Introduction
Nasopharyngeal carcinoma (NPC), originating from the
nasopharyngeal epithelium, is the most prevalent type of head and
neck cancer in Southeast Asian populations (1). It is characterized by local
aggressiveness and early distant metastasis (2). In China, approximately 60,600 new NPC
cases are diagnosed every year, accounting for 40% of the global
cases (3). Currently,
intensity-modulated radiation therapy combined with neoadjuvant
chemotherapy is the primary therapeutic strategy for NPC (4). Efforts focused on cancer diagnosis and
treatment have significantly improved the long-term clinical
outcomes of patients with early-stage NPC. However, numerous
patients are diagnosed with NPC at an advanced stage and have a
poor prognosis (5). Local or regional
relapse and metastasis are the most common reasons for the
management failure (6). Although the
mechanisms of NPC oncogenesis and progression are complicated, they
are mainly attributed to genetics, viral infections, and
environmental factors (7). However,
the detailed underlying mechanisms remain poorly understood.
Therefore, clarification of the mechanisms of NPC pathogenesis is
urgent and necessary for identifying effective anticancer
therapies.
Long noncoding RNAs (lncRNAs), whose length is more
than 200 nucleotides, are emerging as cancer drug targets (8). They belong to a subset of noncoding RNA
molecules that are not translated into proteins but modulate gene
expression (9). Formerly, lncRNAs
were considered transcriptional ‘noise’ or cloning artefacts
(10). However, they have recently
been revealed to be significant regulators of various physical and
pathological phenotypes (11). The
dysregulation of lncRNAs is closely associated with various
processes in NPC oncogenesis and progression (12,13).
Numerous lncRNAs are dysregulated in NPC and exert promoting or
inhibitory effects on cancer regulation (14,15).
MicroRNAs (miRNAs) are a group of short noncoding
transcripts with a length of 17–21 nucleotides (16). They are confirmed as vital gene
modulators and work through complementary pairing with the
3′-untranslated regions (UTRs) of their target genes, ultimately
triggering mRNA degradation or reducing translation (17). Numerous miRNAs are abnormally
expressed in NPC and are essential in inducing malignancy and
tumorigenesis (18–20). The theory of competitive endogenous
RNA (ceRNA) proposed by Salmena et al (21) has accelerated research on lncRNA
mechanisms. LncRNAs function as sponges for miRNA, subsequently
abating the inhibitory effects of miRNAs on their targeted mRNAs
(21).
According to a previous study, SLC9A3 antisense RNA
1 (SLC9A3-AS1) was revealed to play a central role in lung cancer
(22). However, researchers have not
clearly determined whether SLC9A3-AS1 exhibits clinical value in
NPC and how it performs its specific functions in NPC. Its
underlying mechanisms require further study. Thus, SLC9A3-AS1 was
selected as the focus of the present study. SLC9A3-AS1 expression
in NPC was analysed, and roles of SLC9A3-AS1 in NPC were
investigated. Furthermore, mechanistic experiments were used to
dissect the downstream mechanisms of SLC9A3-AS1 in NPC.
Materials and methods
Patient tissues
The present study was approved by the Ethics
Committee of the People's Hospital of Rizhao (Rizhao, China). The
experimental procedures were performed in strict compliance with
the Declaration of Helsinki (2013 version). In addition, all
participants provided written informed consent. A total of 46 NPC
tissues were acquired from patients (28 males and 18 females; age
range, 32–65 years) at our hospital (People's Hospital of Rizhao)
between May 2014 to December 2015. The inclusion criteria of NPC
were as follows: i) Diagnosed as NPC; ii) had not been treated with
preoperative anticancer treatments and iii) agreed to participate
in the study. In addition, 15 normal nasopharyngeal epithelial
tissues were provided by healthy volunteers (9 males and 6 females;
age range, 28–57 years) at our hospital. The inclusion criteria of
the healthy volunteers were as follows: i) Have not been diagnosed
as NPC; and ii) agreed to participate in the study. Patients with
other types of human malignancies and patients who had undergone
any form of anticancer therapy were excluded from this study. All
the excised tissues were immediately flash-frozen and maintained in
liquid nitrogen.
Cell lines
The normal nasopharyngeal epithelial cell line NP69
and three NPC cell lines (SUNE1, C666-1 and 6-10B) were purchased
from the Laboratory of Cell Biology, Modern Analysis and Testing
Center, Central South University (Changsa, China). NP69 cells were
grown in keratinocyte serum-free medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 30 µg/ml bovine pituitary extract (BD
Biosciences). NPC cells were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS), and 1%
penicillin/streptomycin mixture (Gibco; Thermo Fisher Scientific,
Inc.). All the cells were grown in saturated humidity with 5%
CO2 at 37°C.
Cell transfection
Small interfering RNAs (siRNAs) synthesized against
SLC9A3-AS1 (si-SLC9A3-AS1) and a corresponding negative control
(NC) siRNA (si-NC) were obtained from Shanghai GenePharma Co., Ltd.
The si-SLC9A3-AS1#1 sequence was 5′-CACATGTTTTTTATAATAAAACA-3′; the
si-SLC9A3-AS1#2 sequence was 5′-ATGTTTTTTATAATAAAACATAG-3′; and the
si-NC sequence was 5′-CACGATAAGACAATGTATTT-3′. The miR-486-5p
mimic, miR-486-5p inhibitor, and the matching controls (NC mimic
and NC inhibitor, respectively) were prepared by Guangzhou RiboBio
Co., Ltd. The miR-486-5p mimic sequence was
5′-GAGCCCCGUCGAGUCAUGUCCU-3′ and the NC mimic sequence was
5′-UUGUACUACACAAAAGUACUG-3′. The miR-486-5p inhibitor sequence was
5′-CUCGGGGCAGCUCAGUACAGGA-3′ and the NC inhibitor sequence was
5′-ACUACUGAGUGACAGUAGA-3′. The pcDNA3.1-E2F6 plasmid was
constructed by cloning the E2F6 cDNA into pcDNA3.1 (GenScript
Biotech Corporation). The transfection of siRNA (100 pmol), miRNA
oligonucleotides (100 pmol) or plasmids (4 µg) was performed at
room temperature with Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). At 24 h post-transfection, Cell
Counting Kit-8 (CCK-8), colony-forming and cell migration and
invasion assays were performed. Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR), flow cytometry and western
blotting were carried out after 48 h of culture.
RT-qPCR
A mirVana™ miRNA Isolation kit (Ambion; Thermo
Fisher Scientific, Inc.) was employed for isolating small RNA,
which was then reverse transcribed into complementary DNA by
applying miRcute miRNA First-Strand cDNA Synthesis Kit (Tiangen
Biotech Co., Ltd.) according to the manufacturer's instructions.
The expression of miR-486-5p was detected using PCR amplification
with a miRcute miRNA qPCR Detection Kit SYBR-Green (Tiangen Biotech
Co., Ltd.) according to the manufacturer's instructions, with small
nuclear RNA U6 as an internal reference. The thermocycling
conditions were as follows: 95°C for 15 min; and 94°C for 20 sec
and 60°C for 34 sec, for 45 cycles. The extraction of total RNA was
performed using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.). For SLC9A3-AS1 and E2F6 quantification, reverse
transcription was conducted using a PrimeScript reagent Kit with
gDNA Eraser (Takara Biotechnology Co., Ltd.). Then, complementary
DNA was subjected to quantitative PCR by using PrimeScript™ RT
Master Mix (Takara Biotechnology Co., Ltd.). The thermocycling
conditions were as follows: 95°C for 30 sec, 95°C for 3 sec and
60°C for 30 sec, for 40 cycles; 95°C for 15 sec, 60°C for 60 sec
and 95°C for 15 sec. SLC9A3-AS1 and E2F6 levels were normalized to
GAPDH. All gene expression levels were analysed with the
2−ΔΔCq method (23).
The primers were designed as follows: SLC9A3-AS1,
5′-CGAGAGAGGGCAGCGGCTAGT−3′(forward) and
5′-TAACTTTCCAAGGCACCCAGCA-3′ (reverse); E2F6,
5′-TGTGTCCATGAGAAAAGCTCTAAAA-3′ (forward) and
5′-ACCTTGTTTAAGTCAAGAATACCCC-3′ (reverse); GAPDH,
5′-ACCTGACCTGCCGTCTAGAAAA-3′ (forward) and
5′-TTGAAGTCAGAGGAGACCACCTG-3′ (reverse); miR-486-5p,
5′-TCGGCAGGUCCUGUACUGAG-3′ (forward) and 5′-CACTCAACTGGTGTCGTGGA-3′
(reverse); U6, 5′-CTCGCTTCGGCAGCACA-3′ (forward) and
5′-AACGCTTCACGAATTTGCGT-3′ (reverse).
CCK-8 assay
The transfected NPC cells were harvested and counted
separately after being inoculated in 96-well plates. Each well
covered 100 µl cell suspension harbouring 3,000 cells. Cells were
cultured in saturated humidity with 5% CO2 at 37°C. For
cell proliferation measurement, 10 µl of CCK-8 solution (Beyotime
Institute of Biotechnology) were added and incubated at 37°C for 2
h. Subsequently, the cells were counted at 450 nm with an ELISA
plate reader. The assay was executed every 24 h until 72 h, and
acquired data was applied for plotting a growth curve.
Colony-forming assay
The transfected NPC cells were harvested and counted
separately. Then, 2 ml of the cell suspension containing 1,000
cells per well was seeded on 6-well plates. After 2 weeks of
incubation at 37°C, the cells were washed with phosphate-buffered
saline (PBS), and then they were fixed using 4% paraformaldehyde at
room temperature for 30 min and stained using 0.5% crystal violet
at room temperature for 30 min. Ultimately, the formed colonies
(>50 cells) were counted under an inverted light microscope
(Leica Microsystems GmbH).
Flow cytometry
Transfected cells were detached with EDTA-free
trypsin and centrifuged at room temperature with 1,000 × g for 5
min. The percentage of apoptotic cells was determined employing
eBioscience™ Annexin V Apoptosis Detection Kit FITC (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. After one rinse with PBS and one rinse with 1X
binding buffer, the cells were collected and resuspended in 100 µl
of 1X binding buffer. Then, fluorochrome-conjugated Annexin V (5
µl) and propidium iodide (5 µl) was appended to the cell suspension
and cultured for 20 min at 20–25°C under darkness. Finally, the
cell apoptosis rate was assayed with a FACSCalibur flow cytometer
(BD Biosciences). CellQuest software (version 2.9; BD Biosciences)
was utilized for data abalysis.
Cell migration and invasion
assays
Transwell chambers (8 µm; Corning Inc.) were
utilized to uncover the cell migratory and invasive capacities. For
the determination of cell migration, 200 µl of FBS-free medium
carrying 5×104 cells were seeded to the upper chambers.
In the lower chambers, 600 µl of RPMI-1640 medium mixed with 20%
FBS was plated and used as the chemoattractant. Subsequent to 24-h
culture, the non-migrated cells were gently removed from the upper
chamber, while the migrated cells were fixed with 4%
paraformaldehyde at room temperature for 30 min, stained with 0.5%
crystal violet at room temperature for 30 min, and observed and
counted under an inverted light microscope (×200, magnification;
Leica Microsystems GmbH). The invasion test used Matrigel (Corning
Inc.) precoated-Transwell chambers and the same subsequent
experimental procedure as the migration assay.
Lentivirus production and
infection
Lentiviruses were produced using a second-generation
lentiviral system. The short hairpin RNA (shRNA) for SLC9A3-AS1
(sh-SLC9A3-AS1) and NC shRNA (sh-NC; Shanghai GenePharma Co., Ltd.)
were cloned into the pLKO.1 plasmid. The sh-SLC9A3-AS1 sequence was
5′-CCGGATGTTTTTTATAATAAAACATAGCTCGAGCTATGTTTTATTATAAAAAACATTTTTTG-3′
and sh-NC sequence was
5′-CCGGCACGATAAGACAATGTATTTCTCGAGAAATACATTGTCTTATCGTGTTTTTG-3′. The
constructed plasmids were transfected into 293T cells (National
Collection of Authenticated Cell Cultures, Shanghai, China) with
the psPAX2 packaging plasmid and pMD2. G envelope plasmid to
generate lentiviruses. The ratio of lentiviral plasmid: psPAX2:
pMD2.G: pLKO.1 was 1:1:2, and a total of 30 µg plasmids were used
for lentivirus package. After 48 h of incubation at 37°C,
lentiviruses overexpressing sh-SLC9A3-AS1 or sh-NC were collected
and mixed with polybrene (5 µg/ml; Sigma-Aldrich; Merck KGaA) and
culture medium. Then, the mixture was added into SUNE1 cells with
an MOI=5 for lentivirus infection. The infected cells were treated
at 37°C with puromycin (2 µg/ml; Sigma-Aldrich) for 10 days to
obtain SUNE1 cells with stable SLC9A3-AS1 depletion. The
maintenance concentration was 0.3 µg/ml.
Animal studies
All experiments involving animals were approved by
the Animal Ethics Committee of The People's Hospital of Rizhao. A
total of 6 male BALB/c nude mice aged 4–6 weeks (weight, 20 g;
Hunan SJA Laboratory Animal Co., Ltd.) received a subcutaneous
injection of SUNE1 cells with stable SLC9A3-AS1 knockdown to
generate tumours. All mice were housed under specific pathogen-free
conditions at 25°C and 50% humidity, with a 10:14 light/dark cycle
and ad libitum access to food and water. The tumours were
measured with Vernier callipers every 5 days to calculate the
tumour volume using the following formula: Tumour volume=0.5×
length × width2. On day 35, the mice were euthanized by
cervical dislocation, and the tumour xenografts were processed for
weighing, RT-qPCR and western blotting.
Immunohistochemistry
Tumor xenografts were fixed in 4% neutral formalin
at room temperature for 48 h, soaked in 4% paraffin, and were cut
into 4-µm-thick sections. Xylene was applied for deparaffinising.
Next, rehydration was implemented with an ethanol gradient. After
culture with 0.3% H2O2 for 30 min and
blocking with 5% bovine serum albumin (R&D Systems) for 45 min
at 37°C, the slides were incubated all night with E2F6 (product
code ab53061; 1:1,000 dilution) or Ki-67 (product code ab15580;
1:1,000 dilution; both from Abcam) at 4°C, followed by 45 min of
treatment at room temperature with a horseradish
peroxidase-conjugated secondary antibody (cat. no. ab205718; Abcam;
1:500 dilution). Then, 3,3′-diaminobenzidine (DAB) color reagent
was applied to detect the antibody binding, and tumor xenografts
were counterstained with 1% hematoxylin at room temperature for 10
min and dehydrated in ethanol. Finally, image acquisition was
implemented utilizing a light microscope (×200, magnification).
Subcellular fractionation assay
The nuclear and cytoplasmic fractions of NPC cells
were separated using a PARIS kit (Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. Next, total RNA was
isolated from each fraction for RT-qPCR to quantify SLC9A3-AS1 in
the nuclear and cytoplasmic fractions.
Bioinformatics analysis
The binding between SLC9A3-AS1 and miR-486-5p was
predicted applying ENCORI (starBase 3.0; http://starbase.sysu.edu.cn/) and miRDB (http://www.mirdb.org/). Two bioinformatics tools,
ENCORI and TargetScan version 7.2 (http://www.targetscan.org), were adopted to identify
the potential targets of miR-486-5p.
Luciferase reporter assay
The SLC9A3-AS1 and E2F6 fragments harbouring the
predicted wild-type (WT) binding site (Shanghai GenePharma Co.,
Ltd.) were cloned into the pmirGLO luciferase plasmid (Promega
Corporation) to create the reporter plasmids WT-SLC9A3-AS1 and
WT-E2F6. Mutant (MUT) reporter plasmids, MUT-SLC9A3-AS1 and
MUT-E2F6, were also constructed. NPC cells were seeded into 24-well
plates and co-transfected with the reporter plasmids alongside
miR-486-5p mimic or NC mimic applying Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently, 48
h later, the transfected cells were lysed to detect their
luciferase activity employing a dual-luciferase reporter assay
system (Promega Corporation). Renilla luciferase activity
was applied to normalize the firefly luciferase activity.
RNA immunoprecipitation (RIP)
RIP was implemented with an EZ-Magna RIP™
RNA-Binding Protein Immunoprecipitation Kit (cat. no. 03-110; EMD
Millipore) according to the manufacturer's instructions. NPC cells
were lysed with RIP lysis buffer. After centrifugation at 1,000 × g
for 10 min at 4°C, 10% of the cell lysate was regarded as the
positive control (input). The remaining lysate was cultured at 4°C
overnight with magnetic beads, which were already conjugated with
an anti-Argonaute 2 (Ago2) or IgG antibody (negative control) (both
from cat. no. 03-110; dilution, 1:5,000; EMD Millipore).
Subsequently, the immunoprecipitated RNA was purified and analysed
using RT-qPCR.
Western blotting
Total protein was extracted from the cells lysed
with RIPA lysis buffer supplemented with phenylmethylsulfonyl
fluoride (both from Sangon Biotech Co., Ltd.). The quantification
of total protein was conducted with a BCA Protein Assay Kit (Sangon
Biotech Co., Ltd.). Equal amounts of proteins (20 µg) were
separated by 10% SDS-PAGE gels, followed by transferring onto PVDF
membranes. After being blocked with 5% fat-free milk at room
temperature for 2 h, the blots were probed with primary antibodies
targeting E2F6 (product code ab155978; 1:1,000 dilution) or GAPDH
(product code ab181602; 1:1,000 dilution; both from Abcam)
overnight at 4°C. The blots were treated with a horseradish
peroxidase-conjugated secondary antibody (product code ab205718;
1:5,000 dilution; Abcam) at room temperature for 2 h and then
visualized with the Immobilon ECL Ultra Western HRP Substrate
(Merck Millipore; Merck KGaA).
Statistical analysis
The results from three independent experiments were
expressed as the means ± standard deviations. Multigroup
comparisons were conducted applying one-way analysis of variance
(ANOVA), and Tukey's test was used for post hoc comparisons. Both
paired and unpaired Student's t-tests were used to contrast the
differences between the two groups. The survival of patients with
NPC was examined using Kaplan-Meier method, and the overall
survival curves were compared using the log-rank test. Pearson's
correlation analysis was carried out to assess correlations between
the expression of various genes. P<0.05 was considered to
indicate a statistically significant difference.
Results
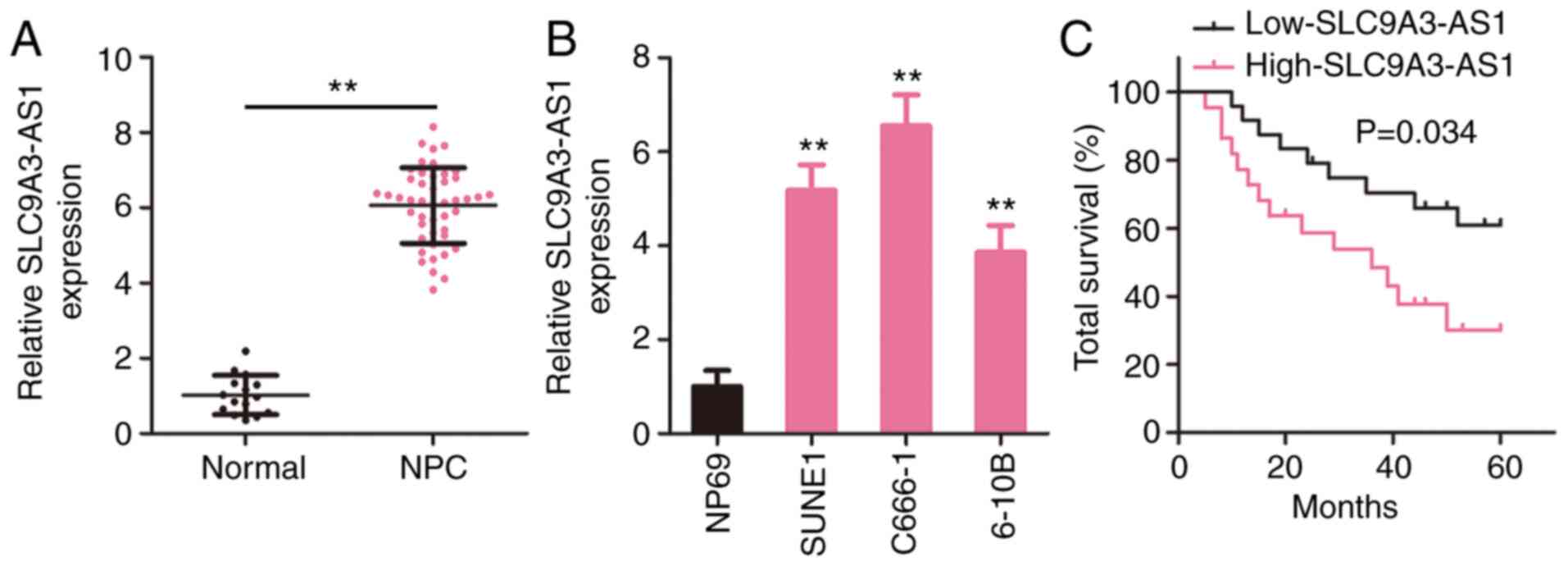
SLC9A3-AS1 is upregulated in NPC and
correlated with a poor clinical outcome
To investigate the pattern of SLC9A3-AS1 expression
in NPC, SLC9A3-AS1 level in 46 NPC tissues and 15 normal
nasopharyngeal epithelial tissues was measured utilizing RT-qPCR.
Compared with normal nasopharyngeal epithelial tissues, SLC9A3-AS1
was expressed at a higher level in NPC tissues (Fig. 1A). Additionally, all three NPC cell
lines exhibited higher SLC9A3-AS1 expression compared with NP69
(Fig. 1B). In addition, all patients
with NPC were assigned to a low-SLC9A3-AS1 (n=23) or
high-SLC9A3-AS1 (n=23) group according to the median value of
SLC9A3-AS1. The overall survival in the low-SLC9A3-AS1 group was
significantly increased compared with that in the high-SLC9A3-AS1
group (Fig. 1C). Based on these data,
SLC9A3-AS1 may participate in the genesis and development of
NPC.
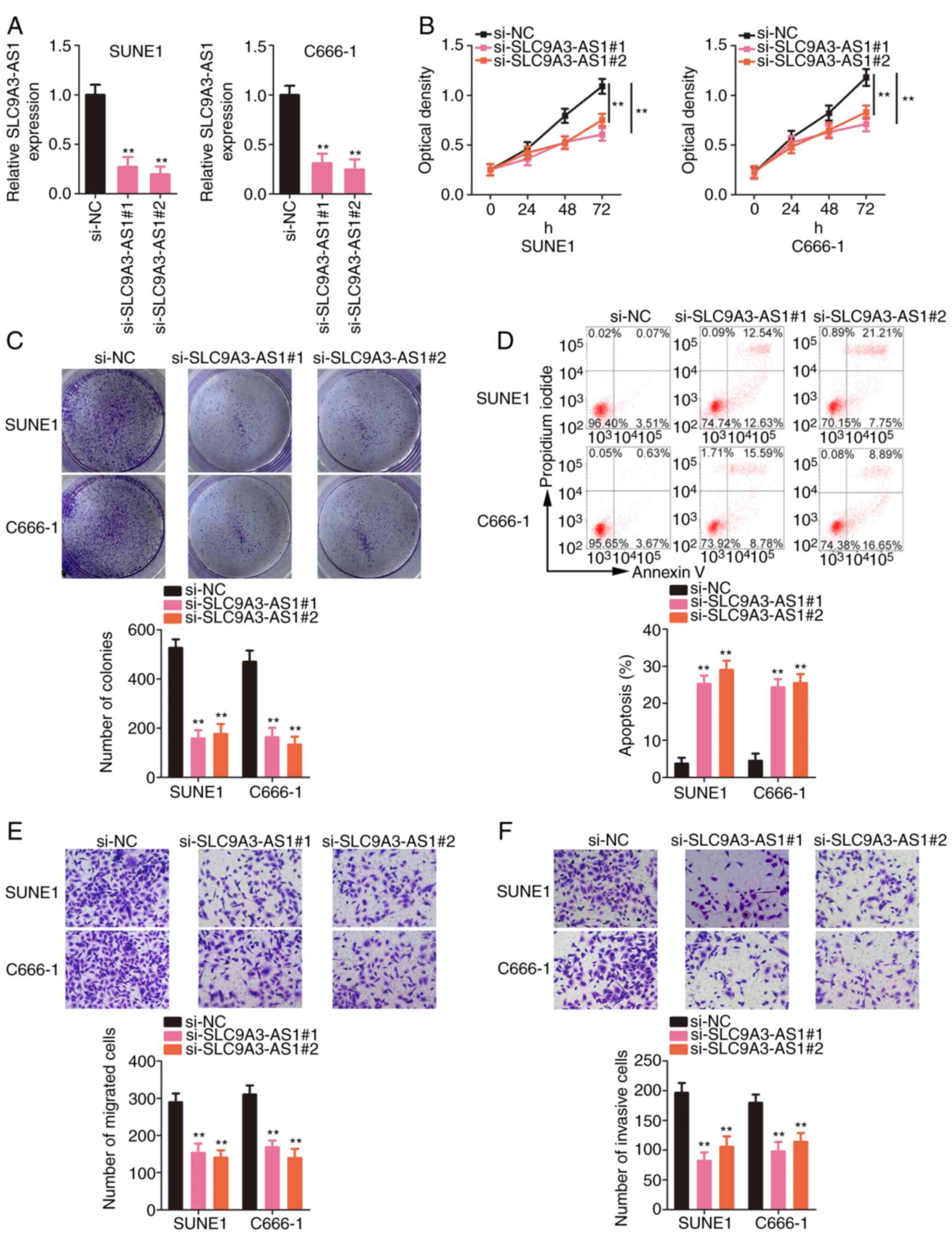
Inhibition of SLC9A3-AS1 supresses the
malignant processes of NPC cells in vitro
Next, the functions of SLC9A3-AS1 in NPC cells were
delineated. SUNE1 and C666-1 cells expressed the highest SLC9A3-AS1
expression level among the three NPC cells; therefore, these cell
lines were selected for functional experiments. First, SLC9A3-AS1
was depleted in SUNE1 and C666-1 cells by transfecting
si-SLC9A3-AS1. Two siRNAs targeting SLC9A3-AS1 were used to avoid
off-target effects; both siRNAs significantly silenced SLC9A3-AS1
expression (Fig. 2A). The
proliferative capacity of NPC cells was suppressed after SLC9A3-AS1
depletion (Fig. 2B). Furthermore,
transfection with si-SLC9A3-AS1 significantly decreased the
formation of NPC cell colonies (Fig.
2C). Additionally, flow cytometric data revealed that the
apoptosis of SLC9A3-AS1-deficient NPC cells was significantly
increased (Fig. 2D). Furthermore,
migration (Fig. 2E) and invasion
(Fig. 2F) activities were inhibited
in NPC cells with SLC9A3-AS1 knockdown. Thus, SLC9A3-AS1 exerted
tumour-promoting effects on NPC cells.
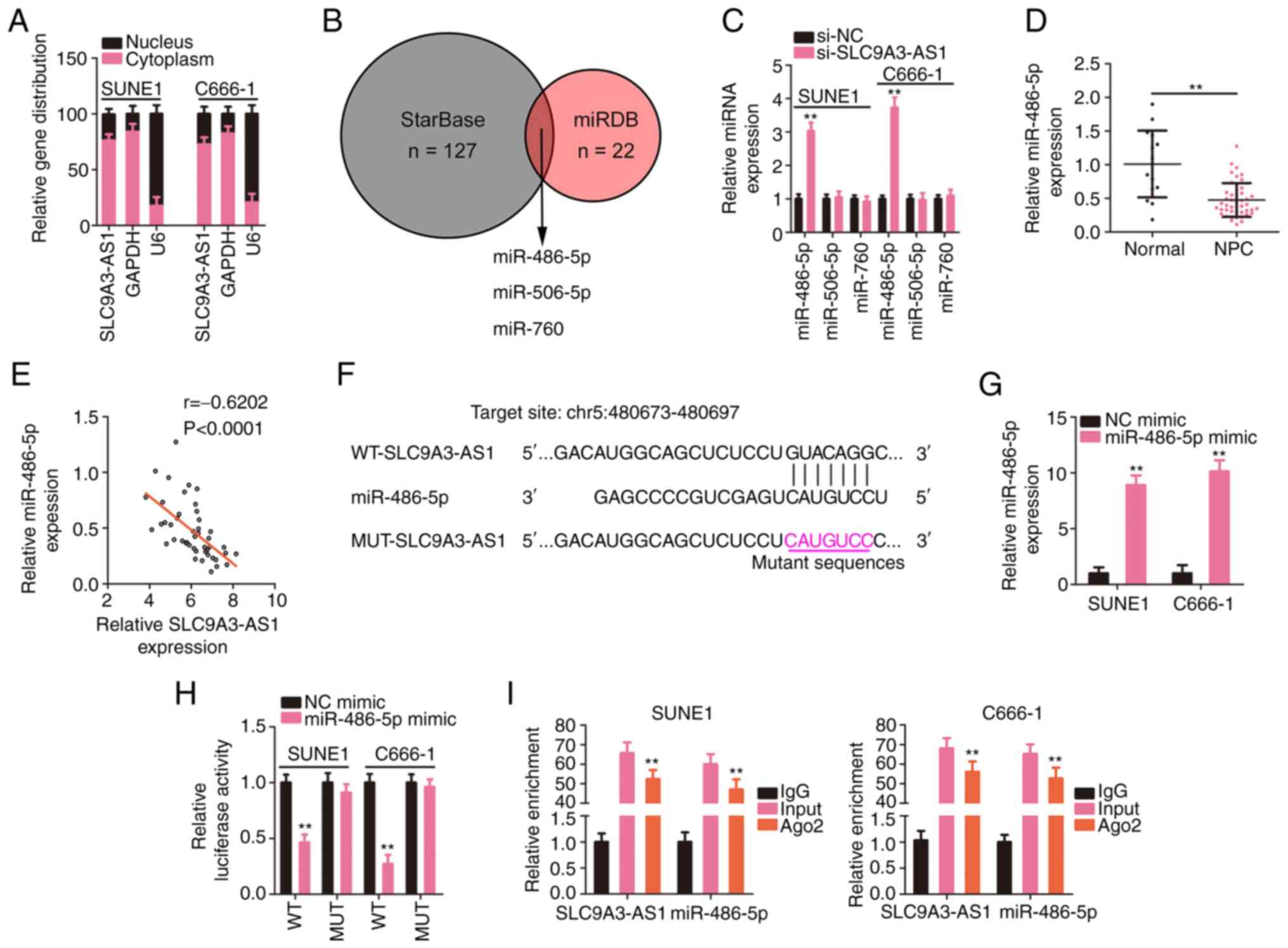
SLC9A3-AS1 directly sponges miR-486-5p
in NPC
The mechanism of SLC9A3-AS1 was further dissected by
investigating the subcellular distribution of SLC9A3-AS1 in NPC
cells. SLC9A3-AS1 was primarily localized in NPC cell cytoplasm
(Fig. 3A), suggesting a role for
SLC9A3-AS1 as a miRNA sponge. The putative target miRNAs of
SLC9A3-AS1 were predicted using starBase and miRDB, and three
overlapping miRNAs from the queries were identified (Fig. 3B). The targeting relationships between
SLC9A3-AS1 and the miRNAs were examined by assessing the levels of
the three overlapping miRNAs in SLC9A3-AS1-silenced NPC cells using
RT-qPCR. Loss of SLC9A3-AS1 expression increased miR-486-5p
expression but exerted no effect on the levels of miR-506-5p and
miR-760 (Fig. 3C). In addition,
miR-486-5p was considerably downregulated in NPC tissues (Fig. 3D), and a modest but significant
inverse relationship was validated between SLC9A3-AS1 and
miR-486-5p expression levels (Fig.
3E). As depicted in Fig. 3F,
miR-486-5p contained sequences complementary to SLC9A3-AS1. Next,
the target interaction between miR-486-5p and SLC9A3-AS1 in NPC
cells was verified employing a luciferase reporter assay.
miR-486-5p upregulation (Fig. 3G)
induced by the miR-486-5p mimic significantly reduced the
luciferase activity driven by WT-SLC9A3-AS1; however, the
modulatory effect was abolished when its binding site for
SLC9A3-AS1 was mutated (Fig. 3H).
Furthermore, the RIP results confirmed that compared with the IgG
group, the enrichment of miR-486-5p and SLC9A3-AS1 was increased in
the Ago2 group (Fig. 3I). Based on
these results, SLC9A3-AS1 was revealed to sponge miR-486-5p in
NPC.
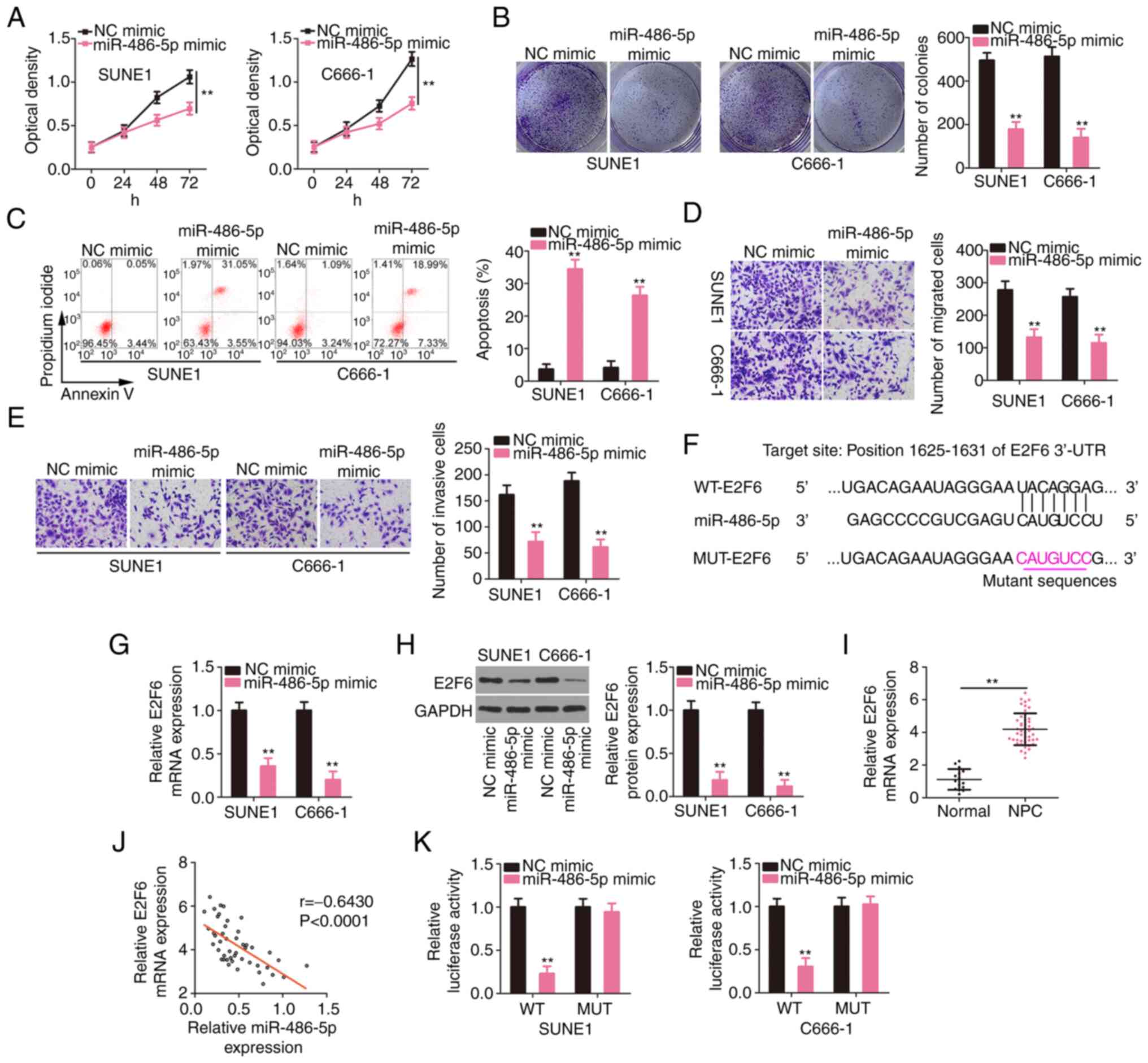
E2F6 is a downstream target of
miR-486-5p in NPC
The roles of miR-486-5p were studied by
overexpressing it in SUNE1 and C666-1 cells and performing
gain-of-function assays. Compared to the NC mimic group, the
proliferative (Fig. 4A) and
colony-forming (Fig. 4B) capacities
of NPC cells were restrained after miR-486-5p overexpression. In
addition, exogenous miR-486-5p caused a significant increase in NPC
cell apoptosis (Fig. 4C). Moreover,
the migration and invasion of NPC cells overexpressing miR-486-5p
were supressed (Fig. 4D and E). Then,
the mechanisms involving miR-486-5p in NPC cells were studied.
According to the bioinformatics prediction, the 3′-UTR of E2F6
contained potential binding sequences for miR-486-5p (Fig. 4F). E2F6 expression levels were reduced
in miR-486-5p overexpressed-NPC cells (Fig. 4G and H). The RT-qPCR data revealed the
upregulation of E2F6 in NPC tissues (Fig.
4I), which presented a negative relationship with miR-486-5p
expression (Fig. 4J). Furthermore,
the luciferase activity driven by WT-E2F6 was downregulated by the
miR-486-5p mimic in NPC cells; but, the luciferase activity of
MUT-E2F6 was unaltered in response to miR-486-5p reintroduction
(Fig. 4K). In summary, miR-486-5p was
revealed to directly target E2F6 in NPC.
SLC9A3-AS1 indirectly regulates E2F6
expression in NPC cells by controlling miR-486-5p
Since the relationships of miR-486-5p with its
upstream regulator SLC9A3-AS1 and its downstream target E2F6 were
confirmed, the regulatory interactions among the three RNAs were
further analysed. E2F6 expression levels (Fig. 5A and B) were significantly suppressed
in NPC cells transfected with si-SLC9A3-AS1. Additionally, a
positive relationship between SLC9A3-AS1 and E2F6 expression levels
were observed in NPC cells (Fig. 5C).
Notably, compared with the IgG group, the enrichment of SLC9A3-AS1,
miR-486-5p, and E2F6 was considerably enhanced in the Ago2 group
(Fig. 5D). Rescue experiments were
implemented to assess whether miR-486-5p elicited the modulatory
roles of SLC9A3-AS1 on E2F6. RT-qPCR verified the efficiency of the
miR-486-5p inhibitor transfection and revealed that it considerably
decreased miR-486-5p expression (Fig.
5E). Then, miR-486-5p inhibitor or NC inhibitor were
transfected into SLC9A3-AS1 depleted-NPC cells, followed by the
detection of E2F6 expression. Treatment of miR-486-5p inhibitor
significantly restored the E2F6 (Fig. 5F
and G) expression, which was previously reduced by SLC9A3-AS1
silencing. Therefore, SLC9A3-AS1 functioned as a ceRNA for
miR-486-5p, thus upregulating E2F6 expression in NPC.
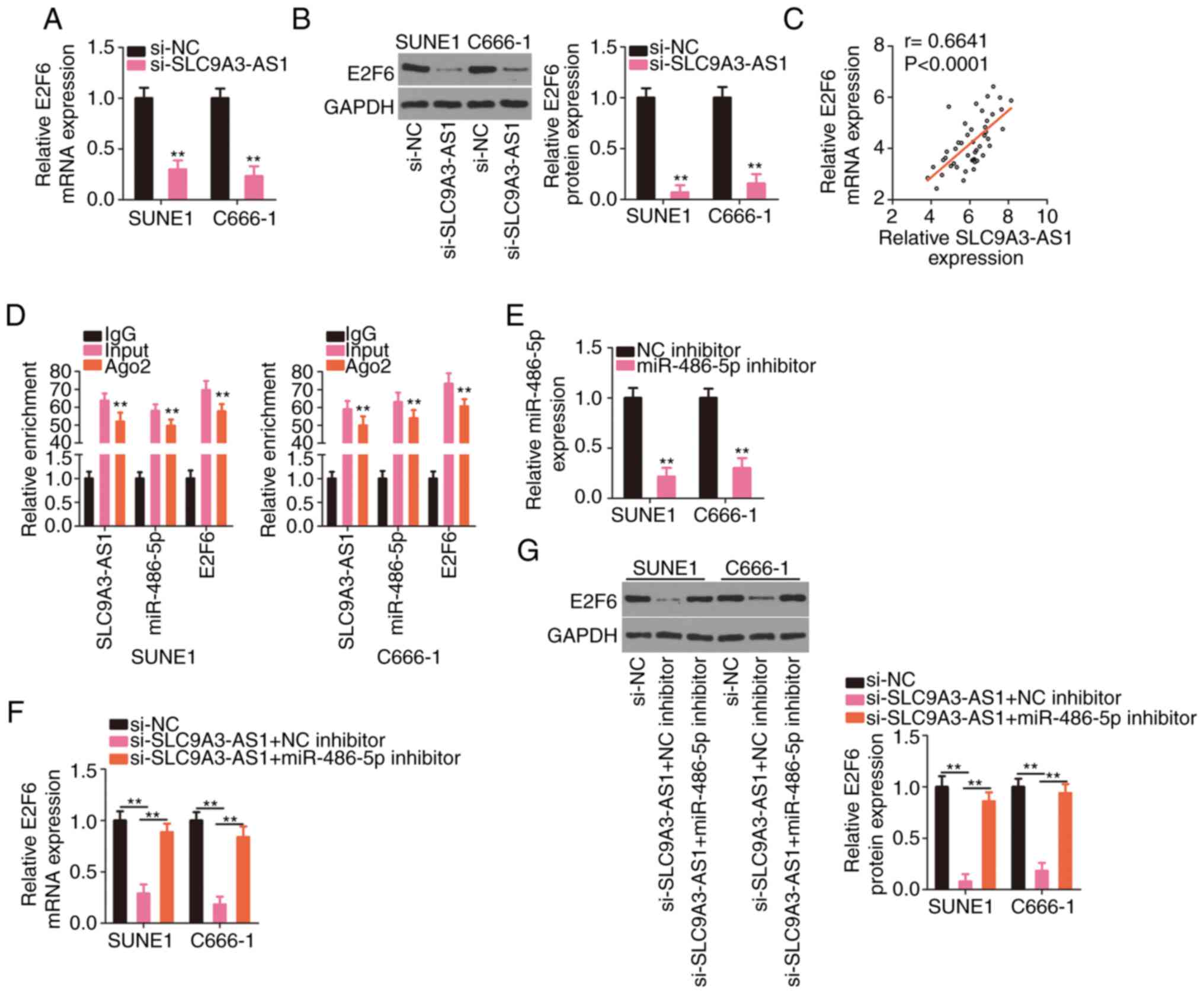 | Figure 5.SLC9A3-AS1 upregulates E2F6 in NPC
cells by sequestering miR-486-5p. (A and B) E2F6 levels were
measured in NPC cells with SLC9A3-AS1 knockdown. (C) The positive
relationship between SLC9A3-AS1 and E2F6 expression in NPC tissues.
(D) The enrichment of SLC9A3-AS1, miR-486-5p, and E2F6 in the IgG,
Ago2, and input groups was assessed with the RIP assay. (E)
miR-486-5p level in NPC cells transfected with the miR-486-5p
inhibitor or NC inhibitor was quantified using RT-qPCR. (F and G)
E2F6 level was quantified in NPC cells co-transfected with
si-SLC9A3-AS1 and miR-486-5p inhibitor or NC inhibitor.
**P<0.01. SLC9A3-AS1, SLC9A3 antisense RNA 1; E2F6, E2F
transcription factor 6; miR, microRNA; NPC, nasopharyngeal
carcinoma; si-, small interfering; NC, negative control. |
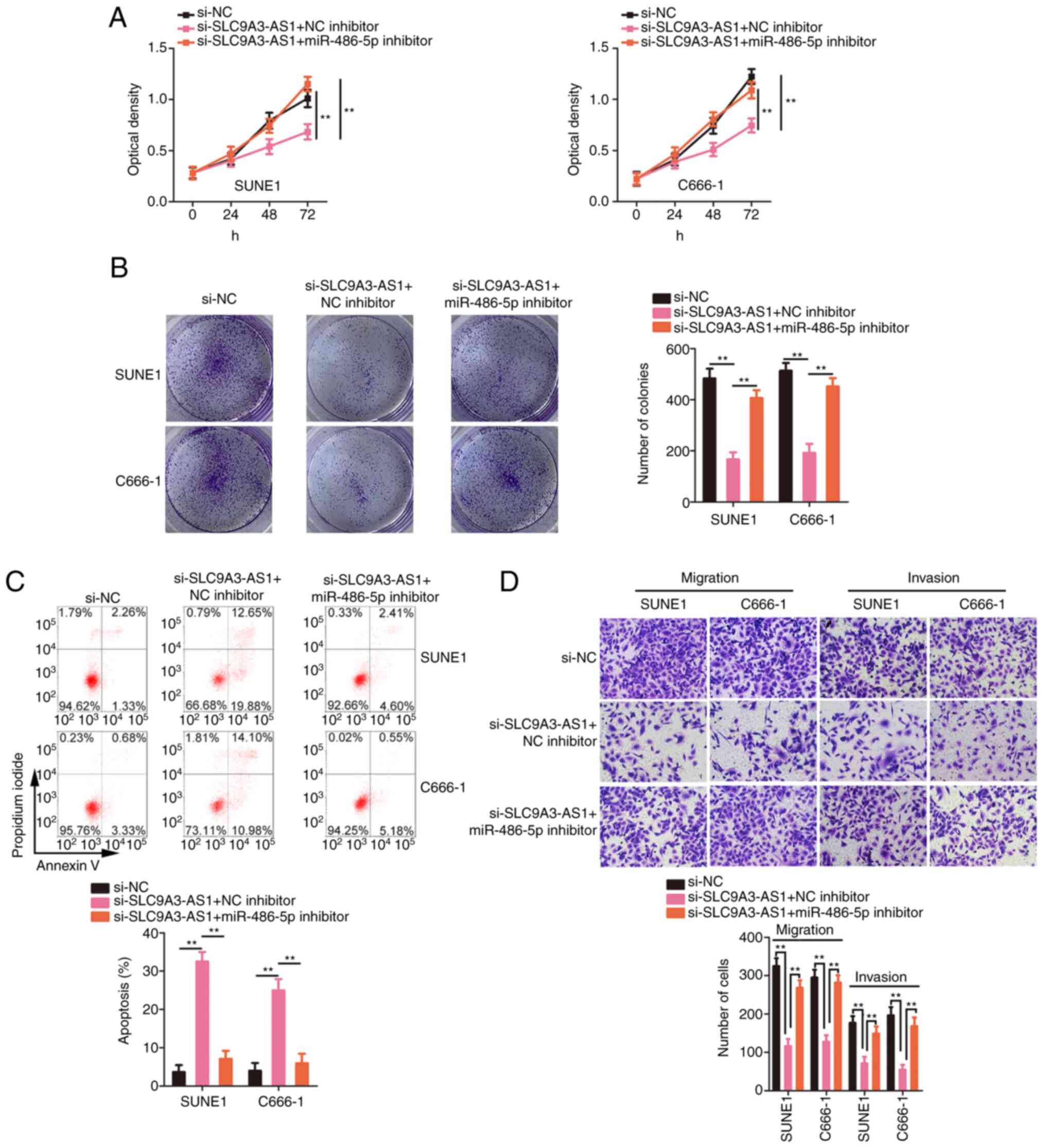
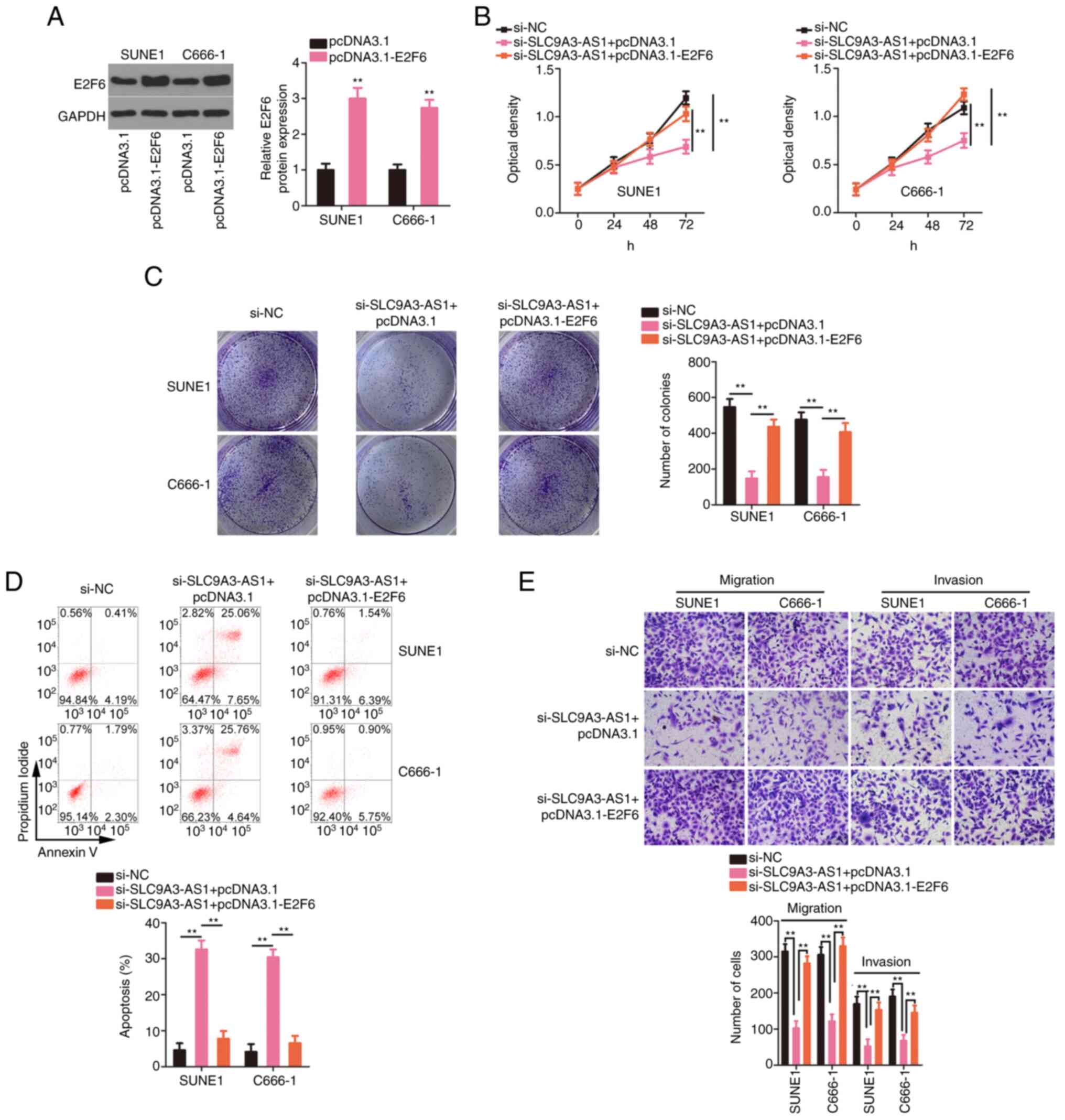
miR-486-5p downregulation or E2F6
overexpression attenuates the influences of si-SLC9A3-AS1 on the
biological functions of NPC cells
Rescue experiments were actualized to ascertain
whether the miR-486-5p/E2F6 axis was indispensable for the
si-SLC9A3-AS1-mediated inhibition of malignancy in NPC. The
inhibition of miR-486-5p reversed the suppressive effect of
si-SLC9A3-AS1 on NPC cell proliferation (Fig. 6A) and colony formation (Fig. 6B). In addition, NPC cell apoptosis was
increased by the silencing of SLC9A3-AS1 but this effect was
reversed after miR-486-5p inhibitor co-transfection (Fig. 6C). Similarly, the migration and
invasion (Fig. 6D) of the
si-SLC9A3-AS1-transfected NPC cells were impaired, but these
changes were abolished upon miR-486-5p downregulation. The
overexpression efficiency of pcDNA3.1-E2F6 was confirmed by western
blotting (Fig. 7A). The inhibition of
proliferation (Fig. 7B) and colony
formation (Fig. 7C) of the cells with
silenced SLC9A3-AS1 was restored by the transfection of
pcDNA3.1-E2F6. Moreover, si-SLC9A3-AS1-induced NPC cell apoptosis
was reversed by E2F6 overexpression (Fig.
7D). Furthermore, migration and invasion (Fig. 7E) were suppressed in NPC cells after
SLC9A3-AS1 interference but recovered after the introduction of
E2F6. In summary, SLC9A3-AS1 promoted carcinogenesis in NPC cells
by targeting the miR-486-5p/E2F6.
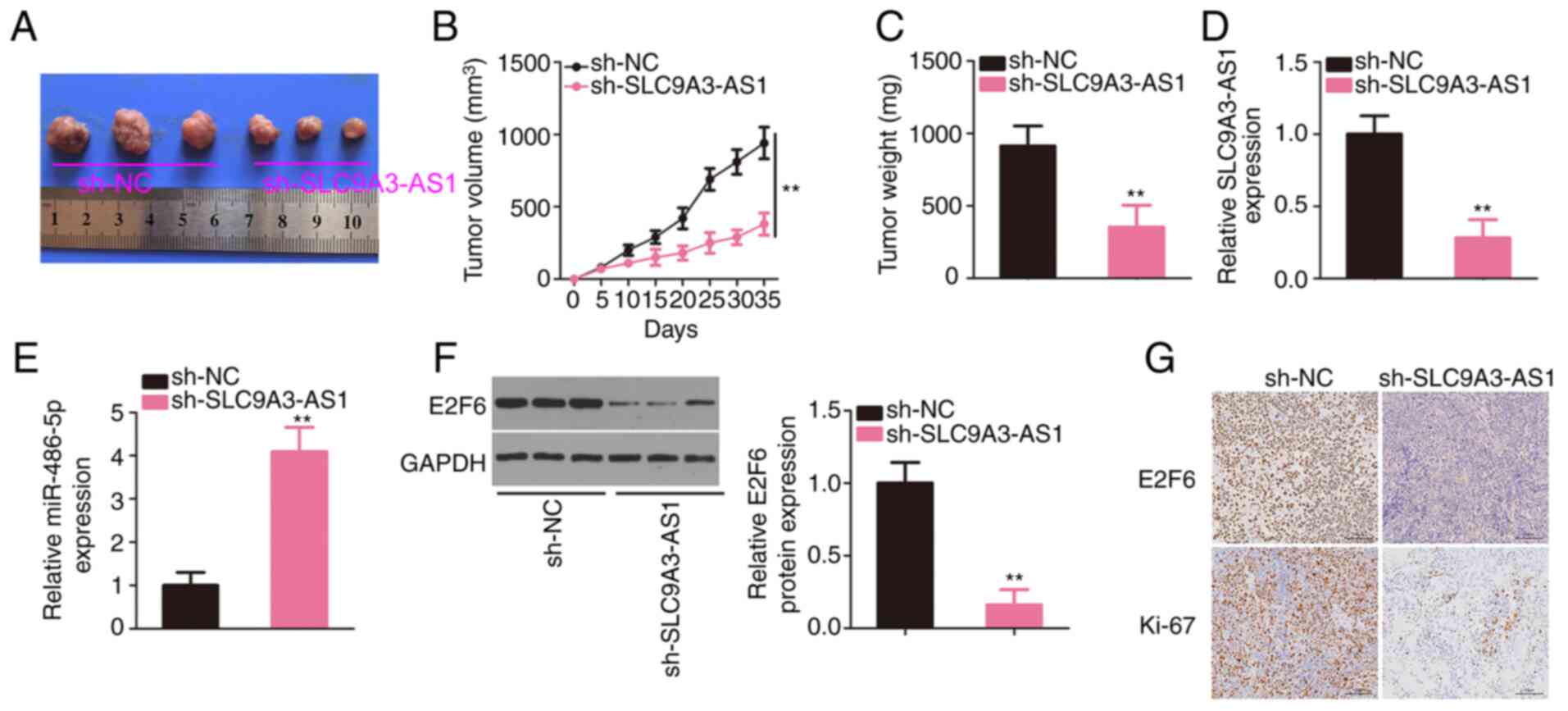
Interference with SLC9A3-AS1 restrains
NPC tumour growth in vivo
SUNE1 cells overexpressing sh-SLC9A3-AS1 were
subcutaneously inoculated into nude mice to establish a mouse
xenograft model. The xenograft tumours originating from
sh-SLC9A3-AS1 were smaller in size (Fig.
8A) and volume (Fig. 8B) than
sh-NC group. The tumours were also lighter in the sh-SLC9A3-AS1
group (Fig. 8C). In addition,
SLC9A3-AS1, miR-486-5p, and E2F6 expression levels in xenograft
tumours were analysed, revealing that SLC9A3-AS1 was expressed at a
low level (Fig. 8D), whereas
miR-486-5p was overexpressed (Fig.
8E) in the tumours injected with sh-SLC9A3-AS1-transfected
cells. Moreover, E2F6 protein level was downregulated in the
SLC9A3-AS1-silencing group (Fig. 8F).
In addition, as demonstrated by immunohistochemistry, xenograft
tumours in the sh-SLC9A3-AS1 group expressed E2F6 and Ki-67 at
lower levels (Fig. 8G). These data
confirmed that the depletion of SLC9A3-AS1 suppressed NPC tumour
growth in vivo.
Discussion
Based on accumulating evidence, aberrantly expressed
lncRNAs in tumours perform significant functions in oncogenesis and
cancer progression (24–26). Extensive lncRNAs are aberrantly
expressed in NPC and participate in the malignant processes of NPC
(27–29). More than 50,000 lncRNAs are present in
the human genome (30); however, most
of them have not been investigated in the context of NPC and thus
require further exploration. To the best of our knowledge, the
present study is the first to analyse the expression and biological
roles of SLC9A3-AS1 in NPC. The related mechanisms underlying the
role played by SLC9A3-AS1 in NPC were also investigated.
SLC9A3-AS1 has been revealed as an overexpressed
lncRNA and has been proposed as a biomarker for lung cancer
(22). However, little is known about
whether SLC9A3-AS1 regulates NPC. The present research revealed
high SLC9A3-AS1 expression in NPC. Patients with NPC with a high
level of SLC9A3-AS1 presented a shorter overall survival than
patients with a low SLC9A3-AS1 level. Functionally, loss of
SLC9A3-AS1 produced anti-carcinogenic effects in NPC, and
participated in the regulation of cell proliferation, colony
formation, apoptosis, migration and invasion in vitro.
Animal experiments further revealed that the interference of
SLC9A3-AS1 hindered the growth of NPC tumours in vivo.
Collectively, the aforementioned observations highlight SLC9A3-AS1
as an effective diagnostic biomarker and treatment target in
NPC.
The localization of lncRNAs determines their
mechanisms of action (31). LncRNAs
that are primarily distributed in the cytoplasm function as miRNA
sponges by directly interacting with miRNAs through miRNA response
elements, subsequently weakening the inhibition of target genes by
miRNAs (32). Numerous lncRNAs are
reported to have roles in NPC as ceRNAs. For instance, a lncRNA
called MEG3 facilitates NPC cell autophagy and apoptosis by working
as a ceRNA for miR-21 and subsequently upregulating PTEN (33).
The molecules involved in the mechanisms of
SLC9A3-AS1 in NPC which have remained largely ambiguous have been
identified in the present study. Subcellular fractionation assays
verified the theoretical basis for SLC9A3-AS1 as a ceRNA in the
present study. According to bioinformatics predictions, SLC9A3-AS1
possessed complementary binding sequences for miR-486-5p. The
direct target interaction between SLC9A3-AS1 and miR-486-5p in NPC
was confirmed by applying luciferase reporter assay. A downstream
target of miRNA is essential for the ceRNA network (34). Further mechanistic investigation
revealed that E2F2 was directly targeted and negatively controlled
by miR-486-5p. SLC9A3-AS1 was revealed to sponge miR-486-5p away
from E2F6; thus, the silencing of SLC9A3-AS1 decreased E2F6
expression in NPC cells. Moreover, the RIP assay indicated that
SLC9A3-AS1, miR-486-5p, and E2F6 all directly interacted with Ago2
in NPC cells, implying the coexistence of the three RNAs in an
RNA-induced silencing complex. Furthermore, a positive expression
relationship between SLC9A3-AS1 and E2F6 and an inverse
relationship between E2F6 and miR-486-5p were observed in NPC
tissues. In other words, the correlations among SLC9A3-AS1,
miR-486-5p and E2F6 have allowed us to propose a new ceRNA pathway
in NPC cells.
Notably, miR-486-5p is dysregulated in numerous
human cancers. For example, miR-486-5p is highly expressed and has
a carcinogenic role in non-small cell lung (35), prostate (36), and pancreatic (37) cancers. Conversely, miR-486-5p is
expressed at low levels in osteosarcoma (38), colorectal cancer (39), and thyroid cancer (40) and is described to have an
anti-oncogenic function. Therefore, miR-486-5p expression and
function exhibit notable tissue specificity in human cancers.
However, further studies are required to understand whether
miR-486-5p contributes to the malignancy of NPC. Decreased
miR-486-5p level was observed in NPC, and overexpressed miR-486-5p
exerted tumour-inhibiting effects during NPC progression.
Furthermore, E2F6, a member of the nuclear transcription factor E2F
family, served as a crucial mediator of miR-486-5p action in NPC.
E2F6 is regulated by multiple miRNAs in human cancer (41–43), and
the present study identified a similar trend for E2F6 in NPC.
Notably, the final rescue experiments revealed that the modulatory
activities of SLC9A3-AS1 silencing on NPC cells could be reversed
by miR-486-5p downregulation or E2F6 reintroduction. In summary,
the miR-486-5p/E2F6 axis is a crucial downstream effector through
which SLC9A3-AS1 exerted oncogenic regulation in NPC.
The phenotypic studies of NP69 compared to NPC cells
with the manipulation of SLC9A3-AS1 expression could help to
further understand the contribution of SLC9A3-AS1 during NPC
progression. However, our study did not execute the phenotypic
studies, and it was a limitation of our research, which will be
addressed in the future.
The present study revealed that SLC9A3-AS1 exerted
carcinogenic effects on NPC cells. SLC9A3-AS1 functioned as a ceRNA
to sequester miR-486-5p, subsequently inducing E2F6 overexpression
and regulating the malignant characteristics of NPC. Therefore, the
newly identified SLC9A3-AS1/miR-486-5p/E2F6 axis may provide novel
targets for therapeutic development in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the People's
Hospital of Rizhao.
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JL and FZ conceived the research. JL, DL, XZ, CL and
FZ conducted the experiments. JL and FZ drafted the manuscript. FZ
acquired, analysed and interpreted the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the People's Hospital of Rizhao (Rizhao, China). All
participants provided written informed consent. All experiments
involving animals were approved by the Animal Ethics Committee of
the People's Hospital of Rizhao.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chua ML, Wee JT, Hui EP and Chan AT:
Nasopharyngeal carcinoma. Lancet. 387:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee AW, Ng WT, Chan YH, Sze H, Chan C and
Lam TH: The battle against nasopharyngeal cancer. Radiother Oncol.
104:272–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wei KR, Zheng RS, Zhang SW, Liang ZH, Ou
ZX and Chen WQ: Nasopharyngeal carcinoma incidence and mortality in
China in 2010. Chin J Cancer. 33:381–387. 2014.PubMed/NCBI
|
|
4
|
Lee AW, Tung SY, Ngan RK, Chappell R, Chua
DT, Lu TX, Siu L, Tan T, Chan LK, Ng WT, et al: Factors
contributing to the efficacy of concurrent-adjuvant chemotherapy
for locoregionally advanced nasopharyngeal carcinoma: Combined
analyses of NPC-9901 and NPC-9902 Trials. Eur J Cancer. 47:656–666.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan WL, Tan EH, Lim DW, Ng QS, Tan DS,
Jain A and Ang MK: Advances in systemic treatment for
nasopharyngeal carcinoma. Chin Clin Oncol. 5:212016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tao Q and Chan AT: Nasopharyngeal
carcinoma: Molecular pathogenesis and therapeutic developments.
Expert Rev Mol Med. 9:1–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lam WK and Chan JY: Recent advances in the
management of nasopharyngeal carcinoma. F1000Res. 7:F1000 Faculty
Rev-1829. 2018. View Article : Google Scholar
|
|
8
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peng WX, Koirala P and Mo YY:
LncRNA-mediated regulation of cell signaling in cancer. Oncogene.
36:5661–5667. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spizzo R, Almeida MI, Colombatti A and
Calin GA: Long non-coding RNAs and cancer: A new frontier of
translational research? Oncogene. 31:4577–4587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanchez Calle A, Kawamura Y, Yamamoto Y,
Takeshita F and Ochiya T: Emerging roles of long non-coding RNA in
cancer. Cancer Sci. 109:2093–2100. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C, Zhang H and Liu H: Long noncoding
RNA UCA1 accelerates nasopharyngeal carcinoma cell progression by
modulating miR-124-3p/ITGB1 axis. Onco Targets Ther. 12:8455–8466.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhong Q, Chen Y and Chen Z: LncRNA MINCR
regulates irradiation resistance in nasopharyngeal carcinoma cells
via the microRNA-223/ZEB1 axis. Cell Cycle. 19:53–66. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao CX, Zhu W, Ba ZQ, Xu HJ, Liu WD, Zhu
B, Wang L, Song YJ, Yuan S and Ren CP: The regulatory network of
nasopharyngeal carcinoma metastasis with a focus on EBV, lncRNAs
and miRNAs. Am J Cancer Res. 8:2185–2209. 2018.PubMed/NCBI
|
|
15
|
Guo H, Huang S, Li S, Yu H, Wu S and Zhou
X: Prognostic significance of the long noncoding RNAs in
nasopharyngeal carcinoma: A systematic review and meta-analysis.
Cancer Manag Res. 10:1763–1779. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Lei C, He Q, Pan Z, Xiao D and Tao
Y: Nuclear functions of mammalian MicroRNAs in gene regulation,
immunity and cancer. Mol Cancer. 17:642018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ji W, Sun B and Su C: Targeting microRNAs
in cancer gene therapy. Genes (Basel). 8:212017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tian Y, Tang L, Yi P, Pan Q, Han Y, Shi Y,
Rao S, Tan S, Xia L, Lin J, et al: MiRNAs in radiotherapy
resistance of nasopharyngeal carcinoma. J Cancer. 11:3976–3985.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang S, Claret FX and Wu W: MicroRNAs as
therapeutic targets in nasopharyngeal carcinoma. Front Oncol.
9:7562019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee KT, Tan JK, Lam AK and Gan SY:
MicroRNAs serving as potential biomarkers and therapeutic targets
in nasopharyngeal carcinoma: A critical review. Crit Rev Oncol
Hematol. 103:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bai Y, Qu Y, Wu Z, Ren Y, Cheng Z, Lu Y,
Hu J, Lou J, Zhao J, Chen C and Mao H: Absolute quantification and
analysis of extracellular vesicle lncRNAs from the peripheral blood
of patients with lung cancer based on multi-colour fluorescence
chip-based digital PCR. Biosens Bioelectron. 142:1115232019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghafouri-Fard S, Shoorei H, Anamag FT and
Taheri M: The role of non-coding RNAs in controlling cell cycle
related proteins in cancer cells. Front Oncol. 10:6089752020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qian Y, Shi L and Luo Z: Long Non-coding
RNAs in cancer: Implications for diagnosis, prognosis, and therapy.
Front Med. 7:6123932020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McCabe EM and Rasmussen TP: LncRNA
involvement in cancer stem cell function and epithelial-mesenchymal
transitions. Semin Cancer Biol. Dec 17–2020.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao J, Liu D, Yang H, Yu S and He H: Long
noncoding RNAs in head and neck squamous cell carcinoma: Biological
functions and mechanisms. Mol Biol Rep. 47:8075–8090. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu J and Hann SS: Functions and roles of
long-non-coding RNAs in human nasopharyngeal carcinoma. Cell
Physiol Biochem. 45:1191–1204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong Q, Zhou L, Liu F, Ao F, Gong X, Jiang
C, Yuan Z and Li J: Long non-coding RNAs in the development,
diagnosis and prognosis of nasopharyngeal carcinoma. Int J Clin Exp
Pathol. 10:8098–8105. 2017.PubMed/NCBI
|
|
30
|
Xu J, Bai J, Zhang X, Lv Y, Gong Y, Liu L,
Zhao H, Yu F, Ping Y, Zhang G, et al: A comprehensive overview of
lncRNA annotation resources. Brief Bioinform. 18:236–249.
2017.PubMed/NCBI
|
|
31
|
Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX
and Hong W: The ways of action of long non-coding RNAs in cytoplasm
and nucleus. Gene. 547:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rashid F, Shah A and Shan G: Long
non-coding RNAs in the cytoplasm. Genomics Proteomics
Bioinformatics. 14:73–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin L, Liu X and Lv B: Long non-coding RNA
MEG3 promotes autophagy and apoptosis of nasopharyngeal carcinoma
cells via PTEN up-regulation by binding to microRNA-21. J Cell Mol
Med. 25:61–72. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sen R, Ghosal S, Das S, Balti S and
Chakrabarti J: Competing endogenous RNA: The key to
posttranscriptional regulation. ScientificWorldJournal.
2014:8962062014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao ZJ, Yuan WD, Yuan JQ, Yuan K and Wang
Y: miR-486-5p functions as an oncogene by targeting PTEN in
non-small cell lung cancer. Pathol Res Pract. 214:700–705. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang Y, Ji C, Guo S, Su X, Zhao X, Zhang
S, Liu G, Qiu X, Zhang Q, Guo H and Chen H: The miR-486-5p plays a
causative role in prostate cancer through negative regulation of
multiple tumor suppressor pathways. Oncotarget. 8:72835–72846.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xia L, Song M, Sun M, Chen W and Yang C:
miR-486 promotes capan-2 pancreatic cancer cell proliferation by
targeting phosphatase and tensin homolog deleted on chromosome 10
(PTEN). Front Genet. 10:5412019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He M, Wang G, Jiang L, Qiu C, Li B, Wang J
and Fu Y: miR-486 suppresses the development of osteosarcoma by
regulating PKC-δ pathway. Int J Oncol. 50:1590–1600. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu C, Li M, Hu Y, Shi N, Yu H, Liu H and
Lian H: miR-486-5p attenuates tumor growth and lymphangiogenesis by
targeting neuropilin-2 in colorectal carcinoma. Onco Targets Ther.
9:2865–2871. 2016.PubMed/NCBI
|
|
40
|
Ma X, Wei J, Zhang L, Deng D, Liu L, Mei
X, He X and Tian J: miR-486-5p inhibits cell growth of papillary
thyroid carcinoma by targeting fibrillin-1. Biomed Pharmacother.
80:220–226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
An Y, Zhang J, Cheng X, Li B, Tian Y,
Zhang X and Zhao F: miR-454 suppresses the proliferation and
invasion of ovarian cancer by targeting E2F6. Cancer Cell Int.
20:2372020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cai Q, Zhao A, Ren LG, Chen J, Liao KS,
Wang ZS and Zhang W: MiR-425 involves in the development and
progression of renal cell carcinoma by inhibiting E2F6. Eur Rev Med
Pharmacol Sci. 22:6300–6307. 2018.PubMed/NCBI
|
|
43
|
Lu Z, Nian Z, Jingjing Z, Tao L and Quan
L: MicroRNA-424/E2F6 feedback loop modulates cell invasion,
migration and EMT in endometrial carcinoma. Oncotarget.
8:114281–114291. 2017. View Article : Google Scholar : PubMed/NCBI
|