Introduction
Liver cancer is one of the most common malignant
tumors worldwide, with the highest incidence in East Asia and
Africa, and with the highest prevalence in China (1,2).
Hepatocellular carcinoma (HCC) is the main histological type,
accounting for approximately 75% of all liver cancers (3). Numerous factors can induce HCC,
including viruses (hepatitis B and hepatitis C virus), alcohol
consumption, and metabolic disorders (4). Treatment of HCC includes
radiotherapy, surgery, and chemotherapy. Even with the use of novel
drugs and improved surgical techniques, the mortality rate of
patients with HCC remains high due to its high recurrence and
metastasis (5). Thus, the
mechanisms of HCC recurrence and metastasis need to be determined
to improve patient outcome.
Cancer stem cells (CSCs) play a decisive role in
tumor initiation and growth, and they are the basis for tumor
recurrence, metastasis, and chemotherapy resistance (6). CSCs are a class of heterogeneous
cells with stem cell properties and strong tumorigenicity. CSCs can
be obtained by screening unique surface markers that include CD133,
epithelial cell adhesion molecule (EpCAM), CD73, Sox-9, Nanog,
Oct-4, and c-Myc (3,7–9).
Previous studies have demonstrated that inhibiting or blocking
surface markers of LCSCs can effectively interfere with the
self-renewal and proliferation of LCSCs, and reverse cell drug
resistance (3,7,8). For
example, knockdown of CD73 was revealed to significantly reduce
lenvatinib resistance and tumorigenicity of LCSCs (9), silencing of CD133 was demonstrated to
reduce the proliferative capacity of LCSCs (10), and downregulation of EpCAM
expression was shown to inhibit the self-renewal and tumorigenicity
of LCSCs (11). Thus, targeting
LCSCs has become a new strategy to treat liver cancer and improve
prognosis (12).
The gap junction is an intercellular protein channel
composed of connexins (Cxs). The channel is used for signal
exchange, which can inhibit tumors and is involved in overcoming
drug resistance in various solid tumors, including breast, lung,
and liver cancers (13). During
the initiation and progression of liver cancer, the expression
level of Cx32 has been revealed to be significantly reduced
(14). Previous studies by the
authors, demonstrated that upregulating the expression of Cx32 in
liver cancer cells could reverse doxorubicin resistance and reduce
invasion and metastasis of liver cancer cells (15,16).
Trosko et al reported that the reason for the emergence of CSCs may
be ascribed to the failure of the transcription of Cxs, or abnormal
gap junction function related to the post-translational
modification of Cxs (17). Based
on published studies and previous research by the authors, it is
inferred that decreased Cx32 expression induces the expansion of
LCSCs, giving rise to enhanced cell invasion, metastasis, and
chemotherapeutic drug resistance.
The phosphoinositide 3-kinase/protein kinase B
(PI3K/Akt) signaling pathway is important in tumor regulation.
Targeting the pathway can effectively inhibit tumor growth.
Numerous drugs targeting PI3K signal transduction have entered
clinical trials (18). Persistent
activation of the PI3K/Akt pathway has been demonstrated to
maintain the stemness of CSCs and promote their expansion (19). For instance, activation of PI3K/Akt
in colorectal cancer stem cells was shown to promote the migration,
invasion, and chemoresistance of spheroid cells (20). In liver cancer, a PI3K/Akt
inhibitor was also revealed to reduce the proportion and weaken the
expansion capacity of LCSCs (21).
In a previous study by the authors, Cx32 was demonstrated to
regulate the activity of the PI3K/Akt signaling pathway in liver
cancer cells. Overexpression of Cx32 inhibited the PI3K/Akt
pathway, while silencing the expression of Cx32 activated the
PI3K/Akt pathway (16). Given that
LCSCs are the source of tumor initiation, metastasis recurrence,
and therapy resistance (22), it
is proposed that Cx32 may affect the expansion of HCC stem cells
through the PI3K/Akt pathway, thus affecting the malignant
phenotypes of HCC cells.
In the present study, 85 patients who underwent
radical surgery for liver cancer were followed-up and the
association between Cx32 expression and patient survival was
analyzed. The Cancer Genome Atlas (TCGA) data was also used to
validate the results of the present study. The role of Cx32 in the
tumorigenicity of LCSCs was studied in nude mice. It was observed
that expansion of LCSCs was altered by modulating the expression of
Cx32 in vitro. By activating and inhibiting the PI3K/Akt pathway,
it was investigated whether the PI3K/Akt pathway mediated the
effects of Cx32 on the expansion of LCSCs. The present study
explored the mechanisms of Cx32 in the invasion, metastasis, and
drug resistance of liver cancer. New targets and prognostic factors
for liver cancer were proposed.
Materials and methods
Reagents
HCCLM3 and HepG2 cells were obtained from Shanghai
TongBai Biological Technology Co., Ltd., and were authenticated by
STR profiling. Dimethyl sulfoxide (DMSO; product no. 276855), SC-79
(product no. 123871) and LY294002 (product no. 528108), antibodies
to Cx32 (product no. MAB3069), and secondary antibodies including
HRP-conjugated goat anti-rabbit IgG (H + L) (cat. AP307P) and
HRP-conjugated goat anti-mouse IgG (H + L) (cat. AP308P) were
purchased from Sigma-Aldrich; Merck KGaA. Antibody to Sox-9 (cat.
no. PA5-81966) was obtained from Invitrogen; Thermo Fisher
Scientific, Inc. Antibodies to phosphorylated (p)-Akt (product code
ab81283), total Akt (product code ab8805), EpCAM (product code
ab223582), CD133 (product code ab222782), Nanog (product code
ab14959), Oct4 (product code ab181557), c-Myc (product code
ab32072), β-actin (product code ab8226) were obtained from Abcam.
Short hairpin RNA (shRNA)-Cx32 and overexpression (OE)-Cx32 plasmid
were obtained from Shanghai GenePharma Co., Ltd.
Patients and tumor samples
The specimens of 85 patients (63 males and 22
females; aged 25–78 years) with liver cancer were collected from
the Department of Hepatobiliary Surgery, the First Affiliated
Hospital of Bengbu Medical College (Bengbu, China) from January
2014 to December 2015. A total of 124 patients were followed-up to
December 2020, but 39 patients were lost to follow-up in the
present study. Inclusion criteria were as follows: A pathological
diagnosis of liver cancer, complete clinical data records, no
history of chemotherapy, radiation, immunotherapy, or other related
treatment, no history of other cancers, and no metastases from
other organs. The collected specimens included HCC tissues and the
corresponding paracancerous tissues. The paracancerous tissues were
non-cancerous liver tissues located at least 5 cm from the lesions.
This study was approved by the Ethics Committee of Bengbu Medical
College (approval no. 2020047).
Western blotting
Proteins of tissues and cells were extracted using a
lysis buffer containing a protease inhibitor (product no. P0013B;
Beyotime Institute of Biotechnology) and quantified using a BCA
protein assay kit (product no. P0012; Beyotime Institute of
Biotechnology). Total proteins (20 µg per lane) were separated
using 10% SDS-PAGE. The resolved proteins were transferred to a
PVDF membrane. The PVDF membrane was then incubated at 4°C for 1 h
with 1X Protein Free Rapid Blocking Buffer (cat. no. PS108P;
Shanghai Epizyme Biomedical Technology Co., Ltd.). Following
sealing, the membrane was incubated at 4°C overnight in the
corresponding primary antibody solution containing EpCAM (1:1,000),
CD133 (1:2,000), Nanog (1:2,000), Oct4 (1:2,000), Sox9 (1:1,000),
c-Myc (1:1,000), Cx32 (1:1,000), p-Akt (1:2,000), total Akt
(1:1,000), or β-actin (1:1,000). The membrane was washed the
following day and incubated with the secondary antibodies,
HRP-conjugated goat anti-rabbit IgG or HRP-conjugated goat
anti-mouse IgG (1:2,000 or 1:4,000), incubated at 4°C for 1 h.
Finally, the membrane was developed in the dark after incubating
with enhanced chemiluminescence reagent (product no. RPN2235;
Cytiva). The gray value of the protein bands was analyzed by ImageJ
software [version 1.46r; National Institutes of Health (NIH)].
Immunohistochemistry (IHC)
The expression of Cx32 and p-Akt was detected by
IHC. Paraffin-embedded specimens were cut into 5 µm-thick slices
using a microtome. The tissue sections were dewaxed and hydrated,
endogenous peroxidase was inactivated with 3% hydrogen peroxide,
antigen repair was performed by microwave irradiation. The tissue
sections were blocked at room temperature in 5% bovine serum
albumin (BSA)/50 mM PBS (pH 7.4) and incubated with the Cx32
antibody (1:100; product no. MAB3069; Sigma-Aldrich; Merck KGaA)
and p-Akt antibody (1:100; product code ab81283; Abcam) at 4°C
overnight. The sections were washed with phosphate-buffered
solution and incubated with secondary antibodies, goat anti-mouse
IgG HRP conjugate (1:200; product no. 12-349) and goat anti-rabbit
IgG HRP conjugate (1:200; product no. 12-348; both from
Sigma-Aldrich; Merck KGaA), followed by color development with 3,3′
diaminobenzidine (DAB) kit (product no. D3939; Sigma-Aldrich, Merck
KGaA), redyeing, dehydration, and sealing. Each section was
observed and images were captured using model IX71 optical
microscope (Olympus Corporation). All imaged sections stained by
immunohistochemistry were analyzed using ImageJ software version
1.46r (NIH). The criteria for staining cells were the same as those
previously reported (15).
Spheroid formation assay
Agarose (1%) was spread on a 10-cm cell culture dish
and allowed to coagulate. A total of 3×105 cells/ml were
plated per well of a gel-coated petri dish and maintained with B27
serum-free microsphere medium (Gibco; Thermo Fisher Scientific,
Inc.). N2 (Gibco; Thermo Fisher Scientific, Inc.), 20 ng/ml
epidermal growth factor, and 20 ng/ml basic fibroblast growth
factor (Invitrogen; Thermo Fisher Scientific, Inc.) were added to
DMEM/F12 basal culture medium (Gibco; Thermo Fisher Scientific,
Inc.). After 3–4 days, the cells were supplemented with microsphere
medium and cultured for another 4–6 days. Images of the spheroids
were captured and counted by fluorescence microscope (Olympus
Corporation).
Mining of the cancer genome atlas
(TCGA) database
TCGA Liver Hepatocellular Carcinoma (LIHC) data were
obtained from UCSC Xena (URL: http://xena.ucsc.edu). Screened samples expressed GJB1
(Cx32) and had sufficient survival information. The R language
Survival package (https://CRAN.R-project.org/package=survival) was used
to evaluate the association between Cx32 expression level and
survival outcomes, such as overall survival (OS), disease-free
interval (DFI), progression-free interval (PFI), and
disease-specific survival (DSS).
Nude mice xenograft tumor model
All animal experiments were conducted in accordance
with the regulations approved by the Laboratory Animal Management
and Ethics Committee of Bengbu Medical College. A total of 64 male
BALB/c mice (4 weeks of age, 20–22 g) were purchased from Beijing
Weitong Lihua Experimental Animal Technology Co., Ltd. They were
bred in individually ventilated cages (IVC) with specific
pathogen-free (SPF) conditions in a 12-h light/dark cycle, 40–70%
relative humidity, and controlled temperature (24±2°C). During the
experiment, all mice had free access to standard mice chow and
water. Following one-week acclimation, the mice were divided
randomly into two groups: HCCLM3 overexpression empty vector
(HCCLM3 EV) group and HCCLM3 overexpression (HCCLM3 OE) group.
Different numbers of HCC cells (1×103, 5×103,
1×104, and 5×104) were suspended in Matrigel
(50%; BD Biosciences) and injected subcutaneously into the nude
mice to observe tumor growth. Finally, the mice were divided into
eight groups, with eight mice in each group. Two months later, the
mice were euthanized by cervical dislocation under anesthesia (1%
pentobarbital sodium, 80 mg/kg, ip) and tumorigenicity of the cells
in vivo was evaluated. The evaluation criterion was negative if no
obvious tumor nodules were found at the injection site of the nude
mice. The animal experiments were approved by the Ethics Committee
of Bengbu Medical College (approval no. 2020090).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA of cells was extracted using
TRIzol® (cat. no. 15596026; Invitrogen; Thermo Fisher
Scientific, Inc.). RNA purity was evaluated by Agilent Bioanalyzer
2100 (Agilent Technologies, Inc.) before RT-qPCR analysis.
Subsequently, the cDNA was reverse-transcribed using TaqMan Reverse
Transcription reagent (cat. no. N8080234; Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol, and was
detected by fluorescence qPCR using SYBR™ Green PCR
Master Mix (cat. no. 4309155; Invitrogen; Thermo Fisher Scientific,
Inc.). PCR amplification conditions were as follows:
Pre-denaturation at 95°C for 15 sec, denaturation at 95°C for 5
sec, and annealing and extension at 62°C for 30 sec. These steps
were repeated for 45 cycles. The absorbance value was read at each
extension stage. β-actin was used as the reference gene. The
relative gene expression levels of each group were calculated
according to quantification cycle (Cq) value (23). Primers of β-actin, Sox-9, CD133,
EpCAM, Nanog, Oct4 and c-Myc were obtained from Shanghai Shenggong
Biological Technology Co., Ltd. Primers for each gene are listed in
Table I.
 | Table I.Primers used for quantitative
PCR. |
Table I.
Primers used for quantitative
PCR.
|
| Primer
sequences(5′-3′) |
|---|
|
|
|
|---|
| Gene name | Forward primer | Reverse primer |
|---|
| β-actin |
CATCCACGAAACTACCTTCAACTCC |
GAGCCGCCGATCCACACG |
| Sox-9 |
AGGAAGTCGGTGAAGAACGG |
AAGTCGATAGGGGGCTGTCT |
| CD133 |
TGGATGCAGAACTTGACAACGT |
ATACCTGCTACGACAGTCGTGGT |
| EpCAM |
CGCAGCTCAGGAAGAATGTG |
TGAAGTACACTGGCATTGACGA |
| Nanog |
AATACCTCAGCCTCCAGCAGATG |
TGCGTCACACCATTGCTATTCTTC |
| Oct4 |
GTGTTCAGCCAAAAGACCATCT |
GGCCTGCATGAGGGTTTCT |
| c-Myc |
CCCTCCACTCGGAAGGACTA |
GCTGGTGCATTTTCGGTTGT |
Cell transfection
Negative control (NC)-shRNA lentivirus and
Cx32-shRNA lentivirus were prepared by inserting NC-shRNA and
Cx32-shRNA into the respective lentivirus vectors. Lentivirus were
produced in 293T cells (Shanghai TongBai Biological Technology Co.,
Ltd.) using a second generation lentiviral system (Invitrogen;
Thermo Fisher Scientific, Inc.). Briefly, 4×106 293T
cells were seeded in a 100-cm culture dish at 24 h before
transfection. Subsequently, 6 µg lentiviral construct, 3 µg psPAX2
(packaging plasmid), and 1.5 µg pDM2.G (envoloping plasmid; both
from Sangon Biotech Co., Ltd.) were co-transfected into 293T cells
(the mixed ratio was pDM2.G: psPAX2: lentivirus, 1:2:4) using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Following transfection for 6 h at 37°C in a
CO2 incubator, the medium was replaced with normal
culture medium. After 48 h, the lentivirus-containing supernatants
were harvested, centrifuged at 800 × g for 4 min to pellet cell
debris. The HepG2 cells were plated at 30–50% confluence,
transfected with lentivirus supernatants at a multiplicity of
infection (MOI) of 20 TU/ml. A total of 24 h after transfection,
the transformed cells were screened by doxycycline (final
concentration, 2 µg/ml) for 7–10 days. The transfection results
were identified by western blotting. The sequences for the shRNA
targeting Cx32 (shCx32) were: shRNA1,
5′-CCGGGCCGTCTTCATGTATGTCTTTCTCGAGCCCTCACTACATGAAGACGGCTTTTTG-3′;
and shRNA2,
5′-CCGGCGTTTGCTATGACCAATTCTTCTCGAGAAGAATTGGTCATAGCAAACGTTTTTG-3′;
and the sequence for the NC was:
5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′.
Statistical analysis
SPSS 23.0 (IBM Corp.) and SigmaPlot 10.0 software
(Jandel Corporation; Systat Software, Inc.) were used for
statistical analysis. Data represented the mean ± SEM from three
independent experiments. Parametric data were analyzed using
unpaired t-test or one-way ANOVA with Tukey's multiple comparison
test for post hoc comparison. The association between Cx32
expression and clinicopathological characteristics was determined
by Pearson χ2 test. The chi-square test and Fisher's
exact probability tests were used as appropriate to evaluate the
significance of differences in data between groups. Survival curves
were analyzed using the Kaplan-Meier method, and significance was
assessed by the Gehan-Breslow-Wilcoxon test or log-rank test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cx32 predicts poor prognosis in HCC
patients
TCGA data analysis showed that the OS of patients
with high expression of Cx32 was significantly higher than those
with low expression of Cx32 (Fig.
1A). The same results were observed for the DSS, DFI and PFI
(Fig. 1B-D). The data of 85
patients with liver cancer was collected and the expression of Cx32
was assessed in cancer tissues and corresponding paracancerous
tissues by IHC. The positive expression rate of Cx32 was 41.2%
(35/85) in cancer tissues and 82.4% (70/85) in paracancerous
tissues (Fig. 1F). The rate in
paracancerous tissue was significantly higher than the rate in
cancer tissues. The 85 patients were followed-up for 5 years
(Table II). Survival analysis
revealed an overall survival rate of 46.39% in the Cx32-positive
group and 10.31% in the Cx32-negative group (Fig. 1G; P=0.007), indicating a
significant association of Cx32 with prognosis of HCC.
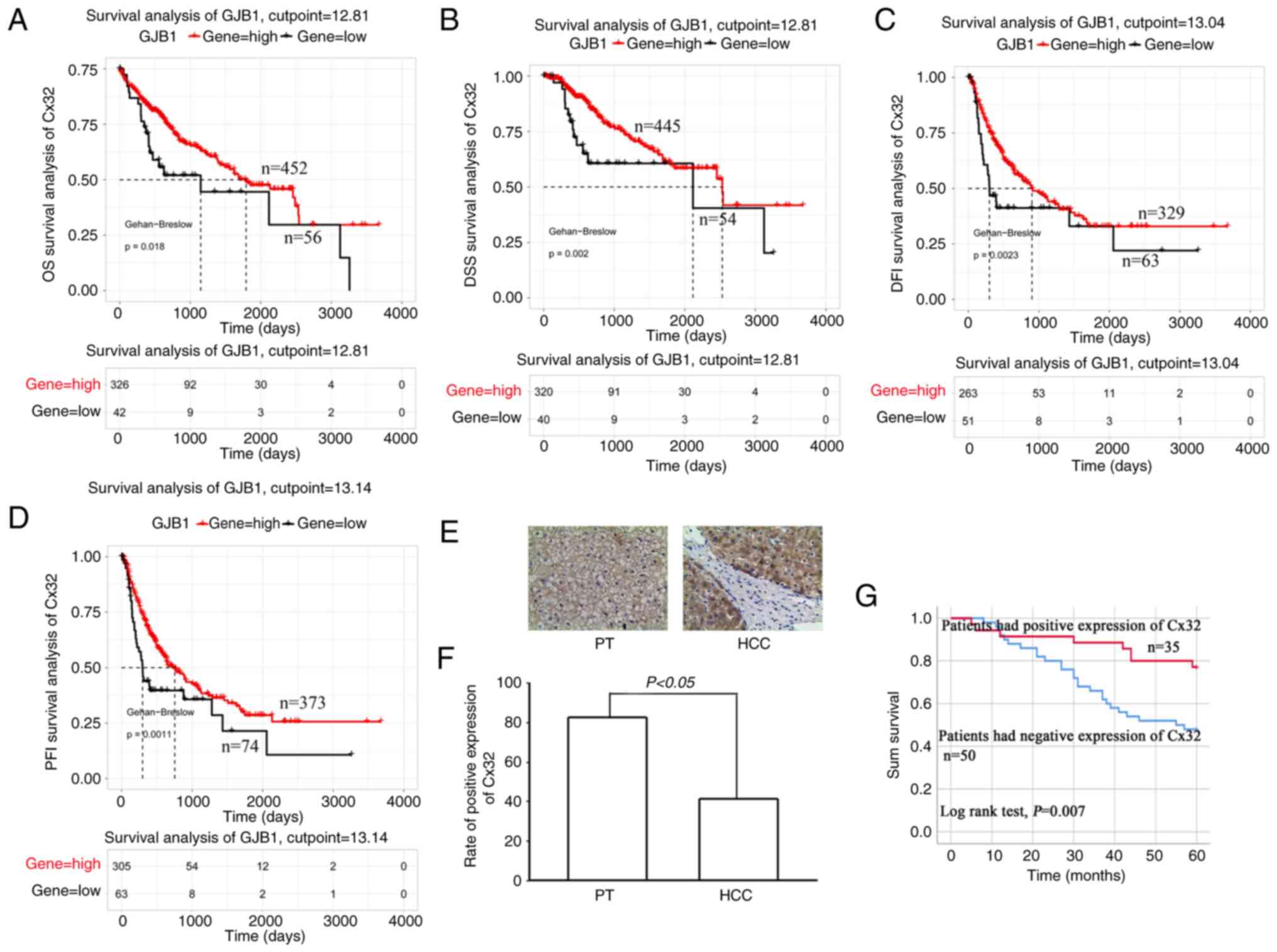 | Figure 1.Cx32 predicts poor prognosis in HCC
patients. (A-D) Survival analysis of the Cx32 gene in patients with
HCC based on TCGA database. Survival is expressed as OS, DSS, DFI
and PFI. (E) Immunohistochemical detection of the expression level
of Cx32 in HCC specimens and corresponding PT. Magnification, ×200.
(F) Rate of positive expression of Cx32 in HCC specimens and PT.
(G) Kaplan-Meier survival plots for all HCC patients with positive
expression of Cx32 and negative expression of Cx32. Log rank
testing was used to analyze the data; P<0.05. Cx32, connexin 32;
HCC, hepatocellular carcinoma; TCGA, The Cancer Genome Atlas; OS,
overall survival; DSS, disease-specific survival; DFI, disease-free
interval; PFI, progression-free interval; PT, paracarcinoma
tissues. |
| Table II.Baseline characteristics of 85
patients with HCC. |
Table II.
Baseline characteristics of 85
patients with HCC.
|
|
|
| Cx32 expression in
HCC |
|
|---|
|
|
|
|
|
|
|---|
| Clinical
characteristics | Variables | No. of
patients | Positive
expression | Negative
expression | P-value |
|---|
| Age (years) | ≤56 | 44 | 22 | 21 | 0.061 |
|
| >56 | 41 | 12 | 29 |
|
| Sex | Female | 22 | 7 | 15 | 0.306 |
|
| Male | 63 | 27 | 35 |
|
| Alpha-fetoprotein
(ng/ml) | ≤400 | 25 | 10 | 15 | 0.0485 |
|
| >400 | 60 | 24 | 35 |
|
| HBV infection | Yes | 51 | 22 | 34 | 0.462 |
|
| No | 28 | 12 | 16 |
|
| Liver
cirrhosis | Yes | 66 | 28 | 37 | 0.325 |
|
| No | 19 | 6 | 13 |
|
| Histological
differentiation | Poorly | 13 | 6 | 7 | 0.559 |
|
| Moderately | 64 | 24 | 39 |
|
|
| Well | 8 | 4 | 4 |
|
| Tumor capsule | Yes | 16 | 3 | 13 | 0.071 |
|
| No | 69 | 31 | 37 |
|
| Tumor size
(cm) | ≤5 | 46 | 21 | 25 | 0.288 |
|
| >5 | 39 | 13 | 25 |
|
| Vital status | Deceased | 34 | 16 | 17 | 0.236 |
|
| Living | 51 | 18 | 33 |
|
| Follow-up time
(months) | ≤60 | 34 | 16 | 17 | 0.229 |
|
| >60 | 51 | 18 | 33 |
|
Cx32 expression is downregulated in
LCSCs
CD133+ and EpCAM+ liver CSCs,
and CD133− and EpCAM− non-liver CSCs were
sorted from primary HCC tissues. RT-qPCR was used to determine the
level of Cx32. Cx32 was significantly reduced in CD133+
and EpCAM+ liver CSCs (Fig.
2A and B). Cx32 mRNA expression was also decreased in HCC
spheres derived from human primary HCC cells, compared with
adherent cells (Fig. 2C).
Consistent with these results, Cx32 was reduced in
CD133+ and EpCAM+ liver CSCs sorted from
spheres of HepG2 and HCCLM3 cells (Fig. 2D and E). Moreover, compared with
the attached cells, Cx32 was obviously decreased in the
self-renewing spheroids (Fig. 2F).
These results indicated that Cx32 regulated the expansion of
LCSCs.
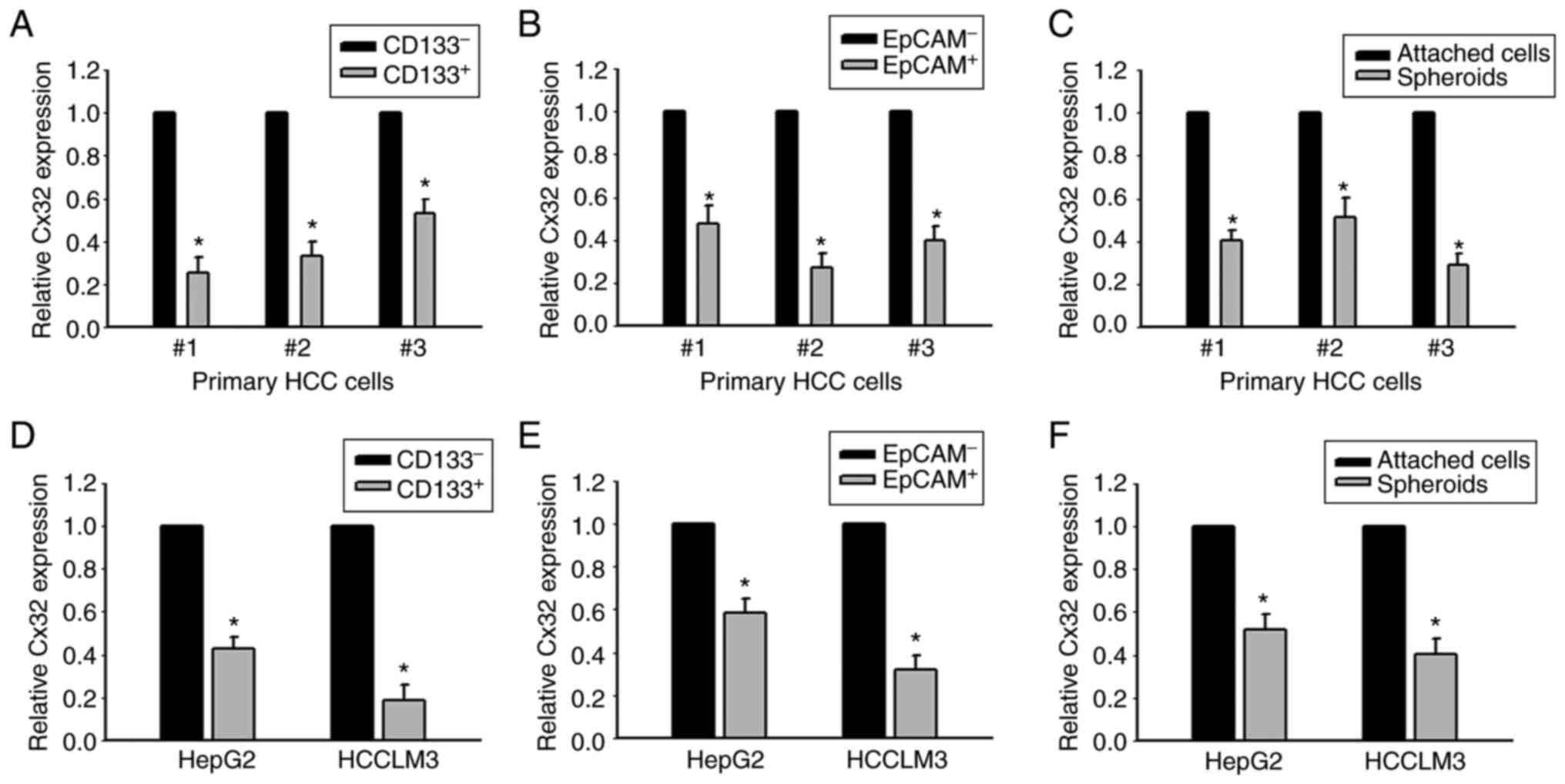 | Figure 2.Expression of Cx32 mRNA is decreased
in HCC cell populations with higher stem cell characteristics. (A
and B) Cx32 mRNA in CD133+, EpCAM+ liver CSCs and CD133-,
EpCAM-non-liver CSCs sorted from primary HCC cells was analyzed by
qPCR. *P<0.05 vs. the CD133- or EpCAM- group. (C) Cx32 mRNA in
HCC spheres and attached cells obtained from primary HCC cells was
detected by qPCR, *P<0.05 vs. the attached cells. (D and E)
Expression level of Cx32 in CD133+, EpCAM+ liver CSCs and CD133-,
EpCA- non-liver CSCs sorted from spheres of HepG2 and HCCLM3 cells
was analyzed by qPCR. *P<0.05 vs. the CD133- or EpCAM- group.
(F) Cx32 mRNA levels were detected in spheres of HepG2 and HCCLM3
cells and corresponding adherent cells. *P<0.05 vs. the attached
cells. Cx32, connexin 32; HCC, hepatocellular carcinoma; EpCAM,
epithelial cell adhesion molecule; CSCs, cancer stem cells; qPCR,
quantitative PCR. |
Cx32 regulates the expansion of
LCSCs
To explore the effect of Cx32 on the expansion of
LCSCs, HepG2 cells (that express a high level of Cx32) and HCCLM3
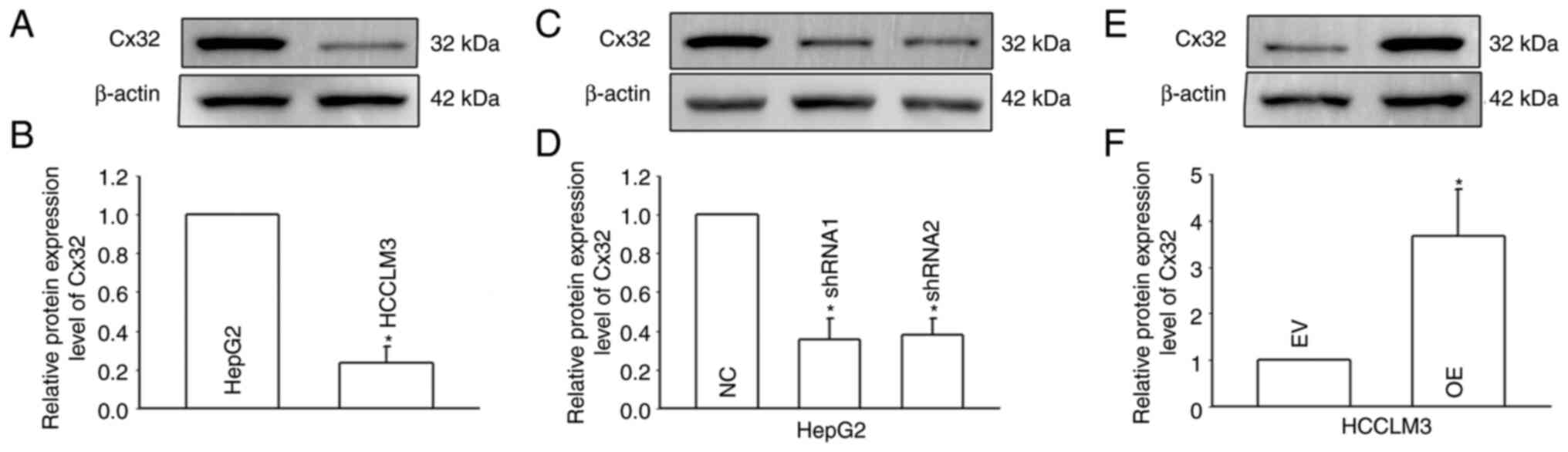
(that express a low level of Cx32) were used (Fig. 3A and B), and Cx32 was silenced in
HepG2 cells and overexpressed in HCCLM3 cells (Fig. 3C-F). RT-qPCR and western blotting
were used to observe the expression levels of stemness-associated
genes, including EpCAM, CD133, Sox9, Nanog, Oct4, and c-Myc.
Self-renewal ability was detected by the spheroid formation assay.
Cx32 knockdown in HepG2 cells significantly enhanced the mRNA
expression of stemness-associated genes (Fig. 4A), and western blotting results
were consistent with those of RT-qPCR (Fig. 4C and D). Moreover, after Cx32 was
silenced in HepG2 cells, the numbers of spheres were significantly
increased (Fig. 4G and H). By
contrast, overexpression of Cx32 in HCCLM3 cells significantly
decreased the expression of stemness-associated genes (Fig. 4B, E, and F) and cells formed
smaller and fewer spheroids than the control cells (Fig. 4I and J). In vivo limiting dilution
assay results showed that overexpression of Cx32 markedly
downregulated the tumorigenicity capacity of HCCLM3 cells (Fig. 4K-N). Collectively, the results
indicated that Cx32 regulated expansion of LCSCs.
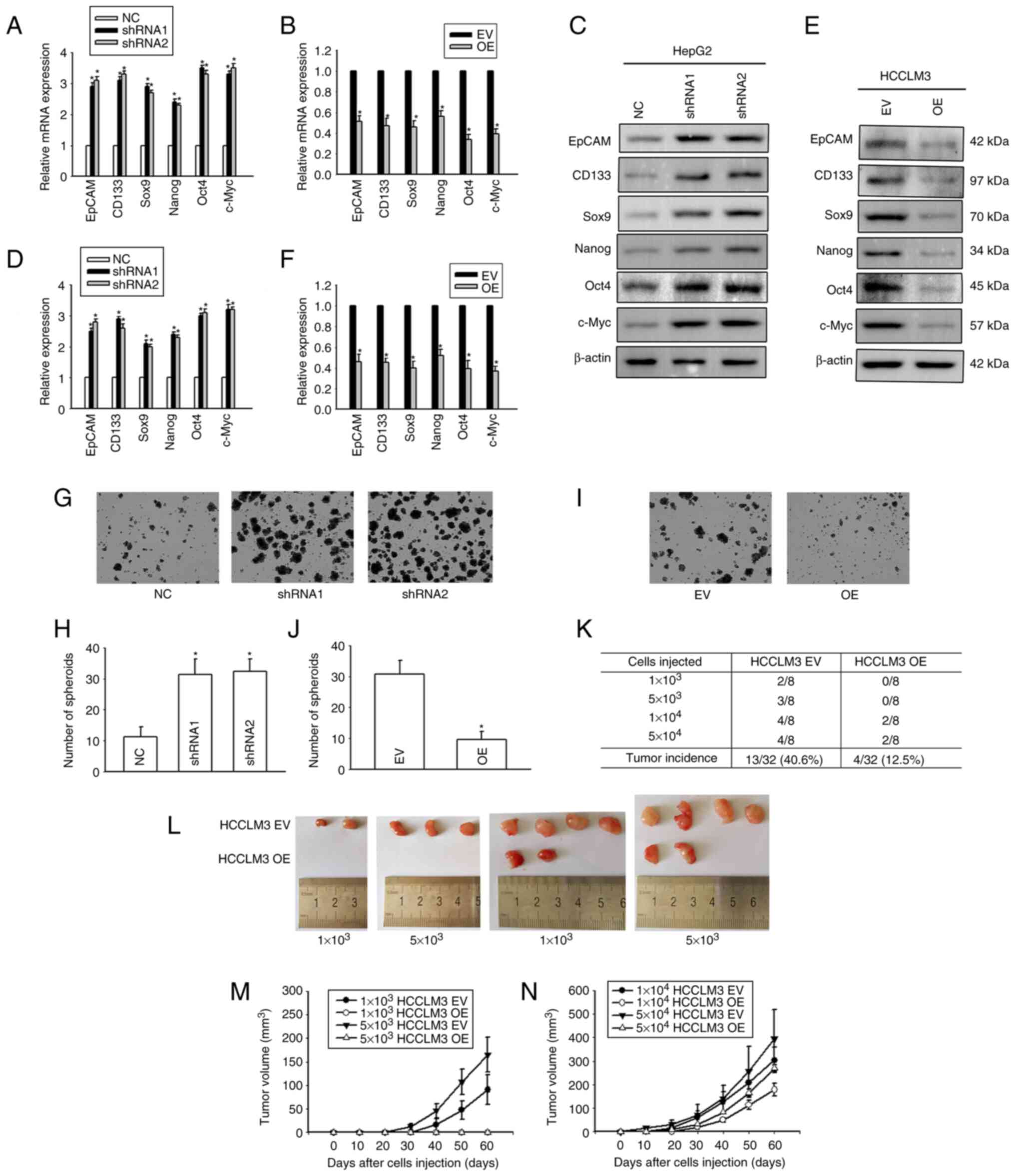 | Figure 4.Cx32 regulates the expansion of
LCSCs. (A) Effect of Cx32 knockdown on the mRNA expression levels
of stemness-associated genes in HepG2 cells. (B) Effect of Cx32
overexpression on the mRNA expression levels of stemness-associated
genes in HCCLM3 cells. (C and D) The expression levels of
stemness-associated genes were examined by western blotting when
HepG2 cells were transfected with shRNA-Cx32. The proteins were
normalized with β-actin. (E and F) The expression levels of
stemness-associated genes were observed when HCCLM3 cells
overexpressed Cx32. The proteins were normalized with β-actin. (G
and H) Effect of Cx32 knockdown on the sphere-forming capacities of
HepG2 cells. Magnification, ×200. The bar graph shows the average
number of spheres >100 µm in diameter. (I and J) Effect of Cx32
overexpression on the sphere-forming capacities of HCCLM3 cells.
Magnification, ×200. The bar graph shows the average number of
spheres >100 µm in diameter. For the aforementioned images, the
error bars represent the mean ± SEM of three independent
experiments; *P<0.05 vs. the NC group; or *P<0.05 vs. the
overexpression EV group. (K and L) Efficiency of tumor formation of
Cx32 overexpression in HCCLM3 cells. (M) Efficiency of tumor
formation of HCCLM3 EV and HCCLM3 OE cells; number of injected
cells: 1×103 and 5×103, n=8. (N) Efficiency of tumor formation of
HCCLM3 EV and HCCLM3 OE cells; number of injected cells: 1×104,
5×104, n=8. Cx32, connexin 32; LCSCs, liver cancer stem cells;
shRNA, small interfering RNA; NC, negative control; EV empty
vector; OE, overexpression; EpCAM, epithelial cell adhesion
molecule. |
Regulation of the PI3K/Akt signaling
pathway by Cx32
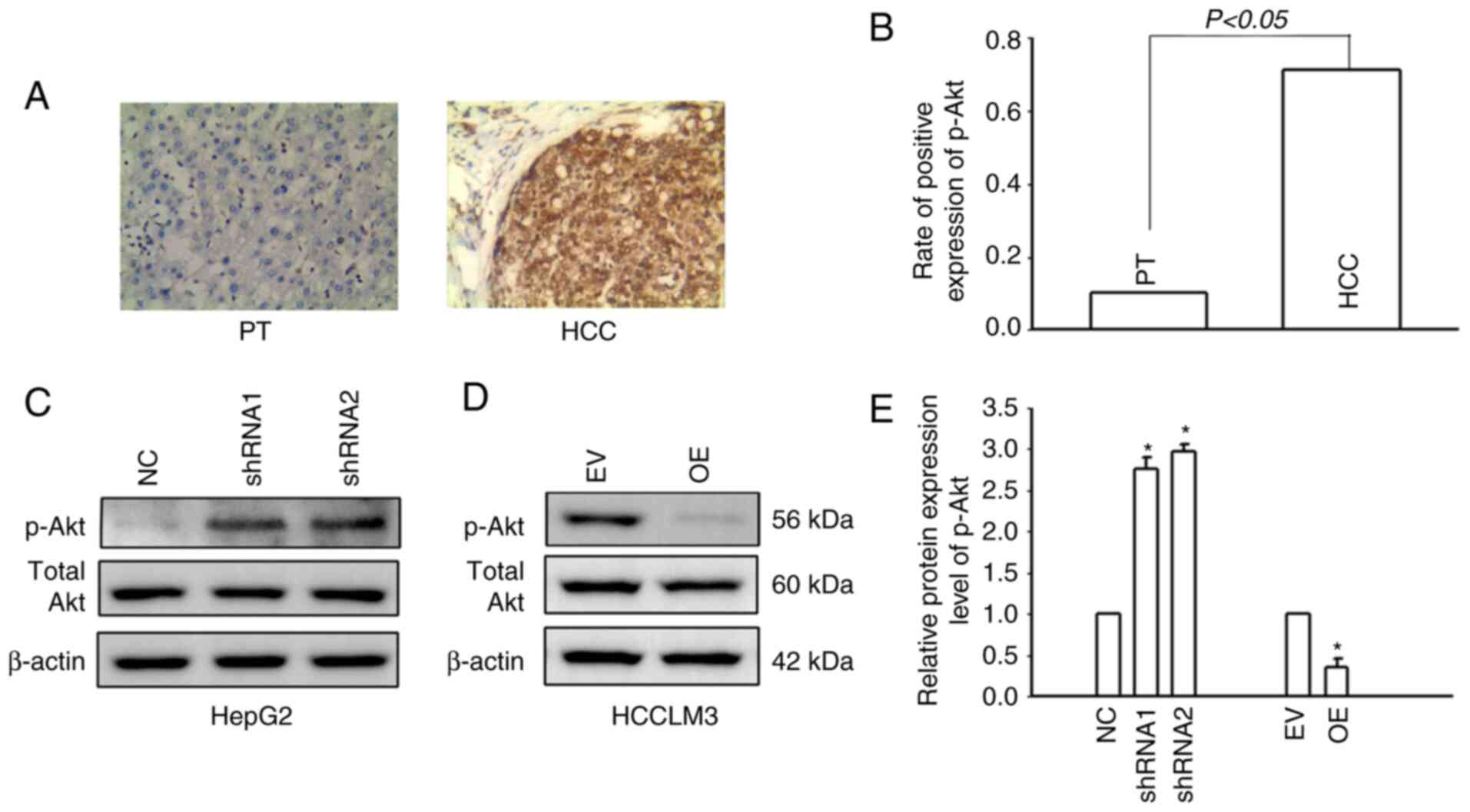
It was previously confirmed by the authors that Cx32
regulates the PI3K/Akt pathway in HCC (16). In the present study, IHC was used
to detect the expression of p-Akt in HCC tissues and corresponding
paracancerous tissues. Only 9.4% (8/85) of patients with HCC had
positive expression of p-Akt in the corresponding paracancerous
tissues, while the positive expression rate of p-Akt in HCC tissues
was 71.8% (61/85) (Fig. 5A and B).
The results indicated that p-Akt was activated in HCC tissues. The
PI3K/Akt signaling pathway was also activated in HCCLM3 cells. Cx32
was silenced in HepG2 cells and overexpressed in HCCLM3 cells.
Western blotting was used to examine PI3K/Akt pathway activity.
Cx32 knockdown in HepG2 cells significantly increased the
expression level of p-Akt, whereas Cx32 overexpression
significantly decreased the expression level of p-Akt (Fig. 5C and D). These findings suggested
that Cx32 regulated the activity of the PI3K/Akt signaling pathway
in HCC cells.
Cx32 regulates LCSC expansion by the
PI3K/Akt signaling pathway
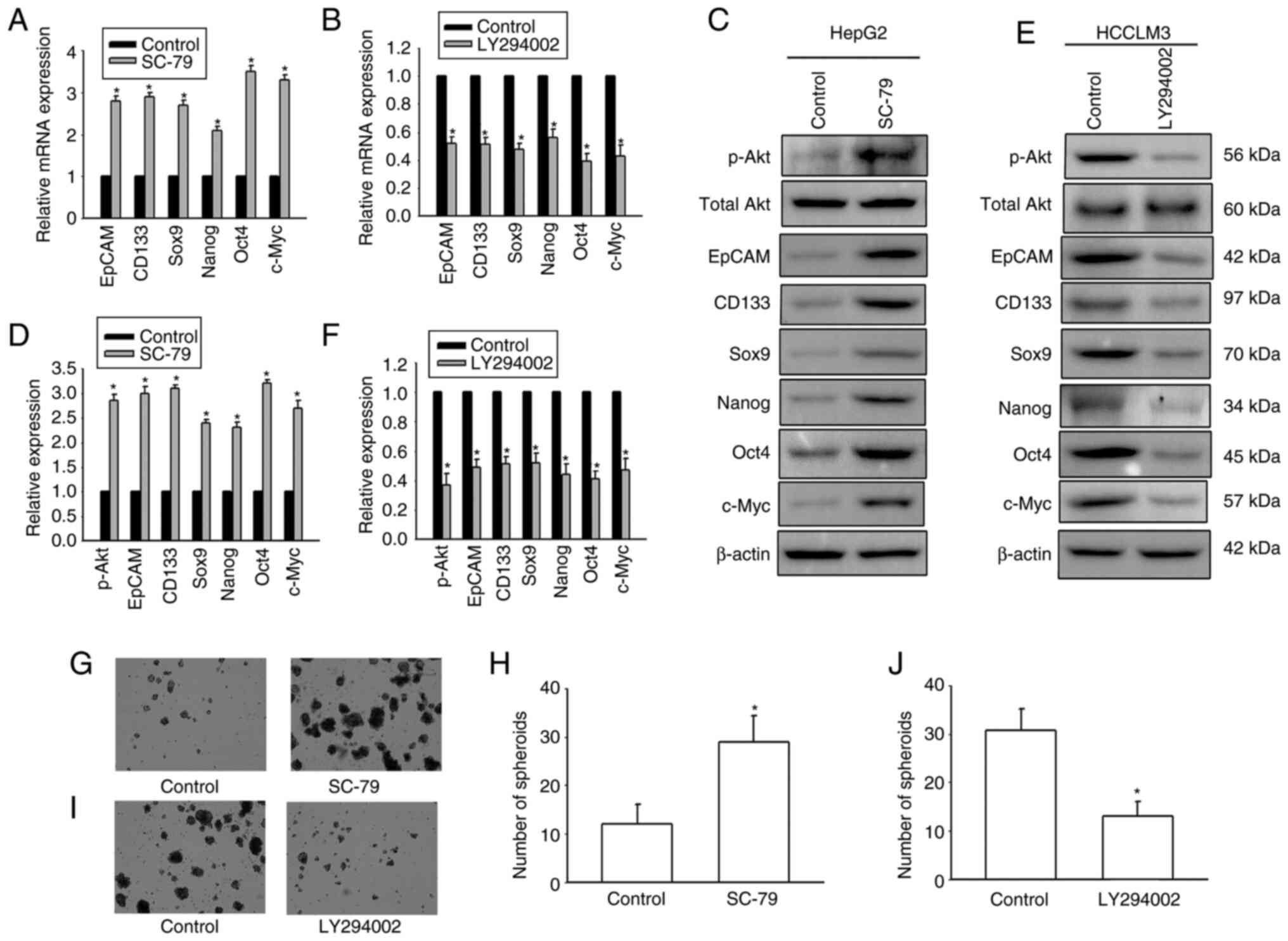
It was next investigated whether Cx32 affected
expansion of LCSCs by regulating the PI3K/Akt signaling pathway.
HepG2 cells were exposed to the AKT agonist SC-79 to activate the
PI3K/Akt signaling pathway. HCCLM3 cells were exposed to the Akt
antagonist LY294002 to inhibit the PI3K/Akt pathway. SC-79
significantly enhanced the expression of stemness-associated genes
and the numbers of spheres were significantly upregulated (Fig. 6). However, LY294002 obviously
decreased the expression of stemness-associated genes, and hepatoma
cells formed small and fewer spheroids. The results suggested that
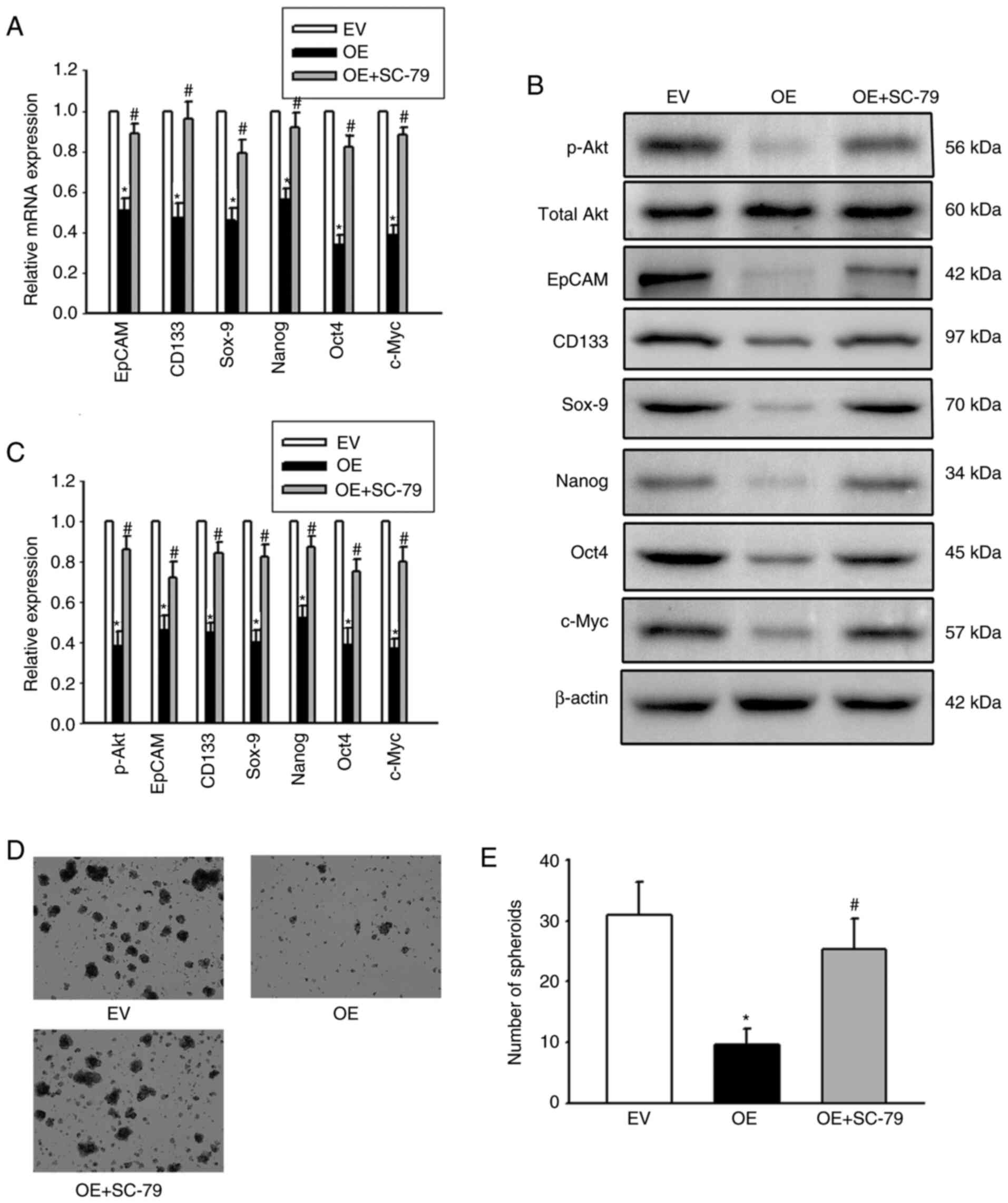
PI3K/Akt regulated expansion of LCSCs. In addition, OE-Cx32-HCCLM
cells that stably expressed Cx32 were stimulated by the AKT agonist
SC-79 and the effect of SC-79 on expansion of LCSCs in these cells
was observed. Overexpression of Cx32 in HCCLM3 cells obviously
decreased the expression of stemness-associated genes, and cells
formed smaller and fewer spheroids, but SC-79 reversed these
effects (Fig. 7). These results
indicated that Cx32 regulated expansion of LCSCs by the PI3K/Akt
signaling pathway.
Discussion
Considering the critical role of LCSCs in tumor
initiation, recurrence, metastasis, and drug resistance (22), the mechanisms underlying the
expansion of LCSCs need to be comprehensively studied and
determined. The present findings from experiments that were
performed and from TCGA data demonstrated the positive association
between the expression level of Cx32 and the prognosis of liver
cancer. Moreover, in vitro and in vivo data indicated that
upregulating the expression of Cx32 inhibited the expansion of
LCSCs, which was mediated by downregulated activity of the PI3K/Akt
pathway. These novel findings revealed a potential mechanism by
which Cx32 regulated the expansion of LCSCs.
A total of 85 patients with liver cancer were
followed-up for 5 years after radical surgery. In these patients,
low expression of Cx32 predicted a worse overall survival. This
finding was validated by TCGA data. In addition, TCGA data revealed
that high expression of Cx32 was associated with improved DSS, DFI,
and PFI compared with low expression of Cx32. These findings
implicated Cx32 as a prognostic biomarker for liver cancer.
Cx32 is a Cx that is mainly expressed in
hepatocytes. A previous study by the authors confirmed that Cx32
reverses doxorubicin resistance by inhibiting the
epithelial-mesenchymal transition (EMT) of HCC cells, and reduces
the invasion and metastatic ability of HCC cells (15). EMT is a process in which epithelial
cells are transformed to mesenchymal cells with a high migration
potential, which promotes the formation of CSCs and maintains their
stemness (24). EMT results in
enhanced tumor cell invasion, metastasis, and acquisition of drug
resistance (25). Dormant tumor
cells undergoing EMT can acquire a CSC-like phenotype and
facilitate metastasis and proliferation (26). The previous study by the authors
revealed that silencing the expression of Cx32 in HepG2 cells
induced EMT of liver cancer cells (15). Thus, it was hypothesized that
downregulation of Cx32 expression may promote the expansion
maintain stemness of LCSCs. In the present study, the expression
level of Cx32 in LCSCs was significantly decreased compared with
non-LCSCs. Furthermore, compared with attached cells, Cx32 was
obviously decreased in the self-renewing spheroids. The previous
and present findings suggest that Cx32 may regulate LCSC
expansion.
Kawasaki et al reported that Cx32 promotes the
expansion of LCSCs when translocated from the cell membrane to the
cytoplasm (27). During the
initiation and development of HCC, the total expression level of
Cx32 is significantly reduced and Cx32 is internalized by being
translocated from the cell membrane to the cytoplasm (14). The results of IHC confirmed that
Cx32 was mainly located in the cytoplasm in liver cancer cells.
When the expression of Cx32 was silenced in HepG2 cells, the mRNA
and protein levels of liver cancer stemness-related genes CD133,
EpCAM, CD73, Sox-9, Nanog, Oct-4, and c-Myc were all significantly
upregulated. In addition, the number of spheroids was also
increased, indicating enhanced expansion of LCSCs. By contrast, the
overexpression of Cx32 in HCCLM3 cells significantly inhibited the
expansion of LCSCs in vitro and in vivo. The present and previous
results by the authors support the theory that Cx32 may regulate
the expansion of LCSCs in either a channel-related, in which Cx32
located in the cell membrane to form a gap junction channel is
downregulated, or in a non-channel-related manner, involving
upregulation of Cx32 located in the cytoplasm (27). The results of the present study
demonstrated that restoring Cx expression and rebuilding the gap
junction is an important strategy to inhibit the expansion of
LCSCs.
Gap junctions are dynamic protein channels, and
functional gap junctions on the membrane are constantly
‘eliminated’ through endocytosis and degradation by lysosomes, and
are replaced by newly synthesized Cxs (28). Cells modulate the rate of Cx
degradation according to the microenvironment, thereby altering the
protein channels in the membrane (28,29).
Acetylation and ubiquitination of Cx32 are the main factors
determining the turnover rate of Cx32. Acetylation stabilizes Cx32
and ubiquitination promotes the degradation of Cx32. Cx32
acetylation may negatively regulate its ubiquitination (30). The effects of acetylation and
ubiquitination of Cx32 on the expansion of LCSCs will next be
studied, to develop new strategies to inhibit the expansion of
LCSCs.
Cx32 regulates the PI3K/Akt signaling pathway in
liver cancer cells (16).
Activation of the PI3K/Akt signaling pathway strengthens the
stemness of CSCs (31–33) and induces drug resistance (32). Therefore, in the present study, the
role of the PI3K/Akt signaling pathway in the regulation of the
expansion of LCSCs by Cx32 was investigated. The expression of
p-Akt in liver cancer tissues was detected and it was determined
that the PI3K/Akt pathway was continuously activated in liver
cancer tissues compared with adjacent tissues. It was further
demonstrated that Cx32 decreased the expansion of LCSCs by
inhibiting the activity of the PI3K/Akt pathway in vitro. These
results shed light on the potential of Cx32 and PI3K/Akt as targets
against liver cancer and drug resistance. PI3K/Akt maintains the
stemness of CSCs and promotes their expansion by mTOR, NF-κB, or
SOX2 (34–36). It was previously confirmed that
Cx32 inhibits PI3K/Akt/NF-κB pathway activity in HCC (16). The investigation of whether
PI3K/Akt/mTOR, PI3K/Akt/NF-κB, and/or PI3K/Akt/SOX2 pathways are
involved in Cx32-regulated expansion of LCSCs, is planned in a
future study.
In conclusion, the low expression of Cx32 was
associated with a worse prognosis for liver cancer. This
association needs to be validated in cohorts involving a larger
number of patients. A strategy to inhibit the expansion of LCSCs by
recovering the expression of Cx32, was also proposed. In addition,
the data illustrated the key role of PI3K/Akt in the regulation of
the expansion of LCSCs by Cx32. The present study provided
experimental evidence that targeting Cx32 potentially inhibits the
invasion and metastasis of liver cancer cells and reverses drug
resistance.
Acknowledgements
We would like to thank Mrs Yingying Huang
(Department of Pharmacy, The First Affiliated Hospital of Bengbu
Medical College) for revising the manuscript.
Funding
The present study was supported by the National Natural Science
Foundation of Anhui (grant nos. 1908085MH293 and 1808085QH269), the
512 Talent Cultivation Plan Foundation of Bengbu Medical College
(grant no. by51201320), the Natural Science Foundation of the
Provincial Education Department of Anhui (grant no. KJ2021A0689),
and the Foundation of Bengbu Medical College (grant no.
2020byzd089).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL performed the in vitro study, collected the liver
cancer tissues and paracancerous tissues, and wrote the manuscript.
BW performed the in vitro study and also collected the liver cancer
tissues and paracancerous tissues. BQ performed the animal study.
GJ analyzed the data and constructed the graphs. MQ designed the
study and revised the manuscript. MY designed the study, wrote the
manuscript, and revised the manuscript. HL and BW confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Ethics approval (approval no. 2020047) was obtained
from the Ethics Committee of Bengbu Medical College (Bengbu, China)
and written informed consent was obtained from each patient. The
animal experiments were approved (approval no. 2020090) by the
Ethics Committee of Bengbu Medical College.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
financial interests or personal relationships that could have
influenced the research reported in the present study.
References
|
1
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McGlynn KA, Petrick JL and El-Serag HB:
Epidemiology of hepatocellular carcinoma. Hepatology. 73 (Suppl
1):S4–S13. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu YC, Yeh CT and Lin KH: Cancer stem
cell functions in hepatocellular carcinoma and comprehensive
therapeutic strategies. Cells. 9:13312020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pinheiro PS, Medina HN, Callahan KE, Jones
PD, Brown CP, Altekruse SF, McGlynn KA and Kobetz EN: The
association between etiology of hepatocellular carcinoma and
race-ethnicity in Florida. Liver Int. 40:1201–1210. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang A, Ju W, Yuan X, Han M, Wang X, Guo
Z, Wei X, Wang D, Zhu X, Wu L and He X: Comparison between liver
resection and liver transplantation on outcomes in patients with
solitary hepatocellular carcinoma meeting UNOS criteria: A
population-based study of the SEER database. Oncotarget.
8:97428–97438. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou G, Latchoumanin O, Bagdesar M,
Hebbard L, Duan W, Liddle C, George J and Qiao L: Aptamer-based
therapeutic approaches to target cancer stem cells. Theranostics.
7:3948–3961. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lan X, Wu YZ, Wang Y, Wu FR, Zang CB, Tang
C, Cao S and Li SL: CD133 silencing inhibits stemness properties
and enhances chemoradiosensitivity in CD133-positive liver cancer
stem cells. Int J Mol Med. 31:315–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karagonlar ZF, Akbari S, Karabicici M,
Sahin E, Avci ST, Ersoy N, Ates KE, Balli T, Karacicek B, Kaplan
KN, et al: A novel function for KLF4 in modulating the
de-differentiation of EpCAM−/CD133-nonStem Cells into
EpCAM+/CD133+ liver cancer stem cells in HCC
cell line HuH7. Cells. 9:11982020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma XL, Hu B, Tang WG, Xie SH, Ren N, Guo L
and Lu RQ: CD73 sustained cancer-stem-cell traits by promoting SOX9
expression and stability in hepatocellular carcinoma. J Hematol
Oncol. 13:112020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jang JW, Song Y, Kim SH, Kim JS, Kim KM,
Choi EK, Kim J and Seo HR: CD133 confers cancer stem-like cell
properties by stabilizing EGFR-AKT signaling in hepatocellular
carcinoma. Cancer Lett. 389:1–10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng Y, Li M, Zhuo M, Guo P, Chen Q, Mo P,
Li W and Yu C: Histone demethylase JMJD2D promotes the self-renewal
of liver cancer stem-like cells by enhancing EpCAM and Sox9
expression. J Biol Chem. 296:1001212021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang B, Yan X and Li Y: Cancer stem cell
for tumor therapy. Cancers (Basel). 13:48142021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu JI and Wang LH: Emerging roles of gap
junction proteins connexins in cancer metastasis, chemoresistance
and clinical application. J Biomed Sci. 26:82019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakashima Y, Ono T, Yamanoi A, El-Assal
ON, Kohno H and Nagasue N: Expression of gap junction protein
connexin32 in chronic hepatitis, liver cirrhosis, and
hepatocellular carcinoma. J Gastroenterol. 39:763–768. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu M, Han G, Qi B and Wu X: Cx32 reverses
epithelial-mesenchymal transition in doxorubicin-resistant
hepatocellular carcinoma. Oncol Rep. 37:2121–2128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu M, Zou Q, Wu X, Han G and Tong X:
Connexin 32 affects doxorubicin resistance in hepatocellular
carcinoma cells mediated by Src/FAK signaling pathway. Biomed
Pharmacother. 95:1844–1852. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trosko JE: Cancer prevention and therapy
of two types of gap junctional intercellular
communication-deficient ‘cancer stem cell’. Cancers (Basel).
11:872019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang J, Nie J, Ma X, Wei Y, Peng Y and Wei
X: Targeting PI3K in cancer: Mechanisms and advances in clinical
trials. Mol Cancer. 18:262019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun S, Xue D, Chen Z, Ou-Yang Y, Zhang J,
Mai J, Gu J, Lu W, Liu X, Liu W, et al: R406 elicits anti-Warburg
effect via Syk-dependent and -independent mechanisms to trigger
apoptosis in glioma stem cells. Cell Death Dis. 10:3582019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mangiapane LR, Nicotra A, Turdo A,
Gaggianesi M, Bianca P, Di Franco S, Sardina DS, Veschi V, Signore
M, Beyes S, et al: PI3K-driven HER2 expression is a potential
therapeutic target in colorectal cancer stem cells. Gut.
71:119–128. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kahraman DC, Kahraman T and Cetin-Atalay
R: Targeting PI3K/Akt/mTOR pathway identifies differential
expression and functional role of IL8 in liver cancer stem cell
enrichment. Mol Cancer Ther. 18:2146–2157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang N, Wang S, Li MY, Hu BG, Liu LP, Yang
SL, Yang S, Gong Z, Lai PBS and Chen GG: Cancer stem cells in
hepatocellular carcinoma: An overview and promising therapeutic
strategies. Ther Adv Med Oncol. 10:17588359188162872018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanabe S, Quader S, Cabral H and Ono R:
Interplay of EMT and CSC in cancer and the potential therapeutic
strategies. Front Pharmacol. 11:9042020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weidenfeld K, Schif-Zuck S, Abu-Tayeh H,
Kang K, Kessler O, Weissmann M, Neufeld G and Barkan D: Dormant
tumor cells expressing LOXL2 acquire a stem-like phenotype
mediating their transition to proliferative growth. Oncotarget.
7:71362–71377. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawasaki Y, Omori Y, Li Q, Nishikawa Y,
Yoshioka T, Yoshida M, Ishikawa K and Enomoto K: Cytoplasmic
accumulation of connexin32 expands cancer stem cell population in
human HuH7 hepatoma cells by enhancing its self-renewal. Int J
Cancer. 128:51–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Totland MZ, Rasmussen NL, Knudsen LM and
Leithe E: Regulation of gap junction intercellular communication by
connexin ubiquitination: Physiological and pathophysiological
implications. Cell Mol Life Sci. 77:573–591. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang JX and Penuela S: Connexin and
pannexin channels in cancer. BMC Cell Biol. 17 (Suppl 1):S122016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alaei SR, Abrams CK, Bulinski JC,
Hertzberg EL and Freidin MM: Acetylation of C-terminal lysines
modulates protein turnover and stability of Connexin-32. BMC Cell
Biol. 19:222018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bamodu OA, Chang HL, Ong JR, Lee WH, Yeh
CT and Tsai JT: Elevated PDK1 expression drives PI3K/AKT/mTOR
signaling promotes radiation-resistant and dedifferentiated
phenotype of hepatocellular carcinoma. Cells. 9:7462020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng J, Bai X, Feng X, Ni J, Beretov J,
Graham P and Li Y: Inhibition of PI3K/Akt/mTOR signaling pathway
alleviates ovarian cancer chemoresistance through reversing
epithelial-mesenchymal transition and decreasing cancer stem cell
marker expression. BMC Cancer. 19:6182019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu Y, Zhang J, Zhang X, Zhou H, Liu G and
Li Q: Cancer Stem Cells: A potential breakthrough in HCC-targeted
therapy. Front Pharmacol. 11:1982020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xia P and Xu XY: PI3K/Akt/mTOR signaling
pathway in cancer stem cells: From basic research to clinical
application. Am J Cancer Res. 5:1602–1609. 2015.PubMed/NCBI
|
|
35
|
Erdogan S, Doganlar O, Doganlar ZB,
Serttas R, Turkekul K, Dibirdik I and Bilir A: The flavonoid
apigenin reduces prostate cancer CD44(+) stem cell survival and
migration through PI3K/Akt/NF-κB signaling. Life Sci. 162:77–86.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park JH, Kim YH, Shim S, Kim A, Jang H,
Lee SJ, Park S, Seo S, Jang WI, Lee SB and Kim MJ:
Radiation-activated PI3K/AKT pathway promotes the induction of
cancer stem-like cells via the upregulation of SOX2 in colorectal
cancer. Cells. 10:1352021. View Article : Google Scholar : PubMed/NCBI
|