Introduction
Adrenocortical carcinoma (ACC) is a rare malignancy
with an incidence of 1–2 per million individuals per year (1). ACC is associated with poor outcomes
due to overproduction of hormones (2). Hypercortisolism is often observed in
patients with ACC. High cortisol levels lead to plethora, diabetes
mellitus, sarcopenia, and osteoporosis. Additionally, excessive
cortisol induces glucocorticoid-mediated mineralocorticoid receptor
activation. Thus, hypokalemia and hypertension are also observed in
ACC. High adrenal androgen levels are also observed in ACC, leading
to rapid-onset male pattern baldness, virilization and menstrual
irregularities in women (3).
Excessive hormones are often recognized late in clinical settings,
which impedes diagnosis and treatment.
Due to the complexity and poor prognosis of ACC,
surgery, radiotherapy and chemotherapies, including etoposide,
doxorubicin, cisplatin and mitotane, are mainly used to treat ACC
(4). Despite a number of efforts to
develop novel therapeutic strategies, it appears that using current
agents is not sufficient to increase the therapeutic responses.
Trials developing novel targeted molecules may be promising
(5).
BI2536 is a polo like kinase 1 (PLK1)-selective
inhibitor. At low concentrations (within the nanomolar range), it
binds to the ATP-binding domain of PLK1 to inhibit the kinase
activity (6). BI2536 treatment
induces apoptosis by activating caspase-3 or caspase-8 in human
cancer cell lines of diverse tissue origins (7,8).
Notably, the antitumor effects of BI2536 have further been
demonstrated using human tumor xenografts in nude mouse models.
Thus, BI2536 has been studied in clinical studies in patients with
locally advanced or metastatic cancer.
The centrosome is the major microtubule organizing
center of mammalian cells. By recruiting γ-tubulin ring complexes
to the pericentriolar materials of the centrosome, the centrosome
nucleates microtubules from the minus-end to the plus-end to
orchestrate the microtubule arrays (9). In addition to organizing the
microtubules, the centrosome also regulates cell cycle progression
during interphase and orchestrates mitotic spindle formation at M
phase (10). Thus, deficiencies in
centrosome structure or function may impede cell cycle progression
or even trigger apoptotic cell death (11). Normally, each cell contains one
centrosome. Coordinated with DNA replication, the centrosome starts
to duplicate at S phase through CDK2 activation. At G2/M
transition, the duplicated centrosomes become mitotic spindle poles
and separate to the opposite of the nucleus to align and segregate
the replicated chromosomes into each daughter cell. Thus, precise
control of centrosome homeostasis is important to maintain genome
integrity.
BI2536 has been tested for the treatment of several
cancer types, including oral, lung and gastric cancer (12–14);
however, to the best of our knowledge, its effects on
adrenocortical tumors have not been tested yet. The present study
revealed that BI2536 inhibited Y1 adrenocortical cell proliferation
by blocking cell cycle progression and inducing apoptosis.
Mechanistically, BI2536 induced centrosome amplification and
aberrant mitotic spindle poles by activating the ATM
serine/threonine kinase (ATM)-ERK cascade. Therefore, the present
study demonstrated that BI2536 could potentially be used for the
treatment of adrenocortical tumors, and the underlying molecular
mechanism was also revealed.
Materials and methods
Cell culture
The Y1 mouse adrenocortical tumor, H295R human
adrenocortical tumor and NIH3T3 mouse embryonic fibroblast cell
lines were cultured in DMEM-F12 full medium containing 10% FBS in a
humidified atmosphere with 5% CO2 at 37°C. Y1, H295R
cell lines and NIH3T3 were kindly provided by Dr Bon-chu Chung
(Academia Sinica, Taipei, Taiwan). Mycoplasma contamination was
regularly checked by DAPI staining.
The following drugs were used in the present
experiments: Vanillin [DNA-dependent protein kinase (DNA-PK)
inhibitor; cat. no. V110-4; 1 mM, (15)], Ku55933 [competitive ATM kinase
inhibitor; cat. no. SML1109; 10 µM, (16)] and U0126 [ERK inhibitor; cat. no.
U120; 20 µM, (17)], which were
purchased from MilliporeSigma. BI2536 (PKL1 inhibitor; cat. no.
S1109; 500 nM) was purchased from Selleck Chemicals. For the drug
treatments, the indicated time points are shown in the figure
legends.
Cell cycle analysis
The cell cycle profiles (106 cells) were
analyzed by FACS according to a previously described methodology
(18). After treating Y1 cells with
BI2536 at 500 nM for 24 h, cells were suspended in PBS containing 1
mM EDTA (PBS-E) buffer. Following centrifugation (1,000 × g for 5
min, 4°C), the supernatant was removed, and the pellets were
suspended with PBS-E buffer. Subsequently, the suspension was
further centrifuged at 1,000 × g for 5 min at 4°C, followed by
removal of the supernatant. The centrifuged cells were suspended
with 0.5 ml PBS-E buffer, followed by fixation of cells with 70%
ethanol at 4°C overnight. Before cell cycle analysis, ethanol was
removed by centrifugation, and cells were washed with 10 ml PBS-E
at least three times. Subsequently, cells were stained with
propidium iodide (SouthernBiotech) for 1 h at room temperature. The
cell cycle profiles were determined and analyzed using a FACScan
flow cytometer (BD Biosciences) and Kaluza software Version 1.5
(Beckman Coulter, Inc.).
5-ethynyl-2′-deoxyuridine (EdU)
incorporation assay
The EdU incorporation assay was performed according
to the manufacturer's instructions (Invitrogen; Thermo Fisher
Scientific, Inc.) and modified according to the previously
described methodology (19). In
brief, the drug-treated cells (5×105) were incubated
with EdU for at least 8 h at 37°C. Subsequently, the cells were
washed with PBS at least three times and fixed with fixation buffer
provided in the commercial kit. To count the EdU-positive cells,
the fixed cells were stained with DAPI (0.5 µg/ml) in PBS at 37°C
for 10 min, and then observed using a fluorescence microscope.
Microtubule nucleation assay
To examine microtubule nucleation, cells were
treated with 25 µM nocodazole in the culture medium to depolymerize
the microtubules. At 1 h after microtubule depolymerization, the
depolymerized cells were washed with PBS three times and then
cultured in drug-free medium for the indicated time points. To
observe the microtubules, the cells were fixed with ice-cold
methanol, followed by staining with antibody against α-tubulin for
further observation.
Comet assay
The comet assay was performed according to the
manufacturer's instructions (cat. no. ab238544; Abcam). Briefly,
the comet agarose (1%) was loaded onto the comet slide to form a
base layer. Then, the cells were mixed with comet agarose and the
mixture was loaded onto the top of the base layer. The cells were
treated with the commercial lysis buffer (2.5 M NaCl, 0.1M EDTA and
10 mM Tris-HCl) and alkaline solution (1% NaOH and 1% 0.5M EDTA),
followed by performing the electrophoresis under alkaline
conditions. The comet tail was observed by staining with DAPI (0.5
µg/ml) at 37°C.
Immunofluorescence microscopy
Y1 cells (105) were plated on coverslips
overnight, followed by treatment of cells with drugs. Subsequently,
the cells were washed with PBS three times and fixed with −20°C
methanol for 6 min. These cells were blocked with 1% normal goat
serum for 1 h at 37°C, followed by incubation with primary
antibodies [anti-α-tubulin and anti-γ-Tubulin, GTU-88 (both at
1:700 dilution in 1% normal goat serum); and anti-γ-H2AX
(phosphorylation at S139, EP854(2)Y; 1:700 dilution in 1% normal
goat serum; cat. no. ab81299; Abcam)] at 4°C overnight.
Subsequently, the cells were washed with PBS with Triton X-100
(PBS-T) three times, followed by incubation with FITC (1:700; cat.
no. F-2761)- or Cy3 (1:700; cat. no. A10521)-conjugated secondary
antibodies (both from Invitrogen; Thermo Fisher Scientific, Inc.)
at 4°C for 1 h. Meanwhile, the nuclei were also co-stained with
DAPI (0.5 µg/ml) at 37°C for 1 h. Next, the cells were washed again
with PBS-T three times and then mounted with 50% glycerol/PBS-T on
microscope slides. The fluorescence signals were examined using an
AxioImager M2 fluorescence microscope (Zeiss AG).
Western blotting
Y1, H295R and NIH3T3 cell extracts were collected by
lysing cells with a lysis buffer containing 0.5% NP-40, 300 mM
NaCl, 1 mM EDTA and a protease inhibitor cocktail (Roche
Diagnostics GmbH). After centrifugation at 15,000 × g at 4°C, the
supernatants were collected and quantified using the Bradford
protein assay kit according to the instruction. Subsequently, equal
amounts of extract (50 µg) were loaded per lane. 12% SDS-PAGE gels
were used to separate the samples. After separation, samples were
transferred to the polyvinylidene difluoride (PVDF) membranes.
Membranes were blocked with 5% skim milk at room temperature for 1
h then incubated with primary antibodies (see below) at 4°C for 12
h. Subsequently, the antibodies were washed out with TBST (TBS with
01% Tween-20) for three times and membranes were incubated with
HRP-conjugated secondary antibodies at room temperature for 1 h.
Finally, after washed by TBST for three times, the protein signal
on membrane were detected by enhanced chemiluminescent (ECL) kit
(cat. no. A38556; Thermo Fisher Scientific, Inc.).
For western blotting, the primary antibodies
(1:2,000 dilution) were incubated at 4°C for 12 h and obtained
commercially: Anti-Ku70 (N3H10; cat. no. GTX23114), anti-ATM (2C1;
cat. no. GTX7010), anti-ATR serine/threonine kinase (ATR; cat. no.
GTX128146), anti-β-actin (AC-15; cat. no. GTX26276), anti-catalytic
subunit of DNA-PK (phosphor-Thr2609; cat. no. GTX24194), anti-Akt
(phosphor-Ser473; cat. no. GTX128414) and anti-p53 (DO1; cat. no.
GTX70214) were purchased from GeneTex, Inc. Anti-acetylated-tubulin
(cat. no. T6793) and anti-α-tubulin (cat. no. T6557) were purchased
from MilliporeSigma. Anti-ATM (phosphor-Ser1981; cat. no. ab81292)
was purchased from Abcam. Anti-ATR (phosphor-Ser428; cat. no.
2853), anti-checkpoint kinase 2 (Chk2; phosphor-Thr68; cat. no.
2661), anti-Chk2 (cat. no. 3440), anti-Akt (phosphor-Ser473; cat.
no. 4060), anti-Akt (cat. no. 4691), anti-p44/42 MAPK (Erk1/2;
phosphor-Thr202/Tyr204; cat. no. 4370), anti-p44/42 MAPK (Erk1/2;
cat. no. 4695), anti-p53 (phosphor-Ser15; cat. no. 9284),
anti-checkpoint kinase 1 (Chk1; phosphor-Ser317; cat. no. 12302)
and anti-Chk1 (cat. no. 2360) were purchased from Cell Signaling
Technology, Inc. Anti-p150glued (cat. no. 610473) was
purchased from BD Biosciences. Goat anti-rabbit (1:7,000; cat. no.
31460) or anti-mouse (1:7,000; cat. no. 31430) IgG (H+L),
HRP-conjugated secondary antibodies (Invitrogen; Thermo Fisher
Scientific, Inc.) were incubated at 37°C for 1 h.
Mitotic index
The proportions of mitotic cells were identified and
quantified according to a previously described protocol (9). In brief, according to the chromosome
alignments, the mitotic cells at different phases of M phase were
quantified. In each independent experiment, >1,000 cells were
counted using an AxioImager M2 fluorescence microscope (Carl Zeiss
AG).
Statistical analysis
The mean ± SD of three independent experiments is
presented for all data. In each individual group, ≥100 cells were
counted. Differences between two individual experiments were
compared using two-tailed Student's t-tests (unpaired) or one-way
ANOVA. Tukey's multiple comparison test was performed as a post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
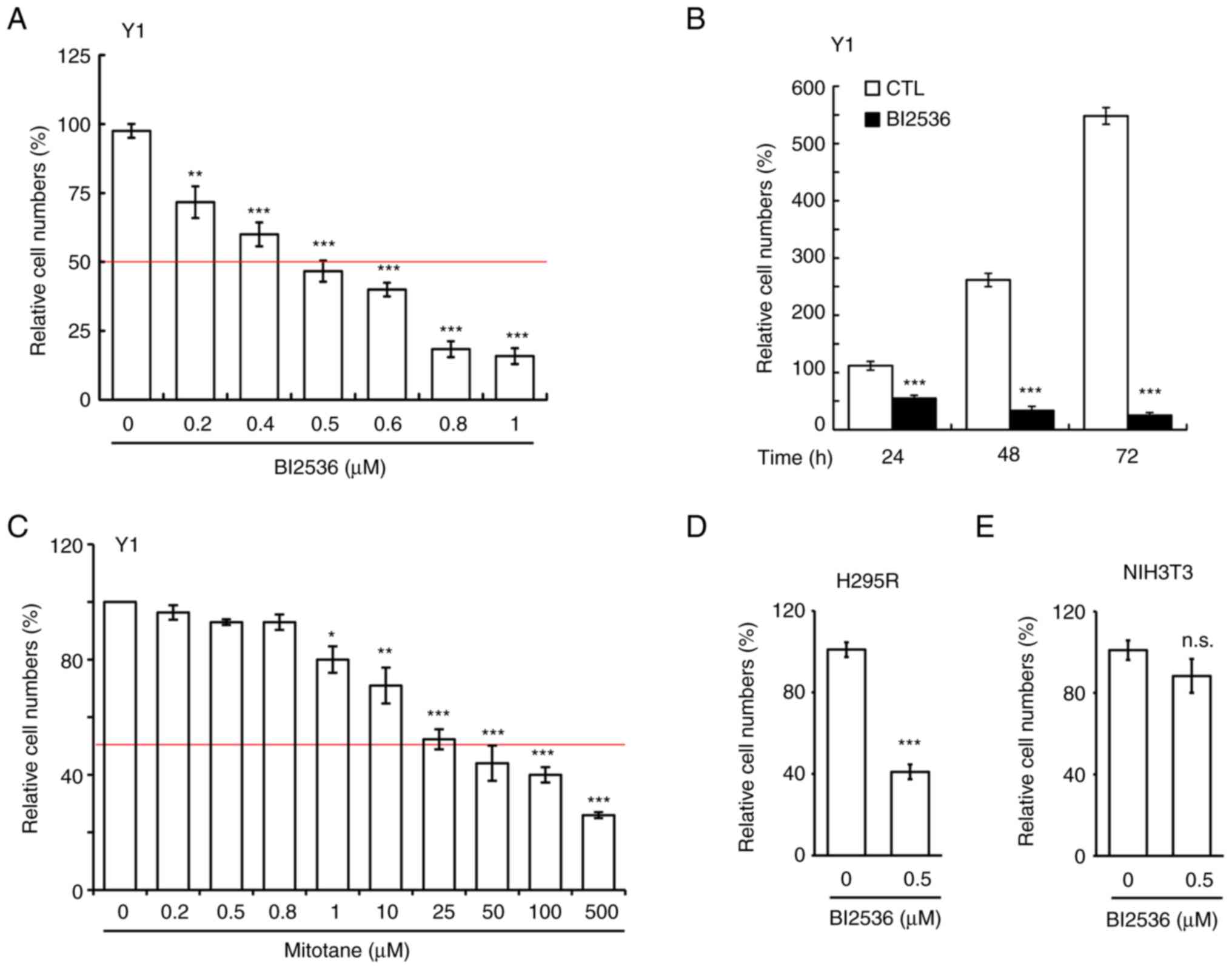
BI2536 inhibits ACC cell growth
BI2536 has been used to treat several cancer types;
however, to the best of our knowledge, its effect on adrenocortical
tumors has not yet been examined. In the present study, it was for
the first time analyzed whether BI2536 inhibited the proliferation
of Y1 mouse adrenocortical cancer cells. BI2536 inhibited Y1 cell
proliferation in a dose-dependent manner (Fig. 1A), and the IC50 of BI2536
in Y1 cells was ~500 nM, thus this concentration was used in the
following experiments. Subsequently, Y1 cells were treated with 500
nM BI2536 for 24, 48 and 72 h, and it was revealed that BI2536
inhibited Y1 cell proliferation in a time-dependent manner
(Fig. 1B). Since mitotane is used
to treat adrenocortical tumors in clinical practice, the present
study examined the IC50 of mitotane on Y1 cell
proliferation. After treating Y1 cells with mitotane at different
concentrations for 24 h, it was demonstrated that the
IC50 of mitotane on Y1 cell proliferation was ~25 µM
(Fig. 1C), which was 50-fold higher
than that of BI2536. The data suggested that BI2536 could be a good
candidate small compound for the treatment of adrenocortical
tumors. The present study also examined the effects of BI2536 on
the H295R human adrenocortical cancer cell line. At a concentration
of 500 nM, BI2536 inhibited H295R cell proliferation (Fig. 1D). The cytotoxic effect of BI2536 on
normal cells was also examined. Treatment of NIH3T3 cells (mouse
embryonic fibroblast cells) with 500 nM BI2536 had no effect on
NIH3T3 cell proliferation (Fig.
1E). Thus, BI2536 reduced adrenocortical tumor cell
proliferation.
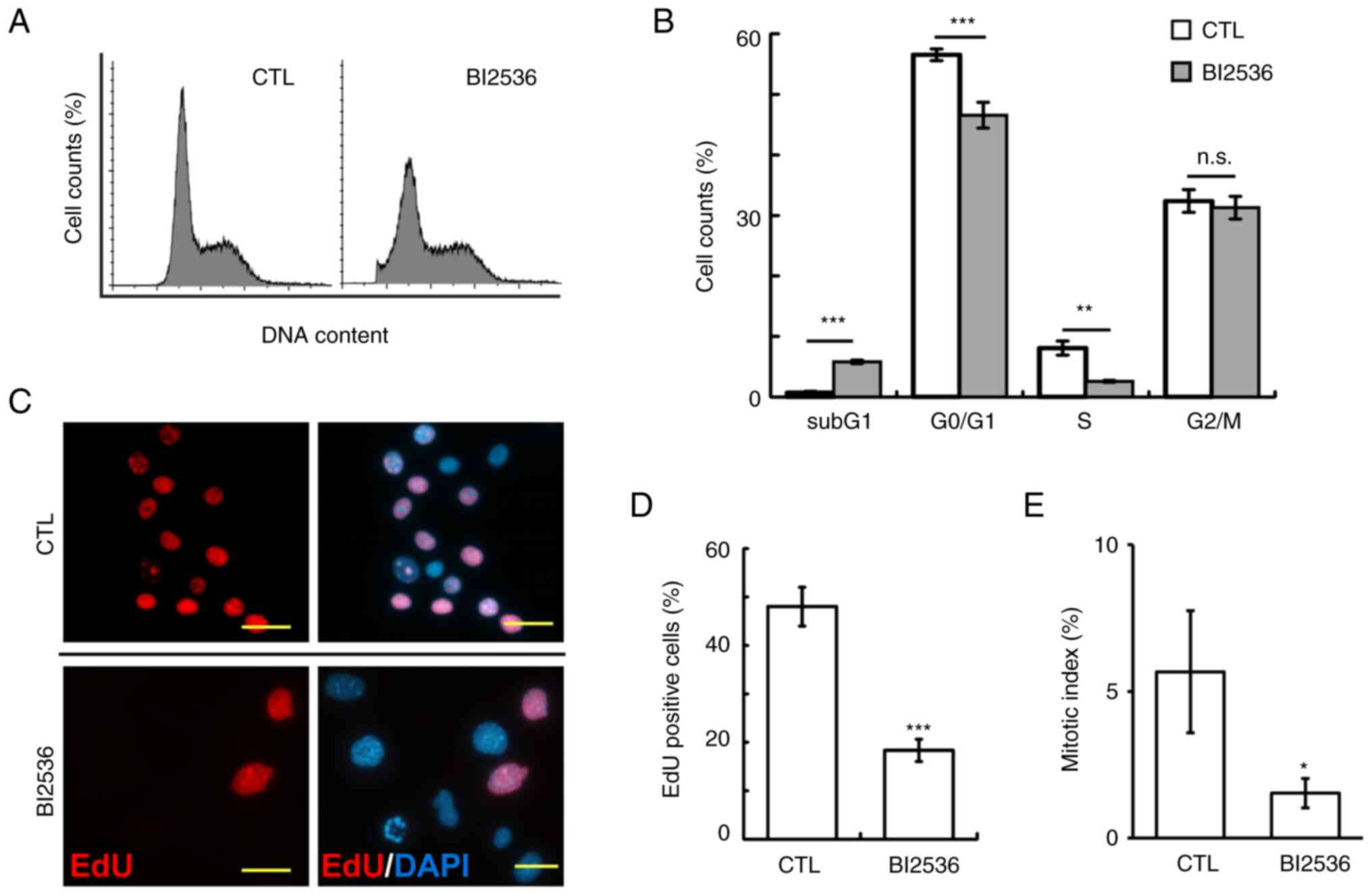
The cell cycle profiles were subsequently analyzed
by flow cytometry. Following 500 nM BI2536 treatment for 24 h, the
proportions of Y1 cells at sub-G1 phase were
significantly increased, while those at G0/G1
and S phase were reduced (Fig. 2A and
B). Subsequently, the ability of cells to enter into S phases
was examined using an EdU incorporation assay. The proportions of
EdU-positive cells were significantly reduced in BI2536-treated
cells (Fig. 2C and D). In addition,
the mitotic index in BI2536-treated Y1 cells was reduced,
suggesting that BI2536 also inhibited entry of cells into M phase
(Fig. 2E). Thus, BI2536 induced
apoptosis and impeded cell cycle progression.
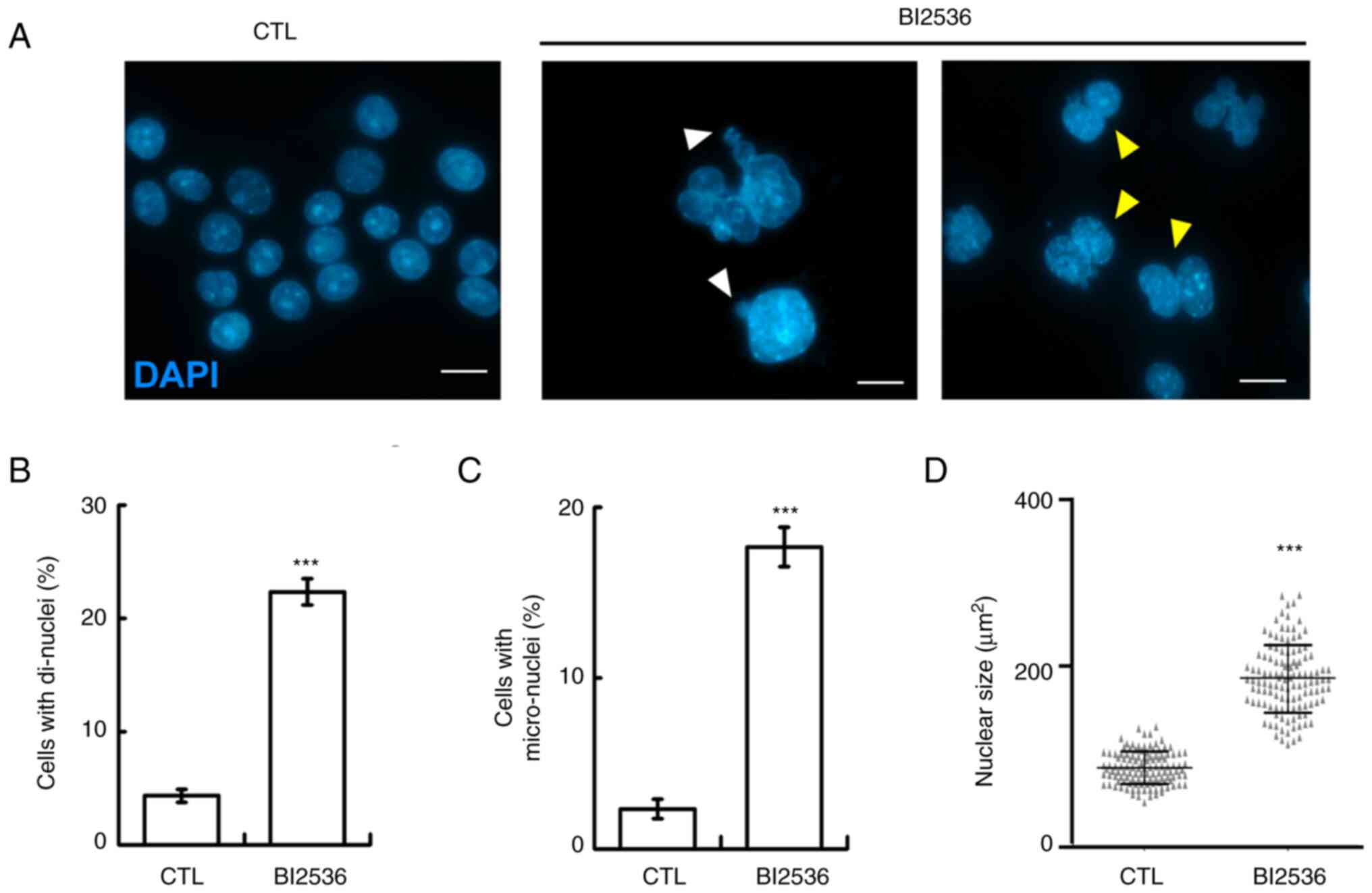
When examining nuclear staining, aberrant nuclear
shapes, including mostly di-nuclei, micro-nuclei and enlarged
nuclei, were observed in BI2536-treated cells, suggesting that
genomic instability occurred (Fig.
3A-D). Genomic instability may be caused by aberrant mitosis,
thus the mitotic spindle poles in the absence or presence of BI2536
were examined. Normally, two mitotic spindle poles (as shown by
γ-tubulin staining) located at the opposite sites of the cell to
align the duplicated chromosomes at the equatorial plate (Fig. 4A, left panel). However, when the
cells were treated with BI2536, aberrant mitotic spindle poles were
observed (Fig. 4A, right panel, and
B), and the chromosomes were not aligned well during mitosis. Thus,
BI2536 induced aberrant mitosis.
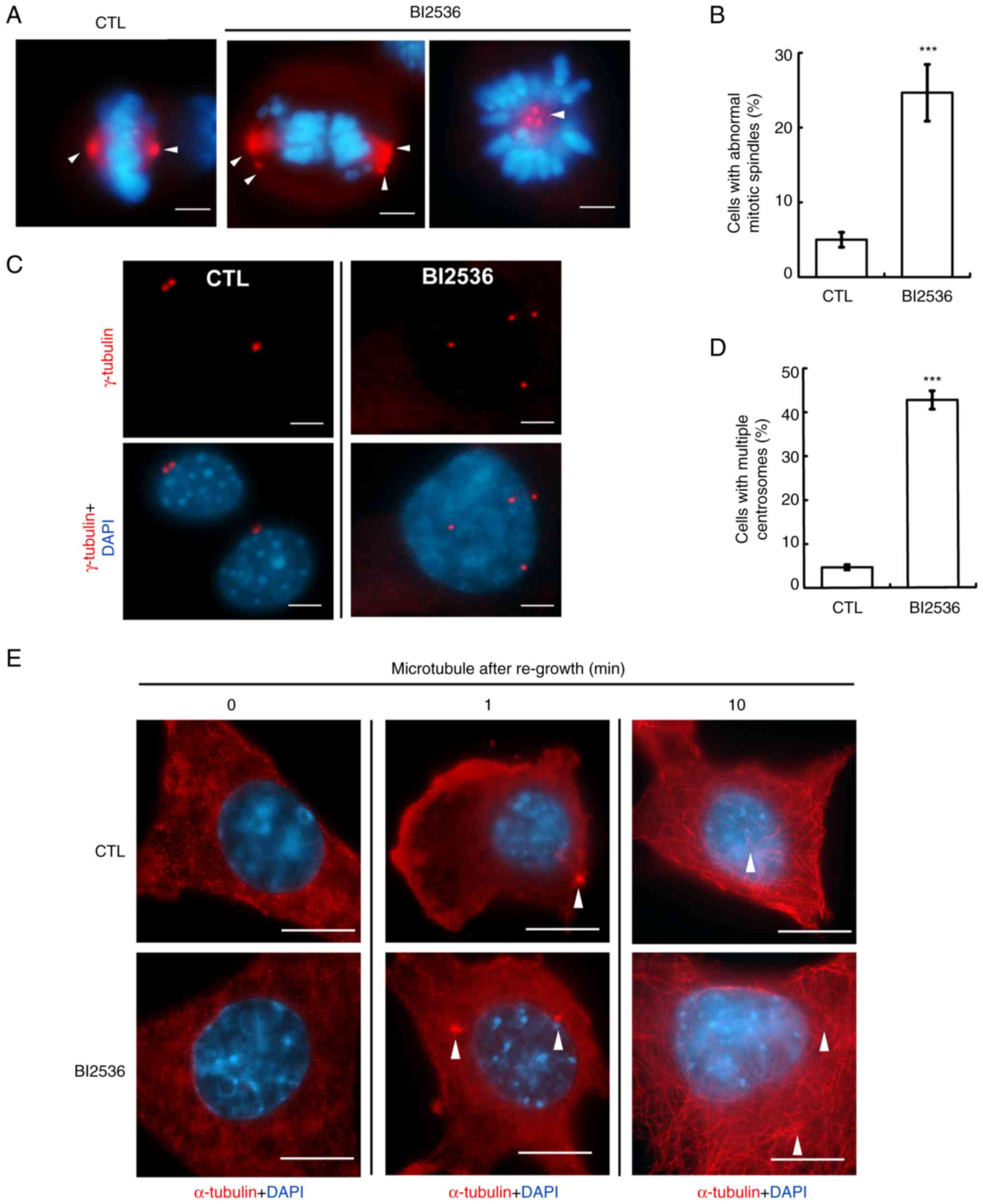
BI2536 induces centrosome
amplification
Mitotic spindle poles were caused by centrosome
amplification (cells with ≥3 centrosomes) (20). The centrosome copy numbers during
interphase were subsequently examined. Normally, cells contained
one (before duplication) or two (after duplication) centrosomes;
however, following BI2536 treatment, centrosome amplification was
observed in both Y1 and H295R cells (Figs. 4C and D, and S1A), suggesting that BI2536 disrupted
centrosome homeostasis. The centrosome is the microtubule
organizing center, and the present study subsequently investigated
whether these amplified centrosomes were functional. The
microtubules were depolymerized by treating cells with nocodazole
for 1 h, followed by washing out the drugs to regrow the
microtubules. After depolymerization, the microtubules were not
observed in the control cells. However, 1 min after drug washout,
the microtubules started to nucleate, and after 10 min, microtubule
arrays were established in the cytoplasm (Fig. 4E, upper panel). The ability of the
microtubules to regrow in BI2536-treated cells was similar to that
in the control cells (Fig. 4E,
lower panel). Thus, BI2536 induced centrosome amplification without
affecting microtubule nucleation.
BI2536 induces centrosome
amplification by activating ATM
DNA damage induces centrosome amplification
(15), thus the present study
investigated whether BI2536 facilitated centrosome amplification
through DNA damage. First, it was examined whether BI2536 induced
DNA damage. The DNA damage marker γ-H2AX was examined using
immunofluorescence staining. Following BI2536 treatment, the
proportion of γ-H2AX-positive cells was markedly increased
(Fig. 5A), suggesting that BI2536
induced DNA damage. To further confirm the present finding, a comet
assay was performed. In the presence of DNA damage, a comet tail
was formed (Fig. 5B). Following
BI2536 treatment, the proportion of comet tail-positive samples was
increased, indicating that BI2536 induced DNA damage (Fig. 5C). Next, it was examined whether the
DNA damage response induced centrosome amplification. The
phosphoinositide 3 kinase-related protein kinases, including ATM,
ATR and DNA-PK, were examined. ATM and DNA-PK, but not ATR, were
activated by BI2536 treatment (Figs. 5D
and E, and S1B). Subsequently,
the effects of these two kinases on centrosome amplification were
examined. Ku55933 inhibited the activation of ATM (Fig. 5F) and centrosome amplification
(Fig. 5G). However, inhibition of
DNA-PK by treating cells with vanillin had no effect on centrosome
amplification (Fig. 5H and I).
Thus, BI2536 induced centrosome amplification by activating
ATM.
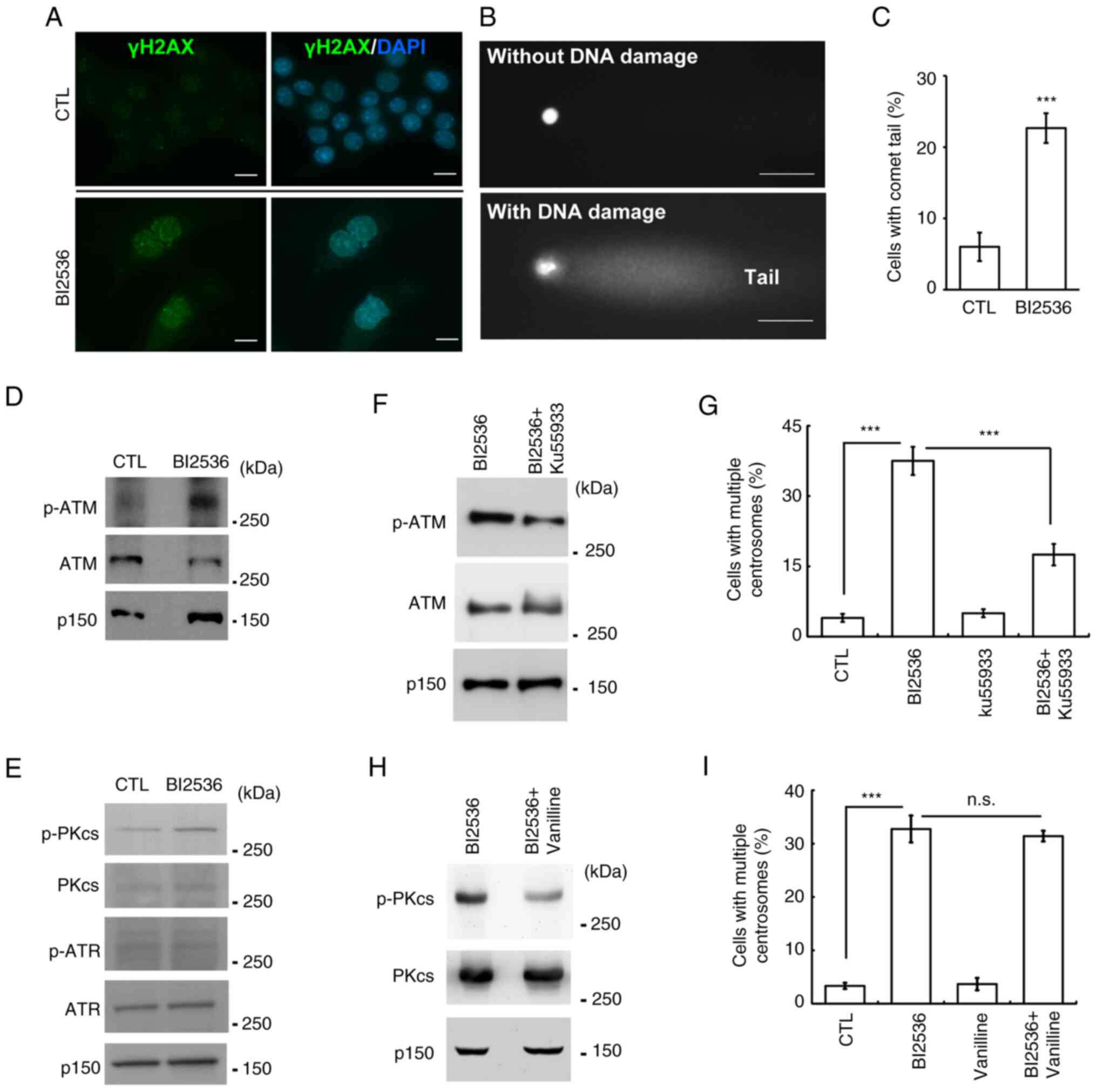 | Figure 5.ATM contributes to centrosome
amplification following BI2536 treatment. (A-C) DNA damage was
induced in BI2536-treated Y1 cells. DNA damage was detected by
immunostaining with an antibody against γ-H2AX. DNA was shown by
DAPI staining. Scale bar, 10 µm. (B) DNA damage was examined using
a comet assay. Representative images of cells without (upper panel)
or with (lower panel) DNA damage. Scale bar, 10 µm. (C)
Quantitative results for (B). (D and E) ATM and DNA-PK were
activated by BI2536 treatment. Cell extracts of CTL or
BI2536-treated cells were analyzed by western blotting with
antibodies against ATM, p-ATM, ATR, p-ATR, DNA-PKcs and p-DNA-PKcs.
P150 was used as a loading control. (F-I) Inhibition of (F and G)
ATM but not (H and I) DNA-PK alleviated BI2536-induced centrosome
amplification. (F and H) ATM and DNA-PK were inhibited by (F)
Ku55933 and (H) vanillin, respectively. Quantitative results of
cells with centrosome amplification in the presence or absence of
(G) ATM inhibitor (Ku55933) or (I) DNA-PK inhibitor (vanillin).
***P<0.001. ATM, ATM serine/threonine kinase; ATR, ATR
serine/threonine kinase; CTL, control; DNA-PK, DNA-dependent
protein kinase; DNA-PKcs, catalytic subunit of DNA-PK; n.s., no
significance; p-, phosphorylated; P150, p150glued. |
BI2536 activates the ATM-ERK axis to
induce centrosome amplification
The downstream effectors of the DNA damage response
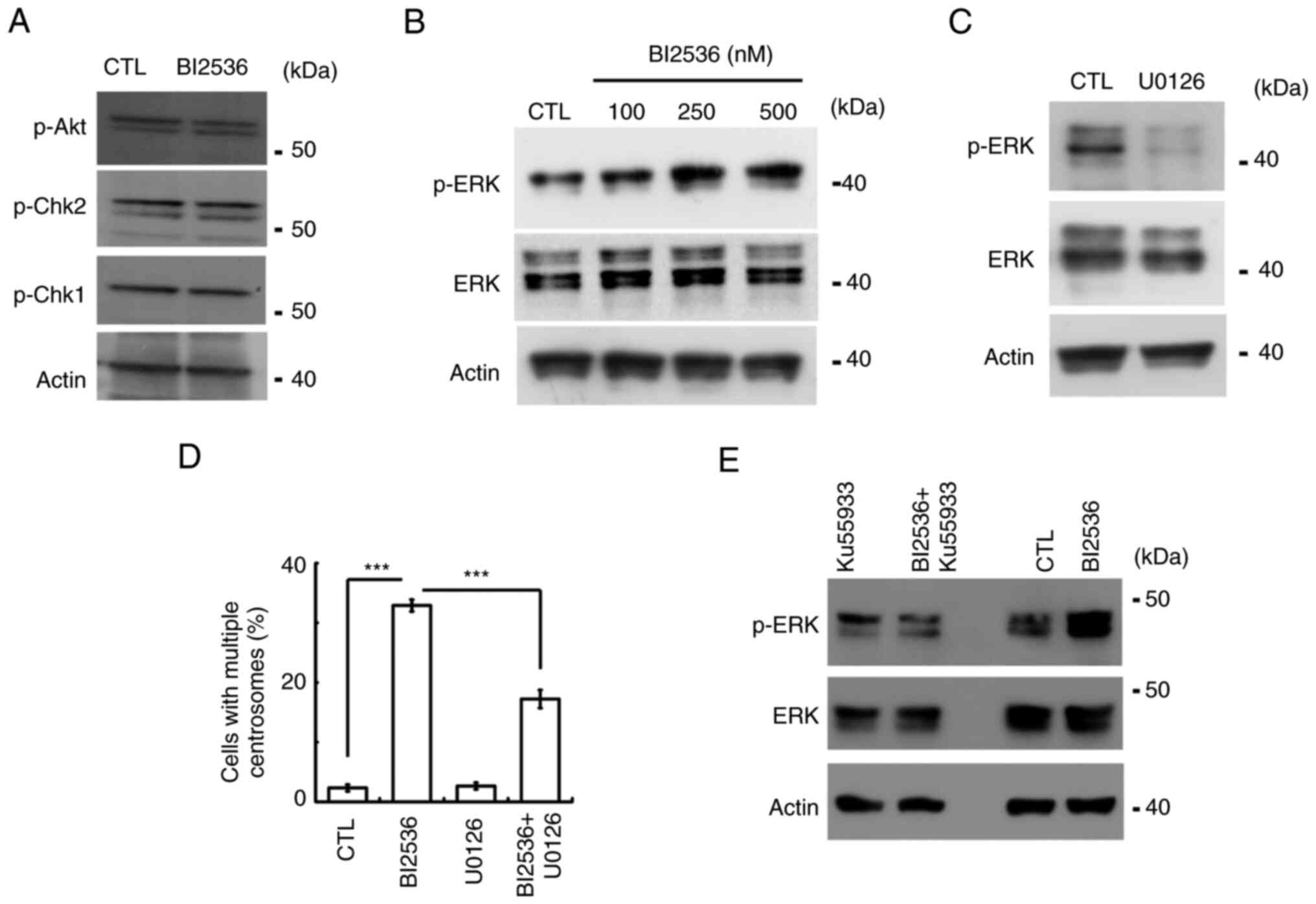
were next examined. Canonical effectors, including Akt, Chk1 and
Chk2, were not activated by BI2536 (Fig. 6A). ERK activation induces centrosome
amplification (3). The present
study then examined whether ERK participated in BI2536-induced
centrosome amplification. Following BI2536 treatment, ERK was
activated in a dose-dependent manner (Figs. 6B and S1C). Treatment with U0126, an
ERK-selective inhibitor, inhibited ERK activation and alleviated
BI2536-induced centrosome amplification (Fig. 6C and D). The data suggested that
BI2536 activated ERK to induce centrosome amplification. The
present study then examined whether ERK was activated by ATM.
Inhibition of ATM reduced ERK activation in both Y1 and H295R cells
(Figs. 6E and S1D). Thus, the present data indicated
that BI2536 activated the ATM-ERK axis to induce centrosome
amplification in adrenocortical tumor cells.
Discussion
The present study revealed that BI2536 inhibited
cell proliferation and induced apoptosis in ACC. Following BI2536
treatment, DNA damage occurred, followed by activation of the
ATM-ERK signaling cascade to induce centrosome amplification.
Aberrant centrosome copy numbers impeded cell cycle progression and
apoptosis, thus reducing adrenocortical tumor cell proliferation.
Therefore, BI2536 could be used as an adjuvant chemotherapeutic
drug for the treatment of ACC.
Mitotane, a derivative of the insecticide
dichlorodiphenyltricholorethane, is approved by the Federal Drug
Administration, USA, to treat ACC in clinical practice. However,
the limitation of this drug, including low efficacy and a narrow
therapeutic window, and its serious toxicity prompt the authors to
identify a novel adjuvant therapeutic molecule (4,5).
BI2536 treatment induces apoptosis in human cancer cell lines of
diverse tissue origins (7,8). Notably, the antitumor effects of
BI2536 have further been demonstrated using human tumor xenografts
in nude mouse models. The present study revealed that BI2536 (~500
nM) inhibited ACC cell proliferation efficiently when compared with
mitotane (~25 µM). In addition, treatment of BI2536 at 500 nM did
not affect the growth of fibroblast cells, suggesting its biosafety
in normal cells. Moreover, BI2536 has been tested in phase II
clinical trials. Thus, the present study suggested that BI2536
could be tested in clinical studies in patients with ACC or used as
an adjuvant therapy to accelerate the chemotherapeutic
efficiency.
The centrosome is not only a microtubule organizing
center but also required for controlling cell cycle progression.
For example, centrosomal targeting of cyclin E or cyclin A is
required for S phase entry (21).
DNA replication-related proteins, including minichromosome
maintenance complex component 5 and origin recognition complex
subunit 1, are also localized to the centrosome to regulate
centrosome duplication (22). Thus,
the crosstalk between the nucleus and centrosome appears to be an
important issue in regulating cell cycle progression properly.
Previous studies have demonstrated that some DNA damage-related
protein also localized to the centrosome to modulate centrosome
copy numbers. Both the regulatory and catalytic subunits of DNA-PK
are recruited to the centrosome and the activated centrosomal
DNA-PK induces centrosome amplification (15,16).
In osteosarcoma, checkpoint protein Chk2 is aberrantly increased in
the centrosome, thus inducing centrosome amplification following
DNA damage. The present study revealed that ATM was activated
following BI2536 treatment. It is known that active ATM is mainly
localized in the nucleus; however, during mitosis, active ATM is
observed in the mitotic spindle poles where it is involved in the
maintenance of mitosis. Thus, it was hypothesized that
BI2536-activated ATM localized to both the nucleus and centrosome.
In the nucleus, ATM initiated the DNA damage response; in the
centrosome, it facilitated centrosome amplification. Thus, distinct
subcellular compartments of ATM may perform different cellular
functions.
BI2536 is a PLK1-selective inhibitor. The present
study revealed that BI2536 induced DNA damage and activated DNA
damage responses. Previous studies have demonstrated that, in
response to high levels of replication stress or DNA damage, PLK1
phosphorylates several targeted proteins such as primase and DNA
directed polymerase, RAD51 recombinase, BRCA2 and MRE11, and is
implicated in the DNA damage response (23–26).
However, how inhibition of PLK1 induces DNA damage under basal
conditions remains less studied. PLK1 interacts with CDK1 to
maintain cell cycle progression (27). CDK1 also participates in homologous
recombination-dependent repair of DNA double-strand breaks, and
couple DNA damage repair to cell cycle progression (28,29).
The present study revealed that ATM and DNA-PK, two kinases
responsible for DNA double-strand breaks, were activated when PLK1
was inhibited. Thus, it was hypothesized that, when PLK1 was
inhibited, the activity of CDK1 was reduced, leading to DNA
double-strand breaks. However, this hypothesis still needs to be
further confirmed in the future.
Supplementary Material
Supporting Data
Acknowledgements
The authors are grateful for the support from the
Core Research Laboratory, College of Medicine, National Cheng Kung
University. The authors also appreciate Dr Bon-chu Chung (Academia
Sinica, Taipei, Taiwan) for kindly providing Y1 and H295R cell
lines.
Funding
The present study was supported by the Ministry of Science and
Technology of Taiwan (grant nos. MOST106-2320-B-006-056-MY3 and
MOST109-2320-B-006-042-MY3) and by An Nan Hospital, China Medical
University (grant no. ANHRF111-17).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
C-YW and S-WT conceptualized the study. R-CL, Y-YC,
W-CL and H-CC implemented methodology. R-CL, Y-YC, W-CL and H-CC
conducted investigation. R-CL, Y-YC, W-CL and H-CC performed
software and formal analysis. S-WT and C-YW wrote, reviewed and
edited the manuscript. S-WT and C-YW supervised the study and
acquired funding. R-CL and C-YW confirm the authenticity of all the
raw data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Long SE and Miller BS: Adrenocortical
cancer treatment. Surg Clin North Am. 99:759–771. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abiven G, Coste J, Groussin L, Anract P,
Tissier F, Legmann P, Dousset B, Bertagna X and Bertherat J:
Clinical and biological features in the prognosis of adrenocortical
cancer: Poor outcome of cortisol-secreting tumors in a series of
202 consecutive patients. J Clin Endocrinol Metab. 91:2650–2655.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen TY, Syu JS, Lin TC, Cheng HL, Lu FL
and Wang CY: Chloroquine alleviates etoposide-induced centrosome
amplification by inhibiting CDK2 in adrenocortical tumor cells.
Oncogenesis. 4:e1802015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fassnacht M, Terzolo M, Allolio B, Baudin
E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A,
Jarzab B, et al: Combination chemotherapy in advanced
adrenocortical carcinoma. N Engl J Med. 366:2189–2197. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Libé R: Adrenocortical carcinoma (ACC):
Diagnosis, prognosis, and treatment. Front Cell Dev Biol. 3:452015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Steegmaier M, Hoffmann M, Baum A, Lénárt
P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant
J, et al: BI 2536, a potent and selective inhibitor of polo-like
kinase 1, inhibits tumor growth in vivo. Curr Biol. 17:316–322.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shin SB, Woo SU and Yim H: Differential
cellular effects of Plk1 inhibitors targeting the ATP-binding
domain or polo-box domain. J Cell Physiol. 230:3057–3067. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matthess Y, Raab M, Knecht R, Becker S and
Strebhardt K: Sequential Cdk1 and Plk1 phosphorylation of caspase-8
triggers apoptotic cell death during mitosis. Mol Oncol. 8:596–608.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen TY, Syu JS, Han TY, Cheng HL, Lu FI
and Wang CY: Cell cycle-dependent localization of dynactin subunit
p150 glued at centrosome. J Cell Biochem. 116:2049–2060. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lai PY, Wang CY, Chen WY, Kao YH, Tsai HM,
Tachibana T, Chang WC and Chung BC: Steroidogenic factor 1 (NR5A1)
resides in centrosomes and maintains genomic stability by
controlling centrosome homeostasis. Cell Death Differ.
18:1836–1844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang CY, Kao YH, Lai PY, Chen WY and Chung
BC: Steroidogenic factor 1 (NR5A1) maintains centrosome homeostasis
in steroidogenic cells by restricting centrosomal DNA-dependent
protein kinase activation. Mol Cell Biol. 33:476–484. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Su S, Chhabra G, Singh CK, Ndiaye MA and
Ahmad N: PLK1 inhibition-based combination therapies for cancer
management. Transl Oncol. 16:1013322022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lian G, Li L, Shi Y, Jing C, Liu J, Guo X,
Zhang Q, Dai T, Ye F, Wang Y and Chen M: BI2536, a potent and
selective inhibitor of polo-like kinase 1, in combination with
cisplatin exerts synergistic effects on gastric cancer cells. Int J
Oncol. 52:804–814. 2018.PubMed/NCBI
|
|
14
|
Choi M, Kim W, Cheon MG, Lee CW and Kim
JE: Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe
following activation of the spindle assembly checkpoint in
non-small cell lung cancer cells. Cancer Lett. 357:591–601. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang CY, Huang EY, Huang SC and Chung BC:
DNA-PK/Chk2 induces centrosome amplification during prolonged
replication stress. Oncogene. 34:1263–1269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen TY, Huang BM, Tang TK, Chao YY, Xiao
XY, Lee PR, Yang LY and Wang CY: Genotoxic stress-activated
DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis
to maintain the DNA damage response. Cell Death Differ.
28:1865–1879. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang CY, Tsai HL, Syu JS, Chen TY and Su
MT: Primary cilium-regulated EG-VEGF signaling facilitates
trophoblast invasion. J Cell Physiol. 232:1467–1477. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Teng YN, Chang HC, Chao YY, Cheng HL, Lien
WC and Wang CY: Etoposide triggers cellular senescence by inducing
multiple centrosomes and primary cilia in adrenocortical tumor
cells. Cells. 10:14662021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chao YY, Huang BM, Peng IC, Lee PR, Lai
YS, Chiu WT, Lin YS, Lin SC, Chang JH, Chen PS, et al: ATM- and
ATR-induced primary ciliogenesis promotes cisplatin resistance in
pancreatic ductal adenocarcinoma. J Cell Physiol. 237:4487–4503.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang CY, Chen WY, Lai PY and Chung BC:
Distinct functions of steroidogenic factor-1 (NR5A1) in the nucleus
and the centrosome. Mol Cell Endocrinol. 371:148–153. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferguson RL and Maller JL: Centrosomal
localization of cyclin E-Cdk2 is required for initiation of DNA
synthesis. Curr Biol. 20:856–860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferguson RL, Pascreau G and Maller JL: The
cyclin A centrosomal localization sequence recruits MCM5 and Orc1
to regulate centrosome reduplication. J Cell Sci. 123:2743–2749.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bailey LJ, Teague R, Kolesar P, Bainbridge
LJ, Lindsay HD and Doherty AJ: PLK1 regulates the PrimPol damage
tolerance pathway during the cell cycle. Sci Adv. 7:eabh10042021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakamura K, Kustatscher G, Alabert C, Hödl
M, Forne I, Völker-Albert M, Satpathy S, Beyer TE, Mailand N,
Choudhary C, et al: Proteome dynamics at broken replication forks
reveal a distinct ATM-directed repair response suppressing DNA
double-strand break ubiquitination. Mol Cell. 81:1084–1099.e6.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee M, Daniels MJ and Venkitaraman AR:
Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated
by DNA damage and mitotic progression. Oncogene. 23:865–872. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Z, Li J, Kong Y, Yan S, Ahmad N and Liu
X: Plk1 phosphorylation of Mre11 antagonizes the DNA damage
response. Cancer Res. 77:3169–3180. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Combes G, Alharbi I, Braga LG and Elowe S:
Playing polo during mitosis: PLK1 takes the lead. Oncogene.
36:4819–4827. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson N, Cai D, Kennedy RD, Pathania S,
Arora M, Li YC, D'Andrea AD, Parvin JD and Shapiro GI: Cdk1
participates in BRCA1-dependent S phase checkpoint control in
response to DNA damage. Mol Cell. 35:327–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ira G, Pellicioli A, Balijja A, Wang X,
Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth
NM, et al: DNA end resection, homologous recombination and DNA
damage checkpoint activation require CDK1. Nature. 431:1011–1017.
2004. View Article : Google Scholar : PubMed/NCBI
|