Introduction
Ubiquitination is a key process of the
post-translational modification of proteins, playing an important
role in the stability of the intracellular environment, the
proliferation and differentiation of cells, and a number of
cellular functions (1), whereas
the imbalance of ubiquitination-mediated protein degradation is a
molecular basis for human diseases. Ubiquitin is activated in an
ATP-dependent reaction catalyzed by the ubiquitin-activating enzyme
(E1). Under the action of the ubiquitin-conjugating enzyme (E2),
the activated ubiquitin is transferred into a specific substrate
along with E3 ubiquitin ligase, which is responsible for the
substrate specificity for ubiquitin ligation (2,3).
Increasing attention has been focused on E3
ubiquitin ligases due to their unique functions compared with E1
and E2. E3 ubiquitin ligases regulate a range of cellular
physiological processes, such as cell proliferation and
differentiation, participate in DNA damage and repair, and control
the cell cycle (1,4–6). In
pathological processes (i.e., cancer and nervous system diseases),
E3 ubiquitin ligases promote or suppress various diseases (7). For example, the speckle-type POZ
protein (SPOP), a typical E3 ubiquitin ligase, is known as a tumor
suppressor protein in prostate cancer (PC) and an oncoprotein in
kidney cancer (8). In addition, E3
ubiquitin ligases with high-frequency mutations (mutations in
SPOP occur in up to 11.13% of PC cases; The Cancer Genome
Atlas, http://portal.gdc.cancer.gov/) in
diseases prove their important role in the development of disease
(4,7).
Currently, E3 ubiquitin ligases can be classified in
three main types [i.e., RING E3s (~600 in humans), homologous to
the E6AP carboxyl terminus (HECT) E3s (~30 in humans) and
RING-between-RING (RBR) E3s (~12 in humans)] depending on the
characteristic domains and the mechanism of ubiquitin transfer to
the substrates (9). Notably, some
E3s, including some RING E3s, all HECT E3s and all RBR E3s,
interact with E2 alone to transfer the ubiquitin to the substrates,
whereas other RING E3s transfer ubiquitin by forming the
ubiquitin-ligase complex (9). The
mechanism of ubiquitin transfer to the substrates gets little
research attention.
The seven in absentia homolog (Siah) family of
proteins, which belong to the RING E3s, are the mammalian homologs
of the Drosophila sina proteins, which are responsible for
the ubiquitination of substrate proteins to promote functional
changes or degradation of substrate proteins through the
proteasomal pathway (10,11). In the human proteome, two proteins
belonging to the Siah family of proteins have been identified and
are known as Siah1 and Siah2. Mice have three Siah family proteins,
termed Siah1a, Siah1b (collectively Siah1 due to their 98%
similarity) and Siah2 (11).
Siah1 and Siah2 share high sequence similarity (86%)
and presumably high structural homology. The difference between
Siah1 and Siah2 is the additional amino acid sequence in the N
terminal of Siah2 (11–14). However, the functions of Siah1 and
Siah2 are almost completely different due to their unique
substrates and different affinity and types of ubiquitination for
the shared substrates (11).
Previous studies focused on the oncoprotein functions of Siah2
(11,15). However, in recent years, a number
of novel substrates of Siah1 have been discovered, and the
functions of Siah1 as an important E3 ubiquitin ligase have been
gradually recognized (10,11,16,17).
A systematic review summarizing the functions of Siah1 in human
diseases, such as cancer and nervous system diseases, is not
available. Siah1 knockout mice exhibit severe growth retardation,
poor bone formation, early lethality and a block in meiotic cell
division during the meiosis I of spermatogenesis (18,19).
However, the Siah2 mutant or knockout mice are fertile and largely
phenotypically normal. Notably, the loss of a single copy of Siah2
enhances the phenotype of early lethality caused by Siah1
homozygous mutation. This phenotype is further enhanced by the
removal of both copies of Siah2, with
Siah1−/−Siah2−/− mice subsequently dying
within hours of birth, showing that Siah1 and Siah2 appear to
perform partially overlapping functions in vivo (18,19).
The functional compensation by Siah1 may maintain the normal
regulation of Siah2 substrate proteins in Siah2−/− mice,
suggesting the critical biological functions of Siah1 (18,19).
In addition, in contrast to that of other E3 ubiquitin ligases (the
regulations of SPOP have rarely been discovered), the regulations
of Siah1 vary greatly (Table I;
Fig. 1A), indicating that Siah1
plays a role as a bridge factor in various signaling pathways and
is a promising therapeutic target (11,18).
The present review will summarize the structure of Siah1 and the
characteristics of its substrates, the regulations of Siah1 in
physiological and pathological conditions, and its function and
clinical significance in cancer and nervous system diseases to
provide help for future researchers to understand Siah1.
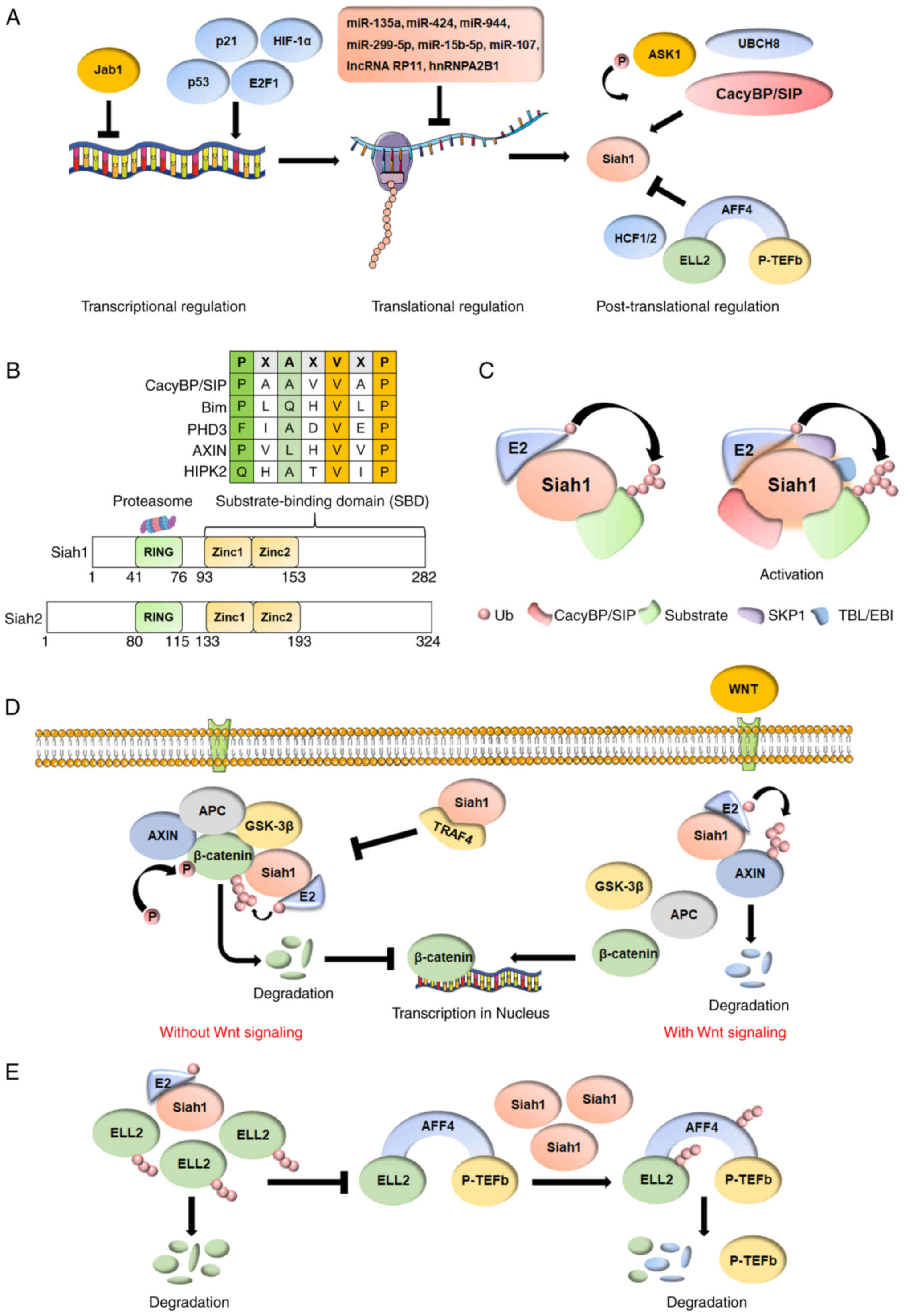 | Figure 1.(A) The regulations of Siah1 vary
greatly. At the transcriptional regulation level, p53, p21, E2F1
and HIF-1α trans-activate the transcription of Siah1. At the
translational regulation level, microRNAs and lncRNAs inhibit the
translation of Siah1 mRNA. At the post-translational regulation
level, ASK1 induces the phosphorylation of Siah1. Overexpression of
CacyBP/SIP promotes the interaction between Siah1 and cytoplasmic
p27, which in turn increases the ubiquitination and degradation of
cytoplasmic p27. Ubiquitin conjugase UbcH8 interacts with Siah1 to
form a complex to ensure the functions of Siah1. HCF1 and HCF2
antagonize the E3 ligase activity of Siah1 through binding and
blocking the substrate-binding domain. The AFF4-ELL2 interaction
sequesters ELL2 away from Siah1, thereby inhibiting Siah1
ubiquitination of ELL2. (B) Siah1 consists of a N-terminal
catalytic RING domain, two zinc finger domains and a C-terminal
substrate binding domain that includes the first two zinc finger
domains. A consensus Pro-X-Ala-X–Val-X-Pro (VxP, core sequence;
where X is not conserved) motif is common to a family of SIPs (for
example, CacyBP/SIP, Bim, PHD3, AXIN, HIPK2). Compared with Siah1,
Siah2 has an additional amino acid sequence (~40 amino acids) at
the N terminal. (C) Siah1 can interact with E2
ubiquitin-conjugating enzyme alone or become an essential part of
the ubiquitin-ligase complex, which includes CacyBP/SIP, SKP1, TBL
or EBI and Siah1. (D) β-catenin, AXIN1, APC and GSK-3β form a
degradation complex without Wnt signaling, inducing the
phosphorylation of β-catenin and finally leading to the degradation
of β-catenin through the ubiquitin-proteasome pathway. The
degradation complex is destroyed in response to Wnt signaling,
releasing β-catenin, and thus activating transcription of
downstream genes to promote the proliferation and survival of
cells. Siah1 also induces the ubiquitination and proteasomal
degradation of AXIN1 to promote the Wnt/β-catenin signaling
pathways with Wnt signaling. TRAF4 protects β-catenin from
Siah1-mediated degradation by competing with β-catenin for binding
to Siah1 and replacing it for degradation. (E) The low protein
level of Siah1 induces the degradation of dissociative ELL2 to
prevent the formation of new SECs. The high protein level of Siah1
degrades all SECs. Siah1, seven in absentia homolog family proteins
1; Jab1, c-Jun activation domain-binding protein 1; ASK1, apoptosis
signal-regulating kinase 1; UBCH8, ubiquitin conjugating enzyme
human 8; Ub, ubiquitin; HCF1/2, host cell factor 1/2; P-TEFb,
positive transcription elongation factor b; TRAF4, TNF
receptor-associated factor 4; APC, adenomatous polyposis coli
protein; GSK-3β, glycogen synthase kinase-3β; ELL2, elongation
factor for RNA polymerase II 2; AFF4, AF4/FMR2 family member 4;
miR, microRNA; lncRNA, long non-coding RNA; CacyBP,
calcyclin-binding protein; Bim, Bcl-2-interacting mediator of cell
death; PHD3, prolyl-hydroxylase protein 3; HIPK2,
homeodomain-interacting protein kinase 2; SIP, Siah1-interacting
protein; TBL/EBI, F-box-like/WD repeat-containing protein TBL1 or
EBI. |
 | Table I.Regulations of Siah1 ubiquitin
ligase. |
Table I.
Regulations of Siah1 ubiquitin
ligase.
| Level of
regulation | Regulator | Mode of
regulation |
|---|
| Transcriptional
regulation | p53 | p53 acts directly
on Siah1, to promote the transcription of Siah1. |
|
| Jab1 | Jab1 inhibit the
expression of p53 to suppress the transcription of
Siah1. |
|
| HIF-1α | HIF-1α
trans-activates the transcription of Siah1 by coordinating
key histone modifications on the Siah1 promoter. |
|
| p21 | The transcription
of Siah1 is directly activated by p21. |
|
| E2F1 | E2F1 can activate
transcription from the Siah1 promoter. |
| Translational
regulation | miR-135a, miR-424,
miR-944, miR-299-5p, miR-15b-5p, miR-107 | MicroRNAs inhibit
the translation of Siah1 mRNA by targeting the 3′UTR. |
|
| lncRNA RP11,
hnRNPA2B1 | RP11 directly binds
to the CDS of Siah1 and significantly downregulates the mRNA
stability of Siah1 by forming the RP11-hnRNPA2B1-mRNA.
complex |
| Post-translational
regulation | ASK1 | Phosphorylation of
Siah1 by ASK1 triggers GAPDH-Siah1 stress signaling. |
|
| CacyBP/SIP | Overexpression of
CacyBP/SIP promotes the interaction between Siah1 and cytoplasmic
p27, which in turn increases the ubiquitination and degradation of
cytoplasmic p27. |
|
| HCF1/2 | HCF1 and HCF2
antagonize the E3 ligase activity of Siah1 by binding and blocking
the substrate-binding domain. |
|
| AFF4 | The AFF4-ELL2
interaction sequesters ELL2 away from Siah1 thereby inhibiting
Siah1 ubiquitination of ELL2. |
|
| UBCH8 | Ubiquitin conjugase
UbcH8 interacts with Siah1 to form a complex to ensure the function
of Siah1. |
Structure of Siah1 and characteristics of
Siah1-interacting proteins (SIPs)
Siah family proteins usually consist of an
N-terminal catalytic RING domain, two zinc finger domains and a
C-terminal substrate-binding domain (SBD) that includes the first
two zinc finger domains (Fig. 1B)
(13). The RING domain is
essential for ubiquitin ligase activity, and the SBD mediates
homodimerization and the interaction with substrates (14). In contrast with other E3 ubiquitin
ligases, Siah1 can interact with E2 alone and become an essential
part of the ubiquitin-ligase complex, which includes
calcyclin-binding protein (CacyBP)/SIP, and adapters SKP1,
F-box-like/WD repeat-containing protein TBL1 or EBI and Siah1
(Fig. 1C) (20). CacyBP/SIP is suggested to position
the substrates and improve the affinity between Siah1 and the
substrate. Thus, Siah1 is most powerful when complexed (21,22).
The characteristics of the SIPs have also been
studied (13). A consensus
Pro-X-Ala-X–Val-X-Pro (VxP, core sequence; where X is not
conserved) motif is common to a family of SIPs (Table I) (11,14,21,23–26).
Some SIPs are also substrates of Siah1 and are targeted by Siah1
for degradation or functional modification. Dysregulation between
Siah1 and substrates will lead to serious human diseases (e.g.,
cancer and nervous system diseases) (Table II) (11,15,16,18,27).
| Table II.SIPs of Siah1 ubiquitin ligase in
human diseases. |
Table II.
SIPs of Siah1 ubiquitin ligase in
human diseases.
| Disease type | Substrate | Function of the
SIPs | Degradation of the
substrate | Physiological
evidence (cell or animal model) |
|---|
| Cancer |
|
|
|
|
| Breast
cancer | JNK | Promotion of cell
apoptosis and inhibition of MAPK signaling pathways. | No | The inhibition of
Siah1 expression promotes human breast cancer cell proliferation,
colony formation, migration and invasion, and inhibits apoptosis
(15,23,57). |
|
| Bim | DNA-binding
transcription factor activity. | No |
|
|
| TRAF4 | Protection of
β-catenin from Siah1-mediated degradation and leading chemotherapy
resistance. | Yes |
|
|
Glioma | HIPK2 | Response to DNA
damage and promotion of cells apoptosis. | Yes | The knockdown of
Siah1 by shRNA severely suppresses the migration and invasion of
human glioma U251 cells under hypoxia, while overexpression of
Siah1 promotes it. However, when Siah1 interacts with CacyBP/SIP,
the overexpression of Siah1 suppresses the migration and invasion
of human glioma U251 and U87 cells (22,26). |
|
| PHD3 | Degradation of the
HIF-1α. | Yes |
|
|
| CacyBP/SIP | The part of Siah1
ubiquitin-ligase complexes. | No |
|
|
| p27/kip1 | Negative regulation
of the cell cycle. | Yes |
|
|
Hepatocellular carcinoma | Axin,
β-catenin | Cell migration and
cell differentiation. | Yes | The overexpression
of Siah1 induces growth arrest and apoptosis in HepG2, SNU475 and
Huh7 cells (30). |
|
| ZEB1 |
Epithelial-mesenchymal transition. | Yes |
|
|
Leukemogenesis | PML-RARa | The fusion protein
of leukemia. | Yes | In the murine
myeloblastic cell line M1 (generated from a spontaneous leukemia),
expression of a stably introduced temperature-sensitive mutant of
the tumor suppressor p53 activates the Siah1 gene (27,74). |
|
| ELL2 | An elongation
factor that modulates gene expression. | Yes |
|
|
| AML1-ETO | The fusion protein
of leukemia. | Yes |
|
|
| AF4-MLL |
|
|
|
|
Colorectal cancer | AKT, YAP | Inhibition of cells
apoptosis | Yes | The knockdown of
Siah1 by shRNA promotes HCT116/SW480 colorectal cancer cell
proliferation and migration, and results in faster tumor growth and
a markedly larger tumor volume in nude mice (108). |
|
| ZEB1 |
Epithelial-mesenchymal transition. | Yes |
|
|
Osteosarcoma | ZEB1 |
Epithelial-mesenchymal transition. | Yes | None. |
| Nervous system
diseases |
|
|
|
|
|
Development delay | Axin | Neuronal
development and cell differentiation. | Yes | The development of
skin and hair follicle development in the angora rabbit is affected
by the level of Siah1 protein (132). |
|
| Akt3 | Neuronal
development. | Yes |
|
|
Neuronal damage | GAPDH | Glycolysis and
promotion of cell apoptosis. | No | Siah1 is
upregulated after spinal cord injury in adult rats (138,139,143). |
|
Parkinson's disease | α-synuclein | The development of
Parkinson's disease and the formation of LBs. | No | Inhibition of Siah1
by siRNA increases cell proliferation and inhibits apoptosis in
SH-SY5Y neuroblastoma cells (113,115). |
|
| Synphilin-1 | The development of
Parkinson's disease and the formation of LBs. | Yes |
|
| Alzheimer's
disease | CacyBP/SIP | Part of the Siah1
ubiquitin-ligase complexes and de-phosphorylation of tau
protein. | No | In tau transgenic
mice, localization of CacyBP/SIP and Siah1 is similar to that
observed for patients with Alzheimer's disease (150). |
Siah1 in cancer
Siah1 in hepatocellular carcinoma
(HCC)
HCC is one of the most common malignancies worldwide
(in 2020, there were 910,000 new cases of HCC worldwide, ranking
sixth among all cancer types; The Cancer Genome Atlas, http://portal.gdc.cancer.gov/), with extremely
high recurrence and metastasis rates (28). Siah1 was identified as one
of the tumor-suppressing genes of HCC in 2003 (29). Siah1 is significantly downregulated
in advanced HCC, including poorly differentiated tumors, large
tumors and tumors in advanced stages (29,30).
Wnt/β-catenin signaling pathways are one of the key
cascades regulating cell growth, cell development and
differentiation of normal stem cells, and have also been tightly
associated with cancers made up of several key proteins, including
Wnt, β-catenin, AXIN1, adenomatous polyposis coli protein (APC) and
glycogen synthase kinase-3β (GSK-3β) (31–33).
β-catenin, AXIN1, APC and GSK-3β form a degradation complex without
Wnt signaling, inducing the phosphorylation of β-catenin and
leading to the degradation of β-catenin through the ubiquitin
proteasome pathway (31–36). The degradation complex is destroyed
in response to Wnt signaling, releasing β-catenin and activating
the transcription of downstream genes to promote the proliferation
and survival of cells (Fig. 1D)
(31–36). β-catenin is widely considered to be
a major oncoprotein in HCC based on the frequency of mutations
[15–33% of patients with HCC carry activating mutations in
ctnnb1 (coding for β-catenin)] associated with aberrant
Wnt/β-catenin signaling pathways in patients with HCC (37). Siah1 functions in the
ubiquitination-dependent degradation of β-catenin, thus inhibiting
the abnormal activation of Wnt/β-catenin signaling pathways
[whether wild-type or mutant Wnt/β-catenin signaling pathways;
HepG2 (β-catenin with activating mutations), SNU475 (AXIN1 with
dysfunctional mutation) and Huh7 (wild-type β-catenin and AXIN1)]
and promoting cell apoptosis and growth arrest of HCC cells
(30).
However, some studies have reported that Siah1 also
promotes Wnt/β-catenin signaling pathways by inducing the
ubiquitination and proteasomal degradation of AXIN1, suggesting the
positive regulation of Siah1 in Wnt/β-catenin signaling pathways
(38). The positive and negative
regulations of Siah1 in Wnt/β-catenin signaling pathways indicate
that Siah1 plays a key role in the dynamic balance of these
pathways, suggesting that targeted therapy of Siah1 for HCC is a
promising but prudent choice (Fig.
1D).
Acquired chemoresistance during long-term
chemotherapy is one of the most important factors to limit the
application of some chemotherapy drugs, such as doxorubicin (Dox),
for the clinical treatment of patients with HCC (39,40).
In addition, chemotherapy-resistant HCC shows a malignant
phenotype, suggesting that the changes in cancer-related factors
occur in the HCC cells (41).
Recent studies have shown that zinc finger E-box-binding homeobox 1
(ZEB1), a powerful epithelial mesenchymal transition-related
transcription factor, is upregulated in Dox-resistant HCC cells and
accompanied by a decrease in the protein expression of Siah1
(41–43). ZEB1 is degraded by the proteasomal
pathway as a substrate for Siah1, but the low protein level of
Siah1 induces the accumulation of ZEB1 (44,45).
The same phenomenon also occurs in colorectal cancer (CRC) cells
and Dox-resistant osteosarcoma cells (44,46).
Some studies have suggested that long non-coding RNA (lncRNA) RP11
accelerates the mRNA degradation of Siah1 in CRC, thus leading to
the accumulation of ZEB1 (46).
Whether the cause of ZEB1 accumulation in HCC is related to RP11
remains unclear, but the lncRNA-Siah1-ZEB1 axis may be an important
target against Dox resistance and ZEB1-related cancer.
Notably, Siah1 in HCC functions as a tumor
suppressor protein and as an oncoprotein (11,15).
Some studies have reported that the nuclear expression of Siah1
induces proliferation and migration and prevents the apoptosis of
HCC cells (47). In addition, in
HCC tissues, the decrease in cytoplasmic Siah1 and the nuclear
accumulation of Siah1 are positively correlated with HCC
progression, suggesting that the functions of Siah1 may be closely
related to subcellular localization (47). The nuclear Siah1 accumulation is
significantly correlated with the expression of the transcription
factor far-upstream element-binding protein 3 (FBP-3). FBP-3
predominantly supports proliferation but cannot explain the reason
for HCC cell migration being affected by Siah1 (47). Thus, the mechanisms for the high
expression of Siah1 in the nucleus of HCC cells and the promotion
of HCC by nuclear Siah1 accumulation are still unclear and should
be studied in the future.
Siah1 in breast cancer (BC)
BC is the most common cancer in women and is
considered the second leading cause of cancer-related death in
women (in 2020, there were 2,260,000 new cases of BC worldwide,
ranking first among all cancer types of women; The Cancer Genome
Atlas, http://portal.gdc.cancer.gov/)
(48–51). BC is a type of cancer with complex
phenotypes and heterogeneity. Thus, traditional pathology has been
unable to meet the needs of BC diagnosis (52,53).
The accurate diagnosis of BC requires entirely new tumor markers,
such as Siah1.
Siah1 was originally identified as a tumor
suppressor gene in BC (54–56).
The expression of Siah1 is downregulated in BC tissues and is
correlated with well to moderately differentiated and early stage
BC (23). In addition, the
inhibition of Siah1 expression promotes human BC cell
proliferation, colony formation, migration and invasion, and
inhibits apoptosis (15,23,57),
suggesting the key role of Siah1 in the occurrence and development
of BC. Some studies have further reported that Siah1 induces
apoptosis, that inhibited invasion in BC cells may by upregulation
of the level of Bcl-2-interacting mediator of cell death through
the activation of the c-Jun NH2-terminal kinase signaling pathway,
and that the suppression of Siah1 expression increases migration
via the activation of the extracellular-regulated protein kinases
signaling pathway (23,57).
The classification of BC is complex. Under the
general trend of the development of precision medicine, oncologists
prefer to classify breast cancer by molecular classification
(51–53). Therefore, based on the expression
of the three key factors for BC [estrogen receptor (ER),
progesterone receptor (PR) and the human epidermal growth factor
receptor 2 (HER2) subtype], oncologists classify BC into six
subtypes: Luminal A (low-grade and ER-positive), luminal B
(high-grade and ER-positive), HER2-overexpressing, triple-negative
BC (TNBC; lacking ER, PR and HER2), normal breast-like tumors and
claudin-low (TNBC with a low expression level of cell adhesion
molecules) (15,49,51,52).
TNBC is the most lethal subtype of BC due to its high
heterogeneity, aggressive nature and lack of treatment options
(58). Notably, the expression of
Siah1 is significantly decreased in TNBC cells (MDA-MB-231), and
the inhibition of Siah1 expression has recently been shown to be
mediated by microRNA (miRNA/miR)-107 (an overexpression miRNA in
BC, especially in TNBC) (59).
miR-107 is considered to be a good predictive parameter of TNBC
recurrence and promotes cell proliferation, colony formation,
migration, invasion and cell cycle progression in human BC cells
(i.e., MCF-7 and MDA-MB-231) through the downregulation of Siah1
expression (59–61). In a previous study, the inhibition
of Siah1 was relieved by the silencing of miR-107, which inhibited
tumor growth in a nude mouse model of TNBC. This phenomenon
suggests that the miR-107-Siah1 axis will be a promising
therapeutic target in TNBC (59).
In addition, miR-944 exhibits a similar function to miR-107,
strongly supporting the importance of the regulation of Siah1
expression by miRNA (62).
Chemotherapy is the basic treatment of BC, and has
made marked progress over the last few decades, with the emergence
of new beneficial treatment methods, such as neoadjuvant
chemotherapy (63–65). However, chemoresistance in BC is
still common, leading to a poor prognosis and high mortality rate
(66). One study has shown that
Siah1 is associated with the chemoresistance of BC, which may be
due to the interaction of Siah1 with tumor necrosis factor receptor
associated-factor 4 (TRAF4) (63).
Siah1 mediates the ubiquitination and degradation of β-catenin,
thus inhibiting the activation of the Wnt/β-catenin signaling
pathways, promoting cell apoptosis and preventing tumor progression
(38). However, TRAF4 protects
β-catenin from Siah1-mediated degradation by competing with
β-catenin for binding to Siah1 and replacing it for degradation
(Fig. 1D) (63). TRAF4 is highly expressed in
chemotherapy-resistant breast cancer cells, and patients with BC
and low TRAF4 expression levels benefit from chemotherapy (67–69).
Notably, the chemoresistance mediated by TRAF4 appears to be
strongest to etoposide (a chemotherapeutic agent that induces
Siah1-mediated degradation of β-catenin), suggesting that the key
role of the Siah1-TRAF4/β-catenin axis in the chemoresistance of BC
and further study of this axis may lead to new treatments (63).
Siah1 in leukemia
The dysregulation of the ubiquitin-proteasome system
(UPS) is observed in solid tumors and leukemia (27). Increased substrates of Siah in
leukemia have been found, suggesting the critical roles of Siah in
leukemogenesis (27,70).
Acute promyelocytic leukemia (APL) is one of the
most characterized forms of acute myeloid leukemia (AML) (71). t(15;17)(q24;q21), generating the
promyelocytic leukemia-retinoic acid receptor α (PML-RARα)
fusion gene, is the hallmark of APL (72,73).
Notably, Pietschmann et al reported that the Siah1/2
cooperates with the E2 ubiquitin conjugase, i.e.,
ubiquitin-conjugating enzyme human 8 (UBCH8), leading to the
proteasomal degradation of PML-RARα (74). In addition, this degradation of
PML-RARα by the UBCH8-Siah1 complex can be significantly enhanced
by all-trans-retinoic acid and sodium valproate (drug combination
against APL), promoting the differentiation and maturation of APL
cells (74,75). Moreover, other leukemia fusion
proteins, including t(8;21)(q22;q22), RUNX family transcription
factor 1 (AML1-ETO) fusion protein, have been identified as
substrates of Siah1, but not Siah2, suggesting the powerful tumor
suppressor effects of Siah1 in leukemia (27,76,77).
Super elongation complexes (SECs) promote the
transcription of normal and leukemia-associated gene expression
(78). SECs contain two different
transcription elongation factors, namely positive transcription
elongation factor b and elongation factor for RNA polymerase II 1/2
(ELL1/2), linked by the scaffolding protein AF4/FMR2 family member
1/4 (AFF1/4) (79). ELL2, a
stoichiometrically limiting protein of SECs and an oncoprotein in
leukemia, is specifically targeted for ubiquitin-mediated
degradation by Siah1, but not by Siah2 (70,80,81).
Notably, when AFF4 interacts with ELL2 to form SECs, the half-life
of ELL2 against the Siah1-mediated ubiquitination is significantly
prolonged (70). Through the
proteasomal degradation induced by Siah1, AFF4 appears to have a
lower affinity for Siah1 than ELL2. Thus, AFF4 is not adequately
degraded by the low protein level of Siah1 (70). Notably, at relatively low protein
levels of Siah1 in cells under physiological conditions, ELL2,
especially the parts of ELL2 outside SECs, is highly sensitive to
Siah1-induced ubiquitin-mediated degradation, suggesting that the
primary effect of Siah1 is to prevent the formation of new SECs
(Fig. 1E) (27,70).
However, when the protein level of Siah1 becomes high, all
remaining SECs are destroyed through Siah1-induced
ubiquitin-mediated degradation (27,70).
The abundance of Siah1 changes rapidly in response to various
stresses, which may be to regulate SECs for the maintenance of a
suitable transcription level of cells to adapt to stresses
(11,71,73,74,77,82).
Therefore, regulating the abundance of Siah1 in leukemia with
disordered SECs may be a feasible method (Fig. 1E).
Siah1 in glioblastoma (GBM)
GBM is the most common brain cancer (48%), with high
tumor heterogeneity and poor survival time (median overall survival
time, 12–14 months) in adults (83–85).
Siah1 is widely regarded as a tumor suppressor in the majority of
cancer types, with the exception of GBM (26,86).
The knockdown of Siah1 by short-hairpin RNA (shRNA) severely
suppresses the migration and invasion of human GBM cells (U251),
whereas the overexpression of Siah1 has the opposite effect
(26). Furthermore, recent studies
have suggested that the tumor promotion of Siah1 in GBM is
associated with hypoxic stress (11,16,26,87,88).
Hypoxia-inducible factor 1α (HIF-1α) is the key transcription
factor that regulates hypoxia-induced genes and enables cells to
adapt to hypoxia (89,90). HIF-1α is extremely unstable under
normal oxygen conditions (21% O2), as the
prolyl-hydroxylation of HIF-1α promotes the binding of von
Hippel-Lindau disease tumor suppressor to HIF-1α, resulting in the
degradation of HIF-1α through the ubiquitin-proteasome pathway
(89,91,92).
The prolyl-hydroxylation of HIF-1α is mediated by the
prolyl-hydroxylase protein (PHD) family, and the activity of PHD is
inhibited in hypoxia (16,89,92).
Notably, PHD3 has been identified as a substrate of Siah1, and
Siah1 mediates the ubiquitination of PHD3 and induces the
degradation of PHD3 through the UPS (11,26,93).
The degradation of PHD3 increases the abundance of HIF-1α, thus
promoting cells to adapt to hypoxia (16,26,87,88,93).
Moreover, HIF-1α trans-activates the transcription of Siah1
by coordinating key histone modifications on the Siah1
promoter to increase its expression continuously and form a
positive feedback loop, thus leading to the progression of GBM via
the tolerance of tumor cells to hypoxia (16,26,87,88,93).
Siah1 may be a potential molecular target for the treatment of GBM
through the interference of the Siah1-PHD3-HIF-1α axis (Fig. 2A).
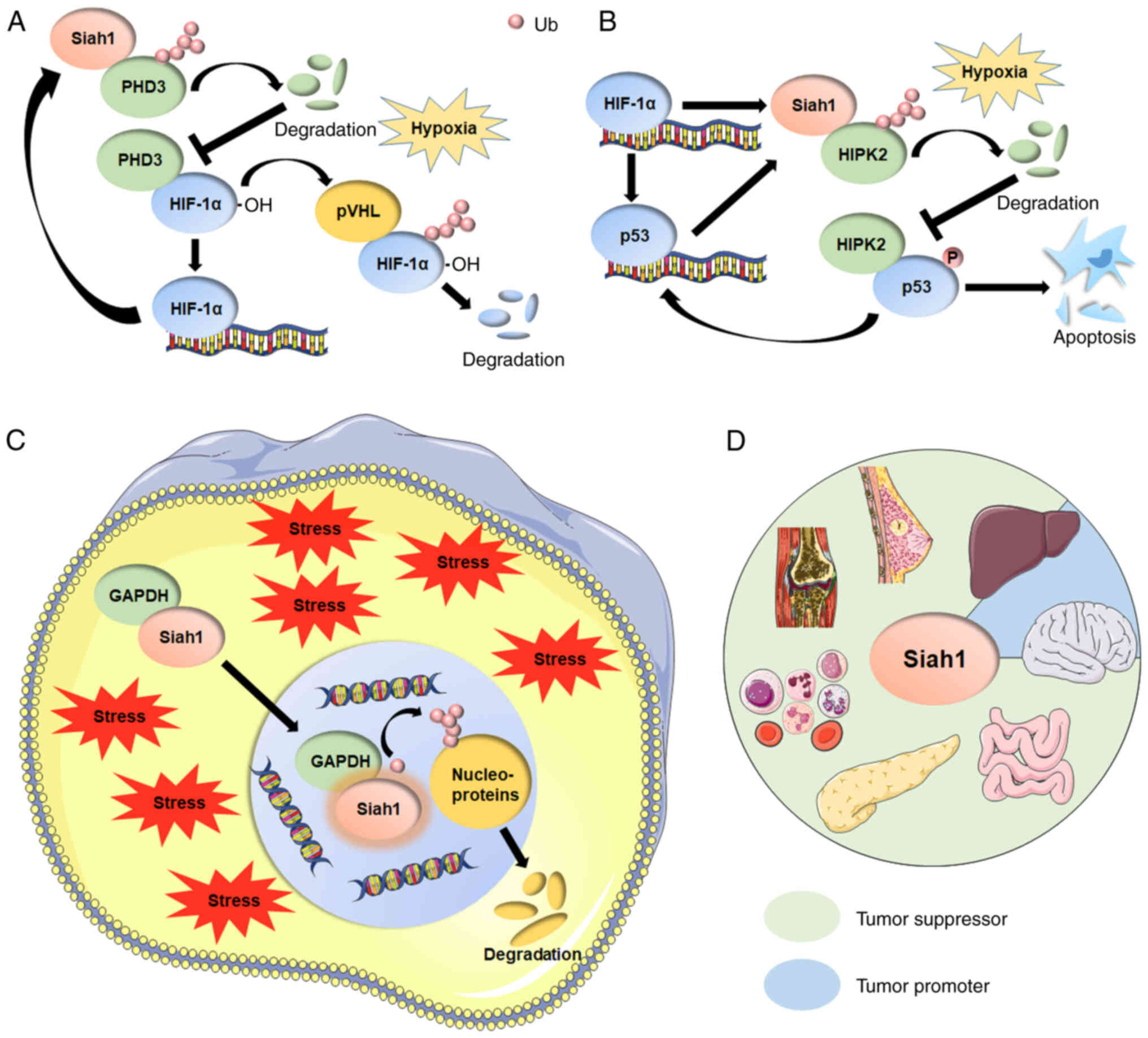 | Figure 2.(A) Siah1 mediates the ubiquitination
of PHD3 and induces the degradation of PHD3, increasing the
abundance of HIF-1α and promoting cells to adapt to hypoxia. HIF-1α
trans-activates the transcription of Siah1 by coordinating key
histone modifications on the Siah1 promoter to continuously
increase HIF-1α expression and form a positive feedback loop. (B)
Siah1 targets HIPK2 for poly-ubiquitination and proteasomal
degradation, thereby inhibiting the phosphorylation of p53 at Ser46
and preventing cell apoptosis. p53 continues to activate the
transcription of Siah1 and forms a positive feedback loop. The
initiation of this positive feedback mechanism may be mediated by
HIF-1α under hypoxic stress. (C) Under cell stress, GAPDH
translocates to the nucleus in a Siah1-dependent manner upon
glutamate stimulation and stabilizes Siah1 to facilitate
degradation of nuclear proteins by Siah1, resulting in cell
apoptosis and neuronal damage. (D) Siah1 functions as a tumor
suppressor in the vast majority of tumors (breast cancer,
hepatocellular cancer, leukemia, colorectal cancer and
osteosarcoma). In some cancer types, such as glioblastoma and a
part of hepatocellular carcinoma (where Siah1 is localized to the
nucleus), Siah1 functions as an oncoprotein. Siah1, seven in
absentia homolog family proteins 1; PHD3, prolyl-hydroxylase
proteins 3; HIF-1α, hypoxia-inducible factor 1α; pVHL, von
Hippel-Lindau disease tumor suppressor protein; HIPK2,
homeodomain-interacting protein kinase 2; p53, tumor suppressor
p53; P, phosphate group; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
Additionally, some studies have suggested that the
Siah1-homeodomain-interacting protein kinase 2 (HIPK2)-p53Ser46
axis plays a key role in the promotion of glioma progression
(86,94). HIPK2 functions as the key factor
that is activated in DNA damage or cell response to stress and that
triggers the phosphorylation of p53 at Ser46 (95,96).
The best-known tumor suppressor gene, TP53, is mutated in
>50% of malignancies and encodes a protein known as p53, a
transcription factor that controls the initiation of the cell cycle
(97–100). However, when p53 is
phosphorylated at Ser46, its functional activity as a transcription
factor is inactivated, whereas the function of apoptosis promotion
is activated, thus leading cells to apoptosis under stress response
(101,102). However, Siah1 has been considered
as the negative regulatory E3 ubiquitin ligase of HIPK2 that
targets HIPK2 for poly-ubiquitination and proteasomal degradation,
thereby inhibiting cell apoptosis by HIPK2 and resulting in the
survival and proliferation of GBM cells (86,94,103,104). Notably, p53 acts directly on
Siah1 to promote its transcription (11). As a tumor suppressor, p53 is
supposed to promote the apoptosis of tumor cells, but the
regulation of p53 on Siah1 in GBM seems to be a contradiction. p53
continuously activates the transcription of Siah1, whereas
the increased abundance of Siah1 targets the degradation of HIPK2
and blocks the phosphorylation of p53 at Ser46, which keeps the
activity of p53 as a transcription factor, continues to activate
the transcription of Siah1 and promotes the progression of
GBM. This phenomenon may also be explained by the hypoxic
microenvironment of GBM, as HIF-1α can promote p53
transcription and stabilize the function of p53 in hypoxic stress
(105,106). Siah1, p53, HIPK2, PHD3 and HIF-1α
constitute an extremely complex network in the process of promoting
the development of GBM (Fig. 2B),
in which Siah1, as a key bridge factor, has the potential to be a
future therapeutic target.
Siah1 may also partially act as a tumor suppressor
in GBM when it interacts with CacyBP/SIP. CacyBP/SIP inhibits the
migration and invasion behaviors of GBM cells by activating
Siah1-mediated ubiquitination and degradation of cytoplasmic
p27/kip1 (a key transcription factor and an oncoprotein highly
expressed in GBM tissues) (22).
Siah1 as an oncoprotein
Studies have shown that Siah1 promotes cancer
progression only in GBM and HCC, and only when Siah1 is localized
in the nucleus (11,47,86,94).
The varying biological functions in these studies are most probably
associated with differences in cell types and the differential
subcellular localization of Siah1.
The functions of Siah1 to induce the proliferation
of cancer cells may be due to increased protein levels of FBP-3
(47). In addition, the
Siah1-PHD3-HIF-1 and HIPK2-p53Ser46 axes explain how Siah1 inhibits
apoptosis (26,86,87,94,107). However, the functions of Siah1 as
an oncoprotein have not been systematically studied and summarized.
Thus, future studies are needed to provide evidence for the
targeted therapy of Siah1 as an oncoprotein.
Siah1 in other cancer types
Siah1 also acts as a tumor suppressor in CRC and
pancreatic carcinogenesis. The knock down of Siah1 by shRNA
promotes HCT116/SW480 CRC cell proliferation and migration, and
results in fast tumor growth and a markedly large tumor volume in
nude mice. Mechanically, Siah1 represses the occurrence and
development of CRC by promoting the ubiquitylation of AKT and
inhibiting the activity of the MAPK, PI3K-AKT and Hippo pathways
(108). Additionally, the
mutations of p53 in pancreatic cancer act in the opposite manner to
the wild-type p53, inhibiting the transcription of Siah1 and
leading to the accumulation of the oncoprotein. This phenomenon
suggests the novel regulation of Siah1 in cancer (109).
Siah1 in nervous system diseases
Siah1 in Parkinson's Disease (PD)
PD, one of the most common neurodegenerative
diseases, is manifested by a series of movement disorders, such as
static tremor, bradykinesia, myotonia, and postural and gait
disorders (110,111). Dopamine neurons and formation of
Lewy bodies (LBs) in the substantia nigra striatum of the midbrain
are regarded as typical pathological features of PD (112). LBs contain misfolded and
abnormally stacked α-synuclein (α-syn), synphilin-1, ubiquitin and
UPS-related enzymes, e.g., Siah1 (113–115), F-box only protein 7 (116), and Parkin (117), suggesting that the disorder of
UPS plays a key role in the pathogenesis of PD (118–120). Notably, Siah1 is reported to
monoubiquitinate or diubiquitinate α-syn, but without degradation,
and is capable of ubiquitination and proteasomal degradation of
synphilin-1, thus limiting the availability of α-syn for binding to
synphilin-1 and the formation of LBs (114,115), suggesting the key pathological
role of Siah1 in the development of PD. In addition, one study
reported the cases of 7 patients with PD and Siah1 mutations
(113), but the function of these
mutations in PD is not fully known yet. Increasing evidence shows
that the PTEN-induced putative kinase 1, synphilin-1 and Siah1
complex constitutes a novel mitophagy pathway (mitochondrial
dysfunction is also considered to be one of the main pathological
features of PD] (7,12,13),
and that the complex has the function of clearing damaged
mitochondria in PD (121),
suggesting that drugs that activate Siah1 provide a novel strategy
to promote the clearance of damaged mitochondria in PD (116,122–127).
Siah1 in developmental delay
Developmental delay is defined as the skills of a
child in one or a number aspects, including physical, motor,
socioemotional, speech and language, and cognitive development,
being significantly slower than those of other children of the same
age (128–131). Developmental delay occurs in up
to 5% of children <5 years of age, and patients can benefit from
the early detection of developmental delay and appropriate
therapeutic measures (129,130). Genetic factors are the main
causes of development delay, and a recent case report showed de
novo monoallelic variants (Cys41Gly, Pro50Leu, Cys128Phe,
Thr168Ala and Gly174Arg) in Siah1 in 5 unrelated patients within a
phenotypic spectrum of developmental delay, infantile hypotonia,
dysmorphism, strabismus and laryngomalacia (128). All patients with Siah1 mutations,
except Pro50Leu, presented with moderate or severe developmental
delay (128).
The overinhibition of the Wnt/β-catenin signaling
pathways is one of the important pathogenesis factors of
developmental delay (34,35). Previous studies have reported that
wild-type Siah1 enhances the Wnt/β-catenin signaling pathways by
mediating the Wnt-induced degradation of Axin (38,132). However, in 293T cells, exogenous
Siah1 mutations (i.e., Cys41Gly, Pro50Leu, Cys128Phe, Thr168Ala and
Gly174Arg) lose the ability to degrade Axin compared with the
wild-type Siah1, resulting in the overinhibition of the
Wnt/β-catenin signaling pathways (128). The best treatment for
developmental delay is early detection. Thus, the prenatal
examination for Siah1 is a highly effective option.
In addition, Siah1 has recently been identified as
an upstream regulator of Akt3 (Akt signaling is an important
regulator of neural development) (133–135), and it is responsible for the
ubiquitination and degradation of Akt3, suggesting that Siah1 may
play a key role in neural development (135).
Siah1 in neuronal damage
Neuronal damage includes a series of diseases,
including spinal cord injury and cerebral ischemia reperfusion, and
the common feature of these diseases is the excessive apoptosis of
nerve cells (136,137). Previous studies have reported
that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is
conventionally considered a critical factor in the process of nerve
cell apoptosis (138–143). GAPDH is important in
glutamate-induced neuronal excitotoxicity, and evidence also
demonstrates that GAPDH nuclear translocation plays a critical role
in cell death (143,144). Notably, recent studies have shown
that GAPDH is translocated to the nucleus in a Siah1-dependent
manner upon glutamate stimulation and that it stabilizes Siah1 to
facilitate the degradation of nuclear proteins by Siah1, resulting
in cell apoptosis and neuronal damage (Fig. 2C) (138,144,145). Notably, the GAPDH/Siah1 cascade
can be inhibited by the administration of the interfering peptide,
the cannabinoid agonist WIN55212-2 and Sivelestat sodium, thus
preventing neuronal damage (138,143,144) and suggesting that the GAPDH/Siah1
cascade can serve as a potential therapeutic target for neuronal
damage treatment.
Siah1 was previously considered to be only a
neuroprotective factor (113,128,128), but in neuronal damage, it appears
to play a role in promoting the progression of neuronal damage
(143,144). This abnormal phenomenon may be
related to the subcellular localization of Siah1, and the function
of nuclear Siah1 seems to be opposite to Siah1 under normal
physiological conditions (114).
Notably, Siah1, a tumor suppressor, functions as an oncoprotein in
some types of liver cancer, and Siah1 in liver cancer is localized
in the nucleus (11,146). Thus, the subcellular localization
of Siah1 is an important focus and may become a novel treatment
strategy for some diseases.
Siah1 in Alzheimer's Disease (AD)
AD, the most common chronic and irreversible
neurodegenerative disease in the world, is characterized by
impaired cognitive function and loss of self-care ability (147). The hyperphosphorylation of Tau is
considered to be one of the main pathological features of AD
(148,149). A recent study showed the
CacyBP/SIP could mediate the dephosphorylation of
phosphorylated-Tau (150),
suggesting the neuroprotective effects of CacyBP/SIP. CacyBP/SIP
has long been identified as a SIP (21,22).
However, the function of Siah1 in AD has been rarely studied and
should be clarified in future studies.
Clinical significance of Siah1 in
human diseases
Siah1 functions as a tumor suppressor in the vast
majority of tumors [e.g., BC (23), HCC (30), leukemia (27), CRC (108) and osteosarcoma (44)]. However, in some cancer types, such
as GBM (86) and a part of HCC
(where Siah1 is localized to the nucleus) (47), Siah1 functions as an oncoprotein
(Table II). The activator lncRNA
SNHG1(151) and inhibitors
miR-135a (152), miR-424
(153), miR-944 (62), miR-299-5p (146), miR-15b-5p (151) and miR-107 (59), are all good choices to regulate the
functions of Siah1 depending on the different cancer contexts
(Table I). Notably, the
oncoprotein functions of Siah1 may be due to the nuclear
localization of Siah1, thus suggesting that the inhibition of the
Siah1 nuclear localization signal is a way to inhibit the
oncoprotein functions of Siah1 but one that does not destroy the
normal physiological function of Siah1 in the cytoplasm (47). This idea is also suitable for the
treatment of nervous system diseases, as the nuclear localization
of Siah1 often promotes nerve apoptosis and leads to the occurrence
of nervous system diseases (138–140,142,143).
Discussion
The functions of a protein depend on a number of
elements, including post-translational modification, cell types,
cellular microenvironment and binding to other proteins. Siah1 was
originally identified as a tumor-suppressing protein for BC
(54–56). However, new functions of Siah1 are
continuously being discovered, suggesting that Siah1 is not only a
tumor-suppressing protein.
As an E3 ubiquitin ligase, the most important
function of Siah1 is the ubiquitination of substrates to promote
their degradation or change their function (11,13).
However, the types of ubiquitin modifications of Siah1 in cancer
and nervous system diseases seem to differ markedly. In cancer,
Siah1 is responsible for the ubiquitination-mediated degradation of
substrates, whether the substrates are oncoproteins or
tumor-suppressing proteins (16,74,76,94,154,155). In nervous system diseases, Siah1
does more to change the function of substrates than to degrade them
via non-degradative ubiquitination (140,142,156). This difference may be related to
the formation of the Siah1 ubiquitin-ligase complex. Siah1 can
interact with E2 alone or become an essential part of the
ubiquitin-ligase complex (13,20).
Notably, CacyBP/SIP, one of the parts of the complex, has the
highest protein levels in the brain and maintains lower protein
levels in other organs, suggesting that the complex may affect the
Siah1-mediated ubiquitination of substrates (21). Siah1 has been reported to
monoubiquitinate or diubiquitinate α-syn. Upon interaction with
CacyBP/SIP, Siah1 can inhibit the development of GBM by inducing
the degradation of p27/kip1, suggesting that the status of Siah1
(interacting with E2 alone or forming the ubiquitin-ligase complex)
significantly affects the functions of Siah1 (22,114,115). Numerous studies are limited to
substrate degradation by Siah1, but they ignore the special status
of Siah1 in this process, thus requiring future supplemental
studies (23,26,57,86).
The studies on the Siah1 ubiquitin-ligase complex may be the key to
study the function of Siah1 thoroughly and for targeting of Siah1
in the future.
The subcellular localization of Siah1 also
determines the function of Siah1. The nuclear localization of Siah1
promotes the occurrence of cancer and nerve apoptosis, suggesting
that the nuclear localization of Siah1 is a pathological phenomenon
(47,138). Although the mechanism that causes
Siah1 to transfer to the nucleus is not clear, we believe that the
inhibition of the Siah1 nuclear localization signal can be
identified as a favorable choice in the treatment of diseases.
The oncoprotein functions of Siah1 in GBM seem to
be closely related to the tumor hypoxic microenvironment (26,93).
However, the function of Siah1 as an oncoprotein in the hypoxic
microenvironment has not been reported in other tumors, especially
in HCC, which is most closely associated with the hypoxic
microenvironment (89). GBM
belongs to the diseases of the nervous system. Thus, studying the
functional differences of Siah1 in cancer and neurological diseases
is a notable and promising topic.
The dysregulation of UPS is usually caused by the
mutations of E3 ubiquitin ligase, such as SPOP (8,157)
and leucine zipper-like transcription regulator 1 (LZTR1) (158). The mutations of SPOP and LZTR1
occur mostly in domains bound to the substrate, severely affecting
their ability to bind to substrates, inhibiting the ubiquitination
and degradation of substrates, and leading to the accumulation of
substrates (4,6,159).
Notably, although one study showed the lack of somatic mutation in
the coding sequence of Siah1 (55), the mutations of Siah1 are rarely
reported. The regulations of SPOP and LZTR1 are rarely recorded,
whereas the regulations of Siah1 are various (Table I), suggesting that Siah1 plays a
core role as a bridge factor in various signaling pathways
(11). Slight changes in Siah1
protein levels and protein localization lead to significant changes
in its functions, which may be as the mutations affect the function
of Siah1 significantly and cause death in the earliest embryos
(knockout of both Siah1 genes is embryonically lethal in mice).
This phenomenon results in little spread of Siah1 mutation heritage
lines (11,27).
Although numerous studies have focused on the
upstream regulations of Siah1 (Table
I), studies on agonists and inhibitors specifically targeting
Siah1 remain lacking. The only drug targeting Siah that has been
described so far is vitamin K3 (menadione), which has been
identified as an inhibitor of Siah2 ubiquitin ligase activity in a
screen of U.S. Food and Drug Administration-approved therapeutic
drugs (158). Therefore, the
future drug research of Siah1 is still needed.
The present review summarized the novel substrates
and complex upstream regulations of Siah1, describes the functions
of Siah1 as a tumor suppressor protein and an oncoprotein, and
discusses the potential mechanisms of the different roles of Siah1
in the nervous system and cancer. In addition, the review focused
on the effects of Siah1 nuclear localization and the special status
of Siah1 (interacting with E2 alone or forming the ubiquitin-ligase
complex). Moreover, it highlighted the clinical significance of
Siah1 in human diseases. This review may provide inspiration for
future Siah1 research.
Acknowledgements
The authors would like to thank Dr Yuqi Wang (West
Lake University, Hangzhou, Zhejiang, China) for the kind help and
good advice provided.
Funding
This study was supported by the Natural Science Foundation of
Zhejiang Province (grant no. LY20C070001), the National Natural
Science Foundation of China (grant no. 31801165), the Graduate
Research Innovation Fund project in Ningbo University (grant no.
IF2021097) and the K.C.Wong Magna Fund in Ningbo University.
Availability of data and materials
Not applicable.
Authors' contributions
XJ conceived the present study. HZ drafted the
manuscript. YG, JW and MY made substantial contributions to the
interpretation and drafting of the study, and revised the
manuscript critically for important intellectual content. All
authors read and approved the final manuscript. Data authentication
is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hershko A and Ciechanover A: The ubiquitin
system. Ann Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mani RS: The emerging role of speckle-type
POZ protein (SPOP) in cancer development. Drug Discov Today.
19:1498–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zou T and Zhang J: Diverse and pivotal
roles of neddylation in metabolism and immunity. FEBS J.
288:3884–3912. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen RH: Cullin 3 and its role in
tumorigenesis. Adv Exp Med Biol. 1217:187–210. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nandi D, Tahiliani P, Kumar A and Chandu
D: The ubiquitin-proteasome system. J Biosci. 31:137–155. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cuneo MJ and Mittag T: The ubiquitin
ligase adaptor SPOP in cancer. FEBS J. 286:3946–3958. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang D, Ma L, Wang B, Liu J and Wei W: E3
ubiquitin ligases in cancer and implications for therapies. Cancer
Metastasis Rev. 36:683–702. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Z, Song Y, Ye M, Dai X, Zhu X and Wei
W: The diverse roles of SPOP in prostate cancer and kidney cancer.
Nat Rev Urol. 17:339–350. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morreale F and Walden H: Types of
ubiquitin ligases. Cell. 165:248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yun S, Möller A, Chae SK, Hong WP, Bae YJ,
Bowtell DD, Ryu SH and Suh PG: Siah proteins induce the epidermal
growth factor-dependent degradation of phospholipase Cepsilon. J
Biol Chem. 283:1034–1042. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi J, Kim H, Scortegagna M and Ronai ZA:
Regulators and effectors of Siah ubiquitin ligases. Cell Biochem
Biophys. 67:15–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garrison JB, Correa RG, Gerlic M, Yip KW,
Krieg A, Tamble CM, Shi R, Welsh K, Duggineni S, Huang Z, et al:
ARTS and Siah collaborate in a pathway for XIAP degradation. Mol
Cell. 41:107–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Santelli E, Leone M, Li C, Fukushima T,
Preece NE, Olson AJ, Ely KR, Reed JC, Pellecchia M, Liddington RC
and Matsuzawa SI: Structural analysis of Siah1-Siah-interacting
protein interactions and insights into the assembly of an E3 ligase
multiprotein complex. J Biol Chem. 280:34278–34287. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Q, Wang Z, Hou F, Harding R, Huang
X, Dong A, Walker JR and Tong Y: The substrate binding domains of
human SIAH E3 ubiquitin ligases are now crystal clear. Biochim
Biophys Acta Gen Subj. 1861:3095–3105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Knauer SK, Mahendrarajah N, Roos WP and
Krämer OH: The inducible E3 ubiquitin ligases SIAH1 and SIAH2
perform critical roles in breast and prostate cancers. Cytokine
Growth Factor Rev. 26:405–413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakayama K and Ronai Z: Siah: New players
in the cellular response to hypoxia. Cell Cycle. 3:1345–1347. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Famulski JK, Trivedi N, Howell D, Yang Y,
Tong Y, Gilbertson R and Solecki DJ: Siah regulation of Pard3A
controls neuronal cell adhesion during germinal zone exit. Science.
330:1834–1838. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wong CS and Möller A: Siah: A promising
anticancer target. Cancer Res. 73:2400–2406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
House CM, Möller A and Bowtell DD: Siah
proteins: Novel drug targets in the Ras and hypoxia pathways.
Cancer Res. 69:8835–8838. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsuzawa S, Li C, Ni CZ, Takayama S, Reed
JC and Ely KR: Structural analysis of Siah1 and its interactions
with Siah-interacting protein (SIP). J Biol Chem. 278:1837–1840.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Topolska-Woś AM, Chazin WJ and Filipek A:
CacyBP/SIP-structure and variety of functions. Biochim Biophys
Acta. 1860:79–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan S, Li A and Liu Y: CacyBP/SIP inhibits
the migration and invasion behaviors of glioblastoma cells through
activating Siah1 mediated ubiquitination and degradation of
cytoplasmic p27. Cell Biol Int. 42:216–226. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wen YY, Yang ZQ, Song M, Li BL, Yao XH,
Chen XL, Zhao J, Lu YY, Zhu JJ and Wang EH: The expression of SIAH1
is downregulated and associated with Bim and apoptosis in human
breast cancer tissues and cells. Mol Carcinog. 49:440–449. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Briant DJ, Ceccarelli DF and Sicheri F: I
Siah substrate! Structure. 14:627–628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Czechowicz JS, Nagel CH, Voges M, Spohn M,
Eibl MM and Hauber J: Interaction between the cellular E3 ubiquitin
ligase SIAH-1 and the viral immediate-early protein ICP0 enables
efficient replication of herpes simplex virus type 2 in vivo. PLoS
One. 13:e02018802018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi H, Zheng B, Wu Y, Tang Y, Wang L, Gao
Y, Gong H, Du J and Yu R: Ubiquitin ligase Siah1 promotes the
migration and invasion of human glioma cells by regulating HIF-1α
signaling under hypoxia. Oncol Rep. 33:1185–1190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Krämer OH, Stauber RH, Bug G, Hartkamp J
and Knauer SK: SIAH proteins: Critical roles in leukemogenesis.
Leukemia. 27:792–802. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sim HW and Knox J: Hepatocellular
carcinoma in the era of immunotherapy. Curr Probl Cancer. 42:40–48.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsuo K, Satoh S, Okabe H, Nomura A,
Maeda T, Yamaoka Y and Ikai I: SIAH1 inactivation correlates with
tumor progression in hepatocellular carcinomas. Genes Chromosomes
Cancer. 36:283–291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yoshibayashi H, Okabe H, Satoh S, Hida K,
Kawashima K, Hamasu S, Nomura A, Hasegawa S, Ikai I and Sakai Y:
SIAH1 causes growth arrest and apoptosis in hepatoma cells through
beta-catenin degradation-dependent and -independent mechanisms.
Oncol Rep. 17:549–556. 2007.PubMed/NCBI
|
|
31
|
Yao H, Ashihara E and Maekawa T: Targeting
the Wnt/β-catenin signaling pathway in human cancers. Expert Opin
Ther Targets. 15:873–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arend RC, Londoño-Joshi AI, Straughn JM Jr
and Buchsbaum DJ: The Wnt/β-catenin pathway in ovarian cancer: A
review. Gynecol Oncol. 131:772–779. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Taciak B, Pruszynska I, Kiraga L, Bialasek
M and Krol M: Wnt signaling pathway in development and cancer. J
Physiol Pharmacol. 69:doi: 10.26402. 2018.PubMed/NCBI
|
|
35
|
Steinhart Z and Angers S: Wnt signaling in
development and tissue homeostasis. Development. 145:dev1465892018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang P, Yan R, Zhang X, Wang L, Ke X and
Qu Y: Activating Wnt/β-catenin signaling pathway for disease
therapy: Challenges and opportunities. Pharmacol Ther. 196:79–90.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kim E, Lisby A, Ma C, Lo N, Ehmer U, Hayer
KE, Furth EE and Viatour P: Promotion of growth factor signaling as
a critical function of β-catenin during HCC progression. Nat
Commun. 10:19092019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ji L, Jiang B, Jiang X, Charlat O, Chen A,
Mickanin C, Bauer A, Xu W, Yan X and Cong F: The SIAH E3 ubiquitin
ligases promote Wnt/β-catenin signaling through mediating
wnt-induced axin degradation. Genes Dev. 31:904–915. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang Y, Zhang J, Wu T, Xu X, Cao G, Li H
and Chen X: Histone deacetylase 2 regulates the doxorubicin (Dox)
resistance of hepatocarcinoma cells and transcription of ABCB1.
Life Sci. 216:200–206. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheng C, Li C, Zhu X, Han W, Li J and Lv
Y: Doxorubicin-loaded Fe(3)O(4)-ZIF-8 nano-composites for
hepatocellular carcinoma therapy. J Biomater Appl. 33:1373–1381.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Long L, Xiang H, Liu J, Zhang Z and Sun L:
ZEB1 mediates doxorubicin (Dox) resistance and mesenchymal
characteristics of hepatocarcinoma cells. Exp Mol Pathol.
106:116–122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li LY, Yang CC, Yang JF, Li HD, Zhang BY,
Zhou H, Hu S, Wang K, Huang C, Meng XM, et al: ZEB1 regulates the
activation of hepatic stellate cells through Wnt/β-catenin
signaling pathway. Eur J Pharmacol. 865:1727872019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qin Y, Yu J, Zhang M, Qin F and Lan X:
ZEB1 promotes tumorigenesis and metastasis in hepatocellular
carcinoma by regulating the expression of vimentin. Mol Med Rep.
19:2297–2306. 2019.PubMed/NCBI
|
|
44
|
Han X, Liu F, Zhang C, Ren Z, Li L and
Wang G: SIAH1/ZEB1/IL-6 axis is involved in doxorubicin (Dox)
resistance of osteosarcoma cells. Biol Chem. 400:545–553. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abshire CF, Carroll JL and Dragoi AM:
FLASH protects ZEB1 from degradation and supports cancer cells'
epithelial-to-mesenchymal transition. Oncogenesis. 5:e2542016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu Y, Yang X, Chen Z, Tian L, Jiang G,
Chen F, Li J, An P, Lu L, Luo N, et al: m6 A-induced
lncRNA RP11 triggers the dissemination of colorectal cancer cells
via upregulation of Zeb1. Mol Cancer. 18:872019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Brauckhoff A, Malz M, Tschaharganeh D,
Malek N, Weber A, Riener MO, Soll C, Samarin J, Bissinger M and
Schmidt J: Nuclear expression of the ubiquitin ligase seven in
absentia homolog (SIAH)-1 induces proliferation and migration of
liver cancer cells. J Hepatol. 55:1049–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Veronesi U, Boyle P, Goldhirsch A,
Orecchia R and Viale G: Breast cancer. Lancet. 365:1727–1741. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Woolston C: Breast cancer. Nature.
527:S1012015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics, 2011. CA Cancer J Clin. 61:409–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ullah MF: Breast cancer: Current
perspectives on the disease status. Adv Exp Med Biol. 1152:51–64.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tsang JYS and Tse GM: Molecular
classification of breast cancer. Adv Anat Pathol. 27:27–35. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Barzaman K, Karami J, Zarei Z,
Hosseinzadeh A, Kazemi MH, Moradi-Kalbolandi S, Safari E and
Farahmand L: Breast cancer: Biology, biomarkers, and treatments.
Int Immunopharmacol. 84:1065352020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bruzzoni-Giovanelli H, Faille A,
Linares-Cruz G, Nemani M, Deist FL, Germani A, Chassoux D, Millot
G, Roperch JP, Amson R, et al: SIAH-1 inhibits cell growth by
altering the mitotic process. Oncogene. 18:7101–7109. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Medhioub M, Vaury C, Hamelin R and Thomas
G: Lack of somatic mutation in the coding sequence of SIAH1 in
tumors hemizygous for this candidate tumor suppressor gene. Int J
Cancer. 87:794–797. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Germani A, Bruzzoni-Giovanelli H, Fellous
A, Gisselbrecht S, Varin-Blank N and Calvo F: SIAH-1 interacts with
alpha-tubulin and degrades the kinesin Kid by the proteasome
pathway during mitosis. Oncogene. 19:5997–6006. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wen YY, Yang ZQ, Song M, Li BL, Zhu JJ and
Wang EH: SIAH1 induced apoptosis by activation of the JNK pathway
and inhibited invasion by inactivation of the ERK pathway in breast
cancer cells. Cancer Sci. 101:73–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nedeljković M and Damjanović A: Mechanisms
of chemotherapy resistance in triple-negative breast cancer-how we
can rise to the challenge. Cells. 8:9572019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang L, Ma P, Sun LM, Han YC, Li BL, Mi
XY, Wang EH and Song M: MiR-107 down-regulates SIAH1 expression in
human breast cancer cells and silencing of miR-107 inhibits tumor
growth in a nude mouse model of triple-negative breast cancer. Mol
Carcinog. 55:768–777. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hong HC, Chuang CH, Huang WC, Weng SL,
Chen CH, Chang KH, Liao KW and Huang HD: A panel of eight microRNAs
is a good predictive parameter for triple-negative breast cancer
relapse. Theranostics. 10:8771–8789. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sahlberg KK, Bottai G, Naume B, Burwinkel
B, Calin GA, Børresen-Dale AL and Santarpia L: A serum microRNA
signature predicts tumor relapse and survival in triple-negative
breast cancer patients. Clin Cancer Res. 21:1207–1214. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Flores-Pérez A, Marchat LA,
Rodríguez-Cuevas S, Bautista VP, Fuentes-Mera L, Romero-Zamora D,
Maciel-Dominguez A, de la Cruz OH, Fonseca-Sánchez M, Ruíz-García
E, et al: Suppression of cell migration is promoted by miR-944
through targeting of SIAH1 and PTP4A1 in breast cancer cells. BMC
Cancer. 16:3792016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ren H, Mi X, Zhao P, Zhao X, Wei N, Huang
H, Meng Z, Kou J, Sun M, Liu Y, et al: TRAF4, a new substrate of
SIAH1, participates in chemotherapy resistance of breast cancer
cell by counteracting SIAH1-mediated downregulation of β-catenin.
Breast Cancer Res Treat. 183:275–289. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Butti R, Gunasekaran VP, Kumar TVS,
Banerjee P and Kundu GC: Breast cancer stem cells: Biology and
therapeutic implications. Int J Biochem Cell Biol. 107:38–52. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fisusi FA and Akala EO: Drug combinations
in breast cancer therapy. Pharm Nanotechnol. 7:3–23. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tang Y, Wang Y, Kiani MF and Wang B:
Classification, treatment strategy, and associated drug resistance
in breast cancer. Clin Breast Cancer. 16:335–343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhou J, Li W, Ming J, Yang W, Lu L, Zhang
Q, Ruan S and Huang T: High expression of TRAF4 predicts poor
prognosis in tamoxifen-treated breast cancer and promotes tamoxifen
resistance. Anticancer Drugs. 31:558–566. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang X, Wen Z and Mi X: Expression and
anti-apoptotic function of TRAF4 in human breast cancer MCF-7
cells. Oncol Lett. 7:411–414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang A, Wang J, Ren H, Yang F, Sun L, Diao
K, Zhao Z, Song M, Cui Z, Wang E, et al: TRAF4 participates in
wnt/β-catenin signaling in breast cancer by upregulating β-catenin
and mediating its translocation to the nucleus. Mol Cell Biochem.
395:211–219. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu M, Hsu J, Chan C, Li Z and Zhou Q: The
ubiquitin ligase Siah1 controls ELL2 stability and formation of
super elongation complexes to modulate gene transcription. Mol
Cell. 46:325–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
de Thé H, Pandolfi PP and Chen Z: Acute
promyelocytic leukemia: A paradigm for oncoprotein-targeted cure.
Cancer Cell. 32:552–560. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
De Braekeleer E, Douet-Guilbert N and De
Braekeleer M: RARA fusion genes in acute promyelocytic leukemia: A
review. Expert Rev Hematol. 7:347–357. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liquori A, Ibañez M, Sargas C, Sanz MA,
Barragán E and Cervera J: Acute promyelocytic leukemia: A
constellation of molecular events around a single PML-RARA Fusion
Gene. Cancers (Basel). 12:6242020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Pietschmann K, Buchwald M, Müller S,
Knauer SK, Kögl M, Heinzel T and Krämer OH: Differential regulation
of PML-RARα stability by the ubiquitin ligases SIAH1/SIAH2 and
TRIAD1. Int J Biochem Cell Biol. 44:132–138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bug G, Ritter M, Wassmann B, Schoch C,
Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer
D, et al: Clinical trial of valproic acid and all-trans retinoic
acid in patients with poor-risk acute myeloid leukemia. Cancer.
104:2717–2725. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bursen A, Moritz S, Gaussmann A, Moritz S,
Dingermann T and Marschalek R: Interaction of AF4 wild-type and
AF4.MLL fusion protein with SIAH proteins: Indication for t(4;11)
pathobiology? Oncogene. 23:6237–6249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Krämer OH, Müller S, Buchwald M, Reichardt
S and Heinzel T: Mechanism for ubiquitylation of the leukemia
fusion proteins AML1-ETO and PML-RARalpha. Faseb J. 22:1369–1379.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Luo Z, Lin C and Shilatifard A: The super
elongation complex (SEC) family in transcriptional control. Nat Rev
Mol Cell Biol. 13:543–547. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Dahl NA, Danis E, Balakrishnan I, Wang D,
Pierce A, Walker FM, Gilani A, Serkova NJ, Madhavan K, Fosmire S,
et al: Super elongation complex as a targetable dependency in
diffuse midline glioma. Cell Rep. 31:1074852020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yu D, Liu R, Yang G and Zhou Q: The
PARP1-Siah1 axis controls HIV-1 transcription and expression of
Siah1 substrates. Cell Rep. 23:3741–3749. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu J, Xue Y, Gao X and Zhou Q: Host cell
factors stimulate HIV-1 transcription by antagonizing
substrate-binding function of Siah1 ubiquitin ligase to stabilize
transcription elongation factor ELL2. Nucleic Acids Res.
48:7321–7332. 2020.PubMed/NCBI
|
|
82
|
Nagel CH, Albrecht N, Milovic-Holm K,
Mariyanna L, Keyser B, Abel B, Weseloh B, Hofmann TG, Eibl MM and
Hauber J: Herpes simplex virus immediate-early protein ICP0 is
targeted by SIAH-1 for proteasomal degradation. J Virol.
85:7644–7657. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Abe T, Umeki I, Kanno SI, Inoue SI,
Niihori T and Aoki Y: LZTR1 facilitates polyubiquitination and
degradation of RAS-GTPases. Cell Death Differ. 27:1023–1035. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Frattini V, Trifonov V, Chan JM, Castano
A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al: The
integrated landscape of driver genomic alterations in glioblastoma.
Nat Genet. 45:1141–1149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wirsching HG, Galanis E and Weller M:
Glioblastoma. Handb Clin Neurol. 134:381–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
He Y, Roos WP, Wu Q, Hofmann TG and Kaina
B: The SIAH1-HIPK2-p53ser46 damage response pathway is involved in
temozolomide-induced glioblastoma cell death. Mol Cancer Res.
17:1129–1141. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fan Z, Li Z, Yang Y, Liu S, Guo J and Xu
Y: HIF-1α coordinates epigenetic activation of SIAH1 in hepatocytes
in response to nutritional stress. Biochim Biophys Acta Gene Regul
Mech. 1860:1037–1046. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Matsui-Hasumi A, Sato Y, Uto-Konomi A,
Yamashita S, Uehori J, Yoshimura A, Yamashita M, Asahara H, Suzuki
S and Kubo M: E3 ubiquitin ligases SIAH1/2 regulate
hypoxia-inducible factor-1 (HIF-1)-mediated Th17 cell
differentiation. Int Immunol. 29:133–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
You L, Wu W, Wang X, Fang L, Adam V,
Nepovimova E, Wu Q and Kuca K: The role of hypoxia-inducible factor
1 in tumor immune evasion. Med Res Rev. 41:1622–1643. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Peng S, Zhang J, Tan X, Huang Y, Xu J,
Silk N, Zhang D, Liu Q and Jiang J: The VHL/HIF axis in the
development and treatment of pheochromocytoma/paraganglioma. Front
Endocrinol (Lausanne). 11:5868572020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gopalsamy A, Hagen T and Swaminathan K:
Investigating the molecular basis of Siah1 and Siah2 E3 ubiquitin
ligase substrate specificity. PLoS One. 9:e1065472014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kim SY, Choi DW, Kim EA and Choi CY:
Stabilization of HIPK2 by escape from proteasomal degradation
mediated by the E3 ubiquitin ligase Siah1. Cancer Lett.
279:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Choi DW and Choi CY: HIPK2 modification
code for cell death and survival. Mol Cell Oncol. 1:e9559992014.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Feng Y, Zhou L, Sun X and Li Q:
Homeodomain-interacting protein kinase 2 (HIPK2): A promising
target for anti-cancer therapies. Oncotarget. 8:20452–20461. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Moll UM and Petrenko O: The MDM2-p53
interaction. Mol Cancer Res. 1:1001–1008. 2003.PubMed/NCBI
|
|
98
|
Krastev DB and Buchholz F: Ribosome
biogenesis and p53: Who is regulating whom? Cell Cycle.
10:3417–3418. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chao CC: Mechanisms of p53 degradation.
Clin Chim Acta. 438:139–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kanapathipillai M: Treating p53 mutant
aggregation-associated cancer. Cancers (Basel). 10:1542018.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Feng L, Hollstein M and Xu Y: Ser46
phosphorylation regulates p53-dependent apoptosis and replicative
senescence. Cell Cycle. 5:2812–2819. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Liebl MC and Hofmann TG: Cell fate
regulation upon DNA damage: p53 serine 46 kinases pave the cell
death road. Bioessays. 41:e19001272019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Winter M, Sombroek D, Dauth I,
Moehlenbrink J, Scheuermann K, Crone J and Hofmann TG: Control of
HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases
ATM and ATR. Nat Cell Biol. 10:812–824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Moehlenbrink J, Bitomsky N and Hofmann TG:
Hypoxia suppresses chemotherapeutic drug-induced p53 Serine 46
phosphorylation by triggering HIPK2 degradation. Cancer Lett.
292:119–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen D, Li M, Luo J and Gu W: Direct
interactions between HIF-1 alpha and Mdm2 modulate p53 function. J
Biol Chem. 278:13595–13598. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wang P, Guan D, Zhang XP, Liu F and Wang
W: Modeling the regulation of p53 activation by HIF-1 upon hypoxia.
FEBS Lett. 593:2596–2611. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Fukuba H, Yamashita H, Nagano Y, Jin HG,
Hiji M, Ohtsuki T, Takahashi T, Kohriyama T and Matsumoto M: Siah-1
facilitates ubiquitination and degradation of factor inhibiting
HIF-1alpha (FIH). Biochem Biophys Res Commun. 353:324–329. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Xiao Z, Wei Z, Deng D, Zheng Z, Zhao Y,
Jiang S, Zhang D, Zhang LJ, Fan M, Chen S, et al: Downregulation of
Siah1 promotes colorectal cancer cell proliferation and migration
by regulating AKT and YAP ubiquitylation and proteasome
degradation. Cancer Cell Int. 20:502020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Wu W, Liu X, Wei L, Li T, Zang Y, Qian Y,
Bai T, Li J, Xie M, Zhu Y, et al: Tp53 mutation inhibits
ubiquitination and degradation of WISP1 via down-regulation of
siah1 in pancreatic carcinogenesis. Front Pharmacol. 9:8572018.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ascherio A and Schwarzschild M: The
epidemiology of Parkinson's disease: Risk factors and prevention.
Lancet Neurol. 15:1257–1272. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bloem BR, Okun MS and Klein C: Parkinson's
disease. Lancet. 397:2284–2303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lees A, Hardy J and Revesz T: Parkinson's
disease. Lancet. 373:2055–2066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Franck T, Krueger R, Woitalla D, Müller T,
Engelender S and Riess O: Mutation analysis of the seven in
absentia homolog 1 (SIAH1) gene in Parkinson's disease. J Neural
Transm (Vienna). 113:1903–1908. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nagano Y, Yamashita H, Takahashi T,
Kishida S, Nakamura T, Iseki E, Hattori N, Mizuno Y, Kikuchi A and
Matsumoto M: Siah-1 facilitates ubiquitination and degradation of
synphilin-1. J Biol Chem. 278:51504–51514. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liani E, Eyal A, Avraham E, Shemer R,
Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R and
Engelender S: Ubiquitylation of synphilin-1 and alpha-synuclein by
SIAH and its presence in cellular inclusions and Lewy bodies imply
a role in Parkinson's disease. Proc Natl Acad Sci USA.
101:5500–5505. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Burchell VS, Nelson DE, Sanchez-Martinez
A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S,
Lewis PA, Houlden H, et al: The Parkinson's disease-linked proteins
Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci.
16:1257–1265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yamamura Y, Kuzuhara S, Kondo K, Yanagi T,
Uchida M, Matsumine H and Mizuno Y: Clinical, pathologic and
genetic studies on autosomal recessive early-onset parkinsonism
with diurnal fluctuation. Parkinsonism Relat Disord. 4:65–72. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Shahnawaz M, Mukherjee A, Pritzkow S,
Mendez N, Rabadia P, Liu X, Hu B, Schmeichel A, Singer W, Wu G, et
al: Discriminating α-synuclein strains in Parkinson's disease and
multiple system atrophy. Nature. 578:273–277. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lövestam S, Schweighauser M, Matsubara T,
Murayama S, Tomita T, Ando T, Hasegawa K, Yoshida M, Tarutani A,
Hasegawa M, et al: Seeded assembly in vitro does not replicate the
structures of α-synuclein filaments from multiple system atrophy.
FEBS Open Bio. 11:999–1013. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Schweighauser M, Shi Y, Tarutani A,
Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T,
Hasegawa K, et al: Structures of α-synuclein filaments from
multiple system atrophy. Nature. 585:464–469. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Szargel R, Shani V, Elghani FA, Mekies LN,
Liani E, Rott R and Engelender S: The PINK1, synphilin-1 and SIAH-1
complex constitutes a novel mitophagy pathway. Hum Mol Genet.
25:3476–3490. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Subramaniam SR and Chesselet MF:
Mitochondrial dysfunction and oxidative stress in Parkinson's
disease. Prog Neurobiol. 106–107. 17–32. 2013.PubMed/NCBI
|
|
123
|
Pickrell AM and Youle RJ: The roles of
PINK1, parkin, and mitochondrial fidelity in Parkinson's disease.
Neuron. 85:257–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ganguly G, Chakrabarti S, Chatterjee U and
Saso L: Proteinopathy, oxidative stress and mitochondrial
dysfunction: Cross talk in Alzheimer's disease and Parkinson's
disease. Drug Des Devel Ther. 11:797–810. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Rocha EM, De Miranda B and Sanders LH:
Alpha-synuclein: Pathology, mitochondrial dysfunction and
neuroinflammation in Parkinson's disease. Neurobiol Dis.
109:249–257. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Macdonald R, Barnes K, Hastings C and
Mortiboys H: Mitochondrial abnormalities in Parkinson's disease and
Alzheimer's disease: Can mitochondria be targeted therapeutically?
Biochem Soc Trans. 46:891–909. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang T, Zhang J, Hu M, Zhang Y, Cui P, Li
X, Li J, Vestin E, Brännström M, Shao LR and Billig H: Differential
expression patterns of glycolytic enzymes and
mitochondria-dependent apoptosis in PCOS patients with endometrial
hyperplasia, an early hallmark of endometrial cancer, in vivo and
the impact of metformin in vitro. Int J Biol Sci. 15:714–725. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Buratti J, Ji L, Keren B, Lee Y, Booke S,
Erdin S, Kim SY, Palculict TB, Meiner V, Chae JH, et al: De novo
variants in SIAH1, encoding an E3 ubiquitin ligase, are associated
with developmental delay, hypotonia and dysmorphic features. J Med
Genet. 58:205–212. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Khan I and Leventhal BL: Developmental
delay. StatPearls StatPearls Publishing. Copyright © 2021,
StatPearls Publishing LLC.; Treasure Island, FL: 2021
|
|
130
|
Oberklaid F and Efron D: Developmental
delay - identification and management. Aust Fam Physician.
34:739–742. 2005.PubMed/NCBI
|
|
131
|
Martiniuk AL, Vujovich-Dunn C, Park M, Yu
W and Lucas BR: Plagiocephaly and developmental delay: A systematic
review. J Dev Behav Pediatr. 38:67–78. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhou T, Chen Y, Zhao B, Hu S, Li J, Liu M,
Liang S, Bao Z and Wu X: Characterization and functional analysis
of SIAH1 during skin and hair follicle development in the angora
rabbit (Oryctolagus cuniculus). Hereditas. 157:102020.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Farmanullah Hosseini SM, Liang A, Hua G,
Rehman ZU, Talpur HS, Salim M, Ahmad S, Abulaiti A, Khan M, et al:
Adaptive molecular evolution of AKT3 gene for positive diversifying
selection in mammals. Biomed Res Int. 2020:25846272020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Alcantara D, Timms AE, Gripp K, Baker L,
Park K, Collins S, Cheng C, Stewart F, Mehta SG, Saggar A, et al:
Mutations of AKT3 are associated with a wide spectrum of
developmental disorders including extreme megalencephaly. Brain.
140:2610–2622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ko HR, Jin E, Lee SB, Kim CK, Yun T, Cho
SW, Park KW and Ahn JY: SIAH1 ubiquitin ligase mediates
ubiquitination and degradation of Akt3 in neural development. J
Biol Chem. 294:15435–15445. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Taoufik E and Probert L: Ischemic neuronal
damage. Curr Pharm Des. 14:3565–3573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Bruce-Keller AJ: Microglial-neuronal
interactions in synaptic damage and recovery. J Neurosci Res.
58:191–201. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Huo J, Zhu XL, Ma R, Dong HL and Su BX:
GAPDH/Siah1 cascade is involved in traumatic spinal cord injury and
could be attenuated by sivelestat sodium. Neuroscience.
330:171–180. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Huo J, Ma R, Chai X, Liang HJ, Jiang P,
Zhu XL, Chen X and Su BX: Inhibiting a spinal cord signaling
pathway protects against ischemia injury in rats. J Thoracic
Cardiovascular Surg. 157:494–503. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Hara MR, Thomas B, Cascio MB, Bae BI,
Hester LD, Dawson VL, Dawson TM, Sawa A and Snyder SH:
Neuroprotection by pharmacologic blockade of the GAPDH death
cascade. Proc Natl Acad Sci USA. 103:3887–3889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Bangaru ML, Park F, Hudmon A, McCallum JB
and Hogan QH: Quantification of gene expression after painful nerve
injury: Validation of optimal reference genes. J Mol Neurosci.
46:497–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Chuang DM and Ishitani R: A role for GAPDH
in apoptosis and neurodegeneration. Nat Med. 2:609–610. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Su BX, Chen X, Huo J, Guo SY, Ma R and Liu
YW: The synthetic cannabinoid WIN55212-2 ameliorates traumatic
spinal cord injury via inhibition of GAPDH/Siah1 in a CB2-receptor
dependent manner. Brain Res. 1671:85–92. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhai D, Chin K, Wang M and Liu F:
Disruption of the nuclear p53-GAPDH complex protects against
ischemia-induced neuronal damage. Mol Brain. 7:202014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li C, Feng JJ, Wu YP and Zhang GY:
Cerebral ischemia-reperfusion induces GAPDH S-nitrosylation and
nuclear translocation. Biochemistry (Mosc). 77:671–678. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Jiang X and Shen X: Knockdown of
miR-299-5p inhibits the progression of hepatocellular carcinoma by
targeting SIAH1. Bull Cancer. 105:873–883. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hui Z, Zhijun Y, Yushan Y, Liping C,
Yiying Z, Difan Z, Chunglit CT and Wei C: The combination of
acyclovir and dexamethasone protects against Alzheimer's
disease-related cognitive impairments in mice. Psychopharmacology
(Berl). 237:1851–1860. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Vogel J, Young A, Oxtoby N, Smith R,
Ossenkoppele R, Strandberg OT, Joie RL, Aksman LM, Grothe MJ,
Iturria-Medina Y, et al: Four distinct trajectories of tau
deposition identified in Alzheimer's disease. Nat Med. 27:871–881.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Knopman DS, Amieva H, Petersen RC,
Chételat G, Holtzman DM, Hyman BT, Nixon RA and Jones DT: Alzheimer
disease. Nat Rev Dis Primers. 7:332021. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Wasik U, Schneider G, Mietelska-Porowska
A, Mazurkiewicz M, Fabczak H, Weis S, Zabke C, Harrington CR,
Filipek A and Niewiadomska G: Calcyclin binding protein and Siah-1
interacting protein in Alzheimer's disease pathology: Neuronal
localization and possible function. Neurobiol Aging. 34:1380–1388.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Chen Y, Lian YJ, Ma YQ, Wu CJ, Zheng YK
and Xie NC: LncRNA SNHG1 promotes α-synuclein aggregation and
toxicity by targeting miR-15b-5p to activate SIAH1 in human
neuroblastoma SH-SY5Y cells. Neurotoxicology. 68:212–221. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Pang RT, Liu WM, Leung CO, Ye TM, Kwan PC,
Lee KF and Yeung WS: miR-135A regulates preimplantation embryo
development through down-regulation of E3 ubiquitin ligase seven in
absentia homolog 1A (SIAH1A) expression. PLoS One. 6:e278782011.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Schafer AR, Smith JL, Pryke KM, DeFilippis
VR and Hirsch AJ: The E3 ubiquitin ligase SIAH1 targets MyD88 for
proteasomal degradation during dengue virus infection. Front
Microbiol. 11:242020. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhou Y, Li L, Liu Q, Xing G, Kuai X, Sun
J, Yin X, Wang J, Zhang L and He F: E3 ubiquitin ligase SIAH1
mediates ubiquitination and degradation of TRB3. Cell Signal.
20:942–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Nagano Y, Fukushima T, Okemoto K, Tanaka
K, Bowtell DD, Ronai Z, Reed JC and Matsuzawa SI: Siah1/SIP
regulates p27(kip1) stability and cell migration under metabolic
stress. Cell Cycle. 10:2592–2602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Hara MR and Snyder SH: Nitric
oxide-GAPDH-Siah: A novel cell death cascade. Cell Mol Neurobiol.
26:527–538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Song Y, Xu Y, Pan C, Yan L, Wang ZW and
Zhu X: The emerging role of SPOP protein in tumorigenesis and
cancer therapy. Mol Cancer. 19:22020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Shah M, Stebbins JL, Dewing A, Qi J,
Pellecchia M and Ronai ZA: Inhibition of Siah2 ubiquitin ligase by
vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and
blocks melanoma tumorigenesis. Pigment Cell Melanoma Res.
22:799–808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Zhang H, Cao X, Wang J, Li Q, Zhao Y and
Jin X: LZTR1: A promising adaptor of the CUL3 family. Oncol Lett.
22:5642021. View Article : Google Scholar : PubMed/NCBI
|














