Introduction
Ovarian cancer, a malignant tumor frequently present
in the reproductive system of women, can be classified based on its
primitive tissue type into epithelial ovarian cancer, germ cell
tumors and stromal tumors. However, the pathogenesis of ovarian
cancer has yet to be fully elucidated. The currently favored
hypotheses include BRCA1 and BRCA2 gene mutations (1), sexual hormone imbalance and prolonged
chronic inflammation stimulation. Concurrently, epidemiologic
studies indicated that the disease prevalence significantly
correlates with ethnicity and country (1). Due to its inconspicuous symptoms at
early stages, it is challenging to identify promptly; although it
can be managed at advanced stages with surgery and treatments such
as chemotherapy and radiotherapy, the outcome remains
unsatisfactory. This, together with the extraordinary incidence and
mortality rate, poses an alarming threat to women's health,
compelling immediate exploration of innovative therapies.
In recent years, mitochondria-associated endoplasmic
reticulum (ER) membrane (MAM), a unique cellular membrane structure
existing between the outer mitochondrial membrane and the ER, has
become implicated in a range of physiological functions such as
mitochondrial biogenesis, function regulation, calcium ion
carriage, lipid metabolism, oxidative stress and autophagy through
this unique position. More importantly, mounting evidence indicates
that MAMs may act as a potential therapeutic target influencing
mitochondria and ER, two vital organelles in the cancer field.
Hence, elucidating the MAMs could potentially provide a novel
strategy for tackling diseases including ovarian cancer in the
future.
However, despite the relatively modest research
conducted on MAMs till date, it is evident from all empirical
findings that MAMs bear a relationship with various disorders
including cancers and neurodegenerative diseases. Regardless of
whether it is basic or clinical research, reports on MAMs are
comparatively scarce. Given its exceptional physiological structure
and distribution, it is conceivable that MAMs hold substantial
research value. Therefore, the impact of MAMs on physiological
function was primarily discussed in the present review and fresh
perspectives on the practical application of MAMs in ovarian cancer
treatment were provided via examining proteins existing on MAMs,
combined with their implementation in the management of ovarian
cancer.
Mitochondrial inner membrane association
with cellular physiological processes
In the appraisal of the profound correlation between
ER and mitochondria, it was found that the dynamic adjustments of
mitochondria and ER directly influence their physiological role and
structure. These phenomena imply the existence of MAMs. Employing
electron microscopy technology, the gap between mitochondria and ER
was observed to be ~10-30 nanometers-an incredibly diminutive
distance facilitating a plethora of protein interactions within
this ambit, thereby constructing the biological grounding to affect
the functions of mitochondria and ER (2). Despite the prevalent perception that
the functions of mitochondria and ER have distinct differences,
they can reciprocally influence each other via MAMs.
While it was previously proposed that MAMs are an
illusion (2), more recent
investigation utilizing cutting-edge scientific tools confirmed:
MAMs indeed exist. Notably, MAMs are not static; their distance
often varies with the presence of ribosomes. For instance, on a
smooth ER (SER), the distance of MAMs is usually 10–15 nanometers,
whereas on a rough ER (RER) it expands to 20–30 nanometers. More
crucially, MAMs serve as pivotal in maintaining cell calcium
homeostasis (3,4), lipid homeostasis (5), mitochondrial dynamic equilibrium
(6) and the stability of
organelles, among other physiological functions, which are critical
for the survival of the organism.
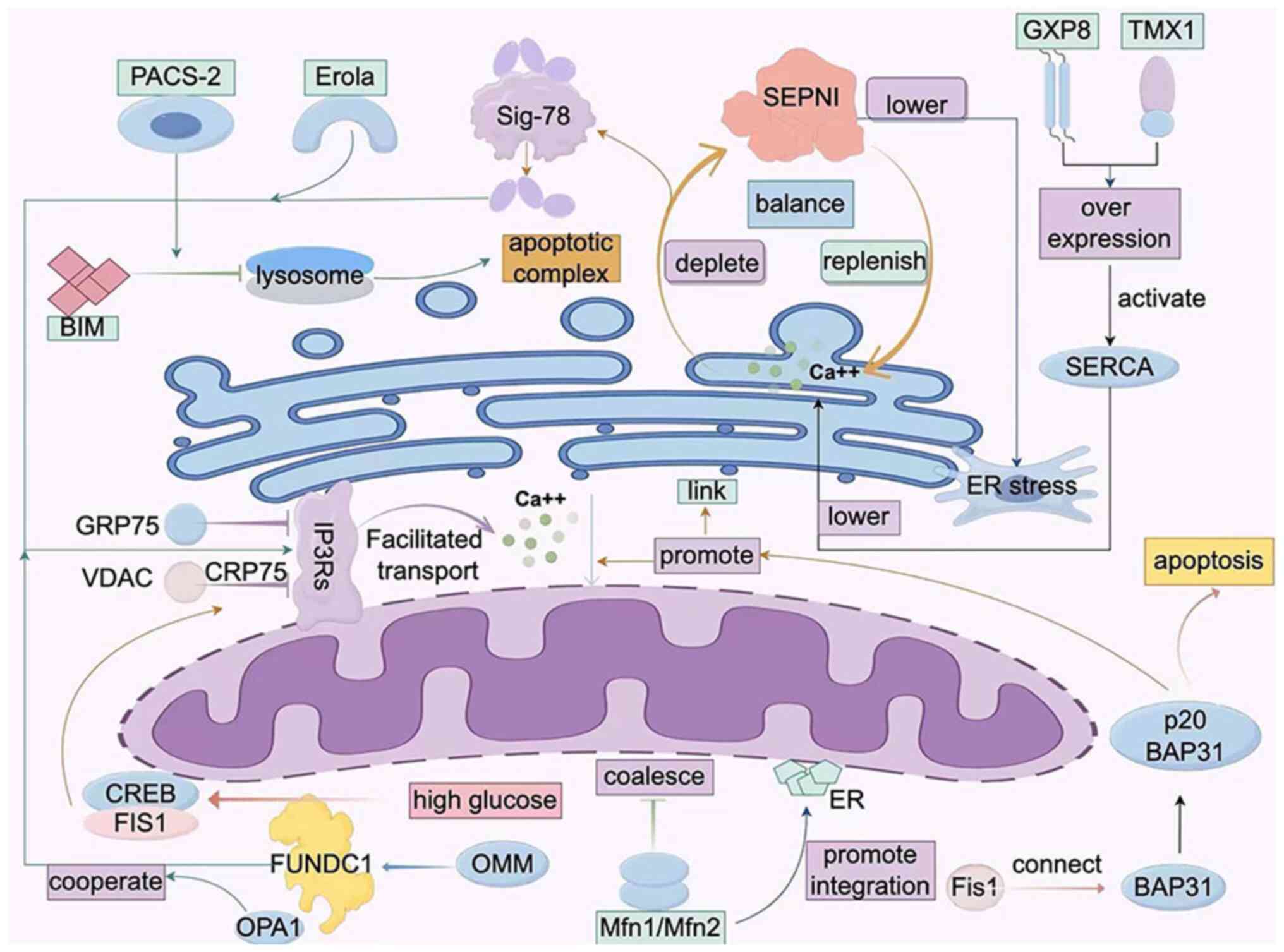
Calcium homoeostasis
Calcium ions (Ca2+) serve as the
secondary messengers within cells, regulating a myriad of cellular
processes. MAMs, significant bridges for Ca2+ transport,
finely tune Ca2+ signal transmission ensuring precise
and flawless execution of cell biological functions. At present,
two primary types of Ca2+ release channels have been
well-understood in MAMs, namely: IP3Rs and RyRs.
The IP3Rs, voltage-dependent anion channels that
dominate Ca2+ release above MAMs, collaborate with GRP75
and VDAC to take on this formidable task. The tripartite protein
complex assists calcium ions in traversing from mitochondria to
ER.
Another primal ER protein SEPN1, abundant in this
type of MAMs, can replenish calcium ions on the ER when it is
depleted, thus circumventing ER stress. Besides protecting IP3R
from oxidative harm by Ero1a, SEPN1 also upholds calcium ion
equilibrium between MAMs, mitochondria and ER (6).
Existing in the same form as a protein complex are
Sig-78 and immunoglobulin. When calcium ions are depleted in the
ER, Sig-78 dissociates from the complex and interacts with IP3Rs,
substantially enhancing Ca2+ influx into mitochondria.
Moreover, Ero1a, an oxidase residing in MAMs, prompts calcium ion
flux towards mitochondria when faced with ER stress by oxidative
binding to IP3R, thereby activating IP3R (7,8).
Lastly, glutathione peroxidase 8 (GXP8), a bipolar
transmembrane protein present in MAMs, can lower calcium level on
the ER by suppressing the activity of SERCA when overexpressed.
Also acting similar to GXP8 is the redox enzyme TMX1 located on the
ER, again by preventing the action of SERCA. SERCA plays a core
role in the transportation process of calcium ions. It regulates
the content of intracellular Ca2+ ions and profoundly
influences the flow of calcium ions in MAMs. When the pressure on
the ER becomes excessive, the activity of SERCA1 dramatically
escalates, thereby propelling calcium ions from the ER to
mitochondria, culminating in mitochondrial apoptosis (7,8).
Moreover, mitochondria demonstrate their unique role
during the transport of calcium ions over a short time period. Upon
the transfer of calcium ions from the ER to MAMs (7,8), high
calcium concentration regions often emerge macroscopically. Under
this state, the speed at which mitochondria absorb calcium ions
markedly accelerates. By contrast, when calcium ion concentration
is low, the efficiency of mitochondria in absorbing calcium ions
noticeably decreases. Therefore, the regulation of calcium ion
concentration is indeed heavily influenced by a profound impact led
by MAMs. Besides, existing research has unveiled that mitofusin 2
(Mfn 2) is a pivotal target for regulating calcium ion
concentration in MAMs; however, some studies have yielded contrary
experimental conclusions (9). Thus,
there still remains considerable research exploration space in
terms of how MAMs affect the degree of influence on the flow of
calcium ions between mitochondria and ER.
Lipid homeostasis
There is robust correlation between MAMs and lipid
homeostasis. Since other organelles lack the capacity to synthesize
phospholipids from their precursor, and the ER is a pivotal site
for regulating cell intrinsic lipid balance, lipids must diffuse
from the ER into other organelles. It has been previously reported
that MAMs are a cholesterol enriched membrane system, predominantly
composed of caveolin proteins, serving an essential function,
contributing towards enriching the related elements of MAMs' lipid
metabolism, thereby underscoring the indispensable role of MAMs in
preserving lipid homeostasis. Phosphatidyl serines (PS), as the
predominant enzyme system generated within the ER, is primarily
localized on MAMs. Subsequently, it translocates to mitochondria to
be converted into phosphatidyl ethanolamines (PE) (10). Some of the PE, upon return to the
ER, transforms into phosphatidyl cholines, which is further
distributed to various other organelles. In this process, the
pro-phospholipid acid of PS is synthesized in the ER, but
necessitates storage in MAMs.
In addition to phospholipid synthesis, MAMs also
perform another crucial element of cholesterol metabolism. The
newly synthesized cholesterol within the ER must be transported to
mitochondria to convert into pregnenolone, the primary substance of
sex steroids. In the rate-limiting step of this process, MAMs serve
a pivotal role. Furthermore, research indicates the abundant
presence of MAMs in cholesterol acyltransferase 1 (ACAT1), an
enzyme responsible for the conversion of free cholesterol into
cholesterol esters (CEs). Such CEs are commonly stored in fat
droplets. Hence, it is feasible to indirectly evaluate the activity
of MAMs by measuring the activity of ACAT1 (11). As such, MAMs have gradually emerged
as the primary activator of mitochondrial steroid synthesis.
Subsequently, MAMs also participate in the generation of ceramide,
regulating cell growth, inflammation response, differentiation and
apoptosis (12,13).
Mitochondrial dynamics
Significantly, it has been previously elucidated
that MAMs are the initial locations of mitochondrial division
(14). For instance, mitochondrial
division proteins 1, mitochondrial division factors, and
mitochondrial dynamics proteins are all sequestered at the MAMs
site and completed prior to mitochondrial division (15). Additionally, processes such as mtDNA
replication, mitochondrial transport and mitochondrial
self-division, all revolve around the MAMs.
Mitochondrial fusion and division, a highly dynamic
network system, maintains a steady state equilibrium of fusion and
division under normal physiological conditions to ensure optimal
cell functioning. Certainly, appropriate stimulation is paramount
for maintaining a high quality of mitochondria. Among key
constituents of the mitochondrial fusion mechanism, Mfn1, Mfn2 and
optic atrophy 1 (OPA1) play pivotal roles. Notably, Mfn1, acting as
a requisite GTPase for outer membrane fusion, primarily restricts
mitochondrial function through its interaction with Mfn1 on the
MAMs (16). In concert with Mfn1
and Mfn2 on the MAMs, Mfn2 helps form the mitochondrial outer
membrane and acquires specific morphology (17). Given the heightened degree of Mfn2
enrichment on the MAMs, it is conjectured that the MAMs hold great
significance during mitochondrial fusion, particularly when
post-MITOL/MARCH5 ubiquitination machinery causes alterations in
the intersection between the ER and mitochondria (18).
Acting as a cytosolic protein, Dlp1 dynamically
congregates to the mitochondrial membrane and substantially
enhances membrane contraction velocity via a GTP-dependent
oligomerization pathway. Under hypoxic circumstances, the
accumulation of the OMM protein FUNDC1 induces ectopic Dlp1
accumulation in the MAMs (19).
Furthermore, as an integral component of the apoptotic process,
Fis1 can bind to BAP31 in the MAMs, transmitting the mitochondrial
apoptotic signals to the ER. BAP31, a critical escort protein on
the ER membrane, participates in the degradation of misfolded
protein and apoptotic signaling within the endoplasmic reticulum
stress pathway. When Fis1 unites with BAP31, it cleaves BAP31 into
p20 BAP31; as an apoptosis-promoting protein, p20 BAP31 activates
procaspase-8 to convert it into a functional form, which can cleave
Bid and initiate cell apoptosis (20,21).
Activation of p20 BAP31 also facilitates Ca2+
translocation from the ER to the mitochondria, indicating the
ability of cellular apoptotic signals to feedback to the
mitochondria (22,23). The stability of calcium ions
similarly directly or indirectly influences the dynamics of the
mitochondria (24).
Apoptosis
The relationship between calcium homeostasis and
tissue structural integrity is exceedingly significant. With the
excessive metabolism of Ca2+, it can induce the opening
of the mitochondrial permeability transition pore, escalating the
permeability of the inner mitochondrial membrane. The mitochondrion
then undergoes depolarization, subsequently impeding ATP synthesis,
leading to an expansion in the mitochondria's molecular form and
ultimately causing the rupture of the outer membrane, thereby
releasing a substantial amount of cytochrome c, culminating in
apoptosis. Noteworthy, previous studies have also elucidated that
apoptotic signaling exists within MAMs (25–27).
The majority of apoptotic signals from the ER converge on the
mitochondria autophagy pathway. This further suggests that the ER
stress situation will directly influence the physiological
alterations of the mitochondria, and the most pivotal factor among
these is the BH3 protein (25),
which is uniquely activated by the ER and transmits autophagy
signaling to the mitochondria (26). However, presently, it remains
elusive whether this protein can be concurrently activated by all
forms of ER damage, or is activated only during specific
pathological processes where the ER is specifically damaged. It is
conceivable that it may emerge as a potential therapeutic drug
target for diseases in the future (27).
Moreover, the mitochondrial division factor Fis1
aids in maintaining the junctions between the ER and mitochondria
through its interaction with Bap31 (28). When procaspase-8 is recruited, the
cleavage of Bap31 will lead to an increase in mitochondrial
permeability and instigate apoptosis (29,30).
In the ordinary state, ceramide is synthesized via the ceramide
synthase pathway. However, under stress conditions (for example,
heat shock, TNF-α, Fas, chemotherapy drugs, toxins, radiation and
other factors), ceramide can be rapidly synthesized in
sphingomyelinase from sphingomyelinase. The accumulation of
ceramide not only modulates various molecules involved in apoptotic
signaling transductions but can interact with them, such as protein
phosphatase 1A/2A, protein kinase C, NF-κB and Ras. A substantial
accumulation of ceramide can lead to the formation of pores on the
external mitochondrial membrane, induce the release of
pro-apoptotic substances (such as cytochrome C) in the
mitochondrial membrane gap, and propagate stress signals from the
ER to the mitochondria (31).
Whereas suppressing the activity of ACS1/4 can reduce the synthesis
of ceramide and deter the occurrence of cell apoptosis (32). Nevertheless, given the intricate
nature and cross-domain relationships of apoptosis itself, markedly
more profound and meticulous discussions are needed to delve deeper
into this complex matter.
Proteins contributing to the function of
MAMs
The unique attributes of MAMs encompass their
protein composition and structure, underscoring their pivotal roles
in numerous physiological processes. Research on MAM proteins has
unveiled a broad spectrum of physiological functions; however,
comprehensive understanding remains to be achieved due to the
intricate diversity present in the richly diverse MAM proteome. For
example, the GTPase Mfn1/Mfn2, which plays an essential role in
mitophagy during mitotic spindle formation, was identified with
seemingly contradictory functional features (33); also present on the surface of MAMs
are the calcium release channels IP3R and VDAC (35), responsible for regulating
mitochondrial calcium homeostasis. Lastly, Grp75 holds a
significant influence on the organizational structure of MAMs
(38). The interactions among these
proteins remain unexplored and hint at the need for further
research.
FUNDC1
Research signifies that FUNDC1 is a ubiquitous
protein within organisms (46).
This protein is composed of 155 amino acids, encompassing three
membrane spanning regions. Notably, its N terminus harbors the LTR
motif, which aids in interaction with LC3 to regulate autophagy in
mitochondria (41,42). The regulation of this activity is
notably prominent under low oxygen environments; the study
predominantly focused on the effects of decreased Src kinase
activity and weaning tyrosine 18 phosphorylation levels, thereby
reinforcing the mutual interplay between LC3 and FUNDC1 (46).
FUNDC1 can also play a pivotal role in calcium
transfer through interaction with IP3R2. Under hyperglycemic
conditions, the activation of FUNDC1 can stimulate the binding of
CREB to the FIS1 promoter, thereby promoting MAM formation. This
indicates that FUNDC1 plays a critical part in the entire process
of MAM formation. Additionally, FUNDC1 also participates in
regulating mitochondrial dynamics, forming a synergistic
relationship with OPA1. It has been discovered that incorporating
change in lysine 10 in FUNDC1 significantly induces mitochondrial
aggregation, reflecting the importance of FUNDC1 in regulating
mitochondrial dynamics (47). NRF1,
as a transcription factor, can stimulate its expression by binding
to the FUNDC1 promoter 186/176 (48).
In HeLa cells, experiments have revealed that
suppressing expression of FUNDC1 propels mitochondrial fusion,
whereas augmenting its expression retards it, probably by inducing
DRP1 to drive mitochondrial proliferation (49). This process also encompasses
phosphorylation at site S13, which attenuates the interplay between
FUNDC1 and Drp1 while amplifying the interaction with OPA1. These
observations signify that alterations in FUNDC1 structure may guide
distinct pathways that culminate in corresponding alterations in
mitochondrial structure. Research along the path of FUNDC1 upstream
regulation and its influence on structural alteration is projected
to be a vital direction of future studies. Within the unique
structure of MAMs, FUNDC1 plays a pivotal role in the evolution of
various pathologies.
Experimental evidence has confirmed that suppression
of FUNDC1 expression fosters the generation of extensive MAMs in
diabetes-induced cardiomyopathy. This morphological and functional
reconfiguration of MAMs initiates cell apoptosis and the
disintegration of abnormal mitochondria, ultimately resulting in
reduced synthesis and disruption of the respiratory chain function
of mitochondria. In both patients afflicted with heart failure and
murine cardiac muscle cells, a decrease was observed in the FUNDC1
protein level (49). Additional
research uncovered that FUNDC1 mainly impairs mitochondrial
function, causing harm to cardiac muscle cells, and the disruption
of MAMs is intricately linked to mitochondrial dysfunction
(50). Similarly, magnesium oxide
and α-lipoic acid can diminish the phenotypic characteristics of
cardiac muscle cells through interaction with FUNDC1 (51). Besides these diseases, FUNDC1 can
also instigate mitochondrial autophagy, promptly triggering cell
apoptosis and myocardial infarction (52). These instances not only underscore
the significance of FUNDC1 but also spotlight the pivotal position
of MAMs within cellular processes.
PACS-2
The PACS family is composed of the two proteins
PACS-1 and PACS-2, which share a similar structure but exhibit
distinct functions. Both proteins contain a furin binding region
(FBR), an intrinsically disordered mid-region (MR), and a
C-terminal region (CTR) (52).
Specifically, the MR domain encompasses a native nuclear
localization signal (NLS) domain and an Akt phosphorylation site,
although the precise function of the CTR domain remains undefined
(53). The prevailing consensus in
academia suggests that the FBR domain plays a crucial role in
recognizing and adhering to cargo proteins.
In 1998, Werneburg et al (54) identified the crucial role of PACS-1
in directing trafficking across the Golgi network (TGN) for the
first time. Subsequently, in 2005, Köttgen et al (55) postulated that both PACS-1 and PACS-2
were involved in the transport process of polycystin-2 mediated by
members of the TRP ion channel family (TRPP2). PACS-2 can precisely
identify and bind cargo proteins with acidic ionization clusters,
facilitating their transportation towards relevant organelles and
thereby influencing cellular activities ranging from calcium
exchange and apoptosis to membrane transport (56).
Numerous studies suggested that PACS-2 influences
immune-induced apoptosis by regulating MAMs and thereby altering
mitochondrial division. Additionally, PACS-2 binds to CNX in the
cytoplasm and regulates the distribution of CNX to MAMs (57). Moreover, PACS-2 can interact with
BIM, inviting the recruitment of BIM to lysosomes in a
mitogen-activated protein kinase-JNK-dependent manner to form the
apoptosis complex (58). Deeper
research indicates that interaction of PACS-2 with ataxia
telangiectasia mutated (ATMs) can activate NF-κB to promote cell
survival (59). Furthermore,
previous evidence demonstrated that PACS-2 participates in DNA
damage-related apoptosis through two independent mechanisms.
Firstly, following DNA damage, PACS-2 employs its NLS domain to
shuttle itself into the nucleus and interact with SIRT1, thus
hindering the deacetylation of p53, leading to cell cycle arrest
(60). Secondly, upon DNA damage,
PACS-2 engages ATM to activate NF-κB, promoting the expression of
the anti-apoptotic protein Bcl-xL (61). Factors such as TRAIL can induce
dephosphorylation of PACS-2 at Ser437 indirectly via Akt, thereby
stimulating mitochondrial membrane permeability increase and
lysosomal membrane permeability increase (62). By contrast, the stable
phosphorylation of PACS-2 is synchronous with the p53/p21 and
NF-κB/Bcl-xL pathways, serving as a cytoprotective mechanism.
Mfn2
Initially recognized as an obligatory dynein-like
protein integral to outer membrane fusion, Mfn2 has recently been
revealed to be localized on the ER membrane, engaging in reciprocal
interactions with the OMM. Research indicated that the expression
of Mfn2 alters the MAM hierarchy and is capable of enhancing the
ER-mitochondria distance within the cell (63), hinting that it plays a pivotal role
in sustaining mitochondrial morphology and ER-mitochondria contact
sites interconnection. Nevertheless, existing findings pose
contradictions to this hypothesis, revealing that cells with Mfn2
variations or deprivation may actually enhance ER-mitochondria
coupling (64,65), suggesting an imperative for further
investigation into the precise impact of Mfn2 on the MAM.
Significantly, Mfn2 serves as a receptor during the
PINK1-Parkin-mediated autophagy process, playing a critical role in
interface management (66).
Moreover, Mfn2 directly interacts with the mitochondrial transport
machinery, encompassing MIRO1/2 transmembrane transporters,
specifically linked to the function of MARC proteins (67). Mfn2 has been proven to influence the
fatty acid metabolism of mitochondria by binding mitochondrial with
lipid particles (68) and
facilitating the transfer of phosphatidylserine from the ER to the
mitochondria.
The downstream repercussions of Mfn2 on
mitochondrial functionality are manifold, including alterations in
the mitochondrial ultrastructure, cristae and supramolecular
structures, affecting the oxidative phosphorylation process
(69,70). Furthermore, the MAM can also
regulate the oxidative phosphorylation process through calcium ion
signal transmission (71). Mfn2 is
also implicated in modulations of mtDNA stability, potentially due
to its fusion effect leading to decreased mtDNA copies and
increased mtDNA deletion frequency (72) which is pivotal for the distribution
of mitochondrial DNA proteins. Additionally, studies have
highlighted the crucial role of Mfn2 in preserving podocyte
apoptosis through regulating the PERK pathway (73). Therefore, the multifaceted functions
of Mfn2 contribute to explaining why cells lacking Mfn2 display
diminished mitochondrial respiration.
Other proteins
Despite the diverse proteins existing on MAMs, the
functional studies of Mfn2, FUNDC1 and PACS-2 are more
comprehensive. Moreover, the influence of these proteins on related
diseases is more profound. In addition to these pivotal proteins,
MAMs also harbor critical participants including DJ-1, TDP-43 and
CypD, although they do not directly affect MAM structure but play a
crucial regulatory role in specific MAM functions. IP3R is an
important Ca2+ efflux channel located at the ER surface,
which mediates the release of Ca2+ from the ER lumen to
the cytoplasm (74). VDAC is an ion
channel located at the outer mitochondrial membrane; it mediates
the movement of various ions and metabolites into and out of
mitochondria and is involved in a series of cellular activities,
including cell apoptosis, metabolism and regulation of
Ca2+ (75). IP3R and
VDAC are linked by Grp75 to maintain MAM structure (76). It is known that overexpression of
VDAC can enhance the connection between ER and mitochondria,
thereby improving the flux of Ca2+ from the ER to the
mitochondria (77); when silencing
VDAC1, the connection between Grp75 and IP3R1 decreases, suggesting
decreased ER-mitochondrial interaction (78). Cells overexpressing Grp75 exhibit
more IP3R1-VDAC1 interaction sites (79). Silence of IP3R1 or Grp75 can also
reduce the connection between VDAC1 and Grp75 or IP3R1 (80).
Moreover, VAPB located within the ER membrane,
participates in the activation of IRE1/XBP1 axis during the
unfolded protein response in the ER (81,82).
VAPB can form a complex with the outer mitochondrial membrane
protein PTPIP51 and help maintain the structure of MAM. The mutant
form of VAPBP56S, showing stronger affinity for PTPIP51, thereby
promotes the transfer of Ca2+ from the ER to the
mitochondria; knocking out any one of these two genes can reduce
the transfer signal of Ca2+ (83) and decrease the level of contact
between the ER and the mitochondria (84,85).
MOSPD2 is another member of the VAP family; it is a
protein located on the surface of the ER membrane which can connect
the ER and other membrane structures, and can also bind to small
proteins that interact with VAP through the MSP (major sperm
protein structural domain), a protein sequence motif called FFAT,
such as PTPIP51 on the outer mitochondrial membrane (86). REEP1 is a protein located on the ER
and the outer mitochondrial membrane. It has been demonstrated that
REEP1 directly connects the ER and mitochondria through
oligomerization and is involved in the formation of the MAM
structure. Furthermore, REEP1 bends the ER membrane so that the ER
can wrap around the mitochondria, which helps form MAMs (86). The complexity they participate in
further highlights the complexity of MAMs, prompting to conduct
more in-depth exploration (Table I)
(Fig. 1).
 | Table I.Functions of proteins on MAMs. |
Table I.
Functions of proteins on MAMs.
| Protein name | Functional
overview | Function | (Refs.) |
|---|
|
IP3Rs-Grp75-VDACs | Calcium
homeostasis | Regulation of
apoptosis, metabolism and Ca2+ | (34) |
| α-Synuclein |
| Promotes
Ca2+ by increasing endoplasmic reticulum and
mitochondrial contact | (39) |
| CypD |
| Affects the
transfer of Ca2+ between the two organelles | (44) |
| FATE1 |
| Downregulation of
the transfer of Ca2+ | (45) |
| Mfn1/MFN2 | The architecture of
MAMs | Maintain the
structural aspects of MAMs and induce endoplasmic Reticulum
stress. | (36) |
| MOSPD2-PTPIP51 |
| Plays a role in
connecting endoplasmic reticulum with other membrane
structures | (37) |
| DJ-1 |
| Enhances the
connection between the ER and mitochondria | (40) |
| TG2 |
| Increase the number
of ER-mitochondrial contacts | (43) |
| NogoB |
| Increase the gap
width of MAMs and affect their function | (45) |
MAMs in ovarian cancer
Cancer cells may modulate the expression of
Ca2+ ion channels, influence signal transduction
mechanisms (87,88), and modify the regulatory capacity of
MAMs on ER-mitochondrial Ca2+ transport to generate a
MDR phenotype (89). This allows
them to evolve continuously and achieve resistance to apoptosis.
Moreover, cancer cells necessitate additional mitochondrial ATP
production to sustain their heightened proliferation activity. As
Ca2+ sustains the activity of various enzymes in the
mitochondrial energy cycle, an appropriate mitochondrial
Ca2+ uptake is crucial for the survival of cancer cells.
However, excessive mitochondrial Ca2+ uptake can lead to
mitochondrial Ca2+ overload, subsequently triggering
mitochondrial dysfunction and cancer cell death. Therefore, how to
balance the control of multiple proteins through MAMs on the
mitochondrial-ER complex mechanism is a pivotal strategy for cancer
cells to acquire resistance to cell death. Currently, the study of
Li et al (90) has
identified GRP75 (a critical chaperone protein) as playing a
pivotal role in the stability of MAMs by forming an
IP3R/GRP75/VDAC1 complex. When GRP75 is absent, it results in a
significant decrease in MAMs' stability, causing Ca2+
disorder between mitochondria and ER, leading to catastrophic
reactive oxygen species (ROS) accumulation, and ultimately
resulting in cell apoptosis. It is evident that targeting proteins
on MAMs can be a key target for improving ovarian cancer
research.
However, due to the particularity of MAMs' location
and structure, there remains a lack of definitive direct
correlation with ovarian cancer. Nevertheless, since MAMs are
located between mitochondria and ER, most current studies primarily
elucidate the impact on ovarian cancer from these two vital
organelles, thereby indirectly indicating the potential application
value of MAMs. Therefore, the following exploration of the
relationship among mitochondria, ER, and ovarian cancer provides
new perspectives and assistance for future direct study of MAMs in
ovarian cancer (Table II).
| Table II.Application of MAMs in ovarian
cancer. |
Table II.
Application of MAMs in ovarian
cancer.
| Author, year | Overall | Pointcut | Outcomes | (Refs.) |
|---|
| Vianello et
al, 2022 | MAMs | Mitochondrial
autophagy | Inhibition of
mitochondrial autophagy can restore response sensitivity in
cancerous cells to cisplatin. | (94) |
| Zhou et al,
2022 |
|
| The
aryl-glycosylated zinc (II)-cryptolepine complex disrupts aspects
of the mitochondrial autophagy pathway to induce programmed cell
death and autophagy in carcinoma cells. | (95) |
| Yu et al,
2017 |
|
| ABT737 can induce
mitochondrial autophagy, resulting in the release of cytochrome
C. | (96) |
| Chen et al,
2021 |
|
| Pardaxin, an
antibacterial peptide, stimulates the process of apoptosis due to
excess ROS production. | (97) |
| Wang et al,
2020 |
|
| Elastin cell growth
factor H propels the demise of cancer cells by catalyzing
mitochondrial autophagy. | (98) |
| Katreddy et
al, 2018 |
|
| Both siRNA or
Herdegradin activate mitochondrial autophagy to selectively
eradicate cancer cells. | (99) |
| Meng et al,
2022 |
|
| Cancer cell
proliferation is suppressed by suppressing the gene chain
CRL4cul4A/DDB1. | (100) |
| Yuan et al,
2023 |
|
| The augmentation
effect of cancer stem cells can be mitigated by judicious
suppression of mitochondrial autophagy. | (101) |
| Martinez-Outschoorn
et al, 2012 |
|
| The absence of
BRCA1 tumor suppressor gene triggers mitochondrial autophagy and
catabolic processes. | (102) |
| Jin et al,
2020 |
|
| The fucosylation of
TGF-β1 has the potential to activate the regulatory function in
ovarian cancer cells. | (103) |
| Yu et al,
2021 |
|
| Enhancing
PINK1/parkin mediated mitochondrial autophagy degrades the
sensitivity of ovarian cancer cells to cisplatin. | (147) |
| Bae et al,
2021 |
| Endoplasmic
reticulum stress | Campesterol can
inhibit ovarian cancer viainducing endoplasmic reticulum stress and
amplify the efficacy of conventional anticancer drugs. | (104) |
| Cheng et al,
2022 |
|
| The interaction
between OMA1 and endoplasmic reticulum stress can deter drug
resistance in cancer cells. | (114) |
| Jung et al,
2020 |
|
| DPP23 can serve to
diminish the resistance of ovarian cancer by invoking endoplasmic
reticulum stress. | (115) |
| Xu et al,
2021 |
|
| The PHLDA1 protein
mediates ovarians cancer cell apoptosis versity through the
endoplasmic reticulum stress response mechanism. | (116) |
| Kim and Lee,
2023 |
|
| The validity of
6-gingerolovercoming resistance towards gefitinib in ovarian cancer
cells is confirmed via stimulating endoplasmic reticulum
stress. | (117) |
| Bahar et al,
2021 |
|
| The combined
administration of PARP inhibitor and cisplatin downregulated
cisplatin-resistant OC cells, thereby mitigating endoplasmic
reticulum stress and overcoming PARP inhibitor cross- resistance in
OC. | (118) |
| Rezghi et
al, 2021 |
|
| Verified
miR-3c-1-1p engages cell apoptosis through endoplasmic reticulum
stress. | (120) |
| Kong et al,
2021 |
|
| The nanoparticle's
reduction-sensitive polymer form can escalate endoplasmic reticulum
stress, thus enhancing therapeutic efficacy. | (121) |
| Wang et al,
2020 |
|
| Epoxyeicosatrienoic
acid H induces endoplasmic reticulum stress and stimulates
apoptosis. | (122) |
| Wang et al,
2020 |
|
| The chemical
reagent DWP05195 triggers cellular apoptosis through the induction
of endoplasmic reticulum stress. | (123) |
| Yart et al,
2022 |
|
| Genomic
reprogramming was accomplished via Activation of UPR subsequent to
cell fusion, augmenting the traits of polytene cancers cells and
in vitro development. | (124) |
| Chen et al,
2022 |
|
| Suppressing ENTPD5
can inhibit the proliferation and migration of PSMA-positive cells,
and impede the activation of the GRP78/p-eIF-2α/CHOP pathway. | (125) |
| Barez et al,
2020 |
|
| It stimulates
intra-endoplasmic reticulum stress of cystathionine-12/3 and
Bax/Bcl-2 proteins, restraining the proliferation of cancerous
cells. | (126) |
| Zundell et
al, 2021 |
|
| Simultaneously, the
IRE1a/XBP1 pathway demonstrates therapeutic efficacy in ovarian
carcinoma har-boring ARID1A mutations. | (127) |
| Ma et al,
2019 |
|
| Hypoglycemia and
metformin induce apoptosis of malignant cells via endoplasmic
reticulum stress. | (128) |
| Lin et al,
2021 |
|
| Suppressing
over-expression of CARM1 exhibits efficacy in ovarian cancer
treatment. | (129) |
| Xiao et al,
2022 |
|
| The WEE1 inhibitor
AZD1775 triggers cell apoptosis, and the combination of AZD1775
with the IRE1α inhibitor MKC8866 synergistically exhibits
anticancer efficacy. | (130) |
| Zhang et al,
2019 |
|
| ANGII exhibits
inhibitory effects on endoplasmic reticulum stress and induces the
morphogenesis and migration of ovarian cancer spheroids. | (131) |
| Singla et
al, 2022 |
|
| Natural compounds
exhibit potent inhibitory properties against endoplasmic reticulum
stress. | (132) |
| Hong et al,
2022 |
|
| The mountain yam
flavonoids can catalyze endoplasmic reticulum stress. | (133) |
| Li et al,
2019 |
|
| Chiwanol I
molecules can induce the death of cancer cells by stimulating the
endoplasmic reticulum stress pathway. | (134) |
| Bae et al,
2020 |
|
| Fucosterol elicits
endoplasmic reticulum stress, which subsequently suppresses the
proliferation of cancer cells. | (135) |
| Zhu et al,
2023 |
|
| Two mechanisms have
been identified to synergistically counteract the resistance of
SKOV3/DDP cells against cisplatin. | (136) |
| Bae et al,
2020 |
|
| The phlorotannins
in Fucus evanescens can inhibit ovarian cancer cell
proliferation by regulating endo-plasmic reticulum stress. | (138) |
| Kim and Ko,
2021 |
|
| In traditional
Chinese medicine, JIO17 induces apoptosis of cancer cells. | (139) |
| Abdullah et
al, 2022 |
|
| The mechanism by
which endoplasmic reticulum stress triggers the release and embrace
of calreticulin in ovarian cancer cells has been scrutinized. | (140) |
| Bi et al,
2021 |
|
|
Methanesulfonylamine can initiate
extranuclear stress to consequently trigger immunogenic cell death
in ovarian cancer. | (142) |
| Lau et al,
2020 |
|
| Taxol elicits
mechanical stress within the endoplasmic reticulum, leading to
immunogenic cell death in malignant cells. | (143) |
| Song et al,
2018 |
|
| Endoplasmic
reticulum stress IRE1a/XBP1 could potentially influence the
antitumor immunity of ovarian cancer patients by manipulating T
cells. | (145) |
| Cao et al,
2019 |
|
| The evidence
underscores the capacity to release T cell-mediated antitumor
immunity by blocking Chop or endoplasmic reticulum stress. | (146) |
Mitochondrial autophagy and ovarian
cancer
Mitochondria are organelles involved in cellular
energy production and biosynthetic processes (91). Both their content of mtDNA and
expression levels of relevant mitochondrial mRNAs are altered
during tumor progression, including the development of ovarian
cancer. Investigations have suggested that suppressing
mitochondrial autophagy may effectively mitigate the disease
progression of cisplatin-resistant ovarian cancer (92,93).
The studies by Yu et al (96) demonstrated that inducing mutations
in the p62 UBA structural domain could regulate subcellular
localization of HK2 in A2780 ovarian cancer cells' mitochondria,
freeing PINK1/parkin-mediated mitochondrial autophagy to decrease
cell sensitivity to cisplatin. Similarly, Vianello et al
(94) observed an increase in the
content of the mitochondrial autophagy receptor BNIP3 in
cisplatin-resistant cells and in ovarian cancers resistant to
platinum chemotherapy. Additionally, Zhou et al (95) revealed that the potent potential of
a complex of glycosylated zinc (II)-cryptolepine could disrupt the
mitochondrial autophagy pathway and induce autophagy and apoptosis
in SKOV-3/DDP cells, highlighting its capability in developing
chemotherapeutic drugs against cisplatin-resistant SKOV-3/DDP
cells. Simultaneously, Yu et al (96) confirmed that ABT737, a potent
inhibitor of BCL2/BCLXL, could considerably elevate the
concentration of DRP1 in mitochondria, thus triggering the release
of Cytochrome C in mitochondria and their autophagy in SKOV3/DDP
cells. This finding underscores the suitability of targeting
antiapoptotic proteins of the BCL2 family as a novel therapeutic
strategy for treating patients with ovarian cancer with cisplatin
resistance.
Inducing mitochondrial autophagy may trigger the
process of programmed cell death in ovarian cancer cells at the
terminal stage. Chen et al (97) discovered that the antimicrobial
peptide pardaxin can stimulate PA-1 and SKOV3 cells to generate
excessive ROS. This surplus ROS accomplishes this by decreasing the
destruction of fusion proteins, declining the expression of
mitochondrial transporters 1/2 and L-/S-OPA1, inducing the
expression of fusion proteins DRP1 and FIS1 to enhance
mitochondrial fission rates, and at the same time initiating the
expression of autophagy-related proteins Beclin, p62 and LC3 to
stimulate the apoptotic pathway. Furthermore, Wang et al
(98) revealed that the
epoxidecolchicine H derived from the metabolism of Phomopsis
was capable of effectively increasing the ROS levels in cells,
weakening the mitochondrial membrane potential, thereby causing
damage to mitochondria, activating the mitochondrial autophagy
process. Concurrently, this compound is also able to mediate
apoptosis pathways associated with ER stress, further propelling
the apoptotic process of ovarian cancer A2780 cells (98). Katreddy et al (99) study identified that high malignant
epidermal growth factor receptors (EGFR) are frequently
overexpressed in solid tumors. The use of siRNA or synthetic EGFR
downregulating peptides (Herdegradin) can effectively downregulate
the expression of EGFR protein. Notably, this downregulation is
achieved by activating the mTORC2/Akt pathway, stimulating
selective mitochondrial autophagy, and achieving the selective
elimination of ovarian cancer cells (100).
Moderate suppression of mitochondrial autophagy may
restrain the tumor's metastatic capacity. Yuan et al
(101) indicated that mice exposed
to BPA/BPS are more susceptible to tumor metastasis, as BPA/BPS can
enhance the stem cell characteristics of ovarian cancer cells via a
non-normal PINK1/p53 mitochondrial autophagy process, and the
moderate suppression of mitochondrial autophagy can restrain this
augmentation effect.
Focusing on the mitochondrial autophagy, researchers
aim to elucidate novel targeted targets in ovarian cancer therapy.
Martinez-Outschoorn et al (102) have elucidated that BRCA1 tumor
suppressor gene deletion can instigate hydrogen peroxide
production, wherein the hydrogen peroxide produced by BRCA1-null
ovarian cancer cells stimulates NFB activation in stromal
fibroblasts, inducing oxidative stress and consequently triggering
mitochondrial autophagy and catabolic processes. Concurrently, MCT4
marker upregulation and Cav-1 expression loss also occur. Jin et
al (103) further discovered
that fucosylation of TGF-β1 augments Trk-like autophosphorylation
via the PI3K/Akt and Ras-Raf-MEK-ERK pathways, thereby enabling
ovarian cancer cell regulatory functions. This provides an
alternate research avenue for TGF-β1 targeting in ovarian cancer
therapy. Bae et al (104)
identified that campesterol influences mitochondrial function,
triggering excessive calcium accumulation and the creation of ROS,
thereby inflicting ER stress and thereby inhibiting ovarian cancer.
They observed in experimental settings that campesterol amplified
the efficacy of conventional anticancer drugs, presenting potential
as a novel drug for future treatment of ovarian cancer; however,
this enhanced anticancer effect requires further study.
In summary, mitochondrial autophagy plays a
substantial role in ovarian cancer therapy. It not only enhances
the sensitivity of ovarian cancer cells to chemotherapy drugs and
reduces drug resistance by inhibiting mitochondrial autophagy; but
also triggers mitochondrial autophagy to drive ovarian cancer cell
apoptosis. Moreover, manipulating mitochondrial dynamics, enhancing
mitochondrial autophagy, or prodigiously initiating ROS generation
and suppressing mitochondrial autophagy can ultimately lead to
ovarian cancer cell demise. Hence, regulating the mechanisms of
mitochondrial autophagy may be conjectured as a potential strategy
for ovarian cancer therapy.
ER stress and ovarian cancer
Based on the presence or absence of ribosomes at the
cisternal face of cell membranes, ER can be classified as RER and
SER. These constitute distinct spatial domains that are
nevertheless integrally related constructs (105). Moreover, the ER can also be
classified based on its membrane structure features. These
structures encompass the outer nuclear membrane, flared ER pools,
and a polygonal pipeline system composed of triangular junction
rings (106).
The ER lies in proximity to numerous other
organelles and boasts extensive volumes within cells, the magnitude
of which is contingent upon the type of cell. Notably, the
interaction with mitochondria merits special mention. Given the
pivotal role of mitochondria in organs (specifically their function
during apoptosis), this association assumes paramount importance
(107,108). In fact, this connection between
the ER and mitochondria is predominantly mediated by the MAM
(109). Additionally, MAM
synergizes with the cell membrane to maintain its stability and
growth. This interplay is controlled by Ca2+ levels and
several proteins, including stromal interaction molecule 1 located
in the ER and calcium release-activated calcium channel protein1
residing in the cell membrane (110). Notably, the ER appears to also
partake in autophagy processes, as observed by linking endocytic
vesicle systems (111). When
contacted with specific ER assemblies known as sub-Golgi bodies,
one of the core structures of autophagosomes-the phagophore will
proliferate and mature into a mature autophagosome (112,113).
Recent scientific literature from recent years
suggests that ER stress can significantly diminish the resistance
of ovarian cancer cells. Cheng et al (114) discovered in their research that
mitochondrial protease OMA1 not only triggers the morphogenesis of
mitochondrial structures by cleaving OPA1 but also significantly
reduces the drug resistance of ovarian cancer cells by integrating
ER stress through its interaction with the PHB2/OMA1/DELE1 pathway.
Additionally, Jung et al (115) identified that DPP23, functioning
as an initiator of ER stress, effectively combats ovarian cancer
resistance. Xu et al (116)
revealed that PHLDA1 fine-tunes the apoptotic sensitivity of
ovarian cancer cells via the ER stress response process. Kim and
Lee (117) corroborated that
6-gingerol induces ER stress in ovarian cancer cells and
successfully overcomes resistance to gefitinib, thus providing a
novel strategy for treating ovarian cancer using the combination of
gefitinib and 6-gingerol. Lastly, Bahar et al (118) suggested that the combination of
PARPis and cis-diminution of cis-OC cells may present an
efficacious method to broaden therapeutic potential in response to
ER stress, thereby overcoming platinum chemotherapy resistance in
OC and PARPi cross-resistance.
ER stress is progressively emerging as a novel
treatment approach for ovarian cancer (119). Rezghi et al (120) demonstrated using experimental
evidence that miR-3c-1-1p regulates XBP30/CHOP/BIM-mediated ER
stress, suppresses transcription of XBP2 in ovarian cancer cells
and consequently triggers the activation of the apoptosis pathway.
Kong et al (121) utilized
platinum (IV) prodrug and near infrared II (NIR II) photothermal
agent IR1048 as the foundation, generating enhanced ER stress
through formation of reduced-sensitive polymer nanoparticles to
amplify therapeutic effects against ovarian cancer. Wang et
al (122) found that during
their research, Erythropoietinaxin 2A9 (EpoxiDRESSIN H), was
capable of triggering apoptosis pathways related to mitochondria
and ER stress, thereby stimulating apoptosis of human ovarian
cancer A2780 cells further. Similarly, Wang et al (123) reported that DWP05195 could induce
ER stress via the ROS-p38-CHOP pathway in human ovarian cancer
cells, triggering cell apoptosis. Yart et al (124) discovered that activation of UPR by
cellular fusions increases the generation of polyploid multi-cancer
cells in vitro and bestows them with new attributes, and
also established that regulation of UPR in patients with ovarian
cancer could represent an intriguing and potent therapeutic
strategy. Chen et al (125)
efficiently suppressed proliferation and migration of prostate
specific membrane antigen (PSMA)-positive cells by inhibiting
ENTPD5 and at the same time, blocked the activation of
GRP78/p-eIF-2α/CHOP pathway, providing potential effective
therapeutic targets for investigating prostate cancer treatment.
Barez et al (126)
confirmed in the context of studying IRE1a inhibitor STF-083010
that suppressing caspases-12/3 and Bax/Bcl-2 proteins could
effectively inhibit ovarian cancer proliferation, revealing
STF-083010 as a novel potential therapeutic target for treating
ovarian cancer cells associated with ER stress. Moreover, in
addition to IRE1a inhibitors, Zundell et al (127) posited that IRE1a/XBP1 pathway
carries innovative therapeutic potential against ARID1A-mutated
ovarian cancers, providing scientific basis for drugs concurrently
utilizing the IRE1a/XBP1 pathway. Ma et al (128) identified that under low glucose
and metformin's effect, ANGII triggers inhibited ER stress,
inducing the formation and migration of ovarian cancer spheroids.
By contrast, Lin et al (129) found that this pathway had
comparable effects on ovarian cancer cells expressing CARM1 and
could achieve the therapeutic objective by decreasing overexpressed
CARM1. Additionally, Xiao et al (130) used WEE1 inhibitor AZD1775 to
stimulate PERK, thereby triggering apoptosis of ovarian cancer
cells, and in clinical practice, combined AZD1775 with IRE1α
inhibitor MKC8866 to achieve synergistic anti-ovarian cancer
effects. Zhang et al (131)
observed that ANGII regulates lipid desaturation, triggering
suppression of ER stress, resulting in the formation and migration
of ovarian cancer spheroids.
Singla et al (132) asserted that naturally occurring
compounds play a pivotal role in regulating ER stress, further
demonstrating that various such natural substances, such as
quercetin, curcumin and resveratrol, exhibit effective ER stress
inhibitor effects in the context of ovarian cancer. Hong et
al (133) employed the action
of mountain heteroflavone complex extracted from tricuspid fruit on
ovarian cancer cells, observing apoptosis of cancer cells;
subsequent studies revealed that mountain heteroflavone can
interfere with mitochondrial co-localization through inhibiting the
PI3K/AKT and MAPK pathways and initiate ER stress. Li et al
(134) identified cucurbitacin I
as inducing death of ovarian cancer cells via the ER stress
pathway. Bae et al (135)
discovered that fucosterol (present in algae) could induce
mitochondrial dysfunction and ER stress, thereby suppressing the
proliferation of ovarian cancer cells. Zhu et al (136) elucidated that naringin could
regulate the PI3K/AKT/mTOR signaling pathway to suppress SKOV3/DDP
cell autophagy, and also facilitate SKOV3/DDP cell apoptosis by
targeting ER stress; these two mechanisms synergistically
counteract tolerance to cisplatin exhibited by SKOV3/DDP cells.
This experimental outcome strongly supports the extensive
application prospects of natural compounds in treating ovarian
cancer therapies (137). Bae et
al (138) discerned the
regulatory effect of fucosanglioside in maqueigao (Centricellularis
longicaudata), a natural compound, on ER stress to restrain the
proliferation of ovarian cancer cells. Kim and Ko (139) also posited that JIO17 from
traditional Chinese medicine can induce ovarian cancer cell
apoptosis through the NOX4/PERK/CHOP pathway.
Abdullah et al (140) investigated the mechanism by which
ER stress induces release and binding of calreticulin in ovarian
cancer cells, hypothesizing that calreticulin has a profound
connection with immunogenic cell death in ovarian cancer cells;
meanwhile, existing experiments have confirmed the value of
calreticulin in prognostic evaluation and prediction (141), and future research remains
necessary. Bi et al (142)
triggered immunogenic cell death in ovarian cancer through
initiating ER stress using a mitochondrial uncoupling agent
benzylamine, and remarkably inhibited tumor progression.
Separately, Lau et al (143) discovered that paclitaxel triggers
generation of ER stress through the TLR4/IKK2/SNARE pathway,
leading to immunogenic cell death in ovarian cancer cells,
suggesting a higher occurrence rate of immunogenic cell death in
ovarian cancer and therefore possessing significant research
significance; the importance of CALR as a crucial indicator for
paclitaxel chemotherapy in ovarian cancer was also underscored.
Beyond addressing mortality, the immune system also
plays a significant role in tumor cells (144,145). For instance, Song et al
(145) discovered that ER stress
IRE1a/XBP1 potentially influences T cell metabolic adaptability and
antitumor potency, thereby influencing the antitumor immunity of
patients with ovarian cancer, providing a novel therapeutic
approach in immunotherapy. Furthermore, Cao et al (146) elucidated the central function of
Chop in tumor-induced dysfunction of CD8 T cells, and the potential
for therapeutic efficacy to release T cell-mediated antitumor
immunity through blocking Chop or ER stress.
Conclusions
In light of the comprehensive factors, MAM carves a
unique functional role as a link between the mitochondria and ER.
It exhibits superior performance during mitochondrial autophagy and
ER stress response. It is capable of sequentially regulating these
two crucial processes to suppress the malignant proliferation,
migration and drug resistance ability of ovarian cancer. Notably,
its intrinsic regulatory mechanism implicates numerous proteins,
some of which may precisely be key players in a certain disease's
progression. Consequently, meticulously exploring its potential
application value and vast research prospects warrants more time
and effort. Nonetheless, the present review elucidated the
correlation between mitochondrial autophagy, ER stress and MAMs,
with an expectation of conducting more thorough research on this
topic in subsequent efforts. Furthermore, given that MAMs have not
been extensively validated in ovarian cancer-related studies, the
integrated analysis related to ovarian cancer outcomes remains
relatively thin. It is expected that in future research endeavors,
such research can be more profoundly conducted.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Hunan Provincial Nature
Foundation (2022JJ30035), Key Guiding Project of Hunan Provincial
Health Commission (202305017379), Key Project of Hunan Provincial
Health Commission (20230589), National College Student Innovation
and Entrepreneurship Program Training Project (S202310541063),
Hunan College Student Innovation (S202310541063) and
Entrepreneurship Program Training Project (S202310541063).
Availability of data and materials
Not applicable.
Authors' contributions
JHZ and YLX were involved in the writing of the
original manuscript. YFD and JZ completed the original manuscript.
HPL completed manuscript revisions. WJP was involved in drawing the
diagram. All authors read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Copeland DE and Dalton AJ: An association
between mitochondria and the endoplasmic reticulum in cells of the
pseudobranch gland of a teleost. J Biophys Biochem Cytol.
5:393–396. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rizzuto R, Pinton P, Carrington W, Fay FS,
Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T: Close contacts with
the endoplasmic reticulum as determinants of mitochondrial Ca2+
responses. Science. 280:1763–1766. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu H, Carvalho P and Voeltz GK: Here,
there, and everywhere: The importance of ER membrane contact sites.
Science. 361:eaan58352018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lev S: Nonvesicular lipid transfer from
the endoplasmic reticulum. Cold Spring Harb Perspect Biol.
4:a0133002012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoppins S and Nunnari J: Cell biology.
Mitochondrial dynamics and apoptosis-the ER connection. Science.
337:1052–1054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Belosludtsev KN, Dubinin MV, Belosludtseva
NV and Mironova GD: Mitochondrial Ca2+ transport: Mechanisms,
molecular structures, and role in cells. Biochemistry (Mosc).
84:593–607. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Filadi R, Greotti E, Turacchio G, Luini A,
Pozzan T and Pizzo P: On the role of mitofusin 2 in endoplasmic
reticulum-mitochondria tethering. Proc Natl Acad Sci USA.
114:E2266–E2267. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gibellini F and Smith TK: The Kennedy
pathway-de novo synthesis of phosphatidylethanolamine and
phosphatidylcholine. IUBMB Life. 62:414–428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Puglielli L, Konopka G, Pack-Chung E,
Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE and Kovacs
DM: Acyl-coenzyme A: Cholesterol acyltransferase modulates the
generation of the amyloid beta-peptide. Nat Cell Biol. 3:905–912.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
El Alwani M, Wu BX, Obeid LM and Hannun
YA: Bioactive sphingolipids in the modulation of the inflammatory
response. Pharmacol Ther. 112:171–183. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nikolova-Karakashian M, Karakashian A and
Rutkute K: Role of neutral sphingomyelinases in aging and
inflammation. Subcell Biochem. 49:469–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedman JR, Lackner LL, West M,
DiBenedetto JR, Nunnari J and Voeltz GK: ER tubules mark sites of
mitochondrial division. Science. 334:358–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murley A, Sarsam RD, Toulmay A, Yamada J,
Prinz WA and Nunnari J: Ltc1 is an ER-localized sterol transporter
and a component of ER-mitochondria and ER-vacuole contacts. J Cell
Biol. 209:539–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ishihara N, Eura Y and Mihara K: Mitofusin
1 and 2 play distinct roles in mitochondrial fusion reactions via
GTPase activity. J Cell Sci. 117:6535–6546. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ainbinder A, Boncompagni S, Protasi F and
Dirksen RT: Role of mitofusin-2 in mitochondrial localization and
calcium uptake in skeletal muscle. Cell Calcium. 57:14–24. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Basso V, Marchesan E, Peggion C,
Chakraborty J, von Stockum S, Giacomello M, Ottolini D, Debattisti
V, Caicci F, Tasca E, et al: Regulation of ER-mitochondria contacts
by Parkin via Mfn2. Pharmacol Res. 138:43–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okamoto K and Shaw JM: Mitochondrial
morphology and dynamics in yeast and multicellular eukaryotes. Annu
Rev Genet. 39:503–536. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Galluzzi L, Kepp O, Trojel-Hansen C and
Kroemer G: Mitochondrial control of cellular life, stress, and
death. Circ Res. 111:1198–1207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwasawa R, Mahul-Mellier AL, Datler C,
Pazarentzos E and Grimm S: Fis1 and Bap31 bridge the
mitochondria-ER interface to establish a platform for apoptosis
induction. EMBO J. 30:556–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Area-Gomez E and Schon EA: On the
pathogenesis of Alzheimer's disease: The MAM hypothesis. FASEB J.
31:864–867. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang B, Nguyen M, Chang NC and Shore GC:
Fis1, Bap31 and the kiss of death between mitochondria and
endoplasmic reticulum. EMBO J. 30:451–452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
MacAskill AF and Kittler JT: Control of
mitochondrial transport and localization in neurons. Trends Cell
Biol. 20:102–112. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Upton JP, Austgen K, Nishino M, Coakley
KM, Hagen A, Han D, Papa FR and Oakes SA: Caspase-2 cleavage of BID
is a critical apoptotic signal downstream of endoplasmic reticulum
stress. Mol Cell Biol. 28:3943–3951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Puthalakath H, O'Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li J, Lee B and Lee AS: Endoplasmic
reticulum stress-induced apoptosis: Multiple pathways and
activation of p53-up-regulated modulator of apoptosis (PUMA) and
NOXA by p53. J Biol Chem. 281:7260–7270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iwasawa R, Mahul-Mellier AL, Datler C,
Pazarentzos E and Grimm S: Fis1 and Bap31 bridge the
mitochondria-ER interface to establish a platform for apoptosis
induction. EMBO J. 30:556–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Area-Gomez E and Schon EA: On the
pathogenesis of Alzheimer's disease: The MAM hypothesis. FASEB J.
31:864–867. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang B, Nguyen M, Chang NC and Shore GC:
Fis1, Bap31 and the kiss of death between mitochondria and
endoplasmic reticulum. EMBO J. 30:451–452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siskind LJ, Kolesnick RN and Colombini M:
Ceramide channels increase the permeability of the mitochondrial
outer membrane to small proteins. J Biol Chem. 277:26796–26803.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Coleman RA, Lewin TM, Van Horn CG and
Gonzalez-Baró MR: Do long-chain acyl-CoA synthetases regulate fatty
acid entry into synthetic versus degradative pathways? J Nutr.
132:2123–2126. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Larsen BD and Sørensen CS: The
caspase-activated DNase: Apoptosis and beyond. FEBS J.
284:1160–1170. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
de Brito OM and Scorrano L: Mitofusin 2
tethers endoplasmic reticulum to mitochondria. Nature. 456:605–610.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mazure NM: VDAC in cancer. Biochim Biophys
Acta Bioenerg. 1858:665–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vinay Kumar C, Kumar KM, Swetha R, Ramaiah
S and Anbarasu A: Protein aggregation due to nsSNP resulting in
P56S VABP protein is associated with amyotrophic lateral sclerosis.
J Theor Biol. 354:72–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Formosa LE and Ryan MT: Mitochondrial
fusion: Reaching the end of mitofusin's tether. J Cell Biol.
215:597–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Di Mattia T, Wilhelm LP, Ikhlef S,
Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G,
Tomasetto C and Alpy F: Identification of MOSPD2, a novel scaffold
for endoplasmic reticulum membrane contact sites. EMBO Rep.
19:e454532018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lim Y, Cho IT, Schoel LJ, Cho G and Golden
JA: Hereditary spastic paraplegia-linked REEP1 modulates
endoplasmic reticulum/mitochondria contacts. Ann Neurol.
78:679–696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Calì T, Ottolini D, Negro A and Brini M:
α-Synuclein controls mitochondrial calcium homeostasis by enhancing
endoplasmic reticulum-mitochondria interactions. J Biol Chem.
287:17914–17929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu Y, Ma X, Fujioka H, Liu J, Chen S and
Zhu X: DJ-1 regulates the integrity and function of ER-mitochondria
association through interaction with IP3R3-Grp75-VDAC1. Proc Natl
Acad Sci USA. 116:25322–25328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stoica R, Paillusson S, Gomez-Suaga P,
Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay-Barrena G, De Vos
KJ, Shaw CE, et al: ALS/FTD-associated FUS activates GSK-3β to
disrupt the VAPB-PTPIP51 interaction and ER-mitochondria
associations. EMBO Rep. 17:1326–1342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thoudam T, Ha CM, Leem J, Chanda D, Park
JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, et al: PDK4
augments ER-mitochondria contact to dampen skeletal muscle insulin
signaling during obesity. Diabetes. 68:571–586. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
D'Eletto M, Rossin F, Occhigrossi L,
Farrace MG, Faccenda D, Desai R, Marchi S, Refolo G, Falasca L,
Antonioli M, et al: Transglutaminase type 2 regulates
ER-mitochondria contact sites by interacting with GRP75. Cell Rep.
25:3573–3581.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X,
Huang K, Xie Z and Zou MH: Binding of FUN14 domain containing 1
with inositol 1,4,5-trisphosphate receptor in
mitochondria-associated endoplasmic reticulum membranes maintains
mitochondrial dynamics and function in hearts in vivo. Circulation.
136:2248–2266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang W, Siraj S, Zhang R and Chen Q:
Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and
protects the heart from I/R injury. Autophagy. 13:1080–1081. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kuang Y, Ma K, Zhou C, Ding P, Zhu Y, Chen
Q and Xia B: Structural basis for the phosphorylation of FUNDC1 LIR
as a molecular switch of mitophagy. Autophagy. 12:2363–2373. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li
Y, Han Z, Chen L, Gao R, Liu L and Chen Q: Mitophagy receptor
FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy.
12:689–702. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu W, Li W, Chen H, Jiang L, Zhu R and
Feng D: FUNDC1 is a novel mitochondrial-associated-membrane (MAM)
protein required for hypoxia-induced mitochondrial fission and
mitophagy. Autophagy. 12:1675–1676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang XL, Feng ST, Wang YT, Yuan YH, Li ZP,
Chen NH, Wang ZZ and Zhang Y: Mitophagy, a form of selective
autophagy, plays an essential role in mitochondrial dynamics of
Parkinson's disease. Cell Mol Neurobiol. 42:1321–1339. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gong Y, Luo Y, Liu S, Ma J, Liu F, Fang Y,
Cao F, Wang L, Pei Z and Ren J: Pentacyclic triterpene oleanolic
acid protects against cardiac aging through regulation of mitophagy
and mitochondrial integrity. Biochim Biophys Acta Mol Basis Dis.
1868:1664022022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu
S, Ren J and Chen Y: NR4A1 aggravates the cardiac microvascular
ischemia reperfusion injury through suppressing FUNDC1-mediated
mitophagy and promoting Mff-required mitochondrial fission by CK2α.
Basic Res Cardiol. 113:232018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Simmen T, Aslan JE, Blagoveshchenskaya AD,
Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM and
Thomas G: PACS-2 controls endoplasmic reticulum-mitochondria
communication and Bid-mediated apoptosis. EMBO J. 24:717–729. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Werneburg NW, Bronk SF, Guicciardi ME,
Thomas L, Dikeakos JD, Thomas G and Gores GJ: Tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) protein-induced
lysosomal translocation of proapoptotic effectors is mediated by
phosphofurin acidic cluster sorting protein-2 (PACS-2). J Biol
Chem. 287:24427–24437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Köttgen M, Benzing T, Simmen T, Tauber R,
Buchholz B, Feliciangeli S, Huber TB, Schermer B, Kramer-Zucker A,
Höpker K, et al: Trafficking of TRPP2 by PACS proteins represents a
novel mechanism of ion channel regulation. EMBO J. 24:705–716.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Myhill N, Lynes EM, Nanji JA,
Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G
and Simmen T: The subcellular distribution of calnexin is mediated
by PACS-2. Mol Biol Cell. 19:2777–2788. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Han S, Zhao F, Hsia J, Ma X, Liu Y, Torres
S, Fujioka H and Zhu X: The role of Mfn2 in the structure and
function of endoplasmic reticulum-mitochondrial tethering in vivo.
J Cel Sci. 134:jcs2534432021. View Article : Google Scholar
|
|
58
|
Leal NS, Schreiner B, Pinho CM, Filadi R,
Wiehager B, Karlström H, Pizzo P and Ankarcrona M: Mitofusin-2
knockdown increases ER-mitochondria contact and decreases amyloid
β-peptide production. J Cel Mol Med. 20:1686–1695. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li J, Qi F, Su H, Zhang C, Zhang Q, Chen
Y, Chen P, Su L, Chen Y, Yang Y, et al: GRP75-faciliated
mitochondria-associated ER membrane (MAM) integrity controls
cisplatin-resistance in ovarian cancer patients. Int J Biol Sci.
18:2914–2931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Barroso-González J, Auclair S, Luan S,
Thomas L, Atkins KM, Aslan JE, Thomas LL, Zhao J, Zhao Y and Thomas
G: PACS-2 mediates the ATM and NF-κB-dependent induction of
anti-apoptotic Bcl-xL in response to DNA damage. Cell Death Differ.
23:1448–1457. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhou H, Zhu P, Wang J, Toan S and Ren J:
DNA-PKcs promotes alcohol-related liver disease by activating
Drp1-related mitochondrial fission and repressing FUNDC1-required
mitophagy. Signal Transduct Target Ther. 4:562019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Filadi R, Greotti E, Turacchio G, Luini A,
Pozzan T and Pizzo P: Mitofusin 2 ablation increases endoplasmic
reticulum-mitochondria coupling. Proc Natl Acad Sci USA.
112:E2174–E2181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li J, Qi F, Su H, Zhang C, Zhang Q, Chen
Y, Chen P, Su L, Chen Y, Yang Y, et al: GRP75-faciliated
mitochondria-associated ER membrane (MAM) integrity controls
cisplatin-resistance in ovarian cancer patients. Int J Biol Sci.
18:2914–2931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen Y and Dorn GW II:
PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling
damaged mitochondria. Science. 340:471–475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cao Y, Chen Z, Hu J, Feng J, Zhu Z, Fan Y,
Lin Q and Ding G: Mfn2 regulates high glucose-induced MAMs
dysfunction and apoptosis in podocytes via PERK pathway. Front Cell
Dev Biol. 9:7692132021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Modi S, López-Doménech G, Halff EF,
Covill-Cooke C, Ivankovic D, Melandri D, Arancibia-Cárcamo IL,
Burden JJ, Lowe AR and Kittler JT: Miro clusters regulate
ER-mitochondria contact sites and link cristae organization to the
mitochondrial transport machinery. Nat Commun. 10:43992019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hernández-Alvarez MI, Sebastián D, Vives
S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR,
Hernández V, Vasconcelos N, et al: Deficient endoplasmic
reticulum-mitochondrial phosphatidylserine transfer causes liver
disease. Cell. 177:881–895. e172019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Baker N, Patel J and Khacho M: Linking
mitochondrial dynamics, cristae remodeling and supercomplex
formation: How mitochondrial structure can regulate bioenergetics.
Mitochondrion. 49:259–268. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Glancy B, Kim Y, Katti P and Willingham
TB: The functional impact of mitochondrial structure across
subcellular scales. Front Physiol. 11:5410402020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gutiérrez T and Simmen T: Endoplasmic
reticulum chaperones tweak the mitochondrial calcium rheostat to
control metabolism and cell death. Cell Calcium. 70:64–75. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rouzier C, Bannwarth S, Chaussenot A,
Chevrollier A, Verschueren A, Bonello-Palot N, Fragaki K, Cano A,
Pouget J, Pellissier JF, et al: The MFN2 gene is responsible for
mitochondrial DNA instability and optic atrophy ‘plus’ phenotype.
Brain. 135:23–34. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Vielhaber S, Debska-Vielhaber G, Peeva V,
Schoeler S, Kudin AP, Minin I, Schreiber S, Dengler R, Kollewe K,
Zuschratter W, et al: Mitofusin 2 mutations affect mitochondrial
function by mitochondrial DNA depletion. Acta Neuropathol.
125:245–256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kawalec M, Boratyńska-Jasińska A,
Beręsewicz M, Dymkowska D, Zabłocki K and Zabłocka B: Mitofusin 2
deficiency affects energy metabolism and mitochondrial biogenesis
in MEF cells. PLoS One. 10:e01341622015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Parys JB and Vervliet T: New insights in
the IP3 receptor and its regulation. Adv Exp Med Biol.
1131:243–270. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mazure NM: VDAC in cancer. Biochim Biophys
Acta Bioenerg. 1858:665–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Szabadkai G, Bianchi K, Várnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rapizzi E, Pinton P, Szabadkai G,
Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE and
Rizzuto R: Recombinant expression of the voltage-dependent anion
channel enhances the transfer of Ca2+ microdomains to mitochondria.
J Cell Biol. 159:613–624. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tubbs E, Theurey P, Vial G, Bendridi N,
Bravard A, Chauvin MA, Ji-Cao J, Zoulim F, Bartosch B, Ovize M, et
al: Mitochondria-associated endoplasmic reticulum membrane (MAM)
integrity is required for insulin signaling and is implicated in
hepatic insulin resistance. Diabetes. 63:3279–3294. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Honrath B, Metz I, Bendridi N, Rieusset J,
Culmsee C and Dolga AM: Glucose-regulated protein 75 determines
ER-mitochondrial coupling and sensitivity to oxidative stress in
neuronal cells. Cell Death Discov. 3:170762017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Thoudam T, Ha CM, Leem J, Chanda D, Park
JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, et al: PDK4
augments ER-mitochondria contact to dampen skeletal muscle insulin
signaling during obesity. Diabetes. 68:571–586. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Vinay Kumar C, Kumar KM, Swetha R, Ramaiah
S and Anbarasu A: Protein aggregation due to nsSNP resulting in
P56S VABP protein is associated with amyotrophic lateral sclerosis.
J Theor Biol. 354:72–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kanekura K, Nishimoto I, Aiso S and
Matsuoka M: Characterization of amyotrophic lateral
sclerosis-linked P56S mutation of vesicle-associated membrane
protein-associated protein B (VAPB/ALS8). J Biol Chem.
281:30223–30233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
De Vos KJ, Mórotz GM, Stoica R, Tudor EL,
Lau KF, Ackerley S, Warley A, Shaw CE and Miller CC: VAPB interacts
with the mitochondrial protein PTPIP51 to regulate calcium
homeostasis. Hum Mol Genet. 21:1299–1311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Stoica R, De Vos KJ, Paillusson S, Mueller
S, Sancho RM, Lau KF, Vizcay-Barrena G, Lin WL, Xu YF, Lewis J, et
al: ER-mitochondria associations are regulated by the VAPB-PTPIP51
interaction and are disrupted by ALS/FTD-associated TDP-43. Nat
Commun. 5:39962014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao
Y, Xu C, Zhao L, Zhu Y, Wang L and Zheng M: PTPIP51 regulates mouse
cardiac ischemia/reperfusion through mediating the mitochondria-SR
junction. Sci Rep. 7:453792017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Di Mattia T, Wilhelm LP, Ikhlef S,
Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G,
Tomasetto C and Alpy F: Identification of MOSPD2, a novel scaffold
for endoplasmic reticulum membrane contact sites. EMBO Rep.
19:e454532018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Szado T, Vanderheyden V, Parys JB, De
Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U,
Söderberg O, et al: Phosphorylation of inositol 1,4,5-trisphosphate
receptors by protein kinase B/Akt inhibits Ca2+ release and
apoptosis. Proc Natl Acad Sci USA. 105:2427–2432. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Prevarskaya N, Ouadid-Ahidouch H, Skryma R
and Shuba Y: Remodelling of Ca2+ transport in cancer: How it
contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol
Sci. 369:201300972014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Monteith GR, Prevarskaya N and
Roberts-Thomson SJ: The calcium-cancer signalling nexus. Nat Rev
Cancer. 17:367–380. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li J, Qi F, Su H, Zhang C, Zhang Q, Chen
Y, Chen P, Su L, Chen Y, Yang Y, et al: GRP75-faciliated
mitochondria-associated ER membrane (MAM) integrity controls
cisplatin-resistance in ovarian cancer patients. Int J Biol Sci.
18:2914–2931. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lim Y, Cho IT, Schoel LJ, Cho G and Golden
JA: Hereditary spastic paraplegia-linked REEP1 modulates
endoplasmic reticulum/mitochondria contacts. Ann Neurol.
78:679–696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zampieri LX, Grasso D, Bouzin C, Brusa D,
Rossignol R and Sonveaux P: Mitochondria participate in
chemoresistance to cisplatin in human ovarian cancer cells. Mol
Cancer Res. 18:1379–1391. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chang CM, Lan KL, Huang WS, Lee YJ, Lee
TW, Chang CH and Chuang CM: 188Re-liposome can induce
mitochondrial autophagy and reverse drug resistance for ovarian
cancer: From bench evidence to preliminary clinical
proof-of-concept. Int J Mol Sci. 18:9032017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Vianello C, Cocetta V, Catanzaro D, Dorn
GW II, De Milito A, Rizzolio F, Canzonieri V, Cecchin E, Roncato R,
Toffoli G, et al: Cisplatin resistance can be curtailed by blunting
Bnip3-mediated mitochondrial autophagy. Cell Death Dis. 13:3982022.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhou Z, Du LQ, Huang XM, Zhu LG, Wei QC,
Qin QP and Bian H: Novel glycosylation zinc(II)-cryptolepine
complexes perturb mitophagy pathways and trigger cancer cell
apoptosis and autophagy in SK-OV-3/DDP cells. Eur J Med Chem.
243:1147432022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Yu Y, Xu L, Qi L, Wang C, Xu N, Liu S, Li
S, Tian H, Liu W, Xu Y and Li Z: ABT737 induces mitochondrial
pathway apoptosis and mitophagy by regulating DRP1-dependent
mitochondrial fission in human ovarian cancer cells. Biomed
Pharmacother. 96:22–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chen YP, Shih PC, Feng CW, Wu CC, Tsui KH,
Lin YH, Kuo HM and Wen ZH: Pardaxin activates excessive mitophagy
and mitochondria-mediated apoptosis in human ovarian cancer by
inducing reactive oxygen species. Antioxidants (Basel).
10:18832021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang J, Xu Z, Hu X, Yang Y, Su J, Liu Y,
Zhou L, Qin J, Zhang D and Yu H: Epoxycytochalasin H: An endophytic
phomopsis compound induces apoptosis in A2780 cells through
mitochondrial damage and endoplasmic reticulum stress. Onco Targets
Ther. 13:4987–4997. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Katreddy RR, Bollu LR, Su F, Xian N,
Srivastava S, Thomas R, Dai Y, Wu B, Xu Y, Rea MA, et al: Targeted
reduction of the EGFR protein, but not inhibition of its kinase
activity, induces mitophagy and death of cancer cells through
activation of mTORC2 and Akt. Oncogenesis. 7:52018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Meng Y, Qiu L, Zeng X, Hu X, Zhang Y, Wan
X, Mao X, Wu J, Xu Y, Xiong Q, et al: Targeting CRL4 suppresses
chemoresistant ovarian cancer growth by inducing mitophagy. Signal
Transduct Target Ther. 7:3882022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yuan X, Chen K, Zheng F, Xu S, Li Y, Wang
Y, Ni H, Wang F, Cui Z, Qin Y, et al: Low-dose BPA and its
substitute BPS promote ovarian cancer cell stemness via a
non-canonical PINK1/p53 mitophagic signaling. J Hazard Mater.
452:1312882023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Martinez-Outschoorn UE, Balliet RM, Lin Z,
Whitaker-Menezes D, Howell A, Sotgia F and Lisanti MP: Hereditary
ovarian cancer and two-compartment tumor metabolism: Epithelial
loss of BRCA1 induces hydrogen peroxide production, driving
oxidative stress and NFκB activation in the tumor stroma. Cell
Cycle. 11:4152–4166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jin S, Gao J, Qi Y, Hao Y, Li X, Liu Q,
Liu J, Liu D, Zhu L and Lin B: TGF-β1 fucosylation enhances the
autophagy and mitophagy via PI3K/Akt and Ras-Raf-MEK-ERK in ovarian
carcinoma. Biochem Biophys Res Commun. 524:970–976. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bae H, Park S, Yang C, Song G and Lim W:
Disruption of endoplasmic reticulum and ROS production in human
ovarian cancer by campesterol. Antioxidants (Basel). 10:3792021.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Borgese N, Francolini M and Snapp E:
Endoplasmic reticulum architecture: Structures in flux. Curr Opin
Cell Biol. 18:358–364. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shibata Y, Voeltz GK and Rapoport TA:
Rough sheets and smooth tubules. Cell. 126:435–439. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lossi L: The concept of intrinsic versus
extrinsic apoptosis. Biochem J. 479:357–384. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hayashi T, Rizzuto R, Hajnoczky G and Su
TP: MAM: More than just a housekeeper. Trends Cell Biol. 19:81–88.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Toulmay A and Prinz WA: Lipid transfer and
signaling at organelle contact sites: The tip of the iceberg. Curr
Opin Cell Biol. 23:458–463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Henne WM, Zhu L, Balogi Z, Stefan C,
Pleiss JA and Emr SD: Mdm1/Snx13 is a novel ER-endolysosomal
interorganelle tethering protein. J Cell Biol. 210:541–551. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hayashi-Nishino M, Fujita N, Noda T,
Yamaguchi A, Yoshimori T and Yamamoto A: Electron tomography
reveals the endoplasmic reticulum as a membrane source for
autophagosome formation. Autophagy. 6:301–303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Uemura T, Yamamoto M, Kametaka A, Sou YS,
Yabashi A, Yamada A, Annoh H, Kametaka S, Komatsu M and Waguri S: A
cluster of thin tubular structures mediates transformation of the
endoplasmic reticulum to autophagic isolation membrane. Mol Cell
Biol. 34:1695–1706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cheng M, Yu H, Kong Q, Wang B, Shen L,
Dong D and Sun L: The mitochondrial PHB2/OMA1/DELE1 pathway
cooperates with endoplasmic reticulum stress to facilitate the
response to chemotherapeutics in ovarian cancer. Int J Mol Sci.
23:13202022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jung E, Koh D, Lim Y, Shin SY and Lee YH:
Overcoming multidrug resistance by activating unfolded protein
response of the endoplasmic reticulum in cisplatin-resistant
A2780/CisR ovarian cancer cells. BMB Rep. 53:88–93. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Xu J, Bi G, Luo Q, Liu Y, Liu T, Li L,
Zeng Q, Wang Q, Wang Y, Yu J and Yi P: PHLDA1 modulates the
endoplasmic reticulum stress response and is required for
resistance to oxidative stress-induced cell death in human ovarian
cancer cells. J Cancer. 12:5486–5493. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kim TW and Lee HG: 6-Shogaol overcomes
gefitinib resistance via ER stress in ovarian cancer cells. Int J
Mol Sci. 24:26392023. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bahar E, Kim JY, Kim DC, Kim HS and Yoon
H: Combination of niraparib, cisplatin and twist knockdown in
cisplatin-resistant ovarian cancer cells potentially enhances
synthetic lethality through ER-stress mediated mitochondrial
apoptosis pathway. Int J Mol Sci. 22:39162021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhang Y, Wang Y, Zhao G, Orsulic S and
Matei D: Metabolic dependencies and targets in ovarian cancer.
Pharmacol Ther. 245:1084132023. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Rezghi Barez S, Movahedian Attar A and
Aghaei M: MicroRNA-30c-2-3p regulates ER stress and induces
apoptosis in ovarian cancer cells underlying ER stress. EXCLI J.
20:922–934. 2021.PubMed/NCBI
|
|
121
|
Kong Q, Wei D, Xie P, Wang B, Yu K, Kang X
and Wang Y: Photothermal therapy via NIR II light irradiation
enhances DNA damage and endoplasmic reticulum stress for efficient
chemotherapy. Front Pharmacol. 12:6702072021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wang J, Xu Z, Hu X, Yang Y, Su J, Liu Y,
Zhou L, Qin J, Zhang D and Yu H: Epoxycytochalasin H: An endophytic
phomopsis compound induces apoptosis in A2780 cells through
mitochondrial damage and endoplasmic reticulum stress. Onco Targets
Ther. 13:4987–4997. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wang YY, Lee KT, Lim MC and Choi JH: TRPV1
antagonist DWP05195 induces ER stress-dependent apoptosis through
the ROS-p38-CHOP pathway in human ovarian cancer cells. Cancers
(Basel). 12:17022020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Yart L, Bastida-Ruiz D, Allard M, Dietrich
PY, Petignat P and Cohen M: Linking unfolded protein response to
ovarian cancer cell fusion. BMC Cancer. 22:6222022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chen X, Zha Z, Wang Y and Chen Y, Pang M,
Huang L and Chen Y: Knockdown of ENTPD5 inhibits tumor metastasis
and growth via regulating the GRP78/p-eIF-2α/CHOP pathway in serous
ovarian cancer. J Ovarian Res. 15:692022. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Barez SR, Atar AM and Aghaei M: Mechanism
of inositol-requiring enzyme 1-alpha inhibition in endoplasmic
reticulum stress and apoptosis in ovarian cancer cells. J Cell
Commun Signal. 14:403–415. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zundell JA, Fukumoto T, Lin J, Fatkhudinov
N, Nacarelli T, Kossenkov AV, Liu Q, Cassel J, Hu CA, Wu S and
Zhang R: Targeting the IRE1α/XBP1 endoplasmic reticulum stress
response pathway in ARID1A-mutant ovarian cancers. Cancer Res.
81:5325–5335. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Ma L, Wei J, Wan J, Wang W, Wang L, Yuan
Y, Yang Z, Liu X and Ming L: Low glucose and metformin-induced
apoptosis of human ovarian cancer cells is connected to ASK1 via
mitochondrial and endoplasmic reticulum stress-associated pathways.
J Exp Clin Cancer Res. 38:772019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Lin J, Liu H, Fukumoto T, Zundell J, Yan
Q, Tang CA, Wu S, Zhou W, Guo D, Karakashev S, et al: Targeting the
IRE1α/XBP1s pathway suppresses CARM1-expressing ovarian cancer. Nat
Commun. 12:53212021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Xiao R, You L, Zhang L, Guo X, Guo E, Zhao
F, Yang B, Li X, Fu Y, Lu F, et al: Inhibiting the IRE1α axis of
the unfolded protein response enhances the antitumor effect of
AZD1775 in TP53 mutant ovarian cancer. Adv Sci (Weinh).
9:e21054692022. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhang Q, Yu S, Lam MMT, Poon TCW, Sun L,
Jiao Y, Wong AST and Lee LTO: Angiotensin II promotes ovarian
cancer spheroid formation and metastasis by upregulation of lipid
desaturation and suppression of endoplasmic reticulum stress. J Exp
Clin Cancer Res. 38:1162019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Singla RK, Sharma P, Kumar D, Gautam RK,
Goyal R, Tsagkaris C, Dubey AK, Bansal H, Sharma R and Shen B: The
role of nanomaterials in enhancing natural product translational
potential and modulating endoplasmic reticulum stress in the
treatment of ovarian cancer. Front Pharmacol. 13:9870882022.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Hong T, Ham J, Song G and Lim W:
Alpinumisoflavone disrupts endoplasmic reticulum and mitochondria
leading to apoptosis in human ovarian cancer. Pharmaceutics.
14:5642022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Li H, Chen H, Li R, Xin J, Wu S, Lan J,
Xue K, Li X, Zuo C, Jiang W and Zhu L: Cucurbitacin I induces
cancer cell death through the endoplasmic reticulum stress pathway.
J Cell Biochem. 120:2391–2403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Bae H, Lee JY, Song G and Lim W:
Fucosterol suppresses the progression of human ovarian cancer by
inducing mitochondrial dysfunction and endoplasmic reticulum
stress. Mar Drugs. 18:2612020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhu J, Lin S, Zou X, Chen X, Liu Y, Yang
X, Gao J and Zhu H: Mechanisms of autophagy and endoplasmic
reticulum stress in the reversal of platinum resistance of
epithelial ovarian cancer cells by naringin. Mol Biol Rep.
50:6457–6468. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Zhao Q, Peng C, Zheng C, He XH, Huang W
and Han B: Recent advances in characterizing natural products that
regulate autophagy. Anticancer Agents Med Chem. 19:2177–2196. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Bae H, Lee JY, Yang C, Song G and Lim W:
Fucoidan derived from fucus vesiculosus inhibits the development of
human ovarian cancer via the disturbance of calcium homeostasis,
endoplasmic reticulum stress, and angiogenesis. Mar Drugs.
18:452020. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kim T and Ko SG: JI017, a complex herbal
medication, induces apoptosis via the Nox4-PERK-CHOP axis in
ovarian cancer cells. Int J Mol Sci. 22:122642021. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Abdullah TM, Whatmore J, Bremer E,
Slibinskas R, Michalak M and Eggleton P: Endoplasmic reticulum
stress-induced release and binding of calreticulin from human
ovarian cancer cells. Cancer Immunol Immunother. 71:1655–1669.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kielbik M, Szulc-Kielbik I and Klink M:
Calreticulin-multifunctional chaperone in immunogenic cell death:
Potential significance as a prognostic biomarker in ovarian cancer
patients. Cells. 10:1302021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Bi F, Jiang Z, Park W, Hartwich TMP, Ge Z,
Chong KY, Yang K, Morrison MJ, Kim D, Kim J, et al: A
Benzenesulfonamide-based mitochondrial uncoupler induces
endoplasmic reticulum stress and immunogenic cell death in
epithelial ovarian cancer. Mol Cancer Ther. 20:2398–2409. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Lau TS, Chan LKY, Man GCW, Wong CH, Lee
JHS, Yim SF, Cheung TH, McNeish IA and Kwong J: Paclitaxel induces
immunogenic cell death in ovarian cancer via
TLR4/IKK2/SNARE-dependent exocytosis. Cancer Immunol Res.
8:1099–1111. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Le HV, Babak MV, Ehsan MA, Altaf M,
Reichert L, Gushchin AL, Ang WH and Isab AA: Highly cytotoxic
gold(i)-phosphane dithiocarbamate complexes trigger an ER
stress-dependent immune response in ovarian cancer cells. Dalton
Trans. 49:7355–7363. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Song M, Sandoval TA, Chae CS, Chopra S,
Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, et
al: IRE1α-XBP1 controls T cell function in ovarian cancer by
regulating mitochondrial activity. Nature. 562:423–428. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cao Y, Trillo-Tinoco J, Sierra RA, Anadon
C, Dai W, Mohamed E, Cen L, Costich TL, Magliocco A, Marchion D, et
al: ER stress-induced mediator C/EBP homologous protein thwarts
effector T cell activity in tumors through T-bet repression. Nat
Commun. 10:12802019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Yu S, Yan X, Tian R, Xu L, Zhao Y, Sun L
and Su J: An experimentally induced mutation in the UBA domain of
p62 changes the sensitivity of cisplatin by up-regulating HK2
localisation on the mitochondria and increasing mitophagy in A2780
ovarian cancer cells. Int J Mol Sci. 22:39832021. View Article : Google Scholar : PubMed/NCBI
|