Introduction
Osteosarcoma (OS) is the most common malignant bone
tumor in children and adolescents (1). The current treatment strategy for
primary OS includes neoadjuvant chemotherapy and definitive
surgery. A methotrexate, adriamycin and cisplatin (CDDP) (MAP)
regimen is administered as a standard neoadjuvant treatment.
Despite this multimodal approach, ~30% of patients with OS die due
to relapse (2,3); this mortality rate has not changed for
>20 years.
Sensitivity to preoperative chemotherapy is a
significant prognostic factor for patients with OS (3–5).
Chemosensitivity of patients with OS is assessed according to the
percentage of tumor necrosis (4,5) or
remaining viable tumor cells in resected tumor specimens (3). It is not specific to chemotherapy
regimen or drug used. Predicting chemosensitivity at an early stage
of diagnosis is a major challenge in OS treatment. Identification
of patients whose tumors have the potential to resist current
preoperative chemotherapy prior to treatment initiation would avoid
ineffective chemotherapy and allow the use of alternative
chemotherapy. Numerous studies employing immunohistochemistry or
reverse transcription-quantitative (RT-q)PCR have proposed
candidate molecules that are associated with outcomes or
chemosensitivity, such as p16 (6),
RUNX family transcription factor 2 (RUNX2) (7), C-X-C motif chemokine receptor 4
(8), MDR1 (P-glycoprotein) (9) and COP9 signalosome subunit 3 (COPS3)
(10). Expression array analysis has
also been employed to define gene profiles for predicting
chemosensitivity of patients with OS; various genes, including
those encoding twist family bHLH transcription factor 1, programmed
cell death 5 (11), desmoplakin,
phospholipase A2 group IIA (12),
aldo-keto reductase family 1 member C4, glutathione peroxidase 1
and glutathione S-transferase ω1 (13), have been reported as chemosensitivity
predictors. However, no overlapping genes were observed between
these aforementioned studies. Therefore, the present study aimed to
identify potential genomic markers for the prediction of
chemosensitivity in patients with OS using a genomic approach on
preoperative biopsy samples.
Materials and methods
Patient eligibility and treatment
The Chiba Cancer Center Tissue Bank includes 124
frozen tumor samples obtained from patients with OS that were
treated at Chiba Cancer Center (Chiba, Japan) since 1993. Eligible
cases were selected for the present study according to the
following criteria: i) Diagnosed with conventional, high-grade OS
occurring in the extremity, ii) pre-therapeutic biopsy samples were
available, iii) age at diagnosis <25 years, and iv) follow-up
was monitored for >5 years for survival cases. Finally, 50 tumor
samples that satisfied these criteria were selected for study,
Chiba OS (ChOS; Table SI). The
present study was approved by the institutional review board of the
Chiba Cancer Center (approval no. CCC19-14) and conformed to the
provisions of the Declaration of Helsinki. Written informed consent
was obtained from patients or legally authorized
representatives.
Preoperative chemotherapy consisting of a MAP
regimen was administered to all patients as described previously
(14). If radiological evaluation by
CT or MRI conducted during preoperative chemotherapy revealed
enlargement of the primary tumor, ifosfamide was added to the MAP
regimen. Definitive tumor resection was performed following
preoperative chemotherapy, and chemotherapy response was evaluated
histologically using resected tumor according to the relative area
of necrosis. A ‘good response’ was defined as a necrotic area
>90% of the tumor; ‘poor response’ was defined as necrotic tumor
area <90%, as previously described (4,14).
Tumor specimen and genomic DNA
extraction
All tumor specimens were obtained from
pre-therapeutic biopsy samples. The tumor content of frozen samples
was histologically confirmed as >80% tumor cellularity using
paraffin sections (4-µm thick) obtained from the same biopsy.
Genomic DNA was extracted using the standard proteinase K digestion
method (15) or AllPrep DNA/RNA Mini
kit (Qiagen KK). DNA concentration and purity were determined using
a NanoDrop 100ND-1000 Spectrometer (Thermo Fisher Scientific,
Inc.).
Array comparative genomic
hybridization (aCGH) and data analysis
For aCGH analysis, 500 ng tumor or control DNA was
fragmented and chemically labeled with Cy3- or Cy5-dye,
respectively. Genomic DNAs extracted from normal placenta tissue
was used as control DNA for aCGH. High-resolution aCGH was
performed using the Agilent Human Genome CGH Microarray kit 4×44K
(Agilent Technologies, Inc.), according to the manufacturers
protocols, and analyzed using Agilent Oligonucleotide Array-Based
CGH for Genomic DNA Analysis (version 3.1; Agilent Technologies,
Inc.). Data extraction from scanned microarray images was performed
using Feature Extraction Software (versions 9.5.3.1, 10.7.3.1 and
11.0.1.1; Agilent Technologies, Inc.). Raw data were subsequently
transferred to Agilent Genomic Workbench Software Version 6.5
(Agilent Technologies, Inc.) and further analyzed using ADM-2
algorithm with a threshold of 5.5. In order to find copy number
variations (CNVs) that discriminated between patients who
demonstrated good and poor responses, differential CNV analyses
were performed with P-value <0.01 using Genomic Workbench
Software. All final retained CNVs were validated using quality
control data of the software. All genomic positions were defined
according to the University of California Santa Cruz Human (version
hg19; Genome Reference Consortium h37;
genome.ucsc.edu/cgi-bin/hgGateway?db=hg19). Microarray data were
deposited in the National Center for Biotechnology Information Gene
Expression Omnibus database (ncbi.nlm.nih.gov/geo/; accession no.
GSE154540).
Targeted exome sequencing
Targeted exome sequencing of 409 cancer-associated
genes in tumor and non-tumor samples was conducted using the Ion
AmpliSeq™ Comprehensive Cancer Panel (CCP) (Thermo Fisher
Scientific, Inc.). For each sample, 40 ng genomic DNA was amplified
and used for library preparation using the Ion AmpliSeq™ Library
2.0–96LV and the Ion Barcode Adaptors 1–96 kits (both Thermo Fisher
Scientific, Inc.). Then, the library was quantified using the Ion
Library Quantification kit (Thermo Fisher Scientific, Inc.) and
diluted to a final concentration of 8 pM. Targeted sequencing was
performed using an Ion Proton sequencer (Thermo Fisher Scientific,
Inc.) with an Ion PI Chip v2 according to the manufacturers
instructions. The sequencing results were analyzed using Torrent
Suite v5.0.4 software (Thermo Fisher Scientific, Inc.) to align the
barcoded reads with the human reference genome (hg19) and to
generate run metrics, including chip loading efficiency, total
reads, and quality. Variant Caller v5.0 and Ion Reporter™ 5.2
software (Thermo Fisher Scientific, Inc.) were used for variant
detection (allele frequency >5%) and annotation (The Single
Nucleotide Polymorphism Database v146; ncbi.nlm.nih.gov/snp/),
respectively. Each variant was confirmed visually using Integrated
Genomics Viewer or validated using Sanger sequencing performed on
an Applied Biosystems 3730 DNA analyzer with Sequencing Analysis
Software v6.0 (both Applied Biosystems; Thermo Fisher Scientific,
Inc.). The CNVs of the 409 genes were calculated by normalizing the
amplicon coverage data from a tumor sample with that of paired
non-tumor tissue. Each copy number ratio (tumor/normal) was
demonstrated as a log2 ratio (log fold change). Sequence data have
been deposited in the Japanese Genotype-phenotype Archive, hosted
by the DNA Data Bank of Japan (accession no. JGAS000282).
Semiquantitative RT-PCR and
RT-qPCR
Total RNA was prepared from human tissue and OS cell
lines using AllPrep DNA/RNA Mini or RNeasy Mini kit (both from
Qiagen KK) and reverse-transcribed using random primers and
SuperScript II (Thermo Fisher Scientific, Inc.), according to the
manufacturers protocols. The synthesized cDNA was subjected to
PCR-based amplification using rTaq DNA polymerase (Takara Bio,
Inc.). Primer sequences are listed in Table SII.
A Veriti PCR system (Thermo Fisher Scientific, Inc.)
was used for PCR amplification. The resultant PCR products were
separated on 2% agarose gel and visualized using a UV
transilluminator (ATTO Corporation) after ethidium bromide
staining. RT-qPCR analysis was conducted using an ABI Prism 7500
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). TaqMan
primers and probes for CDK4 (cat. no. HS00364847_m1) and
β-actin (cat. no. 4310881E) were purchased from Applied
Biosystems (Thermo Fisher Scientific, Inc.). RT-qPCR amplification
was performed in a 20 µl reaction volume containing 10 µl 2X TaqMan
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.), 1
µl 20X TaqMan primers and probes and 2 µl cDNA. The thermocycling
conditions were as follows: Decontamination for 2 min at 50°C,
initial denaturation for 10 min at 95°C and 50 cycles of PCR for 15
sec at 95°C and 1 min at 60°C. The relative standard curve method
was used for data quantification (16). All reactions were performed in
triplicate.
Immunohistochemistry
All tumor samples were routinely decalcified and
fixed with 10% buffered formalin for 12 h at room temperature and
embedded in paraffin. Sections (4-µm thick) were deparaffinized and
antigens were retrieved by autoclaving in 0.01 M citrate buffer
(pH, 6.0) at 97°C for 60 min. Endogenous peroxidase activity was
blocked by exposing the slides to 3% hydrogen peroxide for 15 min
at room temperature. The primary antibody utilized was CDK4 (1:500;
cat. no. AHZ0202; Invitrogen; Themo Fisher Scientific, Inc.). The
slides were incubated for 20 min at room temperature with the
primary antibody and subsequently labeled using the EnVision+/HRP
system (Dako; Agilent Technologies, Inc.). Immunoreactions were
visualized using diaminobenzidine (0.03%, 1 min at room
temperature) by light microscope, and the sections were
counterstained with hematoxylin (FUJIFILM Wako Pure Chemical
Corporation) for 5 min at room temperature.
Cell culture, transfection and drug
treatment
Human OS cell lines U2OS and SJSA1 were obtained
from the American Type Culture Collection and maintained in DMEM or
RPMI-1640 (FUJIFILM Wako Pure Chemical Corporation) supplemented
with 10% heat-inactivated FBS, 100 IU/ml penicillin and 100 µg/ml
streptomycin (all Thermo Fisher Scientific, Inc.) in a humidified
atmosphere of 5% CO2 at 37°C. For transient expression,
CDK4 cDNA was amplified using PCR from human cDNA libraries
incorporating a C-terminal 3X Flag-tag sequence, as described by
Tatsumi et al (17) and
ligated into the EcoRI and XhoI sites of the pCDNA3
vector (Thermo Fisher Scientific, Inc.). For CDK4 overexpression,
OS cells were seeded in 6-well plates (80% confluency) and
transfected with 2.5 µg pCDNA3-CDK4 or empty plasmid (control)
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature. Following 24 h transfection,
DNA damage and apoptosis were introduced by further incubation with
0–60 µM CDDP (Sigma-Aldrich; Merck KGaA) for 24 h in a humidified
atmosphere of 5% CO2 at 37°C. In order to inhibit CDK4
activity, transfected cells were cultured with 0–15 nM PD 00332991
(CDK4/6 inhibitor palbociclib; Santa Cruz Biotechnology, Inc.) for
24 h in a humidified atmosphere of 5% CO2 at 37°C.
Immunoblotting
Whole-cell lysates were separated using SDS-PAGE and
transferred to PVDF membranes (Immobilon-P; EMD Millipore), as
previously described (17). Membranes
were incubated with primary antibodies (all 1:1,000) at room
temperature for 2 h and then treated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:1,500) at room temperature
for 1 h. Finally, signals were detected using a LAS-4000 Image
Analyzer following incubation with ECL reagent (both GE
Healthcare). The following antibodies were purchased from Cell
Signaling Technologies, Inc.: Anti-CDK4 (cat. no. #12790),
anti-Cyclin D1 (cat. no. #2978), anti-Bcl-2 (cat. no. #2870),
anti-caspase-9 (cat. no. #9502), anti-E2F transcription factor 1
(E2F1; cat. no. #3742), anti-retinoblastoma (Rb; cat. no. #9309),
anti-phosphorylated (phospho-)Rb (Ser807/811, cat. no. #9308),
anti-phospho-Rb (Ser780, cat. no. #9307), anti-phospho-Rb
(Ser807/811; cat. no. #9308), HRP-conjugated anti-rabbit (cat. no.
#7074) and HRP-conjugated anti-mouse (cat. no. #7076). Anti-Flag
(M2) antibody (cat. no. F3040) was purchased from Sigma-Aldrich
(Merck KGaA), anti-poly(ADP-ribose) polymerase (PARP)-1 (cat. no.
sc-8007) was purchased from Santa Cruz Biotechnology, Inc. and
anti-actin (cat. no. 013-24553) was purchased from FUJIFILM Wako
Pure Chemical Corporation.
Cell viability and apoptosis
detection
Measurement of cell viability was conducted by
water-soluble tetrazolium salt (WST)-8 assay (Dojindo Molecular
Technologies, Inc.). Apoptosis progression caused by CDDP or
palbociclib treatment was monitored by accumulation of cleaved
caspase-9 and PARP1 via immunoblotting, as previously described
(17).
Statistical analysis
Overall survival (OAS) was calculated using the
Kaplan-Meier method, and differences in survival were assessed
using the log-rank test and Cox proportional-hazards regression
method. OAS was defined as the period from the date of diagnosis to
that of death or the last follow-up and described with 95%
confidence interval (CI). Comparisons were assessed using
χ2 and Fishers exact tests for categorical variables and
Mann-Whitney tests for continuous variables. RT-qPCR data are
presented as the mean (n=3)for triplicate experiments. Cell
viability data are presented as the mean ± SD (n=4). All
statistical tests were two-sided. Statistical analysis was
performed using JMP® version 9.0.2 software (SAS
Institute, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Chemosensitivity is a significant
prognostic factor for patients with OS
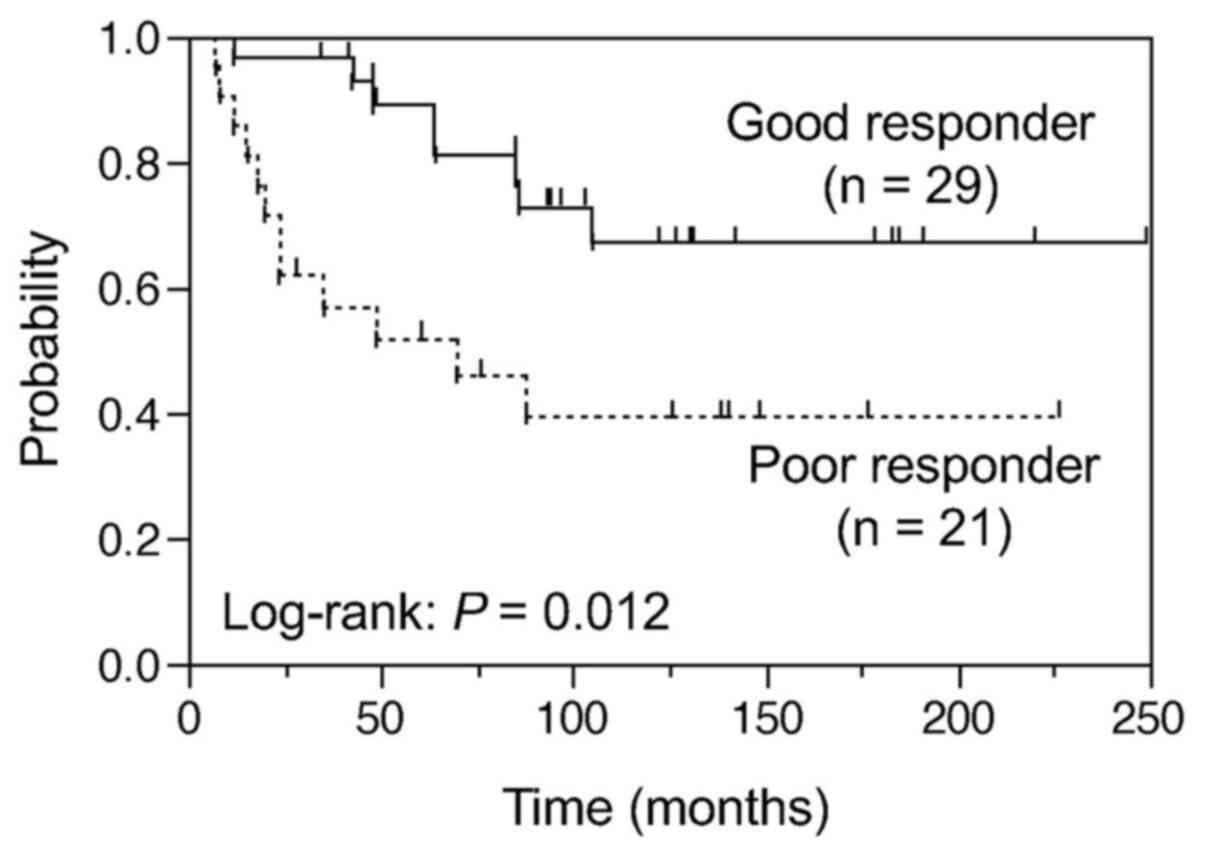
The 5-year OAS for 50 patients with OS was 73% (95%
CI, 59–84%). Univariate analysis of clinical factors revealed that
histological response to preoperative chemotherapy (‘chemotherapy
response’) and metastasis at diagnosis were statistically
significant prognostic factors associated with OAS (P=0.012 and
P=0.0058, respectively), whereas chemotherapy regimen (MAP in the
presence or absence of ifosfamide) was not associated with OAS
(P=0.36) (Table I; Fig. 1). These results were confirmed by
multivariate analysis (Table I).
 | Table I.Univariate and multivariate analysis
of prognostic factors for OS. |
Table I.
Univariate and multivariate analysis
of prognostic factors for OS.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Factor | N (%) | 5-year OAS, % | P-value | 5-year EFS, % | P-value | Risk ratio | 95% CI | P-value |
|---|
| Age, years |
|
|
|
|
|
|
|
|
|
≤20 | 8 (16) | 63 | 0.5400 | 54 | 0.6400 |
|
|
|
|
>20 | 42 (84) | 75 |
| 50 |
|
|
|
|
| Location |
|
|
|
|
|
|
|
|
|
Femur | 30 (60) | 70 | 0.4500 | 56 | 0.9500 |
|
|
|
|
Other | 20 (40) | 79 |
| 50 |
|
|
|
|
| Histological
subtype |
|
|
|
|
|
|
|
|
|
Osteoblastic | 42 (84) | 73 | 0.6200 | 55 | 0.9600 |
|
|
|
|
Other | 8 (16) | 73 |
| 47 |
|
|
|
|
| Chemotherapy
response |
|
|
|
|
|
|
|
|
|
Good | 29 (58) | 89 | 0.0120a | 65 | 0.0200a |
|
|
|
|
Poor | 21 (42) | 52 |
| 38 |
|
|
|
|
| Metastasis at
diagnosis |
|
|
|
|
|
|
|
|
|
Yes | 6 (12) | 33 | 0.0058a | 17 | 0.0078a |
|
|
|
| No | 44 (88) | 79 |
| 59 |
|
|
|
|
| Chemotherapy
regimen |
|
|
|
|
|
|
|
|
|
MAP | 25 (50) | 88 | 0.3600 | 63 | 0.2000 |
|
|
|
|
MAP-I | 25 (50) | 60 |
| 44 |
|
|
|
|
| OAS |
|
|
|
|
|
|
|
|
|
Chemotherapy response, Poor
vs. good |
|
|
|
|
| 3.24 | 1.31–8.50 | 0.011a |
|
Metastasis at diagnosis, Yes
vs. no |
|
|
|
|
| 4.34 | 1.35–12.0 | 0.016a |
|
Chemotherapy regimen, MAP-I
vs. MAP |
|
|
|
|
| 1.11 | 0.44–2.89 | 0.830 |
| EFS |
|
|
|
|
|
|
|
|
| Chemotherapy
response, Poor vs. good |
|
|
|
|
| 2.5 | 1.08–5.96 | 0.032a |
| Metastasis at
diagnosis, Yes vs. no |
|
|
|
|
| 3.48 | 1.11–9.21 | 0.034a |
| Chemotherapy
regimen, MAP-I vs. MAP |
|
|
|
|
| 1.29 | 0.55–3.17 | 0.560 |
Significant enrichment of copy number
gains of 12q14.1 and 19q13.32 in poor responders
Genome-wide aCGH analysis of the pretreatment of OS
tissue samples was performed to identify CNVs associated with tumor
chemosensitivity. Differential analyses of CNV between good (n=29)
and poor responder (n=21) cohorts identified three loci that were
significantly associated with chemosensitivity (P<0.01; Table II). Recurrent gain of chromosomes
12q14.1 and 19q13.32 were identified in 5 of 21 (24%) poor
responders, whereas these gains were not identified in good
responders (0/29, 0%; P=0.0096). A recurrent gain of chromosome
7q36.2-3 was also significantly observed in 8 of 29 (28%) good
responders compared with poor responders (0/21; P=0.008).
Meanwhile, there was no statistically significant recurrent copy
number loss between good and poor responders.
| Table II.Chromosomal alterations significantly
associated with chemosensitivity. |
Table II.
Chromosomal alterations significantly
associated with chemosensitivity.
| Cytogenetic
band | No. of probes | Gain in good
responder (n=29) | Gain in poor
responder (n=21) | P-value | Genes | Cases |
|---|
| 7q36.2–3 | 13 | 8 | 0 | 0.0080 | DPP6, PAXIP1,
HTR5A, INSIG1 | ChOS-5, 25, 27, 31,
39, 63, 76, 82 |
| 12q14.1 | 10 | 0 | 5 | 0.0096 | AGAP2, TSPAN31,
CDK4, MARCH9, CYP27B1 | ChOS-38, 46, 48,
50, 90 |
| 19q13.32 | 12 | 0 | 5 | 0.0096 | BCL3, CBLC,
BCAM, PVRL2, TOMM40, APOE | ChOS-21, 34, 48,
50, 83 |
CDK4 is a key gene associated with
copy number gain of 12q14.1
It was hypothesized that one or multiple genes in
loci with copy number gain, such as 12q14.1 and 19q13.32,
contribute to chemo-resistance in poor responders. In order to
address this, mRNA levels of 11 genes in gained regions 12q14.1 and
19q13.32 were compared between 24 tumors (13 good and 11 poor
responders) with available RNA via semiquantitative RT-PCR.
CDK4 in 12q14.1 was the only gene with higher transcript
levels that were positively associated with copy number gain
(Fig. S1).
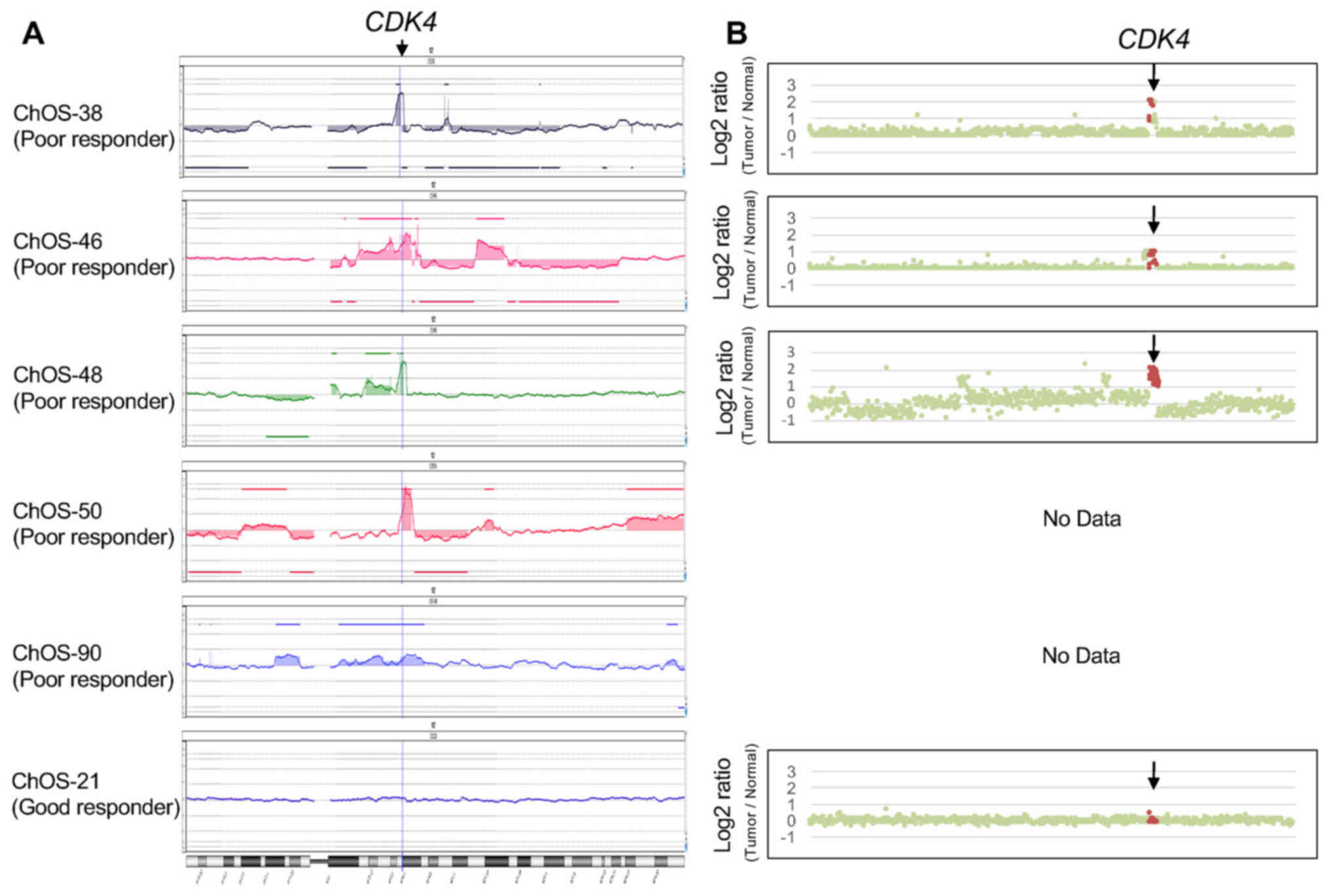
In addition, aCGH confirmed that CDK4 was
located in the minimal region of overlap in the gained region. Copy
number analysis showed amplification or gain of the CDK4
locus in poor responders, which was not observed in good responders
(Fig. 2A). In order to identify
additional markers based on gene mutations, next generation
sequencing (NGS)-based targeted exome sequencing of 409
cancer-associated genes was performed with seven and nine tumors
from good and poor responder cohorts, respectively. Although 16 and
13 non-synonymous somatic gene mutations were found in the
respective cohorts, no recurrent mutation was observed, except for
CDK4 amplification or gain in poor responders (Table SIII). Increased copy number of
CDK4 was also confirmed by calculation of relative read
depth of the target exome sequences in three tumors from poor
responders (Fig. 2B).
CDK4 transcript levels in primary OS tissue
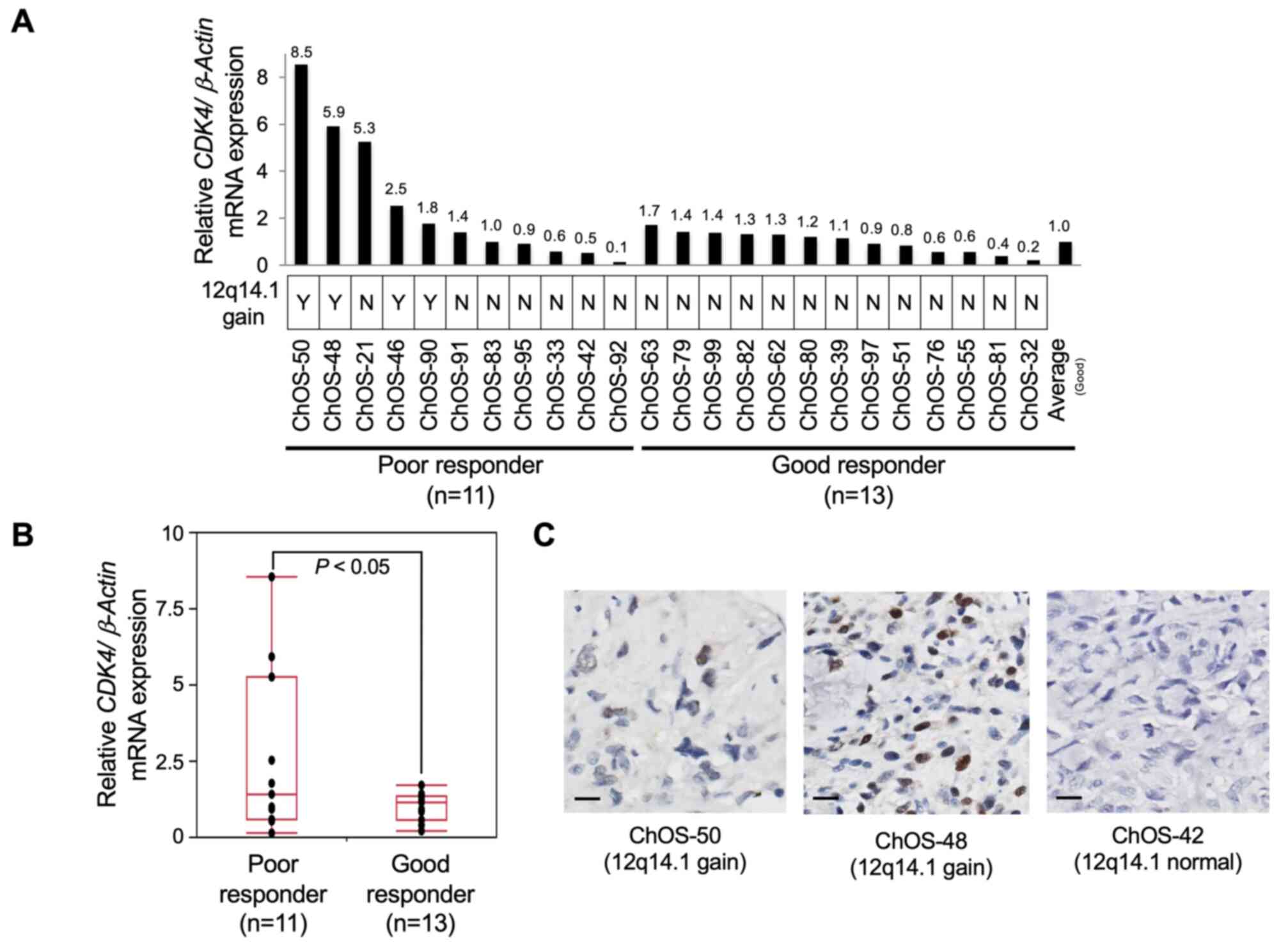
were assessed by RT-qPCR. As expected, higher CDK4
transcript levels were observed in four patients with 12q14.1 gain
compared with those without 12q14.1 gain (Fig. 3A). CDK4 levels were
significantly higher in poor (n=11) than in good responders (n=13;
Fig. 3B; P<0.05).
Immunohistochemistry analysis was employed to confirm positive
nuclear staining of CDK4 in the OS tissue of patients with 12q14.1
gain (Fig. 3C). Thus, CDK4 was
considered a strong candidate genomic prognostic marker and
potential therapeutic target in poor responders.
Overexpression of CDK4 in OS cell
lines attenuates sensitivity to CDDP
Next, it was investigated whether higher expression
of CDK4 in OS cells effectively decreases sensitivity to
chemotherapeutic drugs, such as CDDP. Ectopic expression of CDK4 in
OS (U2OS and SJSA1) cells was induced via pCDNA3-CDK4 transfection.
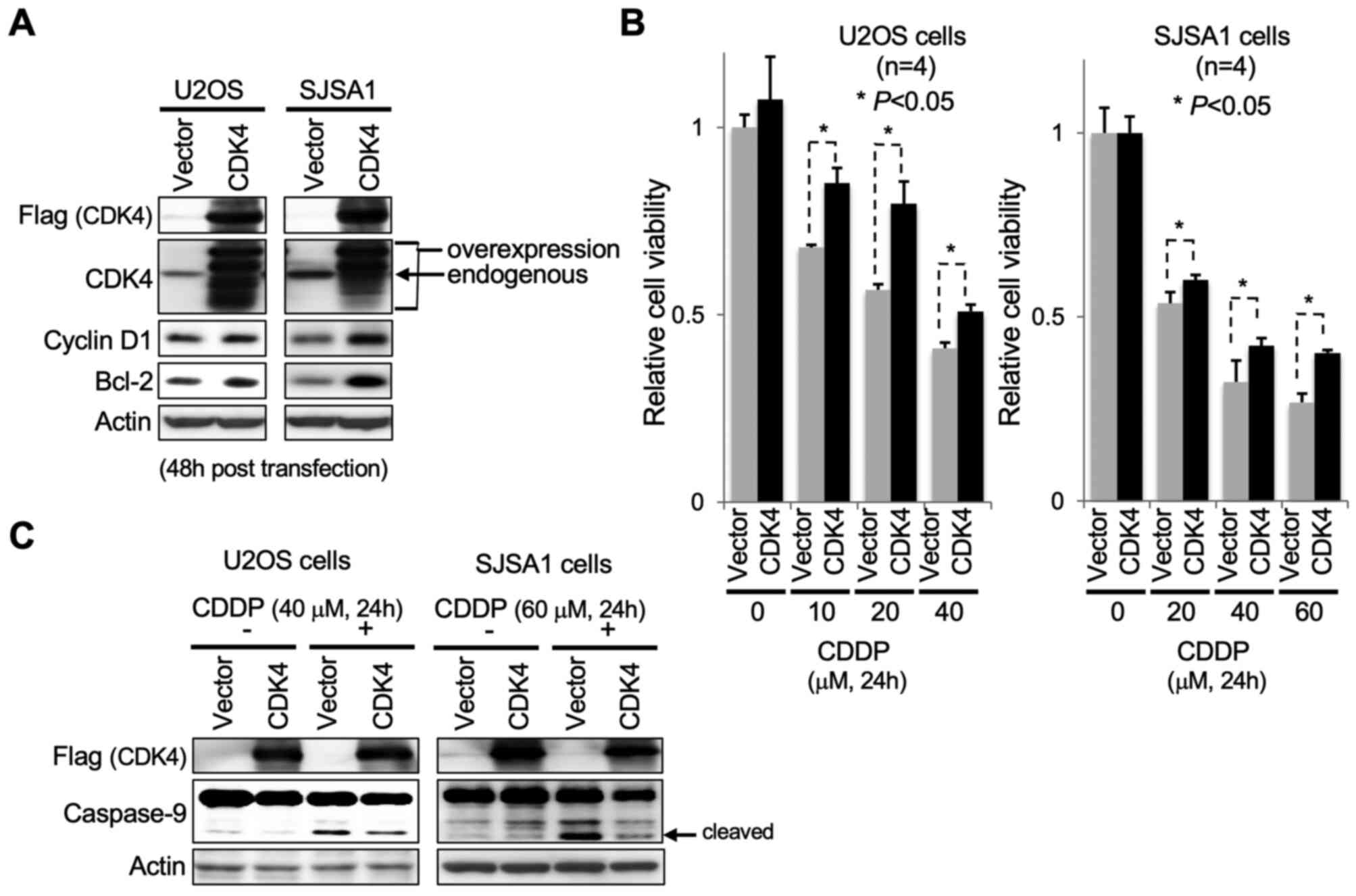
Following 48 h transfection, increased protein levels of cyclin D1,
a binding partner of CDK4, and Bcl-2, a pro-survival factor, were
detected in U2OS and SJSA1 cells, accompanied by increased levels
of exogenously introduced Flag-tagged CDK4 (Fig. 4A). Transfected cells were cultured in
medium containing CDDP for 24 h to induce apoptosis. CDK4
overexpression caused a significant abrogation of the effect of
CDDP on OS cell viability compared with control cells (Fig. 4B). Consistent with this observation,
apoptotic cell death caused by CDDP was markedly decreased in OS
cells overexpressing CDK4, as indicated by immunoblot detection of
cleaved caspase-9 accumulation (Fig.
4C).
Overexpression of CDK4 sensitizes U2OS
cells to a CDK4/6 inhibitor
Finally, it was investigated whether overexpression
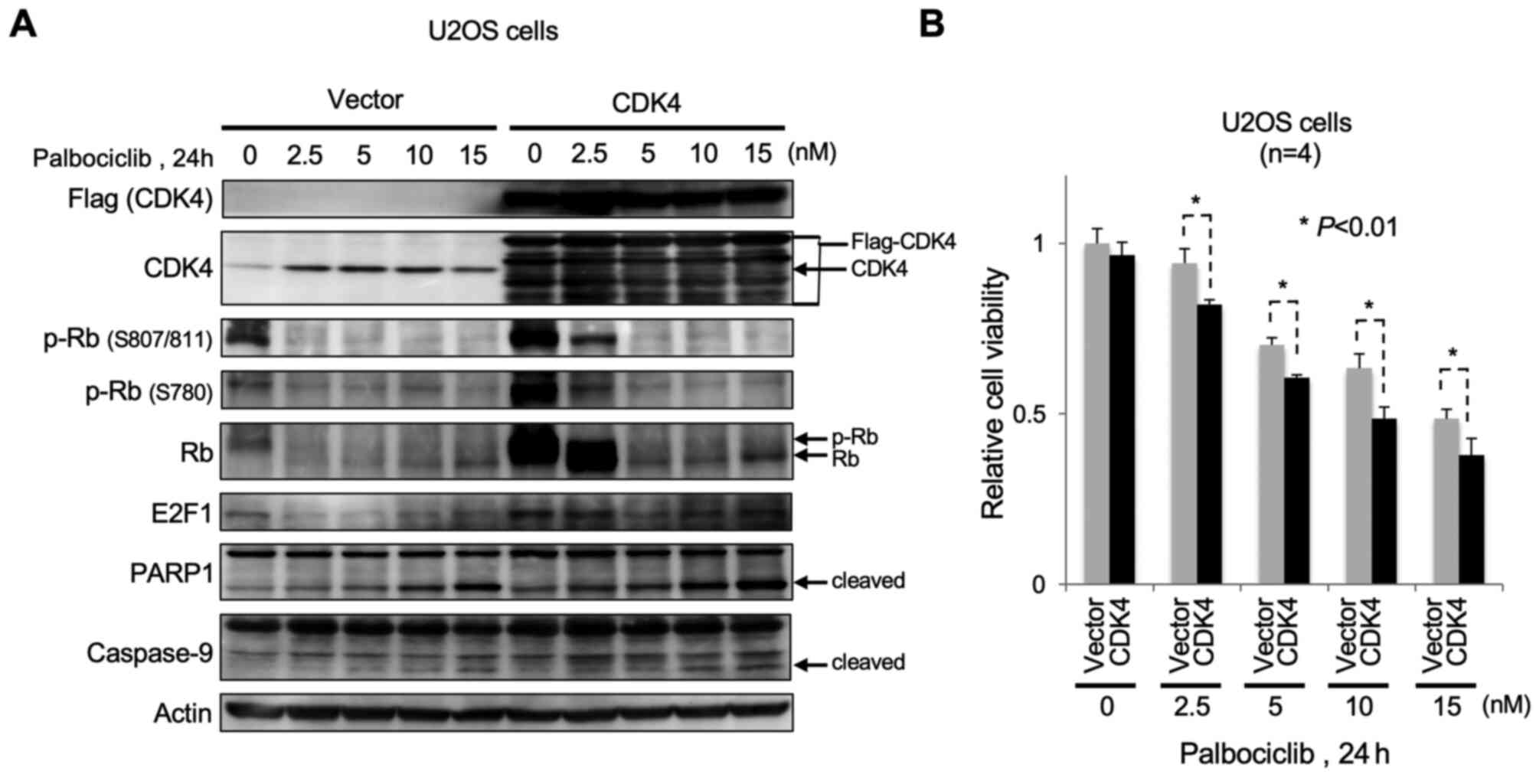
of CDK4 sensitizes OS cells to CDK4/6 inhibitor palbociclib.
When Flag-tagged CDK4 was ectopically overexpressed in U2OS cells,
there was an increased phosphorylation of Rb at S807/811 and S780
under normal culture conditions (0 nM Palbociclib; Fig. 5A). Subsequent treatment with
palbociclib attenuated Rb phosphorylation in a dose-dependent
manner. Treatment of CDK4-overexpressing U2OS cells with
palbociclib facilitated apoptosis in a dose-dependent manner, as
evidenced by cleavage of PARP1 and caspase-9, compared with control
cells. Palbociclib also caused a significant decrease in viability
of CDK4-overexpressing U2OS cells (Fig.
5B).
Discussion
Chemosensitivity to preoperative chemotherapy is a
strong prognostic factor for patients with OS (18). The present study investigated genomic
markers for the prediction of chemosensitivity in patients with
primary OS. Genome-wide CGH array analysis revealed recurrent gain
of chromosomes 12q14.1 and 19q13.32 in the poor responder cohort.
Targeted exome sequence and semiquantitative RT-PCR revealed that
the CDK4 gene was associated with 12q14.1 gain and that
higher CDK4 transcript levels occurred in tumors with 12q14.1 gain.
In functional analysis using OS cell lines, CDK4-overexpressing OS
cells exhibited a significant decrease in sensitivity to CDDP and
subsequent apoptotic cell death. Conversely, overexpression of CDK4
sensitized OS cells to CDK4/6 inhibitor (palbociclib) and was
associated with attenuation of Rb phosphorylation. Based on these
results, patients with OS with high expression levels or gain of
CDK4 may be less sensitive to standard chemotherapy
containing CDDP and may be good candidates for treatment with
CDK4/6 inhibitors.
The present study had two main limitations in the
in vitro experiments. The first limitation is lack of
CDK4 knockdown experiments in OS cells to confirm the
finding that forced overexpression of CDK4 in OS cells attenuated
sensitivity to CDDP. Inhibition of CDK4 in OS cells, such as U2OS
cells, impedes cell proliferation and promotes G1 arrest
(19), whereas CDDP acts on cells in
S phase. Therefore, this could not be assessed using U2OS cells in
the present study and should be addressed in future studies using
other cell lines, such as Rb- and/or p53-deficient OS cells. The
second limitation is that the effects of CDK4 overexpression on
proliferation and differentiation of OS cells were not
investigated. Here, ectopic overexpression of CDK4 in U2OS cells
facilitated Rb phosphorylation. This suggests that CDK4
overexpression promoted proliferation and inhibited differentiation
of OS cells via phosphorylation-dependent inactivation of the
G1 regulator Rb. However, significant changes in the
viability of OS cells were not detected, regardless of enforced
overexpression of CDK4. Future research is necessary to elucidate
the precise underlying molecular mechanisms.
Genetic, genomic and NGS analysis have identified
molecules and genomic alterations associated with OS development
(20,21), although, to the best of our knowledge,
few reports have identified predictive markers of chemosensitivity.
Cheng et al (22) conducted an
integrated analysis utilizing publicly available data sets of copy
number alterations in pediatric sarcoma cell lines, including OS
lines, and identified 33 candidate genes that serve as prognostic
markers. They found that genes encoding MDM2 proto-oncogene (MDM2)
and GLI family zinc finger 1 (GLI1), which are located in 12q13-15,
are predictors of resistance to bleomycin, although this drug is
not a standard treatment for patients with OS. Employing
whole-exome analysis of samples from eight patients with OS (5
chemotherapy responders and 3 non-responders), Chiappetta et
al (23) reported mutations in 15
genes that were observed only in non-responders. Although this was
a small patient cohort, the findings included genes of interest,
such as that encoding Erb-b2 receptor tyrosine kinase 4 (ERBB4),
which serves a role in murine double minute X (MDMX)-MDM2 complex
stability via MDMX Ser-314 phosphorylation, leading to inactivation
of TP53 in poor responders. Zhang et al conducted genotyping
of ERCC excision repair 1, endonuclease non-catalytic subunit
(ERCC1) in 260 OS tumors and indicated that ERCC1
polymorphisms, such as a CC genotype at rs11615 in ERCC1,
were associated with superior chemosensitivity and better outcomes
in patients with OS (24). Although
the clinical impact of this polymorphism remains controversial, it
may be associated with lower ERCC1 expression, which is also
associated with CDDP sensitivity via decreased DNA repair capacity
(25). In the present study, NGS
identified ERBB3 M1055V and ERCC2 A535V mutations in a tumor with
CDK4 amplification (ChOS-50), but no ERBB4 mutation.
Of the 16 tumors investigated by NGS, three exhibited the
ERCC1 TT rs11615 genotype (potential higher risk), although
two were good responders. These candidate markers could not be
evaluated in the present study. Since CDDP is currently a key drug
for OS, genomic factors, such as DNA damage response genes and
CDK4, that affect the CDDP sensitivity of OS tumors should
be considered as a mechanism.
In the present patient cohort, the amplification of
12q14.1, which harbors CDK4, was the most frequent genomic
alteration observed only in poor responders. Previous reports have
suggested that amplification of 12q13-15, harboring CDK4 and
MDM2, may serve an important role in chemoresistance and
lead to poor survival in patients with OS (7,26,27). Amplification of 12q13-15 has been
observed in 9–25% of all patients with OS (26,28), which
is relatively less frequent than aberrations in other loci, such as
17p13.1 (TP53; 70–80%) (29,30), 13q14.2 (Rb1; 43–70%) (26,31),
17p11.2 (peripheral myelin protein 22, COPS3; 20–78%)
(11,32) and 6p21 (RUNX2; 24–75%)
(11,26,32).
Several studies have also reported that amplification of 12q14 is
significantly associated with poor event-free survival of OS
(26,33). Suehara et al (34) reported that pediatric and adult OS
tumors comprise at least three tumor subsets exhibiting 4q12 gain
with platelet-derived growth factor receptor α (PDGFRA) and
kinase insert domain receptor amplifications, 6p12 gain with
VEGFA amplification and 12q13 gain with MDM2 and
CDK4 amplifications. In the present study, 4q12 and 6p12
gain were observed in 19 (38%) and 18 (36%) tumors, respectively,
with no significant enrichment in poor responders. A total of eight
cyclin E1 (CCNE1) (16%), four VEGFA (8%) and four
PDGFRA amplifications (8%) and 10 CDK inhibitor 2A
(CDKN2A) homozygous deletions (20%) in 50 OS tumors were
detected in the present study. CCNE1 amplifications and
CDKN2A homozygous deletions were mutually exclusive in the
sample set, as were VEGFA and PDGFRA amplifications.
Again, CDK4 amplification was only observed in poor
responders, whereas the four aforementioned alterations were also
observed in good responders. Thus, CDK4 may be a good marker
with high specificity to predict poor responders. For clinical use,
further validation with more samples is necessary to determine its
suitable cut-off value (relative expression value of 2.5 in
RT-qPCR, for example), by comparing the distribution in good
responders. A recent report revealed that dedifferentiated OS,
which progresses from low-grade OS, exhibits co-expression of MDM2
and CDK4 (35). However, following
extensive examination of the whole resected specimen by a bone
tumor pathologist, there were no histological low-grade elements in
all four cases with CDK4 amplification in the present study.
Dedifferentiated OS has a relatively good prognosis despite poor
response to chemotherapy; however, the present four cases with
CDK4 amplification demonstrated poor outcomes, and from the
view of clinical behavior, it was hypothesized that these cases
might be different from so-called dedifferentiated OS. Concerning
19q13.32 gain, six genes with University of California, Santa Cruz
identifiers are located in this region; however, they were not
differentially expressed between poor and good responders in the
present study. Unknown microRNAs or long intergenic non-coding RNAs
encoded in this region with higher levels in poor responders may
exist, the effect of 19q13.32 gain on tumor chemosensitivity
remains unknown.
The present study showed that resistance in OS cells
was caused by forced overexpression of CDK4. CDK4 forms a complex
with Cyclin D1 and promotes G1 phase progression via
phosphorylation-dependent-inactivation of Rb. Previous studies have
demonstrated that aberrant activation of Cyclin D1/CDK4 complex
promotes anticancer drug resistance in several types of cancer. For
example, knockdown of CDK4 in nasopharyngeal carcinoma cells
induces sensitivity to CDDP (36) and
overexpression of Cyclin D1 attenuates CDDP sensitivity of
testicular germ cell tumor as well as ovarian and prostate cancer
(37). CDK4 also promotes resistance
to temozolomide (a DNA alkylating drug) in glioma cells (38). Here, OS cells with enforced
overexpression of CDK4 significantly increased phosphorylation
levels of Rb, indicating that CDK4 overexpression increased cyclin
D1/CDK4 activity in OS cells. Thus, it was concluded that CDK4
overexpression promotes CDDP resistance of OS cells, at least in
part, by activation of Cyclin D1/CDK4 complex. Further study will
uncover the mechanism of how CDK4 abrogates drug sensitivity.
It was hypothesized that CDK4/6 inhibitors exhibit
potential efficacy in chemotherapy-resistant OS with 12q14 gain. In
hormone receptor (HR)-positive breast cancer, cyclin D
overexpression, CDK4/6 activation and subsequent promotion of Rb
phosphorylation and E2F release frequently occur, which causes
cells to enter S phase and initiate DNA replication (39). Based on this mechanism, CDK4/6
inhibitors have been utilized to treat breast cancer. A Phase III
clinical trial demonstrated that CDK4/6 inhibitors exhibit
promising antitumor activity and acceptable toxicity for patients
with HR+/human epidermal growth factor receptor
2− advanced breast cancer (39). Moreover, several clinical trials using
various CDK4/6 inhibitors have been conducted for malignant tumors
(40), including non-small-cell lung,
colorectal and pediatric cancer (41), as well as melanoma, glioblastoma and
mantle cell lymphoma (42). Most of
the results of these trials were favorable despite varying
inclusion criteria, including Rb positivity, cyclin D1
overexpression and CDK4 or CDK6 amplification. Also,
most of these studies did not require biomarkers as inclusion
criteria. Precision medicine based on genomic biomarkers has not
been achieved in sarcoma so far. Patients with advanced
well-differentiated and dedifferentiated liposarcoma, another
malignant mesenchymal tumor that harbors CDK4 amplification
at high frequency (>90%), exhibit favorable outcomes with
treatment with a CDK4/6 inhibitor (43). Further studies, including
investigation of drug sensitivity and therapeutic effects of a
CDK4/6 inhibitor alone or in combination with MAP on
CDK4-overexpressing OS cells in vitro and in vivo,
are necessary and assessing CDK4 status in patients with OS may be
valuable for guiding precision medicine with a CDK4/6 inhibitor.
This may be detected by immunohistochemistry for CDK4.
Decalcification, which is required before processing of
osteocartilaginous tissue, is known to negatively affect protein
quality in formalin-fixed paraffin-embedded (FFPE) samples and
subsequent immunohistochemical staining. In order to decrease the
negative effect of decalcification in OS samples, a recent
alternative method (44), which uses
EDTA and short-term formic acid-based decalcification and does not
alter antigenicity of the FFPE, would be a better choice for CDK4
immunostaining. In addition, since most cancer gene panel tests for
personalized cancer genome medicine contain CDK4 gene loci,
CDK4 information in tumors can be obtained simultaneously.
The detection of CDK4 amplification or overexpression by
in situ hybridization, RT-qPCR or immunostaining in biopsy
or resected specimen may support the therapeutic use of CDK4/6
inhibitors.
The molecular mechanism underlying chemo-resistance
in poor responders is still unknown. The present targeted
sequencing data identified several candidate genes that may be
associated with CDDP chemosensitivity in patients with OS. DICER1
is an essential member of the ribonuclease III family, which
controls the maturation of precursor microRNAs, enabling them to
function normally (45). In ovarian
cancer cells, DICER1 downregulation promotes cell proliferation,
migration and cell cycle progression, and Dicer expression is
significantly decreased in CDDP-resistant cells (45). Fms-related receptor tyrosine kinase 1
(FLT1; also known as VEGFR1), a member of the VEGFR family,
promotes endothelial cell proliferation by activating MAPK or PI3K.
Tsuchida et al (46) showed
that CDDP exposure enhances expression of FLT1 by inducing
autocrine signaling, thereby promoting cell survival and expansion
of a drug-resistant side population in OS cell lines. Further
analysis using meta-data may be necessary to identify other
predictive markers and molecular targets for chemoresistance in OS
treatment. Poor responders in the present study also exhibited
CCNE1 (n=2), VEGF (n=1) and PDGFRA
amplification (n=2) and CDKN2A homozygous deletion (n=6).
Although these alterations were not statistically significant
markers for chemosensitivity, cyclin E-CDK2, VEGF, PDGFR and CDK4/6
inhibition may be potential therapeutic strategies for these
tumors.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms Natue
Kitabayashi, Ms Yuki Nakamura and Ms Yasuko Imai (Chiba Cancer
Center Research Institute) for technical assistance. The authors
would also thank Dr Shin-ichiro Tatezaki (Chiba Cancer Center) and
Dr Osamu Shimozato (Chiba Cancer Center Research Institute) for
valuable discussions and suggestions.
Funding
The present study was supported in part by the
Ministry of Health, Labor and Welfare of Japan for the Third Term
Comprehensive 10-year Strategy for Cancer Control, Japan Society
for the Promotion of Science Grants-in-Aid for Scientific Research
(grant nos. 23791673 and 19591742) and Takeda Science
Foundation.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors contributions
SI, YT, HN and MO designed the study, performed the
experiments and analyzed the results. SI, YT and MO wrote the
manuscript. SI, TY, HKa, TT, YH, HKi and TI provided clinical
samples, medical data and survival information. AA and MI performed
immunohistochemical analysis. SI, YT and MO confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from Institutional
Ethics Committee of Chiba Cancer Center (Chiba, Japan; approval no.
CCC19-14). Written informed consent was obtained from each
patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CCP
|
Comprehensive Cancer Panel
|
|
CDDP
|
cisplatin
|
|
CNV
|
copy number variation
|
|
MAP
|
methotrexate, adriamycin and
cisplatin
|
|
OAS
|
overall survival
|
|
OS
|
osteosarcoma
|
References
|
1
|
Ogura K, Higashi T and Kawai A: Statistics
of bone sarcoma in Japan: Report from the bone and soft tissue
tumor registry in Japan. J Orthop Sci. 22:755–764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meyers PA, Schwartz CL, Krailo MD, Healey
JH, Bernstein ML, Betcher D, Ferguson WS, Gebhardt MC, Goorin AM,
Harris M, et al: Osteosarcoma: The addition of muramyl tripeptide
to chemotherapy improves overall survival-A report from the
childrens oncology group. J Clin Oncol. 26:633–638. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bielack SS, Kempf-Bielack B, Delling G,
Exner GU, Flege S, Helmke K, Kotz R, Salzer-Kuntschik M, Werner M,
Winkelmann W, et al: Prognostic factors in high-grade osteosarcoma
of the extremities or trunk: An analysis of 1,702 patients treated
on neoadjuvant cooperative osteosarcoma study group protocols. J
Clin Oncol. 20:776–790. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Picci P, Sangiorgi L, Rougraff BT, Neff
JR, Casadei R and Campanacci M: Relationship of
chemotherapy-induced necrosis and surgical margins to local
recurrence in osteosarcoma. J Clin Oncol. 12:2699–2705. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferrari S, Ruggieri P, Cefalo G, Tamburini
A, Capanna R, Fagioli F, Comandone A, Bertulli R, Bisogno G,
Palmerini E, et al: Neoadjuvant chemotherapy with methotrexate,
cisplatin, and doxorubicin with or without ifosfamide in
nonmetastatic osteosarcoma of the extremity: An Italian sarcoma
group trial ISG/OS-1. J Clin Oncol. 30:2112–2118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Borys D, Canter RJ, Hoch B, Martinez SR,
Tamurian RM, Murphy B, Bishop JW and Horvai A: P16 expression
predicts necrotic response among patients with osteosarcoma
receiving neoadjuvant chemotherapy. Hum Pathol. 43:1948–1954. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sadikovic B, Thorner P, Chilton-Macneill
S, Martin JW, Cervigne NK, Squire J and Zielenska M: Expression
analysis of genes associated with human osteosarcoma tumors shows
correlation of RUNX2 overexpression with poor response to
chemotherapy. BMC Cancer. 10:2022010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laverdiere C, Hoang BH, Yang R, Sowers R,
Qin J, Meyers PA, Huvos AG, Healey JH and Gorlick R: Messenger RNA
expression levels of CXCR4 correlate with metastatic behavior and
outcome in patients with osteosarcoma. Clin Cancer Res.
11:2561–2567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wunder JS, Bull SB, Aneliunas V, Lee PD,
Davis AM, Beauchamp CP, Conrad EU, Grimer RJ, Healey JH, Rock MJ,
et al: MDR1 gene expression and outcome in osteosarcoma: A
prospective, multicenter study. J Clin Oncol. 18:2685–2694. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan T, Wunder JS, Gokgoz N, Gill M,
Eskandarian S, Parkes RK, Bull SB, Bell RS and Andrulis IL: COPS3
amplification and clinical outcome in osteosarcoma. Cancer.
109:1870–1876. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Man TK, Lu XY, Jaeweon K, Perlaky L,
Harris CP, Shah S, Ladanyi M, Gorlick R, Lau CC and Rao PH:
Genome-wide array comparative genomic hybridization analysis
reveals distinct amplifications in osteosarcoma. BMC Cancer.
4:452004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mintz MB, Sowers R, Brown KM, Hilmer SC,
Mazza B, Huvos AG, Meyers PA, Lafleur B, McDonough WS, Henry MM, et
al: An expression signature classifies chemotherapy-resistant
pediatric osteosarcoma. Cancer Res. 65:1748–1754. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ochi K, Daigo Y, Katagiri T, Nagayama S,
Tsunoda T, Myoui A, Naka N, Araki N, Kudawara I, Ieguchi M, et al:
Prediction of response to neoadjuvant chemotherapy for osteosarcoma
by gene-expression profiles. Int J Oncol. 24:647–655.
2004.PubMed/NCBI
|
|
14
|
Iwamoto Y, Tanaka K, Isu K, Kawai A,
Tatezaki S, Ishii T, Kushida K, Beppu Y, Usui M, Tateishi A, et al:
Multiinstitutional phase II study of neoadjuvant chemotherapy for
osteosarcoma (NECO study) in Japan: NECO-93J and NECO-95J. J Orthop
Sci. 14:397–404. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moore DD: Preparation and analysis of DNA.
Current protocols in molecular biology. Ausubel FM, Brent R,
Kingston RF, Moore DD, Seidman JG, Smith JA and Struhl K: 1. units
2.2 and 2.4.Greene Publishing Associates/Wiley-Interscience; New
York, NY: 1989
|
|
16
|
Rutledge RG and Côté C: Mathematics of
quantitative kinetic PCR and the application of standard curves.
Nucleic Acids Res. 31:e932003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tatsumi Y, Takano R, Islam MS, Yokochi T,
Itami M, Nakamura Y and Nakagawara A: BMCC1, which is an
interacting partner of BCL2, attenuates AKT activity, accompanied
by apoptosis. Cell Death Dis. 6:e16072015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Link MP, Goorina AM, Horowitz M, Meyer WH,
Belasco J, Baker A, Ayala A and Shuster J: Adjuvant chemotherapy of
high-grade osteosarcoma of the extremity. Updated results of the
multi-institutional osteosarcoma study. Clin Orthop Relat Res.
8–14. 1991.PubMed/NCBI
|
|
19
|
Zhou Y, Shen JK, Yu Z, Hornicek FJ, Kan Q
and Duan Z: Expression and therapeutic implications of
cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim Biophys
Acta Mol Basis Dis. 1864:1573–1582. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kovac M, Blattmann C, Ribi S, Smida J,
Mueller NS, Engert F, Castro-Giner F, Weischenfeldt J, Kovacova M,
Krieg A, et al: Exome sequencing of osteosarcoma reveals mutation
signatures reminiscent of BRCA deficiency. Nat Commun. 6:89402015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Behjati S, Tarpey PS, Haase K, Ye H, Young
MD, Alexandrov LB, Farndon SJ, Collord G, Wedge DC, Martincorena I,
et al: Recurrent mutation of IGF signalling genes and distinct
patterns of genomic rearrangement in osteosarcoma. Nat Commun.
8:159362017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng L, Pandya PH, Liu E, Chandra P, Wang
L, Murray ME, Carter J, Ferguson M, Saadatzadeh MR,
Bijangi-Visheshsaraei K, et al: Integration of genomic copy number
variations and chemotherapy-response biomarkers in pediatric
sarcoma. BMC Med Genomics. 12 (Suppl 1):S232019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiappetta C, Mancini M, Lessi F, Aretini
P, De Gregorio V, Puggioni C, Carletti R, Petrozza V, Civita P,
Franceschi S, et al: Whole-exome analysis in osteosarcoma to
identify a personalized therapy. Oncotarget. 8:80416–80428. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Q, Lv LY, Li BJ, Zhang J and Wei F:
Investigation of ERCC1 and ERCC2 gene polymorphisms and response to
chemotherapy and overall survival in osteosarcoma. Genet Mol Res.
14:11235–11241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu JJ, Lee KB, Mu C, Li Q, Abernathy TV,
Bostick-Bruton F and Reed E: Comparison of two human ovarian
carcinoma cell lines (A2780/CP70 and MCAS) that are equally
resistant to platinum, but differ at codon 118 of the ERCC1 gene.
Int J Oncol. 16:555–560. 2000.PubMed/NCBI
|
|
26
|
Smida J, Baumhoer D, Rosemann M, Walch A,
Bielack S, Poremba C, Remberger K, Korsching E, Scheurlen W,
Dierkes C, et al: Genomic alterations and allelic imbalances are
strong prognostic predictors in osteosarcoma. Clin Cancer Res.
16:4256–4267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen X, Bahrami A, Pappo A, Easton J,
Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al:
Recurrent somatic structural variations contribute to tumorigenesis
in pediatric osteosarcoma. Cell Rep. 7:104–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wei G, Lonardo F, Ueda T, Kim T, Huvos AG,
Healey JH and Ladanyi M: CDK4 gene amplification in osteosarcoma:
Reciprocal relationship with INK4A gene alterations and mapping of
12q13 amplicons. Int J Cancer. 80:199–204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sandberg AA and Bridge JA: Updates on the
cytogenetics and molecular genetics of bone and soft tissue tumors:
Osteosarcoma and related tumors. Cancer Genet Cytogenet. 145:1–30.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Marina N, Gebhardt M, Teot L and Gorlick
R: Biology and therapeutic advances for pediatric osteosarcoma.
Oncologist. 9:422–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toguchida J, Ishizaki K, Sasaki MS,
Ikenaga M, Sugimoto M, Kotoura Y and Yamamuro T: Chromosomal
reorganization for the expression of recessive mutation of
retinoblastoma susceptibility gene in the development of
osteosarcoma. Cancer Res. 48:3939–3943. 1988.PubMed/NCBI
|
|
32
|
Lau CC, Harris CP, Lu XY, Perlaky L,
Gogineni S, Chintagumpala M, Hicks J, Johnson ME, Davino NA, Huvos
AG, et al: Frequent amplification and rearrangement of chromosomal
bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes
Cancer. 39:11–21. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martin JW, Chilton-MacNeill S, Koti M, van
Wijnen AJ, Squire JA and Zielenska M: Digital expression profiling
identifies RUNX2, CDC5L, MDM2, RECQL4, and CDK4 as Potential
predictive biomarkers for Neo-adjuvant chemotherapy response in
paediatric osteosarcoma. PLoS One. 9:e958432014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Suehara Y, Alex D, Bowman A, Middha S,
Zehir A, Chakravarty D, Wang L, Jour G, Nafa K, Hayashi T, et al:
Clinical genomic sequencing of pediatric and adult osteosarcoma
reveals distinct molecular subsets with potentially targetable
alterations. Clin Cancer Res. 25:6346–6356. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Toki S, Kobayashi E, Yoshida A, Ogura K,
Wakai S, Yoshimoto S, Yonemori K and Kawai A: A clinical comparison
between dedifferentiated low-grade osteosarcoma and conventional
osteosarcoma. Bone Joint J. 101-B:745–752. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Z, Cheng C, Luo X, Xia Q, Zhang Y,
Long X, Jiang Q and Fang W: CDK4 and miR-15a comprise an abnormal
automodulatory feedback loop stimulating the pathogenesis and
inducing chemotherapy resistance in nasopharyngeal carcinoma. BMC
Cancer. 16:2382016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Noel EE, Yeste-Velasco M, Mao X, Perry J,
Kudahetti SC, Li NF, Sharp S, Chaplin T, Xue L, McIntyre A, et al:
The association of CCND1 overexpression and cisplatin resistance in
testicular germ cell tumors and other cancers. Am J Pathol.
176:2607–2615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cao Y, Li X, Kong S, Shang S and Qi Y:
CDK4/6 inhibition suppresses tumour growth and enhances the effect
of temozolomide in glioma cells. J Cell Mol Med. 24:5135–5145.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Finn RS, Martin M, Rugo HS, Jones S, Im
SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al:
Palbociclib and letrozole in advanced breast cancer. N Engl J Med.
375:1925–1936. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Du Q, Guo X, Wang M, Li Y, Sun X and Li Q:
The application and prospect of CDK4/6 inhibitors in malignant
solid tumors. J Hematol Oncol. 13:412020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Edelman MJ, Redman MW, Albain KS, McGary
EC, Rafique NM, Petro D, Waqar SN, Minichiello K, Miao J,
Papadimitrakopoulou VA, et al: SWOG S1400C (NCT02154490)-A Phase II
Study of palbociclib for previously treated cell cycle gene
alteration-positive patients with stage IV squamous cell lung
cancer (Lung-MAP Substudy). J Thorac Oncol. 14:1853–1859. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Leonard JP, LaCasce AS, Smith MR, Noy A,
Chirieac LR, Rodig SJ, Yu JQ, Vallabhajosula S, Schoder H, English
P, et al: Selective CDK4/6 inhibition with tumor responses by
PD0332991 in patients with mantle cell lymphoma. Blood.
119:4597–4607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saâda-bouzid E, Burel-vandenbos F,
Ranchère-vince D, Birtwisle-Peyrottes I, Chetaille B, Bouvier C,
Château MC, Peoch M, Battistella M, Bazin A, et al: Prognostic
value of HMGA2, CDK4, and JUN amplification in well-differentiated
and dedifferentiated liposarcomas. Mod Pathol. 28:1404–1414. 2015.
View Article : Google Scholar
|
|
44
|
Miquelestorena-Standley E, Jourdan ML,
Collin C, Bouvier C, Larousserie F, Aubert S, Gomez-Brouchet A,
Guinebretière JM, Tallegas M, Brulin B, et al: Effect of
decalcification protocols on immunohistochemistry and molecular
analyses of bone samples. Mod Pathol. 33:1505–1517. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kuang Y, Cai J, Li D, Han Q, Cao J and
Wang Z: Repression of Dicer is associated with invasive phenotype
and chemoresistance in ovarian cancer. Oncol Lett. 5:1149–1154.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsuchida R, Das B, Yeger H, Koren G,
Shibuya M, Thorner PS, Baruchel S and Malkin D: Cisplatin treatment
increases survival and expansion of a highly tumorigenic
side-population fraction by upregulating VEGF/Flt1 autocrine
signaling. Oncogene. 27:3923–3934. 2008. View Article : Google Scholar : PubMed/NCBI
|