Introduction
Lung cancer is the most common cause of
cancer-related deaths worldwide (1).
In a study in 2018 in the U.S., lung cancer was the second most
common cancer diagnosis by sex and was newly diagnosed in 14% of
men and 13% of women (2). Typically,
patients with non-small cell lung cancer (NSCLC) are identified
with advanced cancer and approximately 16.43% of patients survive
for five years (3,4). Lung cancer treatments include surgery,
radiotherapy, and chemotherapeutic drugs and their combinations
(5,6).
Specific combination strategies with potent chemotherapeutic drugs
may be a potential approach to cancer treatment (7,8).
Tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) is a highly attractive anticancer treatment that
selectively kills cancer cells without causing toxicity to normal
cells (9). TRAIL binds to death
receptor (DR)4/DR5 to initiate apoptotic cell death. Along with
A549 lung cancer cells, a large number of cancer cells are
resistant to TRAIL due to the insufficient expression of death
receptors DR4/DR5 and the extreme expression of decoy receptors, as
well as the mutation of TRAIL receptors (10–12).
Interestingly, it is possible to overcome TRAIL resistance with
suitable pharmacological agents that can enhance the expression of
TRAIL receptors (13,14).
Autophagy plays an important role in maintaining
cellular homeostasis (15). Autophagy
suppresses tumors by maintaining cellular homeostasis; however,
these tumors can play a survival role when cancer has already
developed (16). Tumor cells fulfill
their energy demands by using autophagy and through this process
develop treatment resistance (17).
Numerous previous studies have demonstrated that blocking autophagy
flux by inhibiting autophagosome-lysosome fusion can be an
encouraging approach for cancer therapy (18,19).
Consequently, pharmacological agents that induce autophagosome
accumulation by inhibiting lysosomal fusion and increase TRAIL
receptors can be an effective approach to overcome TRAIL
resistance.
Depression is a common psychological disorder in
cancer patients. Continuous depression reduces the antitumor immune
response and creates a favorable environment for tumor growth
(20). Animal model studies have
shown that behavioral stress induces the rapid development of
prostate (20), ovarian (21), pancreatic (22), and breast cancer (22), as well as carcinomas and malignant
melanomas (23). Several studies have
recommended that amitriptyline is a productive option to control
cancer-associated depression, anxiety, and pain (24,25).
Amitriptyline is a psychoactive tricyclic
antidepressant (TCA) drug. The drug has been revealed to markedly
exert effective anticancer effects on a large number of cancer cell
types, including colon, prostate, glioma osteosarcoma, skin,
squamous carcinoma, and multiple myeloma (26). Another study revealed that
amitriptyline induced p53 expression, activated caspase-3, and
decreased anti-apoptotic proteins Bcl-2 and Mcl-1 in multiple
myeloma. In combination with bortezomib, amitriptyline induced
apoptosis in multiple myeloma (27).
Amitriptyline has also been studied as a potential candidate for
oxidative therapy for its cytotoxicity in H460 lung cancer cells,
which may be more effective than other chemotherapeutic drugs
(28).
In the present study, it was demonstrated that
amitriptyline could sensitize TRAIL-resistant lung cancer cells to
induce TRAIL-mediated apoptosis. The molecular mechanism underlying
the anticancer effects of amitriptyline in combination with TRAIL
and, specifically, the role of autophagy in lung cancer treatment
was also investigated.
Materials and methods
Cells and culture systems
A549 lung cancer cells were acquired from the
American Type Culture Collection (ATCC). The cells were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco BRL;
Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) fetal
bovine serum (Sigma-Aldrich; Merck KGaA) and antibiotics (100 µg/ml
penicillin-streptomycin; Sigma-Aldrich; Merck KGaA) at 37°C in a 5%
CO2 incubator.
Reagents
Amitriptyline was purchased from Cayman Chemical
Company, and chloroquine (CQ) (20 µM) was obtained from
Sigma-Aldrich; Merck KGaA. Human recombinant TRAIL (100 ng/ml) was
purchased from AbFrontier.
Cell viability assay
Cell viability was assessed with MTT and crystal
violet staining assays. The cells were plated in 12-well plates at
a density of 1.0×104 cells/well and incubated at 37°C
for 24 h. The cells were pretreated with different concentrations
of amitriptyline (0, 10, 20 and 40 µM) or CQ for 12 h and then
exposed to recombinant TRAIL (100 ng/ml) for 3 h. Cell morphology
was observed under an inverted light microscope (magnification,
×100; Nikon Corporation). Cell viability was assessed by adding 50
µl of 5 mg/ml methyl-thiazolyl tetrazolium (MTT) to each well and
incubating them at 37°C for 2 h. After incubation, the MTT solution
was removed and the cells were treated with 500 µl of dimethyl
sulfoxide and the absorbance was measured at 570 nm with a
spectrophotometer (Bio-Rad Laboratories). For the crystal violet
assay, the cells were stained with a staining solution (0.5%
crystal violet in 30% ethanol and 3% formaldehyde) for 10–20 min at
room temperature (RT), washed 3–4 times with phosphate-buffered
saline (PBS), and then imaged.
Lactate dehydrogenase (LDH) assay
Cytotoxicity was analyzed in the collected
supernatant and determined by an LDH cytotoxicity detection kit
(Takara Bio, Inc.) following the manufacturer's protocol. LDH
activity was measured at 490 nm using a microplate reader (Spectra
Max M2; Molecular Devices, LLC).
Colony-formation assay
Cells were plated in 6-well plates at 37°C and
treated with the indicated doses of amitriptyline (40 µM), CQ (20
µM) and TRAIL (100 ng/ml). Two days later, the culture medium was
changed with new medium without amitriptyline, CQ and TRAIL, and
the culture continued for 7 days. Colonies were fixed for 20 min at
RT in 4% paraformaldehyde, stained with 0.05% (w/v) crystal violet
for 10 min at RT, and counted under an inverted light microscope
(Nikon Corporation).
Flow cytometric analysis of
apoptosis
Apoptosis was evaluated cells (50 cells/µl) using
Annexin V-FITC Assay Kit (Santa Cruz Biotechnology, Inc.), for flow
cytometry according to the manufacturer's instructions (Guava
EasyCyte HT System; EMD Millipore). The fluorescence was measured
at 488 nm of excitation and 525/30 emission using Guava®
InCyte and GuavaSuite Software.
Western blot analysis
The cells were lysed in lysis buffer [25 mM HEPES
(pH 7.4), 100 mM ethylenediaminetetraacetic acid (EDTA), 5 mM
MgCl2, 0.1 mM dithiothreitol (DTT), and a protease
inhibitor cocktail], and sonicated to prepare cell lysates. Equal
amounts (40 µg) of proteins were separated by 8–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS) and transferred
onto polyvinylidene fluoride (PVDF) membranes. The membranes were
blocked at 25°C for 1 h, and then incubated with the indicated
concentrations of primary antibodies at 25°C for 1 h, and then they
were blotted with anti-mouse IgG (Alexa Fluor 647 conjugate)
secondary antibodies (product. no. 4410; 1:2,000; Cell Signaling
Technology, Inc.) at 25°C for 1 h. The membranes were developed
with enhanced chemiluminescence reagents (ECL; GE Healthcare Life
Sciences). Primary antibodies used for the immunoblotting included:
DR4 (product. code. ab8414; 1:1,000), DR5 (product. code. ab181846;
1:10,000) (both from Abcam), LC3 (product. no. 3868; 1:1,000), p62
(cat. no. 5114; 1:1,000), cleaved caspase-3 (product. no. 9661;
1:500), p-AMPKα (product. no. 2531; 1:1,000) all from Cell
Signaling Technology, Inc., cleaved caspase-8 (cat. no. 551242;
1:1,000, BD Pharmingen; BD Biosciences), and β-actin (cat. no.
A2228; 1:2,000, Sigma-Aldrich; Merck KGaA). The bands were
visualized and captured with a Fusion-FX7 using easy-to-use
FusionCapt V16.07 Software (both Vilber Lourmat).
Immunocytochemistry
The cells (~1×106 cells) were grown on
glass coverslips, then treated with amitriptyline, washed with 1%
PBS, and fixed with 4% paraformaldehyde in PBS at RT for 15 min.
They were then washed twice with ice-cold PBS and incubated at RT
for 10 min in PBS containing 0.25% Triton X-100. After the
incubation, the cells were washed three times with PBS and blocked
with 1% BSA in PBST for 30 min. The cells were then incubated with
a primary antibody [anti-p62 (1:1,000; product. no. 5114; Cell
Signaling Technology, Inc.) and DR4/5 diluted with 1% BSA in PBST]
in a 5% CO2 incubator for 3 h at 37°C. After incubation,
the cells were washed three times with PBS. Next, the cells were
incubated with a secondary antibody [(Alexa Fluor®
488-conjugate; donkey polyclonal anti-rabbit, 1:500; cat. no.
A-21206; Thermo Fisher Scientific, Inc.), diluted with 1% BSA in
PBST] in the dark for 2 h at RT. The solution was removed and the
cells were washed 3–4 times with PBS. The cells were treated with
DAPI (4′,6-diamidino-2-phenylindole, D9564; Sigma-Aldrich;Merck
KGaA) and incubated for 10 min at 25°C. The cells were washed three
times, then mounted with fluorescent mounting medium and the images
were captured using a fluorescence microscope (Nikon ECLIPSE 80i;
magnification, ×400; Nikon Corporation).
Transmission electron microscopy
Trypsinized cells were fixed with 2% glutaraldehyde
(Electron Microscopy Sciences) for 2 h at 4°C in PBS, followed by
2% osmium tetroxide (Electron Microscopy Sciences), and dehydrated
with an ethanol series (25, 50, 70, 90 and 100%) for 5 min each.
After dehydration, the samples were embedded in epoxy resin (Embed
812; Electron Microscopy Sciences) for 48 h at 60°C according to
the manufacturer's instructions. Ultrathin sections (60 nm) were
prepared using an LKB III ultratome (Leica Microsystems GmbH) and
stained with 0.5% uranyl acetate (Electron Microscopy Sciences) for
20 min and 0.1% lead citrate (Electron Microscopy Sciences) for 7
min at RT. Images were captured on a Hitachi H7650 electron
microscope (magnification ×10,000; Hitachi, Ltd.) installed at the
Center for University-Wide Research Facilities (CURF) at Jeonbuk
National University (JBNU).
RNA interference
The cell line was transfected with small interfering
(si)RNA using Lipofectamine (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Knockdown
proficiency was assessed by immunoblotting and cell viability
tests. DR4 and DR5 siRNA were purchased from Qiagen China Co.,
Ltd., each with mixed two target sequences (forward,
5′-TAGCTCAGCTGCAACCATCAA-3′ and reverse,
5′-CAGGCAATCGACATAATATAT-3′ for DR4; forward,
5′-ACCAGGTGTGATTCAGGTGAA-3′ and reverse,
5′-CCGACTTCACTTGATACTATA-3′ for DR5). The synthetic siRNA and
scramble siRNA [negative control (NC)] (Qiagen) were transfected
using HiPerfect transfection reagent (Qiagen), according to the
manufacturer's protocol. Briefly, each sequence of siRNA (20
µmol/ml) and 10 µl Lipofectamine 2000 was diluted in serum-free
medium (250 µl) at RT for 5 min, mixed together, and incubated for
30 min at RT. The cells were incubated with siRNA or NC siRNA for 6
h and the medium was then changed with 10% FBS for 24 h, followed
by subsequent experimentation.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The DR4 and DR5 mRNA transcripts were measured using
quantitative SYBR Green-based real-time qPCR. Total RNA was
extracted from the A549 cells with RiboEX (GeneAll Biotechnology).
The total RNA extracts were then converted into cDNA by using
reverse transcriptase (TOPscript™ One-step RT PCR kit; Enzynomics
Co., Ltd.) using a CFX96™ Real-PCR Detection system (Bio-Rad
Laboratories, Inc.), following the manufacturer's instructions at
85°C for 5 sec, 37°C for 10 min and 4°C for 15 min. Gene primers (1
µl) and SYBR-Green (Bio-Rad Laboratories, Inc.) contained in a
total reaction volume of 20 µl were used to conduct the RT-qPCR .
The reaction protocols were as follows: Predenaturation at 95°C for
30 sec, 40 cycles of denaturation at 95°C for 5 sec and annealing
at 60°C for 30 sec. GAPDH were used as the respective internal
control. The sequences of the primers used were: DR4 forward,
5′-GGGACAGCACGGACCCAGTG-3′ and reverse, 5′-ATCCTTGACCTTGACCATCC-3′;
DR5 forward, 5′-GCGGTCCTGCTGTTGGTCTC-3′ and reverse,
5-GCTTCTGTCCACACGCTCAG-3′; and GAPDH as an internal control
forward, 5′-TGCACCACCAACTGCTTAG-3′ and reverse,
5′-GGATGCAGGGATGATGTT-3′. All data were evaluated using Bio-Rad CFX
manager, version 2.1 analysis software (Bio-Rad Laboratories,
Inc.). The collected data from three independent experiments were
analyzed using the 2−ΔΔCq method (29).
Statistical analysis
The data are expressed as the mean ± standard
deviation (SD) from three independent experiments. The significance
of the differences between the treatments was analyzed using
one-way analysis of variance (ANOVA), followed by the Tukey-Kramer
post hoc test. Statistical analyses were executed using GraphPad
Prism 7 (GraphPad Software, Inc). P<0.05 was considered to
indicate a statistically significant difference.
Results
Amitriptyline enhances TRAIL-induced
apoptosis in lung cancer cells
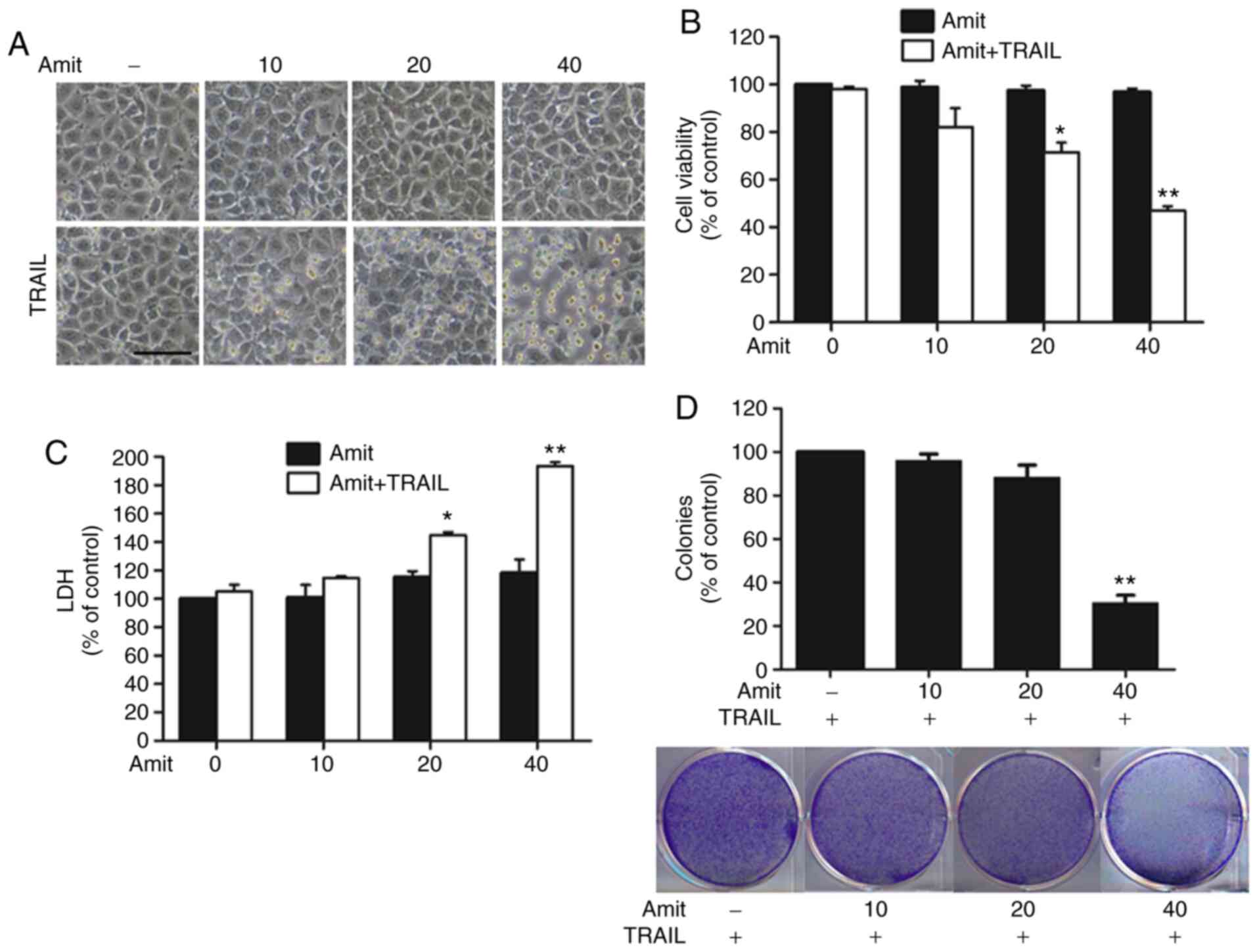
To explore the synergistic effect of amitriptyline
with TRAIL on the inhibition of lung cancer cell viability, the
A549 lung adenocarcinoma cell line was selected. The results
revealed a strong synergistic effect on this cell line. The cells
were pretreated with 40 µM amitriptyline for 12 h, followed by
co-treatment with 100 ng/ml of TRAIL for 3 h. The cell morphologies
were examined under a light microscope. Co-treatment with TRAIL
increased the number of cells undergoing apoptotic death (Fig. 1A). The MTT assay revealed that the
combined treatment triggered significant growth inhibition in a
dose-dependent manner (Fig. 1B). The
LDH levels after combined treatment demonstrated that amitriptyline
induced apoptosis in a dose-dependent manner; however, the
individual use of amitriptyline or TRAIL alone failed to show
similar effects (Fig. 1C).
Additionally, the colony-forming capacity of A549 cancer cells
after combination treatment of amitriptyline and TRAIL was
examined. Amitriptyline alone treatment not shown any inhibition
effects (data now shown), but combine treatment with TRAIL
gradually reduced the colony formation in a dose-dependent manner
(Fig. 1D). These results indicated
that amitriptyline significantly sensitized TRAIL-resistant A549
lung adenocarcinoma cells to TRAIL-mediated apoptosis.
DR4 and DR5 enhancement is required by
amitriptyline for TRAIL-mediated apoptosis
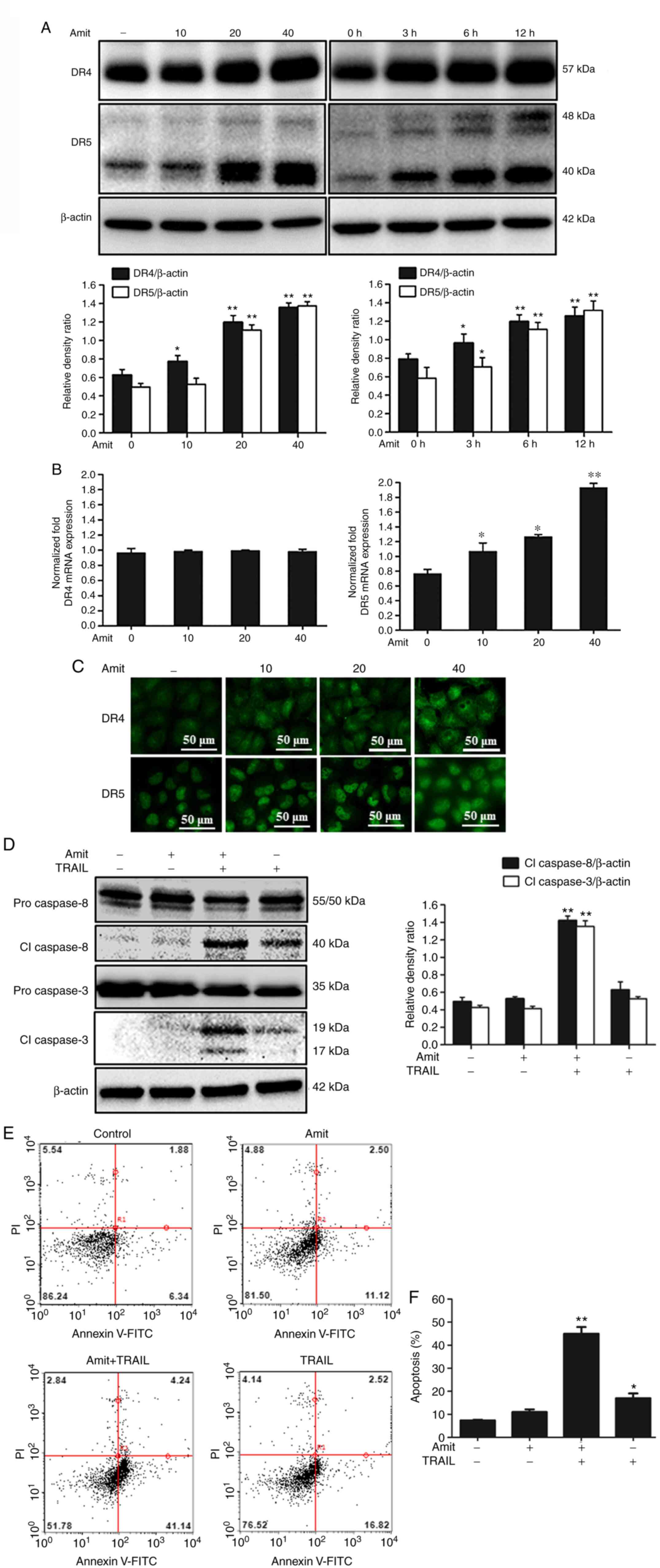
To evaluate the principal mechanism underlying the
apoptosis of A549 cells prompted by the combination of
amitriptyline and TRAIL, the augmented expression of DRs associated
with TRAIL-induced apoptosis was explored. An important reason for
TRAIL resistance in numerous cancer cell lines is associated with
the decreased expression of TRAIL receptors DR4 and DR5 or
upregulation of the decoy receptors DcR1 and DcR2 (30). Western blot analysis demonstrated that
amitriptyline increased DR4 and DR5 expression levels in a
dose-dependent manner and time-dependent manner (Fig. 2A). When assessed via mRNA expression,
amitriptyline treatment increased the transcription of DR5, but not
DR4, (Fig. 2B). These results
indicated that amitriptyline may increase DR5 expression through
transcriptional or post-transcriptional regulation and concurrently
amitriptyline stabilized DR4 protein expression by inhibiting its
degradation through post-translational regulation. Moreover,
immunocytochemistry results demonstrated the significant expression
of DR4 and DR5 in amitriptyline-treated cells compared to
non-treated cells (Fig. 2C). The
apoptosis-indicating proteins cleaved caspase-8 and cleaved
caspase-3 were activated after treatment with amitriptyline and
TRAIL compared to treatments with each individually (Fig. 2D). Furthermore, the apoptosis
percentage by Annexin V assay was measured, which indicated that
amitriptyline and TRAIL in combination enhanced apoptotic cell
death (Fig. 2E and F). Collectively,
these findings indicated that DR4 and DR5 upregulated by
amitriptyline induced TRAIL-mediated apoptosis in TRAIL-resistant
A549 lung cancer cells.
Silencing of DR4 and DR5 expression
negatively controls amitriptyline-induced TRAIL-mediated
apoptosis
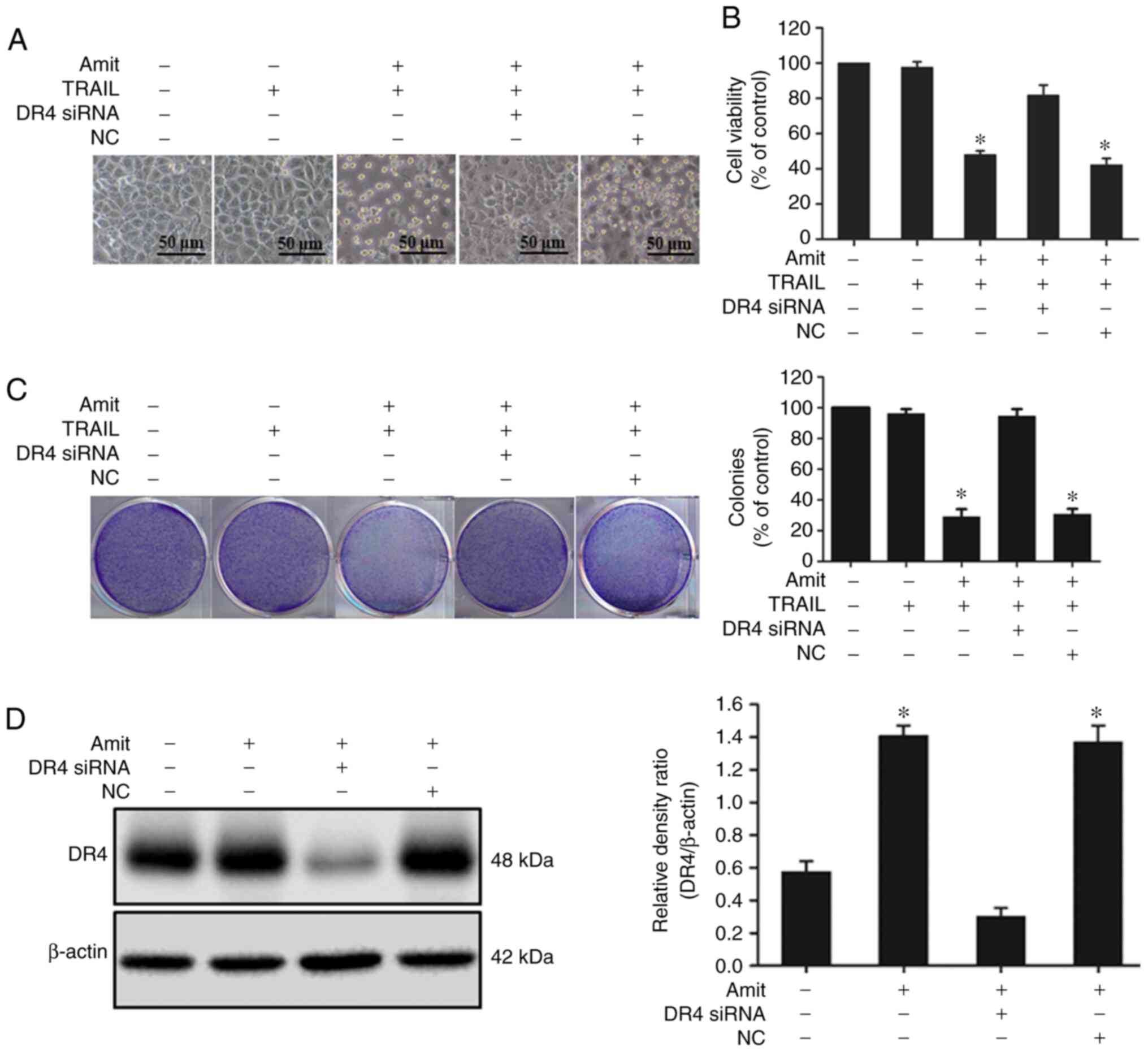
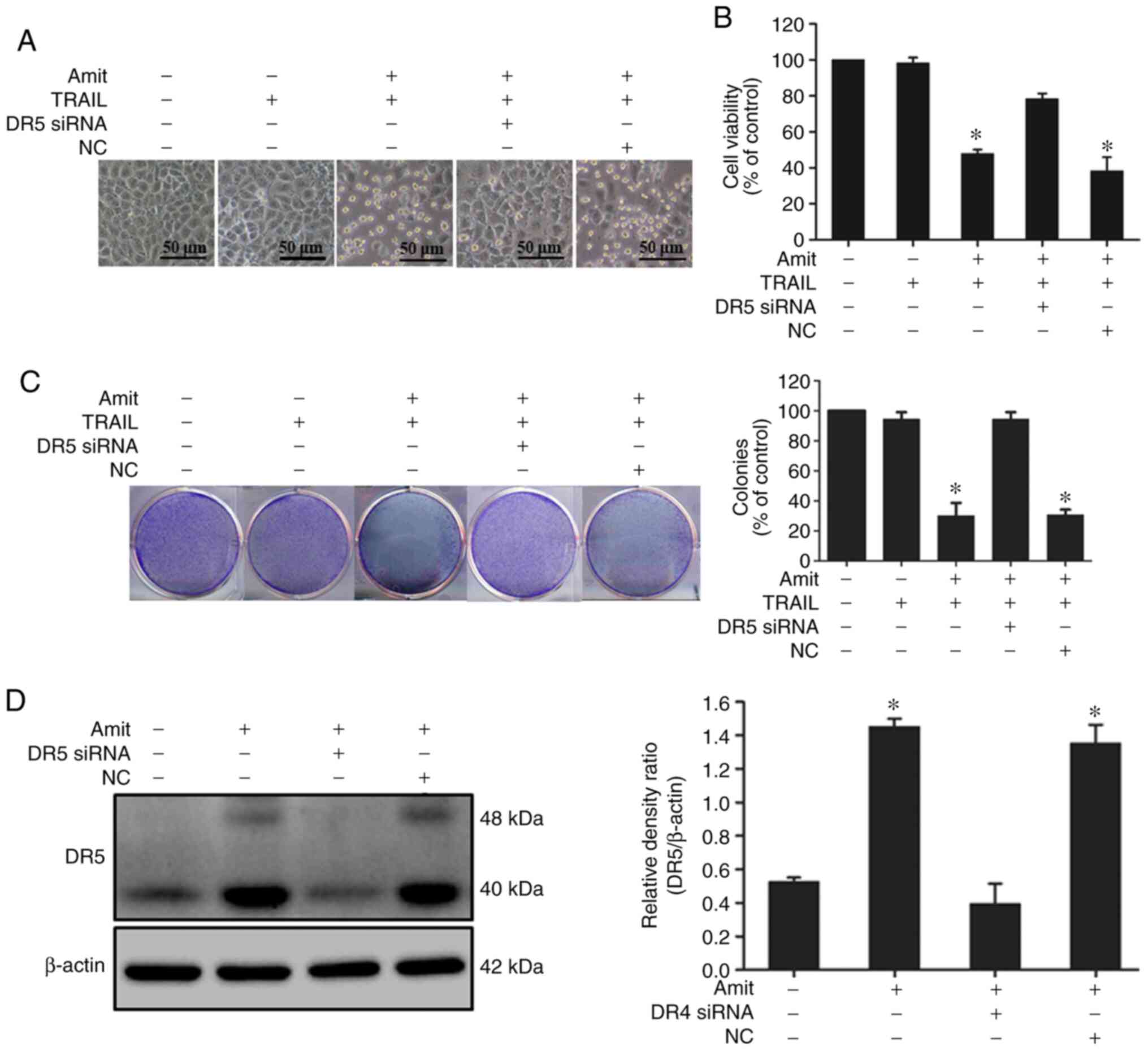
It was hypothesized that DR4 and DR5 played
important roles in amitriptyline-induced TRAIL-mediated apoptosis.
In support of this hypothesis, DR4 and DR5-specific siRNA were
applied to silence DR4 and DR5 expression, respectively. The
silencing of DR4 and DR5 expression with specific siRNA restored
cell viability. These data provided evidence that DR4 and DR5 play
an important role in enhancing the effect of amitriptyline on
TRAIL-induced apoptosis. Cells were transfected with DR4 and
DR5-specific siRNAs or a NC siRNA for 24 h and the cells were
treated with amitriptyline for 12 h, followed by incubation with
TRAIL for an additional 3 h to assess cell viability or for 2 h for
western blot analysis. The cell death induction capacity of
amitriptyline combined with TRAIL significantly decreased after
siRNA transfection. The combined effect of amitriptyline and TRAIL,
however, on viability was similar in the NC siRNA-transfected cells
(Figs. 3A and B, and 4A and B). Moreover, the colony
formation-inhibiting capacity of amitriptyline combined with
TRAIL-treated cells considerably decreased after siRNA
transfection. The colony formation-inhibiting capacity of
amitriptyline combined with TRAIL was similar in the NC control
siRNA-transfected cells (Figs. 3C and
4C). Western blot analysis revealed
that the expression of DR4 and DR5 was blocked after siRNA
transfection compared to the non-transfected cells (Figs. 3D and 4D). These experimental findings confirmed
that the upregulation of DR4 and DR5 is required in attenuating
TRAIL resistance.
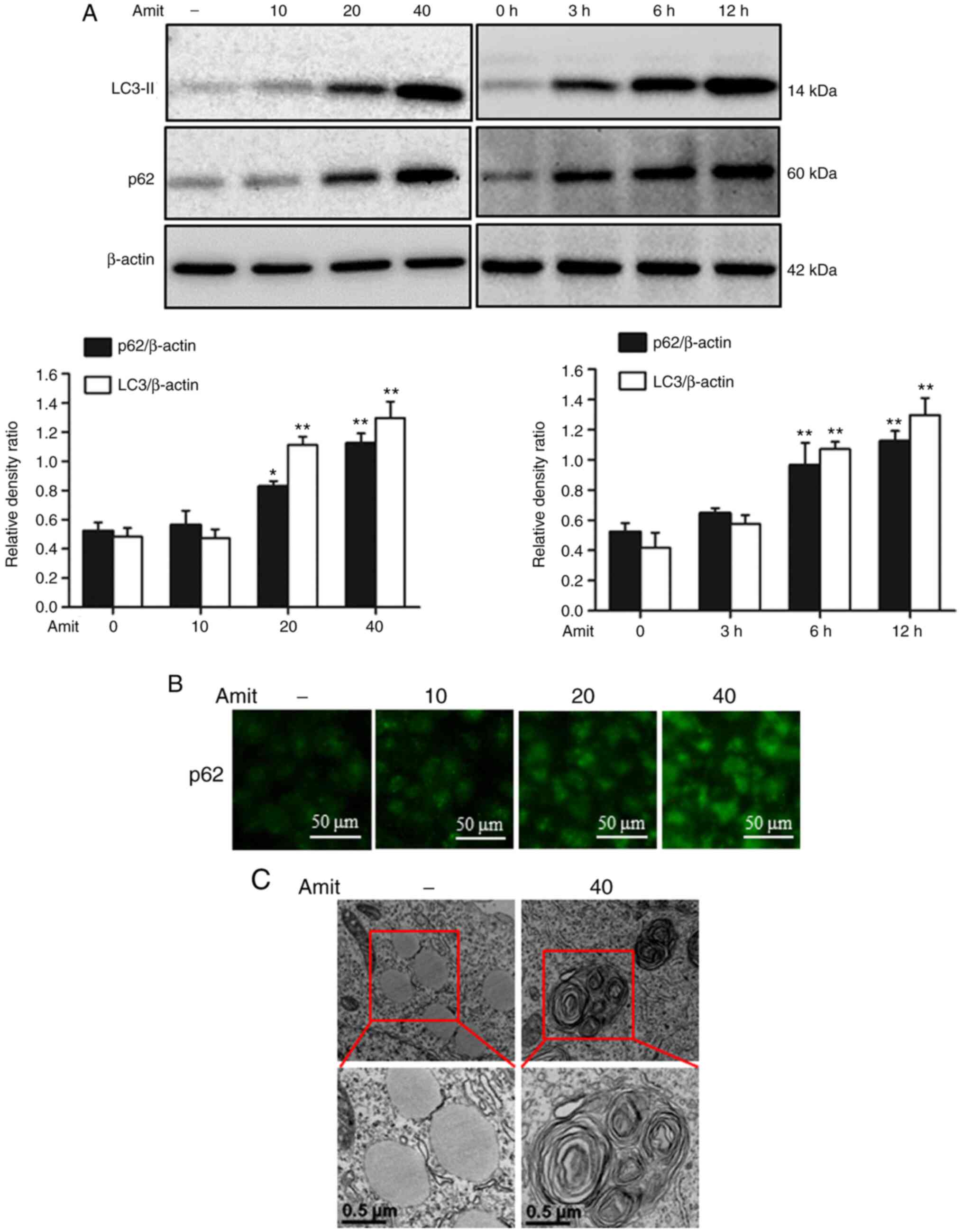
Amitriptyline blocks autophagy by
inhibiting autophagosome-lysosome fusion
To investigate the role of amitriptyline in
autophagy flux, the well-known autophagy markers LC3-II and p62
were analyzed. Western blot analysis revealed the conversion of
LC3I to LC3-II, indicating the formation of complete
autophagosomes. However, p62 is a cargo adaptor protein that
depends on lysosomes or proteasomes for degradation (31). The expression of LC3-II and p62 was
increased following amitriptyline treatment, indicating the
blocking of autophagy flux by inhibiting autophagosome-lysosome
fusion in the late stage of autophagy (Fig. 5A). The immunocytochemistry images also
demonstrated the increased expression of p62 in a dose-dependent
manner (Fig. 5B). Transmission
electron microscopy revealed the higher accumulation of autophagic
vacuoles compared to the control, confirming autophagy flux
inhibition by amitriptyline (Fig.
5C). These results indicated that amitriptyline blocked
autophagy flux at the final stage of autophagy.
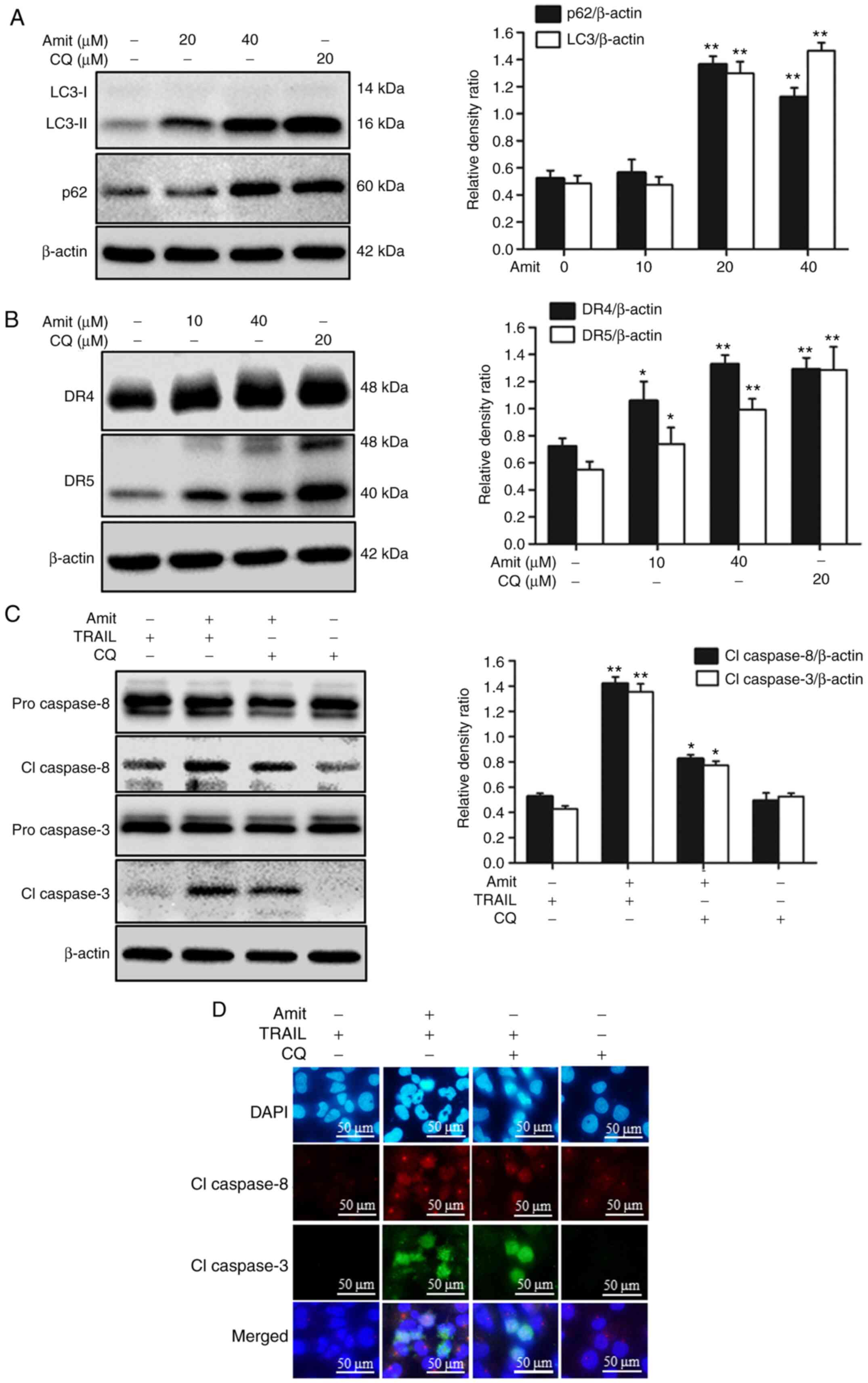
Blocking autophagy induces DR4 and DR5
upregulation and enhances TRAIL-mediated apoptosis
The role of autophagy blocking in death receptor
expression was investigated using an autophagy inhibitor. Blocking
autophagy flux with a final stage autophagy inhibitor CQ
upregulated both DR4 and DR5 expression, leading to an increase in
apoptosis. The cells were treated with or without 20 µM CQ and the
indicated doses of amitriptyline for 12 h. Western blot analysis
revealed that amitriptyline and CQ increased the levels of LC3-II.
Moreover, amitriptyline alone increased p62 levels in a
dose-dependent manner. These results revealed that amitriptyline
blocked autophagy flux to induce apoptosis (Fig. 6A). Furthermore, amitriptyline and the
autophagy inhibitor CQ increased DR4 and DR5 expression (Fig. 6B). After 12 h treatment with CQ and
amitriptyline, along with an additional 2 h TRAIL treatment, the
expression of apoptosis-associated proteins cleaved caspase-8 and
cleaved caspase-3 were observed. Cell lysates analyzed by western
blotting demonstrated that treatment with CQ and TRAIL also
activated caspase-8 and caspase-3 (Fig.
6C). The immunocytochemistry results also revealed that the CQ
and TRAIL co-treatment expressed cleaved caspase-8 and cleaved
caspase-3 compared to treatment with CQ or TRAIL alone (Fig. 6D). Additionally, to investigate the
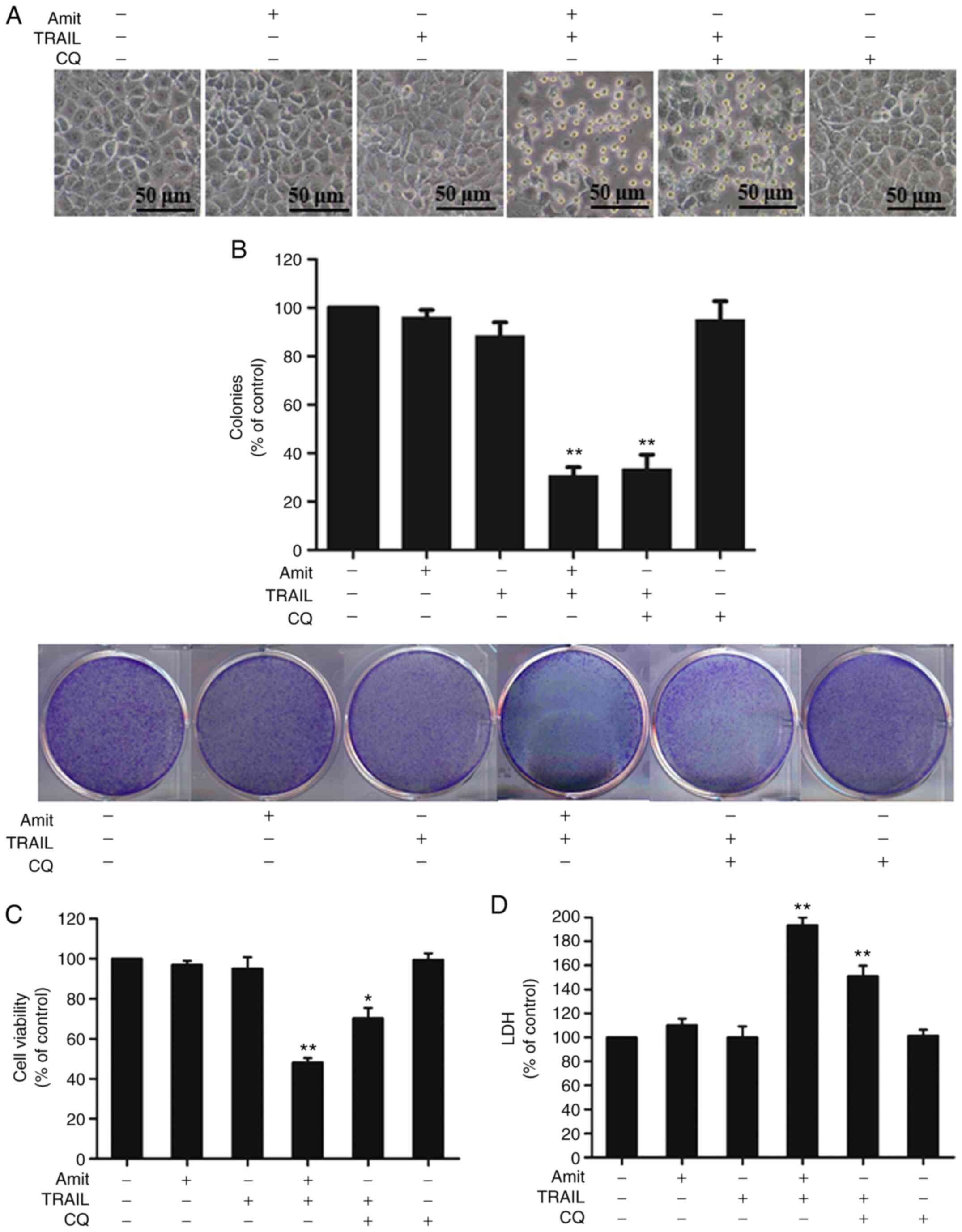
role of autophagy in TRAIL-mediated cell death, the cells were
preincubated with CQ or amitriptyline with the indicated doses for
12 h, and then additionally incubated with TRAIL for 3 h. The cell
morphology analyzed by light microscopy demonstrated slight cell
death of the A549 cells treated with either TRAIL or amitriptyline
alone. TRAIL-mediated cell death, however, was strongly increased
by the combination of amitriptyline or CQ with TRAIL (Fig. 7A). In addition, A549 cells treated
with either TRAIL, amitriptyline or CQ alone slightly reduced
colony formation capacity; but, TRAIL in combination with
amitriptyline or CQ strongly inhibited the colony formation
capacity of A549 cells (Fig. 7B). The
MTT assay showed reduced viability and significantly increased cell
death in cells treated with amitriptyline or CQ plus TRAIL
(Fig. 7C). The LDH assay also showed
that CQ or amitriptyline combined with TRAIL increased apoptotic
cell death (Fig. 7D). Overall, these
results indicated that blocking autophagy-induced DR4 and DR5
upregulation aggravated TRAIL-mediated apoptosis.
Discussion
TRAIL, a member of the tumor necrosis factor (TNF)
ligand superfamily with the exclusive ability to induce
cell-specific apoptosis with negligible or no toxicity to normal
cells, represents a promising approach to treating cancer cells
(32–34). TRAIL binds to DR4 (TRAIL-R1) and DR5
(TRAIL-R1), to form a death-inducing signaling complex (DISC),
which is associated with the adaptor molecule Fas-associated
protein with death domain (FADD), and then recruits pro-caspase-8
and forms a DISC. The recruitment of pro-caspase-8 causes the
activation of DISC and then the consequent cleavage of required
caspases-8/9/7/6. Following this, caspase-3 induces apoptotic cell
death (35–38). The involvement of DRs in
TRAIL-mediated apoptosis enhanced both the intrinsic and extrinsic
apoptosis pathways (39). TRAIL
agonists against TRAIL receptors are actively being developed for
cancer treatment due to their safety and high specificity compared
to other TNF family members (40,41). The
development of resistance toward TRAIL and TRAIL-R agonists,
however, may limit their effectiveness for monotherapy treatment.
Thus, agents that can increase TRAIL-induced apoptosis and
sensitize TRAIL-resistant cancer cells to TRAIL are necessary to
overcome resistance (42,43).
Autophagy involves an alternative cell-death
mechanism, termed programmed cell death type II (44). The main functional role of autophagy
in cells is to eliminate damaged cytosolic organelles and proteins.
In this process, cytosolic components are sequestered into
double-membraned organelles, termed autophagosomes, which
subsequently fuse with lysosomes to form autolysosomes that degrade
internal substances (45,46). A large body of evidence has
demonstrated that autophagy can also play a cell survival role that
delivers energy during metabolic stress and avoids cancer cell
death by several anticancer agents (47,48).
Autophagy inhibition prompts cancer cell death, while autophagy
shows a cell-protective role in anticancer treatments (49,50).
Autophagosome formation is designated by a lipid-conjugated form of
LC3 that is commonly known as an autophagosome marker. The
autophagosome merges with the lysosome where sequestosome-1
(commonly known as p62) incorporates into autophagosomes and
degrades LC3II, along with additional cargo proteins (51). Blocking lysosomal degradation with a
specific lysosomal inhibitor results in the prompt accumulation of
p62, indicating the inhibition of autophagy flux (52). Clinically available autophagy
inhibitors CQ or the related hydroxychloroquine (HCQ) act by
inhibiting lysosomal fusion with autophagosomes. These drugs
prevent cargo degradation by inhibiting the acidification of the
lysosome, subsequently inhibiting the fusion of autophagosomes with
lysosomes (53). Several studies have
suggested that inhibiting autophagy-sensitized cancer cells and
promoting apoptosis is a suitable target for cancer treatment
(54,55). The activation of autophagosome
accumulation and inhibition of its degradation by lysosomes
increase the death of cervical cancer cells and overcomes the
resistance of chemotherapeutic drugs cisplatin and paclitaxel
(56). Previous studies have
demonstrated that inhibition of autophagy by impeding the
acidification of the lysosome could be a possible way to restore
DR5 expression and, in turn, augment the TRAIL-induced apoptosis
(57,58). Shin et al reported that the
hepatitis B virus (HBV) X protein (HBx) inhibited TRAIL signaling
via autophagic removal of DR5 (59).
Another recent study exposed the cause of TRAIL resistance in
circulating tumor cells where DR5 is accumulated in autophagosomes
for lysosomal degradation (60).
Thus, DR5 has been determined to be controlled by the
autophagy-lysosome pathway and inhibiting autophagy may be an
effective option to overcome TRAIL resistance in cancer
therapy.
In the present study, it was determined that small
doses of amitriptyline with TRAIL were effective in increasing the
number of A549 apoptotic cells compared to single treatments. The
combined treatment with amitriptyline and TRAIL attenuated the
TRAIL resistance of lung cancer cells, initiated the expression of
the apoptotic caspase cascade, and, notably, upregulated DR4 and
DR5 expression, leading to apoptosis.
The present study mainly investigated the roles of
DR4 and DR5 in the combination effect and the mechanism of the
upregulation of DR4 and DR5. Agonistic TRAIL-R antibodies are more
attractive than TRAIL because they can target DR4 and DR5 to
initiate TRAIL-induced apoptotic death in several types of tumors
(40,41). The upregulation of DR4 or DR5 by
amitriptyline indicated the potential of a combination of
amitriptyline and TRAIL/TRAIL-R antibodies.
The present findings demonstrated that the genetic
inhibitor of DR4 and DR5 decreased the effect of amitriptyline on
TRAIL-mediated apoptosis. These results indicated that DR4 and DR5
were essential for the combined effect. Additionally, these
findings revealed for the first time that amitriptyline promoted
DR4 and DR5 expression via autophagy inhibition. Cancer cell death
was promoted by autophagy inhibition, while autophagy played a
cell-protective role in anticancer treatment (49,50). Under
such conditions, the aforementioned findings confirmed that
amitriptyline increases autophagosome formation, indicated by
LC3-II accumulation, and inhibits lysosomal fusion resulting in the
accumulation of p62, causing the inhibition of autophagy flux by
blocking autophagosome-lysosome fusion.
The combined effect of TRAIL with amitriptyline or
CQ increased cell death unlike the individual treatments. The
inhibition of autophagy by amitriptyline and the well-known
autophagy inhibitor CQ resulted in DR4 and DR5 upregulation and
improved TRAIL-mediated caspase-dependent cell death confirmed by
the enhanced caspase cascade. Amitriptyline is a psychoactive TCA
drug. In this study, only amitriptyline among the numerous
antidepressant drugs was used to reveal the enhancing effect with
TRAIL. Further studies using other antidepressant drugs are
required to support or demonstrate the sensitization to TRAIL and
anticancer effect by treatment of TCA drugs.
Collectively, these findings contributed to the
mechanistic evidence that amitriptyline sensitized lung cancer
cells to TRAIL and the sensitization was mediated through DR4 and
DR5 upregulation and autophagy inhibition. These results provide an
understanding of the anticancer effect of amitriptyline and suggest
further evaluation is required to develop possible therapeutic
regimens against lung cancer and cancer-associated depression.
Acknowledgements
Not applicable.
Funding
This study was supported by a grant from the
National Research Foundation of Korea (NRF) funded by the Ministry
of Education (grant. no. 2019R1A6A1A03033084).
Availability of data and materials
All datasets generated or analyzed during the
present study are available from the corresponding author upon
reasonable request.
Authors' contributions
KMAZ and SYP designed and performed the study,
analyzed data and wrote the manuscript. Both authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ettinger DS, Akerley W, Borghaei H, Chang
AC, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Ganti AK,
Govindan R, et al: Non-Small cell lung cancer. J Natl Compr Canc
Netw. 10:1236–1271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanitkar AA, Schwartz AG, George J and
Soubani AO: Causes of death in long-term survivors of non-small
cell lung cancer: A regional surveillance, epidemiology, and end
results study. Ann Thorac Med. 13:76–81. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heinzmann K, Nguyen QD, Honess D, Smith
DM, Stribbling S, Brickute D, Barnes C, Griffiths J and Aboagye E:
Depicting changes in tumor biology in response to cetuximab
monotherapy or combination therapy by apoptosis and proliferation
imaging using 18 F-ICMT-11 and 18 F-FLT PET.
J Nucl Med. 59:1558–1565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thomas PA: Stage IIIA N2 non-small-cell
lung cancer: Current controversies in combined-modality therapy.
Eur J Cardiothorac Surg. 36:431–432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nowak-Sliwinska P, Scapozza L and Altaba
AR: Drug repurposing in oncology: Compounds, pathways, phenotypes
and computational approaches for colorectal cancer. Biochim Biophys
Acta Rev Cancer. 1871:434–454. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jia Y, Yun CH, Park E, Ercan D, Manuia M,
Juarez J, Xu C, Rhee K, Chen T, Zhang H, et al: Overcoming
EGFR(T790M) and EGFR(C797S) resistance with mutant-selective
allosteric inhibitors. Nature. 534:129–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nesterov A, Ivashchenko Y and Kraft AS:
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)
triggers apoptosis in normal prostate epithelial cells. Oncogene.
21:1135–1140. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Trivedi R and Mishra DP: Trailing TRAIL
resistance: Novel targets for TRAIL sensitization in cancer Cells.
Front Oncol. 5:692015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marsters SA, Sheridan JP, Pitti RM, Huang
A, Skubatch M, Baldwin D, Yuan J, Gurney A, Goddard AD, Godowski P
and Ashkenazi A: A novel receptor for Apo2L/TRAIL contains a
truncated death domain. Curr Biol. 7:1003–1006. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin CY, Moon DO, Lee JD, Heo MS, Choi YH,
Lee CM, Park YM and Kim GY: Sulforaphane sensitizes tumor necrosis
factor-related apoptosis-inducing ligand-mediated apoptosis through
downregulation of ERK and akt in lung adenocarcinoma A549 cells.
Carcinogenesis. 28:1058–1066. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thorburn A, Behbakht K and Ford H: TRAIL
receptor-targeted therapeutics: Resistance mechanisms and
strategies to avoid them. Drug Resist Updat. 11:17–24. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mérino D, Lalaoui N, Morizot A, Solary E
and Micheau O: TRAIL in cancer therapy: Present and future
challenges. Expert Opin Ther Targets. 11:1299–1314. 2007.
View Article : Google Scholar
|
|
15
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rouschop KM and Wouters BG: Regulation of
autophagy through multiple independent hypoxic signaling pathways.
Curr Mol Med. 9:417–424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zinnah KMA and Park SY: Duloxetine
enhances TRAIL-mediated apoptosis via AMPK-mediated inhibition of
autophagy flux in lung cancer cells. Anticancer Res. 39:6621–6633.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Di Rosso ME, Sterle HA, Cremaschi GA and
Genaro AM: Beneficial effect of fluoxetine and sertraline on
chronic stress-induced tumor growth and cell dissemination in a
mouse model of lymphoma: Crucial role of antitumor immunity. Front
Immunol. 9:13412018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim-Fuchs C, Le CP, Pimentel MA,
Shackleford D, Ferrari D, Angst E, Hollande F and Sloan EK: Chronic
stress accelerates pancreatic cancer growth and invasion: A
critical role for beta-adrenergic signaling in the pancreatic
microenvironment. Brain Behav Immun. 40:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hasegawa H and Saiki I: Psychosocial
stress augments tumor development through beta-adrenergic
activation in mice. Jpn J Cancer Res. 93:729–735. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fann JR, Fan MY and Unützer J: Improving
primary care for older adults with cancer and depression. J Gen
Intern Med. 24 (Suppl 2):S417–S424. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Laird B, Colvin L and Fallon M: Management
of cancer pain: Basic principles and neuropathic cancer pain. Eur J
Cancer. 44:1078–1082. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frick LR and Rapanelli M: Antidepressants:
Influence on cancer and immunity? Life Sci. 92:525–532. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Z, Du X, Zhao C, Cao B, Zhao Y and
Mao X: The antidepressant amitriptyline shows potent therapeutic
activity against multiple myeloma. Anticancer Drugs. 24:792–798.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cordero MD, Sánchez-Alcázar JA,
Bautista-Ferrufino MR, Carmona-López MI, Illanes M, Ríos MJ,
Garrido-Maraver J, Alcudia A, Navas P and de Miguel M: Acute
oxidant damage promoted on cancer cells by amitriptyline in
comparison with some common chemotherapeutic drugs. Anticancer
Drugs. 21:932–944. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan X, Gajan A, Chu Q, Xiong H, Wu K and
Wu GS: Developing TRAIL/TRAIL death receptor-based cancer
therapies. Cancer Metastasis Rev. 37:733–748. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Islam MA, Sooro MA and Zhang P: Autophagic
regulation of p62 is critical for cancer therapy. Int J Mol Sci.
19:14052018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wiley SR, Schooley K, Smolak PJ, Din WS,
Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA,
et al: Identification and characterization of a new member of the
TNF family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Walczak H, Miller RE, Ariail K, Gliniak B,
Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aggarwal BB, Bhardwaj U and Takada Y:
Regulation of TRAIL-induced apoptosis by ectopic expression of
antiapoptotic factors. Vitam Horm. 67:453–483. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang S: TRAIL: A sword for killing tumors.
Curr Med Chem. 17:3309–3317. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chaudhary PM, Eby M, Jasmin A, Bookwalter
A, Murray J and Hood L: Death receptor 5, a new member of the TNFR
family, and DR4 induce FADD-dependent apoptosis and activate the
NF-kappaB pathway. Immunity. 7:821–830. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pan G, O'Rourke K, Chinnaiyan AM, Gentz R,
Ebner R, Ni J and Dixit VM: The receptor for the cytotoxic ligand
TRAIL. Science. 276:111–113. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cretney E, Takeda K and Smyth MJ: Cancer:
Novel therapeutic strategies that exploit the TNF-related
apoptosis-inducing ligand (TRAIL)/TRAIL receptor pathway. Int J
Biochem Cell Biol. 39:280–286. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Danial NN and Korsmeyer SJ: Cell death:
Critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Plummer R, Attard G, Pacey S, Li L, Razak
A, Perrett R, Barrett M, Judson I, Kaye S, Fox NL, et al: Phase 1
and pharmacokinetic study of lexatumumab in patients with advanced
cancers. Clin Cancer Res. 13:6187–6194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hotte SJ, Hirte HW, Chen EX, Siu LL, Le
LH, Corey A, Iacobucci A, MacLean M, Lo L, Fox NL and Oza AM: A
phase 1 study of mapatumumab (fully human monoclonal antibody to
TRAIL-R1) in patients with advanced solid malignancies. Clin Cancer
Res. 14:3450–3455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cheng H, Hong B, Zhou L, Allen JE, Tai G,
Humphreys R, Dicker DT, Liu YY and El-Deiry WS: Mitomycin C
potentiates TRAIL-induced apoptosis through p53-independent
upregulation of death receptors: Evidence for the role of c-Jun
N-terminal kinase activation. Cell Cycle. 11:3312–3323. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dolloff NG, Mayes PA, Hart LS, Dicker DT,
Humphreys R and El-Deiry WS: Off-target lapatinib activity
sensitizes colon cancer cells through TRAIL death receptor
up-regulation. Sci Transl Med. 3:86ra502011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-Eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Amin A, Bajbouj K, Koch A, Gandesiri M and
Schneider-Stock R: Defective autophagosome formation in p53-null
colorectal cancer reinforces crocin-induced apoptosis. Int J Mol
Sci. 16:1544–1561. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong
WY, Liu ZG, Ong CN and Shen HM: Autophagy plays a protective role
during zVAD-induced necrotic cell death. Autophagy. 4:457–466.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
White E: Autophagic cell death unraveled:
Pharmacological inhibition of apoptosis and autophagy enables
necrosis. Autophagy. 4:399–401. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vucicevic L, Misirkic M, Janjetovic K,
Vilimanovich U, Sudar E, Isenovic E, Prica M, Harhaji-Trajkovic L,
Kravic-Stevovic T, Bumbasirevic V and Trajkovic V: Compound C
induces protective autophagy in cancer cells through AMPK
inhibition-independent blockade of Akt/mTOR pathway. Autophagy.
7:40–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shen S, Zhang Y, Wang Z, Zhang R and Gong
X: Bufalin induces the interplay between apoptosis and autophagy in
glioma cells through endoplasmic reticulum stress. Int J Biol Sci.
10:212–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Arozena AA, Adachi H, Adams CM, Adams PD and
Adeli K: Guidelines for the use and interpretation of assays for
monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gómez-Sánchez R, Yakhine-Diop SMS,
Rodríguez-Arribas M, Bravo-San Pedro JM, Martínez-Chacón G,
Uribe-Carretero E, de Castro DC, Pizarro-Estrella E, Fuentes JM and
González-Polo RA: mRNA and protein dataset of autophagy markers
(LC3 and p62) in several cell lines. Data Brief. 7:641–647. 2016.
View Article : Google Scholar
|
|
53
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nordstrøm LU, Sironi J, Aranda E, Maisonet
J, Perez-Soler R, Wu P and Schwartz EL: Discovery of autophagy
inhibitors with antiproliferative activity in lung and pancreatic
cancer cells. ACS Med Chem Lett. 6:134–139. 2015. View Article : Google Scholar
|
|
55
|
Pan H, Wang Y, Na K, Wang Y, Wang L, Li Z,
Guo C, Guo D and Wang X: Autophagic flux disruption contributes to
Ganoderma lucidum polysaccharide-induced apoptosis in human
colorectal cancer cells via MAPK/ERK activation. Cell Death Dis.
10:4562019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gąsiorkiewicz BM, Koczurkiewicz-Adamczyk
P, Piska K and Pękala E: Autophagy modulating agents as
chemosensitizers for cisplatin therapy in cancer. Invest New Drugs.
39:538–563. 2020. View Article : Google Scholar
|
|
57
|
Nazim UM, Yin H and Park SY:
Downregulation of c-FLIP and upregulation of DR-5 by cantharidin
sensitizes TRAIL-mediated apoptosis in prostate cancer cells via
autophagy flux. Int J Mol Med. 46:280–288. 2020.PubMed/NCBI
|
|
58
|
Park EJ, Min Kj, Choi KS, Kubatka P,
Kruzliak P, Kim DE and Kwon TK: Chloroquine enhances TRAIL-mediated
apoptosis through up-regulation of DR5 by stabilization of mRNA and
protein in cancer cells. Sci Rep. 6:229212016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shin GC, Kang HS, Lee AR and Kim KH:
Hepatitis B virus-triggered autophagy targets TNFRSF10B/death
receptor 5 for degradation to limit TNFSF10/TRAIL response.
Autophagy. 12:2451–2466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Twomey JD and Zhang B: Circulating tumor
cells develop resistance to TRAIL-induced apoptosis through
autophagic removal of death receptor 5: Evidence from an in vitro
model. Cancers (Basel). 11:942019. View Article : Google Scholar : PubMed/NCBI
|