Background
Metastasis and therapeutic resistance often limit
survival in cancer (1). These
terminal states are facilitated by the evolution of cancer cells in
primary and distal metastatic sites over extended periods. Cancer
evolution is characterized by the dynamic development of various
cellular subpopulations driven by progressive genetic and/or
non-genetic changes (2). In
response to microenvironmental cues and therapeutic selective
pressures, cancer evolution [which follows the Darwinian theory
(3) and/or Lamarckian theory
(4)] produces tumor heterogeneity
by altering the cellular phenotype. Intratumoral heterogeneity is
responsible for cancer progression, metastasis and therapeutic
failure (5,6). Even if the response to therapy is
clinically complete, adaptive tumor evolution almost inevitably
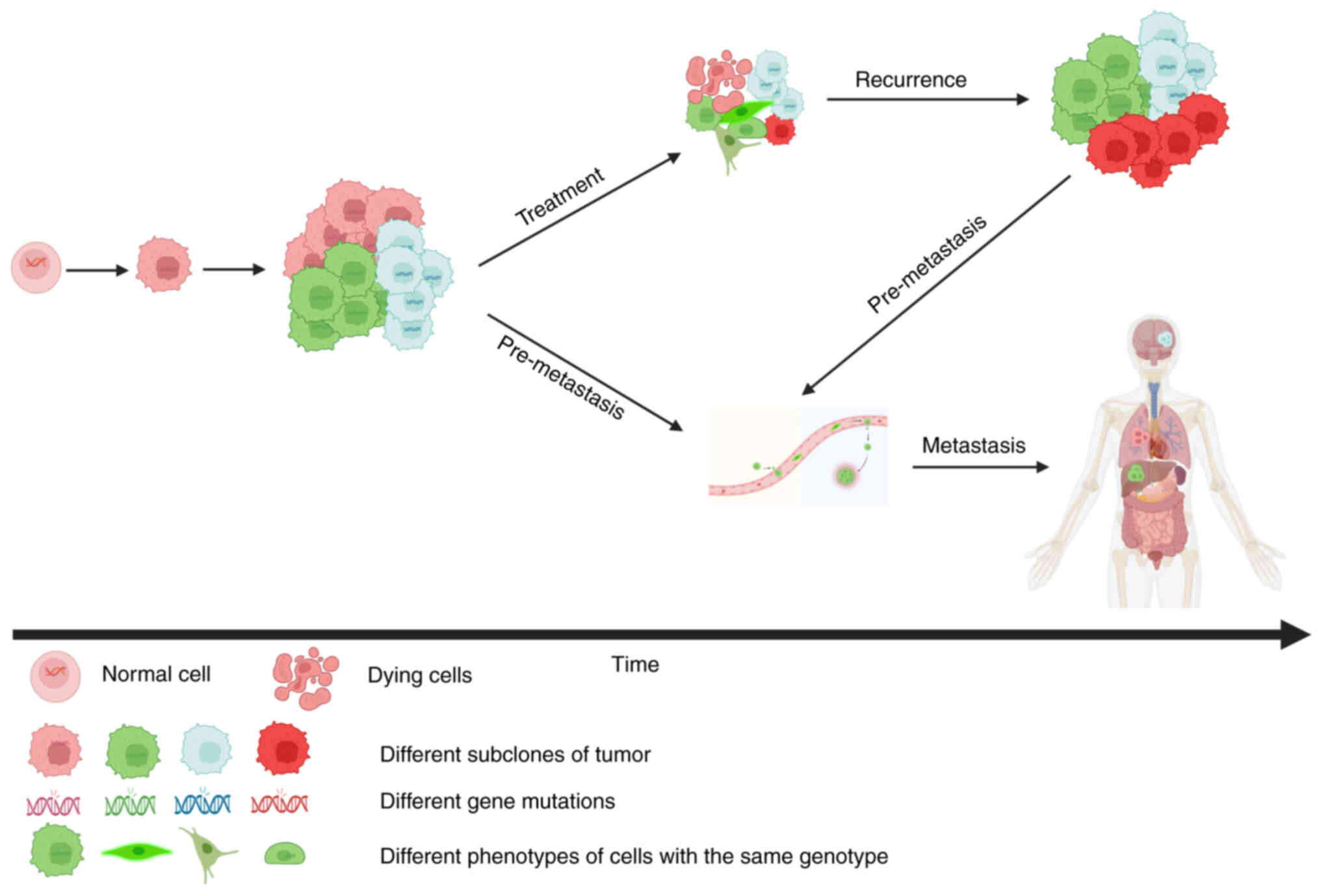
emerges and induces tumor recurrence and metastasis (Fig. 1), which are the primary obstacles to
curing cancer.
Accumulated evidence indicates that individual
cancer genomes have the capacity to generate multiple phenotypic
states. Cancer cells exhibit cellular plasticity, meaning they can
transition between differentiation states without genomic
alterations (7).
Epithelial-to-mesenchymal transition (EMT), which is an archetype
of cancer cell plasticity, facilitates invasion, metastasis and
chemoresistance in malignant epithelial cells via a gradual
transition to the mesenchymal phenotype during tumorigenesis
(8,9). That the phenotypic plasticity of
cancer cells is unlocked has become a new hallmark of cancer
(10). Numerous excellent reviews
have analyzed the role of cell plasticity in tumor development
(8,10). However, the molecular mechanisms of
the phenomenon remain to be fully elucidated. The phenotypic
plasticity of cancer cells is generally underlain by changes in
gene expression, which is a complex process that is regulated at
numerous levels. According to the central dogma, genetic
information flows from DNA to mRNA to protein (11). mRNA, which acts as a bridge between
the genetic information of DNA and protein, has an important role
in translating genotypes into phenotypes. Thus, the regulation of
mRNA, which includes transcription, editing and translation, has a
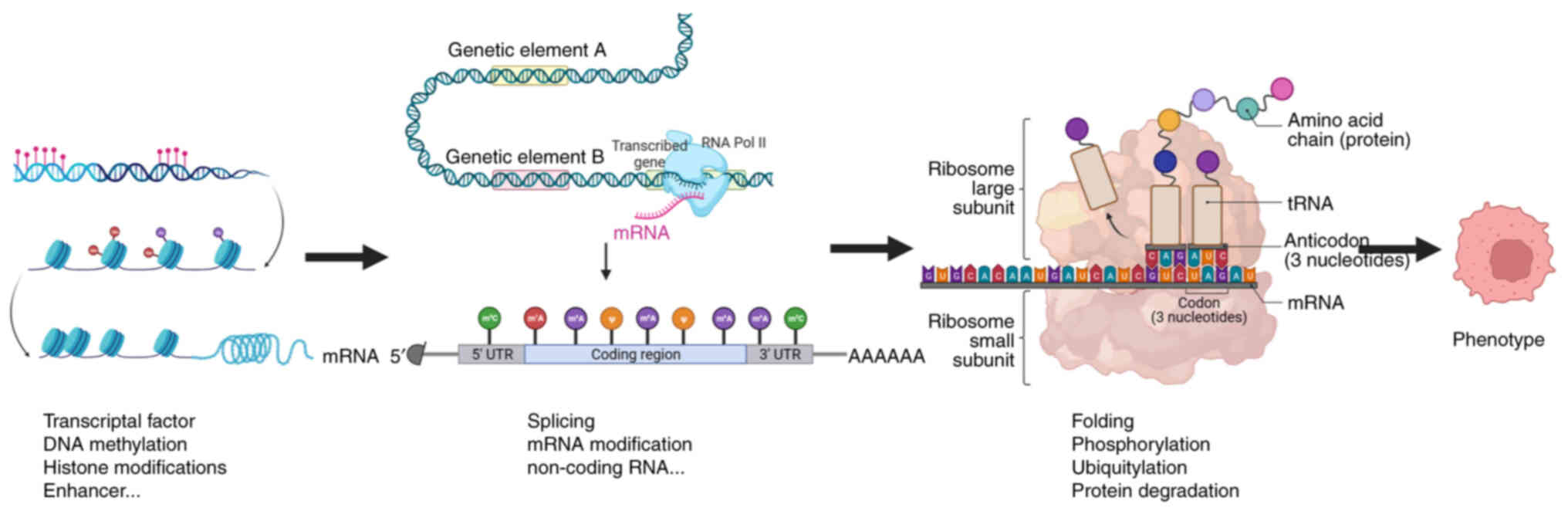
crucial role in mediating gene expression (Fig. 2) (12). The present review aimed to provide
insight into the regulation of cancer plasticity via changes in the
transcription and editing of mRNA. The role of mRNA translation in
the regulation of cancer plasticity has been discussed in other
excellent reviews (13,14) and was therefore not a focus of our
review. Furthermore, relevant therapeutic strategies for cancer
plasticity were also discussed.
Role of transcriptional regulation of mRNA
in cancer cell plasticity
The regulation of gene expression has a key role in
a wide variety of core biological processes ranging from organismal
development and cell differentiation to cellular stress responses
and tissue homeostasis. Much progress has been made in
characterizing the molecular mechanisms of transcription, including
the role of chromatin and its modifications (15). DNA-binding transcriptional factors
(TFs), DNA methylation, histone modifications and enhancers, which
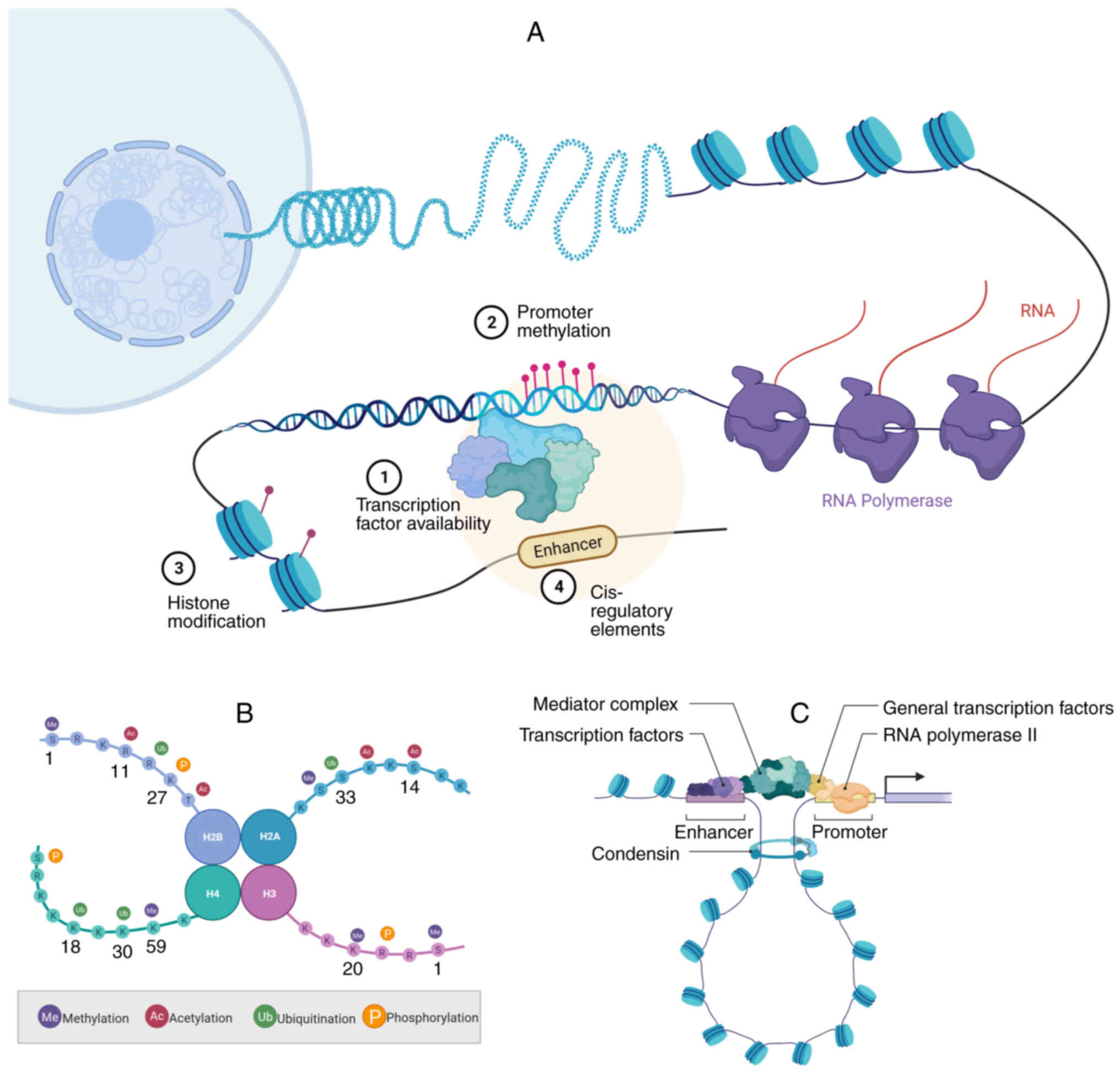
are key regulators of transcription (Fig. 3A) (15,16),
are involved in cancer cell plasticity.
TFs
TFs regulate gene expression and further control
diverse cellular processes and cellular states (17). By regulating dedifferentiation,
transdifferentiation and blocking differentiation, TFs mediate the
plasticity of cancer cells (10).
Dedifferentiation refers to the process by which a
specialized cell is converted to a less differentiated phenotype.
In other cases, incompletely differentiated progenitor cells
maintain a less differentiated phenotype by blocking
differentiation. TF dysregulation is involved in these two modes of
plasticity in various cancers. The TF Sox9, a marker of live
progenitor cells and bile duct lining cells, is a downstream target
of Yes-associated protein (YAP). Liu et al (18) reported that YAP activation in
hepatocytes led to a transition from mature hepatocytes to live
progenitor cells and then the formation of bile duct-lining cells.
By regulating dedifferentiation, Sox9 has an important role in
hepatocarcinogenesis (18).
Furthermore, the TFs transcriptional enhanced associate domain 2
(TEAD2) and transcription factor E2-alpha (E2A) promote oncogenic
dedifferentiation and proliferation in hepatocellular carcinoma
(HCC). Mechanistically, TEAD2 and E2A repress acetyl-CoA synthesis
to induce oncogenic dedifferentiation (19). Other examples include homeobox A5
(20) and SMAD4 (21) in colon cancer, Krüppel-like factor 5
in breast cancer (22) and
microphthalmia-associated transcription factor in melanoma
(23).
In addition, the TF forkhead box M1 (FoxM1) is
highly expressed in breast cancers and is an adverse prognostic
factor. Kopanja et al (24)
found that FoxM1 associates with the retinoblastoma tumor
suppressor (Rb) to repress its transcription. The loss of the
FoxM1-Rb interaction enhances the mammary alveolar differentiation
program by activating Akt signaling. Therefore, the repression of
Rb transcription by FoxM1 is crucial for the plasticity of breast
cancer cells, as it disrupts tumor differentiation (24). Furthermore, in pancreatic ductal
adenocarcinoma (PDAC), the TF hepatocyte nuclear factor 1A (HNF1A)
harbors susceptibility variants, whereas lysine-specific
demethylase 6A (KDM6A) carries somatic mutations. Kalisz et
al (25) show that HNF1A
recruits KDM6A to genomic binding sites in pancreatic acinar cells
and activates differentiated acinar cell programs to indirectly
suppress oncogenic and EMT genes. As other examples, motor neuron
and pancreas homeobox 1 blocks the differentiation of erythroid and
megakaryocytic cells in acute myeloid leukemia (AML) (26), FOXC1 and myocyte enhancer factor 2D
blocks the differentiation of hematopoietic cells in AML (27,28)
and SOX10 blocks melanocyte differentiation in melanoma (29).
Transdifferentiation is a common phenomenon in solid
tumor cells and it is defined as changes in the morphology and
phenotype of a differentiated cell to those of another tissue type
(10). EMT is a quintessence of
transdifferentiation, providing migratory and invasive properties
to cancer cells during tumor progression. The core group of
EMT-associated TFs includes members of the SNAIL family (SNAIL and
SLUG), the TWIST family (TWIST1 and TWIST2) and zinc finger
E-box-binding homeobox (ZEB) factors (8). Xie et al (30) found that inhibitor of NF-κB kinase
subunit epsilon (IKBKE) has an oncogenic role in breast cancer,
with frequent amplification or activation of IKBKE observed in
breast cancer cases. IKBKE controls the stability of SNAIL to
induce EMT and metastasis in breast cancer (30). Yang et al (31) revealed that TWIST has an essential
role in the metastasis of breast cancer. In highly metastatic
breast cancer cells, TWIST suppression specifically inhibits
metastasis from the mammary glands to the lungs by suppressing EMT.
Similar to SNAIL and TWIST, ZEB1 can also induce EMT, promoting the
stemness, invasion and metastasis of pancreatic cancer (32). In addition, transdifferentiation may
occur independently of EMT. Chan et al (33) investigated the molecular mechanisms
of lineage plasticity in prostate cancer and its relationship with
resistance to anti-androgen therapy. They revealed that STAT
upregulation occurs in the mixed luminal-basal phenotype, which is
the beginning of plasticity in an epithelial population and is
responsible for antiandrogen resistance in prostate cancer
(33). In addition, lung
adenosquamous cell carcinoma harbors strong plasticity and carries
a poor prognosis. Tang et al (34) revealed that the dynamic
dysregulation of the counteracting lineage-specific TFs, including
NK2 homeobox 1, forkhead box A2 (FOXA2), tumor protein p63 and
SOX2, finely tunes lineage transition via transdifferentiation. As
other examples, TNF receptor-related factor 3 inactivation promotes
the development of intrahepatic cholangiocarcinoma by
NF-κB-inducing kinase-mediated hepatocyte transdifferentiation
(35), and caudal-type homeobox 1
promotes gastric cancer by inducing intestinal metaplasia (36).
DNA methylation
DNA methylation is a heritable covalent modification
of cytosine nucleotides in CpG dinucleotides during cell division.
It defines cell types and lineages by controlling gene expression
and genome stability (37). DNA
methylation is catalyzed by DNA methyltransferases (DNMTs), which
introduce toxic 3-methylcytosine moieties into DNA. De novo
DNA methylation is mainly catalyzed by DNMT3A and DNMT3B, which
contain a highly conserved DNMT domain and two chromatin reader
domains, in addition to alpha-thalassemia mental retardation
X-linked DNMT3-DNMT3L and proline-tryptophan-tryptophan-proline
(38). DNMT3L, which interacts with
and stimulates the activity of DNMT3A and DNMT3B in the germline,
catalytically inactivates DNMT (38). After de novo DNA methylation,
only symmetrical CpG methylation is maintained during DNA
replication. This is dependent on the methylation of the daughter
DNA strand by DNMT1, representing the mechanism for maintaining DNA
methylation during the cell proliferation process (38,39).
Furthermore, ten-eleven translocation (TET) methylcytosine
dioxygenases, oxidizing 5-methylcytosine to
5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxylcytosine,
demethylate active DNA.
A large number of studies have found abnormal DNA
methylation in various cancers, including lung cancer (40), glioblastoma (41) and breast cancer (42). This aberrant DNA methylation in
cancer cells enhances cellular plasticity and promotes adaptability
and resistance to treatments (43).
Davalos et al (44) reported
that DNA methylation changes accompany the metastasis of melanoma,
and nuclear receptor subfamily 2 group F, member 2 isoform
(NR2F2-Iso2) is a transfer-driven factor involved in epigenetic
regulation. Neural crest cells (NCCs) differentiate into
melanocytes upon the inhibition of NR2F2-Iso2 expression via DNA
methylation, whereas NR2F2-Iso2 is increasingly hypomethylated and
re-expressed in metastatic melanoma. Therefore, it was indicated
that DNA methylation changes allow transformed melanocytes to
acquire NCC- and EMT-like features by controlling NR2F2 activity
(44). In addition, Mancini et
al (45) revealed that the
deregulation of DNMTs and several microRNAs (miRNAs) has a relevant
role in EMT of prostate cancer cells. By targeting DNMT3A, miR-429
modulates the expression of EMT factors, particularly ZEB1
(45). Liu et al (46) demonstrated that miR-135a, in
conjunction with SET and MYND domain-containing 4 (SMYD4),
co-activates Nanog expression, inducing the conversion of
non-cancer stem cells (CSCs) into CSCs. By targeting DNMT1,
miR-135a lowers the methylation level of the CG5 site in the
Nanog promoter. SMYD4 binds to the unmethylated Nanog
promoter to activate Nanog expression in Nanog-negative tumor
cells. These findings indicate that the combination of
miR-135a-DNMT1 with SMYD4 modulates the switch of non-CSCs to CSCs
by regulating DNA methylation of the Nanog promoter
(46). Morinishi et al
(47) demonstrated that TET2
loss-of-function mutations facilitate the reversible switching from
differentiated to stem-like states in AML cells by disturbing DNA
methylation. This leads to increasing numbers of stem-like cells in
AML cell populations. Consequently, AML associated with TET2
loss-of-function mutations is more likely to recur and develop
resistance to drugs (47).
Histone modification
In eukaryotes, chromatin is composed of repeating
units called nucleosomes. Each nucleosome consists of an octamer of
histone proteins and the surrounding DNA fragments (48). The histone octamer forms a spherical
core particle, consisting of an H3-H4 tetramer and two H2A-H2B
dimers, with their N-terminal tails extending outward from the core
particle (49). Over 10
post-translational modifications (PTMs) have been identified on
various amino acid residues of the core histones. These
modifications include acetylation of lysines, methylation of
arginines and lysines, ubiquitination, phosphorylation and
sumoylation (50) (Fig. 3B). These PTMs may occur in the
N-terminal tail as well as in the core domain. Furthermore, PTMs
result in an altered conformational state of chromatin,
consequently regulating gene expression (49).
Mounting evidence has demonstrated that abnormal
PTMs represent a common and pivotal event in a wide range of
cancers. Furthermore, these aberrant PTMs lead to deregulation of
gene expression, further shaping cancer pathogenesis, particularly
cancer plasticity (51,52). In prostate and lung adenocarcinomas,
cancer cell plasticity and neuroendocrine (NE) differentiation are
major causes of resistance to targeted therapy. He et al
(53) reported that the fate
determinant Numb has an important role in mitochondrial autophagy
mediated by Parkin by interacting with Parkin. Numb facilitates
Parkin-mediated mitophagy, significantly contributing to
mitochondrial quality control. Loss of the Numb-Parkin pathway
significantly increases lactic acid production, further leading to
increased histone acetylation and transcription of
neuroendocrine-associated genes (53). Furthermore, epithelial plasticity
describes the reversible regulation of cellular epithelial and
mesenchymal characteristics, and it is associated with tumor
metastasis and chemotherapy resistance. Yuan et al (54) discovered two histone-modifying
enzymes, namely the nuclear SET domain 2 and KDM2A, involved in the
writing and erasing of H3K36me2 that act reciprocally to regulate
epithelial-mesenchymal identity, tumor differentiation and
metastasis. Mechanistically, alteration of histone H3 lysine 36
dimethylation reprograms enhancers associated with the master
regulator of the epithelial-mesenchymal state (54). Histone acetylation, which has key
roles in gene regulation, is highly sensitive to the production and
availability of acetyl-CoA. Carrer et al (55) found that in pancreatic acinar cells
with Kras mutations, histone H4 acetylation increases before the
appearance of precancerous lesions. They observed that acetyl-CoA
levels are elevated in KRAS-mutant acinar cells to support
acinar-to-ductal metaplasia. In PDAC cells, growth factors promote
histone acetylation, resulting in cell proliferation and tumor
growth. Thus, KRAS-driven metabolic alterations promote acinar
plasticity and tumor development by inducing histone acetylation
(55). Furthermore, histone
methylation is involved in the regulation of cancer plasticity.
Liau et al (56)
demonstrated that glioblastoma stem cells (GSCs) can reversibly
transition into a slow-cycling persistent state under the influence
of targeted kinase inhibitors. This transition is involved in the
widespread redistribution of repressive histone methylation. The
upregulation and dependency of persistent GSCs are linked to the
histone demethylases KDM6A and KDM6B. The presence of slow-cycling
cells in primary glioblastomas before treatment, due to high Notch
activity and histone demethylase expression, may contribute to
recurrence (56).
Enhancers
Enhancers are non-coding cis-regulatory elements
bound by TFs, cofactors, mediators and RNA polymerase (Pol) II that
have a central role in precisely regulating spatiotemporal
transcription (Fig. 3C), thereby
participating in development and other biological processes in
eukaryote organisms (57). As a
special cluster of the enhancer family, super-enhancers are more
strongly enriched in TFs, cofactors, mediators, RNA Pol-II and
histone H3 lysine 27 acetylation (H3K27ac) than typical enhancers
(58). Various TFs bind to
enhancers and recruit chromatin-remodeling enzymes, leading to
chromatin opening and the typical pattern of histone modifications
on adjacent nucleosomes, including H3K27ac and histone H3 lysine 4
methylation. Furthermore, via active transcription, certain
enhancers generate non-coding enhancer RNA, which is widely used to
indicate enhancer activity and target gene induction. Numerous
studies confirmed that enhancers have critical roles in cancer
development, therapeutic resistance and cancer cell plasticity
(59,60).
Bi et al (61) showed that endocrine therapy
resistance is related to enhanced phenotypic plasticity, which is
indicated by a general downregulation of luminal-epithelial
differentiation markers and upregulation of basal-mesenchymal
invasive markers. Mechanistically, they reveal that the different
interactions between estrogen receptor α and other oncogenic TFs,
such as GATA binding protein 3 and AP1, driving global enhancer
gain/loss reprogramming that profoundly influences the
transcriptional program in breast cancer. Thus, their study
demonstrated that differential high-order assemblies of TFs on
enhancers triggered genome-wide enhancer reprogramming, resulting
in cancer cell plasticity and therapeutic resistance (61). Similarly, enhancers participate in
cancer metastasis by regulating cancer cell plasticity. Han et
al (62) illustrate that high
expression of quaking (QKI) is related to short overall survival
and metastasis in HCC. The Yin-Yang 1-p65-p300 complex activates
QKI expression via inducing the formation of DNA loops. Aberrant
QKI expression results in the occurrence of EMT and metastasis in
HCC.
Role of mRNA editing in cancer cell
plasticity
All precursor mRNAs (pre-mRNAs) of protein-coding
genes undergo a basal level of RNA processing, including splicing
and polyadenylation. Furthermore, the majority of human genes have
the ability of alternative splicing and selective polyadenylation
sites, leading to the expression of multiple mRNAs (63). In addition to mRNA processing, mRNA
also undergoes another editing mode, namely chemical modification
(64). This mRNA editing has an
important role in translating genotype to phenotype. Therefore,
mRNA editing is also involved in cancer cell plasticity.
mRNA splicing
In eukaryotic cells, the splicing of pre-mRNAs is a
complex and essential step in the flow of information from DNA to
protein (65). Over the past 40
years, research has described the splicing process, including the
detailed characterization of splicing reactions, the definition and
identification of spliceosomes, biochemical analysis of splicing
complexes and the understanding of their regulation (65). The process generates alternatively
spliced mRNAs that produce distinct protein variants, which are
involved in maintaining cellular homeostasis and regulating cell
differentiation and development (66). Dysregulated RNA splicing is a
molecular feature in almost all tumor types. Tumors have up to 30%
more alternative splicing events than normal tissues (67). Recent studies illustrated that
cancer-associated splicing isoforms have critical roles in various
aspects of the biological behavior of cancer cells, such as
increasing cell proliferation, enhancing migration and metastatic
potential, and inducing resistance to therapy (68). In particular, emerging evidence
indicates that cancer-associated splicing isoforms promote a
permissive environment for increasing tumor heterogeneity and
cellular plasticity (69).
Alternative splicing is widely recognized as a key
mechanism for regulating gene expression. Mutations or expression
changes in the components of the splicing machinery or splicing
factors have a crucial role in the plasticity of cancer cells.
Owing to cellular plasticity, NE differentiation is becoming more
prevalent in metastatic castration-resistant prostate cancer
(mCRPC). By analyzing prostate cancer cell lines, mCRPC specimens
and LuCaP patient-derived xenograft models, Labrecque et al
(70) detected alternative splicing
of RE1-silencing transcription factor (REST) to REST4 and reduced
REST activity in mCRPC with NE features. In CRPC cell lines,
serine/arginine repetitive matrix protein 3 (SRRM3) induces
alternative splicing of REST to REST4 and exacerbates the
expression of REST-repressed genes. mCRPC with NE features is
characterized either by REST attenuation and achaete-scute
complex-like 1 activity or the progressive activation of neuronal
transcription factor programs. Therefore, as the principal REST
splicing factor, SRRM3 is expressed in early NE differentiation.
Furthermore, it provides a framework to molecularly classify
diverse NE phenotypes in mCRPC (70). In breast cancer, Li et al
(71) found that QKI and
RNA-binding protein fox-1 homolog 1 coordinately regulate the
splicing and function of the actin-binding protein filamin B
(FLNB), thereby regulating EMT in cancer cells. The skipping of
FLNB exon 30 is strongly associated with EMT gene signatures in
basal-like breast cancer. Furthermore, the skipping of FLNB exon 30
releases the FOXC1 transcription factor to induce EMT. This finding
identified a specific dysregulation of splicing, which regulates
cancer cell plasticity in breast cancer (71). In addition, Xu et al
(72) reported the role of
alternative splicing in eliciting phenotypic plasticity, which is
involved in EMT, in colon cancer. Researchers found that the
differential expression of downstream factors of the EMT master
regulator ZEB1, such as epithelial splicing regulatory protein 1
and other RNA-binding proteins, alters the selective splicing
patterns of a wide range of targets, including CD44 and NUMB. This
resulted in the generation of specific isoforms associated with
increasing invasiveness and metastasis in colon cancer (72).
mRNA modification
RNA epitranscriptomics is a burgeoning field focused
on the study of RNA modifications. Originally, eukaryotic RNA
modifications were primarily identified in transfer RNA and
ribosomal RNA. Over the past decade, they have been identified and
characterized in mRNA and various non-coding RNAs (ncRNAs).
Recently, the significance of mRNA modifications has gained
prominence, as their potential to exert direct functional effects
on gene expression has been recognized (73,74).
Increasing evidence suggests that mRNA modification pathways are
also dysregulated in human cancers (64).
Internal modifications of mRNA include
N6-methyladenosine (m6A), 5-methylcytosine,
N1-methyladenosine and internal 7-methylguanosine
(m7G) (74). The most
characteristic RNA modification is the methylation of adenosine at
the 6th position, resulting in m6A (75). It is involved in multiple aspects of
RNA metabolism, such as RNA stability, translation, splicing,
transport and localization, which have been discovered to affect
various aspects of tumors (76).
Tao et al (77) found that
the m6A levels of RNA were reduced in glioblastoma cells
and glioma tissues. AlkB homolog 5, an eraser of RNA.
m6A enhances the progression of EMT in glioblastoma
cells by decreasing RNA m6A methylation (77). Lin et al (78) illustrated that m6A
modification of mRNAs increased during EMT, representing an
important step in cancer cell metastasis. Downregulation of
m6A induced by the deletion of methyltransferase-like 3
(METTL3) impairs migration, invasion and EMT in cancer cells.
m6A sequencing and functional studies confirmed that the
key transcription factor of EMT SNAIL, involved in EMT, is subject
to m6A regulation. Researchers further demonstrated that
YTH N6-methyladenosine RNA-binding protein 1 mediates the
m6A-induced translation of snail mRNA, thereby
highlighting the critical roles of m6A in the regulation
of EMT in cancer cells (78). In
addition, certain studies indicated that METTL3-mediated
m6A modification is critical for EMT in gastric cancer
(79) and lung cancer (80). Meanwhile, Xia et al (81) verified that the expression of the
m7G methyltransferase WD repeat domain 4 (WDR4) was high
in HCC. WDR4 promotes metastasis and sorafenib resistance through
EMT. Mechanistically, WDR4 enhances cyclin B1 (CCNB1) translation
by promoting the binding of eukaryotic translation initiation
factor 2A to CCNB1 mRNA to increase the progression and metastasis
of HCC (81). Except for
m6A and m7G, the other mRNA modifications,
which include 5-methylcytosine and N1-methyladenosine,
regulate the plasticity of cancer cells, have rarely been reported
and further research is needed.
Role of ncRNAs in cancer plasticity
NcRNAs, which are not translated into proteins,
constitute >90% of RNAs encoded in the human genome. According
to their length, shape and location, ncRNAs may be divided into
different classes, such as miRNA, long nc (lnc)RNA, circular
(circ)RNA and piwiRNA (82).
Growing evidence has facilitated our understanding of the
mechanisms by which ncRNAs perform multiple vital functions in
regulating the expression of genes and communicate with each other.
These ncRNAs perform their function by different molecular
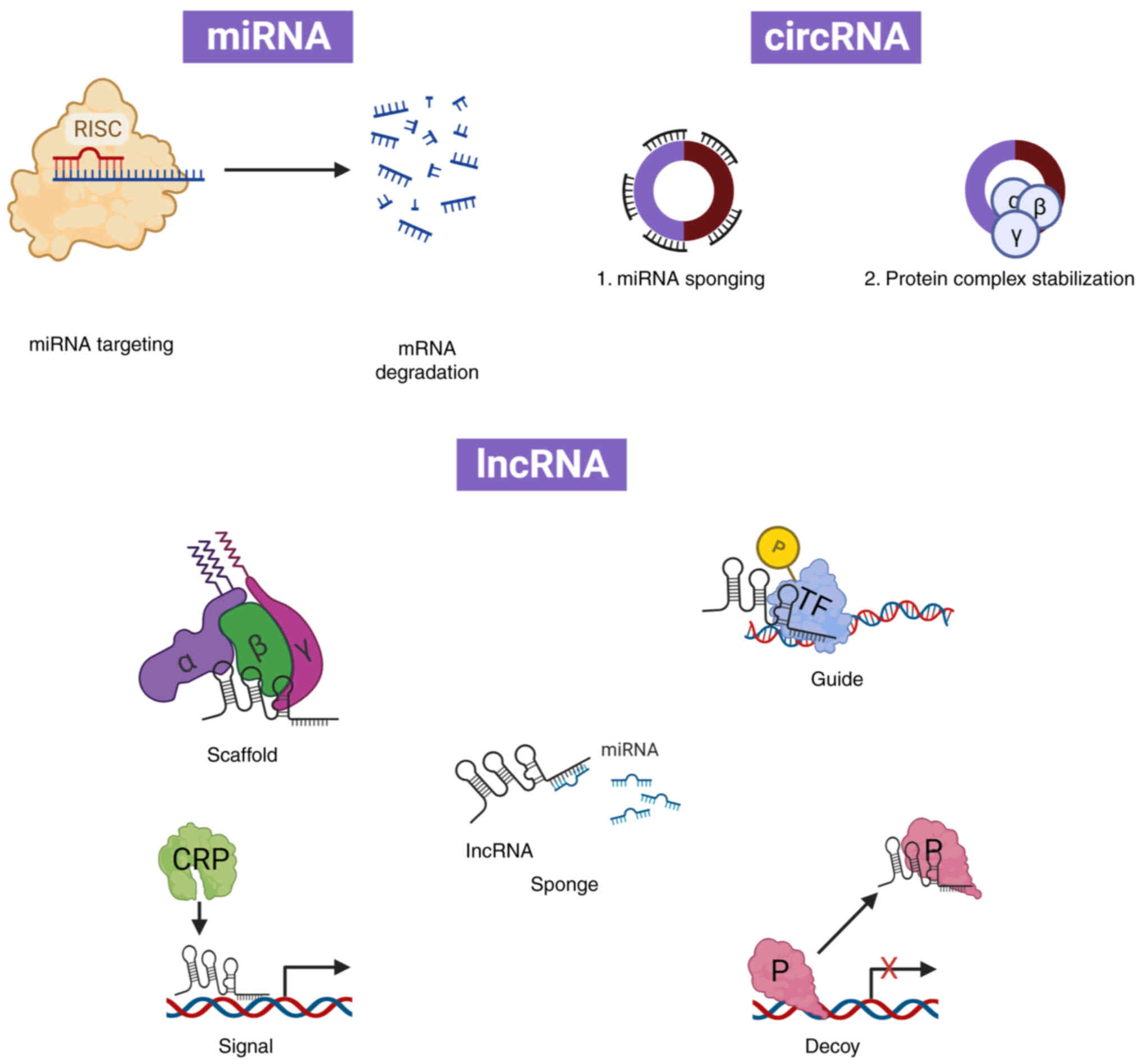
mechanisms (Fig. 4). MiRNAs bind to
complementary sequences in the 3′-UTR of mRNAs, resulting in their
cleavage or translational repression. Compared with miRNAs, lncRNAs
exert their functions via different regulatory models, including
scaffolds, sponges, guides, signals and decoys. Concerning
circRNAs, they perform their functions by sponging with miRNAs or
proteins, thereby translating peptides (83–85).
The dysregulation of ncRNAs has crucial roles in the initiation and
progression of various cancers (82). The discovery of ncRNAs added a new
dimension to understanding the malignant behavior of cancer,
including proliferation, invasion, metastasis and cancer cell
plasticity (86). In regulating
cancer cell plasticity, the roles of ncRNAs are mainly reflected in
the regulation of CSCs and EMT.
miRNA
Concerning miRNAs, Li et al (87) discovered that miR-148/152 family
members are downregulated in gastric CSCs. Integrin α5 is a target
gene of miR-148/152 family members. Their study demonstrated that
miR-148/152 family members inhibit a gastric CSC-like state by
targeting integrin α5 (87). In
addition, Xu et al (88)
found that the miR-119a-5p-SWI-SNF-related, matrix-associated,
actin-dependent regulator of chromatin, subfamily A, member 4 axis
has a role in promoting oral squamous cell carcinoma cell invasion
and metastasis through EMT regulation (88).
LncRNA
lncRNAs are also involved in regulating CSCs and EMT
in cancer. X inactive-specific transcript (XIST) is an lncRNA that
initiates X-chromosome inactivation during early embryonic
development, and its abnormal expression is a common feature in
breast cancer. Ma et al (89) discovered that XIST is a key
regulatory factor in breast cancer stem cells, which exhibit an
aldehyde dehydrogenase-positive (ALDH+) epithelial- and
CD24loCD44hi mesenchymal-like phenotype.
Furthermore, they demonstrated that XIST, acting as a nuclear
sponge for let-7a-2-3p, activates ALDH+ breast cancer
cells to produce IL-6. This, in turn, promotes CSC self-renewal via
STAT3 activation and the expression of key CSC factors. Therefore,
this study concluded that XIST controls paracrine IL-6
pro-inflammatory signaling to promote CSC self-renewal in breast
cancer (89). In addition, Fan
et al (90) reported that
the lncRNA LITATS1 acts as an epithelial gatekeeper in normal
epithelial cells and can inhibit the EMT of breast cancer and
non-small cell lung cancer (NSCLC) cells. Furthermore, they
revealed that LITATS1 enhances the polyubiquitination and
proteasomal degradation of TGF-β1 type receptor (TβR1) and
interacts with TβR1 and the E3 ligase SMAD-specific E3 ubiquitin
ligase 2 (SMURF2), keeping SMURF2 in the cytoplasm. This study
highlighted the function of LITATS1 in epithelial integrity
maintenance via TGF-β-SMAD signaling (90).
CircRNA
Regarding circRNAs, Xiong et al (91) reported that the expression of
circulating Ras-specific GTPase-activating protein 1 (circRACGAP1)
drives the development of NSCLC. CircRACGAP1 is highly expressed in
NSCLC and is associated with the expression of the stemness marker
Sox2. By depleting circRACGAP1, stemness, metastasis and EMT are
repressed in NSCLC cells. Mechanistically, circRACGAP1 recruits the
RNA-binding protein polypyrimidine tract-binding protein 1 to
enhance the stability and expression of sirtuin-3 (SIRT3), leading
to the deacetylation of replication timing regulatory factor 1
(RIF1) and activation of the Wnt-β-catenin pathway. Overexpression
of circRACGAP1 counteracts the SIRT3 or PIF1 knockdown-mediated
inhibition of stemness and metastasis in NSCLC cells. Consequently,
this study uncovered that circRACGAP1 facilitates stemness and
metastasis in NSCLC cells via the recruitment of polypyrimidine
tract-binding protein 1 to promote SIRT3-mediated RIF1
deacetylation (91). CircRNAs also
regulate EMT in cancer cells. Wang et al (92) demonstrated that circZFR sponges
miR-375 to enhance the expression of gremlin 2 (GREM2), which is a
target gene of miR-375. By increasing GREM2 expression, circZFR
enhances the activation of the JNK pathway to promote EMT in
pancreatic cancer cells, facilitating metastasis in pancreatic
cancer (92). Various excellent
reviews have specifically discussed the mechanisms by which ncRNAs
regulate CSCs and EMT in cancer (93–96).
Targeting cancer plasticity
A greater understanding of cancer biology and the
identification of oncogenic drive alterations have markedly altered
the therapeutic landscape, particularly for NSCLC. However, a
phenotypically static cancerous cell state being the driving force
of oncogenesis was an early idea in cancer biology. With the advent
of single-cell multiomics sequencing and sophisticated mathematical
modeling, it was found that cancer cells are heterogeneous, dynamic
entities that evolve over time and change in response to external
cues, including therapy. In the course of cancer evolution, cancer
cell plasticity has a critical role. Most studies on cancer cell
plasticity are motivated by the goal of developing new therapeutic
strategies to cure cancer. The two major contributors to
cancer-related death are metastasis and therapeutic resistance,
both of which are mediated by cancer cell plasticity. Therefore,
cancer cell plasticity both provides therapeutic challenges and
offers novel therapeutic targets that may be exploited to improve
the survival of patients with cancer.
Targeting cancer plasticity to combat
cancer metastasis
Most deaths in patients with cancer are attributable
to metastatic disease opposed to the primary tumor. Cell plasticity
refers to the dynamic non-heritable adaptive capacity of cells in
response to various stressors associated with metastasis and
changes in the tumor microenvironment (TME), is emerging as a
crucial hallmark of metastasis (97). EMT, as a mode of cancer cell
plasticity, is closely related to cancer metastasis. Furthermore,
it was suggested that EMT was dispensable for metastasis in a mouse
model of PDAC (98). During EMT, a
number genetic, epigenomic, transcriptomic and proteomic factors
are involved and interact with each other. These factors have the
potential to be therapeutic targets.
Core EMT-associated TFs, including SLUG, SNAIL,
TWIST and ZEB1, are involved in orchestrating the various
manifestations of EMT. However, the precise role of each
EMT-associated TF depends on the TME and certain EMT-associated TFs
have complimentary and redundant roles. Furthermore, transcription
is a nuclear event not readily accessible to drugs, and
transcriptional factors have been widely considered undruggable
(99). Based on these drawbacks,
targeting EMT-associated TFs is potentially hazardous. Focusing on
their interactions with crucial co-factors would be more
advantageous. Certain clinical trials have investigated the
therapeutic effects on different types of cancer. A phase II
clinical trial examined the Wnt pathway inhibitor LGK974 in
patients with advanced solid tumors. The results indicated good
tolerability of LGK974, but no significant clinical benefit was
observed (100). In addition, the
hedgehog pathway inhibitor vismodegib was investigated in a phase
II trial of patients with metastatic colorectal cancer, but the
results were disappointing (101).
In addition, clinical trials have investigated the effect of
epigenetic regulators to inhibit EMT in cancer therapy. In a phase
I/II clinical trial of the histone deacetylase inhibitor vorinostat
in patients with metastatic breast cancer, the results illustrated
that the combination of vorinostat and paclitaxel was well
tolerated with promising activity in inhibiting EMT in patients
(102). A phase I clinical trial
investigated miRNA mimics, which are used to regulate EMT, in the
treatment of cancer and obtained positive early outcomes. However,
further clinical trials are needed to confirm whether epigenetic
regulators can achieve good clinical outcomes in various
cancers.
In addition, a variety of cues from the TME can
induce EMT in cancer cells, such as growth factors and cytokines.
For instance, the TGF receptor inhibitors galunisertib and
erlotinib have been approved for use in various cancers (103,104). The TGF-β pathway, the most common
inducer of EMT, has been investigated as a potential therapeutic
target for inhibiting EMT in cancer cells. The TGF-β receptor
inhibitor galunisertib was investigated in a phase I/II clinical
trial in patients with advanced HCC. The results demonstrated that
galunisertib was well tolerated, but no clinical benefit was
achieved (105). Table I presents a summary of drugs in
various stages of clinical development that target EMT signaling to
treat metastasis.
 | Table I.Summary of drugs targeting epithelial
to mesenchymal transition signaling to treat metastasis at various
stages of clinical trials. |
Table I.
Summary of drugs targeting epithelial
to mesenchymal transition signaling to treat metastasis at various
stages of clinical trials.
| Drug | Target | Primary tumor
type | FDA approval | Identifier and
status |
|---|
| Galunisertib | TGF-βRI | Pancreatic cancer;
colorectal cancer; breast | Phase 1 and 2 | NCT01373164
(completed); NCT02734160 (completed); NCT01722825 (complete); |
| (LY2157299) |
| cancer; prostate
cancer |
| NCT05700656 (not
yet recruiting); NCT02538471 (terminated); NCT03470350 |
|
|
|
|
| (withdrawn);
NCT02452008 (recruiting); NCT02672475 (active, not
recruiting); |
|
|
|
|
| NCT02154646
(completed) |
| Vactosertib | TGF-βRI | Colorectal cancer;
gastric cancer; pancreatic | Phase 1 and 2 | NCT03844750
(recruiting); NCT03698825 (unknown); (completed) NCT03724851 |
| (TEW-7197) |
| cancer; non-small
cell lung cancer; rectum |
| (active, not
recruiting); NCT04258072 (recruiting); NCT04656002 |
|
|
| cancer; acute
myeloid leukemia; acute |
| (not yet
recruiting); NCT03732274 (unknown); NCT05400122 (recruiting); |
|
|
| lymphoblastic
leukemia; chronic myeloid |
| NCT02160106 |
|
|
| leukemia; chronic
lymphocytic leukemia; |
|
|
|
|
| Hodgkin lymphoma;
non-Hodgkin lymphoma; |
|
|
|
|
| plasma cell
myeloma |
|
|
| Resveratrol | TGF-β1 | Colorectal cancer;
liver cancer | Phase 1 and 2 | NCT00920803
(completed); NCT02261844 (withdrawn) |
| AVID200 | TGF-β | Advanced and
metastatic malignancies | Phase 1 | NCT03834662
(unknown) |
| Trabedersen | TGF-β2 | Pancreatic cancer;
colorectal cancer; melanoma | Phase 1 | NCT00844064
(completed) |
|
| mRNA |
|
|
|
| Regorafenib | RTK | Colorectal cancer;
biliary tract carcinoma; | Phase 1–3 and | NA |
|
|
| pancreatic
adenocarcinoma; gastrointestinal | approved |
|
|
|
| stromal tumors;
sarcoma; esophageal cancer; |
|
|
|
|
| stomach cancer |
|
|
| NIS793 | TGF-β | Pancreatic cancer;
colorectal cancer | Phase 1–3 | NCT04935359
(recruiting); NCT04390763 (active, not recruiting);
NCT04952753 |
|
|
|
|
| (recruiting);
NCT05417386 (recruiting) |
| Fresolimumab | TGF-β | Breast cancer | Phase 2 | NCT01401062
(completed) |
| SAR439459 | TGF-β | Malignant solid
neoplasm | Phase 1 | NCT04729725
(active, not recruiting) |
| Celecoxib | COX-2 | Colorectal cancer;
breast cancer; prostate | Phase 1–3 and | NA |
|
|
| cancer; sarcoma;
melanoma; thyroid cancer; | approved |
|
|
|
| pancreatic cancer;
sarcoma; cholangiocarcinoma; |
|
|
|
|
| renal cell cancer;
esophageal cancer; |
|
|
|
|
| nasopharyngeal
cancer; gastric carcinoma; |
|
|
|
|
| gastroesophageal
junction carcinoma; thoracic |
|
|
|
|
| sarcomas; thoracic
cancers; small cell lung |
|
|
|
|
| cancers; NSCLC;
Ewing's sarcoma |
|
|
| Panitumumab | EGFR | Colorectal cancer;
rectal cancer; breast cancer; | Phase 1–3 and | NA |
|
|
| head and neck
squamous cell carcinoma; | approved |
|
|
|
| esophageal squamous
cell cancer; biliary cancer; |
|
|
|
|
| cholangiocarcinoma;
pancreatic cancer; gastric |
|
|
|
|
| cancer; biliary
tract cancer; gallbladder cancer |
|
|
| Ipatasertib | EGFR | Prostate cancer;
breast cancer; NSCLC; | Phase 1–3 | NCT03072238
(active, not recruiting); NCT04253561 (recruiting);
NCT04920708 |
|
|
| endometrial
adenocarcinoma; head and neck |
| (recruiting);
NCT03853707 (active, not recruiting); NCT04467801
(recruiting); |
|
|
| squamous cell
carcinoma; prostatic cancer; |
| NCT05538897
(recruiting); NCT05172258 (recruiting); NCT04341259
(completed); |
|
|
| gastric cancer;
NSCLC |
| NCT04464174
(active, not recruiting); NCT04404140 (completed); NCT03959891 |
|
|
|
|
| (active, not
recruiting); NCT01896531 (completed); NCT03337724 (completed); |
|
|
|
|
| NCT04060862
(active, not recruiting); NCT04177108 (completed); NCT03800836 |
|
|
|
|
| (completed);
NCT04739202 (recruiting); NCT01562275 (completed); NCT03673787 |
|
|
|
|
| (recruiting);
NCT05554380 (recruiting); NCT02162719 (completed); NCT04632992 |
|
|
|
|
| (active, not
recruiting); NCT034240059 (active, not recruiting);
NCT03337698 |
|
|
|
|
| (recruiting);
NCT04802759 (recruiting); NCT04551521 (recruiting);
NCT03498521 |
|
|
|
|
| (active, not
recruiting); NCT05564377 (recruiting) |
| Tegavivint | β-catenin | NSCLC | Phase 1 | NCT04780568
(recruiting) |
| Cabozantinib | RTK | Renal cell
carcinoma; NSCLC; renal cell | Phase 1–3 | NA |
|
|
| carcinoma; breast
cancer; prostate cancer; | and |
|
|
|
| cervical cancer;
renal cell carcinoma; soft-tissue | approved |
|
|
|
| sarcoma; pancreatic
adenocarcinoma; head |
|
|
|
|
| and neck squamous
cell cancer; adrenal |
|
|
|
|
| cortex carcinoma;
bladder urothelial |
|
|
|
|
| carcinoma;
osteosarcoma; endometrial |
|
|
|
|
| carcinoma;
colorectal cancer; bladder |
|
|
|
|
| urothelial
carcinoma; Merkel cell carcinoma; |
|
|
|
|
| gastrointestinal
stromal tumor; urothelial |
|
|
|
|
| carcinoma;
medullary thyroid cancer; |
|
|
|
|
| hepatocellular
carcinoma; neuroendocrine |
|
|
|
|
| tumors;
osteosarcoma; melanoma; |
|
|
|
|
| adrenocortical
carcinoma; thyroid gland |
|
|
|
|
| carcinoma |
|
|
| Metformin | Snail and | Melanoma; lung
cancer; breast cancer; prostate | Phase 1–3 | NA |
|
| Twist | cancer; pancreatic
adenocarcinoma; colorectal | and |
|
|
|
| cancer; pancreatic
cancer; rectal cancer; head | approved |
|
|
|
| and neck squamous
cell carcinoma; melanoma; |
|
|
|
|
| NSCLC; endometrial
cancer; urothelial cancer |
|
|
| Methotrexate | DHFR | Breast cancer;
osteosarcoma; head and neck | Phase 1–4 and | NA |
|
| and | carcinoma; NSCLC;
colorectal cancer; colorectal | approved |
|
|
| E-cadherin | cancer; head neck
squamous cell cancer; bladder |
|
|
|
|
| cancer; gestational
trophoblastic tumor; renal cell |
|
|
|
|
| carcinoma; sarcoma;
bladder cancer; ureter cancer; |
|
|
|
|
| urethral cancer;
transitional cell carcinoma; |
|
|
|
|
| melanoma; penile
squamous cell carcinoma |
|
|
Targeting cancer cell plasticity to
overcome therapeutic resistance in cancer
One of the current obstacles to curing cancer is the
development of acquired resistance to therapy, which contributes to
~90% of cancer-related deaths (106). With our growing understanding of
the adaptive mechanisms by which cancer evades therapies, the
contribution of cancer cell plasticity to therapeutic resistance
has gained greater and wider recognition in the field. It has been
suggested that adaptation via cancer cell plasticity permits
initial survival under treatment, enabling a small subset of cancer
cells [clinically defined as minimal residual disease (MRD)] to
acquire secondary resistance mutations, leading to disease
progression (107). That is,
drug-tolerant persister (DTP) cells originate from MRD cells
present at the time of clinical remission following initial
therapy. Thus, when the patient who completed therapy and achieved
complete remission enters a convalescent phase of careful
observation, strategies that counter the adaptive mechanisms should
potentially be introduced.
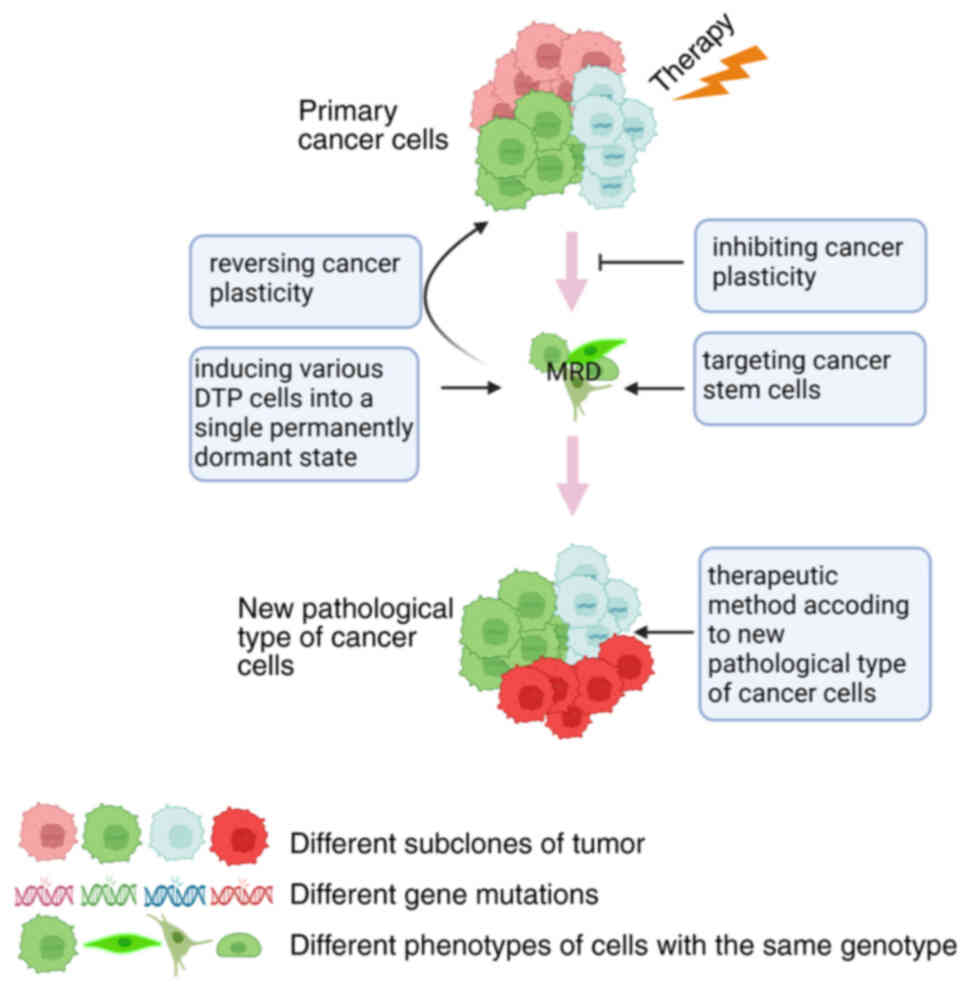
It is conceived that different strategies may target
cancer cell plasticity to resolve the hurdle of DTP (Fig. 5). First, one potential strategy
suppresses the progression of cancer cell plasticity by preventing
its initiation and converting a drug-resistant population to a
drug-sensitive population by leveraging cancer cell plasticity for
therapeutic benefit. However, there is evidence that drug
withdrawal or intermittent drug dosing may overcome DTP cells
(108). However, it is difficult
to prevent the initiation of cancer cell plasticity using this
approach. Targeting the driving factors of cancer cell plasticity
is a potential strategy. For instance, NSCLC PC9 cells express an
activated mutant form of EGFR that drives cellular proliferation,
which is critical for their survival. The cells are highly
sensitive to EGFR inhibitor, resulting in growth arrest and loss of
viability. However, a small population of PC9 cells treated with
EGFR inhibitors may escape cell death during the course of
treatment. After several days of EGFR inhibitor treatment, DTP
cells start proliferating. However, neither acquired EGFR gene
mutations or amplifications, nor expression of the receptor
tyrosine kinase mesenchymal epithelial transition factor receptor,
are observed in DTP PC9 cells. The phenomenon was reversible, as
cells regained drug sensitivity upon discontinuation of treatment.
The DTP state was driven by insulin-like growth factor-1 receptor
tyrosine kinase signaling and the upregulation of histone
demethylase KDM5A. Knockdown of KDM5A was sufficient to restore
drug sensitivity (109).
Similarly, Deng et al (110) revealed that JAK-STAT signaling
pathway is a critical executive factor that drives the plasticity
of the prostate cancer lineage and contributes to androgen receptor
(AR)-targeted therapy resistance. Inhibition of JAK-STAT signaling
may convert AR-targeted therapy-resistant cells to a sensitive
phenotype by leveraging cell plasticity (110). Conversion of the drug-resistant
phenotype is the other strategy for maintaining the sensitivity of
cancer cells. IL-8, a pro-inflammatory cytokine, promotes tumor
cell remodeling and results in the persistence of drug-tolerant
cells. C-X-C chemokine receptor 1 antagonists have been revealed to
reverse IL-8-induced cancer cell plasticity (111). In addition, reversible transition
between EMT and mesenchymal-to-epithelial transition is a key
aspect of cancer cell plasticity. SNAIL is a transcription factor
related to EMT, and revering EMT by inhibiting SNAIL signaling is a
promising strategy to reverse cancer cell plasticity. Qin et
al (112) demonstrated that
SNAIL was upregulated in osimertinib-resistant H1975 cells.
Knockdown of SNAIL restored the sensitivity of
osimertinib-resistant H1975 cells to the drug by reversing cancer
cell plasticity (112). Similarly,
EZH2, which is the central player in epigenetic gene silencing, is
also involved in the regulation of EMT. Overexpression of EZH2 is
related to the conversion of prostate adenocarcinoma to NE prostate
cancer, which is resistant to enzalutamide. Inhibition of EZH2
reverses NE prostate cancer to prostate adenocarcinoma and restores
the sensitivity to enzalutamide (113).
The second strategy involves targeting intermediate
states of cancer plasticity in MRD. It may envisage the other
strategy that diverts the fate of various DTP cells into a single
permanently dormant state. Ideally, DTP cells should be maintained
in a dormant state for a long period to allow them to be eradicated
by taking advantage of their sensitivity to inhibitors or
immune-mediated clearance of the homogeneous dormant cancer cell
population (107). DTP cells are
heterogeneous and not all DTP cells have the ability to contribute
to cancer relapse. After chemotherapy, glioblastoma and
osteosarcoma were demonstrated to relapse from a subset of cancer
cells, namely CSCs overexpressing stem cell genes. Thus, DTP cells
with stemness properties are the main causes of cancer recurrence.
Targeting these CSCs is a potential strategy for various cancers.
Of note, inhibition of crucial CSC regulators, including CSC
markers, epigenetic modifiers and signaling pathways, sensitizes
cancer cells to therapy (114).
For instance, Wang et al (115) revealed that METTL3 induced
m6A methylation of Frizzled 10 (FZD10) mRNA to activate
FZD10 in liver CSCs. FZD10 promotes the self-renewal and
tumorigenicity of liver CSCs by activating β-catenin and YAP1.
Furthermore, the FZD10-β-catenin-c-Jun-MEK-ERK axis determines the
response of hepatoma cells to lenvatinib, and targeting FZD10 or
β-catenin restores sensitivity in lenvatinib in
lenvatinib-resistant HCC (115).
When cancer cell plasticity results in cancer
histological transformation, tumor cells exhibit different
characteristics, necessitating new treatment strategies. After
targeted therapy, histological transformation occurs in up to 10%
of EGFR-mutant lung adenocarcinomas (116) and at least 20% of prostate
adenocarcinomas, leading to acquired resistance to such treatment
(117). When lung adenocarcinomas
transform into SCLC, cancer cells become resistant to EGFR
inhibitors. Transformed SCLC displays greater responsiveness to
platinum-etoposide therapy, similar to primary SCLC. Compared with
primary SCLC, transformed SCLC exhibits higher sensitivity to
taxanes and resistance to immunotherapy, similar to EGFR-mutant
lung adenocarcinomas (118).
Conclusions
Despite substantial progress in the treatment of
cancer, precision therapy based on genomic profiles has produced
mixed clinical outcomes. These sobering results highlight that our
understanding of cancer evolution remains unclear. Although genetic
mutations have a key role in cancer evolution, the importance of
cancer cell plasticity in tumor metastasis and therapeutic
resistance is becoming increasingly apparent. However, numerous
fundamental questions regarding cancer cell plasticity remain
unanswered. First, one key challenge is to determine how to
characterize and define phenotypic states. Even if a phenotypic
state is defined, it is difficult to isolate these specific
phenotypic cells from a given sample. Furthermore, the state is a
continuum rather than a discrete entity because of cancer cell
plasticity. These cells simultaneously or dynamically transition
between different states in response to environmental factors.
Second, genetic mutations and cancer cell plasticity jointly affect
the process of cancer evolution, and the identification and
modeling of these factors remain challenging. Finally, the most
formidable challenge is to construct and identify the
dimensionality of the spatiotemporal state of cancer cells. In
particular, it is difficult to longitudinally collect samples from
individual patients.
To better understand and counteract the molecular
mechanisms of cancer cell plasticity, it will be required to
develop spatiotemporal single-cell multiomics sequencing
technologies, particularly technologies that permit the
simultaneous analysis of the single-cell genomics, epigenomics and
transcriptomics in the same sample. New mathematical theories, such
as statistical techniques, must also be developed to aid in testing
hypotheses regarding the characterization of state, heritability
and transience, dynamics of populations, directionality preferences
and environmental effects in conjunction with experimental and
clinical data. In addition, cancer organoids also provide
assistance for the study of cancer plasticity. Together, advances
in technology and concepts will help us better understand the
mechanisms of cancer cell plasticity and facilitate the development
of innovative therapies to improve outcomes for patients with
cancer.
Acknowledgements
Not applicable.
Funding
This work was supported by Jin Cai Ren (grant no. 2017-18).
Availability of data and materials
Not applicable.
Authors' contributions
CZ and HQ were responsible for collecting studies
from the literature and writing the manuscript. SL and HZ were
responsible for chart editing. RW was responsible for collecting
studies from the literature. All authors have read and approved the
final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lyden D, Ghajar CM, Correia AL,
Aguirre-Ghiso JA, Cai S, Rescigno M, Zhang P, Hu G, Fendt SM, Boire
A, et al: Metastasis. Cancer Cell. 40:787–791. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang D, Jones MG, Naranjo S, Rideout WM
III, Min KHJ, Ho R, Wu W, Replogle JM, Page JL, Quinn JJ, et al:
Lineage tracing reveals the phylodynamics, plasticity, and paths of
tumor evolution. Cell. 185:1905–1923.e25. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vendramin R, Litchfield K and Swanton C:
Cancer evolution: Darwin and beyond. EMBO J. 40:e1083892021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saha S, Pradhan N B N, Mahadevappa R,
Minocha S and Kumar S: Cancer plasticity: Investigating the causes
for this agility. Semin Cancer Biol. 88:138–156. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barkley D, Moncada R, Pour M, Liberman DA,
Dryg I, Werba G, Wang W, Baron M, Rao A, Xia B, et al: Cancer cell
states recur across tumor types and form specific interactions with
the tumor microenvironment. Nat Genet. 54:1192–1201. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Lih TM, Dhanasekaran SM, Mannan R,
Chen L, Cieslik M, Wu Y, Lu RJ, Clark DJ, Kolodziejczak I, et al:
Histopathologic and proteogenomic heterogeneity reveals features of
clear cell renal cell carcinoma aggressiveness. Cancer Cell.
41:139–163.e17. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Drapkin BJ and Minna JD: Studying lineage
plasticity one cell at a time. Cancer Cell. 38:150–152. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lambert AW and Weinberg RA: Linking EMT
programmes to normal and neoplastic epithelial stem cells. Nat Rev
Cancer. 21:325–338. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aggarwal V, Montoya CA, Donnenberg VS and
Sant S: Interplay between tumor microenvironment and partial EMT as
the driver of tumor progression. iScience. 24:1021132021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang HY and Qi LS: Reversing the Central
Dogma: RNA-guided control of DNA in epigenetics and genome editing.
Mol Cell. 83:442–451. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buccitelli C and Selbach M: mRNAs,
proteins and the emerging principles of gene expression control.
Nat Rev Genet. 21:630–644. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fabbri L, Chakraborty A, Robert C and
Vagner S: The plasticity of mRNA translation during cancer
progression and therapy resistance. Nat Rev Cancer. 21:558–577.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee LJ, Papadopoli D, Jewer M, Del Rincon
S, Topisirovic I, Lawrence MG and Postovit LM: Cancer plasticity:
The role of mRNA translation. Trends Cancer. 7:134–145. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pope SD and Medzhitov R: Emerging
principles of gene expression programs and their regulation. Mol
Cell. 71:389–397. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao LY, Song J, Liu Y, Song CX and Yi C:
Mapping the epigenetic modifications of DNA and RNA. Protein Cell.
11:792–808. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joung J, Ma S, Tay T, Geiger-Schuller KR,
Kirchgatterer PC, Verdine VK, Guo B, Arias-Garcia MA, Allen WE,
Singh A, et al: A transcription factor atlas of directed
differentiation. Cell. 186:209–229.e26. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Zhuo S, Zhou Y, Ma L, Sun Z, Wu X,
Wang XW, Gao B and Yang Y: Yap-Sox9 signaling determines hepatocyte
plasticity and lineage-specific hepatocarcinogenesis. J Hepatol.
76:652–664. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park S, Mossmann D, Chen Q, Wang X, Dazert
E, Colombi M, Schmidt A, Ryback B, Ng CKY, Terracciano LM, et al:
Transcription factors TEAD2 and E2A globally repress acetyl-CoA
synthesis to promote tumorigenesis. Mol Cell. 82:4246–4261.e11.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan SH and Barker N: Stemming colorectal
cancer growth and metastasis: HOXA5 forces cancer stem cells to
differentiate. Cancer Cell. 28:683–685. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perekatt AO, Shah PP, Cheung S, Jariwala
N, Wu A, Gandhi V, Kumar N, Feng Q, Patel N, Chen L, et al: SMAD4
Suppresses WNT-Driven dedifferentiation and oncogenesis in the
differentiated gut epithelium. Cancer Res. 78:4878–4890. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu R, Zhou Z, Zhao D and Chen C: The
induction of KLF5 transcription factor by progesterone contributes
to progesterone-induced breast cancer cell proliferation and
dedifferentiation. Mol Endocrinol. 25:1137–1144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thier B, Zhao F, Stupia S, Bruggemann A,
Koch J, Schulze N, Horn S, Coch C, Hartmann G, Sucker A, et al:
Innate immune receptor signaling induces transient melanoma
dedifferentiation while preserving immunogenicity. J Immunother
Cancer. 10:e0038632022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kopanja D, Chand V, O'Brien E,
Mukhopadhyay NK, Zappia MP, Islam A, Frolov MV, Merrill BJ and
Raychaudhuri P: Transcriptional repression by FoxM1 suppresses
tumor differentiation and promotes metastasis of breast cancer.
Cancer Res. 82:2458–2471. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kalisz M, Bernardo E, Beucher A, Maestro
MA, Del Pozo N, Millan I, Haeberle L, Schlensog M, Safi SA, Knoefel
WT, et al: HNF1A recruits KDM6A to activate differentiated acinar
cell programs that suppress pancreatic cancer. EMBO J.
39:e1028082020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nilsson T, Waraky A, Ostlund A, Li S,
Staffas A, Asp J, Fogelstrand L, Abrahamsson J and Palmqvist L: An
induced pluripotent stem cell t(7;12)(q36;p13) acute myeloid
leukemia model shows high expression of MNX1 and a block in
differentiation of the erythroid and megakaryocytic lineages. Int J
Cancer. 151:770–782. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simeoni F, Romero-Camarero I, Camera F,
Amaral FMR, Sinclair OJ, Papachristou EK, Spencer GJ, Lie ALM,
Lacaud G, Wiseman DH, et al: Enhancer recruitment of transcription
repressors RUNX1 and TLE3 by mis-expressed FOXC1 blocks
differentiation in acute myeloid leukemia. Cell Rep. 36:1097252021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao L, Zhang P, Galbo PM, Zhou X, Aryal
S, Qiu S, Zhang H, Zhou Y, Li C, Zheng D, et al: Transcription
factor MEF2D is required for the maintenance of MLL-rearranged
acute myeloid leukemia. Blood Adv. 5:4727–4740. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baggiolini A, Callahan SJ, Montal E, Weiss
JM, Trieu T, Tagore MM, Tischfield SE, Walsh RM, Suresh S, Fan Y,
et al: Developmental chromatin programs determine oncogenic
competence in melanoma. Science. 373:eabc10482021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xie W, Jiang Q, Wu X, Wang L, Gao B, Sun
Z, Zhang X, Bu L, Lin Y, Huang Q, et al: IKBKE phosphorylates and
stabilizes Snail to promote breast cancer invasion and metastasis.
Cell Death Differ. 29:1528–1540. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu M, Zhang Y, Yang J, Zhan H, Zhou Z,
Jiang Y, Shi X, Fan X, Zhang J, Luo W, et al: Zinc-Dependent
regulation of ZEB1 and YAP1 coactivation promotes
Epithelial-Mesenchymal transition plasticity and metastasis in
pancreatic cancer. Gastroenterology. 160:1771–1783.e1. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chan JM, Zaidi S, Love JR, Zhao JL, Setty
M, Wadosky KM, Gopalan A, Choo ZN, Persad S, Choi J, et al: Lineage
plasticity in prostate cancer depends on JAK/STAT inflammatory
signaling. Science. 377:1180–1191. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tang S, Xue Y, Qin Z, Fang Z, Sun Y, Yuan
C, Pan Y, Zhao Y, Tong X, Zhang J, et al: Counteracting
lineage-specific transcription factor network finely tunes lung
adeno-to-squamous transdifferentiation through remodeling tumor
immune microenvironment. Natl Sci Rev. 10:nwad0282023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shiode Y, Kodama T, Shigeno S, Murai K,
Tanaka S, Newberg JY, Kondo J, Kobayashi S, Yamada R, Hikita H, et
al: TNF receptor-related factor 3 inactivation promotes the
development of intrahepatic cholangiocarcinoma through
NF-κB-inducing kinase-mediated hepatocyte transdifferentiation.
Hepatology. 77:395–410. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Choi SI, Yoon C, Park MR, Lee D, Kook MC,
Lin JX, Kang JH, Ashktorab H, Smoot DT, Yoon SS, et al: CDX1
Expression induced by CagA-Expressing helicobacter pylori promotes
gastric tumorigenesis. Mol Cancer Res. 17:2169–2183. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo L, Lee YT, Zhou Y and Huang Y:
Targeting epigenetic regulatory machinery to overcome cancer
therapy resistance. Semin Cancer Biol. 83:487–502. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Greenberg MVC and Bourc'his D: The diverse
roles of DNA methylation in mammalian development and disease. Nat
Rev Mol Cell Biol. 20:590–607. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nishiyama A and Nakanishi M: Navigating
the DNA methylation landscape of cancer. Trends Genet.
37:1012–1027. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liang W, Zhao Y, Huang W, Gao Y, Xu W, Tao
J, Yang M, Li L, Ping W, Shen H, et al: Non-invasive diagnosis of
early-stage lung cancer using high-throughput targeted DNA
methylation sequencing of circulating tumor DNA (ctDNA).
Theranostics. 9:2056–2070. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Maire CL, Fuh MM, Kaulich K, Fita KD,
Stevic I, Heiland DH, Welsh JA, Jones JC, Gorgens A, Ricklefs T, et
al: Genome-wide methylation profiling of glioblastoma cell-derived
extracellular vesicle DNA allows tumor classification. Neuro Oncol.
23:1087–1099. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu Z, Sandler DP and Taylor JA: Blood DNA
Methylation and breast cancer: A prospective Case-Cohort analysis
in the sister study. J Natl Cancer Inst. 112:87–94. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wainwright EN and Scaffidi P: Epigenetics
and cancer stem cells: Unleashing, hijacking, and restricting
cellular plasticity. Trends Cancer. 3:372–386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Davalos V, Lovell CD, Von Itter R,
Dolgalev I, Agrawal P, Baptiste G, Kahler DJ, Sokolova E, Moran S,
Pique L, et al: An epigenetic switch controls an alternative NR2F2
isoform that unleashes a metastatic program in melanoma. Nat
Commun. 14:18672023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mancini M, Grasso M, Muccillo L, Babbio F,
Precazzini F, Castiglioni I, Zanetti V, Rizzo F, Pistore C, De
Marino MG, et al: DNMT3A epigenetically regulates key microRNAs
involved in epithelial-to-mesenchymal transition in prostate
cancer. Carcinogenesis. 42:1449–1460. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu S, Cheng K, Zhang H, Kong R, Wang S,
Mao C and Liu S: Methylation status of the nanog promoter
determines the switch between cancer cells and cancer stem cells.
Adv Sci (Weinh). 7:19030352020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morinishi L, Kochanowski K, Levine RL, Wu
LF and Altschuler SJ: Loss of TET2 Affects proliferation and drug
sensitivity through altered dynamics of Cell-State transitions.
Cell Syst. 11:86–94.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li S, Wei T and Panchenko AR: Histone
variant H2A.Z modulates nucleosome dynamics to promote DNA
accessibility. Nat Commun. 14:7692023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bouyahya A, Mechchate H, Oumeslakht L,
Zeouk I, Aboulaghras S, Balahbib A, Zengin G, Kamal MA, Gallo M,
Montesano D and El Omari N: The role of epigenetic modifications in
human cancers and the use of natural compounds as epidrugs:
Mechanistic pathways and pharmacodynamic actions. Biomolecules.
12:3672022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang Y, Zhang Q, Zhang Y and Han J: The
role of histone modification in DNA Replication-Coupled nucleosome
assembly and cancer. Int J Mol Sci. 24:49392023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao S, Allis CD and Wang GG: The language
of chromatin modification in human cancers. Nat Rev Cancer.
21:413–430. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu Y and Zhu Q: Histone modifications
represent a key epigenetic feature of Epithelial-to-Mesenchyme
transition in pancreatic cancer. Int J Mol Sci. 24:48202023.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
He Y, Ji Z, Gong Y, Fan L, Xu P, Chen X,
Miao J, Zhang K, Zhang W, Ma P, et al: Numb/Parkin-directed
mitochondrial fitness governs cancer cell fate via metabolic
regulation of histone lactylation. Cell Rep. 42:1120332023.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yuan S, Natesan R, Sanchez-Rivera FJ, Li
J, Bhanu NV, Yamazoe T, Lin JH, Merrell AJ, Sela Y, Thomas SK, et
al: Global regulation of the histone mark H3K36me2 underlies
epithelial plasticity and metastatic progression. Cancer Discov.
10:854–871. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carrer A, Trefely S, Zhao S, Campbell SL,
Norgard RJ, Schultz KC, Sidoli S, Parris JLD, Affronti HC, Sivanand
S, et al: Acetyl-CoA metabolism supports multistep pancreatic
tumorigenesis. Cancer Discov. 9:416–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liau BB, Sievers C, Donohue LK, Gillespie
SM, Flavahan WA, Miller TE, Venteicher AS, Hebert CH, Carey CD,
Rodig SJ, et al: Adaptive chromatin remodeling drives glioblastoma
stem cell plasticity and drug tolerance. Cell Stem Cell.
20:233–246.e7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yao J, Chen J, Li LY and Wu M: Epigenetic
plasticity of enhancers in cancer. Transcription. 11:26–36. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li GH, Qu Q, Qi TT, Teng XQ, Zhu HH, Wang
JJ, Lu Q and Qu J: Super-enhancers: A new frontier for epigenetic
modifiers in cancer chemoresistance. J Exp Clin Cancer Res.
40:1742021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen H and Liang H: A High-Resolution map
of human enhancer RNA loci characterizes super-enhancer activities
in cancer. Cancer Cell. 38:701–715.e5. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mirzadeh Azad F and Atlasi Y: Deregulation
of transcriptional enhancers in cancer. Cancers (Basel).
13:35322021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bi M, Zhang Z, Jiang YZ, Xue P, Wang H,
Lai Z, Fu X, De Angelis C, Gong Y, Gao Z, et al: Enhancer
reprogramming driven by high-order assemblies of transcription
factors promotes phenotypic plasticity and breast cancer endocrine
resistance. Nat Cell Biol. 22:701–715. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Han J, Meng J, Chen S, Wang X, Yin S,
Zhang Q, Liu H, Qin R, Li Z, Zhong W, et al: YY1 complex promotes
quaking expression via Super-Enhancer binding during EMT of
hepatocellular carcinoma. Cancer Res. 79:1451–1464. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Manning KS and Cooper TA: The roles of RNA
processing in translating genotype to phenotype. Nat Rev Mol Cell
Biol. 18:102–114. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Barbieri I and Kouzarides T: Role of RNA
modifications in cancer. Nat Rev Cancer. 20:303–322. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shi Y: Mechanistic insights into precursor
messenger RNA splicing by the spliceosome. Nat Rev Mol Cell Biol.
18:655–670. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bonnal SC, Lopez-Oreja I and Valcarcel J:
Roles and mechanisms of alternative splicing in cancer-implications
for care. Nat Rev Clin Oncol. 17:457–474. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kahles A, Lehmann KV, Toussaint NC, Huser
M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C; ancer
Genome Atlas Research Network, ; Rätsch G: Comprehensive Analysis
of Alternative Splicing Across Tumors from 8,705 Patients. Cancer
Cell. 34:211–224.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Bradley RK and Anczukow O: RNA splicing
dysregulation and the hallmarks of cancer. Nat Rev Cancer.
23:135–155. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Other-Gee Pohl S and Myant KB: Alternative
RNA splicing in tumour heterogeneity, plasticity and therapy. Dis
Model Mech. 15:dmm0492332022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Labrecque MP, Brown LG, Coleman IM, Lakely
B, Brady NJ, Lee JK, Nguyen HM, Li D, Hanratty B, Haffner MC, et
al: RNA splicing factors SRRM3 and SRRM4 distinguish molecular
phenotypes of castration-resistant neuroendocrine prostate cancer.
Cancer Res. 81:4736–4750. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li J, Choi PS, Chaffer CL, Labella K,
Hwang JH, Giacomelli AO, Kim JW, Ilic N, Doench JG, Ly SH, et al:
An alternative splicing switch in FLNB promotes the mesenchymal
cell state in human breast cancer. Elife. 7:e371842018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xu T, Verhagen M, Joosten R, Sun W,
Sacchetti A, Munoz Sagredo L, Orian-Rousseau V and Fodde R:
Alternative splicing downstream of EMT enhances phenotypic
plasticity and malignant behavior in colon cancer. Elife.
11:e820062022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Roundtree IA, Evans ME, Pan T and He C:
Dynamic RNA modifications in gene expression regulation. Cell.
169:1187–1200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Frye M, Harada BT, Behm M and He C: RNA
modifications modulate gene expression during development. Science.
361:1346–1349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Penning A, Jeschke J and Fuks F: Why novel
mRNA modifications are so challenging and what we can do about it.
Nat Rev Mol Cell Biol. 23:385–386. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Xu Y, Song M, Hong Z, Chen W, Zhang Q,
Zhou J, Yang C, He Z, Yu J, Peng X, et al: The N6-methyladenosine
METTL3 regulates tumorigenesis and glycolysis by mediating
m6A methylation of the tumor suppressor LATS1 in breast
cancer. J Exp Clin Cancer Res. 42:102023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Tao M, Li X, He L, Rong X, Wang H, Pan J,
Lu Z, Zhang X and Peng Y: Decreased RNA m6A methylation
enhances the process of the epithelial mesenchymal transition and
vasculogenic mimicry in glioblastoma. Am J Cancer Res. 12:893–906.
2022.PubMed/NCBI
|
|
78
|
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J,
Luo G, Tauler J, Du J, Lin S, et al: RNA m6A methylation
regulates the epithelial mesenchymal transition of cancer cells and
translation of Snail. Nat Commun. 10:20652019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yue B, Song C, Yang L, Cui R, Cheng X,
Zhang Z and Zhao G: METTL3-mediated N6-methyladenosine modification
is critical for epithelial-mesenchymal transition and metastasis of
gastric cancer. Mol Cancer. 18:1422019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cheng C, Wu Y, Xiao T, Xue J, Sun J, Xia
H, Ma H, Lu L, Li J, Shi A, et al: METTL3-mediated m6A
modification of ZBTB4 mRNA is involved in the smoking-induced EMT
in cancer of the lung. Mol Ther Nucleic Acids. 23:487–500. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xia P, Zhang H, Xu K, Jiang X, Gao M, Wang
G, Liu Y, Yao Y, Chen X, Ma W, et al: MYC-targeted WDR4 promotes
proliferation, metastasis, and sorafenib resistance by inducing
CCNB1 translation in hepatocellular carcinoma. Cell Death Dis.
12:6912021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Slack FJ and Chinnaiyan AM: The role of
non-coding RNAs in oncology. Cell. 179:1033–1055. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang C and Peng G: Non-coding RNAs: An
emerging player in DNA damage response. Mutat Res Rev Mutat Res.
763:202–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu W, Liang S and Zhang C: Aberrant miRNAs
regulate the biological hallmarks of glioblastoma. Neuromolecular
Med. 20:452–474. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang C, Zhou Y, Gao Y, Zhu Z, Zeng X,
Liang W, Sun S, Chen X and Wang H: Radiated glioblastoma
cell-derived exosomal circ_0012381 induce M2 polarization of
microglia to promote the growth of glioblastoma by CCL2/CCR2 axis.
J Transl Med. 20:3882022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Goodall GJ and Wickramasinghe VO: RNA in
cancer. Nat Rev Cancer. 21:22–36. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li X, Li L and Wu J: The members of the
miR-148/152 family inhibit cancer stem cell-like properties in
gastric cancer via negative regulation of ITGA5. J Transl Med.
21:1052023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xu M, Zhang J, Lu X, Liu F, Shi S and Deng
X: MiR-199a-5p-Regulated SMARCA4 promotes oral squamous cell
carcinoma tumorigenesis. Int J Mol Sci. 24:47562023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ma Y, Zhu Y, Shang L, Qiu Y, Shen N, Wang
J, Adam T, Wei W, Song Q, Li J, et al: LncRNA XIST regulates breast
cancer stem cells by activating proinflammatory IL-6/STAT3
signaling. Oncogene. 42:1419–1437. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Fan C, Wang Q, Kuipers TB, Cats D, Iyengar
PV, Hagenaars SC, Mesker WE, Devilee P, Tollenaar RAEM, Mei H and
Ten Dijke P: LncRNA LITATS1 suppresses TGF-beta-induced EMT and
cancer cell plasticity by potentiating TbetaRI degradation. EMBO J.
42:e1128062023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xiong H, Liu B, Liu XY, Xia ZK, Lu M, Hu
CH and Liu P: circ_rac GTPase-activating protein 1 facilitates
stemness and metastasis of non-small cell lung cancer via
polypyrimidine tract-binding protein 1 recruitment to promote
sirtuin-3-mediated replication timing regulatory factor 1
deacetylation. Lab Invest. 103:1000102023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wang J, Zheng L, Hu C, Kong D, Zhou Z, Wu
B, Wu S, Fei F and Shen Y: CircZFR promotes pancreatic cancer
progression through a novel circRNA-miRNA-mRNA pathway and
stabilizing epithelial-mesenchymal transition protein. Cell Signal.
107:1106612023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mirzaei S, Gholami MH, Hushmandi K,
Hashemi F, Zabolian A, Canadas I, Zarrabi A, Nabavi N, Aref AR,
Crea F, et al: The long and short non-coding RNAs modulating EZH2
signaling in cancer. J Hematol Oncol. 15:182022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
McCabe EM and Rasmussen TP: lncRNA
involvement in cancer stem cell function and epithelial-mesenchymal
transitions. Semin Cancer Biol. 75:38–48. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Pan G, Liu Y, Shang L, Zhou F and Yang S:
EMT-associated microRNAs and their roles in cancer stemness and
drug resistance. Cancer Commun (Lond). 41:199–217. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kristensen LS, Jakobsen T, Hager H and
Kjems J: The emerging roles of circRNAs in cancer and oncology. Nat
Rev Clin Oncol. 19:188–206. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gerstberger S, Jiang Q and Ganesh K:
Metastasis. Cell. 186:1564–1579. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Pastushenko I and Blanpain C: EMT
Transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang N, Ng AS, Cai S, Li Q, Yang L and
Kerr D: Novel therapeutic strategies: Targeting
epithelial-mesenchymal transition in colorectal cancer. Lancet
Oncol. 22:e358–e368. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Liu Y, Qi X, Donnelly L,
Elghobashi-Meinhardt N, Long T, Zhou RW, Sun Y, Wang B and Li X:
Mechanisms and inhibition of Porcupine-mediated Wnt acylation.
Nature. 607:816–822. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zarzosa P, Garcia-Gilabert L, Hladun R,
Guillen G, Gallo-Oller G, Pons G, Sansa-Girona J, Segura MF,
Sanchez de Toledo J, Moreno L, et al: Targeting the hedgehog
pathway in rhabdomyosarcoma. Cancers (Basel). 15:7272023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bondarev AD, Attwood MM, Jonsson J,
Chubarev VN, Tarasov VV and Schioth HB: Recent developments of HDAC
inhibitors: Emerging indications and novel molecules. Br J Clin
Pharmacol. 87:4577–4597. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Tian C, Liu Y, Xue L, Zhang D, Zhang X, Su
J, Chen J, Li X, Wang L and Jiao S: Sorafenib inhibits ovarian
cancer cell proliferation and mobility and induces radiosensitivity
by targeting the tumor cell epithelial-mesenchymal transition. Open
Life Sci. 17:616–625. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chan CY, Hong SC, Chang CM, Chen YH, Liao
PC and Huang CY: Oral squamous cell carcinoma cells with acquired
resistance to erlotinib are sensitive to Anti-Cancer effect of
quercetin via pyruvate kinase M2 (PKM2). Cells. 12:1792023.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ali S, Rehman MU, Yatoo AM, Arafah A, Khan
A, Rashid S, Majid S, Ali A and Ali MN: TGF-β signaling pathway:
Therapeutic targeting and potential for anti-cancer immunity. Eur J
Pharmacol. 947:1756782023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Dhanyamraju PK, Schell TD, Amin S and
Robertson GP: Drug-Tolerant persister cells in cancer therapy
resistance. Cancer Res. 82:2503–2514. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
De Conti G, Dias MH and Bernards R:
Fighting drug resistance through the targeting of Drug-Tolerant
persister cells. Cancers (Basel). 13:11182021. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kim HD, Yoo C, Ryu MH and Kang YK: A
randomised phase 2 study of continuous or intermittent dosing
schedule of imatinib re-challenge in patients with tyrosine kinase
inhibitor-refractory gastrointestinal stromal tumours. Br J Cancer.
129:275–282. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
East MP and Johnson GL: Adaptive chromatin
remodeling and transcriptional changes of the functional kinome in
tumor cells in response to targeted kinase inhibition. J Biol Chem.
298:1015252022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Deng S, Wang C, Wang Y, Xu Y, Li X,
Johnson NA, Mukherji A, Lo UG, Xu L, Gonzalez J, et al: Ectopic
JAK-STAT activation enables the transition to a stem-like and
multilineage state conferring AR-targeted therapy resistance. Nat
Cancer. 3:1071–1087. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Matsushima K, Yang D and Oppenheim JJ:
Interleukin-8: An evolving chemokine. Cytokine. 153:1558282022.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Qin Q, Li X, Liang X, Zeng L, Wang J, Sun
L and Zhong D: Targeting the EMT transcription factor Snail
overcomes resistance to osimertinib in EGFR-mutant non-small cell
lung cancer. Thorac Cancer. 12:1708–1715. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Shi ZD, Pang K, Wu ZX, Dong Y, Hao L, Qin
JX, Wang W, Chen ZS and Han CH: Tumor cell plasticity in targeted
therapy-induced resistance: mechanisms and new strategies. Signal
Transduct Target Ther. 8:1132023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Garcia-Mayea Y, Mir C, Masson F, Paciucci
R and ME LL: Insights into new mechanisms and models of cancer stem
cell multidrug resistance. Semin Cancer Biol. 60:166–180. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang J, Yu H, Dong W, Zhang C, Hu M, Ma W,
Jiang X, Li H, Yang P and Xiang D: N6-methyladenosine-mediated
up-regulation of FZD10 regulates liver cancer stem cells'
properties and lenvatinib resistance through WNT/β-Catenin and
hippo signaling pathways. Gastroenterology. 164:990–1005. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hao L, Chen H, Wang L, Zhou H, Zhang Z,
Han J, Hou J, Zhu Y, Zhang H and Wang Q: Transformation or tumor
heterogeneity: Mutations in EGFR, SOX2, TP53, and RB1 persist in
the histological rapid conversion from lung adenocarcinoma to
small-cell lung cancer. Thorac Cancer. 14:1036–1041. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Quintanal-Villalonga A, Chan JM, Yu HA,
Pe'er D, Sawyers CL, Sen T and Rudin CM: Lineage plasticity in
cancer: A shared pathway of therapeutic resistance. Nat Rev Clin
Oncol. 17:360–371. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Marcoux N, Gettinger SN, O'Kane G, Arbour
KC, Neal JW, Husain H, Evans TL, Brahmer JR, Muzikansky A, Bonomi
PD, et al: EGFR-Mutant adenocarcinomas that transform to small-cell
lung cancer and other neuroendocrine carcinomas: Clinical outcomes.
J Clin Oncol. 37:278–285. 2019. View Article : Google Scholar : PubMed/NCBI
|