Introduction
In 1990, mitochondria (Mt)-associated membranes
(MAMs) were first discovered (1) as
the communication network between Mt and endoplasmic reticulum
(ER), acting via proteins expressed on the lipid membranes of these
organelles (2). To date, >1,000
proteins rest in Mt-ER contact sites (MERCSs) each associated with
one or even a variety of cellular biochemical functions, including
calcium (Ca2+) homeostasis, lipid metabolism, apoptosis,
autophagy and tumour growth (3,4). The
integration and coordination between these two organelles in a
well-orchestrated network crucial for maintaining homeostasis and
serves as a regulatory, signalling and external protein interaction
point. Disturbance to the configuration of MERCSs results in
miscommunication among the Mt-ER linkage, which leads to a plethora
of pathological conditions, such as Alzheimer's disease (5), Parkinson's disease (6,7),
lysosomal storage diseases (8),
inflammation (9) and cancer
(10,11).
The length of MERCSs, depending on cell type, varies
from 10–100 nm; 10–15 nm in the smooth ER and 20–30 nm in the rough
ER, with 15–20% of their total surface being juxtaposed to the ER
(12). The proteins found in the
MERCSs are divided into two types: Connective proteins (CPs) that
participate in the physical connection between ER and Mt, and
interfering proteins that can alter the distance between the two
organelles and decrease the contact sites (13). Through MERCSs, the ER and Mt
exchange signals and stress stimuli, as well as chemical responses
and cell death/survival events, a fact that has numerous times been
investigated and the ‘mitochondrial-associated membrane structure’
considered as an independent sub-organelle (13,14).
Evidence supports the fact that the loss of optimal Mt-ER organelle
communication affects MERCS activities involving cellular processes
such as apoptosis (Bcl2), regulation of cell growth
[serine/threonine-specific protein kinase (Akt)], senescence and
metabolism (Ras), but also tumour suppression [breast cancer type 1
susceptibility protein, and phosphatase and tensin homolog (PTEN)]
(Fig. 1). Dysregulation in the
function of these proteins results in multiple pathologies,
including cancer (12).
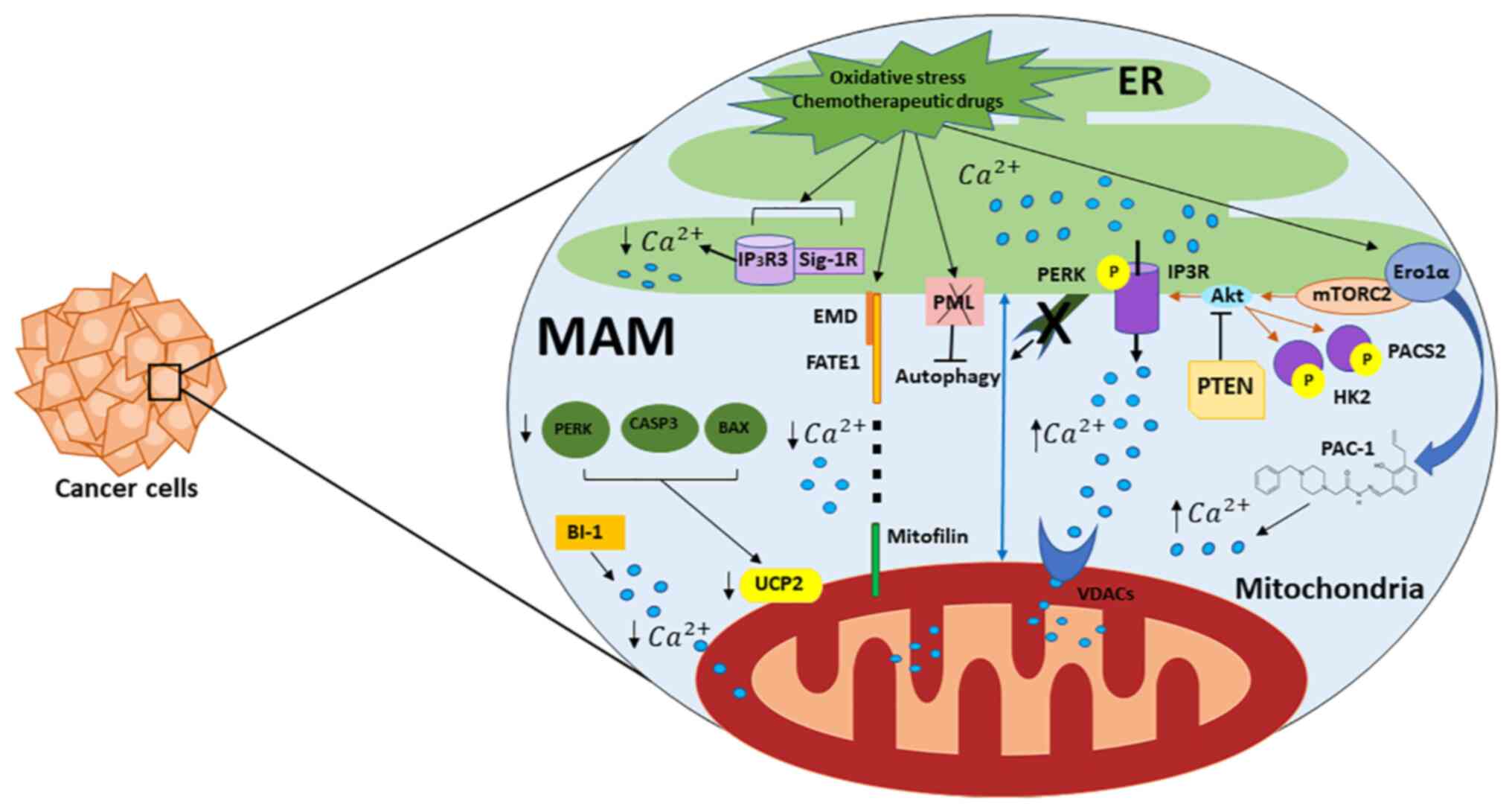 | Figure 1.Role of Mt-associated ER membranes in
calcium regulation and cancer. Important proteins present in MERCSs
used as Ca2+ transfer systems at the ER and Mt and
contributing to cell death and survival are shown. Regulators of ER
Ca2+ release machinery are IP3Rs that act as
ligand-gated channels and facilitate Ca2+ release from
the ER to the Mt through VDACs. A fraction of PTEN proteins,
localized to the ER and MERCSs, regulate Ca2+ release by
preventing the activating phosphorylation of Akt, which reduces
Ca2+ release via IP3Rs. Mt, mitochondria; ER,
endoplasmic reticulum; MERCS, Mt-ER contact site; IP3R, inositol
1,4,5-trisphosphate receptor type 3; VDAC, mitochondrial
voltage-dependent anion channel; PTEN, phosphatase and tensin
homolog; Akt, serine/threonine-specific protein kinase; MAM,
Mt-associated membrane; p, phosphate. |
Autophagosome formation by autophagy-related
proteins, e.g., Vps34 and Beclin 1, is a cellular activity affected
by Mt-ER interaction, along with anti- and pro-apoptotic proteins
(12,15). Should the two organelles not
maintain Ca2+ homeostasis and result in an Mt
Ca2+ overload, the permeability transition pore (PTP)
will be unlocked, paving the way for the activation of caspases and
the release of pro-apoptotic factors (16). This activity also releases
cytochrome c resulting in apoptotic cell death. MERCSs also
promote apoptosis or ferroptosis via reactive oxygen species (ROS)
and lipid peroxidation products in cells (10). Targeting MERCSs can aid in cancer
therapeutics, such as activating signal transducer and activator of
transcription 3 (STAT3), located in this region, resulting in
apoptotic resistance due to Ca2+ balancing of inositol
1,4,5-trisphosphate receptor type 3 (IP3R3) degradation mediated by
IP3R3/STAT3 interaction (17).
In addition, the transfer of Ca2+ from
the ER to the Mt is crucial for the regulation of several oncogenes
and tumour suppressors (18). One
of the key activators of IP3R is Akt, which activates IP3R isoform
3 in MERCSs and inhibits apoptosis (19). Akt activity is regulated by its
inhibitors, PTEN, the tumor suppressor promyelocytic leukemia
protein (PML) and the activator mechanistic target of rampamycin
kinase complex 2 (mTORC2), all of which are found to be enriched in
MERCSs (20). One of the major
tumour suppressors is p53. ER-MAM localization allows tumour
suppressor p53 to facilitate Ca2+-dependent apoptosis,
via the sarco/ER Ca2+-ATPase (SERCA), which is expressed
on the ER membrane (21). Overall,
Mt-ER linkage tightening increases Ca2+ uptake and
induces apoptosis, while loosening of ER-Mt
connections/interactions promotes cell survival and Mt respiration
(22). The following report
summarizes the role proteins expressed in MERCSs play in
pharmacological inhibition/activation in cancer environments.
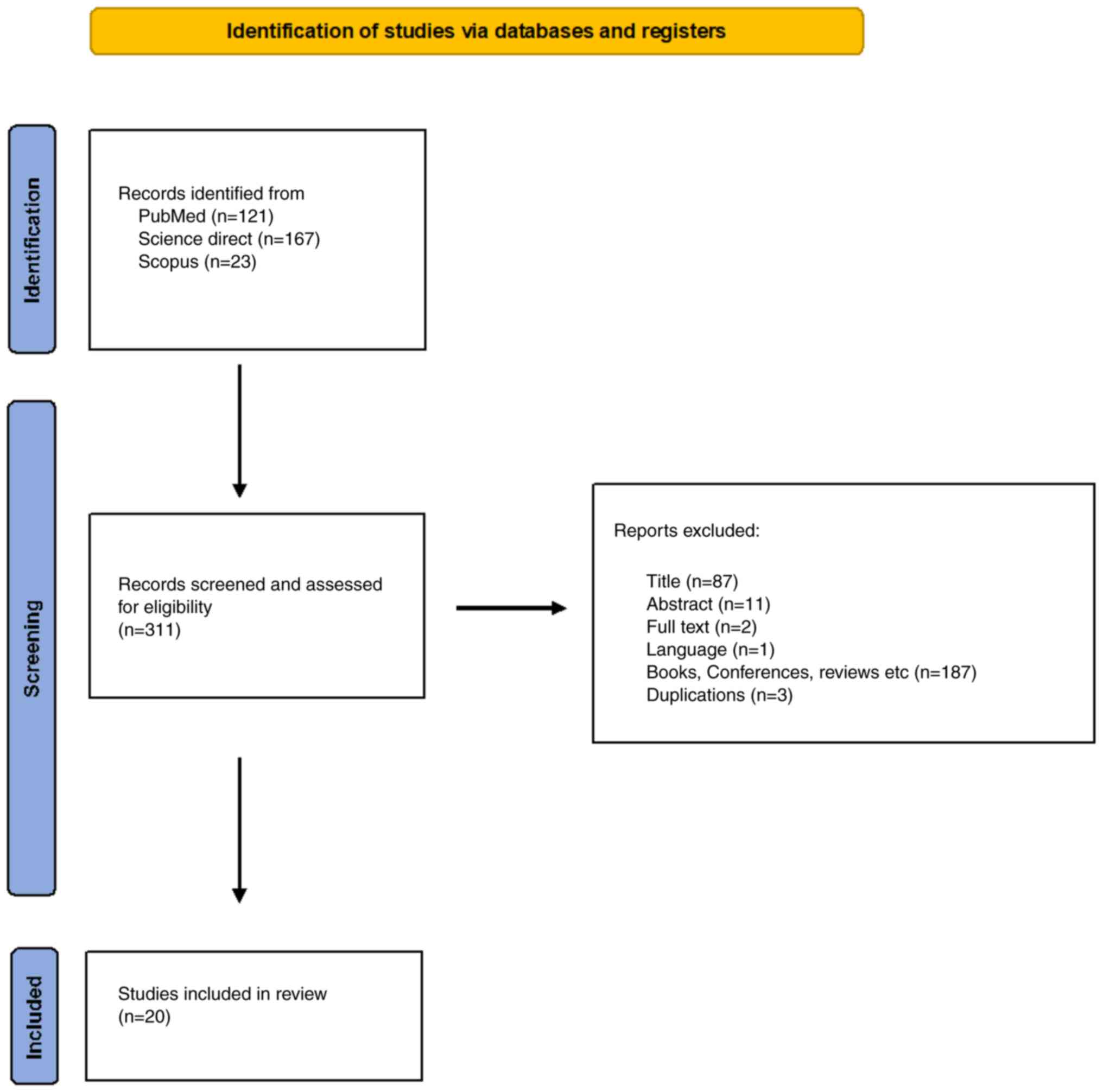
Findings have been collaborated using PRISMA guidelines.
Key features of MERCSs
Lipid synthesis
Enzymes within the ER are responsible for the
synthesis of cellular lipids; however, the activity of these
enzymes is affected by alterations to the status quo of both the
organelle and cytosol (23). The Mt
also act as a factory for lipid membrane components such as
cardiolipin, synthesised solely within the Mt, and
phosphatidylethanolamine (PE), synthesised within Mt but requiring
intervention from the cytosol and ER (24). Phosphatidylserine (PS) is a lipid
metabolising protein synthesised by PS synthases 1 and 2 (PSS1 and
PSS2) in the ER; it is then stored in MERCSs until it is signalled
to enter the Mt for its conversion to PE via carboxylases.
Phosphatidylcholine (PC) is another lipid metabolism protein
recruited within MERCSs. In cancer cells, lipid membrane integrity
and function are affected as the Kennedy pathway (synthesis of PC
and PE) is disrupted (25), thus
explaining why PE is utilised as a diagnostic marker, as its
concentration increases as excess cell proliferation occurs in
cancer environments. Interrupting the alignment between the Mt and
ER dysregulates lipid synthesis and the transfer of lipids to
target organelles, and hence results in damage/loss of lipid
membranes (9).
MERCSs are cholesterol-rich membranes carrying a
sterol interacting protein, known as caveolin, which is involved in
sterol metabolism (26). Especially
during acute stress, cholesterol is transported to the Mt for
steroidogenesis. An additional protein found in MERCSs that is
involved in steroidogenesis is ATPase family AAA domain-containing
protein 3B; this protein is involved in transporting cholesterol
from the ER to the Mt during MERCS formation, an action that
expresses chemoresistance and anti-proliferative abilities through
mechanisms that remain unclear (27). Finally, MERCSs play a central role
in the formation of ceramide, a sphingolipid product that is also
involved in apoptotic cell death, inflammation, cell growth and
differentiation (9,19,28,29).
Ca2+ signalling
SERCA pumps regularly supply the ER with
Ca2+ from the cytosol (30). A SERCA pump has three isoforms,
SERCA1, SERCA2 and SERCA3, of which SERCA2 is expressed in MERCSs.
An array of different mechanisms and pathways are triggered by
Ca2+ signalling in the Mt (31). For example, the presence of
Ca2+ increases the activity of the tricarboxylic acid
cycle enzymes, and stimulates the electron transport chain and
oxidative phosphorylation (32).
Overall, MERCSs are responsible for the regulation of cellular
metabolism through Ca2+ signalling driving ATP
production, metabolism, gene activation and cell survival pathways
(31). While the ER is the main
Ca2+ storage organelle, the presence of MERCSs is
responsible for the transport and accumulation of Ca2+
in Mt, which further affects crucial cellular activities, as
mentioned throughout this review.
At the same time, the accumulation/overload of
Ca2+ to the Mt can cause swelling and cell death
(33). Apart from SERCA pumps,
multiple ER-Ca2+ proteins, each with a different role,
are found in MERCSs, including, but not limited to, IP3R and SERCA.
IP3R regulates Ca2+ transmission via mitochondrial
voltage-dependent anion channel 1 (VDAC1), which is found at the
outer mitochondrial membrane and is connected to the MERCSs. The
molecular chaperone glucose-regulated protein 75 succeeds the
connection that regulates the IP3R-VDAC1 interaction. When the
Ca2+ flux from the ER to the Mt declines, cells become
more resistant to apoptosis, whereas overexpression of
Ca2+ results in apoptosis, as seen in vascular smooth
muscle cells and epithelial cancer cells, due to the ion's
relationship with the Mt-ER associated membrane fusion mediator,
Mitofusin 2 (Mfn2) (34). Transfer
of this ion across MAMs is also regulated through a supplementary
pathway involving PML, Akt and IP3R3, where Ca2+ flux
from the ER to Mt is increased, thus increasing apoptosis and
acting against cancer.
Methodology
Evidence regarding Mt-associated ER membranes and
cancer was reviewed for the present study. A literature search was
performed in PubMed (https://pubmed.ncbi.nlm.nih.gov/), Science Direct
(https://www.sciencedirect.com/) and
Scopus (https://scopus.com) to identify relevant
studies that were published up to and including March 4, 2022, or
within the last 10 years of this search date.. The search was based
on the following key words and terms in these databases: i) PubMed:
((((((((mitochondria-associated ER membranes[Title/Abstract]) OR
(MERCSs[Title/Abstract])) AND (cancer[Title/Abstract])) OR
(tumor[Title/Abstract])) OR (tumour[Title/Abstract])) OR
(carcinoma[Title/Abstract])) OR (malignancy[Title/Abstract])) OR
(proliferation[Title/Abstract])) OR (onco*[Title/Abstract]); ii)
Science direct: (mitochondria associated ER membranes, MERCSs,
cancer, tumor, tumour, carcinoma, malignancy, proliferation); iii)
Scopus: TITLE-ABS-KEY (‘mitochondria associated ER membrane’ AND
‘cancer’ OR ‘tumor’ OR ‘carcinoma’ OR ‘malignancy’ OR
‘proliferation’) AND [LIMIT-TO (PUBSTAGE, ‘final’)] AND [LIMIT-TO
(DOCTYPE, ‘ar’)] AND [LIMIT-TO (LANGUAGE, ‘English’)]. Abstracts
were studied to identify papers on MERCSs in cancer following the
PRISMA guidelines (Fig. 2).
Publications describing the role of MERCS on cellular integrity and
effects on cancer were accessed and reviewed meticulously, and
those that did not refer to activity in MERCS space were excluded
from the review. The references of the relevant articles were also
accessed and reviewed. Finally, expression/alteration data were
collected from cBioPortal (https://www.cbioportal.org/) and the University of
Alabama at Birmingham Cancer data analysis portal (UALCAN;
http://ualcan.path.uab.edu/).
Association of key genes/proteins expressed
in MERCSs with cell fate
As aforementioned, MAMs within MERCS include an
assortment of proteins (Fig. 1)
that compose the MERCSs and regulate cellular activity. Silencing
the expression of some of these proteins will subsequently affect
Mt-ER Ca2+ flow, instigate/stifle ER stress and/or
threaten cell integrity. Altering Ca2+ flow into the Mt
affects mitochondrial performance, such as ATP production and
autophagy activity. Autophagy is known to provide alternative
sources of carbon, e.g., through fatty acid oxidation, so the cell
can meet the higher metabolic demands of tumour microenvironments
and permit the cell to elude cell toxicity. For instance, silencing
PML within MERCSs results in reduced Ca2+ transfer
between the ER and Mt via IP3R, and subsequently, ATP production
decreases while AMPK activity increases, setting off the
AMPK/mTOR/UIK1 pathway that prolongs autophagy. Similarly these
effects are recorded in p53-/- cells where delocalisation of PML
occurs (34). AMPK interacts with
the vesicle-trafficking mediator Beclin-1 following reduced
Ca2+ transfer to the Mt across MAM, which actuates
autophagosome formation, whereas silencing the Beclin-1 gene,
BECN1, has the opposite effect (35).
Proteins that complement one another's functionality
may compartmentalise across membranes, forming raft-like lipid
microdomains ensuring protein interactions take place effortlessly,
as is the case with autophagy and Beclin 1 regulator 1 (AMBRA1), ER
lipid raft associated 1 (ERLIN1) and mitofusin 2 (MFN2) proteins
and ganglioside GD3 (gGD3). AMBRA1 and ERLIN1 interact with support
from MFN2 and gGD3 within MERCSs to stimulate the formation of
autophagosomes (36). One study
showed that knockdown of ST8SIA1 (gGD3) or MFN2 halted
autophagosome development, as did the silencing of ERLIN1 (36). This suggests that all proteins are
required in MERCSs for autophagy to occur. Thapsigargin (Tg) is an
ER stressor drug affecting Ca2+ homeostasis. In another
study, in both HeLa and Du145 cells, unphosphorylated ER stress
sensor IRE was dominant, while following treatment of cells with
Tg, readings of phosphorylated IRE (pIRE) increased (37). Knockdown of sigma non-opioid
intracellular receptor 1 dephosphorylates IRE, returning cells to
an unstressed stage, an action significant for counteracting
autophagy, indicating the need of the chaperone to protect IRE from
ER-associated degradation. Following Sig-1R knockdown, XBP1 protein
splicing is also reduced, as XBP1 cannot bind to fluorescent
protein sites due to the absence of the pIRE responsible for its
activation (37). Tg was also used
to treat AR42J and Neuro2A cells producing excessive
phosphorylation of IRE1 (38).
Overall, these results present the crucial role of Sig-1Rs in the
IRE1-XBP1 signalling pathway involved in cell survival. In the
research analysed so far, the IRE1-XBP1 pathway does not induce
apoptosis or autophagy as such, but it promotes cell survival by
acting against ER stress through Sig-1R chaperones and IRE in
MAMs.
The flow of Ca2+ into the Mt can also be
affected by the distance between the Mt and ER or altering the
positioning of the organelles, consequently affecting cell
proliferation, integrity and apoptosis. Adrenocortical carcinoma
(ACC), for example, is deemed resistant to mitotane drug treatment,
as upregulation of fetal and adult testis-expressed 1 (FATE1)
disturbs the flow of Ca2+ into the Mt as it uncouples
the Mt and ER, consequently increasing the resistance of tumour
cells to oxidative stress and chemotherapeutic drugs (39). Knockdown of FATE1 reverses these
effects and allows ACC to respond to treatment and undergo
apoptosis during treatment. Similarly, high FATE1 expression has
also been associated with reduced survival time in patients with
breast cancer (Fig. S1).
Furthermore, knockdown of the ER stress sensor PERK
(PERK−/−) in mouse embryonic fibroblasts (MEFs)
distorted ER morphology, increasing the distance between the Mt and
ER, shielding the Mt from ROS-mediated effector build-up, and
subsequently avoiding ER-stress and apoptosis. Cells expressing
PERK regulated Mt-ER defense responses against ROS, but also
expressed high levels of pro-apoptotic C/EBP homologous protein
(CHOP) triggering apoptosis more effectively than Tg. The effects
of PERK−/− were also evaluated in CT26 and MDA-MB468
cell lines that were resistant to cell death due to increased
expression of GRP78 chaperone, therefore initiating
Ca2+overflow outside the ER, low caspase activation and
depletion of CHOP (40).
Alterations in ER morphology, as well as a weaker Mt-ER
association, were also perceived in MEFs deficient in MFN2. CHOP
and Ero1α are upregulated by procaspase-activating compound-1
(PAC-1), a direct caspase-activator, triggering ER stress. To
evaluate these effects, both proteins were silenced in HeLa D1ER
cells, reducing Ca2+ release from the ER and decreasing
apoptosis. Furthermore, PAC-1 expression induced GRP78 and GRP94
chaperone activity causing Ca2+ leakage subsequent to
Ero1a-dependent ER luminal hyper-oxidation in MCF7 and MCF7C3
cells. As a result, ER stress and Mt-mediated apoptosis were
recorded (41).
As seen so far, MERCSs may be affected directly or
indirectly. Uncoupling protein 2 (UCP2), for example, enhances
protein arginine N-methyltransferase 1 activity, which in turn
methylates mitochondrial calcium uptake 1, a Ca2+
protein pump located in the MAM. Madreiter-Sokolowski et al
(2021) (42) reported that there
was an inverse correlation between UCP2 and the proteins
responsible for stabilizing Mt-ER association, in prostate
adenocarcinoma tissues, breast invasive cancer, cervical squamous
cell carcinoma and multiple other cancer types, affecting cell
viability. For example, an IP correlation was observed in HeLa
cells where upregulation of Rab32, an A-anchoring protein fostering
Mt-ER tethering, resulted in a downregulation of UCP2 and
consequently permitted the cancer cells to escape mitochondrial
Ca2+ overload-induced cell death. Mondet et al
(2021) (43) reasoned that Mt in
acute myeloid leukemia (AML) can be targeted to regulate the
proliferation and chemosensitivity within these cells. Assembly of
the Mt ultrastructure and its influence on cellular integrity were
evaluated in multiple leukaemia cell lines (HEL, HL60, K562, KG1
and OCI-AML3). Alterations were unearthed on a molecular level
between the different cell lines. In the case of K562 cells, an
ASXL1 gene mutation was recorded, affecting the Mt shape within
MERCSs and creating a distance between the Mt and ER, allowing the
cells to resist chemotherapy drugs and continue proliferating. This
mutation also affected genes expressing MAM complexes, such as
VAMP-associated protein B and C (VAPB) and oxysterol binding
protein like 5 (OSBPL5), affecting the physiology and integrity of
the organelle (43). HL60 cells,
where the mutation is lacking, displayed sensitivity to drugs as
the distance between the Mt and ER was unchanged.
The physiology of the Mt and ER is greatly dependent
on the integrity and expression of proteins within MERCSs.
Transient receptor potential melastatine 8 (TRPM8) channel isoforms
are expressed on the ER lipid membrane, whose knockdown initiates
apoptosis in cancer cells (44). It
is understood that the 4TM-TRPM8 isoform aids the survival of
prostate cancer epithelial cells by actively regulating
Ca2+ trafficking from the ER into the cytosol, and
subsequently Mt uptake, which as a result protects the cells from
ER stress. Alternatively, survival of tumor cells is affected by
the expression of NLRX1, coding for a NOD-like receptor immune
system regulator, where expression of this gene regulates
TNF-α-induced metabolism and cell death (45). Overexpression of Bax inhibitor-1
(BI-1) in HT1080 cells (HT1080/BI-1) permitted them to avert
apoptosis by leaking Ca2+ out of the ER, so no ER stress
resulted, and reducing mitochondrial Ca2+ intake,
therefore reducing cytochrome c release (involved in
apoptosis) and PTP opening. To maintain mitochondrial homeostasis
in HT1080/BI-1, mitoKca channels open, permitting an
influx of K+, further protecting the cell against ER
stress and its consequences (46).
Silencing BI-1 decreased Ca2+ in the ER and
simultaneously increased mitochondrial Ca2+, permitting
the cell to respond to drugs and undergo apoptosis (46). Additionally, the crucial role of
FUNDC1 in angiogenesis has been investigated both in vitro
and in vivo. Disruption of FUNDC1-related MAM formation
contributed to intracellular Ca2+ dys-homeostasis,
resulting in decreased levels of pSRF and VEGFR2, and subsequent
reduction of VEGF-induced angiogenesis (12). A recent study supported that chronic
increases in MAM formation resulted in mitochondrial
Ca2+ overload, impairing mitochondrial bioenergetics
function and increasing ROS production in vivo, also leading
to aging-associated pathologies, including Alzheimer's disease,
Parkinson's disease and amyotrophic lateral sclerosis (47). The expression levels of IP3R, FUNDC1
and MFN2 were significantly elevated in MAM fractions isolated from
VEGF-treated endothelial cells. The interaction between FUNDC1 and
IP3R1 in MAMs mediated the changes in Ca2+ levels. IP3R1
knockdown significantly inhibited vascular angiogenesis in
vivo (48).
ER stress, Ca2+ regulation and gene
knockdown all influence MAM behavior, and these in turn are
dependent on the length of MERCSs to ensure optimum interplay to
succeed in affecting the vulnerability of cancer cells (40). Separation of the two organelles can
result in resistance to chemotherapeutic drugs, ER stress and/or
apoptosis. This is supported by the study by Doghman-Bouguerra
et al (2016) (39), where
increased FATE1 expression in ACC cells caused the Mt and ER to
detach and apoptosis to be eliminated. FATE1 protein localised near
calreticulin and HSP60 (ER and Mt markers, respectively) in MERCSs.
In the same study, FATE1 knockdown caused a significant increase in
angiotensin II-stimulated aldosterone levels, which resulted in the
increase of blood pressure, dehydroepiandrosterone and cortisol, as
well as aldosterone, with multiple biological pathways
affected.
The mTORC2 protein facilitates the phosphorylation
of IP3R, hexokinase 3 and phosphofurin acidic cluster sorting
protein 2 (PACS2) via Akt in non-small cell lung carcinomas
(NSCLC). Triggering PACS2 with a compound such as oxyphyllanene B,
increases its activity that, as a result, distorts Mt-ER
communication and allows cancer cells, such as glioblastoma, to
overcome chemotherapy resistance (49). At the same time, pharmacologically
inhibiting mTOR decreases cell proliferation and increases
apoptosis (50). Alternatively,
tyrosine kinase inhibitors (TKIs) relocate the expression of mTORC2
from MERCSs to the cytoplasm, assisting cancer cells such as H1299
and H1975 to overcome CD20 mono-antibody EGFR-TKI resistance
(39). To overcome the resistance
of NSCLC to EGFR-TKIs, the drug rituximab was administered in
synergy with erlotinib to move the expression of the mTORC2 protein
from MERCS region to the cytoplasm. Consequently, both H1299 and
H1975 cells overcame EGFR-TKI resistance (51). PTEN expresses multiple actions and
is localised in the ER and MAM region, affecting Ca2+
transport and apoptosis induction. Stimulation of the silenced PTEN
cells with ATP led to IP3 expression, meaning greater interaction
with IP3Rs, as aforementioned, resulting in the ensuing cell
apoptotic activity. High localization of PTEN in the ER during
ArA-mediated apoptosis supports the involvement of PTEN in
Ca2+ mediated apoptosis via IP3Rs (52).
Finally, the key MAM proteins reported by the
literature that affect cancer outcomes are summarized in the
present review, and using bioinformatics analysis, their levels of
expression in different cancer types, including their role in cell
outcome, are reviewed. Gene expression analysis using The Cancer
Genome Atlas data from UALCAN indicated that MERCS genes (Table I) are differentially expressed in
respect to cancer type. Consequently, genes regularly expressing
proteins in MERCSs affect patient survival, including FATE1,
EIF2AK3 and TRPM8 in breast cancer, AMBRA1 and ERLIN1 in pancreatic
cancer, mTORC2 in ovarian cancer, PML and PRKAA2 in kidney renal
clear cell carcinoma, MFN2 in thyroid cancer, UCP2 in sarcoma,
BECN1 in liver hepatocellular carcinoma and ASXL1 in adenoid cystic
carcinoma.
 | Table I.Data extracted from the literature
search indicating type of study, cancer type and the gene or
protein studied and findings. |
Table I.
Data extracted from the literature
search indicating type of study, cancer type and the gene or
protein studied and findings.
| First author,
year | Type of
cancer/cells | Aim | Gene | Protein |
Findings/results | (Refs.) |
|---|
| Bidaux et
al, 2018 | Prostate
cancer | Explore the
molecular properties of the TRPM8 channel subfamily and their
function | TRPM8 | Transient receptor
potential cation channel subfamily M (melastatin) member 9 | Ca2+
flow into the Mt and ER is also affected by 4TM-TRPM8. | (44) |
| Mori et al,
2013 | Prostate
cancer | Assess the role of
Sig-1R chaperones at MAM when cells are under oxidative/ER
stress | SIGMAR,
ERN1 | Sigma-1,
Serine/threonine-protein kinase/endoribonuclease IRE1 | IRE1 is chaperoned
by Sig-1Rs in MAM when the cell is under ER stress. The cell avoids
apoptosis via this physical support but also through the
transmission of stress signals from ROS from the Mt to the nucleus
via MAM | (37) |
| Wang et al,
2021 | Lewis lung
carcinoma | Determine how to
expression of FUNDC1 affects the ER-Mt association and consequently
how it affects MAM-related proteins such as VEGFR2 | FUNDC1 | FUN14
domain-containing protein 1 | Deleting FUNDC1
gene results in decreased ER-Mt co-localisation, consequently
decreasing VEGFR2 expression. VEGFR2 plays a critical role in
angiogenesis; therefore, in its absence, angiogenesis is disrupted
thus limiting the progress of cancer | (12) |
| Lee et al,
2014 | Fibrosarcoma | This study
considers regulatory effects of the concentration of
Ca2+ ions in the Mt on cell death. Also how BI-1 reduces
[Ca2+] mito | TMBIM6 | Bax
inhibitor-1 | Both
Ca2+ uniporter and Ca2+-dependent
K+ channels are regulated by BI-1, which causes them to
open. As a result, the mitochondrial permeability transition pore
is inhibited preventing the inflow of Ca2+ and
mitochondrial swelling. Through this mechanism the cell can escape
cell death | (46) |
| Manganelli et
al, 2020 2020 | Fibrosarcoma | Understand the
interaction between ERLIN1 and AMBRA1, and how MFN2 and ganglioside
GD3 affect autophagy activity in MERCSs | ERLIN1 | ER lipid raft
associated 1 | Upon analysis of
2FTGH cells, a physical relationship is presented between ERLIN1
and AMBRA1. This relationship is severed if MFN2 or ST8SIA1 are
knocked down. For autophagy to occur, assembly of AMRA1 with
additional molecules is necessary in MERCSs
(AMBRA1/BECN1/WIPI1) | (36) |
| Xu et al,
2017 | Non-small cell lung
cancer (T790M EGFR mutation) | To evaluate if
erlotinib can inhibit cell proliferation in NSCLC cells positive
for EGFR T790M mutation or whether it requires the addition of
rituximab | RICTOR | mTORC2 | NSCLC with the
T790M mutation is resistant to erlotinib (EGFR-TKI), as EGFR
phosphorylation is prevented; hence, cancer cells resist apoptosis.
However, administration of a CD20 mono-antibody, such as rituximab,
inhibits the expression of rictor, an essential mTORC2 protein, in
MERCSs, thus allowing greater EGFR kinase activity. As a
consequence, EGFR-TKI resistance is reversed, suggesting that
synergistic administration of both drugs can act as a targeted
therapy for T790M-mutated NSCLC. | (57) |
| Verfaillie et
al, 2012 | Undifferentiated
colon carcinoma and breast cancer | Identify the role
of PERK in ROS signalling | EIF2AK3 | PERK | Silencing of
EIF2AK3 results in deformed ER morphology and disrupted
Ca2+ signalling. This absence of PERK increases the
distance between the two organelles, hence shielding Mt from
ROS-mediated events triggering apoptosis. BAX-mediated
mitochondrial apoptosis is also suspended in PERK silenced cells as
cytosolic Ca2+ and other components are delayed. | (40) |
| Shioda et
al, 2012 | Neuroblastoma | Determine whether
σ1SR (isoform of chaperone σ1R) overexpression initiates neuro-2a
cell differentiation and what affect it has on cell survival | Sigma-1 | Sigma-1
receptor | Autophagic response
by σ1SR when under ER stress and also decrease mitochondrial ATP
levels, as it destabilizes IP3R by inhibiting σ1R. | (38) |
| Çoku et al,
2022 | Neuroblastoma | Understanding the
role of MERCSs in therapy resistance | BCL2L11 | Bcl-2-like protein
11 | Analysis of
relapsed neuroblastoma samples after they have undergone treatment,
revealing a mitigated response to stress (via MERCSs) compared with
the same cells analysed at diagnosis prior to treatment. Samples
from relapsed cases are more resistant to stress-induced MOMP, as
supported by their shape and size, while resistance to MOMP is
expressed in tumour cells where ceramide is naturally reduced. | (59) |
| Bononi et
al, 2013 | Human embryonic
kidney cell line [transfected with PTEN (C124S) and PTEN
(G129E)] | Investigate the
effect of PTEN activity on Ca2+ signalling and
apoptosis. | PTEN | Phosphatase and
tensin homolog deleted on chromosome 10 | Cancer cells can
avoid cell death through apoptosis-inducing Ca2+ signals
by driving deletion or mutation of PTEN; hence, the protein cannot
be expressed in MERCSs or ER. PTEN shuts down Akt-mediated
phosphorylation of IP3R3; therefore, the cell is exposed to
Ca2+-mediated apoptosis. | (52) |
| Doghman-Bouguerra
et al, 2016 | Adrenocortical
carcinoma | Investigate, in
cancer cells, the function of FATE1 in regulating
Ca2+-dependent apoptosis but also drug-dependent
apoptosis | FATE1 | Steroidogenic
factor 1 | In adrenocortical
carcinoma, ER-Mt uncoupling occurs due to increased expression of
FATE1, by effect of SF-1, permitting the cells to resist
chemotherapeutic drugs but also escape apoptosis. | (39) |
| Ciscato et
al, 2020 | Chronic lymphocytic
leukaemia | Provide a detailed
presentation of the molecular mechanisms of HK2 that lead to cell
damage and express anti-neoplastic characteristics. All of this
supported by a detailed comprehension of HK2 mechanisms | HK2 | Hexokinase 2 | Silencing of HK2
prevents cells from proliferating and finally induces death in
B-chronic lymphatic leukaemia cells via calpain-dependent
events. | (60) |
| Missiroli et
al, 2016 | Promyelotic
leukaemia | To investigate the
rold of PML in in certain molecular pathways which affect cell
death and proliferation of cancer | Pml | Promyelocytic
leukemia protein | Autophagosome
formation is suppressed when PML is localised within MAM; hence,
autophagy induction does not occur. However, p53 is also involved
in ensuring the placement of PML from MERCSs. ER-mitochondrial
Ca2+ transfer via IP3R is reduced if PML is
displaced. | (34) |
| Mondet
et al, 2021 | Leukaemia | Analyse five
different subtypes of acute myeloid leukaemia to explore the
mitochondrial ultrastructure and its function | ASXL1 | Polycomb group
protein ASXL1 | Molecular
variations are expressed in the different leukemic cells, which
also present with altered mitochondrial functions. HL60 cells
express more ER-mitochondrial contact sites. Cells carrying the
ASXL1 mutation have significantly depleted expression of MAM
genes. | (43) |
| Ciscato et
al, 2020 | Neurofibroma | Define the
mechanism by which HK2 triggers cell death in cancer cell
lines | CAPN1 | Calpain | With pre-prepared
cleaved HK2-peptides, damage can be avoided to secondary targets
and still be tailored to multiple malignancies. HK2-inactivation
delays mitochondrial polarisation followed by cell death mediated
via Ca2+-dependent protease calpains when displaced from
MERCSs. | (60) |
| Ciscato et
al, 2020 | Colorectal
cancer | Evaluate the
outcome of silencing HK2 in solid tumours | HK2 | Hexokinase 2 | Displacing of HK2
further impedes colorectal cancer cell growth in solid tumours,
while injection in allografts of colon cancer significantly
decreases proliferation of cells. | (60) |
| Ciscato et
al, 2020 | Breast cancer | Evaluate the
outcome of silencing HK2 in solid tumours | HK2 | Hexokinase 2 | A cleaved peptide
that displaces HK2 from MERCSs causing an overload of calcium ions
in Mt decreases colony formation and kills the tumour cells. | (60) |
| Seervi et
al, 2013 | Breast cancer | Recognise the
effects PAC-1 has on cancer cells lines | ERO1A | ERO1-like protein
α | Systematic action
of PAC-1 results in G1 arrest and autophagy of cells as
a response to stress. Cytochrome c release and cell death in
MCF7 cells is reduced by PAC-1 when PUMA is silenced | (41) |
| Singh et al,
2015 | Breast cancer | Does NLRX1 regulate
ATP production by Mt. | NRLX1 | NLR family member
X1 | Downregulation of
NLRX1 permits cells to grow in the presence of TNF-α, and ATP is
increased. Expression of NLRX1 accompanied by TNF-α/CHX increases
mitochondrial ROS levels and consequently induces cell death by
activating caspase-8 | (45) |
| Seervi et
al, 2013 | Cervical
cancer | Provide a breakdown
of the mechanisms involved in PAC-1 cell death | ERO1A | ERO1-like protein
α | Silencing of Ero1a
results in decreased Ca2+ release from ER and PAC-1
activates cell death. Therefore, PAC-1 and Ero1a affect ion
movement between ER and Mt via MERCSs | (41) |
| Ciscato et
al, 2020 | Cervical
cancer | Define the
mechanism by which HK2 triggers cell death in cancer cell
lines | CAPN1 | Calpain | With pre-prepared
cleaved HK2-peptides, damage to secondary targets can be avoided
and still be tailored to multiple malignancies. HK2-inactivation
delays mitochondrial polarisation followed by cell death mediated
via Ca2+−dependent protease calpains when displaced from
MERCSs. | (60) |
| Singh et al,
2015 | Cervical
cancer | Determine the role
of NLRX1 expression on migration and growth in response to
physiological stress such as cancer | NLRX1 | NLR family member
X1 | Following treatment
with TNF-α/CHX, cells transfected with NLRX1, which localizes in
the Mt, are sensitized to TNF-α-induced death. Knockdown of NLRX1
results in decreased ROS stimulation and reverses acidification by
tumour cells. | (45) |
|
Madreiter-Sokolowski et al,
2021 | Cervical
cancer | Define the role of
UCP2 in cancer cell viability through its involvement in
MERCSs | UCP2 | Uncoupling Protein
2 | In response to
histamine-induced ER Ca2+, mitochondrial Ca2+
uptake is depleted upon UCP2 knockdown whereas the opposite occurs
with UCP2 overexpression. Administration of tunicamycin to HeLa
cells upregulates proteins known to be involved in MAM
stabilization. | (42) |
| Ahumada-Castro
et al, 2018 | Cervical
cancer | Identify the
mechanism that interrupts mTORC1-dependent autophagy | PRKAA2 and
mTOR | AMP-activated
protein kinase and mammalian target of rapamycin complex 1 | If MAM is disrupted
and communication between the ER and MERCSs is lost, AMPK
approaches the lysosomal membrane where it directly or indirectly
phosphorylates the mTORC1 binding complex consequently inhibiting
mTORC1. | (35) |
| Cui et al,
2022 | Glioblastoma | Analyse the SAR of
56 sesquiterpenes and confirm whether OLB is cytotoxic in
pa-resistant glioblastoma cells | PACS2 | Phosphofurin acidic
cluster sorting protein 2 | MAM and ER-specific
chaperone concentration is increased and ER stress is induced via
OLB. OLB further causes morphological alterations in the MAM
network and induces glioblastoma cell apoptosis. OLB activity is
reduced following PACS2 deletion. | (49) |
Adjusting the MT-ER microenvironment
determining cell fate
Cancer cells require excessive amounts of energy to
grow, proliferate and migrate (53). This energy demand is attained when
adequate Ca2+ uptake in Mt occurs and is processed by
the Krebs cycle and oxidative phosphorylation. The current review
presents evidence emphasizing that the association between Mt and
ER, across MERCSs, influences cancer cell proliferation and
migration, and induces cancer cell death (Table I) activities that are determined by
which proteins are expressed (37).
Further analysis of FATE1 behaviour in additional
cancer cell lines would be appropriate to determine its potential
as a cancer treatment target, given its ability to decrease
caspase-3/7 activity and increase H2O2, both
elements amplified in cellular stress environments (39,40).
The aforementioned evidence supports the fact that regulating
Ca2+ homeostasis in cancer cells aids in the
perseverance of cellular stress and the prevention of autophagy, as
indicated by increasing expression of 4TM-TRPM8 isoforms in
prostate epithelial cells. Therefore, 4TM-TRPM8 channels, partially
localized in MERCSs, are ‘new gatekeepers’ for the regulation of
Ca2+ and any complementary outcomes expressed (44). Alternatively, Mt-ER coupling could
be increased by prescribing tunicamycin in HeLa cells, which
upregulated the concentration of three proteins (Rab32, PEMT1 and
GPR75), responsible for stabilizing MAM, and decreased UCP2, thus
effectively decreasing cancer cell viability (37).
ER stress on the other hand occurs in concentrated
environments of ROS, a characteristic common in tumours. Therefore,
determining the association between mitochondrial apoptosis and
oxidative stress is detrimental. NLR Family Member X1 (NLRX1)
protein, expressed in the Mt, activates Caspase-8, which allows
TNF-α/cycloheximide to reduce ROS assembly, and overall neutralises
the acidity expressed by tumour cells. This regulation marks NLRX1
as a tumor suppressor (45). A
study by Verfaillie et al (2012) (40) illustrated the expression of PERK,
through unfolded protein response mechanisms, in MAM, and described
its essential role in coupling the Mt and ER, therefore regulating
ROS-induced cell death. Additional effects included reduced
caspase-3 activity, prolonged XBP1 accumulation and consequent IRE1
activation, which together with CHOP facilitated apoptosis. These
characteristics were not expressed in wild-type cells. Furthermore,
treating PERK−/− cells with Tg led to ER Ca2+
store depletion, due to the inhibition of SERCA, thus abolishing
the survival of clonogenic cells and stimulating cell death. The
decline in Ca2+ signalling by PERK−/− MEFs
following treatment with Tg can be caused by IP3 (40).
Numerous MAM proteins determine whether the cell
will undergo apoptosis or survive. In the case of BI-1, mostly
localized in the ER with a smaller portion expressed in the Mt,
Ca2+ uptake by the Mt is reduced, affecting the
regulation of cytochrome c and the mitochondrial
permeability transition pore. This consequently protects the cell
against cell death. Analysing the differences between HT1090/BI-1
and HT1080/Neo cells, HT1090/BI-1 showed a lower calcium ion
capacity, allowing these cells to close the permeability transition
pore, preventing ion evacuation and avoiding stress-induced
apoptosis (46). Leakage of
mitochondrial Ca2+ can affect the physiology of the
organelle and hence its activities. Having said that, alternative
channels, such as mitoKCa, can be opened to maintain
homeostasis within the organelle by drawing K+ into the
Mt, increasing water uptake and thus preventing cell destruction.
Additional information is necessary to determine the relationship
of BI-1 with other proteins within MERCSs and to analyse the
mechanisms involved.
Targeting MERCSs
Chemotherapy drugs are designed to target cancer
cells with the sole purpose of inducing death. However, once at its
target site, drug activity may be obstructed by multiple
mechanisms, including ion imbalances, cellular activity (e.g.,
triggering autophagy), gene expression and other factors (e.g.,
drugs) (34,54). The following proteins indicating
autophagy activity in MERCSs can be utilized as diagnostic tools
for PML levels: Microtubule-associated protein 1A/1B-light chain 3,
autophagy-related 14 and syntaxin-17 (34). Administering an autophagy inhibitor
in synergy with 5-FU (Fluorouracil), a chemotherapeutic drug,
reduces solid tumour size in mice transfected with acute
promyelocytic leukemia (34).
Patients with the ASXL1 mutation in AML presented
with downregulation of VAPB and PTPIP51, which collaborate to
assist delivery of Ca2+ via IP3R (55). Two more proteins affected by this
mutation include ITPR1, which is also involved in regulating
Ca2+, while the effects of OSBL5 on cholesterol
expression in the region could explain why leukaemia cells have
previously been described as being sensitive to cytostatic agents
that inhibit cell growth or induce cell death (56). This sensitivity, however, can be
hindered via morphological alterations and the integrity of ER
communication.
In MCF7, MCF7C3, SiHa and HeLa cells, PAC-1 arrests
the cell cycle at the G1 phase, ultimately inducing cell
death. PAC-1 is a caspase-activating compound released in response
to cellular stress and regulating autophagosome activity, while as
aforementioned, Ero1a favors the environment of the cell by
hyper-oxidizing the ER lumen, admitting Ca2+, which is
followed by PAC-1 signaling and ER stress, causing the MAM to
become narrower. Using PAC-1 as the objective treatment in multiple
cell lines (breast, cervical, ovarian and colon cancer cells)
resulted in upregulation of Ero1α, increasing ER calcium release
and cell death (41). ER stress
upregulated PUMA in the MAM region, which in turn activated
pro-apoptotic Bax/Bak proteins. The product of these events was
calcium leakage at the MAM, and ultimately the induction of
autophagy and Mt-mediated apoptosis (46).
A crucial mechanism responsible for regulating
eukaryotic cellular growth is the serine/threonine protein kinase
known as mTOR. One of the complexes formed by mTOR is mTORC1.
Blocking ER-Mt Ca2+ flow recruits AMPK for the
consumption of glucose, as higher ATP levels are required, while
simultaneously inhibiting mTORC1, which induces anabolic pathways,
to cutback energy consumption. Eventually AMPK activates autophagy
via BECN1, a class III pshosphodylinositol 3-kinase member
expressed in the main site where autophagosome fragments assemble
(35). A subsequent mTOR complex is
mTORC2, which phosphorylates Akt and further phosphorylates PACS2,
IP3R and HK2 proteins, any of which can act as a drug target for
regulating calcium flux, cell metabolism, integrity of the space
and overall cell survival (50).
The involvement of IP3R in Ca2+-mediated apoptosis was
also supported by the study by Bononi et al (2013) (52), which presented PTEN as a compound
expressed in the MAM and ER and is capable of neutralising Akt
activation. Inactivity of Akt signifies that IP3R is not
phosphorylated and hence Ca2+ apoptosis occurs due to
unregulated ER-Ca2+ release (43).
Finally, multiple mutations of NSCLC responsible for
affecting behaviour and treatment have been identified over the
years (57). One genetic alteration
that occurs in NSCLC cell lines (H1975 and H1299) is the
substitution of a threonine by methionine at amino acid position
790 (T790M) resulting in cancer cell lines expressing resistance to
tyrosine kinase inhibitors, such as erlotinib. This is an
impediment often encountered in chemotherapy treatments. Possible
solutions involve reversing resistance to anticancer drugs or
selecting a different treatment target that will require the use of
other drugs. Xu et al (2017) (57) chose to evaluate the first option, as
EGFR TKI-resistant cells are known to be directly linked to mTORC2,
and determined that prescribing rituximab, a CD20 monoclonal
antibody, adjusted mTORC2 expression in MERCSs and allowed
resistance to erlotinib to be reversed (54). Supplementary evidence on mTORC2
analysis is essential as to its behavior in cancer cells and role
in treatments (58).
Conclusion
Evaluation of the literature supports the fact that
disruption of protein expression within MERCSs, affecting
Ca2+ influx/efflux, can also impact the physiology of
the Mt and ER, inducing autophagy or the apoptosis of cells. Given
the vast number of proteins expressed in MERCSs, further analysis
is necessary to define protein function, as well as for biomarkers
and their potential use as targets for treatment. Multiple examples
mentioned in the present review show promising results and
insinuate broader exploration is necessary. Finally, the genes
highlighted in the review are all expressed within MERCSs and are
capable of affecting cellular survival, and furthermore, the
clinical attitude of the patients with different cancer types.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
ST and AY were responsible for the study concept.
Data collection and interpretation was performed by ST, PC, TK, AZ
and AY. The manuscript was drafted by ST, CF and AY. All authors
have read and approved the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vance JE: Phospholipid synthesis in a
membrane fraction associated with mitochondria. J Biol Chem.
265:7248–7256. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wieckowski MR, Giorgi C, Lebiedzinska M,
Duszynski J and Pinton P: Isolation of mitochondria-associated
membranes and mitochondria from animal tissues and cells. Nat
Protoc. 4:1582–1590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sala-Vila A, Navarro-Lérida I,
Sánchez-Alvarez M, Bosch M, Calvo C, López JA, Calvo E, Ferguson C,
Giacomello M, Serafini A, et al: Interplay between hepatic
mitochondria-associated membranes, lipid metabolism and caveolin-1
in mice. Sci Rep. 6:273512016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Missiroli S, Danese A, Iannitti T,
Patergnani S, Perrone M, Previati M, Giorgi C and Pinton P:
Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of
the tumor cell fate. Biochim Biophys Acta Mol Cell Res.
1864:858–864. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Area-Gomez E, de Groof A, Bonilla E,
Montesinos J, Tanji K, Boldogh I, Pon L and Schon EA: A key role
for MAM in mediating mitochondrial dysfunction in Alzheimer
disease. Cell Death Dis. 9:3352018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gómez-Suaga P, Pedro JM, González-Polo RA,
Fuentes JM and Niso-Santano M: ER-mitochondria signaling in
Parkinson's disease. Cell Death Dis. 9:3372018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Annunziata I, Sano R and d'Azzo A:
Mitochondria-associated ER membranes (MAMs) and lysosomal storage
diseases. Cell Death Dis. 9:3282018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lau DHW, Hartopp N, Welsh NJ, Mueller B,
Glennon EB, Mórotz GM, Annibali A, Gomez-Suaga P, Stoica R,
Paillusson S and Miller CCJ: Disruption of ER-mitochondria
signalling in fronto-temporal dementia and related amyotrophic
lateral sclerosis. Cell Death Dis. 9:3272018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Missiroli S, Patergnani S, Caroccia N,
Pedriali G, Perrone M, Previati M, Wieckowski MR and Giorgi C:
Mitochondria-associated membranes (MAMs) and inflammation. Cell
Death Dis. 9:3292018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sassano ML, van Vliet AR and Agostinis P:
Mitochondria-associated membranes as networking platforms and
regulators of cancer cell fate. Front Oncol. 7:1742017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marchi S and Pinton P: Alterations of
calcium homeostasis in cancer cells. Curr Opin Pharmacol. 29:1–6.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang N, Wang C, Zhao H, He Y, Lan B, Sun L
and Gao Y: The MAMs structure and its role in cell death. Cells.
10:6572021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Csordás G, Renken C, Várnai P, Walter L,
Weaver D, Buttle KF, Balla T, Mannella CA and Hajnóczky G:
Structural and functional features and significance of the physical
linkage between ER and mitochondria. J Cell Biol. 174:915–921.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu H, Sun C, Gong Q and Feng D:
Mitochondria-associated endoplasmic reticulum membranes in breast
cancer. Front Cell Dev Biol. 9:6296692021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szabadkai G and Duchen MR: Mitochondria:
The hub of cellular Ca2+ signaling. Physiology (Bethesda).
23:84–94. 2008.PubMed/NCBI
|
|
17
|
Avalle L, Camporeale A, Morciano G,
Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P
and Poli V: STAT3 localizes to the ER, acting as a gatekeeper for
ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death
Differ. 26:932–942. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rimessi A, Pedriali G, Vezzani B, Tarocco
A, Marchi S, Wieckowski MR, Giorgi C and Pinton P: Interorganellar
calcium signaling in the regulation of cell metabolism: A cancer
perspective. Semin Cell Dev Biol. 98:167–180. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Danese A, Patergnani S, Bonora M,
Wieckowski MR, Previati M, Giorgi C and Pinton P: Calcium regulates
cell death in cancer: Roles of the mitochondria and
mitochondria-associated membranes (MAMs). Biochim Biophys Acta.
1858:615–627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Patergnani S, Missiroli S, Marchi S and
Giorgi C: Mitochondria-associated endoplasmic reticulum membranes
microenvironment: Targeting autophagic and apoptotic pathways in
cancer therapy. Front Oncol. 5:1732015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Giorgi C, Bonora M, Sorrentino G,
Missiroli S, Poletti F, Suski JM, Ramirez FG, Rizzuto R, Di
Virgilio F, Zito E, et al: p53 at the endoplasmic reticulum
regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci
U S A. 112:1779–1784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pinton P: Mitochondria-associated
membranes (MAMs) and pathologies. Cell Death Dis. 9:4132018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacquemyn J, Cascalho A and Goodchild RE:
The ins and outs of endoplasmic reticulum-controlled lipid
biosynthesis. EMBO Rep. 18:1905–1921. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Watson H: Biological membranes. Essays
Biochem. 59:43–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gibellini F and Smith TK: The Kennedy
pathway-De novo synthesis of phosphatidylethanolamine and
phosphatidylcholine. IUBMB Life. 62:414–428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sano R, Annunziata I, Patterson A,
Moshiach S, Gomero E, Opferman J, Forte M and d'Azzo A:
GM1-ganglioside accumulation at the mitochondria-associated ER
membranes links ER stress to Ca(2+)-dependent mitochondrial
apoptosis. Mol Cell. 36:500–511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li S and Rousseau D: ATAD3, a vital
membrane bound mitochondrial ATPase involved in tumor progression.
J Bioenerg Biomembr. 44:189–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rieusset J: The role of endoplasmic
reticulum-mitochondria contact sites in the control of glucose
homeostasis: An update. Cell Death Dis. 9:3882018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nikolova-Karakashian M, Karakashian A and
Rutkute K: Role of neutral sphingomyelinases in aging and
inflammation. Subcell Biochem. 49:469–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dang D and Rao R: Calcium-ATPases: Gene
disorders and dysregulation in cancer. Biochim Biophys Acta.
1863:1344–1350. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Patergnani S, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S,
Poletti F, et al: Calcium signaling around mitochondria associated
membranes (MAMs). Cell Commun Signal. 9:192011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Glancy B and Balaban RS: Role of
mitochondrial Ca2+ in the regulation of cellular energetics.
Biochemistry. 51:2959–2973. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giorgi C, Romagnoli A, Pinton P and
Rizzuto R: Ca2+ signaling, mitochondria and cell death. Curr Mol
Med. 8:119–130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Missiroli S, Bonora M, Patergnani S,
Poletti F, Perrone M, Gafà R, Magri E, Raimondi A, Lanza G,
Tacchetti C, et al: PML at mitochondria-associated membranes is
critical for the repression of autophagy and cancer development.
Cell Rep. 16:2415–2427. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahumada-Castro U, Silva-Pavez E, Lovy A,
Pardo E, Molgό J and Cárdenas C: MTOR-independent autophagy induced
by interrupted endoplasmic reticulum-mitochondrial Ca2+
communication: A dead end in cancer cells. Autophagy. 15:358–361.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manganelli V, Matarrese P, Antonioli M,
Gambardella L, Vescovo T, Gretzmeier C, Longo A, Capozzi A,
Recalchi S, Riitano G, et al: Raft-like lipid microdomains drive
autophagy initiation via AMBRA1-ERLIN1 molecular association within
MAMs. Autophagy. 17:2528–2548. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mori T, Hayashi T, Hayashi E and Su TP:
Sigma-1 receptor chaperone at the ER-mitochondrion interface
mediates the mitochondrion-ER-nucleus signaling for cellular
survival. PLoS One. 8:e769412013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shioda N, Ishikawa K, Tagashira H,
Ishizuka T, Yawo H and Fukunaga K: Expression of a truncated form
of the endoplasmic reticulum chaperone protein, σ1 receptor,
promotes mitochondrial energy depletion and apoptosis. J Biol Chem.
287:23318–23331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Doghman-Bouguerra M, Granatiero V, Sbiera
S, Sbiera I, Lacas-Gervais S, Brau F, Fassnacht M, Rizzuto R and
Lalli E: FATE1 antagonizes calcium- and drug-induced apoptosis by
uncoupling ER and mitochondria. EMBO Rep. 17:1264–1280. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Verfaillie T, Rubio N, Garg AD, Bultynck
G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A
and Agostinis P: PERK is required at the ER-mitochondrial contact
sites to convey apoptosis after ROS-based ER stress. Cell Death
Differ. 19:1880–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Seervi M, Sobhan PK, Joseph J, Mathew KA
and Santhoshkumar TR: ERO1α-dependent endoplasmic
reticulum-mitochondrial calcium flux contributes to ER stress and
mitochondrial permeabilization by procaspase-activating compound-1
(PAC-1). Cell Death Dis. 4:e9682013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Madreiter-Sokolowski CT, Gottschalk B,
Sokolowski AA, Malli R and Graier WF: Dynamic control of
mitochondrial Ca2+ levels as a survival strategy of cancer cells.
Front Cell Dev Biol. 9:6146682021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mondet J, Lo Presti C, Chevalier S,
Bertrand A, Tondeur S, Blanchet S, Leer AM, Pernet-Gallay K and
Mossuz P: Mitochondria in human acute myeloid leukemia cell lines
have ultrastructural alterations linked to deregulation of their
respiratory profiles. Exp Hematol. 98:53–62.e3. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bidaux G, Gordienko D, Shapovalov G,
Farfariello V, Borowiec AS, Iamshanova O, Lemonnier L, Gueguinou M,
Guibon R, Fromont G, et al: 4TM-TRPM8 channels are new gatekeepers
of the ER-mitochondria Ca2+ transfer. Biochim Biophys Acta Mol Cell
Res. 1865:981–994. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Singh K, Poteryakhina A, Zheltukhin A,
Bhatelia K, Prajapati P, Sripada L, Tomar D and Singh R, Singh AK,
Chumakov PM and Singh R: NLRX1 acts as tumor suppressor by
regulating TNF-α induced apoptosis and metabolism in cancer cells.
Biochim Biophys Acta. 1853:1073–1086. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee GH, Lee HY, Li B, Kim HR and Chae HJ:
Bax inhibitor-1-mediated inhibition of mitochondrial Ca2+ intake
regulates mitochondrial permeability transition pore opening and
cell death. Sci Rep. 4:51942014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Elfawy HA and Das B: Crosstalk between
mitochondrial dysfunction, oxidative stress, and age related
neurodegenerative disease: Etiologies and therapeutic strategies.
Life Sci. 218:165–184. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang C, Dai X, Wu S, Xu W, Song P and
Huang K: FUNDC1-dependent mitochondria-associated endoplasmic
reticulum membranes are involved in angiogenesis and
neoangiogenesis. Nat Commun. 12:26162021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cui P, Chen F, Ma G, Liu W, Chen L, Wang
S, Li W, Li Z and Huang G: Oxyphyllanene B overcomes temozolomide
resistance in glioblastoma: Structure-activity relationship and
mitochondria-associated ER membrane dysfunction. Phytomedicine.
94:1538162022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Betz C, Stracka D, Prescianotto-Baschong
C, Frieden M, Demaurex N and Hall MN: mTOR complex 2-Akt signaling
at mitochondria-associated endoplasmic reticulum membranes (MAM)
regulates mitochondrial physiology. PNAS. 110:12526–12534. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fei SJ, Zhang XC, Dong S, Cheng H, Zhang
YF, Huang L, Zhou HY, Xie Z, Chen ZH and Wu YL: Targeting mTOR to
overcome epidermal growth factor receptor tyrosine kinase inhibitor
resistance in non-small cell lung cancer cells. PLoS One.
8:e691042013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bononi A, Bonora M, Marchi S, Missiroli S,
Poletti F, Giorgi C, Pandolfi PP and Pinton P: Identification of
PTEN at the ER and MAMs and its regulation of Ca2+ signaling and
apoptosis in a protein phosphatase-dependent manner. Cell Death
Differ. 20:1631–1643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Han T, Kang D, Ji D, Wang X, Zhan W, Fu M,
Xin HB and Wang JB: How does cancer cell metabolism affect tumor
migration and invasion? Cell Adh Migr. 7:395–403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Marchi S, Patergnani S, Missiroli S,
Morciano G, Rimessi A, Wieckowski MR, Giorgi C and Pinton P:
Mitochondrial and endoplasmic reticulum calcium homeostasis and
cell death. Cell Calcium. 69:62–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gómez-Suaga P, Pérez-Nievas BG, Glennon
EB, Lau DHW, Paillusson S, Mórotz GM, Calì T, Pizzo P, Noble W and
Miller CCJ: The VAPB-PTPIP51 endoplasmic reticulum-mitochondria
tethering proteins are present in neuronal synapses and regulate
synaptic activity. Acta Neuropathol Commun. 7:352019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Koczian F, Nagło O, Vomacka J, Vick B,
Servatius P, Zisis T, Hettich B, Kazmaier U, Sieber SA, Jeremias I,
et al: Targeting the endoplasmic reticulum-mitochondria interface
sensitizes leukemia cells to cytostatics. Haematologica.
104:546–555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xu ZH, Liu CH, Hang JB, Gao BL and Hu JA:
Rituximab effectively reverses Tyrosine kinase inhibitors (TKIs)
resistance through inhibiting the accumulation of rictor on
mitochondria-associated ER-membrane (MAM). Cancer Biomarkers.
20:581–588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chiang CT, Demetriou AN, Ung N, Choudhury
N, Ghaffarian K, Ruderman DL and Mumenthaler SM: mTORC2 contributes
to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor
resistant cells in non-small cell lung cancer. Cancer Lett.
434:152–159. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Çoku J, Booth DM, Skoda J, Pedrotty MC,
Vogel J, Liu K, Vu A, Carpenter EL, Ye JC, Chen MA, et al: Reduced
ER-mitochondria connectivity promotes neuroblastoma multidrug
resistance. EMBO J. 41:e1082722022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ciscato F, Filadi R, Masgras I, Pizzi M,
Marin O, Damiano N, Pizzo P, Gori A, Frezzato F, Chiara F, et al:
Hexokinase 2 displacement from mitochondria-associated membranes
prompts Ca2+ -dependent death of cancer cells. EMBO Rep.
21:e491172020. View Article : Google Scholar : PubMed/NCBI
|