine 2000 (cat.
no. 11668019; Invitrogen; Thermo Fisher Scientific, Inc.) for 24 h
at 37°C followed by subsequent experiments such as being cultured
another 24 h for whole-cell protein preparation or RNA
purification, or reseeded to 96-well plates in an equal amount for
SRB assay. The sequences of siRNAs were as follows: siRNA#1 (for
BRD4), 5′-CUCCCUGAUUACUAUAAGATT-3′, 5′-GCACAAUCAAGUCUAAACUTT-3′ and
5′-GGAGAUGACAUAGUCUUAATT-3′; siRNA#2 (for BRD3),
5′-GUGCAAGCGAAUGUAUGCATT-3′, 5′-CGGAUGUUCUCGAAUUGCUTT-3′ and
5′-GUAGUGCACAUCAUCCAAUTT-3′; siRNA#3 (for BRD2),
5′-CAGCUGCAAUACCUACACATT-3′, 5′-GACUUCUCAAGUCCUUGCATT-3′ and
5′-GGACAGCUCAAUUCUACUATT-3′.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Cells were harvested for RT-qPCR as previously
described (30). The primer sequences
used for the present study are as follows: BRD4 forward (F),
5′-AGCAGCAACAGCAATGTGAG-3′ and reverse (R),
5′-GCTTGCACTTGTCCTCTTCC-3′; BRD3 F, 5′-CGGAAGCTCCAGGACGTGTT-3′ and
R, 5′-GGAGCCACCTTGGCCTTCTT-3′; BRD2 F, 5′-CAGGAACAGCTTCGGGCAGT-3′
and R, 5′-TCATGGGCCTGCTCTCTTCC-3′; GAPDH F,
5′-ATGGGGAAGGTGAAGGTCG-3′ and R, 5′-GGGGTCATTGGCAACAACAATA-3′.
Western blots analysis
Cells were subjected to western blot analysis as
previously described (31). The
chemiluminescent signal was collected and analyzed using the
ChemiScope 3400 Mini by Clinx Science Instruments Co., Ltd..
ELISA assay
Cells were treated with either 4 µmol/l of JQ1 or 10
ng/ml of PTX for 24 h. For the JQ1 and PTX co-treatment, cells were
pre-treated with 4 µmol/l of JQ1 for 2 h before PTX was added to
the medium at a final concentration of 10 ng/ml. Cells were then
cultured for another 24 h. The concentration of caspase-3 in the
cells was determined using a human caspase-3 ELISA kit (product no.
E-EL-H0017c; Elabscience) according to the manufacturer's
instructions.
Statistical analysis
All results are expressed as the mean ± SD of at
least three independent experiments. For statistical analysis,
one-way analysis of variance (ANOVA) with Sidak's correction for
multiple comparisons or two-tailed unpaired t-test with GraphPad
Prism 7.0 (GraphPad Software, Inc.) were used. P<0.05 was
considered to indicate a statistically significant difference.
Kaplan-Meier database (www.kmplot.com) was used to analyze the association
between the expression of BET and overall patient survival using a
hazard ratio (HR) with 95% confidence intervals (CI) and log rank
P-value. Parameters were set as follows: Set ‘Survival’ as ‘Overall
Survival (OS)’, set ‘Follow up threshold’ as ‘all’ and set all of
the ‘Restrict analysis to subtypes’ as ‘all’.
Results
Association between BET and NSCLC
Increased expression of BRD4 has been reported in
the cancerous tissues from patients with NSCLC when compared to
peripheral normal tissues. A negative association has also been
revealed between BRD4 expression and the 5-year survival rate of
patients with NSCLC (16). However,
the expression and the role of BRD2 and BRD3 in NSCLC has not been
investigated. Therefore, the association between BET mRNA
expression levels and NSCLC was evaluated according to the datasets
in the TCGA database using the UALCAN website (http://ualcan.path.uab.edu), and significant increases
in BRD2, BRD3, and BRD4 in lung squamous cell carcinoma, but not in
adenocarcinoma (Fig. S1) were
observed.
The association between the expression of BET and
overall patient survival based on datasets in the Kaplan-Meier
database (www.kmplot.com) (32) were then analyzed. The patients were
divided into two groups based on their median mRNA expression
levels (high vs. low expression) and assessed by a Kaplan-Meier
survival plot using a hazard ratio (HR) with 95% confidence
intervals (CI) and log rank P-value. The expression of BRD2, BRD3,
and BRD4 was negatively associated with the overall survival (OS)
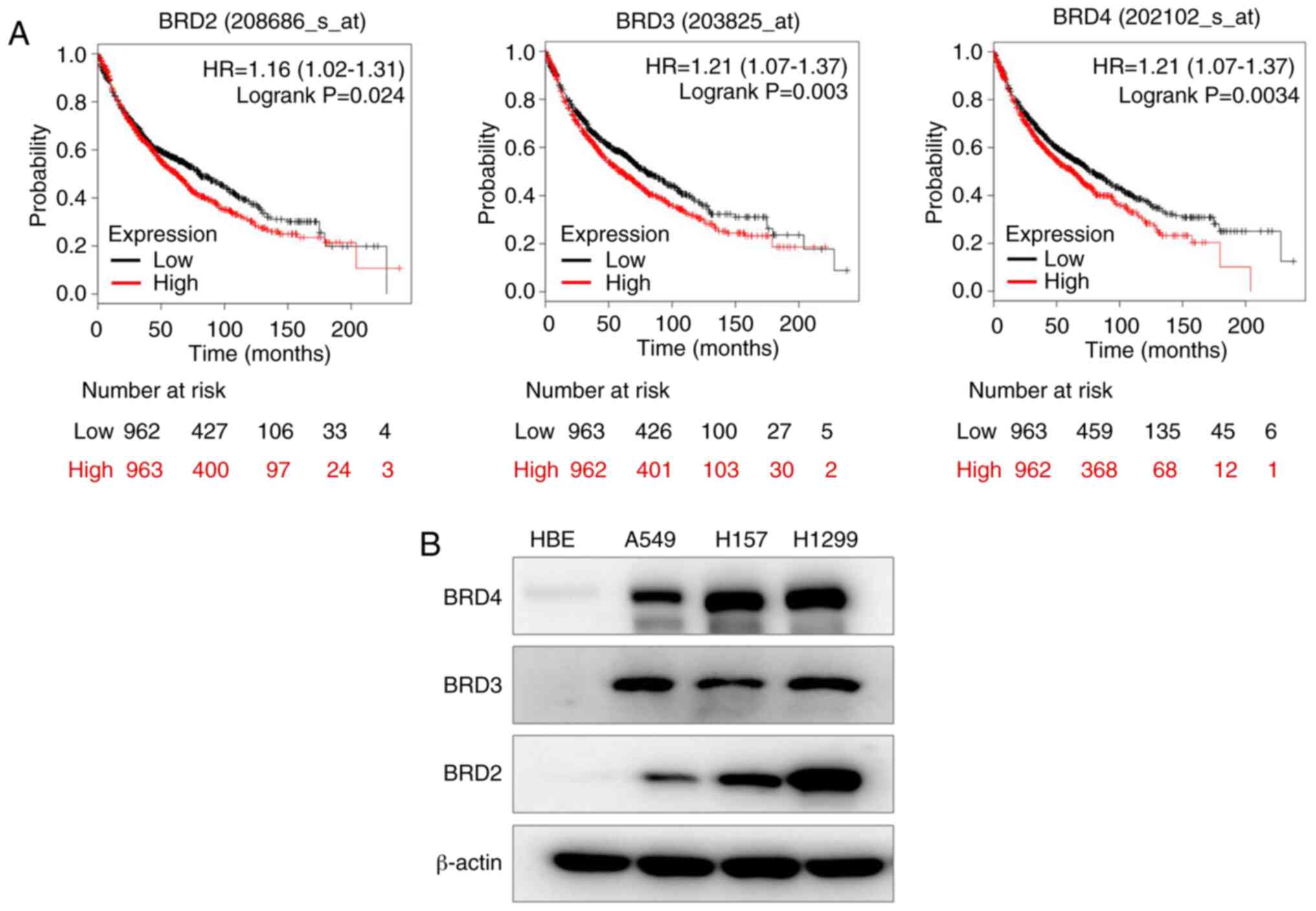
probability of the patients with NSCLC (Fig. 1A). Then, the BET protein levels in
four NSCLC cell lines were determined, and it was revealed that BET
expression was higher in A549, H157, and H1299 cells than in normal
HBE cells (Fig. 1B). Therefore, all
three BET may have important roles in NSCLC.
Targeting BET inhibits the growth of
NSCLC
The function of BET (especially of BRD2 and BRD3, as
these have not been evaluated previously) was explored by knocking
down their expression in A549 and H157 cells using siRNAs. The SRB
assay results revealed that silencing of each individual BET
gene significantly inhibited cell growth compared to control cells
(Fig. 2A). Western blotting confirmed
successful knockdown of all three proteins in both A549 and H157
cell lines (Fig. 2B).
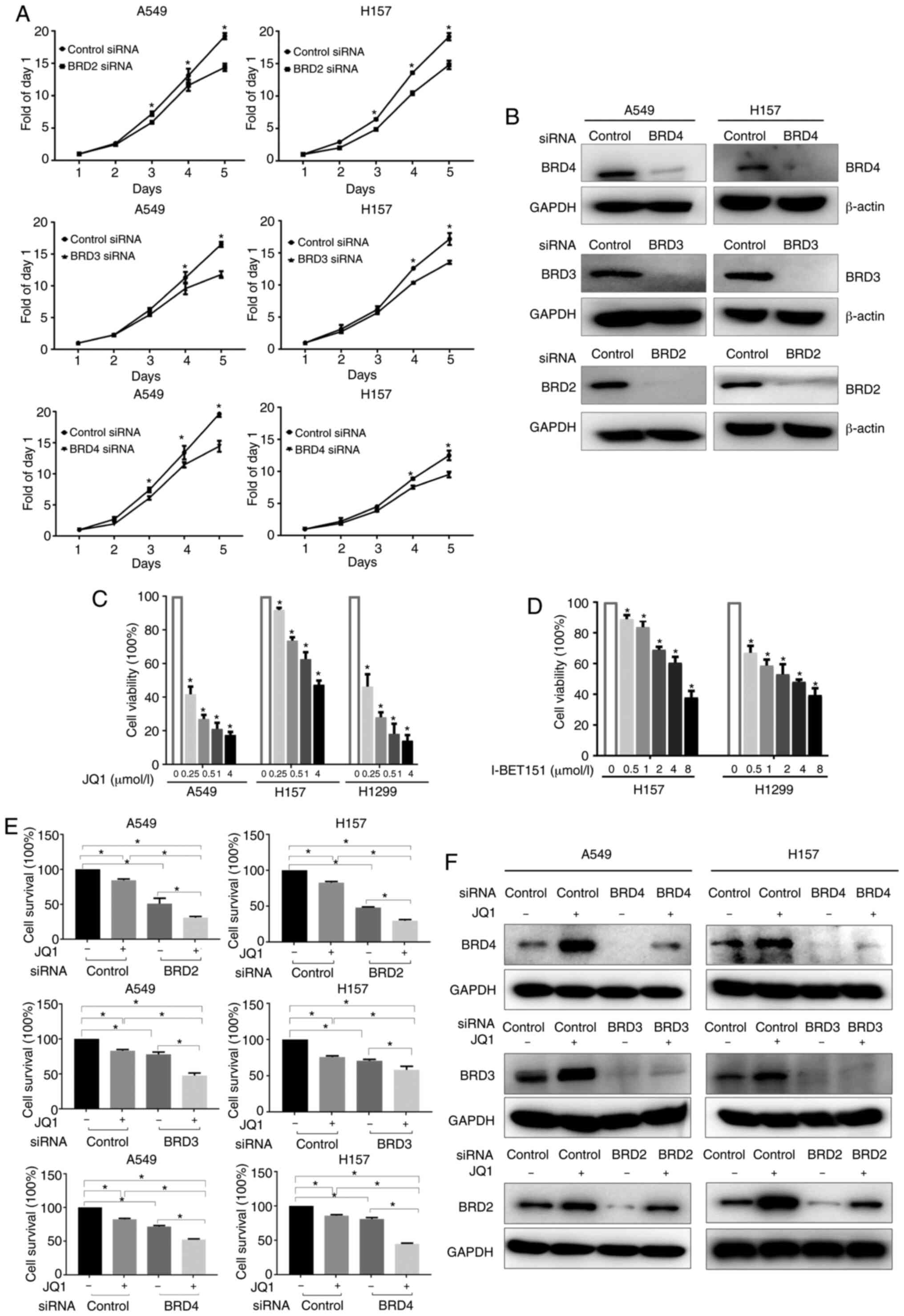 | Figure 2.Targeting BET inhibits the growth of
NSCLC cells. (A and B) NSCLC cells were transfected with siRNAs (a
pool containing 3 sequences of siRNA) targeting BRD2, BRD3, BRD4,
or control siRNA for 24 h, cells were then re-seeded into 96-well
plates, cultured for 5 days, and subjected to either (A) SRB assay
or (B) western blot analysis. (C and D) NSCLC cells were treated
with either JQ1 or I-BET151 for 3 days and subjected to SRB assay.
Data are presented as the mean ± SD (n=4). (E) NSCLC cells were
transfected with BRD2, BRD3, BRD4 or control siRNAs for 24 h,
re-seeded into 96-well plates, and treated with JQ1 (2 µmol/l) on
the second day for another 72 h. The cell survival rate was
evaluated by SRB assay. (F) Twenty-four hours after BET
siRNA-transfection, cells were treated with or without JQ1 for
another 24 h, and subjected to western blot analysis. *P<0.05.
BET, bromodomain and extra-terminal domains; NSCLC, non-small cell
lung cancer; SRB, sulforhodamine B. |
The effect of BET inhibitors on the growth of NSCLC
cells was then evaluated. Treatment with JQ1, a BET family protein
inhibitor that mainly blocks BRD4, dose-dependently suppressed the
growth of A549, H157, and H1299 cells, with IC50 values
of 0.06, 4.625, and 0.11 µmol/l, respectively (Fig. 2C). Another BET family protein
inhibitor, I-BET151, also dose-dependently inhibited the growth of
NSCLC cells (Fig. 2D).
The effect of BET silencing on cell survival in
combination with JQ1 treatment was also assessed. A549 and H157
cells were transfected with BRD2, BRD3, or BRD4 siRNAs and then
treated with or without JQ1 for another 24 h. As revealed in
Fig. 2E, JQ1 treatment enhanced the
growth inhibitory effects triggered by silencing of each individual
BET gene. Western blotting confirmed the successful
knockdown of each individual BET (Fig.
2F). It was also observed that knockdown of one BET had
negligible effects on the expression level of the other two BET,
indicating an absence of any compensatory expression among the BET
(data not shown). It was also observed that JQ1 treatment alone
induced a significant increase in the expression of BET compared
with that in the control group without JQ1 treatment (Fig. 2F). Therefore, BET inhibitor treatments
inhibited the protein function of BET by binding competitively to
the BET bromodomains, as previously reported (33), but they also concurrently upregulated
BET expression.
BET inhibitors upregulate both the
mRNA and protein expression of BET
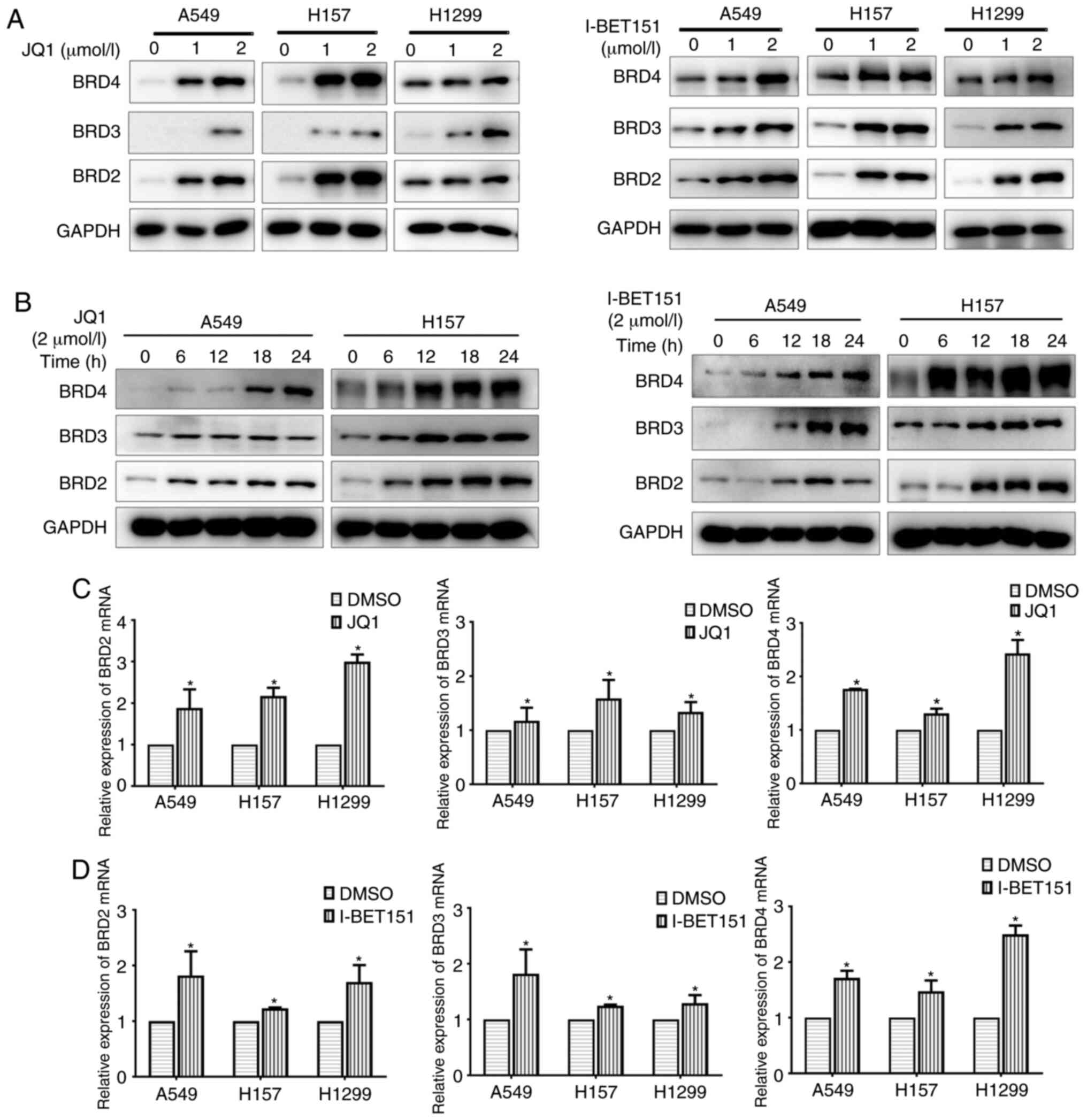
The regulation of BET was examined by the BET
inhibitors, JQ1 and I-BET151. Both JQ1 and I-BET151 upregulated BET
expression in a dose- and time-dependent manner (Fig. 3A and B). A significant increase in BET
expression was also observed after a 6-h JQ1 treatment, indicating
a relatively rapid regulation (Fig.
3B). The regulatory pattern of I-BET151 on BET was similar to
that of JQ1, except that a significant increase in BRD2 and BRD3
expression was only observed after a 12-h treatment, rather than
6-h (Fig. 3B). Both BET inhibitors
upregulated the mRNA expression levels of BET (Fig. 3C and D). The upregulation of BET
expression by BET inhibitors could therefore contribute to the
limited efficacy of BET inhibitors observed for NSCLC compared to
leukemia or glioblastoma (34).
Chemotherapeutic medicines paclitaxel
and cisplatin downregulate BET protein levels and enhance the
growth inhibitory effect of JQ1
Paclitaxel and cisplatin are classical anticancer
medicines that are recommended as first-line chemotherapies for
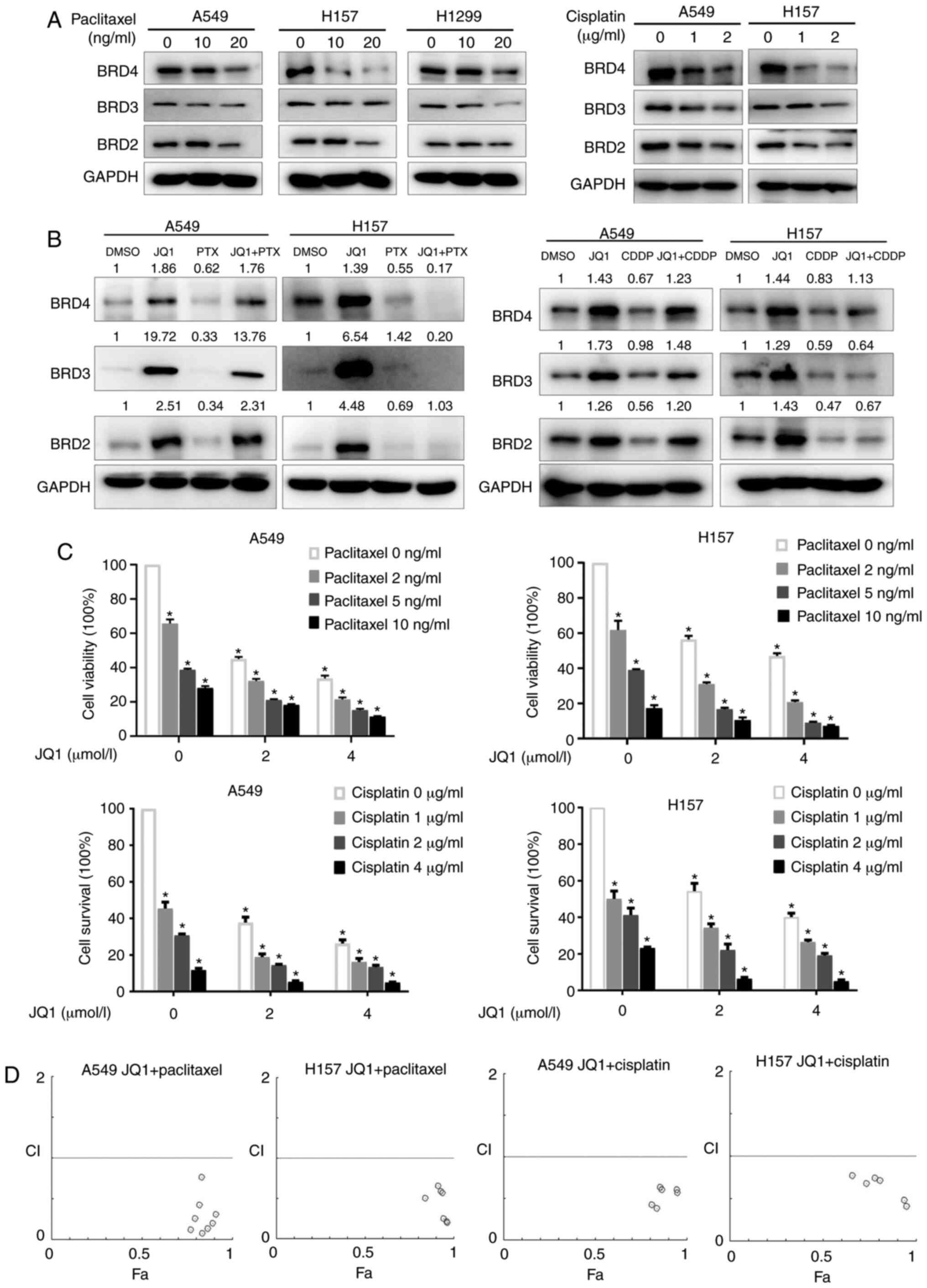
NSCLC. Their effects on the expression of BET were explored.
Western blotting revealed a dose-dependent downregulation of BET by
either paclitaxel or cisplatin (Fig.
4A). Both paclitaxel and cisplatin partially reversed the
JQ1-induced increase in BET expression in A549 and H157 cells
(Fig. 4B). The combination of JQ1
with paclitaxel or cisplatin revealed synergistic growth-inhibitory
effects on A549 and H157 cells in SRB assays, with CIs <1
(Fig. 4C and D). The combination of
JQ1 and chemotherapeutic medicine therefore synergistically
inhibited the growth of NSCLC cells. This synergistic effect may
involve the downregulation of BET by chemotherapeutic drugs.
Combination of JQ1 and paclitaxel
inhibits protective autophagy and promotes apoptosis
Autophagy and apoptosis both play important roles in
lung cancer progression. JQ1 induces both autophagy and apoptosis
in acute myeloid leukemia (AML), and protective autophagy is known
to promote cancer cell survival by allowing escape from
chemotherapy-induced apoptosis (35–37).
Microtubule-associated protein light chain 3 (LC3 II) is a standard
marker for autophagosomes and is generated by the conjugation of
cytosolic LC3 I to phosphatidylethanolamine (PE) on the surface of
nascent autophagosomes (38).
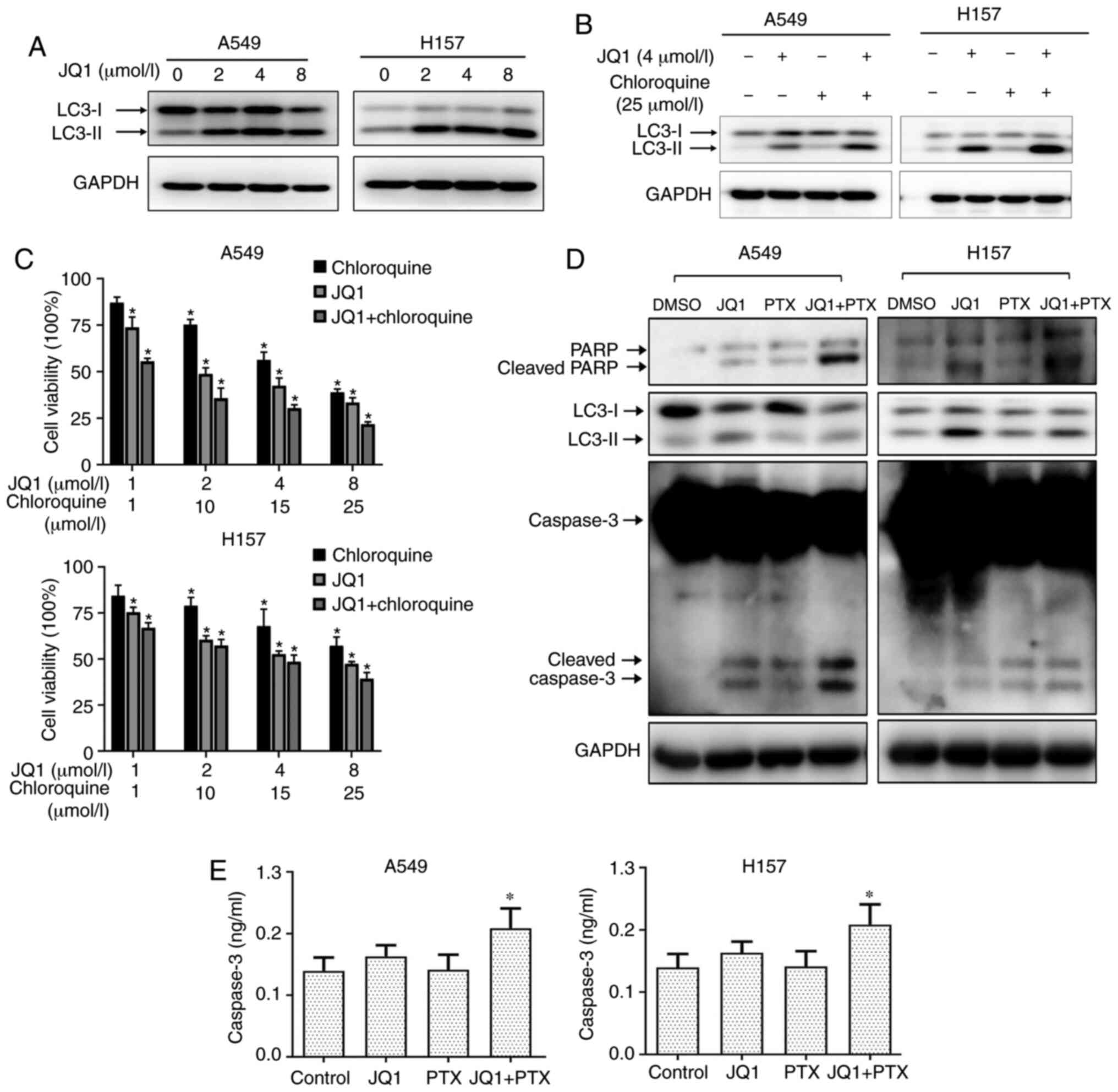
The effect of JQ1 on cell autophagy was evaluated by
examining LC3 II expression in NSCLCs. JQ1 dose-dependently
increased the expression of LC3 II, indicating an induction of
autophagy (Fig. 5A). The combined use
of JQ1 and CQ, a late-stage autophagy inhibitor, further
upregulated LC3 II levels compared to either JQ1 or CQ treatment
alone, suggesting an inhibition of autophagic flux (Fig. 5B). Furthermore, CQ significantly
enhanced the growth-inhibitory effects of JQ1 in A549 and H157
cells, suggesting that the protective autophagy induced by JQ1
impeded its inhibitory efficacy (Fig.
5C). JQ1 treatment also increased the levels of cleaved PARP
and cleaved caspase-3 in both A549 and H157 cells, suggesting an
induction of apoptosis. The combination of PTX and JQ1 partially
attenuated the JQ1-induced increase in LC3 II and further increased
the levels of cleaved PARP and cleaved caspase-3, suggesting an
inhibition of autophagy and enhancement of apoptosis (Fig. 5D).
Enzyme-linked immunoassays of caspase-3 confirmed
the enhancement of apoptosis observed with the combination of JQ1
and PTX (Fig. 5E). Collectively, the
results of combined treatments using a BET inhibitor and a
chemotherapeutic drug indicate a synergistic suppression of NSCLC
cell growth through the promotion of apoptosis and suppression of
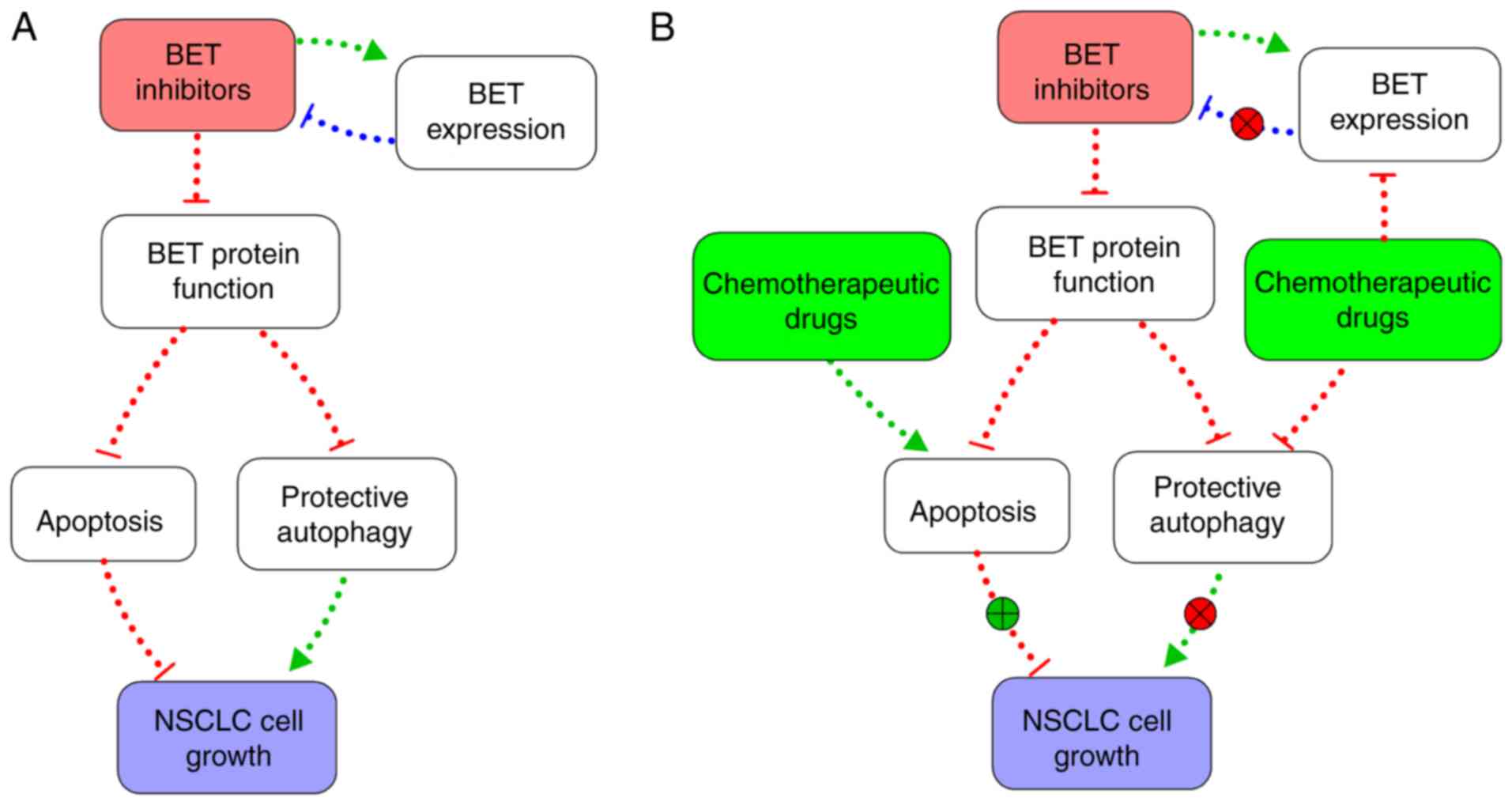
autophagy (Fig. 6).
Discussion
The antitumor effects of BET inhibitors have been
reported in benign tumors and in malignant diseases, such as
glioblastoma (39),
castration-resistant prostate (40),
breast (41), epithelial ovarian
(42) and lung cancers (43). In a previous study, we revealed that
targeting BRD4 inhibited the growth of NSCLC by downregulating
eIF4E expression (17). Liao et
al reported that BRD4 expression was increased in tissue
samples from patients with NSCLC and negatively correlated with the
5-year survival rate (16). In the
present study, higher expression of BET was observed in several
lung cancer cell lines than in normal bronchial epithelial cells.
Bioinformatics analysis of the BET expression revealed that
individual BRD2, BRD3, and BRD4 expression was
significantly increased in squamous cell carcinoma but not
adenocarcinoma. Our KM-plot analysis also revealed that increased
expression of BRD2, BRD3, and BRD4 was individually
associated with low overall survival of patients with lung cancer,
indicating an oncogenic function of these genes.
Targeting these three BET, either by administering
the BET family protein inhibitors (JQ1 and I-BET151) or by
silencing BET expression, suppressed the growth of NSCLC cells. The
inhibitory effect on cell growth in different cell lines using BET
inhibitors varied, which may due to the different basal expression
levels of BET among cell lines. JQ1 and I-BET151 were more
selective for BRD4, but they also affected BRD2 and BRD3. The use
of a BET inhibitor along with silencing the expression of
BET revealed more potent inhibitory effects than was
achieved with a single agent treatment. These findings indicated
that inhibiting the activity of BET proteins using small molecular
inhibitors or suppressing protein expression by gene silencing
could suppress the growth of NSCLC. Conversely, silencing the
expression of all three BET spontaneously using techniques such as
CRISPR-Cas9 system or shRNAs would be a promising research
direction in the future.
In the present study, it was also revealed that
I-BET151 or JQ1 treatment upregulated the expression of BET at both
the mRNA and protein levels, which suggested that inhibition of the
BET function in cancer cells triggered a compensatory upregulation
of BET expression. The mechanisms underlying this increase in BET
expression are unknown, however it is theorized that they may
involve either epigenetic regulation or a decrease in protein
degradation through E3 ligase-mediated ubiquitination signaling
(44). BET inhibitors may bind to E3
ligase, thereby preventing the binding of BET with E3 ligase due to
steric hindrance. It was also observed that JQ1-induced expression
pattern of BET varied between different NSCLC cell lines. This
could be caused by the heterogeneity of different cell lines, and
deserves future investigation.
ubiBrowser (45) was
used to predict the possible E3 ligase associated with BET and E3
ubiquitin-protein ligase Mdm2 (MDM2), which is reported to
facilitate BRD4 degradation, was identified to be a potential BET
binding partner (18). However,
proteolysis-targeting chimaeras (PROTACs) are known to bridge one
target protein with an E3-ubiquitin ligase and subsequently trigger
protein degradation (46). The
BET-protein PROTACs, ARV-771 and ARV-825, can degrade BET
effectively by recruiting an E3-ubiquitin ligase (47). Therefore, induction of BET expression
by BET inhibitors may attenuate BET anticancer efficacy. Clarifying
the mechanism underlying BET inhibitor effects may therefore help
to enhance the efficacy of BET inhibitors as treatment for NSCLC,
and would be an interesting future research direction.
The efficacy of numerous anticancer drugs is limited
by drug resistance, one of the substantial challenges in cancer
therapeutics. The resistance to epigenetic-targeting drugs is now
attracting great attention, however the mechanism for this
resistance remains poorly understood (48). Iniguez et al revealed that
forced expression of PI3K promoted the resistance to BET inhibitors
in neuroblastoma and that PI3K inhibitors synergized with BET
inhibitors to suppress tumor progression (49). Similarly, Hishiki et al
reported that resistance to I-BET151 in the U937R histiocytic
lymphoma cell line was related to the constitutive activation of
NF-κB signaling that increased both BRD2 and BRD4 expression
(50). Zong et al revealed
that BRD4 expression levels determined the sensitivity of the BET
degraders, ZBC260, but they did not correlate this with a response
to JQ1 (51). In the present study,
it was determined that BET inhibitors induced the expression of
BET, which may weaken the efficiency and confer resistance to BET
inhibitors in NSCLC. Whether this induction of expression of BET by
BET inhibitors is specific for NSCLC cells warrants further study.
The effects of combination therapies using BET inhibitors along
with the chemotherapeutic drugs paclitaxel and cisplatin in two of
the NSCLC cell lines were assessed. Both drugs downregulated BET
expression and they both reversed the upregulation of BET
expression caused by JQ1. Notably, in certain cell lines, such as
in A549, the reversal of the expression of BET induced by
inhibitors using chemotherapeutic drugs was not markedly evident.
The effect of knockdown of BET in parallel with chemotherapeutics
would be one of the goals for next-step research. Furthermore, the
detailed mechanism through which the chemotherapeutic agents induce
the inhibition of the expression of BET also warrants further
study. The combination of JQ1 with either paclitaxel or cisplatin
significantly delayed the growth of cancer cells when compared with
treatment with a single agent. Consistent with our study, JQ1 was
revealed to synergize with a PD-1 blockade to promote an antitumor
response in lung cancer (52).
Overall, the combination of chemotherapy and BET inhibitors could
reveal synergistic anticancer effects that could represent a potent
NSCLC therapy.
The anticancer effects of most drug treatments rely
on effects on autophagy, a self-degradative system that represents
a double-edged sword in the cancer research field. Interventions
that stimulate or inhibit autophagy have been suggested in
different cancer therapies (53,54).
Drug-activated autophagy can protect cancer cells against
apoptosis, and this contributes to the multi-drug resistance of
cancer cells. Therefore, the effect of chemotherapeutic drugs could
be improved by suppressing autophagy in certain cases (55). Li et al, using both in
vivo and in vitro experiments, reported a link between
osimertinib resistance and enhanced autophagy in lung cancer
(56). In the present study, we found
that JQ1 induced a protective autophagy, as inhibition of autophagy
by CQ suppressed NSCLC cell growth. This JQ1-induced autophagy may
in turn confer resistance to JQ1 in NSCLC. JQ1 treatment stimulated
cell apoptosis, whereas paclitaxel treatment suppressed JQ1-induced
autophagy and promoted cell apoptosis.
Mechanistically, it was reported recently that
knockdown of BRD4 could inhibit NSCLC cell growth by inhibiting
eIF4E expression. JQ1 treatment greatly reduced the binding of BRD4
with eIF4E promoter, which led to the abatement of the mRNA level
of eIF4E (17). Inhibition of BET by
BET degraders, particularly ZBC260, promoted apoptosis in sensitive
NSCLC cells in parallel with the reduction of c-FLIP and Mcl-1
levels (51). In other types of
cancer, studies revealed that suppression of BET could lead to the
downregulation of MYC expression (57,58).
Inhibition of BET by JQ1 was revealed to be more effective in
MYC-amplified medulloblastoma cell lines (57). In glioblastoma, p21 and Bcl-xL were
revealed to play key roles in BET-mediated cell growth and
apoptosis, while p21CIP1/WAF1 partially contributed to
JQ1-induced cell growth inhibition (33). Tumor cell growth could be influenced
by chromatin accessibility (57). A
recent study reported that BET inhibitor JQ1 could reduce chromatin
accessibility, which resulted in the suppression of RUNX2/NID1
signaling (59). βIII-tubulin (TUBB3)
has been revealed to play an important role in breast cancer
metastasis. BET inhibition could increase TUBB3 expression
by suppressing the expression of transcription factor myeloid zinc
finger-1 (MZF-1), the upstream regulator of TUBB3 (60). BET bromodomain inhibitors promote
apoptosis and cell cycle arrest resulting from the reduction of the
KIT (a receptor tyrosine kinase) enhancer domain and decreased
KIT expression in gastrointestinal stromal tumor (61). While in multiple myeloma, the function
of BET inhibitor lies in its ability to inhibit myeloid ecotropic
insertion site 2 (MEIS2) expression (62). All these mechanisms, where the
efficacy of BET and BET inhibitors lies, deserve further
exploration in our combined treatment system described in this
study.
In a clinic-related study, Liao et al
revealed increased expression of BRD4 in human NSCLC tissues and
increased BRD4 expression was correlated with the poor prognosis of
NSCLC patients (16). In the present
study, bioinformatics analysis of the datasets in TCGA database was
performed. Further validation is required in vivo and in the
clinic in the future. Another aspect that requires consideration
are the side effects of BET inhibitors, as summarized by Doroshow
et al (15). It was reported
that the side effects for MK-8628/OTX015, a BET inhibitor, included
thrombocytopenia, anemia and fatigue in NSCLC (15), indicating that the choice of a
specific type of inhibitor for each particular type of cancer
varies and requires individual assessment.
The aforementioned results indicated that a
combination of chemotherapy and BET inhibitors could give rise to
synergistic anticancer effects and could represent a promising
therapeutic strategy for treating NSCLC in the clinic.
Additionally, these synergistic anticancer effects may be even more
evident in BET-overexpressing cell lines and NSCLC cases compared
to those in BET medium or low-expressing cells and cases. Further
studies are required to verify this point.
In conclusion, the present study revealed that
targeting BET with small molecular inhibitors or gene silencing can
suppress NSCLC cell growth. However, BET inhibitors also increased
the expression of BET and induced a protective autophagy, which in
turn compromised the efficacy of BET inhibitors as anticancer
agents. Chemotherapeutic medicines, such as paclitaxel and
cisplatin, can enhance the effects of JQ1 by downregulating BET
expression, thereby inhibiting this autophagy while also promoting
apoptosis. The present findings indicated a potential novel
combination therapy for NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr Jialiang Wang at
Vanderbilt University (Nashville, USA) for the insightful
suggestions.
Funding
The present study was supported by the National
Natural Science Foundation of China under grant no. 81372395 for
ZC; grant nos. 81473241, 81102458 and 81172004 for XW; grant no.
81702882 for TS; and The Natural Science Foundation of Jiangsu
Province under grant no. BK20171056 for TS.
Availability of data and materials
The datasets analyzed during the present study are
available from the corresponding authors upon reasonable
request.
Authors' contributions
XZ, YM, JL, JC, BX, and ZZ conducted the
experiments. XZ, TS, and ZC were involved in study design and data
analysis. XW and ZC were responsible for study design, data
analysis, and manuscript writing. All authors reviewed and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Sun K, Zheng R, Zeng H, Zhang S,
Xia C, Yang Z, Li H, Zou X and He J: Cancer incidence and mortality
in China, 2014. Chin J Cancer Res. 30:1–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun KX, Zheng RS, Zeng HM, Zhang SW, Zou
XN, Gu XY, Xia CF, Yang ZX, Li H, Chen WQ and He J: The incidence
and mortality of lung cancer in China, 2014. Zhonghua Zhong Liu Za
Zhi. 40:805–811. 2018.(In Chinese). PubMed/NCBI
|
|
5
|
Gamerith G, Kocher F, Rudzki J and Pircher
A: ASCO 2018 NSCLC highlights-combination therapy is key. Memo.
11:266–271. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blandin Knight S, Crosbie PA, Balata H,
Chudziak J, Hussell T and Dive C: Progress and prospects of early
detection in lung cancer. Open Biol. 7:1700702017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong J, Li B, Lin D, Zhou Q and Huang D:
Advances in targeted therapy and immunotherapy for non-small cell
lung cancer based on accurate molecular typing. Front Pharmacol.
10:2302019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kelly AD and Issa JJ: The promise of
epigenetic therapy: Reprogramming the cancer epigenome. Curr Opin
Genet Dev. 42:68–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schiffmann I, Greve G, Jung M and Lübbert
M: Epigenetic therapy approaches in non-small cell lung cancer:
Update and perspectives. Epigenetics. 11:858–870. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mehta A, Dobersch S, Romero-Olmedo AJ and
Barreto G: Epigenetics in lung cancer diagnosis and therapy. Cancer
Metastasis Rev. 34:229–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
French CA: Small-molecule targeting of BET
proteins in cancer. Adv Cancer Res. 131:21–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pervaiz M, Mishra P and Günther S:
Bromodomain drug discovery-the past, the present, and the future.
Chem Rec. 18:1808–1817. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Doroshow DB, Eder JP and LoRusso PM: BET
inhibitors: A novel epigenetic approach. Ann Oncol. 28:1776–1787.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liao YF, Wu YB, Long X, Zhu SQ, Jin C, Xu
JJ and Ding JY: High level of BRD4 promotes non-small cell lung
cancer progression. Oncotarget. 7:9491–9500. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao Z, Yuan T, Zhou X, Ni P, Sun G, Li P,
Cheng Z and Wang X: Targeting BRD4 proteins suppresses the growth
of NSCLC through downregulation of eIF4E expression. Cancer Biol
Ther. 19:407–415. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hines J, Lartigue S, Dong H, Qian Y and
Crews CM: MDM2-recruiting PROTAC offers superior, synergistic
antiproliferative activity via simultaneous degradation of BRD4 and
stabilization of p53. Cancer Res. 79:251–262. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu Y and Vakoc CR: Targeting cancer cells
with BET bromodomain inhibitors. Cold Spring Harb Perspect Med.
7:a0266742017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lam LT, Lin X, Faivre EJ, Yang Z, Huang X,
Wilcox DM, Bellin RJ, Jin S, Tahir SK, Mitten M, et al:
Vulnerability of small-cell lung cancer to apoptosis induced by the
combination of BET bromodomain proteins and BCL2 inhibitors. Mol
Cancer Ther. 16:1511–1520. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu C, Buczkowski KA, Zhang Y, Asahina H,
Beauchamp EM, Terai H, Li YY, Meyerson M, Wong KK and Hammerman PS:
NSCLC driven by DDR2 mutation is sensitive to dasatinib and JQ1
combination therapy. Mol Cancer Ther. 14:2382–2389. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong Y, Huang BY and Yin JY:
Pharmacogenomics of platinum-based chemotherapy in non-small cell
lung cancer: Focusing on DNA repair systems. Med Oncol. 34:482017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang C, Wang H, Zhang B, Chen Y, Zhang Y,
Sun X, Xiao G, Nan K, Ren H and Qin S: LCL161 increases
paclitaxel-induced apoptosis by degrading cIAP1 and cIAP2 in NSCLC.
J Exp Clin Cancer Res. 35:1582016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu J, Su C, Zhao F, Tao J, Hu D, Shi A,
Pan J and Zhang Y: Paclitaxel promotes lung cancer cell apoptosis
via MEG3-P53 pathway activation. Biochem Biophys Res Commun.
504:123–128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ruiz-Ceja KA and Chirino YI: Current
FDA-approved treatments for non-small cell lung cancer and
potential biomarkers for its detection. Biomed Pharmacother.
90:24–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan H, Hu J and Liu S: Efficacy and safety
of nanoparticle albumin-bound paclitaxel in non-small cell lung
cancer: A systematic review and meta-analysis. Artif Cells Nanomed
Biotechnol. 47:268–277. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma Z, Zhu L, Luo X, Zhai S, Li P and Wang
X: Perifosine enhances mTORC1-targeted cancer therapy by activation
of GSK3β in NSCLC cells. Cancer Biol Ther. 13:1009–1017. 2014.
View Article : Google Scholar
|
|
29
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu S, Yang L, Wu D, Gao Z, Li P, Huang W
and Wang X: AEG-1 induces gastric cancer metastasis by upregulation
of eIF4E expression. J Cell Mol Med. 21:3481–3493. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang W, Yang L, Liang S, Liu D, Chen X,
Ma Z, Zhai S, Li P and Wang X: AEG-1 is a target of perifosine and
is over-expressed in gastric dysplasia and cancers. Dig Dis Sci.
58:2873–2880. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Filippakopoulos P, Qi J, Picaud S, Shen Y,
Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et
al: Selective inhibition of BET bromodomains. Nature.
468:1067–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce
LA, Thompson RC, Muller S, Knapp S and Wang J: Inhibition of BET
bromodomain targets genetically diverse glioblastoma. Clin Cancer
Res. 19:1748–1759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jang JE, Eom JI, Jeung HK, Cheong JW, Lee
JY, Kim JS and Min YH: AMPK-ULK1-mediated autophagy confers
resistance to BET inhibitor JQ1 in acute myeloid leukemia stem
cells. Clin Cancer Res. 23:2781–2794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin JF, Lin YC, Tsai TF, Chen HE, Chou KY
and Hwang TI: Cisplatin induces protective autophagy through
activation of BECN1 in human bladder cancer cells. Drug Des Devel
Ther. 11:1517–1533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu
LZ and Jiang BH: Downregulation of ATG14 by EGR1-MIR152 sensitizes
ovarian cancer cells to cisplatin-induced apoptosis by inhibiting
cyto-protective autophagy. Autophagy. 11:373–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma Y, Cheng Z, Liu J, Torre-Healy L,
Lathia JD, Nakano I, Guo Y, Thompson RC, Freeman ML and Wang J:
Inhibition of farnesyltransferase potentiates NOTCH-targeted
therapy against glioblastoma stem cells. Stem Cell Reports.
9:1948–1960. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Welti J, Sharp A, Yuan W, Dolling D, Nava
Rodrigues D, Figueiredo I, Gil V, Neeb A, Clarke M, Seed G, et al:
Targeting bromodomain and extra-terminal (BET) family proteins in
castration-resistant prostate cancer (CRPC). Clin Cancer Res.
24:3149–3162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shu S, Lin CY, He HH, Witwicki RM,
Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et
al: Response and resistance to BET bromodomain inhibitors in
triple-negative breast cancer. Nature. 529:413–417. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Momeny M, Eyvani H, Barghi F, Ghaffari SH,
Javadikooshesh S, Hassanvand Jamadi R, Esmaeili F, Alishahi Z,
Zaghal A, Bashash D, et al: Inhibition of bromodomain and
extraterminal domain reduces growth and invasive characteristics of
chemoresistant ovarian carcinoma cells. Anticancer Drugs.
29:1011–1020. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Klingbeil O, Lesche R, Gelato KA, Haendler
B and Lejeune P: Inhibition of BET bromodomain-dependent XIAP and
FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic
agents. Cell Death Dis. 7:e23652016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zengerle M, Chan KH and Ciulli A:
Selective small molecule induced degradation of the BET bromodomain
protein BRD4. ACS Chem Biol. 10:1770–1777. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z,
Guo F, He Y, Liu Y, Huang Q, et al: An integrated bioinformatics
platform for investigating the human E3 ubiquitin ligase-substrate
interaction network. Nat Commun. 8:3472017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gadd MS, Testa A, Lucas X, Chan KH, Chen
W, Lamont DJ, Zengerle M and Ciulli A: Structural basis of PROTAC
cooperative recognition for selective protein degradation. Nat Chem
Biol. 13:514–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sun B, Fiskus W, Qian Y, Rajapakshe K,
Raina K, Coleman KG, Crew AP, Shen A, Saenz DT, Mill CP, et al: BET
protein proteolysis targeting chimera (PROTAC) exerts potent lethal
activity against mantle cell lymphoma cells. Leukemia. 32:343–352.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
To KKW, Tong WS and Fu LW: Reversal of
platinum drug resistance by the histone deacetylase inhibitor
belinostat. Lung Cancer. 103:58–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Iniguez AB, Alexe G, Wang EJ, Roti G,
Patel S, Chen L, Kitara S, Conway A, Robichaud AL, Stolte B, et al:
Resistance to epigenetic-targeted therapy engenders tumor cell
vulnerabilities associated with enhancer remodeling. Cancer Cell.
34:922–938.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hishiki K, Akiyama M, Kanegae Y, Ozaki K,
Ohta M, Tsuchitani E, Kaito K and Yamada H: NF-κB signaling
activation via increases in BRD2 and BRD4 confers resistance to the
bromodomain inhibitor I-BET151 in U937 cells. Leuk Res. 74:57–63.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zong D, Gu J, Cavalcante GC, Yao W, Zhang
G, Wang S, Owonikoko TK, He X and Sun SY: BRD4 levels determine the
response of human lung cancer cells to BET degraders that potently
induce apoptosis through suppression of Mcl-1. Cancer Res.
80:2380–2393. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Adeegbe DO, Liu S, Hattersley MM, Bowden
M, Zhou CW, Li S, Vlahos R, Grondine M, Dolgalev I, Ivanova EV, et
al: BET bromodomain inhibition cooperates with PD-1 blockade to
facilitate antitumor response in Kras-mutant non-small cell lung
cancer. Cancer Immunol Res. 6:1234–1245. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Levy JMM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li L, Wang Y, Jiao L, Lin C, Lu C, Zhang
K, Hu C, Ye J, Zhang D, Wu H, et al: Protective autophagy decreases
osimertinib cytotoxicity through regulation of stem cell-like
properties in lung cancer. Cancer Lett. 452:191–202. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Henssen A, Thor T, Odersky A, Heukamp L,
El-Hindy N, Beckers A, Speleman F, Althoff K, Schäfers S, Schramm
A, et al: BET bromodomain protein inhibition is a therapeutic
option for medulloblastoma. Oncotarget. 4:2080–2095. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al:
RNAi screen identifies Brd4 as a therapeutic target in acute
myeloid leukaemia. Nature. 478:524–528. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhou S, Zhang S, Wang L, Huang S, Yuan Y,
Yang J, Wang H, Li X, Wang P, Zhou L, et al: BET protein inhibitor
JQ1 downregulates chromatin accessibility and suppresses metastasis
of gastric cancer via inactivating RUNX2/NID1 signaling.
Oncogenesis. 9:332020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kanojia D, Panek WK, Cordero A, Fares J,
Xiao A, Savchuk S, Kumar K, Xiao T, Pituch KC, Miska J, et al: BET
inhibition increases βIII-tubulin expression and sensitizes
metastatic breast cancer in the brain to vinorelbine. Sci Transl
Med. 12:eaax28792020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hemming ML, Lawlor MA, Andersen JL, Hagan
T, Chipashvili O, Scott TG, Raut CP, Sicinska E, Armstrong SA,
Demetri GD and Bradner JE: Enhancer domains in gastrointestinal
stromal tumor regulate KIT expression and are targetable by BET
bromodomain inhibition. Cancer Res. 79:994–1009. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Abruzzese MP, Bilotta MT, Fionda C,
Zingoni A, Soriani A, Petrucci MT, Ricciardi MR, Molfetta R,
Paolini R, Santoni A and Cippitelli M: The homeobox transcription
factor MEIS2 is a regulator of cancer cell survival and IMiDs
activity in multiple myeloma: Modulation by bromodomain and
extra-terminal (BET) protein inhibitors. Cell Death Dis.
10:3242019. View Article : Google Scholar : PubMed/NCBI
|