Introduction
Autophagy can remove and degrade damaged organelles
and macromolecular substances, it participates in the basic
transformation of cell components, so that nutrients can be
recycled and cells can self-renew, thereby providing cells with
nutrients and energy to maintain cell homeostasis (1–4). In
recent years, numerous studies have shown that abnormality of
autophagy is related to the occurrence of numerous kinds of tumors
(4,5). Autophagy can promote cell survival by
removing damaged organelles and macromolecules in cells and
maintain cellular homeostasis. Nevertheless, autophagy can also
promote cell death through its connection with apoptosis. There is
a strong link between autophagy and apoptosis: Autophagy and
apoptosis often interact with each other, autophagy can promote or
inhibit apoptosis and apoptosis can also promote or inhibit
autophagy (6–8). The disruption of the dynamic balance
between autophagy and apoptosis may be one of the important reasons
for tumorigenesis (9). However,
the functions of autophagy and apoptosis in different types of
tumors development are still up for debates. Since autophagy can
influence both cell's death and survival, so it is very important
to research. Under what circumstances autophagy can promote cell's
death and under what circumstances autophagy can promote cell's
survival. Only by truly grasping the relationship between autophagy
and cell's fate, the study of autophagy can start to develop
effective drugs for autophagy-related tumor diseases. In the
present review, the process of autophagy and the role of
interaction between autophagy and apoptosis in tumorigenesis shall
be described.
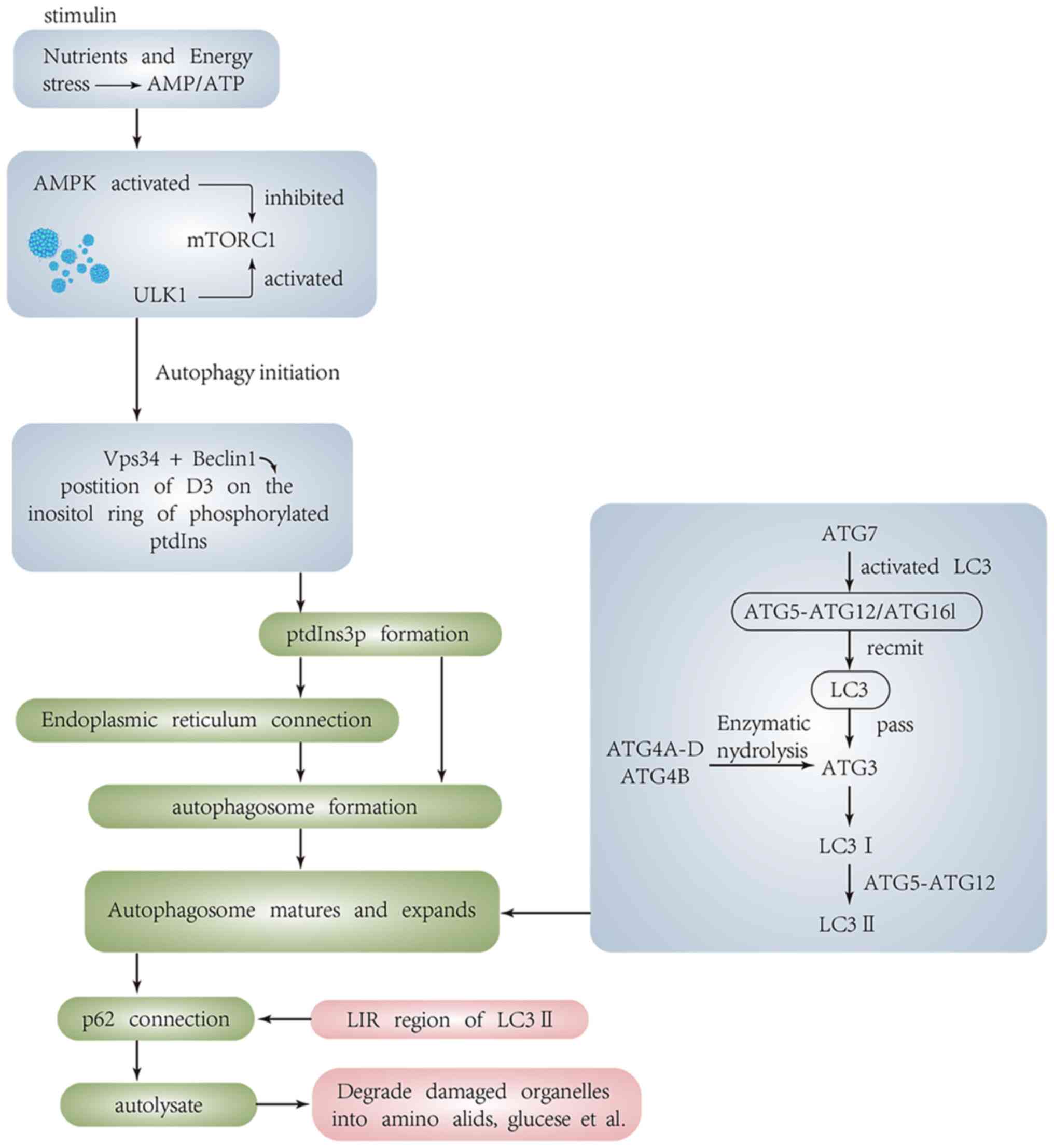
Molecular mechanisms of autophagy
(Fig. 1)
Mammalian target of rapamycin (mTOR) is a central
regulator of cell growth and proliferation (10). It consists of mTORC1 and mTORC2,
among which mTORC1 is regulated by signaling factors such as
cellular energy and amino acids (11). Under normal circumstances,
autophagy is inhibited by mTOR. When cells are subjected to
nutrient, oxidative and endoplasmic reticulum (ER) stress, the
activity of mTOR C1 is inhibited and autophagy is initiated by
phosphorylating unc-51-like kinase 1 (ULK1)-2 (ULK2) (12–15).
The formation of a separation membrane (i.e., phagocytes) marks the
beginning of autophagy. Under the action of autophagy-related
proteins, the detached double-layered membrane at the ribose-free
attachment zone of the rough ER will wrap the organelles or
macromolecules in the cytoplasm that need to be degraded (16). Then the membrane elongates and
self-encloses to form autophagosomes (17). The autophagosome and lysosome fuse
to form autophagolysosome, and the relevant hydrolases in the
lysosome decompose the encapsulated organelles or macromolecules
into amino acids, fatty acids and free nucleotides, then release
them back into the cytoplasm to realize the self-renewal of cell
components (18).
Formation of autophagosomes
Formation of the initial phagocyte membrane relies
on the class III phosphatidylinositol 3-kinase (PI3K) complex.
VPS34 is the only class III [(PI3K) in mammals)], which binds to a
coiled-coil protein encoded by beclin-1 to generate
phosphatidylinositol 3-phosphate (PtdIns 3P) through
phosphorylation at D-3 position of inositol ring (19,20).
The autophagosome is formed in the cup-shaped chamber of PtdIns 3P,
which is dynamically connected to the ER. When cells are under
stress, this compartment and the ER membrane are rearranged under
the action of autophagy-related proteins, thereby forming
phagocytes (21–23). Beclin-1 is the mammalian homolog of
the yeast autophagy-related protein Atg6, which is part of the
Vps34 complex and plays an important regulatory role in the
regulation of autophagy (24). The
composition of autophagy-specific Vps34 complex is very complex,
and its components include Vps34, Vps15, beclin-1 and Atg14L,
UVRAG. In addition, VMP1, Ambra-1, Bif-1 and Rubicon have also been
reported as components of the Vps34 complex (20,25,26).
Each component of the VPS34 complex has broad regulatory roles in
autophagy, with the Atg14L complex in autophagosome formation, the
UVRAG complex in autophagosome maturation, and the Rubicon complex
considered to inhibit autophagosome maturation (27–29).
As a positive regulator of beclin-1, Ambra-1 plays an important
role in the activation of beclin-1; while Dapper-1 promotes the
formation of autophagosomes by enhancing the formation of
beclin-1-Vps34-Atg14L complex (30), Bif-1 can regulate autophagosome
maturation by interacting with UVRAG and beclin-1 (31).
AMPK is a well-known energy sensor which maintains
cellular energy homeostasis when cells are in nutrient deprivation
(32). AMPK consists of a
catalytic α subunit and two regulatory β and γ subunits. Under
energy stress, the AMP/ATP ratio increases, in which case the gamma
subunit of AMPK binds directly to AMP (33). Afterwards, the AMPK complex
undergoes morphological changes and allosteric activation, during
which LKB1 leads to AMPK activation by promoting the
phosphorylation of Thr172 in the AMPKα subunit and inhibiting its
dephosphorylation (34,35). Activated AMPK can inhibit the
activity of mTORC1, thereby activating the ULK complex. ULK1
induces autophagosome formation by phosphorylating beclin-1 and
activating VPS34 lipid kinase (36–38).
ULK can be directly activated by AMPK upon cell starvation
(39,40). In addition, AMPK can directly
regulate the VPS34 complex through phosphorylation. Activated AMPK
can directly phosphorylate T163/s165 of VPS34 and s91/s94 of
beclin-1 through ATG14L (41).
Mammalian ULK1, 200-kDa focal adhesion kinase family interacting
protein (FIP200) and autophagy-related protein Atg13 interact to
form a stable complex ULK1-Atg13-FIP200 (42,43).
This complex is localized to the phagosome during starvation and
inhibits the dephosphorylation of mTOR-dependent sites, resulting
in enhanced activity of ULK1 and interaction with the
vertebrate-specific autophagy protein ATG101 (44), which plays an important regulatory
role in autophagosome formation.
Maturation and elongation of
autophagosomes
Beclin-1 binds to the UVRAG-targeted Class C Vps
complex and recruits the mammalian homology of yeast Atg8,
microtubule-associated protein-1 light chain kinase 3 (LC3), via
the ATG5-ATG12/ATG16L multimeric complex, assisting in the
maturation and elongation of autophagosomes. In addition, the
formation and maturation of autophagosomes is also closely related
to phosphatidylethanolamine (PE) conjugates (45,46).
The location of the Atg16L complex on phagosome determines the
location of LC3 binding reaction (47). ATG7 can activate LC3 and transmit
it to ATG3 (48–50), and convert Pro-LC3 into its active
cytoplasmic isomer LC3 I by enzymatically degrading a small segment
of polypeptides from Atg4A-D and Atg4B in the Atg4 protein family.
With the help of the ATG5/12 conjugate, glycine residues are left
at C-terminus of LC3 I and binds to the polar head of PE, a
component of the phospholipid bilayer, and converts to the
autophagosome membrane type, LC3-II. The LC3-II/LC3I ratio is often
considered the gold standard for macroautophagy. LC3-II wraps
around the inner and outer surfaces of autophagosomes and, together
with ATG5, acts as a discrete marker for autophagosomes and
autophagosome precursors, respectively, until they fuse with
lysosomes (51). Notably, ATG12 is
also required to be activated by ATG7 to bind to an isopeptide bond
on an internal lysine on ATG5.
Degradation of autophagosomes
After the maturation and elongation of
autophagosomes, the selective autophagy adaptor protein p62/SQSTM1
can bind to LC3 through LC3 interacting region (LIR). P62 binds to
ubiquitinated proteins at the C-terminus and to LC3-II at
N-terminus. Therefore, p62 acts as a bridge between ubiquitinated
proteins and LC3; p62 is degraded by autolysosomes after
autophagosomes fuse with lysosomes to form autolysosomes (52,53).
Simultaneous detection of LC3 and p62 can reflect the integrity of
autophagic flux. The accumulation of p62 protein represents the
impaired degradation process of autophagosome, thus p62 is often
regarded as a negative indicator of autophagy.
Mechanism of apoptosis
Death receptor (DR) pathway
The three main apoptotic pathways are DR pathway,
mitochondrial damage pathway and ER stress initiation pathway. The
extrinsic pathway is DR, which is mainly through the binding of
Fas/FasL, tumor necrosis factor receptor 1 (TNFR1) and related
death domain (TRADD), or TNF-related Apoptosis-inducing ligand
receptor (TRAILR). The caspase protease caspase-8 is activated by
dimerization following ligand/receptor binding. For example, FasL
binds to its receptor Fas and induces Fas molecules to aggregate to
form dimers. Through the binding of the death domain in the
cytoplasm to the adaptor protein FADD, the FADD effector domain
binds to caspase-8 to form a death signal complex DISC. When a
large amount of DISC is generated, activated caspase-8 can bypass
mitochondria and directly activate other proteins of the caspase
family such as caspase-3, caspase-7, caspase-6, thereby cleaving
hundreds of different substrates, including cytoskeletal proteins,
nuclear structural proteins, lipid metabolism and endonucleases.
Cleavage-dependent activation or inactivation of specific proteins
leads to changes in the morphological and biochemical
characteristics of apoptosis, including phosphatidylserine
exposure, plasma membrane blebbing, and DNA fragmentation, thereby
inducing apoptosis (54,55).
Mitochondrial apoptosis pathway
Mitochondrial apoptosis pathway is mainly marked by
mitochondrial outer membrane permeability (MOMP). When cells are
induced by apoptotic signals such as DNA damage and cytokine
extraction, the permeability of mitochondrial membrane is opened in
an irreversible manner, releasing cytochrome c (Cyt c) from the
mitochondrial intermembrane space; Cyt c and the adaptor protein
APAF-1 binds to form a complex of apoptosomes and activates
caspase-9. After caspase-9 is activated, caspase-3 and caspase-7
are cleaved and activated, thereby initiating caspase-level
reaction and completing apoptosis. The aforementioned process is
maintained by a delicate balance between BCL-2 protein family
(56), which consists of
anti-apoptotic proteins and pro-apoptotic proteins. Bcl-2 protein
family is divided into three categories: i) Pro-apoptotic effector
proteins (including BAX, BAK, Bik), which promote apoptosis and
irreversibly change MOMP after being activated by pro-apoptotic
signals; ii) Anti-apoptotic Bcl-2-like proteins (including Bcl-2,
Bcl-xL and MCL-1) are mainly distributed in mitochondrial membrane
and cytoplasm, bind and inhibit BH3 with pro-apoptotic activity
protein and effector protein, block the occurrence of MOMP, thereby
inhibiting cell death; iii) Pro-apoptotic BH3 pure proteins
(including BID, BIM and PUMA). BID, located in the cytoplasm, is
cleaved into truncated (t)BID by caspase-8, and tBID has strong
pro-apoptotic activity, which can transmit apoptotic signals to
mitochondria and induce the release of Cyt c. It promotes apoptosis
by inhibiting anti-apoptotic Bcl-2 protein and directly activating
effector proteins to signal MOMP activation (57). The exogenous and endogenous
apoptotic pathways do not act on their own, but cross-talk each
other through the activation and cleavage of pro-apoptotic protein
BID mediated by caspase-8, produced BID cleavage product (58,59).
ER stress pathway
The ER stress pathway is a newly discovered
apoptotic pathway in recent years, and ER stress is a very
important trigger for the activation of autophagy. When ER is
stimulated by hypoxia, starvation, infection and other factors, the
homeostasis of ER is disrupted, resulting in the accumulation of
unfolded or misfolded proteins, which induces ER stress. Sustained
ER stress activates the ATF4/CHOP and IRE1/TRAF2/ASK/JNK pathways.
Activation of both JNK and CHOP attenuates the function of
anti-apoptotic protein Bcl-2, while enhancing the activity of
pro-apoptotic proteins such as Bim, Bax and PUMA, leading to
mitochondrial dysfunction and Cyt c release. ER stress activates
IRE1 to recruit and activate necrotic tumor receptor-associated
factor 2 (TRAF2), which further activates JNK and leads to
apoptosis (60,61). CHOP can regulate the expression of
Bcl-2, GADD34 and TRB3 (62).
Firstly, CHOP downregulates Bcl-2 expression, but upregulates the
pro-apoptotic gene Bim, and promotes the translocation of Bax to
mitochondria to promote apoptosis (63). Secondly, CHOP can directly bind to
the promoter of TRB3 gene and upregulate its expression (64), thereby inhibiting the activation of
AKT and leading to apoptosis. Notably, TRB3 can regulate the
expression of CHOP through a negative feedback mechanism.
Overexpressed TRB3 inhibits the transcriptional induction of CHOP,
whereas silencing TRB3 leads to the upregulation of CHOP under
normal and stress conditions (64–66).
During ER stress, p53 induces the activation of another BH3-only
protein and promotes the expression of a regulator of apoptosis
(PUMA). PUMA-deficient cells reduce ER stress-induced apoptosis.
Activation of multiple apoptotic pathways during ER stress jointly
induces apoptosis (67).
Inhibition and promotion of autophagy on
apoptosis
Autophagy functions in both pro-survival and
pro-death ways within the same cell. Autophagy can promote the
survival of normal cells during nutrient starvation, when cells are
in a state of stress, including oxidative, ER, nutrient and energy
stress. Autophagy can maintain cell homeostasis by removing damaged
organelles and can also provide nutrients for cell survival by
degrading macromolecular substances in cells. Therefore, from this
perspective, elevated levels of autophagy contribute to cell
survival, and the lack of autophagy increases cell death
susceptibility when cells are under stress (68,69).
Another important mechanism by which autophagy inhibits apoptosis
is that it can engulf damaged mitochondria. When mitochondria are
damaged, various death signals will be released that cause the
transmembrane potential within the mitochondria to dissipate,
resulting in cell death. Autophagy can also reduce apoptosis by
selectively reducing the abundance of pro-apoptotic proteins in
cells. For example, autophagy can selectively remove active
caspase-8. When the autophagy gene Atg7 is knocked out, the
activity of caspase-8 is increased, indicating that autophagy
deficiency can promote apoptosis (68).
Autophagy can promote apoptosis and induce cell
death in cells with damaged apoptotic mechanisms, and excessive
levels of autophagy also promote cell death. It was found that
numerous components of autophagy are necessary for apoptotic
factors to mediate cell death (69). In addition, numerous autophagy
proteins can induce apoptosis. For example, Atg5 and Atg12 proteins
can activate caspases through the mitochondrial pathway. When Atg5
and Atg12 are knocked out, the activity of caspases is
significantly reduced (68–70).
The increase in Atg12 mRNA expression promotes cell death (71,72);
reducing the expression of Atg12 or other autophagy genes
effectively inhibited cell death (73–76).
Furthermore, autophagy can promote apoptosis by degrading
anti-apoptotic and cell survival factors and depleting endogenous
inhibitors of intracellular death pathways. For example, autophagy
can degrade inhibitor of apoptosis proteins (IAPs) (77). When autophagy is activated, the
accumulated autophagosomes can irreversibly open the mitochondrial
membrane when they accumulate in the cell body, leading to
apoptosis. Fap-1 is an inhibitor of Fas-mediated apoptosis, and its
autophagic degradation sensitizes type I cells to Fas-induced
apoptosis (78). In proliferating
cell populations, different levels of autophagy within a single
cell lead to different cell fates. Autophagy proteins, on the other
hand, promote cell death by providing scaffolds for cell death
complexes and signaling molecules. In mouse embryonic fibroblasts
treated with sphingosine kinase inhibitor (SKI), SKI promotes cell
death by inhibiting sphingosine 1-phosphate (79). SKI induces the translocation of
caspase-8 homologous complex and Fas-associated protein and death
domain (FADD) to Atg5- and Atg16L-positive autophagosome membranes,
which provides a scaffold for efficient formation of intracellular
death induction signaling complex (iDISC) (54,79).
Promotion and inhibition of apoptosis on
autophagy
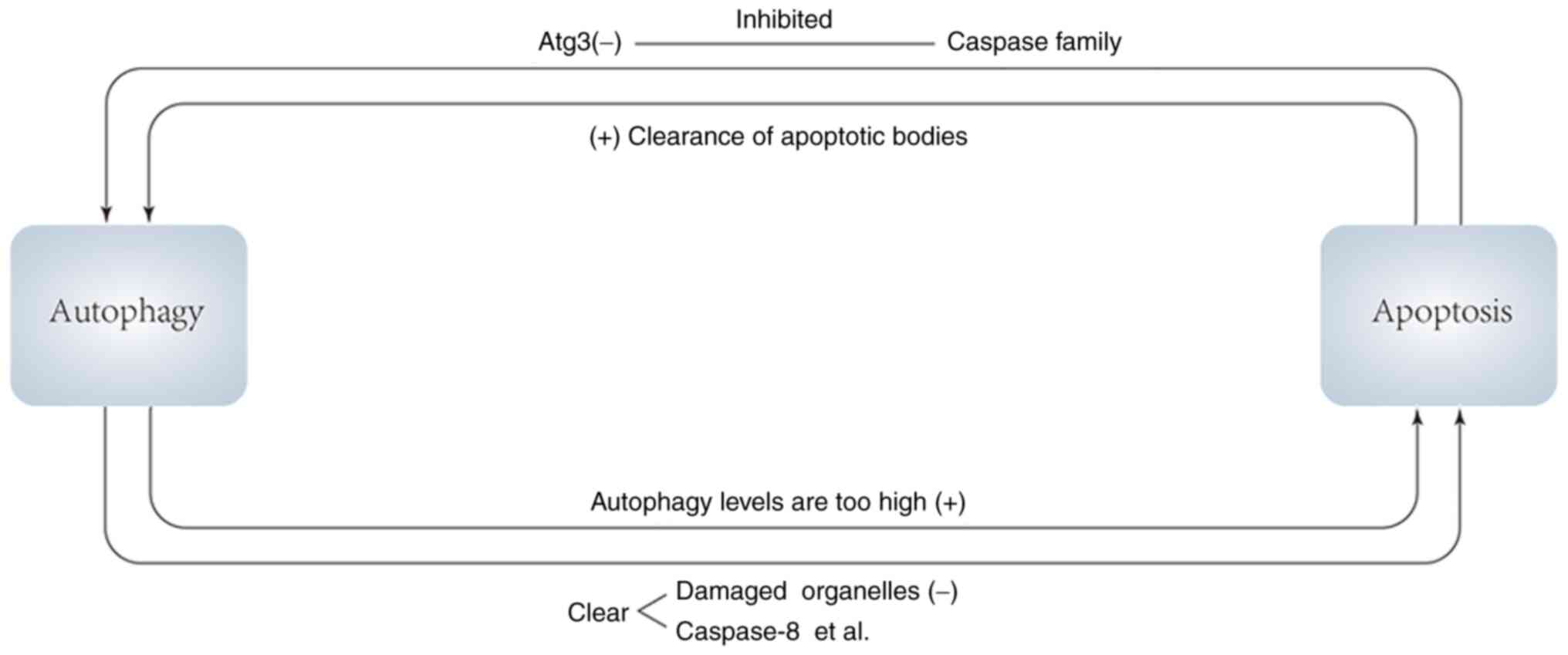
(Fig. 2)
Autophagy promotes or inhibits apoptosis, and in
turn apoptotic signals and apoptotic products promote or inhibit
autophagy. On the one hand, apoptosis can inhibit autophagy, and
caspases, a key role in apoptosis, can digest several essential
autophagy proteins, resulting in the inactivation of the autophagy
program. For example, caspases can target Atg3 to cause its
inactivation to inhibit the level of autophagy. In addition, the
autophagy protein AMBRA1 can be irreversibly degraded under the
combined action of caspases and calpains, and autophagy is thus
inhibited (80,81).
In the process of mammalian embryonic development,
autophagy neither delays nor promotes apoptosis, but the clearance
of apoptotic bodies requires autophagy, thus apoptosis promotes
autophagy in a sense (82). From
this, it is hypothesized that in mammals, if impaired autophagy
cannot effectively remove apoptotic corpses, the accumulation of
apoptotic bodies may induce gene changes, or there may be a
negative feedback mechanism in apoptosis. When apoptotic bodies
accumulate, they will negatively feed back to the pro-apoptotic
signaling pathway and the anti-apoptotic signaling pathway,
resulting in the inhibition of the pro-apoptotic signaling pathway
and the activation of the anti-apoptotic signaling pathway, thereby
reducing apoptosis (80). When
autophagy is defective, apoptotic bodies cannot be removed by
autophagy, and their accumulation induces cell mutation. On the
other hand, the accumulation of apoptotic bodies inhibits
apoptosis, so that cells that should be apoptotic continue to
survive, leading to tumorigenesis over time. A recent study
identified that under aggressive tumorigenic conditions associated
with metabolic stress, autophagy prevents genomic destabilization,
thereby suppressing tumorigenesis (82). The aforementioned study also
verifies our conjecture to certain extent.
There are intricate connections between autophagy
and apoptosis. Numerous autophagy and apoptosis factors can
directly interact with each other through specific domains to
affect the expression of each other. The relationship between them
cannot be simply judged. It may be different due to different
environments or stimulatory signals, so maintaining autophagy and
apoptosis in a relatively stable state is of great significance for
maintaining the physiological functions of cells. When the balance
between autophagy and apoptosis is disturbed, it will lead to
diseases like neurodegenerative diseases or cancer. Next, the
possible mechanism of the imbalance between autophagy and apoptosis
in pathogenesis of cancer will be explored.
Effects of autophagy and apoptosis on
tumorigenesis
Cancer cells can proliferate due to their ability to
avoid apoptosis or death. This is why inducing apoptosis in cancer
has been identified as a target of cancer therapy, as numerous
cancer patients have been found to have mutations in
apoptosis-related genes (83,84).
The inhibition of pro-apoptotic genes and the activation of
anti-apoptotic genes are considered to be closely related to the
occurrence of cancer. Numerous studies have found that excessive
apoptosis may also have oncogenic functions (54,85,86),
and that higher levels of apoptosis may be associated with poor
prognosis in cancer patients (87). It has also been revealed that the
abnormal level of autophagy is also one of the important mechanisms
leading to cancer. Beclin-1 has been identified to be a tumor
suppressor that inhibits tumorigenesis. Beclin-1 can inhibit
tumorigenesis, and its expression level is reduced in human breast
cancer. These findings suggested that reduced expression of
autophagy proteins may contribute to the development and
progression of breast cancer and other human malignancies (88). Autophagy can not only prevent
tumorigenesis by removing abnormal cells, but also reduces the
oncogenic mutation of cells by removing abnormally large amounts of
DNA-damaging reactive oxygen species released from damaged
mitochondria by removing damaged mitochondria (89). However, after the occurrence of
tumors, autophagy can provide nutritional support for tumor cells
by degrading macromolecular substances and damaged organelles, and
promote the survival of tumor cells. When the expression level of
autophagy in tumor cells is elevated, tumor cells antagonize
anticancer drugs due to autophagy. The balance is the key to
maintaining the biological function of cells. When this balance is
disrupted, the apoptosis of cells is abnormal and cell homeostasis
is disrupted, which in turn promotes tumorigenesis.
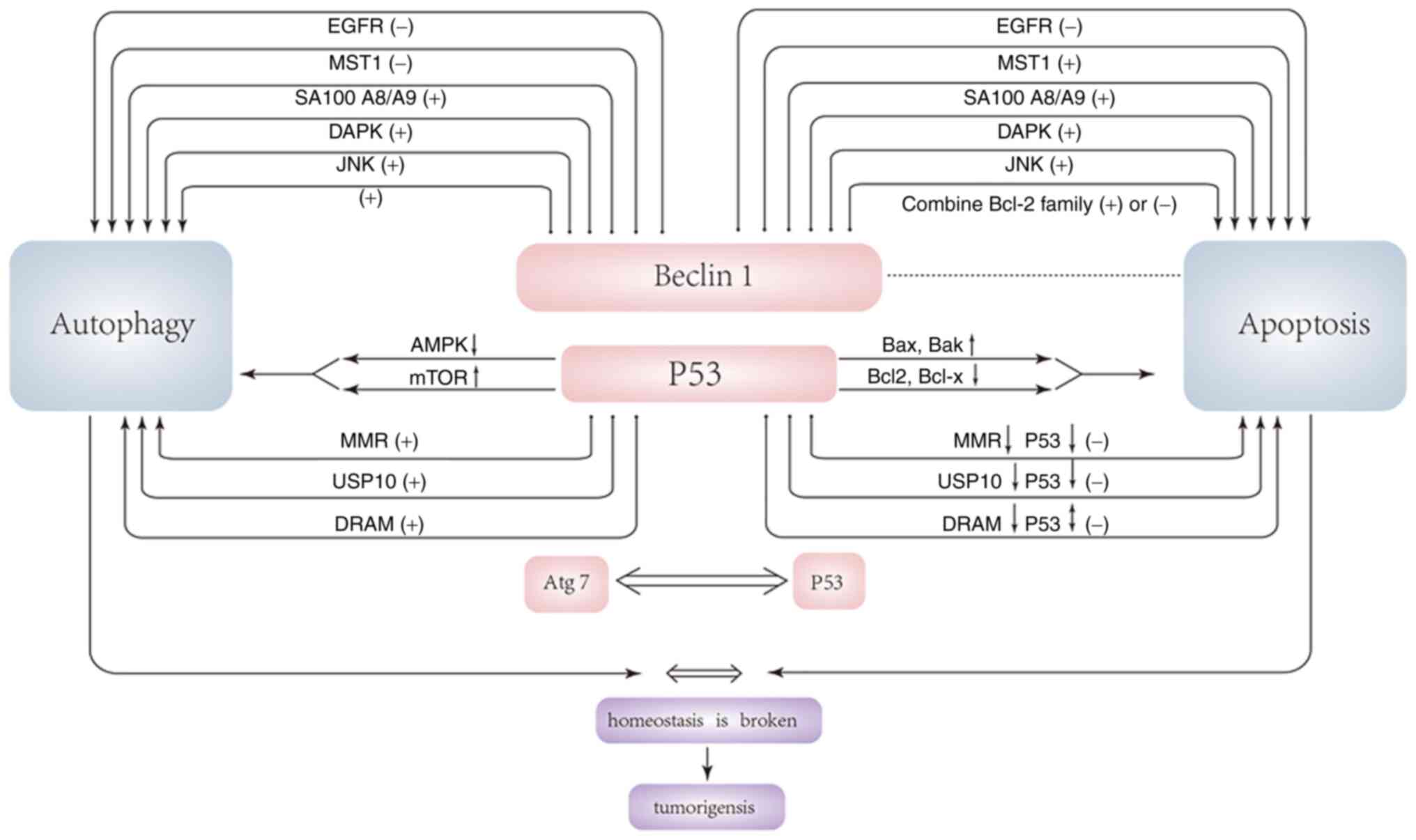
Effects of p53-related autophagy on
tumorigenesis
P53 is considered a BH3-only protein that acts both
as a direct activator of Bax and as a de-repressor. Under
pro-apoptotic conditions, p53 co-immunoprecipitates with Bcl2,
Bcl-XL and Bak (90). p53
suppresses oncogenic potential by mediating irreversible cell cycle
arrest or triggering apoptotic cell death. Under the induction of
various apoptotic stimuli, p53 moves to the mitochondria, and after
reaching the mitochondria, p53 induces MOMP, thereby triggering the
release of pro-apoptotic factors in the mitochondrial intermembrane
space (91,92). Due to its pro-apoptotic effect, p53
is considered an important tumor suppressor gene, as ~half of human
types of cancer have p53-inactivating mutations. Most of the
remaining malignancies are caused by the inhibition of the
pro-apoptotic function of p53 by increasing its inhibitors,
decreasing its activators, or inactivating its downstream targets
(93). A recent study found that
p53 has a significant inhibitory effect on autophagy, and the loss
of autophagy makes cells sensitive to TRAIL through up-regulation
of PUMA (p53 upregulated modulator of apoptosis). Autophagy
counteracts the lethal effect of MOMP by removing damaged and
permeable mitochondria. The inhibition of autophagy by p53 reduced
this effect and further promoted cell death via the MOMP pathway.
Previous studies have found that the inhibition of autophagy is
mediated by p53 in the cytoplasm rather than the nucleus, and
physiological inducers of autophagy (such as nutrient depletion)
must destroy the p53 pool in the cytoplasm to induce autophagy.
Thus, inhibition of ubiquitin E3 ligaseMdm2 targeting p53
disruption can inhibit starvation, rapamycin, lithium or ER
stress-induced autophagy (94).
Another study demonstrated that p53 in the cytoplasm can inhibit
the expression of AMPK and activate the activity of mTOR, thereby
inhibiting autophagy. However, how these effects are achieved
remains a puzzle. Certain studies are contradictory to the
aforementioned research conclusions, considering that p53 can
prevent the occurrence of tumors by inhibiting mTOR and increasing
autophagy and promoting apoptosis (9,94).
A previous study also found the close relation
between Atg7 and apoptosis that p53 and Atg7 exist in a single
complex (9). Abnormal expression
of Atg7 is closely related to the occurrence of rectal cancer.
Cells lacking Atg7 impair p53-mediated cell cycle arrest. When
cells are under nutrient stress, endogenous Atg7 exists in the
promoter region of p21 together with p53, and cells lacking Atg7
cannot properly induce the expression of p21 (95). The p53 tetramer domain mediates the
interaction with Atg7, and Atg7 promotes p53 tetramer formation.
Atg7 and p53 can directly bind to each other, and this binding is
facilitated when cells are under nutrient stress. In the case of
Atg7 deficiency, the pro-apoptotic activity of p53 induced by
metabolic stress also changes, and the enhancement of the
pro-apoptotic activity of p53 can also regulate autophagy through
Atg7. Summing up, the correlation between the two maintains the
balance between autophagy and apoptosis, which plays an important
role in tumor suppression (9,95).
DNA mis-match repair (MMR) is one of several DNA
repair processes critical for maintaining genome stability
(96). MMR is an important tumor
suppressor mechanism, and MMR deficiency contributes to the
development of human rectal cancer and solid tumors (96,97).
As a response to DNA damaging agents such as 6-thioguanine (6-TG)
and 5-fluorouracil (5-FU), MMR plays an important role in cell
cycle arrest, autophagy and apoptosis. 6-TG induces an
MMR-dependent autophagic response, and autophagic flux in cells is
upregulated after 6-TG induction. MMR initiates 6-TG-induced
autophagy in a p53- and mTOR-dependent manner (98–100). A recent study revealed that
adenovirus E1B 19 kilodalton interacting protein (BNIP3) was also
required for induction of autophagy after DNA MMR treatment of 6-TG
and 5-FU. BNIP3 is a Bcl-2 homeodomain protein of the Bcl-2 protein
family, which can cause autophagy, apoptosis and necrosis depending
on the type and nature of cell stimuli (101). A previous study found that BNIP3
plays an important role in mediating 6-TG- and 5-FU-induced
autophagy (102). Reactive oxygen
species are considered to be key triggers for the activation of
autophagy, which are abundantly produced during BNIP3-mediated
apoptosis. After being driven by reactive oxygen species signaling,
mTOR activity is inhibited and autophagy is initiated. Notably, the
mTOR-S6K1 axis regulates BNIP3 protein translation (S6K1 is one of
the mTOR downstream effectors) and plays an active role in
regulating 6-TG-induced autophagy. During apoptosis, overexpression
of BNIP3 induces apoptosis. Upon initiation by inducible MMR
treatment with 6-TG and 5-FU, p53 was activated and acted as a
transcription factor to upregulate BNIP3 transcription. Inhibition
of p53 expression impairs BNIP3 upregulation. It is hypothesized
that the role of BNIP3 in apoptosis may be related to the
interaction of P53. Furthermore, BNIP3 is localized to the
mitochondrial outer membrane through its transmembrane domain,
which leads to the loss of mitochondrial membrane potential and the
opening of the mitochondrial permeability transition pore,
promoting apoptosis (103,104).
The mechanism by which BNIP3 and MMR inhibit tumorigenesis may be
related to the regulation of autophagy and apoptosis.
Two ubiquitin-specific peptidases, USP10 and USP13,
were found to regulate the deubiquitination of beclin-1 in the
Vps34 complex. Decreased expression of USP10 significantly reduced
the levels of ubiquitinated beclin-1. Similarly, the removal of
beclin-1 or Vps34 also significantly reduces the levels of USP10
and USP13. As a deubiquitinating enzyme of p53, USP10 and USP13 can
regulate p53 levels through p53 ubiquitination and degradation
(105). The abnormal expression
of USP10 is closely related to the occurrence of breast cancer. A
recent study found that the lack of USP13 and USP10 also reduces
the expression level of p53. Decreased expression of beclin-1 leads
to a decrease in the expression level of p53 by increasing its
ubiquitination. In addition to beclin-1, inhibition of the
expression of Vps34 complexes such as Vps34, p150, UVRAG and Atg14L
leads to a decrease in the level of p53 (106). Vps34 complexes may regulate the
cellular level of p53 through deubiquitinating enzymes such as
USP10 and USP13, in which beclin-1 may be the target of the
interaction of Vps34 complex with USP10 and USP13. While the
removal of other Vps34 complex components (such as Vps34, p150,
UVRAG and Atg14L) may result in decreased p53 levels due to the
codependent regulation of stability of the core components of the
Vps34 complex, beclin-1 deubiquitination may be sufficient to
control levels of the entire complex (45).
DRAM is a lysosomal protein that regulates
autophagy. A previous study found that DRAM is significantly
downregulated in certain epithelial malignancies, and the p53/DRAM
axis plays a very important role in the treatment of breast cancer
(107). The mechanism by which
DRAM causes tumorigenesis may be related to the ability of DRAM to
regulate autophagy and apoptosis. DRAM is induced by DNA damage and
is a direct target of p53. Even in the presence of inhibitors of
protein synthesis, DRAM-induced RNA damage does not require the
synthesis of intermediate proteins and can therefore be considered
a major target of p53. DRAM knockdown reduces p53-mediated
apoptosis, indicating that DRAM is directly involved in
p53-mediated apoptosis. p53 has been shown to regulate autophagy
(108), and p53 induces autophagy
in a DRAM-dependent manner, indicating that DRAM plays an important
regulatory role in autophagosomes. DRAM is a regulator of
p53-induced autophagy, and DRAM-dependent induction of autophagy is
required and critical for p53-mediated apoptosis. Therefore, the
decreased expression of DRAM leads to the decrease of autophagy and
apoptosis levels, which may be an important mechanism of
tumorigenesis.
Effect of beclin-1-related autophagy
and apoptosis on tumorigenesis
Beclin-1, the mammalian homolog of yeast ATG6, is a
mammalian tumor suppressor (109,110). The beclin-1 gene is
monoallelic, deleted in up to 75% of ovarian, 50% of breast and 40%
of prostate cancers (111).
Reduced expression of beclin-1 is also observed in other types of
cancer such as human brain tumors and cervical cancers (112,113). Beclin-1 acts as an important
confluence of autophagy and apoptosis through its interaction with
the apoptotic protein family Bcl-2 (114). It was found that Bcl-2 interacts
with the BH3 domain of beclin-1, UVRAG interacts with the CCD of
beclin-1, and class III PtdIns 3-kinase interacts with the ECD and
CCD of beclin-1. Beclin-1 acts as a platform or scaffold for the
formation of complexes during autophagy, and is also a bridge for
the interaction between autophagy and apoptosis. Previous studies
have shown that synthetic peptides containing the BH3 domain of
beclin-1 induce apoptosis (73,109–115), beclin-1 function can be regulated
by other BH3-only proteins, such as Bad. In addition to its
pro-apoptotic effects, Bad induces autophagy by competitively
disrupting the interaction between beclin-1 and Bcl-2/Bcl-X. The
interaction between Bcl-2 and beclin-1 is greatly reduced after
starvation, suggesting that the segregation of Bcl-2 and beclin-1
is of great importance for the activation of autophagy, which
contributes to the protection of cells upon starvation (73,115). Since autophagy and apoptosis are
interconnected, and their relationship may vary in specific
contexts, beclin-1 may play a regulatory role in apoptosis and
other related cellular events. In a mouse model, it was found that
beclin-1 is not required for apoptotic cell death, but for the
generation of signals that allow phagocytes to clear apoptotic
corpses (116). However, in
another study, there was a different opinion, that the autophagy
genes ATG7 and beclin-1 are required for apoptosis (117). Therefore, the regulatory
mechanism of beclin-1 in autophagy and apoptosis still needs to be
further explored.
JNK is an important pro-apoptotic component; a
recent study found that the activation of JNK signaling plays an
important role in inhibiting the occurrence of lung cancer
(118). It has been revealed that
JNK can reduce the mutual inhibition of beclin-1 and Bcl-2
pro-apoptotic family members by phosphorylating Bcl-2 in the
flexible loop between its BH4 and BH3 domains, thus inducing
autophagy and apoptosis. Besides, reactive oxygen species can
inhibit DNA repair and promote cell cycle arrest by activating JNK
signaling, thereby promoting the occurrence of autophagy and
apoptosis.
DAP kinase (DAPK), a death-associated protein kinase
with important pro-apoptotic effect (89,119), is an important tumor suppressor.
Recent studies found that the activation of DAPK has a very
important inhibitory effect on thyroid cancer and small cell lung
cancer (120,121). DAPK can activate protein
phosphatase 2A (PP2A), a form of cell death caused by shedding of
adherent cells from their substrates, to promote cell death in the
presence of dysregulated ceramide-induced apoptosis. Meanwhile,
DAPK is also an autophagy stimulator and is involved in the
regulation of autophagy and apoptosis. DAPK phosphorylates beclin-1
within its BH3 domain (Thr119), which prevents beclin-1 from
binding to its inhibitor Bcl-2/Bcl-x, thereby promoting its
autophagic activity (122). In
addition, DAPK can activate protein kinase D (PKD), and PKD
activates VPS34 through phosphorylation and degradation (123), which promotes the occurrence of
autophagy. As the binding between beclin-1 and the pro-apoptotic
protein family BCL-2 is inhibited, not only the level of autophagy
but also the level of apoptosis is increased, thereby inhibiting
tumorigenesis.
S100A8/A9 are two members of the S100
calcium-binding protein family, and their abnormal expression is
related to the occurrence of various cancers. S100A8/A9 can
effectively inhibit the occurrence of head and neck squamous cell
carcinoma (124). Its complex can
promote the apoptosis of various cells, and S100A8/A9 is also
closely related to the occurrence of autophagy (125). S100A8/A9 was revealed to activate
caspase-9, caspase-3 and caspase-7, leading to cleavage of poly
(ADP-ribose) polymerase-1 in cells, thereby promoting apoptosis.
The mechanism of S100A8/A9-induced apoptosis may also be related to
BNIP3 and reactive oxygen species. BNIP3 has a single BH3 domain
and a C-terminal trans-membrane (TM) domain. As can be observed
from the previous description, BNIP3 is an atypical pro-apoptotic
Bcl2 family member with strong pro-apoptotic activity.
Transient-transfected BNIP3 showed mitochondrial damage and
mitochondrial autophagy, accompanied by the opening of
mitochondrial permeability transition pore and increased production
of reactive oxygen species, leading to mitochondrial dysfunction,
which in turn results in cell apoptosis (126,127). A very important process in
BNIP3-induced apoptosis is that BNIP3 needs to be integrated into
the mitochondrial outer membrane to induce cell death. A rapid
decrease in mitochondrial membrane potential was identified when
cells were treated with S100A8/A 9, triggering the pro-apoptotic
mechanism of Bak and promoting the translocation of BNIP3 to
mitochondria. The association of BNIP3 with mitochondria is
enhanced after S100A8/A9-induced apoptosis (104,128). The damage of mitochondria is
accompanied by the release of a large amount of reactive oxygen
species, which is an important signal for the activation of
autophagy. In the meantime, it was found that after S100A8/A9
treatment, the number of autophagosomes and apoptotic bodies
increased significantly; the formation of LC3-II protein,
Atg12-Atg5 and the expression level of beclin-1 were both increased
under transmission electron microscope. This indicated that
S100A8/A9 can promote both apoptosis and autophagy. In
S100A8/A9-treated cells, LC3-II co-localized with mitochondria and
lysosomes, and the increased autophagy induced by S100A8/A9 may be
related to the induction of mitochondrial damage (125).
The pro-apoptotic kinase Mst1 is a serine-threonine
kinase with strong pro-apoptotic activity. Studies have found that
the expression of Mst1 in cancer tissues of patients with cervical
and lung cancer is significantly lower than that in adjacent
tissues, which may be related to the pro-apoptotic activity of
Mst1. Mst1 is a component of the Hippo signaling pathway, and
previous studies have found that the Hippo pathway is a tumor
inhibiting signaling pathway (129), which indicates the importance of
Mst1 in inhibiting tumors. With the in-depth study of Mst1, it was
found that Mst1 can inhibit autophagy by promoting the interaction
between beclin-1 and Bcl-2. Mst1 phosphorylates the Thr108 residue
in the BH3 domain of beclin-1, and phosphorylated beclin-1
co-locates with ER marker motifs, but not with mitochondrial or
Golgi markers, suggesting that Mst1 phosphorylates ER beclin-1
(130). The interaction between
beclin-1 and Bcl-2 and Bcl-xL is enhanced upon phosphorylation by
Mst1, which disrupts the interaction between beclin-1 and Vps34,
while attenuating the binding of Atg14L to beclin-1.
Phosphorylation of Mst1 directly inhibits the formation of the
beclin-1-VPS34 complex, leading to the formation of beclin-1
homologous dimer and thus inhibiting autophagosome formation. When
Bcl-2 and Bcl-xL were downregulated, the inhibition of autophagy by
Mst1 was abolished, indicating that the inhibitory effect of Mst1
on autophagy occurs mainly by enhancing the mutual binding between
beclin-1 and Bcl-2. MST1-induced Bcl-2 and Bcl-xL are sequestered
by beclin-1, which activates Bax, thereby stimulating apoptosis and
inhibiting tumorigenesis. The ability of Mst1 to regulate autophagy
and apoptosis may help suppress tumorigenesis and progression by
eliminating adaptive mechanisms for tumor cells to survive in
hypoxic environments and promoting cell death (129,130).
Epidermal growth factor receptor (EGFR) is a
receptor tyrosine kinase whose upregulation has been implicated in
the development of cancers (131). Studies have found that EGFR is an
important target for the treatment of non-small cell lung, breast
and gastroesophageal cancer (132). Active EGFR blocks autophagy and
active EGFR inhibits autophagy by activating the PI3K/AKT/mTOR
signaling pathway and inhibiting beclin-1 (133). Active EGFR could
co-immunoprecipitate with amino acids 1–135 of beclin-1 but not
with amino acids 1–115, suggesting that amino acids 115–135
containing the BH3 domain contribute to the interaction between
beclin-1 and EGFR. Furthermore, EGFR can bind to the amino acid
244–377 domain of beclin-1 (i.e., the evolutionarily-conserved
domain ECD), thus beclin-1 has at least two domains of BH3 and ECD
that can bind to EGFR (132). As
aforementioned, Bcl-2 interacts with the BH3 domain of beclin-1,
and the ECD domain of beclin-1 can bind to the autophagy inhibitor
Rubicon. Notably, study demonstrated that active EGFR also helps
with the initiation of autophagy while inhibiting it (131,132). Inactive EGFR promotes the
separation of the Rubicon-beclin-1 complex through its interaction
with Rubicon, thus initiating autophagy. There is an ECD region
between EGFR, Rubicon and beclin-1, and the three can be combined
with each other. Therefore, the binding of EGFR and Rubicon may
also be through the ECD region. Active EGFR selectively binds to
the BH3 domain of beclin-1 through the BH3 region, resulting in the
inability of beclin-1 to bind to the BH3 domain of Bcl-2 through
the BH3 domain. Bcl-2 is separated from beclin-1, and the release
of Bcl-2 leads to increased anti-apoptotic ability, which in turn
leads to tumorigenesis (131).
However, inactive EGFR selectively binds to the ECD domain of
Rubicon through the ECD region, resulting in the release of
beclin-1 and thus promoting autophagy. When cells are not
stimulated by external stimuli, EGFR activity is inhibited and the
binding of the ECD domain of EGFR is active, and the BH3 domain is
inhibited. At this time, autophagy proceeds normally and
tumorigenesis is inhibited. When cells are stimulated by external
stimuli, EGFR activity increases so that the binding of the ECD
domain is inhibited while the BH3 domain is activated, thereby
inhibiting autophagy and stimulating tumorigenesis (131,133).
Inhibition of autophagy is considered to be an
important mechanism of tumorigenesis, but autophagy can instead
promote cancer cell survival when cells are under metabolic stress
(134). Lysosome-associated
trans-membrane 4B (LAPTM4B) is a 4-transmembrane protein localized
in late endosomes and lysosomes (135). Studies have found that abnormal
elevation of LAPTM4B is closely related to liver cancer. Moreover,
elevated LAPTM4B has also been observed in breast, lung, ovarian
and colon cancers (132),
suggesting that abnormal expression of LAPTM4B stimulates normal
cell mutation, and LAPTM4B also promotes cancer cell proliferation,
migration and invasion (136,137). LAPTM4B is important to
EGFR-mediated cell survival. It is suggested that LAPTM4B promotes
cancer cell proliferation by upregulating PI3K/AKT signaling
(136), and promotes active EGFR
signaling by blocking EGF-stimulated EGFR intraluminal sorting and
lysosomal degradation (132).
Serum starvation increases EGFR endosomal accumulation and enhances
the correlation between LAPTM4B and EGFR, whereas EGF stimulation
reduces the interaction between the two, suggesting that LAPTM4B
preferentially interacts with inactive EGFR and that EGFR-dependent
autophagy initiation may be associated with LAPTM4B-mediated
endosome localization of EGFR. Upon serum starvation, LAPTM4B
senses EGFR inactivation on endosomes and selectively complexes
with inactive EGFR, and recruit the Sec5 exocyst subcomplex
(137). This EGFR complex binds
to the autophagy inhibitor Rubicon, causing it to dissociate from
beclin-1, releasing the Rubicon-free beclin-1 complex to initiate
autophagy. Under nutrient-rich conditions, activated EGFR inhibits
autophagy through agonist-stimulated EGFR signaling. Under
metabolic stress conditions, inactivated EGFR promotes the survival
of cancer cells under starvation by activating autophagy (138–140). Therefore, inactivated EGFR may
have dual roles in tumorigenesis, that is, to promote autophagy to
inhibit tumorigenesis when tumorigenesis has not yet occurred,
while its autophagy activation promotes tumor cell survival when
tumorigenesis occur (Fig. 3).
Effects of other types of autophagy and
apoptosis on tumorigenesis
Glycolysis is a major determinant of mitotic
survival. PFKFB3 is a key regulator of the glycolytic kinase
phosphofructokinase-1 (PFK1), is often overexpressed in cancer
cells leading to the Warburg effect, a metabolic shift from
oxidative stress to rapid glucose extraction, glycolysis and
lactate export. This is characteristic of numerous tumor cells
(141,142). A recent study found that the high
expression of PFKFB3 is closely related to the incidence and poor
prognosis of nasopharyngeal carcinoma (143). PFKFB3 is known to be regulated by
AMPK and p38 MAPK in a phosphorylation-dependent manner in cancer.
AMPK-dependent phosphorylation of PFKFB3 may help enhance
glycolysis during mitosis to promote cell survival. AMPK is a major
homeostatic regulator of cellular ATP levels, and its activity is
enhanced during mitosis (144),
and an increase in the AMP/ATP ratio during mitotic arrest may
contribute to AMPK activation (145). Mitotic cells may be more
sensitive to AMPK due to the absence of the nuclear envelope. AMPK
activation can significantly increase glucose uptake and
glycolysis, and can promote more energy-efficient oxidative
metabolism by upregulating mitochondrial biogenesis and oxidase
expression (146). Activation of
AMPK and subsequent glycolysis switches observed in mitotic cells
increase the possibility of energy-dependent pathway survival in
mitosis. In the context of mitochondrial dysfunction, AMPK triggers
glycolysis and further promotes autophagy during mitotic delay.
From this perspective, activation of AMPK and autophagy appears to
contribute to tumor cell survival, but there is ample evidence that
AMPK activation inhibits the development of numerous cancers and
tumors (147). AMPK activity is
necessary and sufficient for activation of pro-apoptotic proteins
(such as Bim or Bmf) and for cell death. Studies have found that
autophagy can regulate cell survival during mitosis. Since Raptor
is a member and active regulator of mTORC1 that inhibits autophagy,
knockdown of Raptor resulted in early mitotic death, whereas
downregulation of Ulk1, Vps34, or beclin-1 prevented cell death
during mitosis.
Atg5 plays a very important role in autophagy. In
addition to promoting autophagy, studies have also found that it
has important pro-apoptotic properties. A previous study found loss
of Atg5 associated with melanoma development and poorer overall
survival (148). The Atg5-Atg12
complex triggers autophagic cell death through the interaction of
Atg5 with FADD, which has been shown to have a critical role in
interferon-γ-induced cell death. Atg5 is also a substrate for
calpain, and the truncated form of Atg5 is generated by
calpain-dependent cleavage at the Thr 193 position, a pro-apoptotic
molecule that translocates to mitochondria. The pro-apoptotic
activity of the truncated form of Atg5 can be attributed to
inactivation of Bcl-x binding, as the truncated calpain-cleavage
form of Atg5 can not only induce apoptosis but also sensitize tumor
cells to anticancer drug treatment (149). In addition, studies have found
that other autophagy proteins also have pro-apoptotic functions
after being cleaved by caspases. For example, the autophagy protein
beclin-1 is cleaved by caspases to generate a pro-apoptotic BH3
domain, and BH3 localizes to mitochondria in cells, leading to
increased mitochondrial permeability and promoting the release of
Cyt c. Similarly, ATG4D acquired pro-apoptotic ability after being
cleaved by caspase 3. The aforementioned increase in the expression
level of autophagy proteins not only promotes autophagy, but also
produces apoptotic protein precursors for apoptosis, which can
accelerate apoptosis upon activation of caspases (17).
Abnormalities of the PI3K-AKT
signaling axis
Abnormal activation of PI3K-AKT signaling axis is
closely related to the occurrence of breast cancer (150), and it was found that PI3K/AKT
signaling, a signaling pathway that inhibits apoptosis, also
inhibits autophagy. AKT inhibits autophagy in a PI3K-dependent
manner, AKT phosphorylates Bcl-1-related agonist of death,
apoptosis signal-regulating kinase 1 (ASK1 also known as MAP3K5),
human caspase 9, and E3 ubiquitin ligase MDML, thereby inhibiting
apoptosis. AKT also inhibits apoptosis by promoting the degradation
of TKB, thereby activating NF-kB and inhibiting apoptosis by
transcribing anti-apoptotic genes. It was previously revealed that
the inhibition of autophagy by Akt can be mediated not only by
activating mTOR (151), but
active Akt can also inhibit autophagy through an mTOR-independent
mechanism. Studies also found that beclin-1 is phosphorylated by
Akt at residues 295 (and possibly 234) in an mTOR-independent
manner. Beclin-1 interacts with 14-3-3 protein via phosphorylation
sites S234 and S295, and this interaction is negatively regulated
by starvation and Akt inhibition. Active Akt promotes the
interaction of beclin-1 with vimentin via the 14-3-3 protein
(151,152). And the mechanism through which
active Akt regulates the interaction of beclin-1 with 14-3-3
vimentin is through the intermediate filament to inhibit autophagy
and Akt-mediated transformation. Expression of beclin-1 mutants
resistant to Akt-mediated phosphorylation increases autophagy and
inhibits Akt-driven tumorigenesis. Akt signaling, intermediate
filaments, and 14-3-3 proteins may be involved in autophagy
inhibition and tumorigenesis mechanisms through regulation of the
beclin-1 complex (152). AKT1
inhibits autophagy in fibroblasts, reduces co-immunoprecipitation
of class III PI3K Vps34 with beclin-1 and reduces
beclin-1-associated lipid kinase activity, which all suggest that
the inhibition of autophagy may be a mechanism of tumorigenesis.
The interaction between oncogenic factors and autophagy may be a
key factor regulating carcinogenesis (152,153).
A previous study found that knockout of the Bif-1
gene in mice promotes tumorigenesis, and the expression of Bif-1 is
reduced in gastric cancer. The mechanism of the inhibition of Bif-1
gene expression to promote tumorigenesis may be related to the
interaction of Bif-1 in autophagy and apoptosis. It was previously
described that Bif-1 is an important regulator involved in
autophagy: Bif-1 contains a C-terminal SH3 domain that forms a
complex with beclin-1, thereby participating in the formation of
autophagosomes (154); during
starvation, Bif-1 forms a complex with beclin-1 via UVRAG to
enhance PI3KC3 lipid kinase activity and induce autophagosome
formation. Bif-1 plays a key role in vesicle formation for coat
protein I (COPI)-mediated retrograde transport from the trans-Golgi
network to the ER, whereas beclin-1 has been shown to be localized
to the Golgi (155–158), thus Bif-1 may act as a bridge in
the formation of autophagy. In addition to being an important
regulator of autophagy, A previous study found that Bif-1 plays a
very important role in caspase-independent cell death, that is,
when the activity of Bif-1 is inhibited and the expression of
autophagy decreases, it also promotes activation of caspase-3.
Initiation of autophagy inhibits apoptosis while activating
non-caspase cell death pathways. During apoptosis induced by
endogenous death stimuli, Bif-1 localizes to mitochondria and
regulates the activation of pro-apoptotic Bax and Bak proteins
(159). The activation of Bif-1
plays a very important role in both autophagy and apoptosis, thus
the loss of Bif-1 can induce tumorigenesis.
It was recently revealed that long non-coding RNAs
(lncRNAs) can regulate a variety of cellular processes, which play
important biological functions. A recent study found that lncRNAs
NBR2 can inhibit the occurrence of liver cancer (160). Previously, it was shown that the
downregulation of lncRNAs NBR2 expression is related to the
occurrence of various tumors (161). Deficiency of NBR2 can lead to
unexamined cell cycling under energy stress conditions and promote
cell proliferation, thereby promoting tumorigenesis (162). The reason why NBR2 inhibits tumor
may be related to the fact that NBR2 can promote autophagy. A study
found that NBR2 can directly bind to the α subunit of AMPK, thus
NBR2 may promote AMPK kinase activity through the interaction with
the AMPK kinase domain as all three splicing isoforms in the NBR2
gene can induce AMPK activation. During glucose starvation, the
binding of NBR2 to AMPK is significantly enhanced, and the binding
can directly promote the activation of AMPK (161). NBR2 deficiency leads to decreased
AMPK activity, rendering AMPK unable to be activated under energy
stress conditions, resulting in enhanced mTORC1 activity, thereby
suppressing autophagy levels. When autophagy is inhibited, damaged
organelles and macromolecular substances cannot be removed by
autophagy, and harmful substances such as reactive oxygen species
produced by these substances will induce gene mutations, thereby
inducing tumorigenesis (160–162).
Impaired autophagy can lead to the accumulation of
p62, and p62 overexpression can stimulate the production of
reactive oxygen species and enhance genomic instability, thereby
promoting tumorigenesis. It was recently revealed that autophagy
defects in apoptosis-impaired tumor cells lead to increased p62
accumulation, which is necessary for tumor inducers to induce
tumorigenesis in vitro and in vivo (163). The oncogenic potential of
p62-deficient cells is reduced, thus the elimination of p62 by
autophagy can inhibit the tumorigenesis (164,165). P62 is a protein with multiple
domains involved in the activation of transcription factor NF-κB
(163). The domains of P62
include LIR, TRAF6 binding (TB) domain, PB1 domain,
ubiquitin-associated domain (UBA) and ZZ-type zinc finger domain.
P62 interacts with LC3 through the LIR signaling domain to allow
itself to be cleared by autophagy, and binds through the TB domain
to TRAF6, a lysine 63 (K63) E3 ubiquitin ligase involved in NF-κB
activation (166). After p62
binds to TRAF6, it activates TRAF6 by promoting its
oligomerization, and then induces K63 polyubiquitination of TRAF6,
which leads to the activation of NF-κB (167,168). The UBA domain of p62 helps to
improve the efficiency of p62 catalyzing TRAF6. In addition, p62
can also induce the activation of NF-κB signaling pathway through
other pathways. For example, p62 can interact with APKC signaling
molecules through the PB1 domain, which is related to interleukin-1
(IL-1), RANK ligand (RANKL) or nerve growth factor (NGF) activation
of cells to stimulate the downstream transcription factor NF-κB
signaling pathway. Activation of the NF-κB signaling pathway leads
to the abnormal expression of a series of tumor-related genes and
inhibits the apoptosis of tumor cells. Impaired autophagy leads to
accumulation of P62 to activate the NF-κB signaling pathway,
thereby inhibiting apoptosis-inducing tumor (163,168).
In conclusion, autophagy and apoptosis interact with
each other, either promoting or inhibiting. The mechanism of their
link in tumorigenesis is still being explored. Certain scholars
consider that autophagy can reduce tumorigenesis by promoting
apoptosis (17,109–113,141–143,148,149): When autophagy is inhibited, cell
apoptosis decreases, resulting in abnormal cell growth and
tumorigenesis. Other scholars consider that autophagy and apoptosis
are mutually inhibitory (68–70,73–76):
Numerous cellular molecules often promote apoptosis by inhibiting
autophagy, thereby reducing tumorigenesis. Nevertheless, more
scholars tend to consider that autophagy plays a double-edged sword
role in tumors: On the one hand, autophagy inhibits tumorigenesis
by promoting apoptosis. On the other hand, autophagy provides
energy and material support for the growth of tumor cells through
its own catabolic ability after tumorigenesis. Not only that, but
elevated levels of autophagy also lead cancer cells to develop
resistance to antitumor drugs. Besides, there is no doubt that
autophagy is of great importance in pathogenesis. It would be
interesting to investigate how to regulate the relationship between
autophagy and apoptosis, thereby inhibiting tumorigenesis. As
aforementioned, it was found that numerous protein molecules
related to tumor pathogenesis affect tumorigenesis by regulating
autophagy and apoptosis, most of which are through autophagy
protein beclin-1, apoptosis protein family Bcl-2 or interaction
between the two. The key link is the BH3 domain, which is also the
direct link between the two. The interaction between P53 in the
apoptosis family and Atg5, Atg7 and beclin-1 in autophagy protein
family is also an important connection point for regulating the
interaction between autophagy, apoptosis and tumorigenesis.
Therefore, the link between P53 and autophagy, beclin-1 and Bcl-2
may serve as one of the targets for the treatment of cancer in the
future. It is hypothesized that the dual role of autophagy in
tumors can be attributed to the different expression levels of
autophagy before and after tumorigenesis. When the tumor does not
occur and the expression of autophagy is reduced, necrotic cells
and unfolded or misfolded proteins cannot be cleared by autophagy.
The accumulation of these harmful substances in the human body
leads to gene mutation, which leads to a decrease in the expression
of apoptosis-related molecules, which leads to impaired apoptosis
and induces tumorigenesis. After apoptosis is impaired, the
interaction between autophagy proteins and apoptotic proteins is
weakened, and the level of autophagy increases. At this time,
autophagy provides conditions for cancer cells to survive under
hypoxic conditions. Therefore, there may be a possibility that the
expression level of autophagy varies greatly before tumorigenesis,
during precancerous lesions and different tumor stages. In the
future, it may be able to design an experiment to detect the levels
of autophagy and apoptosis in cells with different pathological
morphologies in tumor patients, which may provide us with a greater
understanding of the role of autophagy in tumor pathogenesis. Only
by improved understanding of the expression levels of autophagy in
different stages of tumors, new treatment and tumor prevention
solutions for autophagy can be developped. Perhaps one day in the
future, autophagy can become an important target for the treatment
of tumors. It is known that the fragment of autophagy protein
cleaved by caspases has a pro-apoptotic effect. From this
perspective, autophagy promotes apoptosis while apoptosis inhibits
autophagy. Therefore, the interesting question is, since autophagy
is inhibited by apoptosis, that is, the apoptosis promoted by
autophagy in turn inhibits itself, how does autophagy proceed? Is
there a negative feedback mechanism between autophagy and
apoptosis? Autophagy proteins are cleaved by apoptosis, and
autophagy is inhibited, which in turn stimulates the activation of
autophagy to generate more autophagic signals to promote the
synthesis of autophagic proteins. Then, the question is what are
the stimulatory signals in this negative feedback mechanism of
autophagy. Is it as it was hypothesized before? Autophagy is
required to remove apoptotic bodies, and the autophagy pathway is
thereby activated, resulting in the production of autophagic
proteins. On the one hand, autophagy proteins play the function of
autophagy to remove abnormal cells and harmful substances in the
body, thereby maintaining cell survival. On the other hand,
autophagy proteins are cleaved by caspases and play a pro-apoptotic
function. This mechanism keeps autophagy and apoptosis in a dynamic
balance to maintain cell life and death. This mechanism enables
cells in the human body to survive and die normally, thereby
preventing the occurrence of cancer. When cells are stimulated by
external stimuli, this mechanism can keep the survival and death of
cells at a normal level, so as to reduce the damage caused by
external stimulation to human body. However, when the intensity of
external stimulation exceeds this regulatory mechanism of cells, it
leads to tumorigenesis. The interaction between autophagy and
apoptosis is a very delicate process. Only by studying this
mechanism more thoroughly, a greater chance of success in future
research on anticancer drugs can be achieved.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
HX wrote the manuscript. QD took charge of the
examination and modification of manuscript. SW, BW and XH
participated in the induction of literature and part of the
examination and modification of manuscript. RS and ML participated
in the literature searching and the examination of manuscript. XL
revised the manuscript. All authors listed have made a substantial,
direct and intellectual contribution to the work and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang X, Wu D, Wang C, Luo Y, Ding X, Yang
X, Silva F, Arenas S, Weaver JM, Mandell M, et al: Sustained
activation of autophagy suppresses adipocyte maturation via a
lipolysis-dependent mechanism. Autophagy. 16:1668–1682. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Varusai TM, Jupe S, Sevilla C, Matthews L,
Gillespie M, Stein L, Wu G, D'Eustachio P, Metzakopian E and
Hermjakob H: Using reactome to build an autophagy mechanism
knowledgebase. Autophagy. 17:1543–1554. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu K, Guo C, Lao Y, Yang J, Chen F, Zhao
Y, Yang Y, Yang J and Yi J: A fine-tuning mechanism underlying
self-control for autophagy: deSUMOylation of BECN1 by SENP3.
Autophagy. 16:975–990. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thorburn A: A new mechanism for autophagy
regulation of anti-tumor immune responses. Autophagy. 16:2282–2284.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saleem M, Asif J, Asif M and Saleem U:
Amygdalin from apricot kernels induces apoptosis and causes cell
cycle arrest in cancer cells: An updated review. Anticancer Agents
Med Chem. 18:1650–1655. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang JY, Zhang YF, Meng XP and Kong XF:
Delayed effects of autophagy on T-2 toxin-induced apoptosis in
mouse primary leydig cells. Toxicol Ind Health. 35:256–263. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Girardot T, Rimmelé T, Venet F and
Monneret G: Apoptosis-induced lymphopenia in sepsis and other
severe injuries. Apoptosis. 22:295–305. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu Z, Song Y and Wang F: Rational design
of genetically encoded reporter genes for optical imaging of
apoptosis. Apoptosis. 25:459–473. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee IH, Kawai Y, Fergusson MM, Rovira II,
Bishop AJ, Motoyama N, Cao L and Finkel T: Atg7 modulates p53
activity to regulate cell cycle and survival during metabolic
stress. Science. 336:225–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dossou AS and Basu A: The emerging roles
of mTORC1 in macromanaging autophagy. Cancers (Basel). 11:14222019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bodemann BO, Orvedahl A, Cheng T, Ram RR,
Ou YH, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva
M, et al: RalB and the exocyst mediate the cellular starvation
response by direct activation of autophagosome assembly. Cell.
144:253–267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang JF, Mei ZG, Fu Y, Yang SB, Zhang SZ,
Huang WF, Xiong L, Zhou HJ, Tao W and Feng ZT: Puerarin protects
rat brain against ischemia/reperfusion injury by suppressing
autophagy via the AMPK-mTOR-ULK1 signaling pathway. Neural Regen
Res. 13:989–998. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen WR, Yang JQ, Liu F, Shen XQ and Zhou
YJ: Melatonin attenuates vascular calcification by activating
autophagy via an AMPK/mTOR/ULK1 signaling pathway. Exp Cell Res.
389:1118832020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu X, Liu JM, Song HH, Yang QK, Ying H,
Tong WL, Zhou Y and Liu ZL: Aurora-B knockdown inhibits
osteosarcoma metastasis by inducing autophagy via the mTOR/ULK1
pathway. Cancer Cell Int. 20:5752020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang H, Wen Y, Zhang M, Liu Q, Zhang H,
Zhang J, Lu L, Ye T, Bai X, Xiao G and Wang M: MTORC1 coordinates
the autophagy and apoptosis signaling in articular chondrocytes in
osteoarthritic temporomandibular joint. Autophagy. 16:271–288.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das S, Shukla N, Singh SS, Kushwaha S and
Shrivastava R: Mechanism of interaction between autophagy and
apoptosis in cancer. Apoptosis. 26:512–533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song Y, Quach C and Liang C: UVRAG in
autophagy, inflammation, and cancer. Autophagy. 16:387–388. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhong Y, Morris DH, Jin L, Patel MS,
Karunakaran SK, Fu YJ, Matuszak EA, Weiss HL, Chait BT and Wang QJ:
Nrbf2 protein suppresses autophagy by modulating Atg14L
protein-containing beclin 1-Vps34 complex architecture and reducing
intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem.
289:26021–26037. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Itakura E, Kishi C, Inoue K and Mizushima
N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell.
19:5360–5372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuijpers M and Haucke V: Neuronal
autophagy controls the axonal endoplasmic reticulum to regulate
neurotransmission in healthy neurons. Autophagy. 17:1049–1051.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayashi-Nishino M, Fujita N, Noda T,
Yamaguchi A, Yoshimori T and Yamamoto A: A subdomain of the
endoplasmic reticulum forms a cradle for autophagosome formation.
Nat Cell Biol. 11:1433–1437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ylä-Anttila P, Vihinen H, Jokitalo E and
Eskelinen EL: 3D tomography reveals connections between the
phagophore and endoplasmic reticulum. Autophagy. 5:1180–1185. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim J, Kim YC, Fang C, Russell RC, Kim JH,
Fan W, Liu R, Zhong Q and Guan KL: Differential regulation of
distinct Vps34 complexes by AMPK in nutrient stress and autophagy.
Cell. 152:290–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Munson MJ, Allen GF, Toth R, Campbell DG,
Lucocq JM and Ganley IG: mTOR activates the VPS34-UVRAG complex to
regulate autolysosomal tubulation and cell survival. EMBO J.
34:2272–2290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM,
Chait BT, Heintz N and Yue Z: Distinct regulation of autophagic
activity by Atg14L and Rubicon associated with beclin
1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 11:468–476.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Funderburk SF, Wang QJ and Yue Z: The
beclin 1-VPS34 complex-at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation-the role of ULK1 and beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma B, Cao W, Li W, Gao C, Qi Z, Zhao Y, Du
J, Xue H, Peng J, Wen J, et al: Dapper1 promotes autophagy by
enhancing the beclin1-Vps34-Atg14L complex formation. Cell Res.
24:912–924. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takahashi Y, Coppola D, Matsushita N,
Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et
al: Bif-1 interacts with beclin 1 through UVRAG and regulates
autophagy and tumorigenesis. Nat Cell Biol. 9:1142–1151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hardie DG, Schaffer BE and Brunet A: AMPK:
An energy-sensing pathway with multiple inputs and outputs. Trends
Cell Biol. 26:190–201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hardie DG: AMPK-sensing energy while
talking to other signaling pathways. Cell Metab. 20:939–952. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung TY, Ryu JE, Jang MM, Lee SY, Jin GR,
Kim CW, Lee CY, Kim H, Kim E, Park S, et al: Naa20, the catalytic
subunit of NatB complex, contributes to hepatocellular carcinoma by
regulating the LKB1-AMPK-mTOR axis. Exp Mol Med. 52:1831–1844.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li N, Wang Y, Neri S, Zhen Y, Fong LWR,
Qiao Y, Li X, Chen Z, Stephan C, Deng W, et al: Tankyrase disrupts
metabolic homeostasis and promotes tumorigenesis by inhibiting
LKB1-AMPK signalling. Nat Commun. 10:43632019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen W, Pan Y, Wang S, Liu Y, Chen G, Zhou
L, Ni W, Wang A and Lu Y: Cryptotanshinone activates AMPK-TSC2 axis
leading to inhibition of mTORC1 signaling in cancer cells. BMC
Cancer. 17:342017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gao X and Locasale JW: Serine metabolism
links tumor suppression to the epigenetic landscape. Cell Metab.
24:777–779. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheong H, Nair U, Geng J and Klionsky DJ:
The Atg1 kinase complex is involved in the regulation of protein
recruitment to initiate sequestering vesicle formation for
nonspecific autophagy in saccharomyces cerevisiae. Mol Biol Cell.
19:668–681. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ganley IG, Lam du H, Wang J, Ding X, Chen
S and Jiang X: ULK1.ATG13.FIP200 complex mediates mTOR signaling
and is essential for autophagy. J Biol Chem. 284:12297–12305. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Corona Velazquez A, Corona AK, Klein KA
and Jackson WT: Poliovirus induces autophagic signaling independent
of the ULK1 complex. Autophagy. 14:1201–1213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu Y, Nguyen PT, Wang X, Zhao Y, Meacham
CE, Zou Z, Bordieanu B, Johanns M, Vertommen D, Wijshake T, et al:
TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in
exercise. Nature. 578:605–609. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mochida K, Oikawa Y, Kimura Y, Kirisako H,
Hirano H, Ohsumi Y and Nakatogawa H: Receptor-mediated selective
autophagy degrades the endoplasmic reticulum and the nucleus.
Nature. 522:359–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujita N, Itoh T, Omori H, Fukuda M, Noda
T and Yoshimori T: The Atg16L complex specifies the site of LC3
lipidation for membrane biogenesis in autophagy. Mol Biol Cell.
19:2092–2100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vujić N, Bradić I, Goeritzer M, Kuentzel
KB, Rainer S, Kratky D and Radović B: ATG7 is dispensable for
LC3-PE conjugation in thioglycolate-elicited mouse peritoneal
macrophages. Autophagy. 17:3402–3407. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Frudd K, Burgoyne T and Burgoyne JR:
Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat
Commun. 9:952018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nath S, Dancourt J, Shteyn V, Puente G,
Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B and Melia TJ:
Lipidation of the LC3/GABARAP family of autophagy proteins relies
on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol.
16:415–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kuehl WM and Bergsagel PL: Molecular
pathogenesis of multiple myeloma and its premalignant precursor. J
Clin Invest. 122:3456–3463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
He JP, Hou PP, Chen QT, Wang WJ, Sun XY,
Yang PB, Li YP, Yao LM, Li X, Jiang XD, et al: Flightless-I blocks
p62-mediated recognition of LC3 to impede selective autophagy and
promote breast cancer progression. Cancer Res. 78:4853–4864. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu G, Wang X, Yu H, Wang C, Liu Y, Zhao R
and Zhang G: Beclin 1, LC3, and p62 expression in paraquat-induced
pulmonary fibrosis. Hum Exp Toxicol. 38:794–802. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cao K and Tait SWG: Apoptosis and cancer:
Force awakens, phantom menace, or both? Int Rev Cell Mol Biol.
337:135–152. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Boege Y, Malehmir M, Healy ME, Bettermann
K, Lorentzen A, Vucur M, Ahuja AK, Böhm F, Mertens JC, Shimizu Y,
et al: A dual role of caspase-8 in triggering and sensing
proliferation-associated DNA damage, a key determinant of liver
cancer development. Cancer Cell. 32:342–359.e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
D'Orsi B, Mateyka J and Prehn JHM: Control
of mitochondrial physiology and cell death by the Bcl-2 family
proteins Bax and Bok. Neurochem Int. 109:162–170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sunilkumar D, Drishya G, Chandrasekharan
A, Shaji SK, Bose C, Jossart J, Perry JJP, Mishra N, Kumar GB and
Nair BG: Oxyresveratrol drives caspase-independent apoptosis-like
cell death in MDA-MB-231 breast cancer cells through the induction
of ROS. Biochem Pharmacol. 173:1137242020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Roumane A, Berthenet K, El Fassi C and
Ichim G: Caspase-independent cell death does not elicit a
proliferative response in melanoma cancer cells. BMC Cell Biol.
19:112018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chang WL, Cui L, Gu Y, Li M, Ma Q, Zhang
Z, Ye J, Zhang F, Yu J and Gui Y: TBC1D20 deficiency induces
Sertoli cell apoptosis by triggering irreversible endoplasmic
reticulum stress in mice. Mol Hum Reprod. 25:773–786. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiong Y, Jin E, Yin Q, Che C and He S:
Boron attenuates heat stress-induced apoptosis by inhibiting
endoplasmic reticulum stress in mouse granulosa cells. Biol Trace
Elem Res. 199:611–621. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bromati CR, Lellis-Santos C, Yamanaka TS,
Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF
and Bordin S: UPR induces transient burst of apoptosis in islets of
early lactating rats through reduced AKT phosphorylation via
ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr
Comp Physiol. 300:R92–R100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chen J, Xie JJ, Shi KS, Gu YT, Wu CC, Xuan
J, Ren Y, Chen L, Wu YS, Zhang XL, et al: Glucagon-like peptide-1
receptor regulates endoplasmic reticulum stress-induced apoptosis
and the associated inflammatory response in chondrocytes and the
progression of osteoarthritis in rat. Cell Death Dis. 9:2122018.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ohoka N, Yoshii S, Hattori T, Onozaki K
and Hayashi H: TRB3, a novel ER stress-inducible gene, is induced
via ATF4-CHOP pathway and is involved in cell death. EMBO J.
24:1243–1255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Aimé P, Karuppagounder SS, Rao A, Chen Y,
Burke RE, Ratan RR and Greene LA: The drug adaptaquin blocks
ATF4/CHOP-dependent pro-death Trib3 induction and protects in
cellular and mouse models of Parkinson's disease. Neurobiol Dis.
136:1047252020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hu F, Duan M and Peng N: Knockdown of TRB3
improved the MPP+/MPTP-induced Parkinson's disease
through the MAPK and AKT signaling pathways. Neurosci Lett.
709:1343522019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Han CW, Lee HN, Jeong MS, Park SY and Jang
SB: Structural basis of the p53 DNA binding domain and PUMA
complex. Biochem Biophys Res Commun. 548:39–46. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rubinstein AD, Eisenstein M, Ber Y, Bialik
S and Kimchi A: The autophagy protein Atg12 associates with
antiapoptotic Bcl-2 family members to promote mitochondrial
apoptosis. Mol Cell. 44:698–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kessel DH, Price M and Reiners JJ Jr: ATG7
deficiency suppresses apoptosis and cell death induced by lysosomal
photodamage. Autophagy. 8:1333–1341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lin TY, Chan HH, Chen SH, Sarvagalla S,
Chen PS, Coumar MS, Cheng SM, Chang YC, Lin CH, Leung E and Cheung
CHA: BIRC5/Survivin is a novel ATG12-ATG5 conjugate interactor and
an autophagy-induced DNA damage suppressor in human cancer and
mouse embryonic fibroblast cells. Autophagy. 16:1296–1313. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rami A and Benz A: Exclusive activation of
caspase-3 in mossy fibers and altered dynamics of autophagy markers
in the mice hippocampus upon status epilepticus induced by kainic
acid. Mol Neurobiol. 55:4492–4503. 2018.PubMed/NCBI
|
|
72
|
Shravage BV, Hill JH, Powers CM, Wu L and
Baehrecke EH: Atg6 is required for multiple vesicle trafficking
pathways and hematopoiesis in Drosophila. Development.
140:1321–1329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Qiu Y, Li C, Wang Q, Zeng X and Ji P:
Tanshinone IIA induces cell death via beclin-1-dependent autophagy
in oral squamous cell carcinoma SCC-9 cell line. Cancer Med.
7:397–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chang CH, Lee CY, Lu CC, Tsai FJ, Hsu YM,
Tsao JW, Juan YN, Chiu HY, Yang JS and Wang CC: Resveratrol-induced
autophagy and apoptosis in cisplatin-resistant human oral cancer
CAR cells: A key role of AMPK and Akt/mTOR signaling. Int J Oncol.
50:873–882. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Dagdeviren Ozsoylemez O and Ozcan G:
Effects of colchicum baytopiorum leaf extract on cytotoxicity and
cell death pathways in C-4 I and Vero cell lines. J BUON.
26:1135–1137. 2021.PubMed/NCBI
|
|
76
|
Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li
T, Li H, Li Q, Wang X, Uchiyama Y, et al: Neuroprotection by
selective neuronal deletion of Atg7 in neonatal brain injury.
Autophagy. 12:410–423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nezis IP, Shravage BV, Sagona AP, Lamark
T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH and
Stenmark H: Autophagic degradation of dBruce controls DNA
fragmentation in nurse cells during late Drosophila melanogaster
oogenesis. J Cell Biol. 190:523–531. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gump JM, Staskiewicz L, Morgan MJ, Bamberg
A, Riches DW and Thorburn A: Autophagy variation within a cell
population determines cell fate through selective degradation of
Fap-1. Nat Cell Biol. 16:47–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Doherty J and Baehrecke EH: Life, death
and autophagy. Nat Cell Biol. 20:1110–1117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Pagliarini V, Wirawan E, Romagnoli A,
Ciccosanti F, Lisi G, Lippens S, Cecconi F, Fimia GM, Vandenabeele
P, Corazzari M and Piacentini M: Proteolysis of Ambra1 during
apoptosis has a role in the inhibition of the autophagic
pro-survival response. Cell Death Differ. 19:1495–1504. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Toton E, Lisiak N, Sawicka P and
Rybczynska M: Beclin-1 and its role as a target for anticancer
therapy. J Physiol Pharmacol. 65:459–467. 2014.PubMed/NCBI
|
|
82
|
Miller DR and Thorburn A: Autophagy and
organelle homeostasis in cancer. Dev Cell. 56:906–918. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Marei HE, Althani A, Afifi N, Hasan A,
Caceci T, Pozzoli G, Morrione A, Giordano A and Cenciarelli C: p53
signaling in cancer progression and therapy. Cancer Cell Int.
21:7032021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wilson AJ, Gupta VG, Liu Q, Yull F,
Crispens MA and Khabele D: Panobinostat enhances olaparib efficacy
by modifying expression of homologous recombination repair and
immune transcripts in ovarian cancer. Neoplasia. 24:63–75. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lopez J and Tait SW: Mitochondrial
apoptosis: Killing cancer using the enemy within. Br J Cancer.
112:957–962. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang K, Zhan Y, Chen B, Lu Y, Yin T, Zhou
S, Zhang W, Liu X, Du B, Wei X and Xiao J: Tubeimoside I-induced
lung cancer cell death and the underlying crosstalk between
lysosomes and mitochondria. Cell Death Dis. 11:7082020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Feng X, Yu Y, He S, Cheng J, Gong Y, Zhang
Z, Yang X, Xu B, Liu X, Li CY, et al: Dying glioma cells establish
a proangiogenic microenvironment through a caspase 3 dependent
mechanism. Cancer Lett. 385:12–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Song P, Li Y, Dong Y, Liang Y, Qu H, Qi D,
Lu Y, Jin X, Guo Y, Jia Y, et al: Estrogen receptor β inhibits
breast cancer cells migration and invasion through CLDN6-mediated
autophagy. J Exp Clin Cancer Res. 38:3542019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bhutia SK, Mukhopadhyay S, Sinha N, Das
DN, Panda PK, Patra SK, Maiti TK, Mandal M, Dent P, Wang XY, et al:
Autophagy: Cancer's friend or foe? Adv Cancer Res. 118:61–95. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang F, Gómez-Sintes R and Boya P:
Lysosomal membrane permeabilization and cell death. Traffic.
19:918–931. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Timofeev O, Schlereth K, Wanzel M, Braun
A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsässer HP and Stiewe
T: p53 DNA binding cooperativity is essential for apoptosis and
tumor suppression in vivo. Cell Rep. 3:1512–1525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Della-Fazia MA, Castelli M, Piobbico D,
Pieroni S and Servillo G: HOPS and p53: Thick as thieves in life
and death. Cell Cycle. 19:2996–3003. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lévy J, Cacheux W, Bara MA, L'Hermitte A,
Lepage P, Fraudeau M, Trentesaux C, Lemarchand J, Durand A, Crain
AM, et al: Intestinal inhibition of Atg7 prevents tumour initiation
through a microbiome-influenced immune response and suppresses
tumour growth. Nat Cell Biol. 17:1062–1073. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mardenborough YSN, Nitsenko K, Laffeber C,
Duboc C, Sahin E, Quessada-Vial A, Winterwerp HHK, Sixma TK, Kanaar
R, Friedhoff P, et al: The unstructured linker arms of MutL enable
GATC site incision beyond roadblocks during initiation of DNA
mismatch repair. Nucleic Acids Res. 47:11667–11680. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
de Rosa N, Rodriguez-Bigas MA, Chang GJ,
Veerapong J, Borras E, Krishnan S, Bednarski B, Messick CA, Skibber
JM, Feig BW, et al: DNA mismatch repair deficiency in rectal
cancer: Benchmarking its impact on prognosis, neoadjuvant response
prediction, and clinical cancer genetics. J Clin Oncol.
34:3039–3046. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zeng X, Yan T, Schupp JE, Seo Y and
Kinsella TJ: DNA mismatch repair initiates 6-thioguanine-induced
autophagy through p53 activation in human tumor cells. Clin Cancer
Res. 13:1315–1321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zeng X and Kinsella TJ: A novel role for
DNA mismatch repair and the autophagic processing of chemotherapy
drugs in human tumor cells. Autophagy. 3:368–370. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jacob S, Aguado M, Fallik D and Praz F:
The role of the DNA mismatch repair system in the cytotoxicity of
the topoisomerase inhibitors camptothecin and etoposide to human
colorectal cancer cells. Cancer Res. 61:6555–6562. 2001.PubMed/NCBI
|
|
101
|
Kang C, Kang M, Han Y, Zhang T, Quan W and
Gao J: 6-Gingerols (6G) reduces hypoxia-induced PC-12 cells
apoptosis and autophagy through regulation of miR-103/BNIP3. Artif
Cells Nanomed Biotechnol. 47:1653–1661. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zeng X and Kinsella TJ: BNIP3 is essential
for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy
following DNA mismatch repair processing. Cell Res. 20:665–675.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Burton TR and Gibson SB: The role of Bcl-2
family member BNIP3 in cell death and disease: NIPping at the heels
of cell death. Cell Death Differ. 16:515–523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen BC, Weng YJ, Shibu MA, Han CK, Chen
YS, Shen CY, Lin YM, Viswanadha VP, Liang HY and Huang CY: Estrogen
and/or estrogen receptor α inhibits BNIP3-induced apoptosis and
autophagy in H9c2 cardiomyoblast cells. Int J Mol Sci. 19:12982018.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Yuan J, Luo K, Zhang L, Cheville JC and
Lou Z: USP10 regulates p53 localization and stability by
deubiquitinating p53. Cell. 140:384–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yang R, Chen H, Xing L, Wang B, Hu M, Ou
X, Chen H, Deng Y, Liu D, Jiang R and Chen J: Hypoxia-induced
circWSB1 promotes breast cancer progression through destabilizing
p53 by interacting with USP10. Mol Cancer. 21:882022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cui L, Song Z, Liang B, Jia L, Ma S and
Liu X: Radiation induces autophagic cell death via the p53/DRAM
signaling pathway in breast cancer cells. Oncol Rep. 35:3639–3647.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Robin M, Issa AR, Santos CC, Napoletano F,
Petitgas C, Chatelain G, Ruby M, Walter L, Birman S, Domingos PM,
et al: Drosophila p53 integrates the antagonism between autophagy
and apoptosis in response to stress. Autophagy. 15:771–784. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jiang Y, Woosley AN, Sivalingam N,
Natarajan S and Howe PH: Cathepsin-B-mediated cleavage of
disabled-2 regulates TGF-β-induced autophagy. Nat Cell Biol.
18:851–863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang J, Xie S, Yang J, Xiong H, Jia Y,
Zhou Y, Chen Y, Ying X, Chen C, Ye C, et al: The long noncoding RNA
H19 promotes tamoxifen resistance in breast cancer via autophagy. J
Hematol Oncol. 12:812019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Miracco C, Cosci E, Oliveri G, Luzi P,
Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M,
Malagnino V, et al: Protein and mRNA expression of autophagy gene
beclin 1 in human brain tumours. Int J Oncol. 30:429–436.
2007.PubMed/NCBI
|
|
113
|
Wang ZH, Xu L, Duan ZL, Zeng LQ, Yan NH
and Peng ZL: Beclin 1-mediated macroautophagy involves regulation
of caspase-9 expression in cervical cancer HeLa cells. Gynecol
Oncol. 107:107–113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhang J, Zhang S, Shi Q, Allen TD, You F
and Yang D: The anti-apoptotic proteins Bcl-2 and Bcl-xL suppress
beclin 1/Atg6-mediated lethal autophagy in polyploid cells. Exp
Cell Res. 394:1121122020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ma R, Yu D, Peng Y, Yi H, Wang Y, Cheng T,
Shi B, Yang G, Lai W, Wu X, et al: Resveratrol induces AMPK and
mTOR signaling inhibition-mediated autophagy and apoptosis in
multiple myeloma cells. Acta Biochim Biophys Sin (Shanghai).
53:775–783. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng
P, Hogan RN, Gilpin C and Levine B: Autophagy gene-dependent
clearance of apoptotic cells during embryonic development. Cell.
128:931–946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yu L, Alva A, Su H, Dutt P, Freundt E,
Welsh S, Baehrecke EH and Lenardo MJ: Regulation of an ATG7-beclin
1 program of autophagic cell death by caspase-8. Science.
304:1500–1502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Almasi S, Long CY, Sterea A, Clements DR,
Gujar S and El Hiani Y: TRPM2 silencing causes G2/M arrest and
apoptosis in lung cancer cells via increasing intracellular ROS and
RNS levels and activating the JNK pathway. Cell Physiol Biochem.
52:742–757. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jing ZF, Bi JB, Li Z, Liu X, Li J, Zhu Y,
Zhang XT, Zhang Z, Li Z and Kong CZ: Inhibition of miR-34a-5p can
rescue disruption of the p53-DAPK axis to suppress progression of
clear cell renal cell carcinoma. Mol Oncol. 13:2079–2097. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wei F, Wu Y, Wang Z, Li Y, Wang J, Shao G,
Yang Y and Shi B: Diagnostic significance of DNA methylation of
PTEN and DAPK in thyroid tumors. Clin Endocrinol (Oxf). 93:187–195.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chen Z, Fan Y, Liu X, Shang X, Qi K and
Zhang S: Clinicopathological significance of DAPK gene promoter
hypermethylation in non-small cell lung cancer: A meta-analysis.
Int J Biol Markers. 37:47–57. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zalckvar E, Berissi H, Mizrachy L,
Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R
and Kimchi A: DAP-kinase-mediated phosphorylation on the BH3 domain
of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and
induction of autophagy. EMBO Rep. 10:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Eisenberg-Lerner A and Kimchi A: PKD is a
kinase of Vps34 that mediates ROS-induced autophagy downstream of
DAPk. Cell Death Differ. 19:788–797. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Argyris PP, Slama Z, Malz C, Koutlas IG,
Pakzad B, Patel K, Kademani D, Khammanivong A and Herzberg MC:
Intracellular calprotectin (S100A8/A9) controls epithelial
differentiation and caspase-mediated cleavage of EGFR in head and
neck squamous cell carcinoma. Oral Oncol. 95:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ghavami S, Eshragi M, Ande SR, Chazin WJ,
Klonisch T, Halayko AJ, McNeill KD, Hashemi M, Kerkhoff C and Los
M: S100A8/A9 induces autophagy and apoptosis via ROS-mediated
cross-talk between mitochondria and lysosomes that involves BNIP3.
Cell Res. 20:314–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Tang Z, Chen J, Zhang Z, Bi J, Xu R, Lin Q
and Wang Z: HIF-1 α activation promotes luteolysis by enhancing ROS
levels in the corpus luteum of pseudopregnant rats. Oxid Med Cell
Longev. 2021:17649292021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Abdrakhmanov A, Yapryntseva MA, Kaminskyy
VO, Zhivotovsky B and Gogvadze V: Receptor-mediated mitophagy
rescues cancer cells under hypoxic conditions. Cancers (Basel).
13:40272021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhang J and Ney PA: Role of BNIP3 and NIX
in cell death, autophagy, and mitophagy. Cell Death Differ.
16:939–946. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Roßwag S, Sleeman JP and Thaler S:
RASSF1A-mediated suppression of estrogen receptor alpha
(ERα)-driven breast cancer cell growth depends on the hippo-kinases
LATS1 and 2. Cells. 10:28682021. View Article : Google Scholar
|
|
130
|
Maejima Y, Kyoi S, Zhai P, Liu T, Li H,
Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al: Mst1
inhibits autophagy by promoting the interaction between beclin1 and
Bcl-2. Nat Med. 19:1478–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Friedlaender A, Subbiah V, Russo A, Banna
GL, Malapelle U, Rolfo C and Addeo A: EGFR and HER2 exon 20
insertions in solid tumours: From biology to treatment. Nat Rev
Clin Oncol. 19:51–69. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Wei Y, Zou Z, Becker N, Anderson M,
Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et
al: EGFR-mediated beclin 1 phosphorylation in autophagy
suppression, tumor progression, and tumor chemoresistance. Cell.
154:1269–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Zhou K, Dichlberger A, Martinez-Seara H,
Nyholm TKM, Li S, Kim YA, Vattulainen I, Ikonen E and Blom T: A
ceramide-regulated element in the late endosomal protein LAPTM4B
controls amino acid transporter interaction. ACS Cent Sci.
4:548–558. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Li L, Shan Y, Yang H, Zhang S, Lin M, Zhu
P, Chen XY, Yi J, McNutt MA, Shao GZ and Zhou RL: Upregulation of
LAPTM4B-35 promotes malignant transformation and tumorigenesis in
L02 human liver cell line. Anat Rec (Hoboken). 294:1135–1142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yang H, Xiong F, Wei X, Yang Y, McNutt MA
and Zhou R: Overexpression of LAPTM4B-35 promotes growth and
metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer
Lett. 294:236–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Singh AB: EGFR-signaling and autophagy:
How they fit in the cancer landscape. J Adenocarcinoma. 1:92016.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yu JJ, Zhou DD, Cui B, Zhang C, Tan FW,
Chang S, Li K, Lv XX, Zhang XW, Shang S, et al: Disruption of the
EGFR-SQSTM1 interaction by a stapled peptide suppresses lung cancer
via activating autophagy and inhibiting EGFR signaling. Cancer
Lett. 474:23–35. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zhang P, Holowatyj AN, Roy T, Pronovost
SM, Marchetti M, Liu H, Ulrich CM and Edgar BA: An SH3PX1-dependent
endocytosis-autophagy network restrains intestinal stem cell
proliferation by counteracting EGFR-ERK signaling. Dev Cell.
49:574–589.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Birkeland ES, Koch LM and Dechant R:
Another consequence of the Warburg effect? Metabolic regulation of
Na+/H+ exchangers may link aerobic glycolysis
to cell growth. Front Oncol. 10:15612020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wang L, Lin Y, Zhou X, Chen Y, Li X, Luo
W, Zhou Y and Cai L: CYLD deficiency enhances metabolic
reprogramming and tumor progression in nasopharyngeal carcinoma via
PFKFB3. Cancer Lett. 532:2155862022. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhao H, Li T, Wang K, Zhao F, Chen J, Xu
G, Zhao J, Li T, Chen L, Li L, et al: AMPK-mediated activation of
MCU stimulates mitochondrial Ca2+ entry to promote
mitotic progression. Nat Cell Biol. 21:476–486. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Hardie DG, Ross FA and Hawley SA:
AMP-activated protein kinase: A target for drugs both ancient and
modern. Chem Biol. 19:1222–1236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Doménech E, Maestre C, Esteban-Martínez L,
Partida D, Pascual R, Fernández-Miranda G, Seco E, Campos-Olivas R,
Pérez M, Megias D, et al: AMPK and PFKFB3 mediate glycolysis and
survival in response to mitophagy during mitotic arrest. Nat Cell
Biol. 17:1304–1316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Galluzzi L, Bravo-San Pedro JM and Kroemer
G: Organelle-specific initiation of cell death. Nat Cell Biol.
16:728–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Codogno P and Meijer AJ: Atg5: More than
an autophagy factor. Nat Cell Biol. 8:1045–1047.
2006.García-Fernández M, Karras P, Checinska A, Cañón E, Calvo GT,
Gómez-López G, Cifdaloz M, Colmenar A, Espinosa-Hevia L, Olmeda D
and Soengas MS: Metastatic risk and resistance to BRAF inhibitors
in melanoma defined by selective allelic loss of ATG5. Autophagy
12. 1776–1790. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Oh DS and Lee HK: Autophagy protein ATG5
regulates CD36 expression and anti-tumor MHC class II antigen
presentation in dendritic cells. Autophagy. 15:2091–2106. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Martinez JD, Mo Q, Xu Y, Qin L, Li Y and
Xu J: Common genomic aberrations in mouse and human breast cancers
with concurrent P53 deficiency and activated PTEN-PI3K-AKT pathway.
Int J Biol Sci. 18:229–241. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Wang RC, Wei Y, An Z, Zou Z, Xiao G,
Bhagat G, White M, Reichelt J and Levine B: Akt-mediated regulation
of autophagy and tumorigenesis through beclin 1 phosphorylation.
Science. 338:956–959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Jian M, Yunjia Z, Zhiying D, Yanduo J and
Guocheng J: Interleukin 7 receptor activates PI3K/Akt/mTOR
signaling pathway via downregulation of Beclin-1 in lung cancer.
Mol Carcinog. 58:358–365. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Yang Z, Wu Y, Wang L, Qiu P, Zha W and Yu
W: Prokineticin 2 (PK2) rescues cardiomyocytes from high
glucose/high palmitic acid-induced damage by regulating the
AKT/GSK3β pathway in vitro. Oxid Med Cell Longev.
2020:31636292020.PubMed/NCBI
|
|
154
|
Liang C, Feng P, Ku B, Dotan I, Canaani D,
Oh BH and Jung JU: Autophagic and tumour suppressor activity of a
novel beclin1-binding protein UVRAG. Nat Cell Biol. 8:688–699.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wechman SL, Pradhan AK, DeSalle R, Das SK,
Emdad L, Sarkar D and Fisher PB: New insights into beclin-1:
Evolution and pan-malignancy inhibitor activity. Adv Cancer Res.
137:77–114. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Ren T, Takahashi Y, Liu X, Loughran TP,
Sun SC, Wang HG and Cheng H: HTLV-1 Tax deregulates autophagy by
recruiting autophagic molecules into lipid raft microdomains.
Oncogene. 34:334–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Xie T, Li SJ, Guo MR, Wu Y, Wang HY, Zhang
K, Zhang X, Ouyang L and Liu J: Untangling knots between autophagic
targets and candidate drugs, in cancer therapy. Cell Prolif.
48:119–139. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Lee JW, Jeong EG, Soung YH, Nam SW, Lee
JY, Yoo NJ and Lee SH: Decreased expression of tumour suppressor
Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric
carcinomas. Pathology. 38:312–315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Takahashi Y, Karbowski M, Yamaguchi H,
Kazi A, Wu J, Sebti SM, Youle RJ and Wang HG: Loss of Bif-1
suppresses Bax/Bak conformational change and mitochondrial
apoptosis. Mol Cell Biol. 25:9369–9382. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Sheng JQ, Wang MR, Fang D, Liu L, Huang
WJ, Tian DA, He XX and Li PY: LncRNA NBR2 inhibits tumorigenesis by
regulating autophagy in hepatocellular carcinoma. Biomed
Pharmacother. 133:1110232021. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Ulitsky I and Bartel DP: lincRNAs:
Genomics, evolution, and mechanisms. Cell. 154:26–46. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Manirujjaman M, Ozaki I, Murata Y, Guo J,
Xia J, Nishioka K, Perveen R, Takahashi H, Anzai K and Matsuhashi
S: Degradation of the tumor suppressor PDCD4 is impaired by the
suppression of p62/SQSTM1 and autophagy. Cells. 9:2182020.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Cui YH, Yang S, Wei J, Shea CR, Zhong W,
Wang F, Shah P, Kibriya MG, Cui X, Ahsan H, et al: Autophagy of the
m6A mRNA demethylase FTO is impaired by low-level
arsenic exposure to promote tumorigenesis. Nat Commun. 12:21832021.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Li P, He J, Yang Z, Ge S, Zhang H, Zhong Q
and Fan X: ZNNT1 long noncoding RNA induces autophagy to inhibit
tumorigenesis of uveal melanoma by regulating key autophagy gene
expression. Autophagy. 16:1186–1199. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Yamamoto M, Gohda J, Akiyama T and Inoue
JI: TNF receptor-associated factor 6 (TRAF6) plays crucial roles in
multiple biological systems through polyubiquitination-mediated
NF-κB activation. Proc Jpn Acad Ser B Phys Biol Sci. 97:145–160.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Kim MJ, Min Y, Kwon J, Son J, Im JS, Shin
J and Lee KY: p62 negatively regulates TLR4 signaling via
functional regulation of the TRAF6-ECSIT complex. Immune Netw.
19:e162019. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Xu Y, Liao C, Liu R, Liu J, Chen Z, Zhao
H, Li Z, Chen L, Wu C, Tan H, et al: IRGM promotes glioma M2
macrophage polarization through p62/TRAF6/NF-κB pathway mediated
IL-8 production. Cell Biol Int. 43:125–135. 2019. View Article : Google Scholar : PubMed/NCBI
|