Introduction
The interconnected network of extracellular matrix
(ECM) proteins that help comprise the non-cellular stroma, not only
provides a mechanical scaffold for cells and tissues, but can also
help facilitate the ability of cells to fine tune their capacity to
sense and respond to a variety of soluble external cues (1–3). The
remarkable diversity of signaling cascades regulated by the ECM
suggests that discrete receptor-mediated contact sites between
cells and ECM proteins may function as signaling hubs that helps
integrate the transfer of both solid-phase and soluble information
from outside the cell to the inside (3,4). These
functional signaling hubs may provide cells with the needed
signaling flexibility to survive and grow in diverse tissue
microenvironments (1–4).
The collagen family of molecules is a large group of
triple helical matrix proteins that represent a major component of
the ECM (3,5). A specific subset of the integrin
family of ECM receptors are known to bind to discrete regions of
intact triple helical collagen and facilitate signaling (3,6,7).
Evidence indicated that while the intact triple helical
conformation of collagen can stimulate signaling cascades
predominately through β1-integrins, cryptic collagen elements
exposed following structural remodeling can be recognized by a
different subset of integrins including αvβ3, which may in turn
play roles in governing cellular processes during pathological
events (3,4). These cryptic collagen elements may be
defined as either conformationally altered regions that are exposed
within the immobilized collagen fibers, or they may be released as
soluble peptide fragments (3,4). The
functional importance of these cryptic elements in biological
processes are beginning to emerge (3,4).
While proteolytic cleavage of pre-existing collagen
allows structural remodeling of the local ECM to help create less
restrictive physical barriers for migration and invasion, less is
known concerning whether soluble collagen fragments bind to cell
surface receptors and stimulate signaling cascades that potentiate
tumor growth in vivo. To this end, estimates suggested that
up to 20% of newly synthesized collagen can be degraded
intracellularly (8), however,
little is known concerning whether these degraded collagen
fragments are released from the cell and play a functional role in
regulating tumor growth. Previously, an endogenously generated
small bioactive collagen fragment that can be secreted by M2-like
macrophages was identified (9).
This 16 kDa RGDKGE-containing collagen fragment binds to and
activates integrin αvβ3 expressed on vascular endothelial cells
leading to enhanced nuclear accumulation of the Yes-associated
protein (YAP) (9). Recently, it was
also demonstrated by the authors that ovarian carcinoma cell
interactions with this collagen fragment suppressed phosphorylation
of the hippo effector kinase LATS1, and reduced the inhibitory
phosphorylation of YAP which ultimately led to its nuclear
accumulation (10). Selectively
targeting this collagen fragment in ovarian carcinoma cells
inhibited YAP nuclear accumulation and suppressed tumor growth
in vivo (10).
Among the numerous factors considered to be
controlled by YAP, is the immune checkpoint molecule programed
death ligand-1 (PD-L1), which is expressed in a number of cell
types (11–13). While the levels of soluble collagen
peptides have been correlated with resistance to immune checkpoint
inhibitors in humans (14,15), it is not known whether these
collagen peptides play a functional role in immune suppression or
directly regulate the levels of immune checkpoint molecules. Given
the importance of targeting the PD-1/PD-L1 pathway to control tumor
growth (16–18), obtaining a more in-depth
understanding of the different mechanisms by which the levels of
PD-L1 is regulated is of particular importance. Among the most
well-studied mechanisms by which the levels of PD-L1 can be
controlled include transcription, translation, post-translational
modifications and protein stability (19–21).
Studies have shown that PD-L1 can be regulated by molecules such as
growth factors and cytokines including EGF, TGFβ, IL-6 and INF-γ to
name just a few (19–21). Signaling stimulated through binding
of these factors to their respective cell surface receptors leads
to the activation of numerous pathways such as MAP/Erk, PI3K/Akt,
and JAK/Stat cascades that can ultimately contribute to the
differential control of PD-L1 (19–21).
In addition, the levels of PD-L1 can also be controlled by
alterations of protein stability through mechanism associated with
glycosylation, lysosomal-mediated degradation, ER-associated
degradation (ERAD) and proteasomal-mediated degradation (21–26).
New evidence is now emerging that in addition to growth factor and
cytokine receptor signaling, integrin receptors may also play a
role in regulating PD-L1. In fact, integrins such as α2β1 and αvβ3
have been suggested to contribute to the regulation of this
important immune checkpoint molecule (27–29).
In the present study, evidence was provided for the
first time for a previously unappreciated signaling cascade in
which β3-integrin mediated cellular interaction with the secreted
RGDKGE-containing collagen fragment stimulates an autocrine-like
signaling pathway that helps control PD-L1 by a proteasome
dependent mechanism. Inhibiting this novel autocrine-like signaling
cascade led to enhanced (protein kinase-A) PKA activity and reduced
PD-L1 levels by a proteasome-dependent process. These findings are
consistent with the concept that certain tumor cells have the
capacity to regulate β3 integrin signaling in an autocrine-like
manner by binding to this secreted soluble fragment of collagen.
This autocrine-like signaling cascade may provide tumor cells with
the ability to regulate PD-L1 stability by controlling PKA, YAP and
proteasome function. Selectively targeting the secreted
RGDKGE-containing collagen fragment inhibited tumor growth in
vivo. Taken together, our new findings may help in establishing
a novel strategy to control tumor growth by selectively disrupting
an autocrine-like signaling pathway that regulates proteasome
dependent control of PD-L1 levels.
Materials and methods
Cells and cell culture
B16F10 murine melanoma cells, 4T1 murine mammary
carcinoma cells, HUVEC human endothelial cells, RAW264.7 murine
macrophages, YUMM1.7 murine melanoma cells, A375 human melanoma
cells, C32 human melanoma cells and murine Lewis Lung Carcinoma
(LLC) cells designated LL/2 (LLC1) were obtained from the American
Type Culture Collection (ATCC; cat. no. CRL-1642). Human Dermal
Fibroblast (HDF) were obtained from ScienCell Research
Laboratories, Inc. and cultured in 2% FBS in growth medium
supplemented with Fibroblast Growth Supplements from ScienCell
Research Laboratories, Inc. YUMM1.7 cells were cultured in DMEM
F-12 medium in presence of 10% FBS, 1% NEAA and 1% Pen-Strep. 4T1
cells were cultured in RPMI-1640 medium in the presence of 5% FBS,
1% Pen-Strep and 1% sodium pyruvate. HUVECs were cultured in VCBM
media plus Endothelial Cell Growth Kit. Β16F10, RAW264.7, C32, A375
and LLC cells were cultured in DMEM plus 10% FBS 1.0% Pen-Strep and
1.0% sodium pyruvate. Human primary melanocytes were obtained from
ScienCell Research Laboratories, Inc. and cultured in Melanocyte
Media containing 0.5% FBS and Melanocyte Growth Supplement.
Animals
C57BL/6J (4–11 mice per experiment, 6–8 weeks old
and 18–20 g) and Balb/CJ female mice (7 mice per experiment; 6–8
weeks old; weight 18–20 g) were obtained from the Jackson
Laboratory (Bar Harbor, ME). Mice were housed in MaineHealth
Institute for Research pathogen-free air barrier facility, which
has a temperature range from 20–24°C, a humidity range from 30–70%
and a light/dark cycle of 14/10 h, respectively. All animal had
access to food and water continuously, with no restrictions, and
all handling and procedures were approved by the Maine Medical
Center Institutional Animal Care and Use Committee (Scarborough,
USA) under approved IACUC protocol number 2206.
Reagents, chemicals and
antibodies
Bovine serum albumin (BSA), Proteasome 20S activity
assay kit, MG132 proteasome inhibitor, Cathepsin inhibitor-I, DMSO
and crystal violet were obtained from MilliporeSigma. Anti-YAP1
(cat. no. 14074), anti-integrin β3 (cat. no. 4702s), anti-β-tubulin
(cat. no. 2145s), anti-actin (cat. no. 4907s) and anti-TATA-binding
protein (TBP) (cat. no. 44059s) antibodies were all obtained from
Cell Signaling Technology, Inc. Anti-PD-L1 antibody (cat. no.
ab26974), the PKA kinase activity assay kit and PKA inhibitor
KT5720 were all obtained from Abcam. Brefeldin-A and Super Signal
Pico Plus were obtained from Thermo Fisher Scientific, Inc. Mab
XL313 (cat. no. BE0324) and non-specific control antibody (cat. no.
BP0083) was obtained from Bio-X Cell. P2 peptide (CQGPRGDKGEC) and
control peptide CP (CQGPGGAAGGC) were from QED Bioscience. Alexa
594 labeled secondary antibody (cat. no. A11005) was from Thermo
Fisher Scientific, Inc. The PKA inhibitor H89 was obtained from
Selleck Chemicals. Function blocking β3 antibody (cat. no. 104310)
was obtained from BioLegend, Inc. Mouse PD-L1 specific short
hairpin (sh)RNAs, Coll-1α2 chain shRNAs and control non-targeting
shRNAs were obtained from OriGene Technologies, Inc.
Cytotoxicity and cell growth
assays
Sub-confluent Β16F10 melanoma cells were washed and
then resuspended in serum-free medium (DMEM) in the presence of
non-specific control antibody or anti-XL313 Mab (100 µg/ml) for 1
h. Next cells were spun down and resuspended in 2.5% growth medium
in the presence of non-specific control or anti-XL313 Mab (100
µg/ml) and added (2×103/well) to 96 well culture plates
and allowed to proliferate for 24 h. Cell growth was quantified
using Cell Proliferation Kit I (MTT) according to the
manufacturer's instructions (MilliporeSigma), with formazan
solubilized with 10% SDS and 0.01 M HCL solution provided with the
kit. The absorbance was measured at a wavelength of 600 nm. For
cytotoxicity assays, sub-confluent Β16F10 cells were washed and
resuspended in serum-free medium (DMEM) in suspension for 1 h in
the presence of non-specific control antibody or anti-XL313 Mab
(100 µg/ml). Next cells were spun down and resuspended in 1.0%
serum containing medium containing 1 mM MgCl2, 0.2 mM
MnCl2 in the presence of non-specific control or
anti-XL313 Mab (100 µg/ml) and added (2×103/well) to 96
well culture plates and allowed to incubate for 24 h. Cell
cytotoxicity was quantified using CytoTox 96 Non-radioactive
cytotoxicity assay kit according to manufactures instructions
(Promega Corporation). Cell growth and cytotoxicity was quantified
from triplicate wells and assays carried out 3 independent
times.
Western blot analysis
To determine if tumor cells could express the
RGDKGE-containing collagen fragment, cell lysates from growing
conditions and 24 h serum-free conditioned medium (CM) were
collected from equal number of Β16F10, C32, YUMM1.7, A375, 4T1,
LLC, Raw 264.7, HUVEC, human melanocytes and HDFs. Β16F10 cells
(0.5×106) were serum-starved for 1 h in suspension at
37°C. Next, cells were resuspended in 500 µl of serum-free media
and incubated over a time course (0–180 min). CM and lysates were
prepared at 0, 15, 60 and 180 min. To confirm that the RGDKGE
containing collagen fragment was secreted, Β16F10, YUMM1.7, and C32
cells were serum-starved for 1 h in suspension at 37°C with
Brefeldin-A (10 µM) or equivalent control. Next, cells were
resuspended in serum-free DMEM with Brefeldin-A (10 µM) or control
(methanol) for 15 min or 1 h and lysate and CM were prepared. In
experiments to study the impact of cathepsin on the expression of
the low molecular weight RGDKGE containing collagen fragment,
sub-confluent cultures of cells were serum-starved in suspension
for 1 h at 37°C in the presence of 25 µM of Cathepsin inhibitor or
control. Next, cells were resuspended in serum-free DMEM media
containing cathepsin inhibitor (25 µM) or control for 3 h and
lysates were prepared.
In experiments to study the mechanism by which Mab
XL313 may regulate PD-L1, cells were serum-starved in suspension
for 1 h at 37°C with 10 µM MG132, 10 µM H89, 5 µM KT5720, or the
equivalent control. Subsequently, cells were resuspended in
serum-free DMEM with the indicated inhibitors in the presence of
Mab XL313 or control antibody (100 µg/ml) for 15 min or 1 h and
cell lysates were prepared.
To examine the effects of Mab XL313 on PD-L1 levels,
cells from sub-confluent cultures were serum-starved in suspension
for 1 h at 37°C, then resuspended in serum-free DMEM media
containing Mab XL313 or control antibody (100 µg/ml) for 15 min or
1 h and cell lysates were prepared. To examine whether
RGDKGE-containing collagen fragment could counteract the effects of
Brefeldin-A on PD-L1, Β16F10 cells were serum-starved in suspension
for 1 h with Brefeldin-A (10 µM) or control. Next, cells were
resuspended in the presence of Brefeldin-A (10 µM) or control along
with P2 or CP (100 ng/ml) for 15 min and lysates were then
prepared. All cells were lysed in RIPA lysis buffer with 1X
protease inhibitor cocktail, 2 mM of PMSF, 1 mM of sodium
orthovanadate (Santa Cruz Biotechnology, Inc.). Equal amounts of
cell lysates (5–30 µg per lane), determined by BCA assay, were
separated by SDS PAGE (using 10 or 15% polyacrylamide gels).
Membranes (0.45-µM nitrocellulose) were blocked with 10% milk-TBST
for 1 h at room temperature. Primary antibodies were diluted in 5%
BSA-TBST. Primary antibodies were incubated (Mab XL313, 5 µg/ml),
anti-PD-L1 (2 µg/ml), anti YAP (1:1,000) anti-Tubulin (1:5,000),
anti-Actin (1:1,000) with membranes overnight at 4°C. Secondary
antibodies (anti-mouse HRP-labeled, cat. no. W4021; and anti-rabbit
HRP-labeled, cat. no. W4011; both diluted at 1:10,000 from Promega
Corporation) were diluted in 5% milk-TBST and incubated for 1 h at
room temperature. Western blots were performed at least 3 to 4
times and visualized by chemiluminescence detection using Super
Signal Pico Plus obtained from Thermo Fisher Scientific, Inc.
Quantification of the mean relative changes in proteins was carried
out using ImageJ software (version 5.0; National Institutes of
Health).
Preparation of tissue lysates
Female C57BL/6J or Balb/CJ mice (6–8 weeks old) were
injected with Β16F10 or 4T1 cells, respectively. Individual tumors
(n=4) were harvested and snap frozen on dry ice and ground in a
cold mortar. Ground up tissues from individual tumors were mixed
with RIPA lysis buffer, and whole tumor tissue lysates were
generated for western blot analysis. Fully de-identified snap
frozen human melanoma biopsy tissues were obtained from MaineHealth
Institute for Research Bio Bank and tissue lysates were generated
as aforementioned.
Cell line generation
Cd274 mouse shRNA plasmid (TL503436) specific for
Pd-l1 or scrambled non-targeting (NT) shRNA plasmid (TL30021)
containing the backbone cassette in PGFP-C-shLenti shRNA vectors
(2.5 µg of plasmids per 6-well plates) were transfected to Β16F10
cells at 37°C with TurboFectin™ 8.0 transfection reagent (cat. no.
TF81001) from OriGene Technologies, Inc. according to the
manufacturer's instructions. After 24 h of transfection, the cells
were selected with 3 µg/ml of puromycin containing growth medium to
generate stably expressing shRNA cell lines. The effective
targeting sequence of Pd-l1 gene is
5′-GTGGTGGAGTATGGCAGCAACGTCACGAT-3′. The Col-1α2 shRNA plasmid
(TL500407) or scrambled negative control non-targeting shRNA
plasmid (TR30021) containing the backbone cassette in
PGFP-C-shLenti shRNA vectors, were obtained from OriGene
Technologies, Inc. Β16F10 cells were transfected with lipofectamine
3000 transfection kit (L3000) from Invitrogen by Thermo Fisher
Scientific, Inc. After 24 h of transfection, the cells were
selected with 3 µg/ml of puromycin containing growth medium. The
effective targeting sequence of Coll-1 α2 gene is
5′-AGTGGTCCTCAAGGCATCCGAGGTGACAA-3′. Next, the same number of
actively growing cells were seeded for determining knock down
efficiency by reverse transcription-quantitative (RT-q)PCR and the
RGDKGE-containing collagen fragment expression by western blotting.
The murine RT-qPCR primers specific for collagen-1 α2 chain were
forward, 5′-CTAGCCAACCGTGCTTCTCA-3′ and reverse, 5′
TCTCCTCATCCAGGTACGCA-3′. For the housekeeping gene of mouse
Cyclophilin, the primers were forward, 5′-CCACCGTGTTCTTCGACAT-3′
and reverse, 5′-CAGTGCTCAGAGCTCGAAAG-3′. RT-qPCR assays were
repeated at least 3 times.
Nuclear protein extraction
To examine the effects of blocking cellular
interactions with the secreted RGDKGE-containing collagen fragment
on nuclear accumulation of YAP, sub-confluent Β16F10 cells were
washed and serum-starved for 1 h in suspension at 37°C and cells
were then resuspended in media containing Mab XL313 or nonspecific
control antibody (100 µg/ml) for 15 min. The nuclear and
cytoplasmic proteins were extracted using NE-PER nuclear and
cytoplasmic extraction reagents from Thermo Fisher Scientific, Inc.
according to the manufacturer's instructions. The nuclear
extractions were examined by western blot analysis with anti-YAP or
anti-TBP (TATA binding protein) antibody.
RNA isolation, cDNA synthesis and
RT-qPCR
Sub-confluent Β16F10 cells were washed and
serum-starved in suspension for 1 h at 37°C. Next, cells were
resuspended in media containing Mab XL313 or control antibody (100
µg/ml). To examine the effects of Mab XL313 on PD-L1 levels, cells
were lysed after incubation over a time course of 15 min, 1 and 3 h
using RNA isolation lysis buffer. RNA was isolated by using RNase
plus mini kit from Qiagen following the manufacturer's protocol.
Total RNA (1.0 µg) was synthesized with iScript cDNA synthesis kit
from Bio-Rad Laboratories, Inc according to the manufacturer's
instruction. Equivalent amount of cDNA was used for RT-qPCR, and
the murine RT-qPCR primers used for mouse Pd-l1 were
forward, 5′-TGCGGACTACAAGCGAATCACG-3′ and reverse,
5′-CTCAGCTTCTGGATAACCCTCG-3′ and for the housekeeping gene mouse
b2m, the primers were forward, 5′-CTGACCGGCCTGTATGCTAT-3′ and
reverse, 5′-CCGTTCTTCAGCATTTGGAT-3′. RT-qPCR assays were repeated 5
times. The expression of Pd-l1 genes was examined by RT-qPCR using
Bio-Rad CFX connect real time systems with a SYBR green-based 3
step amplification protocol. The reaction was set up by incubation
at 95°C for 3 min for initial denaturation, followed by 45 cycles
of denaturation, annealing, and extension steps (95°C for 10 sec,
57°C for 30 sec and 72°C for 30 sec), with data collection at each
annealing step. To ensure the specificity of the amplifications,
each amplification reaction was followed by a melting phase,
according to the default settings of the CFX connect instrument
(from 65 to 95°C), and each melting curve was assessed for the
presence of a single peak. The transcript level of the Pd-l1 gene
was normalized to the transcript level of house-keeping gene b2m.
The method of quantification used was 2−ΔΔCq (30).
Immunofluorescence staining
Sub-confluent Β16F10 melanoma cells were harvested
from culture and washed. Β16F10 cells were serum-starved for 1 h in
suspension and plated on 10% serum-coated slides and allowed to
attach for 1 h. Cells were washed and fixed with 50% acetone and
50% methanol for 10 min. Next cells were washed and incubated for 1
h at room temperature with anti-PD-L1 antibody (cat. no.
NBP1-76769; Novus Biologicals, LLC) (10 µg/ml diluted in 1.0% BSA
in PBS). Next, cells were washed 3 times in PBS and incubated for
30 min with both alexa-594 secondary antibody (cat. no. A11005)
(1:5,000) and DAPI (1:3,000) from Thermo Fisher Scientific, Inc.
Finally, cells were washed and mounted for analysis.
Proteasome 20S activity assay
To examine whether the effects of Mab XL313 on PD-L1
were due to proteasome-mediated degradation, a proteasome activity
assay kit from MilliporeSigma was used. Briefly, Β16F10,
Β16F10-YAP-K/D (Yap knock down), and Β16F10-NT (non-targeting
vector) cells were serum-starved for 1 h at 37°C in suspension.
Afterwards, cells were resuspended in serum-free DMEM containing
Mab XL313, non-specific control IgG (100 µg/ml) or anti-β3 antibody
(50 µg/ml) for 15 min. For stimulation with control peptide CP or
collagen peptide P2, cells were stimulated with peptides (100
ng/ml) for 5 min. Next, cells were spun down, and media was
replaced with 300 µl of Proteasome Loading Solution and 100 µl was
added to a 96 well flat bottom black plate (in triplicate)
according to the manufacturer's protocol. The plate was incubated
at 37°C for 1 h and then the fluorescence intensity was measured
using an excitation wavelength at 490 nm and emission wavelength at
525 nm. Proteasome activity assays were performed in triplicate 3
to 4 times.
PKA activity assay
To examine whether the Mab XL313 or the collagen
peptide P2 impacts PKA activity, a PKA activity assay obtained from
Abcam was performed using cell lysates. Briefly, Β16F10, Β16F10-NT
and Β16F10-YAP-K/D cell lysates were prepared by serum-starving
cells in suspension for 1 h at 37°C. Next, cells were resuspended
in serum-free DMEM containing Mab XL313, non-specific control IgG
or function blocking anti-β3 antibody for 15 min. In similar
studies, Β16F10 cells were serum-starved in suspension for 1 h at
37°C. Next, cells were resuspended in serum-free DMEM containing
either control peptide CP (100 ng/ml) or collagen peptide P2 (100
ng/ml) for 15 min. Cells were then lysed in RIPA lysis buffer with
1X protease inhibitor cocktail, 2 mM of PMSF, 1 mM of sodium
orthovanadate and stored at −80°C. The PKA activity assay was
performed according to the manufacturer's instructions. PKA
activity assays were performed in triplicate 3 to 4 times.
Cell binding assays
A total of 48 well non-tissue culture plates were
coated with 100 µg/ml of control peptide (CP) or RGDKGE-containing
peptide (P2). Tumor cells were suspended in adhesion buffer (DMEM)
containing 1 mM MgCl2, 0.2 mM MnCl2 and 0.5%
BSA, and 1×105 cells were added to the wells in the
presence of 100 µg/ml of Mab XL313, anti-β3 integrin antibody,
anti-β1 integrin antibody or non-specific control IgG and allowed
to bind for 10–20 min at 37°C. Non-attached cells were removed and
attached cells were stained with crystal violet as previously
described (10,31). Cell binding was quantified by
measuring the optical density of eluted dye as previously described
(10,31). Cell binding assays were performed at
least 3 times with triplicate wells per condition.
Viral vectors for cell line
generation
Lentiviral vectors (pLKO.1 based) encoding short
hairpin RNAs (shRNAs) specific for YAP or a control
non-targeting construct developed by the RNAi Consortium were
obtained from GE Healthcare Life Science. The effective targeting
sequence of YAP gene was 5′-TTCTTTATCTAGCTTGGTGGC-3′.
Lentivirus was packaged in the recombinant viral vector core
facility at MaineHealth Institute for Research. Multiplicity of
infection (MOI) was 1. Β16F10 cells were transduced with shRNA
overnight in a viral culture room incubator. The next day,
transduced cells were washed twice with PBS and fresh growth medium
was added in the presence of 3 µg/ml of puromycin (Invitrogen;
Thermo Fisher Scientific, Inc.) and brought to the lab cell culture
incubator. After 7–10 days of selection, the established stable
cell pool was used to determine the knockdown efficiency by western
blot. Once knockdown efficiency was confirmed, transduced cells
were used for further experiments within approximately 2 to 3
weeks. Cells were periodically tested prior to carrying out
experiments to confirm knockdown efficiency by western blot.
Tumor growth assays
Tumor growth assays were performed as previously
described (10,30). Briefly, mice were injected
subcutaneously with either 3.5×105 Β16F10 cells,
non-target Β16F10 cells, 3.5×106 PD-L1 knock-down Β16F10
(N=8–10 C57BL/6 mice per condition) or 3×105 4T1 mammary
carcinoma cells resuspended in 100 µl of sterile PBS. Mice were
allowed to form palpable tumors for 3 or 7 days, then the mice were
intraperitoneally injected with Mab XL313 or nonspecific control
antibody (0–250 µg/mouse) 3X times per week for 14 or 21 days.
Tumor size was measured with caliper and the tumor volume was
calculated using the formula V=L2 × W/2, where V=volume,
L=length, W=width. All mice were euthanized by inhalant isoflurane
to effect by administering isoflurane from a 100% stock solution as
a vapor in a closed chamber until lack of breathing and heartbeat
was observed, which was followed by cervical dislocation. Death was
verified by lack of breathing and lack of heartbeat determined by
palpation in accordance with MaineHealth IACUC approved protocol
number 2206.
Statistical analysis
Statistical analysis was performed using the
Prism/Graph Pad (version 6; GraphPad Software, Inc.) statistical
program for Macintosh computers. Data were analyzed for statistical
significance using unpaired Student's t-test with comparison to the
control group, performed separately at each time point. For
experiments in which there were more than two groups, we performed
a one-way ANOVA and then used a Dunnett's multiple comparison test
to adjust for comparison of each level to the control group.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Detection of the RGDKGE-containing
collagen fragment in malignant tumor cells and effects of targeting
it on tumor growth in vivo
While it is well-accepted that cellular interactions
with triple helical collagen can regulate the behavior of distinct
tumors (31–34), less is known concerning the impact
of endogenously secreted soluble collagen fragments have on
malignant cells. While stromal cells such as fibroblasts represent
a major source of collagen, other cells types within tumor lesions
also express collagen, including alternatively activated M2-like
macrophages (9,35,36).
We identified a bioactive 16 kDa RGDKGE-containing collagen
fragment that can be secreted from M2-like macrophages (9). To determine the expression of this
collagen fragment within different tumor cells, we analyzed
serum-free CM and whole cell lysates from a panel of different
tumor cell lines. To facilitate these studies, Mab XL313 was used,
which specifically recognizes the RGDKGE sequence within
proteolyzed collagen, but does not bind to intact collagen or other
RGD-containing ECM molecules (9).
The low molecular weight 16 kDa RGDKGE-containing collagen fragment
was detected within whole cell lysates and in serum-free CM from
several tumor cell lines including Β16F10 melanoma, 4T1 mammary
carcinoma and LLC (Fig. 1A). By
contrast, while this low molecular weight collagen fragment was
detected in RAW 264.7 macrophages, it was not detected in normal
human melanocytes, endothelial cells (HUVEC) or HDF (Fig. 1B). The RGDKGE-containing collagen
fragment was also detected within both whole cell lysates and
serum-free CM in additional tumor cell lines including C32 and
YUMM1.7 cells, while A375 cells expressed minimal if any detectable
levels (Fig. 1C). In addition, the
RGDKGE-containing collagen fragment was also detected in whole
tissue lysates of 4T1 mammary carcinomas and Β16F10 melanomas
growing in vivo (Fig. 1D and
E), as well as in malignant human melanoma biopsies (Fig. 1F). Collectively, these data
indicated that the RGDKGE-containing collagen fragment can be
differentially generated by several different tumor cell lines and
malignant human melanomas.
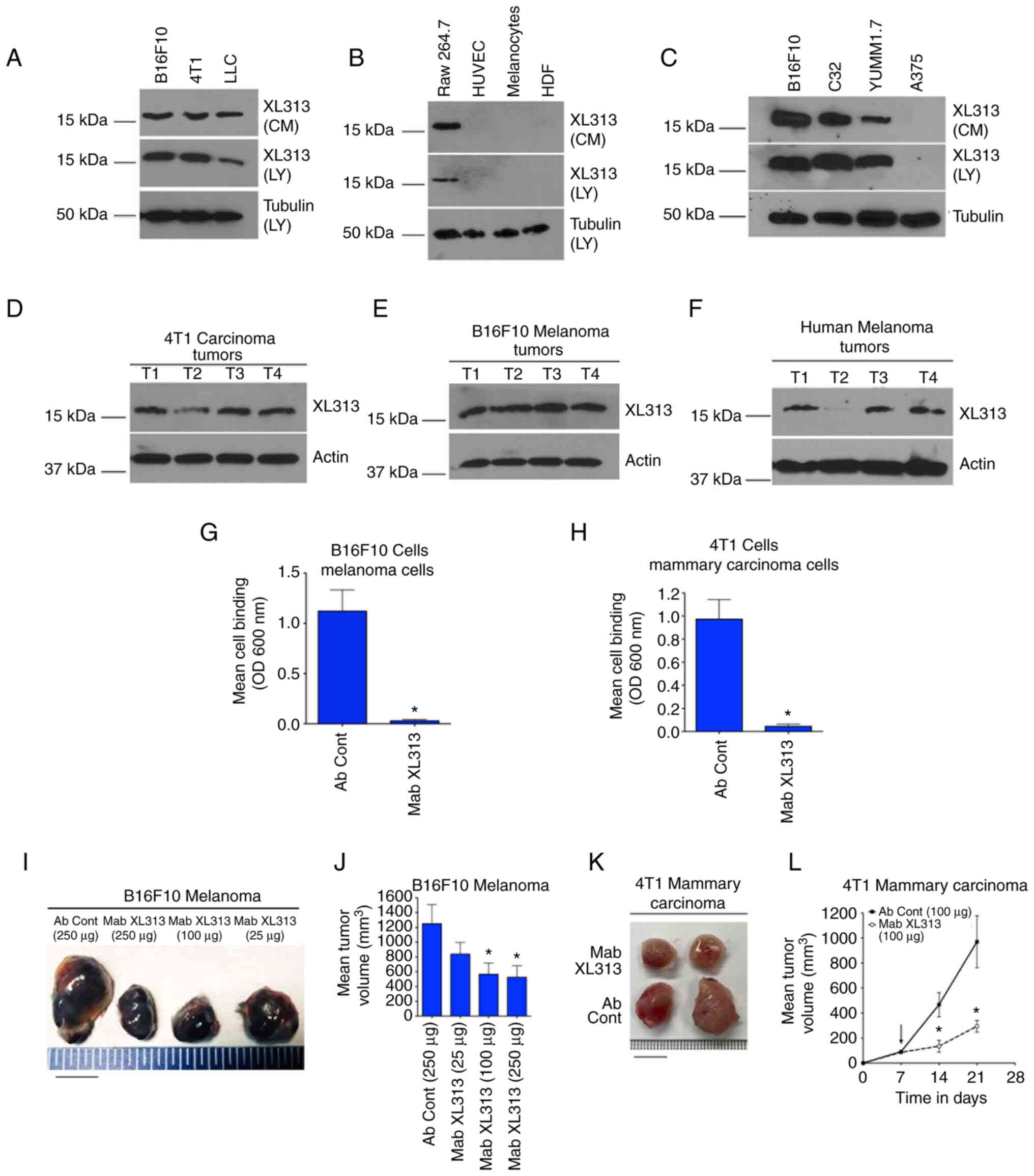 | Figure 1.RGDKGE-containing collagen fragment
in distinct tumor types. (A-C) Western blot analysis of LY and
serum-free CM for the 16 kDa RGDKGE-containing collagen fragment or
loading control tubulin in (A) Β16F10 melanoma, 4T1 mammary
carcinoma and LLC; in (B) macrophages (RAW 264.7), endothelial
cells (HUVEC), normal human melanocytes and HDF and in (C) Β16F10
melanoma, C32 melanoma, YUMM1.7 melanoma and A375 melanoma cells.
(D and E) Western blot analysis of lysates from individual solid
(D) 4T1 and (E) Β16F10 tumors growing in mice for the 16 kDa
RGDKGE-containing collagen fragment or loading control actin. (F)
Western blot analysis of lysates from individual biopsies of
malignant human melanoma tumors for the 16 kDa RGDKGE-containing
collagen fragment or loading control actin. (G and H)
Quantification of (G) Β16F10 and (H) 4T1 cell binding to collagen
peptide P2 in the presence of non-specific control antibody (Ab
Cont) or anti-RGDKGE antibody (Mab XL313). Data bars represent the
mean ± SEM cell binding from triplicate wells. (I) Example of
changes in Β16F10 tumor size following treatment of mice with
different doses of Mab XL313 or control antibody (Ab Cont) (scale
bar, 1 cm). (J) Quantification of the dose-dependent effects of
anti-RGDKGE collagen fragment antibody (Mab XL313) or non-specific
control antibody (Ab Cont) on the growth Β16F10 tumors in
vivo. Data bars indicate the mean ± SEM tumor volumes (n=4–6
per group). P-value represents comparison of each group to antibody
control. (K) Example of changes in 4T1 tumor size following
treatment of mice with Mab XL313 or control antibody (Ab Cont)
(scale bar, 1 cm). (L) Quantification of the effects of anti-RGDKGE
collagen fragment antibody (Mab XL313) or non-specific control
antibody (Ab Cont) on the growth 4T1 tumors in vivo. Data
bars indicate the mean ± SEM tumor volumes (n=7 per group).
*P<0.05 vs. control at each time point. LY, whole cell lysates;
CM, conditioned medium; LLC, Lewis Lung Carcinoma; HDF, human
dermal fibroblasts; Mab, monoclonal antibody. |
Taking into consideration previous studies by the
authors (9,10), it was sought to confirm that
different tumor cell types also have the capacity to directly
interact with the RGDKGE-containing collagen fragment. Similar to
previous studies (9,10), a synthetic RGDKGE-containing
collagen fragment termed P2, which is specifically recognized by
anti-XL313 Mab (9), can bind to a
variety of distinct tumor cell lines, including 4T1 mammary
carcinomas and a number of melanoma cell lines including Β16F10,
C32 and YUMM1.7 (Fig. S1A-D). To
confirm the function blocking activity of anti-XL313 Mab, the
ability of Mab XL313 to block tumor cell interaction with the
RGDKGE-containing collagen fragment P2 was examined. As revealed in
Fig. 1G and H anti-XL313 Mab
inhibited Β16F10 and 4T1 tumor cell binding to this
RGDKGE-containing collagen fragment. Similar results were observed
with C32 and YUMM1.7 melanoma cells (Fig. S1E and F).
Given the expression of the RGDKGE-containing
collagen fragment in tumor tissues, it was next sought to examine
the therapeutic impact of selectively targeting the bioactive
RGDKGE-containing collagen fragment. To begin these studies, the
dose-dependent impact of Mab XL313 on Β16F10 tumors growing in
vivo was examined. As demonstrated in Fig. 1I and J and Table SI, treatment of mice with
established Β16F10 tumors with Mab XL313 resulted in a
dose-dependent inhibitory effect, with a significant inhibition of
~50% achieved at doses of 100 µg per each mouse 3 times per week.
To confirm the antitumor activity of Mab XL313 in a second
independent in vivo tumor model, 4T1 mammary carcinomas
growing in Balb/c mice were used. As shown in Fig. 1K and L and Table SII, anti-XL313 Mab also
significantly inhibited 4T1 mammary carcinoma tumor growth.
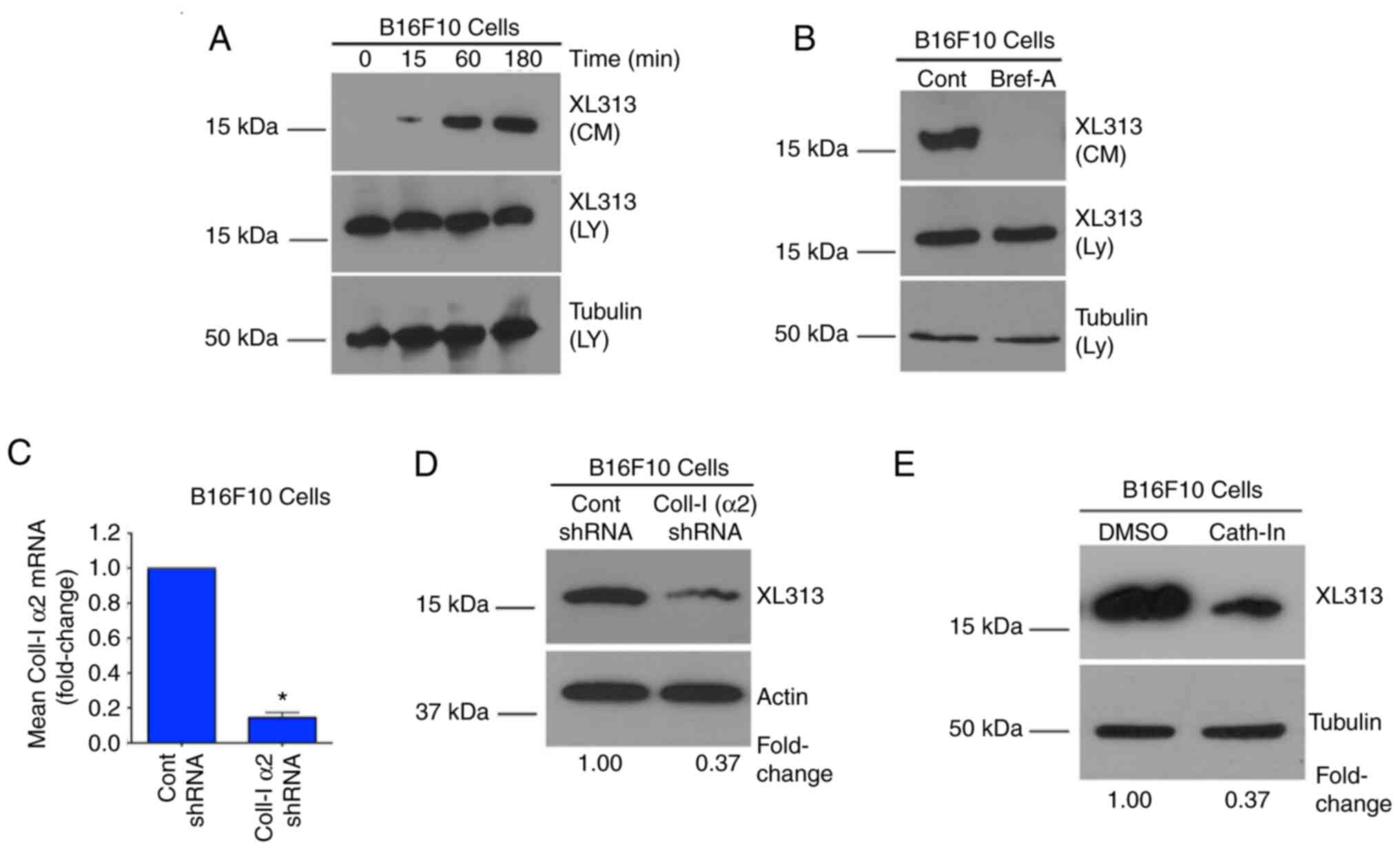
The RGDKGE-containing collagen
fragment can be generated by a cathepsin-dependent process
To further study the RGDKGE-containing collagen
fragment, it was chosen to focus primarily on Β16F10 melanoma cells
and started by examining the kinetics of the release of this
collagen fragment. To facilitate these studies, Β16F10 cells were
serum-starved in suspension for 1 h and then washed and resuspended
in serum-free medium and CM was collected over a time course of 3
h. Beginning as early as 15 min, a time-dependent increase in the
accumulation of the low molecular weight 16 kDa RGDKGE collagen
fragment was observed in the CM (Fig.
2A). To confirm that the collagen fragment detected within the
CM was actively secreted, CM was collected from cells that were
first pre-treated with the ER/Golgi inhibitor Brefeldin-A, which
inhibits protein secretion. As revealed in Fig. 2B, secretion of this collagen
fragment was inhibited by Brefeldin-A treatment. Similar findings
were also observed with C32 and YUMM1.7 melanoma cells (Fig. S2A and B). These data are consistent
with the ability of cells to secrete the soluble 16 kDa
RGDKGE-containing collagen fragment by an ER/Golgi dependent
mechanism.
Previous studies indicated that knocking down
collagen could reduce the levels of the RGDKGE-containing collagen
fragment in macrophages (9). To
confirm these findings, collagen was knocked down in Β16F10 cells
(Fig. 2C) and the levels of the
RGDKGE-containing collagen fragment were examined. As expected,
knocking down the a2 chain of collagen-1 reduced the
RGDKGE-containing collagen fragment (Fig. 2D). Importantly, up to 20% of newly
synthesized collagen can be proteolyzed intracellularly (8). Cathepsins represent a major family of
intracellular enzymes capable of cleaving collagen. Therefore, it
was examined whether cathepsins played a role in the generation of
the low molecular weight RGDKGE-containing collagen fragment. To
test this possibility, Β16F10 cells were incubated with a cell
permeable cathepsin inhibitor. As anticipated, treatment of
melanoma cells with the cathepsin inhibitor reduced the levels of
the RGDKGE-containing collagen fragment (Fig. 2E). Similar results were also
observed following treatment of C32 and YUMM1.7 melanoma cells
(Fig. S2C and D). Collectively,
these findings are consistent with a role for cathepsin in
generating the low molecular weight RGDKGE-containing collagen
fragment in these cells.
Selectively targeting the secreted
RGDKGE-containing collagen fragment reduces PD-L1 levels
Previous studies indicated that blocking β3
integrin-mediated interactions with the RGDKGE-containing collagen
fragment inhibits YAP nuclear accumulation in endothelial cells as
well as ovarian carcinoma cells (9,10).
Notably, both β3 integrin and YAP have been shown to regulate the
levels of the immune checkpoint molecule PD-L1 (11–13).
Thus, it was sought to determine whether the secreted
RGDKGE-containing collagen fragment may regulate PD-L1. To begin
these studies, the expression of PD-L1 in Β16F10 cells was first
confirmed. Consistent with previous studies (37), Β16F10 cells readily expressed PD-L1
(Fig. S3A). Next, it was confirmed
that Β16F10 cells use β3 integrin to bind to the RGDKGE-containing
collagen fragment. As expected, Β16F10 melanoma cells bind to the
RGDKGE-containing collagen fragment in a β3 integrin-dependent
manner as Β16F10 cell interaction with P2 could be inhibited by
anti-β3 integrin antibody, but not by anti-β1 integrin antibody or
non-specific control antibody (Fig.
S3B).
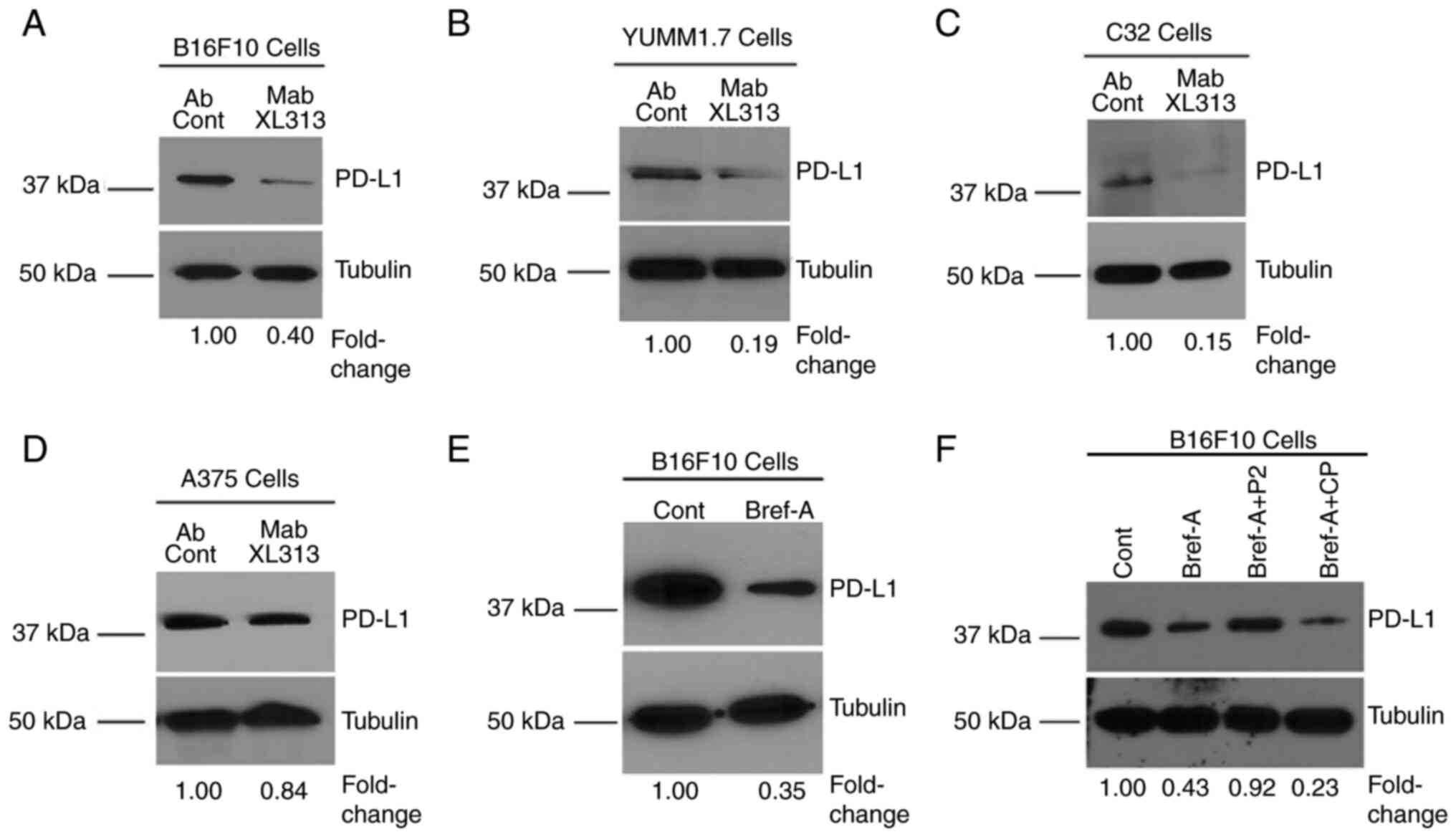
Next, it was determined whether blocking the
RGDKGE-containing collagen fragment may affect PD-L1 levels. As
revealed in Fig. 3A-C, targeting
the secreted collagen fragment with Mab XL313 reduced PD-L1 by
~60–80% in Β16F10, C32 and YUMM1.7 cells, while treatment of A375
cells, which express little if any of the RGDKGE-containing
collagen fragment (Fig. 1C) had
minimal impact on PD-L1 levels (Fig.
3D). Notably, PD-L1 was also reduced in cells in which the
secretion of the RGDKGE collagen fragment was inhibited with
Brefeldin-A (Fig. 3E), and the
level of PD-L1 in Brefeldin-treated cells could be restored to near
control levels by the addition of exogenous RGDKGE-containing
collagen peptide P2, but not by treatment with control peptide CP
(Fig. 3F). Previous studies have
suggested that reduction of PD-L1 in some tumor cell lines may
affect cell growth and survival (38–40).
To this end, the effects of Mab XL313 on Β16F10 cell cytotoxicity
were first examined in vitro. Incubation of Β16F10 cells
with Mab XL313 failed to exhibit any significant effect on Β16F10
cell cytotoxicity compared with non-specific control antibody
(Fig. S3C). Next, the effects of
anti-XL313 Mab on cell growth were examined. Incubation of Β16F10
cells with Mab XL313 showed a small but significant inhibitory
effect on cell growth (Fig. S3D),
which was consistent with previously published results suggesting
that reduction of PD-L1 may impact Β16F10 tumor cell growth in
vitro (38). Since β3 integrin
can serve as a receptor for the collagen fragment, experiments were
carried out to assess the effects of directly inhibiting β3
integrin may have on PD-L1. Selectively targeting β3-integrin with
a function-blocking antibody also reduced the levels of PD-L1
(Fig. S3E). Collectively, these
data are consistent with an autocrine-like pathway involving the
secreted RGDKGE collagen fragment binding to β3-integrin that
regulates the levels of PD-L1.
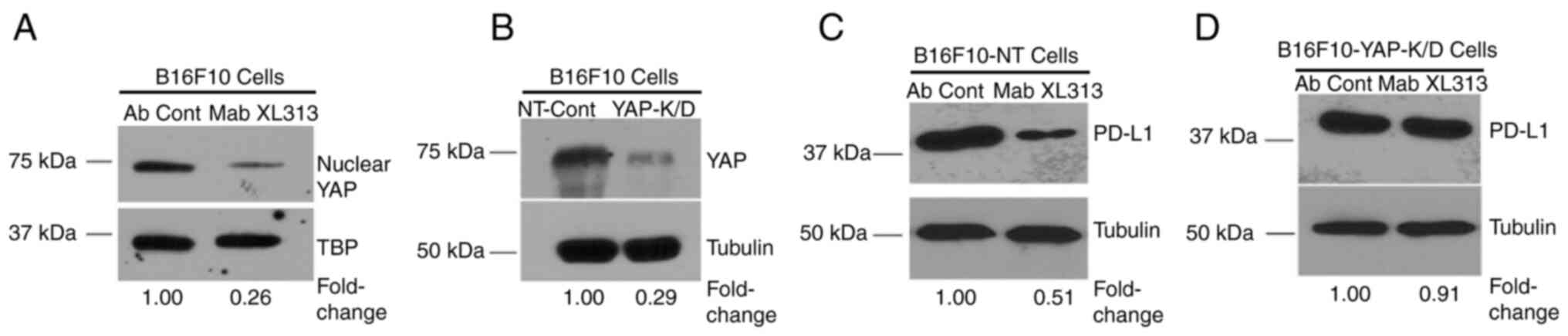
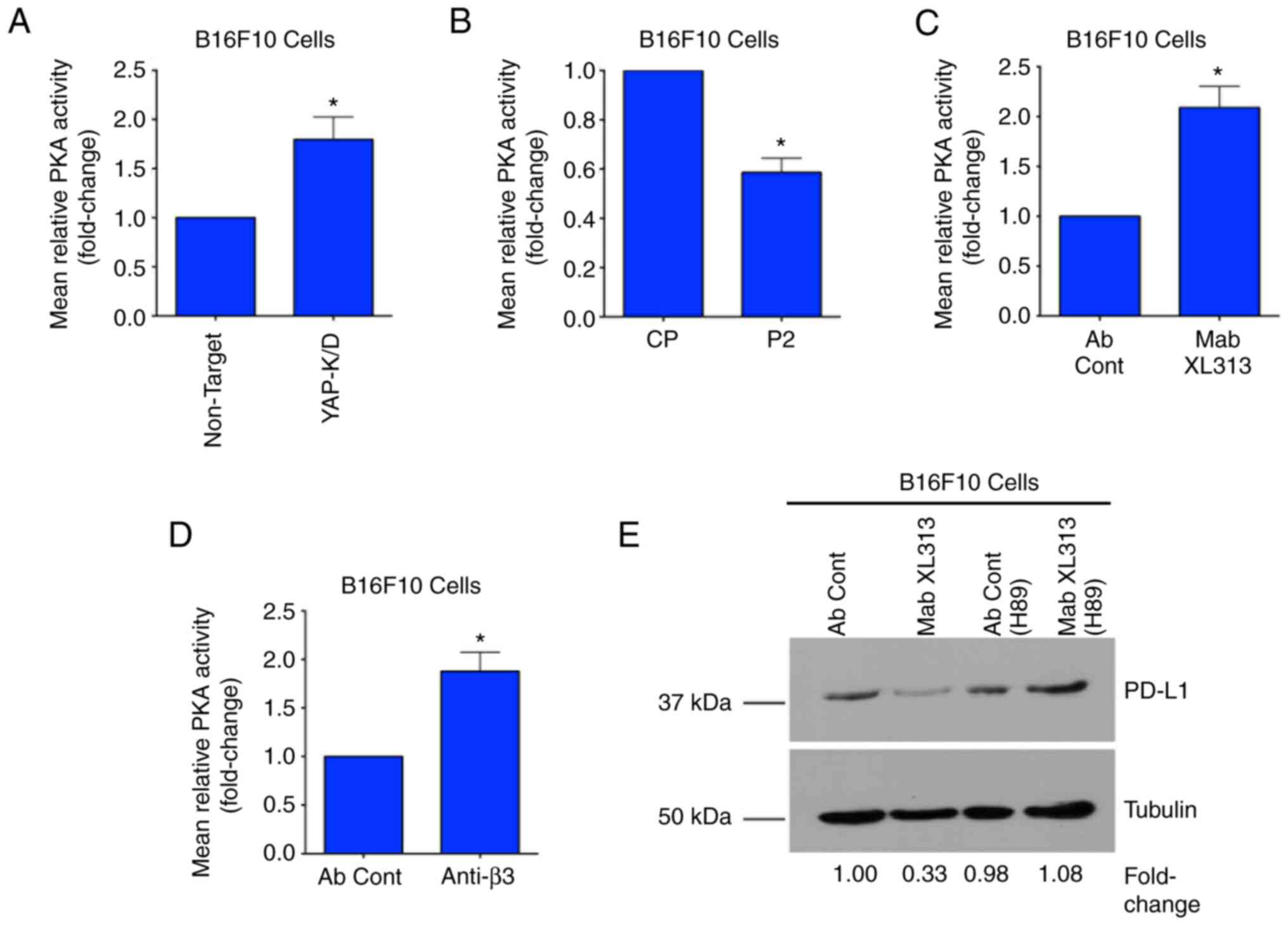
The ability of Mab XL313 to regulate
PD-L1 in Β16F10 cells depends on YAP
Given our previous findings that the
RGDKGE-containing collagen fragment can regulate nuclear
accumulation of YAP (9,10), it was assessed whether the ability
of this secreted collagen fragment to control PD-L1 depends on YAP.
First, it was confirmed that blocking the RGDKGE collagen fragment
with Mab XL313 reduced nuclear YAP levels in Β16F10 cells. As
expected, treatment of Β16F10 cells with anti-XL313 Mab reduced
nuclear YAP levels (Fig. 4A). Next,
yap gene expression was knocked down in Β16F10 cells (Fig. 4B), and the ability of Mab XL313 to
reduce PD-L1 was compared in either control-transfected (Β16F10-NT)
or in YAP knock-down cells (Β16F10-YAP-K/D). While specifically
targeting the RGDKGE collagen fragment with Mab XL313 reduced PD-L1
levels in control transfected cells (Fig. 4C), Mab XL313 failed to significantly
alter PD-L1 levels in YAP knock-down cells (Fig. 4D). These data are consistent with a
role for YAP in mediating the ability of this collagen fragment to
regulate PD-L1.
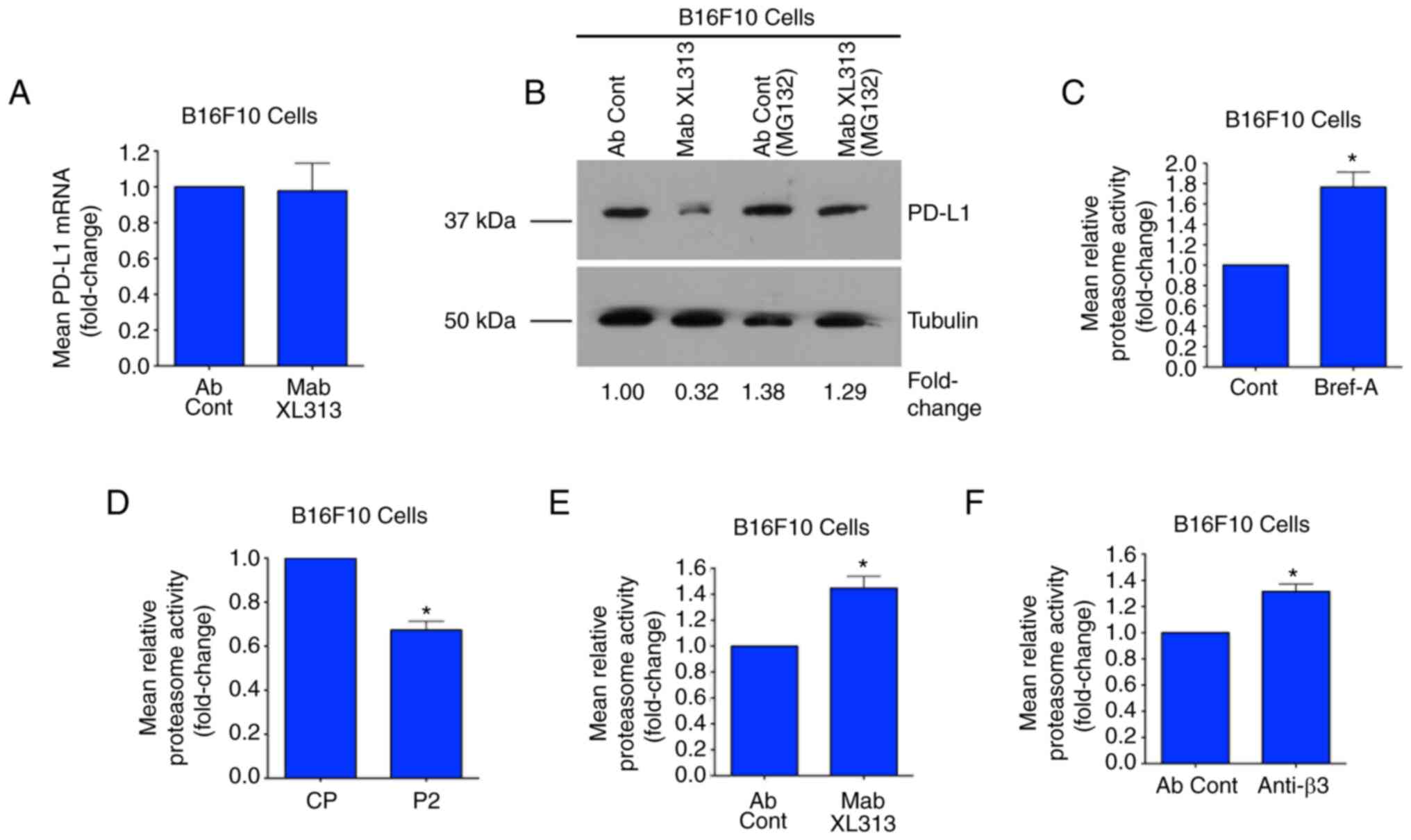
Selectively targeting the
RGDKGE-containing collagen fragment reduces PD-L1 levels by a
proteasome-dependent mechanism
Studies have suggested that YAP can contribute to
the control of PD-L1 by regulating its transcription or indirectly
by regulating the expression of cytokines and growth factors known
to modulate PD-L1 expression (11–13,19,20).
Given that the secreted RGDKGE-containing collagen fragment can
regulate PD-L1, it was sought to examine possible mechanisms to
account for this effect. Given the relatively rapid change in
levels of PD-L1 that can be observed within 15 min of treating
cells with Mab XL313 (Fig. S4A),
it appeared unlikely that this change was associated with
alterations in transcription. Consistent with this notion, RT-qPCR
analysis of cells treated with either control antibody or Mab XL313
for 15 min resulted in no significant change in the levels of PD-L1
mRNA (Fig. 5A). Moreover, targeting
the RGDKGE-containing collagen fragment with Mab XL313 also failed
to significantly affect PD-L1 mRNA at later time-points including 1
and 3 h (Fig. S4B and C),
suggesting that the reduction of PD-L1 protein was not related to
changes in mRNA levels.
Changes in PD-L1 protein can occur as a result of
proteasome-mediated degradation (21–23).
Thus, it was examined whether the ability of Mab XL313 to alter
PD-L1 was associated with proteasome activity. First, the ability
of the XL313 antibody to reduce PD-L1 levels was assessed in the
presence or absence of the proteasome inhibitor MG132. As
demonstrated in Fig. 5B, Β16F10
cells that were first pre-treated with control DMSO and then
treated with Mab XL313, exhibited reduced levels of PD-L1. By
contrast, no significant changes in PD-L1 were detected following
treatment of cells with Mab XL313 that had been pre-treated with
the proteasome inhibitor MG132 (Fig.
5B). Similar results were also observed with C32 and YUMM1.7
cells (Fig. S5A and B). These
observations are consistent with the possibility that this collagen
fragment may affect proteasome activity. The effects on proteasome
activity of Brefeldin-A, which inhibits secretion of the RGDKGE
collagen fragment, were next examined. Notably, Brefeldin-A
treatment of Β16F10 cells significantly enhanced proteasome
activity by nearly 1.7-fold compared with control treatment
(Fig. 5C). Given that Brefeldin-A
can inhibit secretion of this collagen fragment, the effects on
proteasome activity of treating Β16F10 cells with exogenously added
RGDKGE-containing collagen peptide (P2) were next assessed. As
revealed in Fig. 5D, treatment of
cells with the RGDKGE-containing collagen peptide P2 reduced
proteasome activity compared with control peptide CP. Next, it was
sought to determine the effects of selectively targeting the
collagen fragment or its cell surface receptor integrin β3 may have
on proteasome activity. As demonstrated in Fig. 5E and F, targeting either the RGDKGE
collagen fragment with Mab XL313 or its cell surface receptor with
anti-β3 antibody enhanced the relative proteasome activity compared
with controls. Collectively, these new findings are consistent with
the notion that inhibiting the RGDKGE-containing collagen fragment
controls PD-L1 levels through modulation of proteasome
activity.
The ability of the RGDKGE-containing
collagen fragment to control PD-L1 depends on regulation of PKA
activity
Prior evidence indicated that proteasome activity
can be controlled in part, by PKA (41–44).
Previous studies also suggested that integrin dependent signaling
may suppress PKA activity in certain cell types (45,46).
These observations coupled with studies indicating cross talk
signaling between PKA and YAP (47,48),
prompted the authors to examine whether PKA plays a role in the
ability of the RGDKGE-containing collagen fragment to control PD-L1
in our model. First, it was examined whether YAP may affect the PKA
activity. As revealed in Fig. 6A,
the levels of PKA activity were elevated in cells in which YAP was
knocked down. Since the RGDKGE-containing collagen fragment can
control YAP nuclear localization, the impact this collagen peptide
had on PKA activity was assessed. As shown in Fig. 6B, stimulation of Β16F10 cells with
exogenously added RGDKGE collagen peptide P2, reduced the levels of
PKA activity compared with control peptide CP. Taking into
consideration these results, it was sought to determine whether the
ability of Mab XL313 to control the levels of PD-L1 may depend on
PKA. Treatment of Β16F10 cells with Mab XL313 enhanced the relative
levels of PKA activity compared with control antibody-treated cells
(Fig. 6C). In similar experiments,
a function blocking antibody directed to β3 integrin, a receptor
for the RGDKGE collagen fragment, also enhanced the relative levels
of PKA activity in Β16F10 cells (Fig.
6D).
Finally, it was sought to determine whether the
ability of Mab XL313 to reduce PD-L1 depended on PKA by carrying
out experiments in the presence or absence of PKA inhibitor H89. As
demonstrated in Fig. 6E, while
treatment of Β16F10 cells with Mab XL313 under control
(DMSO)-treated conditions reduced the levels of PD-L1, similar
experiments performed with cells pre-treated with the PKA inhibitor
failed to demonstrate significantly altered PD-L1 levels (Fig. 6E). Similar studies were carried out
with a second PKA inhibitor (KT5720) which demonstrated similar
results (Fig. S6A). Moreover,
blocking PKA activity also prevented the ability of Mab XL313 to
alter PD-L1 in C32 and YUMM1.7 cells (Fig. S6B and C, respectively).
Collectively, these novel findings are consistent with a role for
the RGDKGE-containing collagen fragment in controlling PD-L1 by a
mechanism that involves PKA.
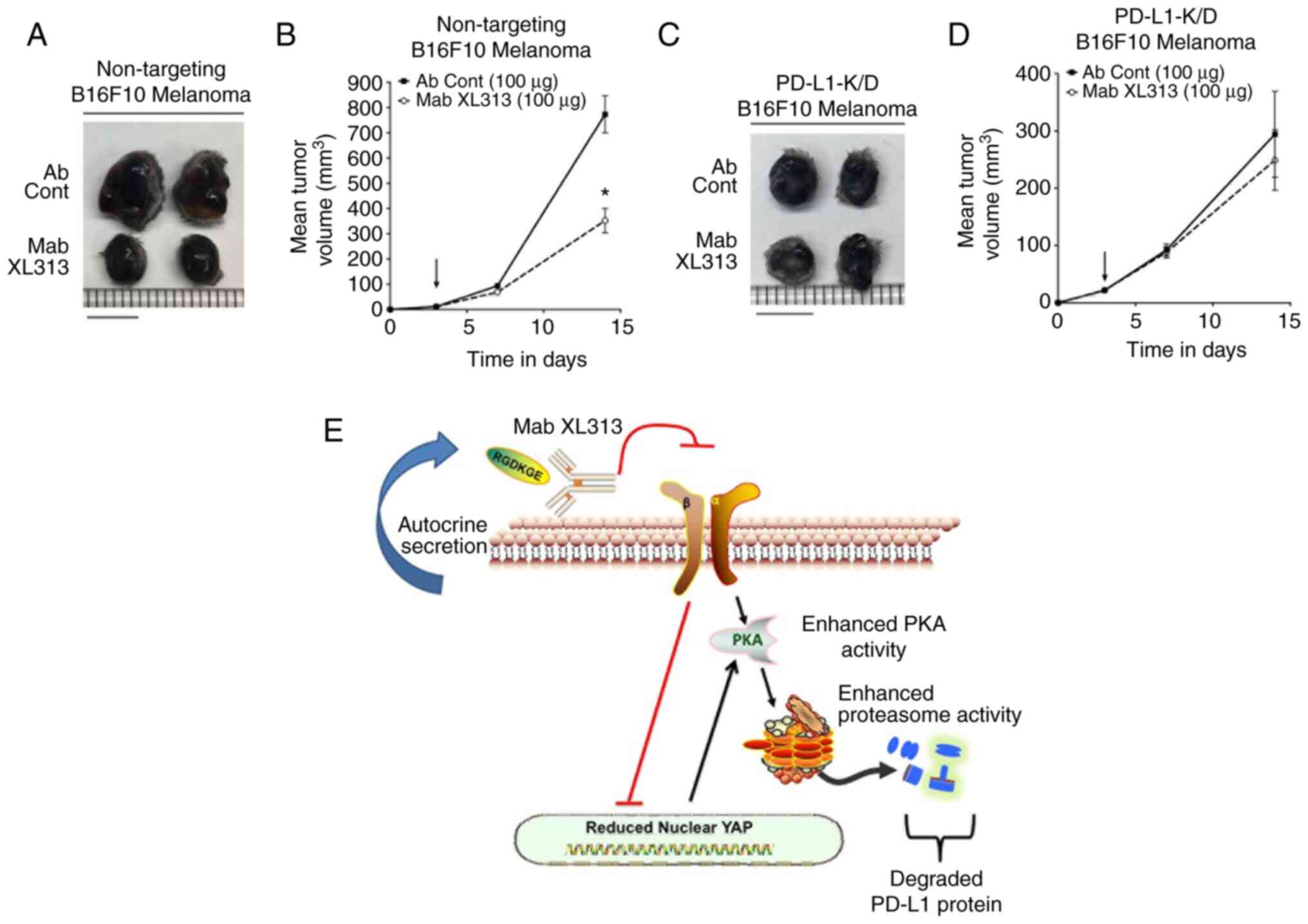
The in vivo inhibitory activity of Mab
XL313 depends on levels of tumor-associated PD-L1
The functional role of integrin-mediated signaling
in controlling angiogenesis and tumor growth is well established
(49–52). However, while the overall
therapeutic strategy of directly targeting integrins such as αvβ3
to control tumor growth has shown activity in animal models and in
human clinical studies (49–52),
the overall impact of this approach has been limited (52). Among some of the possible reasons
for the modest therapeutic benefit include the complexity of the
cellular processes controlled by integrins such as αvβ3, which
include both pro-tumorigenic and anti-tumorigenic signaling
(53–58). Importantly, the ability of integrin
signaling to control tumors depends on the functional contributions
of the distinct cell types that express the integrin and the nature
of the signaling pathways disrupted by directly targeting the
integrins. Thus, a therapeutic strategy that selectively targets a
pro-tumorigenic ligand of the integrin rather than the integrin
receptor itself, may provide a therapeutically effective
alternative strategy.
Taking into consideration the present studies
indicating that disrupting cellular interactions with the secreted
RGDKGE-containing collagen fragment could reduce PD-L1 and inhibit
tumor growth, it was sought to determine whether the antitumor
activity of Mab XL313 depended on the levels of PD-L1. To this end,
the antitumor effects of Mab XL313 on the growth of Β16F10 tumors
that had reduced levels of PD-L1 were examined (Fig. S7). As revealed in Fig. 7A and B and Table SIII, Mab XL313 inhibited the growth
of control-transfected Β16F10 tumors. By contrast, treatment of
mice with Mab XL313 failed to significantly alter Β16F10 tumor
growth in which PD-L1 was knocked down (Fig. 7C and D and Table SIV). These findings are consistent
with the capacity of Mab XL313 to inhibit tumor growth that depends
on levels of PD-L1. Taken together, the present studies are
consistent with a working model by which selectively targeting the
secreted RGDKGE-containing collagen fragment prevents it from
binding and signaling through integrin β3, thereby reducing the
levels of nuclear localized YAP and enhancing PKA activity, which
ultimately contributes to the reduction of PD-L1 by controlling its
proteasomal-mediated degradation (Fig.
7E).
Discussion
It has been suggested that alterations in collagen
density, fiber orientation as well as its molecular architecture
correlates with phenotypic changes in different malignant tumors
(59–63). In fact, second harmonic generation
imaging of tumor biopsies has revealed unique collagen
organizational signatures between normal tissues and tumors
(59–63). Biophysical alterations in collagen
structure can alter signaling that controls cell adhesion,
migration and proliferation, and selective targeting of certain
cryptic collagen elements can inhibit angiogenesis, tumor growth
and metastasis in animal models (3,9,10,31,64).
These and other findings not only implicate collagen as an
important regulator of malignant tumors, but also suggests that
discrete changes in its geometrical configuration play distinct
roles in stimulating signaling cascades that control tumor
progression.
In addition to alterations in the biophysical
features of insoluble collagen fibers, evidence suggested that
certain circulating fragments of collagen correlate with aggressive
behavior of a variety of histologically different tumor types,
including, colon, breast and pancreatic carcinomas, as well as
melanomas (65–68). High levels of circulating collagen
fragments not only correlate with more aggressive disease, but also
correlate with poor clinical outcomes following therapeutic
intervention (65–68). For example, studies have correlated
poor clinical responses in humans treated with immune checkpoint
inhibitors targeting CTLA-4 and PD-1/PD-L1, with high levels of
circulating collagen fragments (14,15).
While these studies provided evidence of the differential levels of
soluble collagen peptides during tumor progression and treatment,
little evidence is available as to whether these fragments play
direct functional roles in controlling tumor growth. Evidence
suggested that soluble collagen-derived fragments may not only
represent useful biomarkers, but may also have biological activity.
In this regard, an MMP-2 generated proteolytic fragment of the
a1chain of collagen type-I (C-1158/59) has been detected in human
plasma and a synthetic peptide corresponding to this fragment
demonstrated the ability to alter fibroblast migration,
angiogenesis, fibrosis, wound healing, and ventricle dilation
(69). Moreover endotrophin, a
bioactive fragment of the a3 chain of collagen type-VI, modulates
recruitment of macrophages and endothelial cells and regulates
angiogenesis and tumor growth in vivo (70). These and other studies suggested
that soluble collagen fragments indeed play roles in regulating
tumor growth in vivo.
Our laboratory identified an RGDKGE-containing
collagen peptide that was initially discovered as an endogenously
secreted bioactive collagen fragment from M2-like macrophages
(9). This collagen fragment was
shown to regulate nuclear accumulation of YAP in endothelial cells
and control cytokine-induced angiogenesis in vivo (9). The expression of this collagen
fragment was not simply restricted to activated M2-like
macrophages, but could also be expressed by tumors such as ovarian
carcinomas (10). In fact, this
collagen peptide was capable of regulating nuclear accumulation of
YAP in ovarian carcinoma cells by a mechanism associated with
modulating the phosphorylation of the hippo effector kinase LATS1
(10). Selectively blocking binding
of the RGDKGE collagen fragment led to enhanced LATS1
phosphorylation, and increased phosphorylation of YAP, which
ultimately reduced the levels of nuclear YAP, and inhibited ovarian
tumor growth in vivo (10).
While YAP is considered to regulate cell growth, it
can also differentially control cell migration by modulating
cytoskeletal dynamics as well as the expression and activity of
small GTPases including ARHGAP29 (71–73).
In certain cell types, YAP can also control the expression of the
immune checkpoint molecule PD-L1 (11–13).
While evidence indicated that targeting PD-L1 can help to control
the growth of a variety of different tumor types, only a fraction
of subjects treated with current anti-PD-L1 antagonists show
significant and durable responses (14–17,19,20,39).
Thus, it is important to develop a more in-depth understanding of
the various mechanisms by which the levels of PD-L1 are controlled,
as well as the unique functional impact of this immune checkpoint
molecule has in different cell types. Recent evidence continues to
provide new understanding of the complicated roles played by PD-L1
during tumor growth. For example, beyond the well characterized
effects of PD-L1 in regulating immune evasion through binding to
PD-1, studies have also uncovered evidence for tumor cell intrinsic
signaling by PD-L1, thereby helping to control multiple processes
such as regulating gene expression, proliferation, apoptosis and
cell cycle control (74–77). In addition to cell surface
expression of PD-L1, the release of soluble forms of PD-L1 can also
contribute to regulating growth of tumors (78). Moreover, a recent study revealed
that differential sub-cellular localization of PD-L1 in the nucleus
of certain cell types, may help actively control cell survival and
apoptosis (79). Given the
diversity of functions of cell-associated and soluble forms of
PD-L1, uncovering new mechanisms by which PD-L1 expression can be
controlled may help in designing new strategies to limit
therapeutic resistance and enhance the antitumor activity of immune
checkpoint inhibition.
In this regard, unique mechanisms by which PD-L1
can be regulated continue to be uncovered as investigators have
provided evidence for the ability of Fusobacterium nucleatum
to regulate PD-L1 in colorectal cancer (80). In addition, a recent study also
demonstrated a novel pathway by which RNA-RNA cross-talk can
function in-conjunction with RICTOR to control PD-L1 expressing
exosomes that affect hepatocellular carcinoma tumor growth
(81). In further studies,
metformin has been shown to help control PD-L1 levels by a unique
mechanism involving ERAD-associated proteasome-mediated degradation
of this immune checkpoint molecule (23). Notably, recent studies have also
shown that metformin can affect YAP which is also known to regulate
PD-L1 (23,82). In fact, the ability of metformin to
control PD-L1 in some cell types may depend at least in part, on
the activation of the Hippo signaling leading to enhanced
phosphorylation of YAP and reduced levels of nuclear YAP (82). These studies provided additional
understanding of the complicated roles played by YAP in its ability
to control both PD-L1 expression as well as tumor growth.
Adding to the complexity by which YAP controls
cellular behavior, YAP may exhibit opposing functions on tumor
growth depending on the cell type it is functioning in. While
enhanced YAP activity has been shown to promote the growth of
several tumor types such as ovarian carcinomas and melanomas, other
studies have implicated YAP in suppressing breast and liver cancers
(10,11,31,83,84).
These seemingly contradictory findings may be explained at least in
part, by the differential roles that YAP plays in different cell
types, as well as the different functional contributions of each
unique cell type has on tumor growth. While YAP has been shown to
play a role in regulating cytoskeletal dynamics required for
efficient tumor cell motility (71,73) it
may have the opposite effects in certain immune cell subsets
(85,86). In this regard, reducing YAP activity
can inhibit tumor growth and reduce T-Reg infiltration of tumors,
while by contrast, blocking YAP activity may enhance
CD8+ T-cell motility and tumor infiltration (85–87).
Moreover, previous studies have also demonstrated a role for YAP in
regulating the pro-tumorigenic effects of macrophages as well as
promoting the secretion of chemotactic factors that help recruit
myeloid-derived suppressor cells (88–90).
Therefore, a more in-depth analysis of the differential roles of
YAP within distinct cellular compartments that control tumor growth
is of critical importance as it relates to developing novel YAP
based therapeutic strategies for the treatment of cancer (91).
In the present study, evidence was provided that
the RGDKGE-containing cryptic collagen fragment can be secreted in
a soluble form by an ER/Golgi-dependent mechanism. Additionally,
this secreted collagen fragment can bind to β3 integrin in melanoma
cells in an autocrine-like manner. Selectively targeting this
collagen fragment can regulate nuclear accumulation of YAP that is
independent from mechanisms associated with cell adhesion and
spreading, as Mab XL313 treatment reduced nuclear YAP levels in
melanoma cells held in suspension. These observations are
consistent with previous studies indicating that YAP activation and
its nuclear accumulation can occur by multiple mechanisms,
including adhesion independent pathways (78).
As aforementioned, both YAP as well as integrin
signaling has been implicated in regulating the levels of the
immune-checkpoint molecule PD-L1. The present studies suggested for
the first time, to the best of our knowledge, that specifically
targeting the RGDKGE collagen fragment can lead to a reduction in
the levels of PD-L1. The reduction in PD-L1 observed following
treatment with Mab XL313 depended on YAP, as Mab XL313 failed to
reduce PD-L1 in cells in which YAP was knocked down. Moreover,
targeting β3 integrin with a function blocking antibody also
reduced PD-L1 levels. These observations are consistent with the
concept that blocking binding of this collagen peptide to β3
integrin results in suppressing an autocrine-like signaling cascade
that controls PD-L1 levels. While studies have implicated β3
integrin signaling in regulating PD-L1 gene expression (29), the ability of Mab XL313 to reduce
PD-L1 levels was not associated with significant changes in PD-L1
mRNA.
Notably, while targeting the secreted RGDKGE
collagen fragment with Mab XL313 inhibited PD-L1 levels, Mab Xl313
failed to significantly reduce PD-L1 in cells pre-treated with the
proteasome inhibitor, indicating that the ability of Mab XL313 to
reduce PD-L1 in these cells required proteasome activity. The
proteasome functions as a major regulator of signaling cascades by
rapidly controlling the levels of effector molecules (92). For example, cell attachment to the
ECM protein fibronectin induced alterations in the small GTPases
Rac and CDC42, ultimately leading to enhance proteasome activity
and enhanced degradation of the cyclin dependent kinase
P21CIP1 (93). Notably,
members of both the β1 and β3 integrin subfamilies can bind to
fibronectin and mediate downstream signaling from this ECM
molecule. These studies are consistent with the possibility that
integrin-mediated signaling can differentially affect proteasome
function. Consistent with this notion, treatment of neuroblastoma
cells with a β1 integrin binding fibronectin peptide resulted in
inactivation of β1 integrin, which enhanced proteasome dependent
degradation of Myc proteins (94).
These observations are similar to the present studies, in which the
selective blockade of the RGDKGE-containing collagen peptide
binding to β3 integrin resulted in enhanced proteasome-mediated
degradation of PD-L1.
While the relative levels of cellular proteasome
activity can be affected by multiple mechanisms, notably, previous
studies have implicated PKA in regulating proteasome activity
(41–44). Integrin signaling can differentially
regulate PKA activity (45,46) and play roles in regulating YAP
activity, angiogenesis, tumor growth and metastasis (47,48).
Given the ability of Mab XL313 to reduce the levels of nuclear YAP,
it was examined whether reducing YAP may affect the levels of PKA
activity. The findings of the present study suggested that reducing
the levels of YAP can enhance PKA activity in Β16F10 cells.
Moreover, stimulation with the synthetic RGDKGE-containing collagen
peptide (P2) reduced PKA activity, while direct targeting of this
collagen fragment or its cell surface receptor β3 integrin,
enhanced PKA activity. Currently, the exact molecular mechanism by
which the RGDKGE collagen fragment functions through β3 integrin to
control PKA activity, and how YAP and PKA contributes to the
control of proteasome activity are not completely understood. To
this end, it would be interesting in the future to make use of
FRET-based PKA sensors to study how the RGDKGE collagen fragment
may function through β3 integrin in cells to regulate PKA and
proteasome activity in order to define this signaling pathway in
more detail.
Direct targeting of β3 integrin has demonstrated
antitumor activity in animal models as well as in human subjects,
however the overall therapeutic activity was limited. Among the
possible explanations for this limited antitumor activity, the
complex and opposing roles played by β3 integrin in distinct
stromal cell compartments are included (50–53).
The opposing roles played by different stromal cell types on tumor
progression have been illustrated by studies focusing on the impact
of differentially polarized stromal cells on tumor growth. For
example, in the case of cancer associated fibroblast (CAFs),
conflicting outcomes using a strategy to target CAFs to inhibit
tumor growth have been observed, as depleting fibroblast activation
protein expressing fibroblasts resulted in reduced tumor growth in
pancreatic cancer, while depletion of αSMA-expressing fibroblasts
resulted in more aggressive disease (95,96).
In addition, macrophages can also have opposing roles in tumor
progression, as demonstrated by the fact that while M1-like
macrophages help eliminate tumor cells, M2-like macrophage, as well
as myeloid-derived suppressor cells, are considered to promote
tumor growth (57,58,89).
Finally, while infiltration of CD8+ cytotoxic T-cells
play roles in inhibiting tumor growth, regulatory T-cells can help
facilitate tumor progression (85–87).
Given the fact that these stromal cell types may differentially
express integrins including β3 integrin (85–90,92,93), a
clinical strategy that seeks to directly target the global function
of β3 integrin in all cell types including stromal cells such as
M1-like macrophages and CD8+ effector T-cells, may
result in suboptimal clinical outcomes, and actually limit the
overall effectiveness of this therapeutic strategy. Developing
novel approaches to control β3 integrin signaling within the
pro-tumorigenic cellular compartments without directly targeting β3
integrin itself, may allow for a new alternative approach to
selectively control pro-tumorigenic signaling from β3 integrin
without disrupting its anti-tumorigenic activity (57,58,97,98).
While our previous studies have shown that
anti-XL313 antibody can inhibit the growth of ovarian tumors, which
was associated with altered regulation of nuclear YAP, the ability
of YAP to affect the growth of different types of tumor can vary
considerably (10,11,31,83,84).
In the present study, previous studies by the authors were extended
and several new experimental findings that substantially improve
our current understanding of how the RGDKGE collagen fragment may
function were provided. First, it was demonstrated that the ability
of the anti-XL313 Mab to inhibit tumor growth is not restricted to
ovarian tumors, but can also affect the growth of other tumor types
such as melanoma and mammary carcinomas in vivo. In
addition, it was revealed that the RGDKGE collagen fragment can be
generated inside cells by a cathepsin-associated mechanism and can
be released extracellularly in a soluble form by an ER/Golgi
dependent process. Moreover, evidence was provided for the first
time, for a previously unappreciated ability of the RGDKGE collagen
fragment to function in an autocrine-like manner to regulate the
levels of PD-L1 by a PKA and proteasome-dependent mechanism.
Collectively, these novel findings provided important new insight
into the roles by which the soluble RGDKGE collagen fragment may
affect tumor growth.
In summary, new evidence was provided that
selectively targeting a secreted pro-tumorigenic ligand of β3
integrin, instead of directly targeting β3 integrin itself, can
inhibit Β16F10 and 4T1 tumor growth in vivo. While Mab XL313
significantly inhibited control transfected Β16F10 tumors, this
antibody failed to inhibit the growth of Β16F10 tumors in which
PDL-1 was knocked down, indicating an important role for PD-L1 in
the ability of the RGDKGE collagen fragment to control Β16F10 tumor
growth in vivo. Taken together, our studies provided
evidence for an autocrine-like signaling pathway involving β3
integrin that provides tumor cells with the ability to regulate
proteasome-mediated control of PD-L1 by modulating the activity of
PKA and YAP. Moreover, our new findings may help in establishing a
selective new alternative strategy to control pro-tumorigenic
activity of β3-integrin signaling without directly disrupting β3
integrin's tumor suppressing functions in other cell
populations.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported in part from NIH (grant no.
CA196739). Further support was from the Northern New England
Clinical and Translational Research Center (CTR; grant no.
U54GM115516), and the Mesenchymal and Neural Regulation of
Metabolic Networks (grant no. P20GM121301. Partial support was also
provided by NIH (grant nos. RO1 CA238237, U54 CA224070 and PO1
CA114046) and the Dr. Miriam and Sheldon G. Adelson Medical
Research Foundation. Additional support was provided by the Maine
Medical Center and the Cross Insurance Agency, John Benoit and
Thomas Holden.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JMC helped conceive, design, coordinate studies,
performed and analyzed experiments and helped write, review and
edited the manuscript. XH helped conceive, design, coordinate
studies, performed and analyze experiments and helped write, review
and edited the manuscript. CWL, PS, SCR and MSE assisted in
analysis and interpretation of the data and writing, reviewing and
editing the manuscript. MH helped coordinate studies, assisted in
analysis and interpretation of the data and in writing, reviewing
and editing of the manuscript. PCB helped conceive, design,
coordinate studies, analyzed experiments, and interpreted the data,
and helped write, review and edited the manuscript. All authors
read and approved the final version of the manuscript and agree to
be accountable for all aspects of the work. JMC, XH and PCB confirm
the authenticity of the raw data.
Ethics approval and consent to
participate
Anonymous de-identified human tumor samples were
used. Use of the human tumor samples were considered exempt as it
relates to human subjects' research by MaineHealth IRB and Biobank
as all tissues were anonymous and de-identified. Animal studies
were approved by the Maine Medical Center Institutional Animal Care
and Use Committee (Scarborough, USA).
Patient consent for publication
Not applicable.
Competing interests
PCB holds an equity position in CryptoMedix, Inc.
The rest of the authors declare that they have no competing
interests.
References
|
1
|
Brassart-Pasco S, Brezillon S, Brassart B,
Ramont L, Oudart JB and Monboisse JC: Tumor microenvironment:
Extracellular matrix alterations influence tumor progression. Front
Oncol. 10:3972020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ruiter D, Bogenrieder T, Elder D and
Herlyn M: Melanoma-stroma interactions: Structural and functional
aspects. Lancet Oncol. 3:35–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Han X, Caron JM and Brooks PC: Cryptic
collagen elements as signaling hubs in the regulation of tumor
growth and metastasis. J Cell Physiol. 235:9005–9020. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Contois L, Akalu A and Brooks PC:
Integrins as ‘functional hubs’ in the regulation of pathological
angiogenesis. Semin Cancer Biol. 19:318–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ricard-Blum S: The collagen family. Cold
Spring Harb Perspect Biol. 3:a0049782011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zeltz C and Gullberg D: The
integrin-collagen connection-a glue for tissue repair? J Cell Sci.
129:12842016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leitinger B: Transmembrane collagen
receptors. Annu Rev Cell Dev Biol. 27:265–290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bienkowski RS, Curran SF and Berg RA:
Kinetics of intracellular degradation of newly synthesized
collagen. Biochemistry. 25:2455–2459. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ames JJ, Contois L, Caron JM, Tweedie E,
Yang X, Friesel R, Vary C and Brooks PC: Identification of an
endogenously generated cryptic collagen epitope (XL313) that may
selectively regulate angiogenesis by an integrin yes-associated
protein (YAP) mechano-transduction pathway. J Biol Chem.
291:2731–2750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han X, Caron JM, Lary CW, Sathyanarayana
P, Vary C and Brooks PC: An RGDKGE-containing cryptic collagen
fragment regulates phosphorylation of large tumor suppressor
kinase-1 and controls ovarian tumor growth by a YAP-associated
protein-dependent mechanism. Am J Pathol. 191:527–544. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA,
Park SH, Shin EC and Kim J: YAP-induced PD-L1 expression drives
immune evasion in BRAFi-resistant melanoma. Cancer Immunol Res.
6:255–266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsu PC, Miao J, Wang YC, Zhang WQ, Yang
YL, Wang CW, Yang CT, Huang Z, You J, Xu Z, et al: Inhibition of
yes-associated protein down-regulates PD-L1 (CD274) expression in
human malignant pleural mesothelioma. J Cell Mol Med. 22:3139–3148.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee BS, Park DI, Lee DH, Lee JE, Yeo MK,
Park YH, Lim DS, Choi W, Lee DH, Yoo G, et al: Hippo effector YAP
directly regulates the expression of PD-L1 transcripts in
EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophy Res Commun.
491:493–499. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jensen C, Madsen DH, Hansen M, Schmidt H,
Svane IM, Karsdal MA and Willumsen N: Non-invasive biomarkers
derived from the extracellular matrix associate with response to
immune checkpoint blockade (anti-CTLA-4) in metastatic melanoma
patients. J Immunother Cancer. 6:1522018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hurkmans DP, Jensen C, Koolen SLW, Aerts
J, Karsdal MA, Mathijssen RHJ and Willumsen N: Blood-based
extracellular matrix biomarkers are correlated with clinical
outcome after PD-1 inhibition in patients with metastatic melanoma.
J Immunother Cancer. 8:e0011932020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abdou Y, Pandey M, Sarma M, Shah S, Baron
J and Ernstoff MS: Mechanism-based treatment of cancer with immune
checkpoint inhibitor therapies. Br J Clin Pharmacol. 86:1690–1702.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gandhi S, Pandey MR, Attwood K, Ji W,
Witkiewicz AK, Knudsen ES, Allen C, Tario JD, Wallace PK, Cedeno
CD, et al: Phase I clinical trial of combination propranolol and
pembrolizumab in locally advanced and metastatic melanoma: Safety,
tolerability, and preliminary evidence of antitumor activity. Clin
Cancer Res. 27:87–95. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Somasundaram R, Connelly T, Choi R, Choi
H, Samarkina A, Li L, Gregorio E, Chen Y, Thakur R, Abdel-Mohsen M,
et al: Tumor-infiltrating mast cells are associated with resistance
to anti-PD-1 therapy. Nat Commun. 12:3462021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cha JH, Chan LC, Li CW, Hsu JL and Hung
MC: Mechanisms controlling PD-L1 expression in cancer. Mol Cell.
76:359–370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun C, Mezzadra R and Schumacher TN:
Regulation and function of the PD-L1 checkpoint. Immunity.
48:434–452. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gou Q, Dong C, Xu H, Khan B, Jin J, Liu Q,
Shi J and Hou Y: PD-L1 degradation pathway and immunotherapy for
cancer. Cell Death Dis. 11:9552020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo
CW, Khoo KH, Chang SS, Cha JH, Kim T, et al: Glycosylation and
stabilization of programmed death ligand-1 suppresses T-cell
activity. Nat Commun. 7:126322016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cha JH, Yang WH, Xia W, Wei Y, Chan LC,
Lim SO, Li CW, Kim T, Chang SS, Lee HH, et al: Metformin promotes
antitumor immunity via endoplasmic-reticulum-associated degradation
of PD-L1. Mol Cell. 71:606–620.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Burr ML, Sparbier CE, Chan YC, Williamson
JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg
S, et al: CMTM6 maintains the expression of PD-L1 and regulates
anti-tumour immunity. Nature. 549:101–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang H, Yao H, Li C, Shi H, Lan J, Li Z,
Zhang Y, Liang L, Fang JY and Xu J: HIP1R targets PD-L1 to
lysosomal degradation to alter T cell-mediated cytotoxicity. Nat
Chem Biol. 15:42–50. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Coelho MA, de Carné Trécesson S, Rana S,
Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E,
Barnouin K, et al: Oncogenic RAS signaling promotes tumor
immunoresistance by stabilizing PD-L1 mRNA. Immunity.
47:1083–1099.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin HY, Chin YT, Nana AW, Shih YJ, Lai HY,
Tang HY, Leinung M, Mousa SA and Davis PJ: Actions of l-thyroxine
and Nano-diamino-tetrac (Nanotetrac) on PD-L1 in cancer cells.
Steroids. 114:59–67. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ren D, Zhao J, Sun Y, Li D, Meng Z, Wang
B, Fan P, Liu Z, Jin X and Wu H: Overexpressed ITGA2 promotes
malignant tumor aggression by up-regulating PD-L1 expression
through the activation of the STAT3 signaling pathway. J Exp Clin
Cancer Res. 38:4852019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vannini A, Leoni V, Barboni C, Sanapo M,
Zaghini A, Malatesta P, Campadelli-Fiume G and Gianni T:
αvβ3-integrin regulates PD-L1 expression and is involved in cancer
immune evasion. Proc Natl Acad Sci USA. 116:20141–20150. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Caron JM, Han X, Contois L, Vary CPH and
Brooks PC: The HU177 collagen epitope controls melanoma cell
migration and experimental metastasis by a CDK5/YAP-dependent
mechanism. Am J Pathol. 188:2356–2368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Provenzano PP, Inman DR, Eliceiri KW,
Knittel JG, Yan L, Rueden CT, White JG and Keely PJ: Collagen
density promotes mammary tumor initiation and progression. BMC Med.
6:112008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vellinga TT, den Uil S, Rinkes IH, Marvin
D, Ponsioen B, Alvarez-Varela A, Fatrai S, Scheele C, Zwijnenburg
DA, Snippert H, et al: Collagen-rich stroma in aggressive colon
tumors induces mesenchymal gene expression and tumor cell invasion.
Oncogene. 35:5263–5271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Kempen LC, Rijntjes J,
Mamor-Cornelissen I, Vincent-Naulleau S, Gerritsen MJ, Ruiter DJ,
van Dijk MC, Geffrotin C and van Muijen GN: Type I collagen
expression contributes to angiogenesis and the development of
deeply invasive cutaneous melanoma. Int J Cancer. 122:1019–1029.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Noel A, Munaut C, Boulvain A, Calberg-Bacq
CM, Lambert CA, Nusgens B, Lapiere CM and Foidart JM: Modulation of
collagen and fibronectin synthesis in fibroblasts by normal and
malignant cells. J Cell Biochem. 48:150–161. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Afik R, Zigmond E, Vugman M, Klepfish M,
Shimshoni E, Pasmanik-Chor M, Shenoy A, Bassat E, Halpern Z, Geiger
T, et al: Tumor macrophages are pivotal constructors of tumor
collagenous matrix. J Exp Med. 213:2315–2331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gupta HB, Clark CA, Yuan B, Sareddy G,
Pandweswara S, Padron AS, Hurez V, Conejo-Garcia J, Vadlamudi R, Li
R and Curiel TJ: Tumor cell-intrinsic PD-L1 promotes
tumor-initiating cell generation and functions in melanoma and
ovarian cancer. Signal Transduct Target Ther. 1:160302016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Clark CA, Gupta HB, Sareddy G, Pandeswara
S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K,
et al: Tumor-intrinsic PD-L1 signals regulate cell growth,
pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer
Res. 76:6964–6974. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hudson K, Cross N, Jordan-Mahy N and
Leyland R: The extrinsic and intrinsic roles of PD-L1 and its
receptor PD-1: Implications for immunotherapy treatment. Front
Immunol. 11:5689312020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xue Z, Zheng S, Linghu D, Liu B, Yang Y,
Chen MK, Huang H, Song J, Li H, Wang J, et al: PD-L1 deficiency
sensitizes tumor cells to DNA-PK inhibition and enhances cGAS-STING
activation. Am J Cancer Res. 12:2363–2375. 2022.PubMed/NCBI
|
|
41
|
VerPlank JJS, Lokireddy S, Zhao J and
Goldberg AL: 26S Proteasomes are rapidly activated by diverse
hormones and physiological states that raise cAMP and cause Rpn6
phosphorylation. Proc Natl Acd Sci USA. 116:4228–4237. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asai M, Tsukamoto O, Minamino T, Asanuma
H, Fujita M, Asano Y, Takahama H, Sasaki H, Higo S, Asakura M, et
al: PKA rapidly enhances proteasome assembly and activity in in
vivo canine hearts. J Mol Cell Cardiol. 46:452–462. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lokireddy S, Kukushkin NV and Goldberg AL:
cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11
enhances their activity and the degradation of misfolded proteins.
Proc Natl Acad Sci USA. 112:E7176–E7185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qin XY, Zhang YL, Chi YF, Yan B, Zeng XJ,
Li HH and Liu Y: Angiotensin II regulates Th1 T cell
differentiation through angiotensin II type 1 receptor-PKA-mediated
activation of proteasome. Cell Physiol Biochem. 45:1366–1376. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim S, Harris M and Varner JA: Regulation
of integrin alpha vbeta 3-mediated endothelial cell migration and
angiogenesis by integrin alpha5beta1 and protein kinase A. J Biol
Chem. 275:33920–33928. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Whelan MC and Senger DR: Collagen I
initiates endothelial cell morphogenesis by inducing actin
polymerization through suppression of cyclic AMP and protein kinase
A. J Biol Chem. 278:327–334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim M, Kim M, Lee S, Kuninaka S, Saya H,
Lee H, Lee S and Lim DS: cAMP/PKA signalling reinforces the
LATS-YAP pathway to fully suppress YAP in response to actin
cytoskeletal changes. EMBO J. 32:1543–1555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yu FX, Zhang Y, Park HW, Jewell JL, Chen
Q, Deng Y, Pan D, Taylor SS, Lai ZC and Guan KL: Protein kinase A
activates the hippo pathway to modulate cell proliferation and
differentiation. Genes Devel. 27:1223–1232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brooks PC, Clark RA and Cheresh DA:
Requirement of vascular integrin alpha v beta 3 for angiogenesis.
Science. 264:569–571. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brooks PC, Montgomery AM, Rosenfeld M,
Reisfeld RA, Hu T, Klier G and Cheresh DA: Integrin alpha v beta 3
antagonists promote tumor regression by inducing apoptosis of
angiogenic blood vessels. Cell. 79:1157–1164. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Delbaldo C, Raymond E, Vera K,
Hammershaimb L, Kaucic K, Lozahic S, Marty M and Faivre S: Phase I
and pharmacokinetic study of etaracizumab (Abegrin), a humanized
monoclonal antibody against alphαvβeta3 integrin receptor, in
patients with advanced solid tumors. Invest New Drugs. 26:35–43.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hersey P, Sosman J, O'Day S, Richards J,
Bedikian A, Gonzalez R, Sharfman W, Weber R, Logan T, Buzoianu M,
et al: A randomized phase 2 study of etaracizumab, a monoclonal
antibody against integrin alpha(v)beta(3), + or -dacarbazine in
patients with stage IV metastatic melanoma. Cancer. 116:1526–1534.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Petitclerc E, Strömblad S, von Schalscha
TL, Mitjans F, Piulats J, Montgomery AM, Cheresh DA and Brooks PC:
Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin
by regulating tumor cell survival. Cancer Res. 59:2724–2730.
1999.PubMed/NCBI
|
|
54
|
Natali PG, Hamby CV, Felding-Habermann B,
Liang B, Nicotra MR, Di Filippo F, Giannarelli D, Temponi M and
Ferrone S: Clinical significance of alpha(v)beta3 integrin and
intercellular adhesion molecule-1 expression in cutaneous malignant
melanoma lesions. Cancer Res. 57:1554–1560. 1997.PubMed/NCBI
|
|
55
|
Kanamori M, Vanden Berg SR, Bergers G,
Berger MS and Pieper RO: Integrin beta3 overexpression suppresses
tumor growth in a human model of gliomagenesis: Implications for
the role of beta3 overexpression in glioblastoma multiforme. Cancer
Res. 64:2751–2758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jinushi M, Chiba S, Baghdadi M, Kinoshita
I, Dosaka-Akita H, Ito K, Yoshiyama H, Yagita H, Uede T and Takaoka
A: ATM-mediated DNA damage signals mediate immune escape through
integrin-αvβ3-dependent mechanisms. Cancer Res. 72:56–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Su X, Esser AK, Amend SR, Xiang J, Xu Y,
Ross MH, Fox GC, Kobayashi T, Steri V, Roomp K, et al: Antagonizing
integrin β3 increases immunosuppression in cancer. Cancer Res.
76:3484–3495. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Reynolds LE, Wyder L, Lively JC, Taverna
D, Robinson SD, Huang X, Sheppard D, Hynes RO and Hodivala-Dilke
KM: Enhanced pathological angiogenesis in mice lacking beta3
integrin or beta3 and beta5 integrins. Nature Med. 8:27–34. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xi G, Guo W, Kang D, Ma J, Fu F, Qiu L,
Zheng L, He J, Fang N, Chen J, et al: Large-scale tumor-associated
collagen signatures identify high-risk breast cancer patients.
Theranostics. 11:3229–3243. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Drifka CR, Loeffler AG, Mathewson K,
Keikhosravi A, Eickhoff JC, Liu Y, Weber SM, Kao WJ and Eliceiri
KW: Highly aligned stromal collagen is a negative prognostic factor
following pancreatic ductal adenocarcinoma resection. Oncotarget.
7:76197–76213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wu PC, Hsieh TY, Tsai ZU and Liu TM: In
vivo quantification of the structural changes of collagens in a
melanoma microenvironment with second and third harmonic generation
microscopy. Sci Rep. 5:88792015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Birk JW, Tadros M, Moezardalan K,
Nadyarnykh O, Forouhar F, Anderson J and Campagnola P: Second
harmonic generation imaging distinguishes both high-grade dysplasia
and cancer from normal colonic mucosa. Dig Dis Sci. 59:1529–1534.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Burke K, Smid M, Dawes RP, Timmermans MA,
Salzman P, van Deurzen CH, Beer DG, Foekens JA and Brown E: Using
second harmonic generation to predict patient outcome in solid
tumors. BMC Cancer. 15:9292015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xu J, Rodriguez D, Petitclerc E, Kim JJ,
Hangai M, Moon YS, Davis GE and Brooks PC: Proteolytic exposure of
a cryptic site within collagen type IV is required for angiogenesis
and tumor growth in vivo. J Cell Biol. 154:1069–1079. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Willumsen N, Ali SM, Leitzel K, Drabick
JJ, Yee N, Polimera HV, Nagabhairu V, Krecko L, Ali A, Maddukuri A,
et al: Collagen fragments quantified in serum as measures of
desmoplasia associate with survival outcome in patients with
advanced pancreatic cancer. Sci Rep. 9:197612019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kehlet SN, Sanz-Pamplona R, Brix S,
Leeming DJ, Karsdal MA and Moreno V: Excessive collagen turnover
products are released during colorectal cancer progression and
elevated in serum from metastatic colorectal cancer patients. Sci
Rep. 6:305992016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lipton A, Leitzel K, Ali SM, Polimera HV,
Nagabhairu V, Marks E, Richardson AE, Krecko L, Ali A, Koestler W,
et al: High turnover of extracellular matrix reflected by specific
protein fragments measured in serum is associated with poor
outcomes in two metastatic breast cancer cohorts. Int J Cancer.
143:3027–3034. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hamilton HK, Rose AE, Christos PJ, Shapiro
RL, Berman RS, Mazumdar M, Ma MW, Krich D, Liebes L, Brooks PC and
Osman I: Increased shedding of HU177 correlates with worse
prognosis in primary melanoma. J Transl Med. 8:192010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lindsey ML, Iyer RP, Zamilpa R,
Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA,
Bratton D, Flynn ER, et al: A novel collagen matricryptin reduces
left ventricular dilation post-myocardial infarction by promoting
scar formation and angiogenesis. J Am Coll Cardiol. 66:1364–1374.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang J and Pan W: The biological role of
the collagen alpha-3 (VI) chain and its cleaved C5 domain fragment
endotrophin in cancer. Onco Targets Ther. 13:5779–5793. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jang JW, Kim MK and Bae SC: Reciprocal
regulation of YAP/TAZ by the hippo pathway and the small GTPase
pathway. Small GTPases. 11:280–288. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Feng X, Degese MS, Iglesias-Bartolome R,
Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino
G, Sodhi A, et al: Hippo-independent activation of YAP by the GNAQ
uveal melanoma oncogene through a trio-regulated rho GTPase
signaling circuitry. Cancer Cell. 25:831–845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Qiao Y, Chen J, Lim YB, Finch-Edmondson
ML, Seshachalam VP, Qin L, Jiang T, Low BC, Singh H, Lim CT and
Sudol M: YAP regulates actin dynamics through ARHGAP29 and promotes
metastasis. Cell Rep. 19:1495–1502. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kornepati AVR, Boyd JT, Murray CE,
Saifetiarova J, de la Peña Avalos B, Rogers CM, Bai H, Padron AS,
Liao Y, Ontiveros C, et al: Tumor intrinsic PD-L1 promotes DNA
repair in distinct cancers and suppresses PARP inhibitor-induced
synthetic lethality. Cancer Res. 82:2156–2170. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lee JJ, Kim SY, Kim SH, Choi S, Lee B and
Shin JS: STING mediates nuclear PD-L1 targeting-induced senescence
in cancer cells. Cell Death Dis. 13:7912022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yu J, Qin B, Moyer AM, Nowsheen S, Tu X,
Dong H, Boughey JC, Goetz MP, Weinshilboum R, Lou Z and Wang L:
Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res.
30:590–601. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ghebeh H, Lehe C, Barhoush E, Al-Romaih K,
Tulbah A, Al-Alwan M, Hendrayani SF, Manogaran P, Alaiya A,
Al-Tweigeri T, et al: Doxorubicin downregulates cell surface B7-H1
expression and upregulates its nuclear expression in breast cancer
cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer
Res. 12:R482010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ye L, Zhu Z, Chen X, Zhang H, Huang J, Gu
S and Zhao X: The importance of exosomal PD-L1 in cancer
progression and its potential as a therapeutic target. Cells.
10:32472021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gao Y, Nihira NT, Bu X, Chu C, Zhang J,
Kolodziejczyk A, Fan Y, Chan NT, Ma L, Liu J, et al:
Acetylation-dependent regulation of PD-L1 nuclear translocation
dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol.
22:1064–1075. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gao Y, Bi D, Xie R, Li M, Gau J, Liu H,
Guo X, Fang J, Ding T, Zhu H, et al: Fusobacterium nucleatum
enhances the efficacy of PD-L1 blockade in colorectal cancer.
Signal Transduct Target Ther. 6:3982021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wei Y, Tang X, Ren Y, Yang Y, Song F, Fu
J, Liu S, Yi M, Chen J, Wang S, et al: An RNA-RNA crosstalk network
involving HMGΒ1 and RICTOR facilitates hepatocellular carcinoma
tumorigenesis by promoting glutamine metabolism and impedes
immunotherapy by PD-L1+ exosomes activity. Signal Transduct Target
Ther. 6:4212021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang JJ, Zhang QS, Li ZQ, Zhou JW and Du
J: Metformin attenuates PD-L1 expression through activating hippo
signaling pathway in colorectal cancer cells. Am J Transl Res.
11:6965–6976. 2019.PubMed/NCBI
|
|
83
|
Moya IM, Castaldo SA, Van den Mooter L,
Soheily S, Sansores-Garcia L, Jacobs J, Mannaerts I, Xie J,
Verboven E, Hillen H, et al: Peritumoral activation of the hippo
pathway effectors YAP and TAZ suppresses liver cancer in mice.
Science. 366:1029–1034. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yuan M, Tomlinson V, Lara R, Holliday D,
Chelala C, Harada T, Gangeswaran R, Manson-Bishop C, Smith P,
Danovi SA, et al: Yes-associated protein (YAP) functions as a tumor
suppressor in breast. Cell Death Differ. 15:1752–1759. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lebid A, Chung L, Pardoll DM and Pan F:
YAP attenuates CD8 T cell-mediated anti-tumor response. Front
Immunol. 11:5802020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Stampouloglou E, Cheng N, Federico A,
Slaby E, Monti S, Szeto GL and Varelas X: Yap suppresses T-cell
function and infiltration in the tumor microenvironment. PLoS Biol.
18:e30005912020. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ni X, Tao J, Barbi J, Chen Q, Park BV, Li
Z, Zhang N, Lebid A, Ramaswamy A, Wei P, et al: YAP is essential
for Treg-mediated suppression of antitumor immunity. Cancer Discov.
8:1026–1043. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yang W, Yang S, Zhang F, Cheng F, Wang X
and Rao J: Influence of the Hippo-YAP signalling pathway on tumor
associated macrophages (TAMs) and its implications on cancer
immunosuppressive microenvironment. Ann Transl Med. 8:3992020.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang
S, Fang Z, Zhao K, Konaparthi R, Hua S, et al: Targeting
YAP-dependent MDSC infiltration impairs tumor progression. Cancer
Discov. 6:80–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Shibata M, Ham K and Hoque MO: A time for
YAP1: Tumorigenesis, immunosuppression and targeted therapy. Int J
Cancer. 143:2133–2144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lee JY, Dominguez AA, Nam S, Stowers RS,
Qi LS and Chaudhuri O: Identification of cell context-dependent
YAP-associated proteins reveals β1 and β4
integrin mediate YAP translocation independently of cell spreading.
Sci Rep. 9:171882019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lecker SH, Goldberg AL and Mitch WE:
Protein degradation by the ubiquitin-proteasome pathway in normal
and disease states. J Am Soc Nephrol. 17:1807–1819. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Bao W, Thullberg M, Zhang H, Onischenko A
and Strömblad S: Cell attachment to the extracellular matrix
induces proteasomal degradation of p21(CIP1) via Cdc42/Rac1
signaling. Mol Cell Biol. 22:4587–4597. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Sasada M, Iyoda T, Asayama T, Suenaga Y,
Sakai S, Kase N, Kodama H, Yokoi S, Isohama Y and Fukai F:
Inactivation of beta1 integrin induces proteasomal degradation of
Myc oncoproteins. Oncotarget. 10:4960–4972. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kraman M, Bambrough PJ, Arnold JN, Roberts
EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA and Fearon DT:
Suppression of antitumor immunity by stromal cells expressing
fibroblast activation protein-alpha. Science. 330:827–830. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Özdemir BC, Pentcheva-Hoang T, Carstens
JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C,
Novitskiy SV, et al: Depletion of carcinoma-associated fibroblasts
and fibrosis induces immunosuppression and accelerates pancreas
cancer with reduced survival. Cancer Cell. 25:719–734. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Legler DF, Johnson-Léger C, Wiedle G, Bron
C and Imhof BA: The alpha v beta 3 integrin as a tumor homing
ligand for lymphocytes. Eur J Immunol. 34:1608–1616. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Larochelle C, Uphaus T, Broux B, Gowing E,
Paterka M, Michel L, Dudvarski Stankovic N, Bicker F, Lemaître F,
Prat A, et al: EGFL7 reduces CNS inflammation in mouse. Nature
Commun. 9:8192018. View Article : Google Scholar : PubMed/NCBI
|