Introduction
Testicular germ cell tumors (TGCTs) are the most
common solid tumors in men aged 15 to 35 years and account for
>90% of primary malignant tumors of the testes (1). TGCTs can be clinically classified into
two main categories: seminoma and non-seminoma. The latter can be
further subdivided into embryonic cell carcinoma, choriocarcinoma,
yolk sac tumor or teratoma (2).
Non-seminomas have a high degree of differentiation but can have
multiple morphological phenotypes, since they contain both stem
cells and cells that have differentiated to varying degrees towards
somatic lineages. Seminomas have a lower level of differentiation
and resemble primordial germ cells and/or intratubular germ cell
neoplasia of unclassified type cells (3–5). Seminomas
can be staged according to the degree of proliferation and
metastasis. Stage I seminomas can be completely treated by surgery
and additional interventions, such as close follow-up,
drug-assisted chemotherapy and adjuvant radiotherapy (6). However, stage II/III seminomas have a
lower survival rate following surgery and postoperative
radiotherapy and chemotherapy (7).
Recent studies have shown that immunotherapy and gene therapy are
effective in treating seminomas (8,9).
Therefore, exploring the gene expression profile of seminomas is
essential to improve the understanding of its oncogenesis and
progression.
DNA methylation is an important epigenetic
modification that changes genomic expression without altering DNA
sequences. DNA methyltransferase 3α (DNMT3A), encodes a DNA
methyltransferase that is thought to function in methylation.
Several studies have shown that DNA methylation plays an important
role in the development and progression of tumors, mainly through
the changes in DNA methylation patterns and DNA methyltransferase
expression levels (10). Evidence has
shown that hypermethylation can inhibit the expression of
tumor-suppressor genes, thereby promoting tumor progression. For
example, in sporadic retinoblastoma (RB), RB tumor-suppressor gene
functions may become inactivated due to methylation, with its
expression levels reduced to 8%, as compared with unmethylated RB
(11,12).
Potassium voltage-gated channel subfamily C member 1
(KCNC1) encodes a member of the family of membrane proteins
that mediate voltage-dependent potassium ion permeability in
excitable membranes (13). The
abnormal expression and activity patterns of K+ channels
on the surface of cancer cells can drive tumor transformation,
malignant progression and metastasis, or drug resistance, through
Ca2+ activated IK or BK channels (14,15). Other
studies have shown that K+ channels can affect cell
proliferation and cell cycle progression in breast cancer cells
(16). However, K+
channels are poorly studied in seminoma, and their role in seminoma
progression remains unclear. Previous studies have found that
microRNA (miRNA/miR)-199a-3p, as a tumor-suppressing miRNA,
negatively regulates the expression of methylation marker
DNMT3A and impacts the level of glycometabolism in
testicular germ cells, ultimately inhibiting the progression of
seminomas (17,18).
In the present study, the methylation sites and
RNA-seq data collected from The Cancer Genome Atlas (TCGA) were
integrated to identify KCNC1 as a methylation-regulated gene
that plays an important role in the progression of malignant
seminoma. Furthermore, the expression of KCNC1 in seminoma tissues
and cell lines was verified by immunohistochemical staining,
western blot analysis and reverse transcription-quantitative
(RT-q)PCR. In the present study, we hypothesized that
hypermethylation can affect the expression of KCNC1, thereby
promoting seminoma progression. The objective was to discover novel
target genes for the treatment of seminoma in order to inhibit its
progression and improve the prognosis of patients.
Materials and methods
Data sourcing and analysis
RNA-seq data from 26 patients with stage I seminoma
and 106 patients with stage II/III seminoma were downloaded from
The Cancer Genome Atlas (TCGA) database https://www.cancer.gov/tcga (19). GDCRNATools (version 1.10.1) (20) in the R package (version 4.0.3)
(https://cran.r-project.org/) was used to
analyze differentially expressed genes (DEGs). |Fold-change| >2
and false discovery rate (FDR) <0.05 were set as the screening
thresholds. Differential methylation sites of 156 seminoma
specimens in the TCGA database were analyzed using ELMER software
package (version 2.14.0) (21). A
total of 162 differential hypermethylation sites between stage
II/III and stage I seminomas were obtained, and the regulated genes
around these methylation sites were then confirmed. The genes
associated with differential hypermethylation sites were
cross-referenced with the DEGs, and correlation analysis was
performed. A total of 14 methylation-DEGs were obtained and
survival analysis was performed. The results confirmed that
KCNC1, a gene regulated by hypermethylation, affected the
overall survival of seminoma patients. Correlation analysis between
KCNC1 and methylation and demethylation markers was
performed using the GEPIA online tool (http://gepia.cancer-pku.cn/) (22).
KCNC1 gene expression analysis
Public data were analyzed using cBioPortal
(http://www.cbioportal.org/) and GEPIA.
The expression of KCNC1 in the normal tissue was compared to that
in TGCTs, as well as in metastatic and non-metastatic TGCTs. All
data originated from the TGCT dataset of TCGA. Disease-free
survival analysis based on KCNC1 expression was obtained
from GEPIA.
Cell culture and immunohistochemical
staining
Tissue specimens (collected from 2015 to 2019 from
patients 30–40 years of age) were obtained from the Pathology
Department of The Second Hospital of Tianjin Medical University,
Tianjin, China. The use of tumor tissue for immunohistochemistry
was approved by the Second Hospital of Tianjin Medical University
Ethics Committee (Tianjin, China). The family members of the
patients were informed that the tumor tissue removed during surgery
would be used for further scientific research, and the approval of
the family members of the patients and their informed consent was
obtained.
Immunohistochemical staining of formalin-fixed,
paraffin-embedded seminoma and adjacent non-tumor tissues was
performed. These specimens were preserved by our hospital, and were
approved by a pathologist prior to use. Tissues were classified
according to normal testicular tissue, localized seminoma and
metastatic seminoma (3 cases per group). A total of 9 tissue
sections from the following pathological groups were prepared:
normal, stage I and stage II/III. Immunohistochemical staining
using 0.4-µm thick sections was performed using a KCNC1 antibody
(rabbit; dilution, 1:500; Abcam) and stained with hematoxylin. The
staining was performed according to normal immunohistochemical
procedures. The human Ntera-2 testicular tumor (NT2), normal human
testis Hs1.Tes (HT) and Tcam-2 cell lines (human testicular
seminoma cells) were purchased from the American Type Culture
Collection. They were cultured at 37°C with 5% CO2 in
RPMI-1640 medium (Thermo Fisher Scientific, Inc.) containing 10%
FBS (Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin.
Western blot analysis and RT-qPCR
Protease inhibitors (Beyotime Institute of
Biotechnology) were mixed with total protein extraction reagent
(Beijing Solarbio Science & Technology Co., Ltd.) at a ratio of
1:100, and then mixed with cultured cells. The protein
concentration was determined using BCA. Protein samples were mixed
with loading buffer and loaded onto a 10% SDS-PAGE for western blot
analysis. The protein expression levels of KCNC1, DNMT3A and GAPDH,
as well as epithelial-mesenchymal transition (EMT)-related markers
and apoptotic markers were detected by western blot analysis. The
antibodies used are listed in Table
I. The primary antibody was incubated at 4°C overnight, the
secondary antibody was incubated at room temperature for 1 h, and
ECL (Biochannel Co., Ltd.) was used for exposure.
 | Table I.Antibodies used in the western blot
analysis. |
Table I.
Antibodies used in the western blot
analysis.
| Antibodies | Company | Dilution ratio | Secondary
species | Molecular
weight |
|---|
| KCNC1 | Abcam | 1:500 | Rabbit | 58 |
| DNMT3A | Proteintech | 1:500 | Rabbit | 120 |
| Zeb1 | Proteintech | 1:500 | Rabbit | 170 |
| N-cadherin | Proteintech | 1:500 | Rabbit | 130 |
| Vimentin | Proteintech | 1:500 | Rabbit | 54 |
| Casepase-3 | Proteintech | 1:1,000 | Mouse | 30 |
| BCL2 | Proteintech | 1:500 | Mouse | 26 |
| BAX | Proteintech | 1:500 | Mouse | 21 |
| GAPDH | Proteintech | 1:1,500 | Mouse | 37 |
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.), and the Reverse
Transcription Kit (Thermo Fisher Scientific, Inc.) was used for
reverse transcription. FastStart Universal SYBR-Green Master Mix
(Hoffmann-La Roche Ltd.) was used for RT-qPCR following the
manufacturer's instructions (Table
II). GAPDH mRNA expression was used as an endogenous
control for normalization in each sample. The specific steps are as
follows: reaction temperature and reaction time: Pre denaturation
at 94°C for 3 min, denaturation for 0.5 min at 94°C, annealing at
58°C for 0.5 min, extension at 72°C for 1 min and final extension
at 72°C for 5 min. The melting curve and amplification curve of
qPCR were observed. The experimental data were calculated using the
2−ΔCq formula (23).
| Table II.Primer list. |
Table II.
Primer list.
| Gene | Forward
primers | Reverse
primers |
|---|
| KCNC1 |
CGCTCTTCGAGGACCCGTA |
CGTCTTGTTCACGATGGGGT |
| DNMT3A |
TACTTCCAGAGCTTCAGGGC |
ATTCCTTCTCACAACCCGC |
| GAPDH |
GGATTTGGTCGTATTGGG |
GGAAGATGGTGATGGGATT |
Small interfering RNA (siRNA)
transfection and plasmid construction
Following the manufacturer's instructions, siRNA and
negative control siRNA (Suzhou GenePharma Co., Ltd.) were
transfected into HT cells with X-treme gene siRNA transfection
reagent (Suzhou GenePharma Co., Ltd.). NT2 cells were transduced
with a lentivirus encoding KCNC1 overexpression or control
plasmid (Beijing Syngentech Co., Ltd.). The knockdown of
KCNC1 expression following siRNA transfection was verified
by RT-qPCR and western blot analysis. The siRNA sequence used was
5′-CCGGGCCCGTCATCGTGAACAATTTCTCGAGAAATTGTTCACGATGACGGGCTTTTTG-3′.
The negative control sequence used was:
5′-GTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAACTTTTTTG-3′. Six
hours after siRNA transfection, the transfection medium was removed
and the new medium was added.
Transwell and Cell Counting Kit
(CCK)-8 cell viability assays
In the Transwell assay, 2–3,000 transfected NT2 and
HT cells were transferred to the upper Transwell chamber. RPMI-1640
medium (200 µl) was added to the upper chamber and full medium (600
µl) to the lower chamber. Then, 1 day later, DAPI staining was used
to observe cell membrane permeability under a fluorescence
microscope. HT and NT2 cells were inoculated in a 96-well culture
plate. When the cells reached 30–50% confluence, lv-KCNC1 and
KCNC1-siRNA or negative control was used for transfection. At 24,
48 and 72 h after transfection, CCK-8 solution was added to 96-well
plates at a ratio of 1:9. The optical density value at a 450 nm
wavelength was measured by a microplate reader.
Flow cytometry
Flow cytometry was performed 48 h after the
transfection of NT2 and HT cells. A total of 1×105
transfected NT2 and HT cells were resuspended in 500 µl PI/RNase
staining solution (Sungene Biotech) 15 min before flow cytometry,
and Annexin V-FITC/PI kit (US Everbright, Inc.) was used for cell
apoptosis detection.
Dot blot analysis
Genomic DNA was extracted from NT2 and HT cells,
using a DNA isolation kit, following the manufacturer's
instructions (Qiagen AB). DNA samples were dropped on the
corresponding spots of the nitrocellulose membrane soaked in sodium
citrate. The nitrocellulose membrane was baked in the oven at 80°C
for 1 h, and then exposed to ultraviolet light for 30–45 min for
sealing. Finally, the nitrocellulose membrane was incubated with a
5-mc and anti-5-hmc antibody (dilution, 1:1,000; Abcam) in a
refrigerator at 4°C overnight.
Statistical analysis
All experiments were carried out at least three
times. All data are expressed as the mean ± standard deviation.
ANOVA was performed to evaluate the difference of three or more
groups by Turkey post hoc test, and the statistical analysis was
carried out by using GraphPad Prism 8.0 software (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference. SPSS version 22 was used for
statistical analysis (IBM, Corp.).
Results
KCNC1 participates in the malignancy
and prognosis of seminomas
RNA-seq data were collected from TCGA-TGCT datasets.
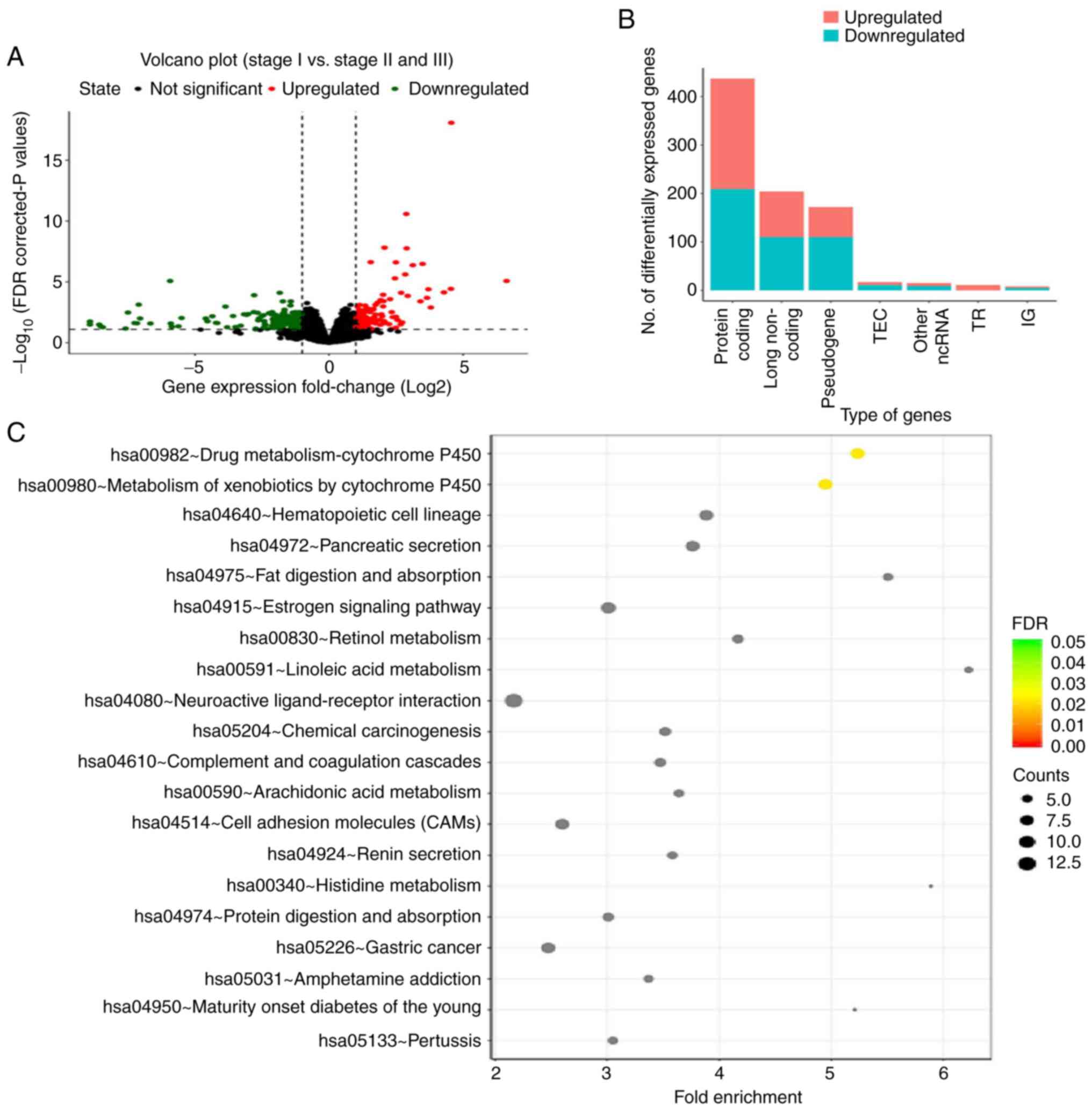
|Fold-change| >2 and FDR <0.05 were set as the screening
thresholds. A total of 864 DEGs were identified, with 410
upregulated and 456 downregulated genes (Fig. 1A). Gene Ontology and Kyoto
Encyclopedia of Genes and Genomes signaling pathway enrichment
analysis was performed (Fig. 1B and
C). The two major signaling pathways associated with the DEGs
were found to be ‘drug metabolism-cytochrome P450’ and ‘metabolism
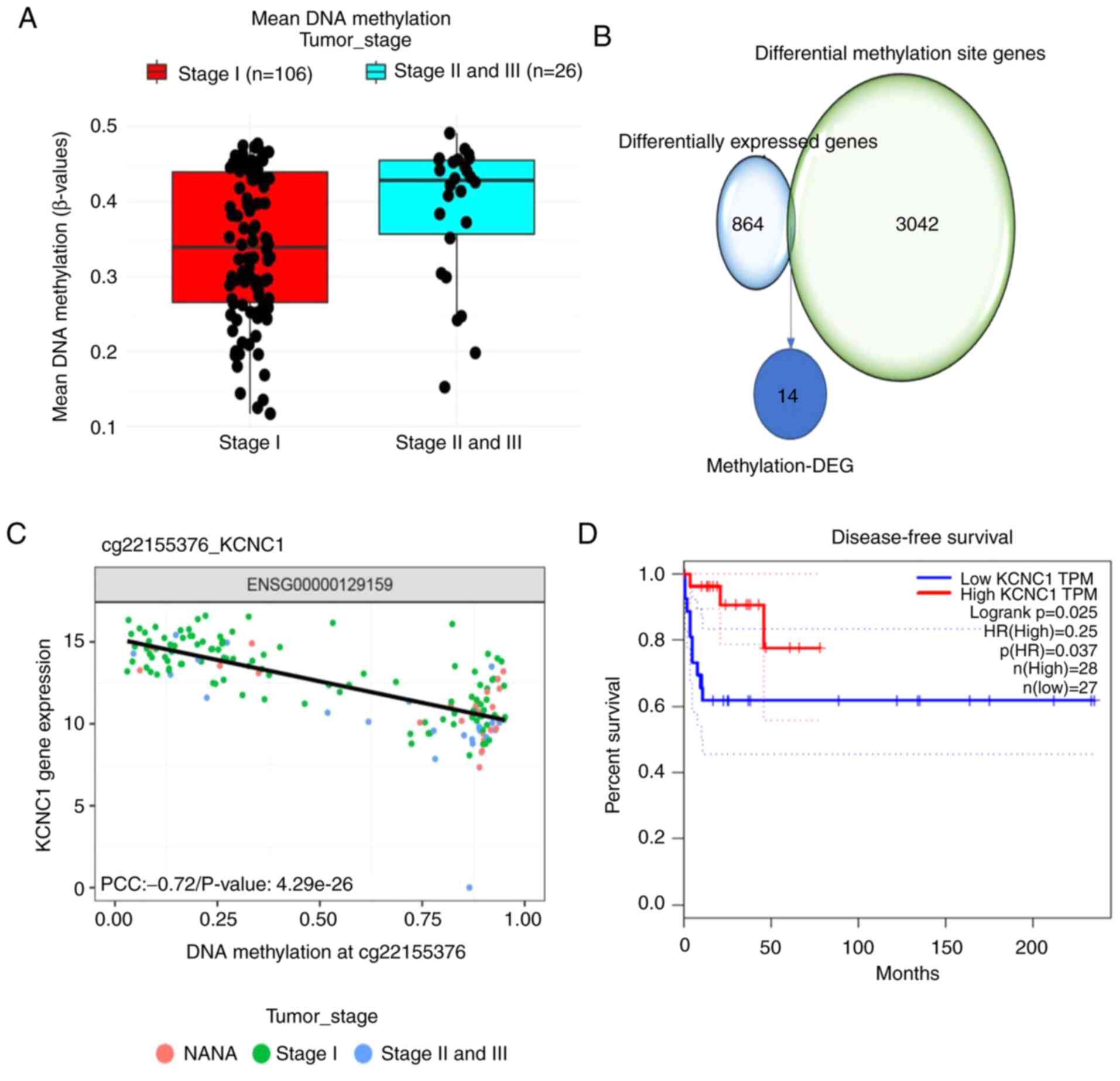
of xenobiotics by cytochrome P450’. The average methylation level
of stage II/III seminomas was higher than that of stage I
seminomas, with 162 elevated methylation sites in stage II/III
seminomas (Fig. 2A). A total of 20
genes in close proximity to the differential methylation sites were
selected, and their correlation with the DEGs was assessed. A total
of 14 differentially methylated sites and DEGs were paired up
(Fig. 2B). Pearson's correlation
analysis was carried out at the level of paired methylation and
gene expression. Survival analysis of 14 methylation-DEGs revealed
that only KCNC1 expression affected disease-free survival in
patients with seminomas (Fig. 2C and
D; Table III). These results
indicate methylation-DEG gene KCNC1 expression in stage II/III
seminomas is significantly lower than that in stage I seminomas and
lower KCNC1 expression reduces disease-free survival in patients
with seminomas.
| Table III.Disease-free survival of the
differentially methylated genes. |
Table III.
Disease-free survival of the
differentially methylated genes.
| Gene | P-value |
|---|
| KCNC1 | 0.025a |
|
KIAA0513 | 0.24 |
| LRMP | 0.88 |
| PTPRC | 0.53 |
| FMN2 | 0.16 |
| FMO3 | 0.44 |
| PIK3CG | 0.50 |
|
KIAA0513 | 0.24 |
| SLC18A2 | 0.43 |
| TUBB8 | 0.43 |
| SNORD37 | 0.88 |
| NKPD1 | 0.15 |
| BLK | 0.27 |
High KCNC1 expression of mRNA is found
in normal tissues and localized seminomas
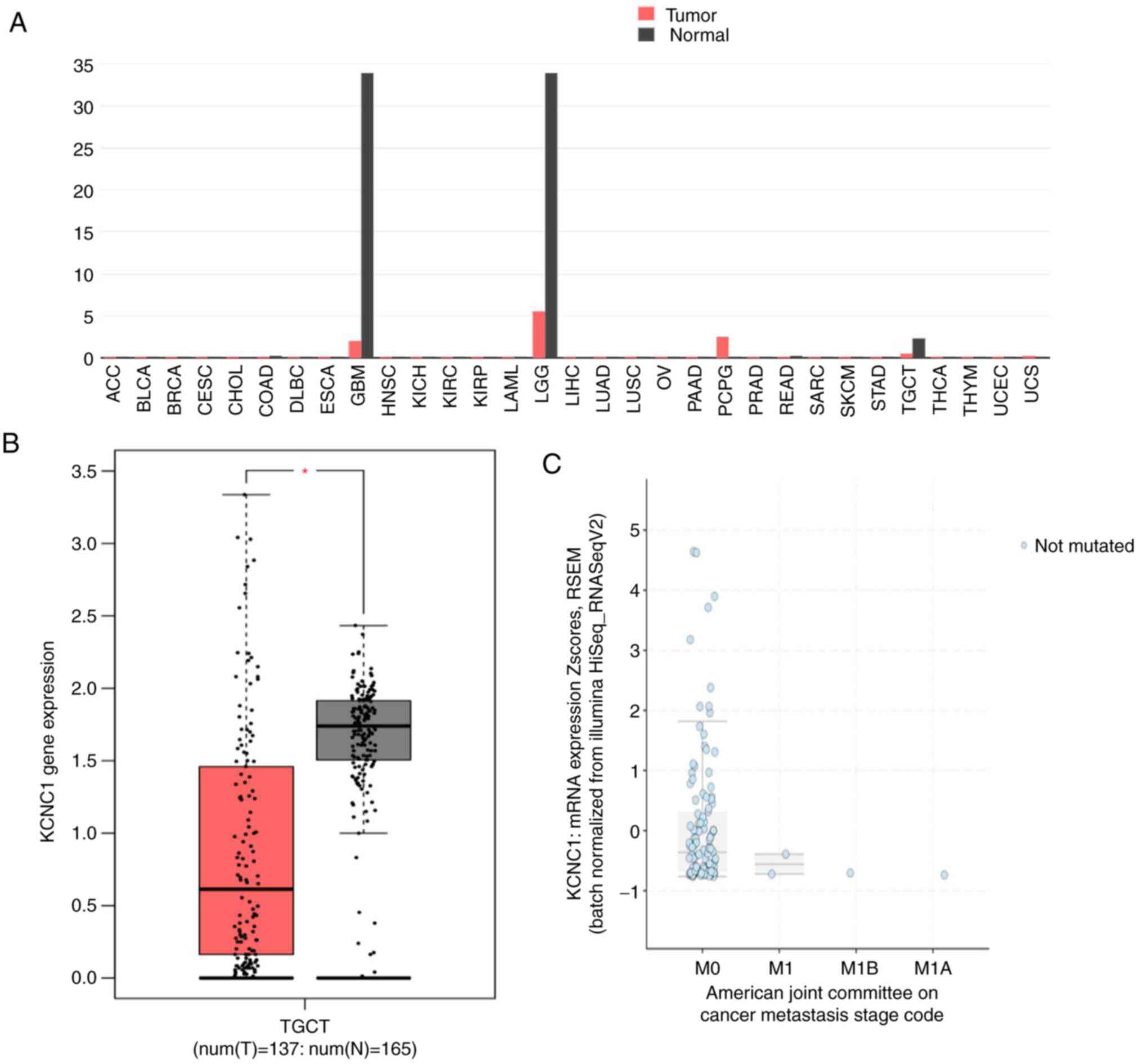
KCNC1 gene expression across all tumor
samples and paired normal tissues was analyzed using the GEPIA
online tool. The expression of KCNC1 in glioblastoma
multiforme (GBM), brain lower grade glioma (LGG) and TGCTs was
significantly lower than that in normal tissues (Fig. 3A and B). The expression of KCNC1 in
localized seminoma was also analyzed by cBioportal, which was
significantly higher than that in metastatic seminoma (Fig. 3C). In addition, there was no mutation
of DNMT3A in seminoma at present by analyzing of cBioportal. These
results further confirmed the expression of KCNC1 in different
stages of seminoma
Low KCNC1 expression is associated
with malignant seminomas
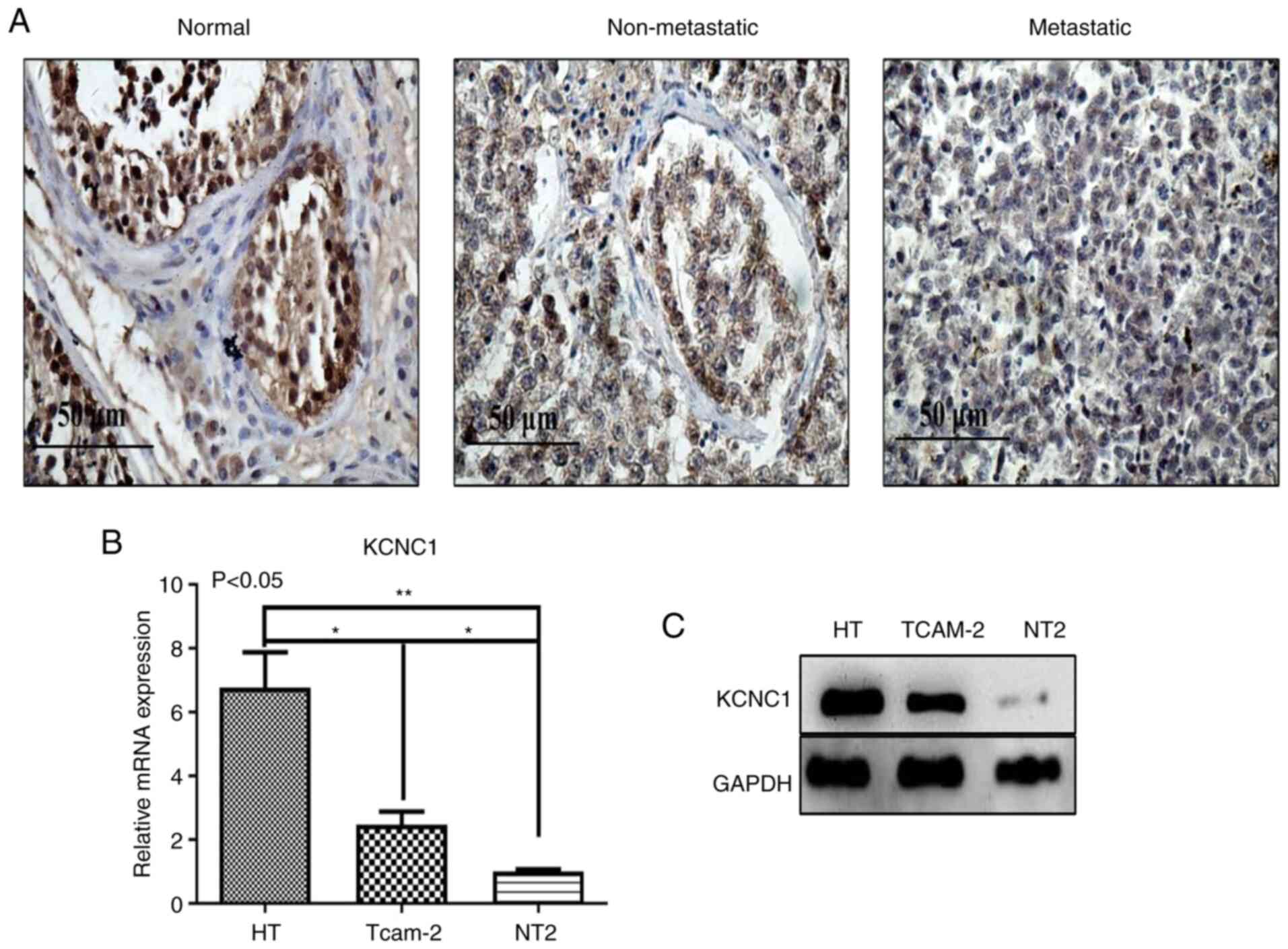
Immunohistochemical analysis showed that the
expression of KCNC1 protein was the highest in normal tissues,
significantly higher than that in non-metastatic and metastatic
seminoma tissues (Fig. 4A). KCNC1
expression was further verified in three cell lines. The expression
of KCNC1 was the highest in the normal HT cell line and the lowest
in the metastatic NT2 cell line (Fig. 3B
and C). The results above indicated that KCNC1 is negatively
correlated with the malignancy of seminoma, and as a
tumor-suppressor gene, KCNC1 plays an important role in tumor
progression.
Silencing and overexpression of KCNC1
alters seminoma cell invasion and metastasis, respectively
To determine whether KCNC1 plays a functional role
in seminoma cells, KCNC1-specific siRNAs were transiently
transfected into HT cells, and a lentivirus encoding KCNC1
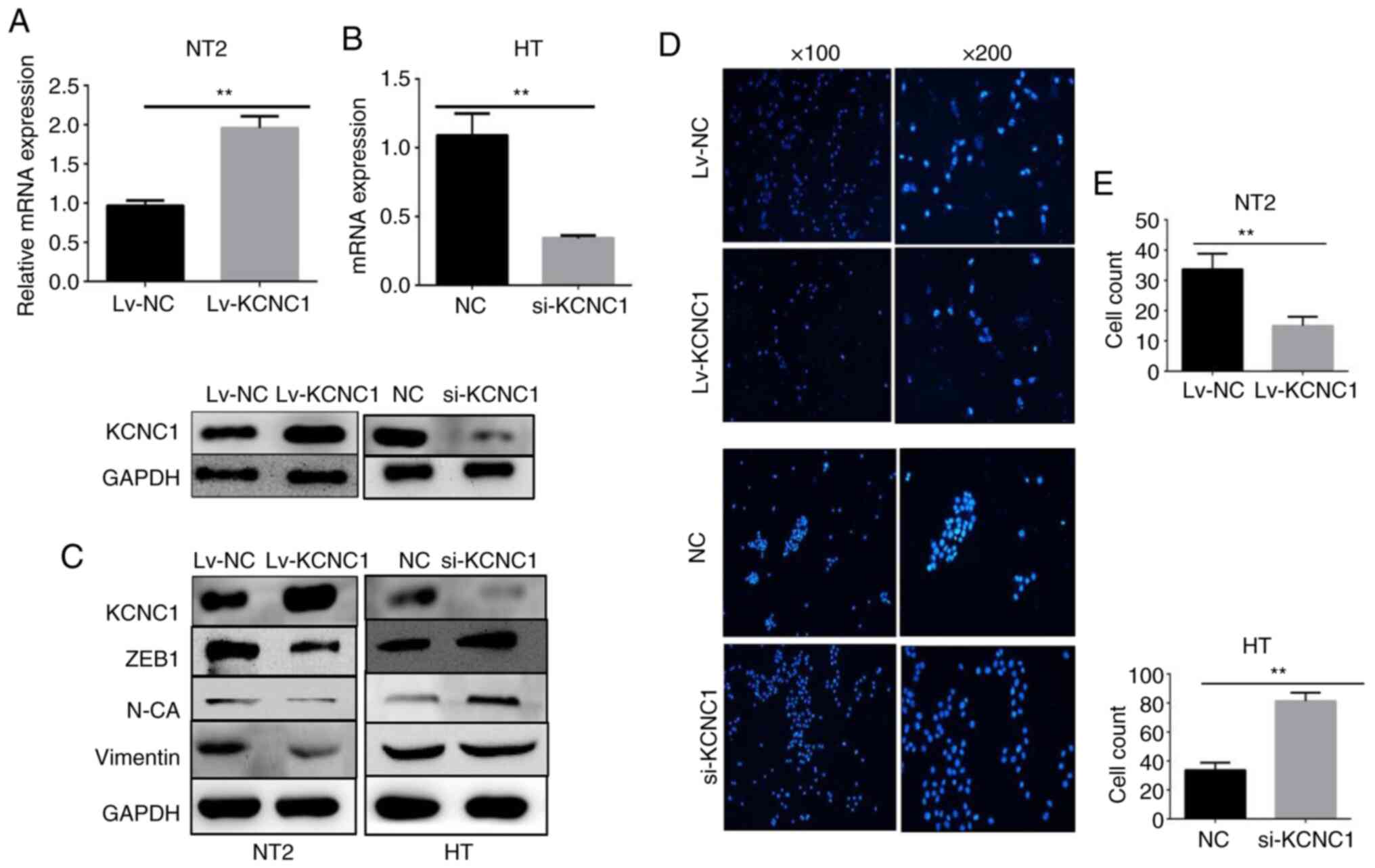
into NT2 cells. Changes in the KCNC1 mRNA and protein expression
were confirmed by RT-qPCR and western blot analysis (Fig. 5A and B). The expression of EMT-related
markers was verified following knockdown and overexpression of
KCNC1 in the HT and NT2 cells by western blot analysis. The
expression of vimentin, ZEB1 and N-cadherin was significantly
altered, which means that the metastatic ability of the
KCNC1-overexpressing NT2 cells and KCNC1-silenced HT cells were
altered accordingly (Fig. 5C). A
Transwell invasion assay showed that the invasion ability of HT
cells was significantly enhanced following KCNC1 knockdown.
However, overexpression of KCNC1 attenuated the invasion ability of
NT2 cells (Fig. 5D). Fig. 5E shows the quantification of invaded
cells. The invasion ability of tumor cells allows them to
metastasize to distant organs and thus increases its
malignancy.
Changes in the apoptosis and
proliferation of seminoma cells are observed following the aberrant
expression of KCNC1
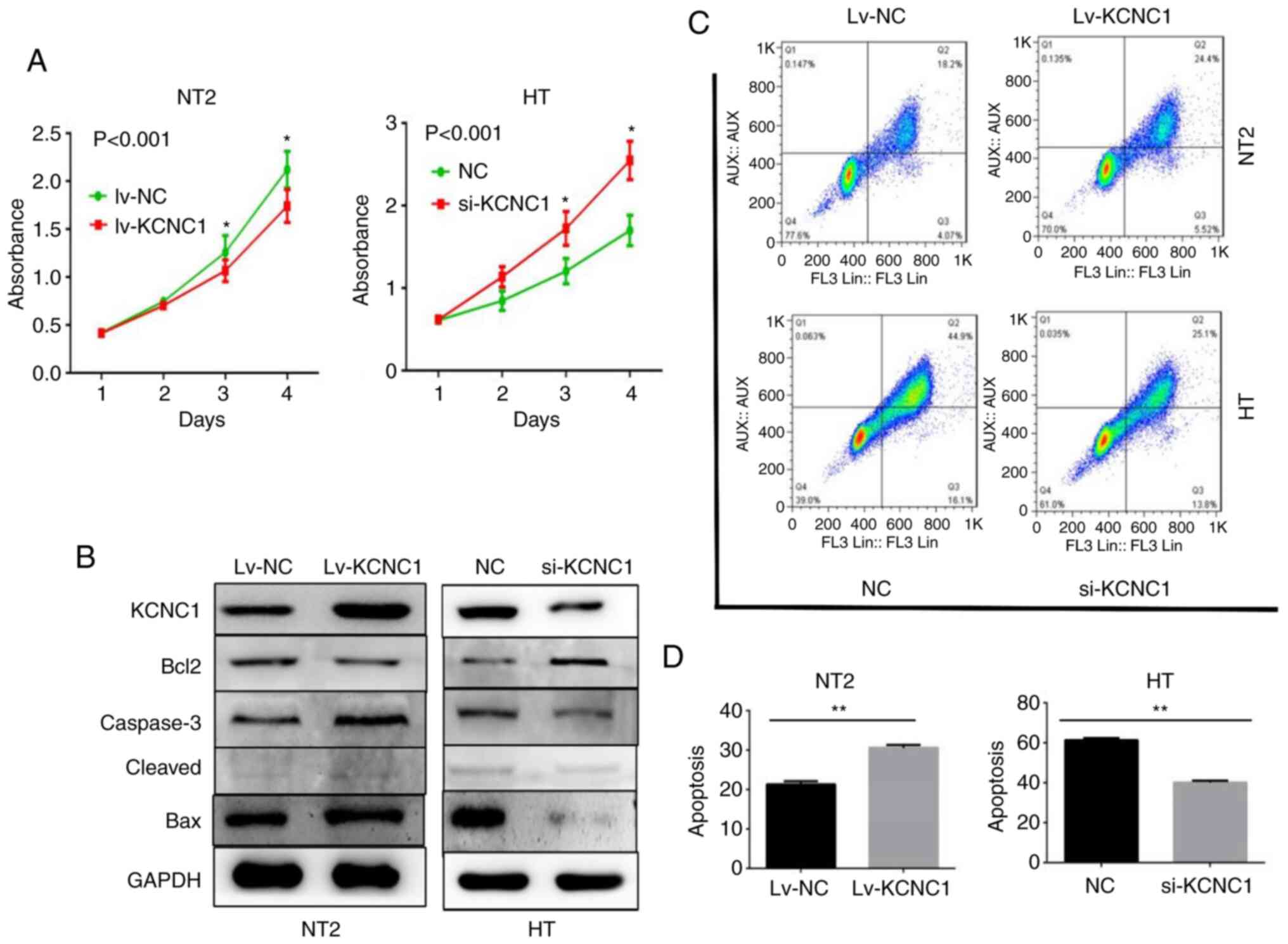
CCK-8 proliferation assay showed that KCNC1
knockout in HT cells and overexpression in NT2 cells significantly
increased and decreased cell proliferation at 48, 72 and 96 h,
respectively (Fig. 6A). The
apoptosis-related markers casepase-3 and Bax (associated with the
promotion of apoptosis) and Bcl-2 (associated with the inhibition
of apoptosis) expression levels also changed accordingly (Fig. 6B). The flow cytometry results
demonstrated that adjustments in KCNC1 induced an apparent change
in the number of early- (Annexin V signal only) and late- (Annexin
V plus PI signal) apoptotic cells. Annexin V/PI staining (Fig. 6C) and quantitative processing were
performed as shown in Fig. 6D.
KCNC1 knockout in HT cells and overexpression in NT2 cells
significantly decreased and increased cell apoptosis, respectively.
The above results demonstrated that HT and NT2 cell proliferation
and viability changed markedly following the aberrant expression of
KCNC1.
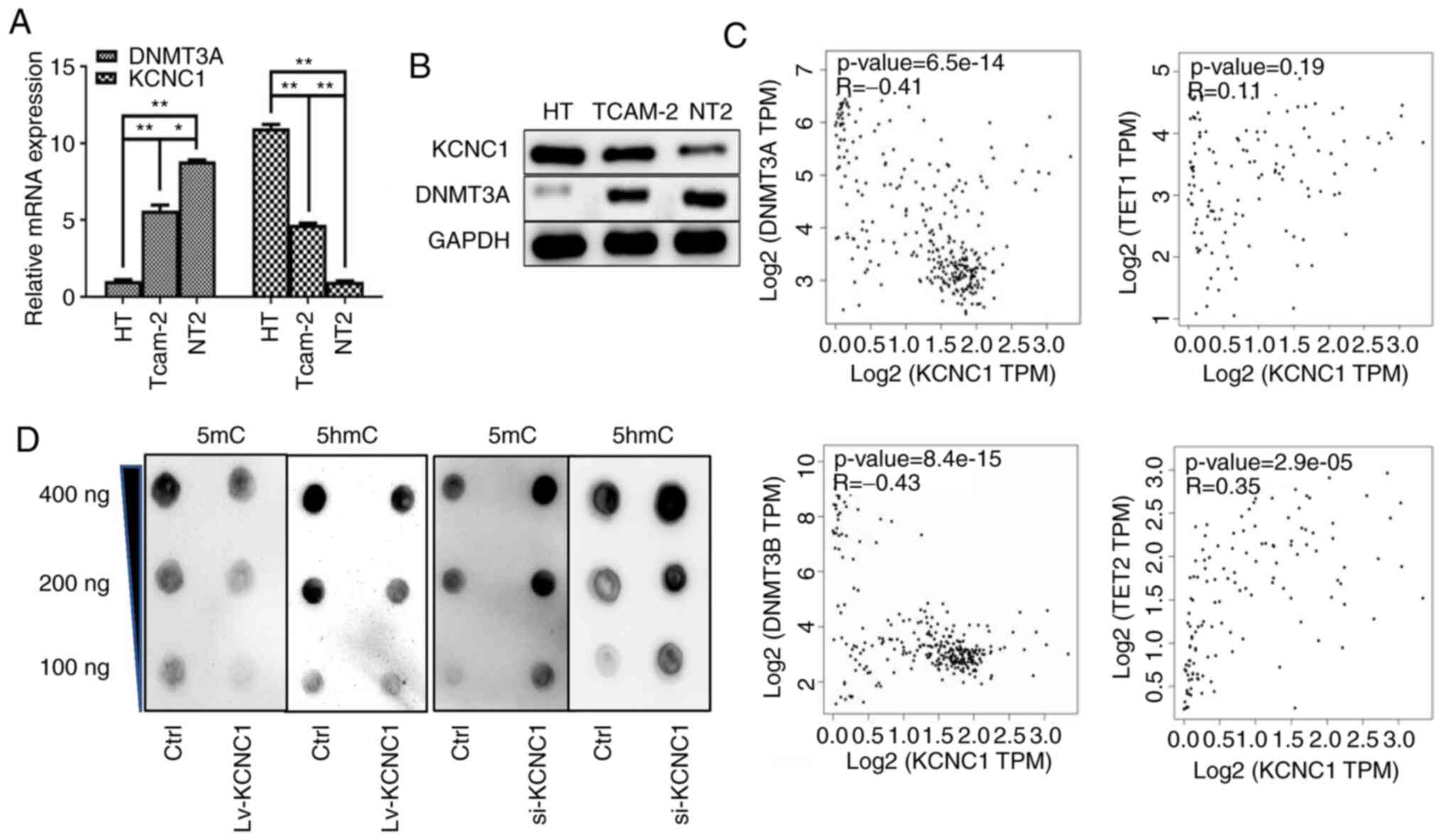
KCNC1 is negatively correlated with
methylation
The DNA methylation pattern in a genome is realized
by a DNA methyltransferase. DNMT3 is a DNA methyltransferase that
can be divided into DNMT3A and DNMT3B, and play a role in
maintaining methylation. The association between KCNC1 and DNMT3A
was performed in seminoma cells, and the result indicated that
KCNC1 is lowly expressed in metastatic seminoma cells with
hypermethylation (Fig. 7A and B). The
GEPIA online tool was used to confirm the correlation between
KCNC1, DNMT3a/DNMT3b and TET1/TET2 (Fig.
7C). Dot blot analysis was performed to determine the level of
methylation after the alterations in KCNC1 expression (Fig. 7D).
Discussion
The presence of seminoma poses a serious threat to
the health of men aged 15–35 years. Its biology and treatment
remain an active area of research worldwide. For early-stage
seminoma, surgical treatment can achieve a curative effect.
However, when tumors progress and metastasize, treatment options
are limited and patient prognosis is poor. Therefore, novel and
more effective therapeutic targets for seminoma are urgently
required. Public databases, such as TCGA and Gene Expression
Omnibus databases, contain a wealth of valuable transcriptome
sequencing data. Fully utilizing these public databases provides a
more in-depth understanding of the biomarkers and therapeutic
targets of seminoma, as well as the mechanisms underlying their
development and progression. In the present study, the RNA-seq data
from TCGA-TGCT dataset were analyzed, to screen for DEGs between
stage II/III and stage I seminomas. Methylation data of seminoma
specimens was also analyzed using the Elmer package. Corresponding
methylation-regulated DEGs were thus obtained, and a new
seminoma-related gene, KCNC1, was identified.
Immunohistochemical staining, western blot analysis and RT-qPCR
confirmed the expression of KCNC1 in seminoma tissues and cells.
The results showed that hypermethylation could inhibit the
expression of KCNC1, promoting seminoma progression and adversely
affecting the disease-free survival of seminoma patients. Following
the aberrant expression of KCNC1 in HT and NT2 cells, their
invasion, metastasis and proliferation abilities were significantly
altered, which influenced the progression of seminoma malignancy.
This suggested that KCNC1 can be used as a potential clinical
therapeutic target, and that the overexpression of KCNC1 can
effectively inhibit the progression of seminoma.
Normal body fluid volume, osmotic pressure and
electrolyte content are crucial to maintaining a normal metabolism,
stable internal environment and normal function of various organs.
When tumors occur, the tumor cells and surrounding environment
create the tumor microenvironment (TME) (25). In the TME, the opening and exchange of
ion channels on the surface of tumor cells also change accordingly,
which has a certain impact on the activity, invasion and
proliferation of tumor cells, and plays a role in the occurrence
and development of tumors (24,26). The
Kv channel on the plasma membrane is involved in several cellular
processes, including cell proliferation, migration, invasion and
apoptosis. KCNC1 is a subunit of the Kv3 potassium channel
(27). Voltage gated K+
channels are critically involved in the proliferation of tumor
cells. Moreover, in certain cells, the inhibition of the
K+ channel has been shown to be beneficial to apoptosis,
whereas the activation of the K+ channel can prevent
apoptosis (28). It was found herein
that hypermethylation can regulate the expression of KCNC1, and
then affect the proliferation, invasion and metastasis of seminoma
cells. By changing the expression of KCNC1, the metastasis ability
of the seminoma cell line was significantly altered, which was
mainly reflected in the level of EMT-related markers. At present,
research on the associated mechanism has not been elucidated, and
no relevant literature Is available. Further research would
therefore be beneficial.
In conclusion, the present study revealed that KCNC1
is associated with seminoma progression and is regulated by
methylation. The abnormal expression of KCNC1 may alter the number
of K+ channels on the surface of cancer cells,
potentially promoting tumor transformation, malignant progression
and metastasis. Based on the present findings, this may be a
potential mechanism of seminoma progression, and overexpression of
KCNC1 may be an innovative strategy for the treatment of seminomas.
The mechanism of KCNC1 remains unclear. The present study
demonstrated that the expression of KCNC1 can affect the expression
of DNMT3A/DNMT3B and TET1/TET2, and then change the methylation
level of seminoma cells. Therefore, it needs to be explored whether
KCNC1 can affect the expression of a promoter by regulating their
promoter sequence by double luciferase reporter gene detection
experiment and Chip experiment. In addition, the effects of DNMT3A
on the methylation of the promoter lesion of KCNC1 is also a
research direction worthy of further study, because at present, the
regulation mode between many genes is bidirectional. If we can
confirm that DNMT3A regulates the promoter of KCNC1, it will deepen
our understanding of the function of KCNC1 in the progression of
seminoma. Only through further study of the mechanism is it
possible to fully identify the important role of KCNC1 in the
progression of seminoma.
Acknowledgements
The authors would like to thank all members of the
laboratory staff for their great assistance with the
experiments.
Funding
This work was supported by the Tianjin Municipal
Science and Technology Bureau Project (grant no.
18ZXDBSY00020).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SC conducted the project development, data
collection, data analysis, and manuscript writing. LX carried out
the data analysis. HP performed the data collection. ZW assisted in
the experimental procedures. JB conducted the project development,
and coordinated financial support. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The use of tumor tissue for immunohistochemistry was
approved by the Second Hospital of Tianjin Medical University
Ethics Committee (Tianjin, China). The family members of the
patients were informed that the tumor tissue removed during surgery
would be used for further scientific research, and the approval of
the family members of the patients and their informed consent was
obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Gonzalez-Exposito R, Merino M and Aguayo
C: Molecular biology of testicular germ cell tumors. Clin Transl
Oncol. 18:550–556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
di Pietro A, de Vries EG, Gietema JA,
Spierings DC and de Jong S: Testicular germ cell tumours: The
paradigm of chemo-sensitive solid tumours. Int J Biochem Cell Biol.
37:2437–2456. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meyts ER, McGlynn KA, Okamoto K, Jewett MA
and Bokemeyer C: Testicular germ cell tumours. Lancet.
387:1762–1774. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chaganti RS and Houldsworth J: Genetics
and biology of adult human male germ cell tumors. Cancer Res.
60:1475–1482. 2000.PubMed/NCBI
|
|
5
|
Jørgensen N, Meyts ER, Graem N, Müller J,
Giwercman A and Skakkebaek NE: Expression of immunohistochemical
markers for testicular carcinoma in situ by normal human fetal germ
cells. Lab Invest. 72:223–231. 1995.PubMed/NCBI
|
|
6
|
Richie JP: Re: Management of seminomatous
testicular cancer: A binational prospective population-based study
from the Swedish norwegian testicular cancer study group. J Urol.
186:2255–2256. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chung PW, Gospodarowicz MK, Panzarella T,
Jewett MA, Sturgeon JF, Tew-George B, Bayley AJ, Catton CN,
Milosevic MF, Moore M and Warde PR: Stage II testicular seminoma:
Patterns of recurrence and outcome of treatment. Eur Urol.
45:754–759; discussion 759-60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fankhauser CD, Curioni-Fontecedro A,
Allmann V, Beyer J, Tischler V, Sulser T, Moch H and Bode PK:
Frequent PD-L1 expression in testicular germ cell tumors. Br J
Cancer. 113:411–413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamada Y, Takayama KI, Fujimura T,
Ashikari D, Obinata D, Takahashi S, Ikeda K, Kakutani S, Urano T,
Fukuhara H, et al: A novel prognostic factor TRIM44 promotes cell
proliferation and migration, and inhibits apoptosis in testicular
germ cell tumor. Cancer Sci. 108:32–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costello JF and Plass C: Methylation
matters. J Med Genet. 38:285–303. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Beta M, Chitipothu S, Khetan V, Biswas J
and Krishnakumar S: Hypermethylation of adenomatosis polyposis
coli-2 and its tumor suppressor role in retinoblastoma. Curr Eye
Res. 40:719–728. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ohtani-Fujita N, Dryja TP, Rapaport JM,
Fujita T, Matsumura S, Ozasa K, Watanabe Y, Hayashi K, Maeda K,
Kinoshita S, et al: Hypermethylation in the retinoblastoma gene is
associated with unilateral, sporadic retinoblastoma. Cancer Genet
Cytogenet. 98:43–49. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perney TM, Marshall J, Martin KA,
Hockfield S and Kaczmarek LK: Expression of the mRNAs for the Kv3.1
potassium channel gene in the adult and developing rat brain. J
Neurophysiol. 68:756–766. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stegen B, Klumpp L, Misovic M, Edalat L,
Eckert M, Klumpp D, Ruth P and Huber SM: K(+) channel signaling in
irradiated tumor cells. Eur Biophys J. 45:585–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Edalat L, Stegen B, Klumpp L, Haehl E,
Schilbach K, Lukowski R, Kühnle M, Bernhardt G, Buschauer A, Zips
D, et al: BK K+ channel blockade inhibits radiation-induced
migration/brain infiltration of glioblastoma cells. Oncotarget.
7:14259–14278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ouadid-Ahidouch H and Ahidouch A: K+
channel expression in human breast cancer cells: involvement in
cell cycle regulation and carcinogenesis. J Membr Biol. 221:1–6.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen BF, Gu S, Suen YK, Li L and Chan WY:
MicroRNA-199a-3p, DNMT3A, and aberrant DNA methylation in
testicular cancer. Epigenetics. 9:119–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Duan H, Zhou S, Liu Z, Wu D, Zhao
T, Xu S, Yang L and Li D: MicroRNA-199a-3p functions as tumor
suppressor by regulating glucose metabolism in testicular germ cell
tumors. Mol Med Rep. 14:2311–2320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li R, Qu H, Wang S, Wei J, Zhang L, Ma R,
Lu J, Zhu J, Zhong WD and Jia Z: GDCRNATools: An R/Bioconductor
package for integrative analysis of lncRNA, miRNA and mRNA data in
GDC. Bioinformatics. 34:2515–2517. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoadley KA, Yau C, Wolf DM, Cherniack AD,
Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V,
et al: Multiplatform analysis of 12 cancer types reveals molecular
classification within and across tissues of origin. Cell.
158:929–944. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silva TC, Coetzee SG, Gull N, Yao L,
Hazelett DJ, Noushmehr H, Lin DC and Berman BP: ELMER v.2: An
R/Bioconductor package to reconstruct gene regulatory networks from
DNA methylation and transcriptome profiles. Bioinformatics.
35:1974–1977. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schulz M, Salamero-Boix A, Niesel K,
Alekseeva T and Sevenich L: Microenvironmental regulation of tumor
progression and therapeutic response in brain metastasis. Front
Immunol. 10:17132019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Palma M, Biziato D and Petrova TV:
Microenvironmental regulation of tumour angiogenesis. Nat Rev
Cancer. 17:457–474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ehnman M and Larsson O: Microenvironmental
targets in sarcoma. Front Oncol. 5:2482015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rudy B and McBain CJ: Kv3 channels:
Voltage-Gated K+ channels designed for high-frequency repetitive
firing. Trends Neurosci. 24:517–526. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lang F and Stournaras C: Ion channels in
cancer: Future perspectives and clinical potential. Philos Trans R
Soc Lond B Biol Sci. 369:201301082014. View Article : Google Scholar : PubMed/NCBI
|