Introduction
On a global scale, breast cancer ranks as the second
most commonly occurring malignant neoplasm, and the leading cause
of cancer-associated mortality among women (1). Despite substantial advancements in
molecularly targeted interventions and immunotherapeutic
approaches, therapeutic outcomes and prognostic indicators for
patients with breast cancer continue to demonstrate suboptimal
efficacy (2,3). Within the multimodal therapeutic
framework for breast carcinoma, radiation therapy serves as a
crucial component, demonstrating efficacy in managing localized
tumor advancement, reducing the probability of recurrence and
enhancing patient survival outcomes (4–6).
However, the clinical efficacy of radiotherapy is frequently
compromised by inherent or progressively acquired radioresistance
mechanisms (7). Among the various
factors influencing radiosensitivity, dysregulation of DNA repair
mechanisms and cell cycle progression have been identified as
pivotal elements underlying therapeutic resistance in neoplastic
cells (8). Therefore, revealing the
radiation therapy resistance mechanisms in breast cancer may
provide improved opportunities to overcome tumor resistance.
DNA is vulnerable to multiple types of damage; such
damage encompasses harm to the nucleotides (comprising bases and
sugars) that constitute the DNA framework, the formation of
crosslinks, and the occurrence of single-stranded breaks and
double-stranded breaks (DSBs) within the DNA molecule (9). Among them, DSBs represent the most
cytotoxic form of radiation-induced damage, initiating a cascade of
molecular events within the cellular DNA damage response (DDR)
network. This intricate process involves DNA damage perception,
signal transduction pathway activation, DNA repair machinery
engagement and cell cycle regulation (7,10,11).
The defense mechanism of the cell utilizes a variety of strategies
to handle DSBs, with the two primary repair routes being homologous
recombination (HR) and non-homologous end joining (NHEJ) (12,13).
The NHEJ mechanism, predominantly active during the G1
phase, exhibits rapid repair kinetics but is associated with
reduced fidelity. By contrast, HR-mediated repair, which utilizes
undamaged sister chromatids as templates, demonstrates superior
accuracy in damage correction (14,15).
While DNA damage repair occurs, cell cycle checkpoints are
activated to trigger cell cycle arrest, which provides a critical
repair time for cancer cells and prevents them from entering
mitosis without completing repair (8,16). In
normal cells, these mechanisms act synergistically to maintain
genome stability; however, in tumor cells, aberrant activation of
the DDR often leads to radioresistance. Consequently, one of the
most crucial methods for overcoming tumor radioresistance is to
target the DDR signaling pathway (7,17).
An important part of the spindle assembly checkpoint
system is BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B)
(18), which serves a key role in
maintaining chromosome stability by interacting with Bub3, Mad2 and
Cdc20 to form a mitotic checkpoint complex. Emerging evidence from
numerous previous studies has established a strong association
between dysregulated BUB1B expression and the development of
malignancies, including extrahepatic cholangiocarcinoma, lung
adenocarcinoma and thyroid carcinoma, with its upregulation
consistently linked to unfavorable clinical outcomes (19–21).
Furthermore, BUB1B is associated with chemoradiotherapy resistance
in cancer, such as bladder cancer, glioblastoma and multiple
myeloma (22–24).
In the present study, the extent of BUB1B expression
in breast cancer and its association with patient prognosis was
assessed. Subsequently, the MDA-MB-231 cell line, which shows a
high level of BUB1B expression, was selected to conduct a more
in-depth exploration of its biological functions during tumor
development and its response to radiation. The present study
underscores the crucial role that BUB1B assumes in breast cancer
and identifies a potential therapeutic target for enhancing the
sensitivity of breast cancer to radiotherapy.
Materials and methods
Bioinformatics analysis
The UALCAN (The Cancer Genome Atlas module,
http://ualcan.path.uab.edu/) and Gene
Expression Omnibus (GEO, (https://www.ncbi.nlm.nih.gov/geo/) databases were
employed to investigate the expression state of BUB1B in breast
cancer samples. The Mann-Whitney U test was used to analyze the GEO
datasets GSE38959 (25) and
GSE65194 (26). Subsequently,
receiver operating characteristic (ROC) curve analysis was
conducted to assess the clinical diagnostic capability of BUB1B.
Survival analysis related to BUB1B expression was analyzed using
the bc-GenExMiner website (http://bcgenex.ico.unicancer.fr). Gene Set Enrichment
Analysis (GSEA) was performed on data from the GEO database using R
language (version 4.3.1; R Foundation for Statistical Computing).
Breast cancer samples were categorized into two groups based on the
median expression level of BUB1B mRNA: High-expression and
low-expression cohorts. Subsequently, GSEA was performed to
identify distinct molecular pathways between these two groups.
Cell lines
The normal mammary epithelial cell line MCF-10A, and
breast cancer cell lines MDA-MB-231, BT-549, MCF-7 and BT-474 cell
lines were procured from Procell Life Science & Technology Co.,
Ltd. Different culture media were used for each cell line. MCF-10A
cells were cultured in MCF 10A Cell Complete Medium (cat. no.
CM-0525; Procell Life Science & Technology Co., Ltd.). To
culture MDA-MB-231 and MCF-7 cells, high-glucose DMEM (cat. no.
C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; cat. no. C04001; Shanghai
VivaCell Biosciences, Ltd.) was employed, whereas BT-549 and BT-474
cells were cultured in RPMI 1640 medium (cat. no. C11875500BT;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
All cells were cultured at 37°C in a 5% CO2
incubator.
Lentivirus infection
The expression of BUB1B was first examined in normal
mammary epithelial cells and different breast cancer cell lines,
and, based on the experimental results, the MDA-MB-231 cell line
was selected for lentiviral infection in subsequent experiments, as
it had the highest expression of BUB1B. The lentiviral short
hairpin RNA (shRNA) constructs, comprising non-targeting nonsense
negative control (NC) sequences and specific shBUB1B1/2/3 targeting
human genes, were acquired from Shanghai Hanhang Technology Co.,
Ltd. The shRNA sequences were as follows: NC: Top strand
5′-GATCCGTTCTCCGAACGTGTCACGTAATTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTC,
bottom strand
5′-AATTGAAAAAATTCTCCGAACGTGTCACGTAATCTCTTGAATTACGTGACACGTTCGGAGAACG;
shBUB1B-1, top strand
5′-GATCCGAGACAACTAAACTGCAAATTCTCGAGAATTTGCAGTTTAGTTGTCTCTTTTTTG,
bottom
5′-AATTCAAAAAAGAGACAACTAAACTGCAAATTCTCGAGAATTTGCAGTTTAGTTGTCTCG-3′;
shBUB1B-2, top strand
5′-GATCCGCCAGTTCTGTTTGTCAAGTAACTCGAGTTACTTGACAAACAGAACTGGTTTTTTG-3′,
bottom strand
5′-AATTCAAAAAACCAGTTCTGTTTGTCAAGTAACTCGAGTTACTTGACAAACAGAACTGGCG-3′;
shBUB1B-3, top strand
5′-GATCCGCTGTATTGTTTGGCACCAATACTCGAGTATTGGTGCCAAACAATACAGTTTTTTG-3′,
bottom strand
5′-AATTCAAAAAACTGTATTGTTTGGCACCAATACTCGAGTATTGGTGCCAAACAATACAGCG-3′.
The second-generation lentiviral packaging system was used. All
vectors, reagents, and cell lines utilized for lentiviral packaging
were provided by Shanghai Hanhang Technology Co., Ltd. The plasmids
used for transfection included: pSPAX2: 10 µg, pMD2G: 5 µg,
pHBLV-U6-MCS-CMV-ZsGreen-PGK-PUROplasmid (carrying shRNA): 10 µg.
These plasmids were co-transfected into the 293T packaging cells
using a transfection reagent (Lipofiter™, 75 µl), and
the cells were incubated at 37°C for 72 h, after which, the viral
supernatant was collected by ultracentrifugation. The lentivirus
was then infected into MDA-MB-231 cells using the half-volume
infection method at a multiplicity of infection of 30, followed by
incubation in a 37°C incubator for 24 h. After replacing the
medium, the cells were cultured for a further 48 h and fluorescence
was observed under a fluorescence microscope. Stable infected cell
lines were selected by culturing the cells in medium containing 4
µg/ml puromycin (Beyotime Institute of Biotechnology) for 7
days.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from the MCF-10A,
MDA-MB-231, BT-549, MCF-7 and BT-474 cell using the Total RNA
Isolation Kit (cat. no. M5105; New Cell & Molecular Biotech).
Subsequently, the concentration and purity of the extracted RNA
were assessed with a NanoDrop ND-1000 Spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). The extracted RNA
was stored at −80°C. cDNA was obtained from the extracted RNA using
the Script Reverse Transcription Supermix Kit (cat. no. RR047A;
Takara Bio, Inc.) according to the manufacturer's instructions.
Subsequently, qPCR was performed using the TB Green®
Fast qPCR Mix (cat. no. RR430A; Takara Bio, Inc.). The gene
expression levels were measured using the 2−ΔΔCq method
(27), with GAPDH serving as the
internal reference gene. The primer sequences utilized for
amplification were as follows: BUB1B, forward
5′-AAATGACCCTCTGGATGTTTGG-3′ and reverse
5′-GCATAAACGCCCTAATTTAAGCC-3′; and GAPDH, forward
5′-CAGGAGGCATTGCTGATGAT-3′ and reverse
5′-GAAGGCTGGGGCTCATTT-3′.
Western blot analysis
Cells were lysed with RIPA lysis solution (cat. no.
P0013B; Beyotime Institute of Biotechnology) and protein
supernatants were collected. Protein concentration was determined
using a NanoDrop spectrophotometer (Thermo Fisher Scientific,
Inc.). The proteins (20 µg) were denatured by boiling and were
separated by SDS-PAGE on a 4.5% stacking gel and 7.5% resolving
gel. The proteins were subsequently transferred to a PVDF membrane,
which was blocked with 5% skimmed milk for 1 h at room temperature.
The membrane was then incubated with primary antibodies at 4°C
overnight. On the next day, secondary antibody incubation was
performed, using HRP-labeled secondary antibodies (cat. nos. 511203
and 511103; 1:5,000; Chengdu Zen-Bioscience Co., Ltd.) for 1 h at
room temperature. After immersing the membrane in BeyoECL Plus
(cat. no. P0018S; Beyotime Institute of Biotechnology) working
solution for 1 min, the bands were detected using a
chemiluminescence imaging system (Bio-Rad Laboratories, Inc.).
Semi-quantitative analysis was performed using ImageJ software
(version 1.54g; National Institutes of Health). The primary
antibodies used were against BUB1B (cat no. ab183496; 1:20,000;
Abcam), β-actin (cat. no. AB0035; 1:10,000; Shanghai Abways
Biotechnology Co., Ltd.), E-cadherin (cat. no. 60335-1-Ig; 1:2,000;
Proteintech Group, Inc.), N-cadherin (cat. no. R380671; 1:500;
Chengdu Zen-Bioscience Co., Ltd.), vimentin (cat. no. R22775;
1:500; Chengdu Zen-Bioscience Co., Ltd.), RAD51 (cat. no. R27223;
1:500; Chengdu Zen-Bioscience Co., Ltd.), PI3K (cat. no.
60225-1-Ig; 1:2,000; Proteintech Group, Inc.), AKT (cat. no.
60203-1-Ig; 1:2,000; Proteintech Group, Inc.), phosphorylated
(p)-PI3K (cat. no. 341468; 1:500; Chengdu Zen-Bioscience Co., Ltd.)
and p-AKT (cat. no. R381555; 1:500; Chengdu Zen-Bioscience Co.,
Ltd.).
Cell counting kit-8 (CCK-8) assay
A total of 5,000 cells was added to each well of a
96-well plate, and incubated in a 37°C incubator for 24, 48, 72 or
96 h. Subsequently, CCK-8 reagent (cat. no. A311-01; Vazyme Biotech
Co., Ltd.) was added to each well and incubated at 37°C for 1 h.
Next, a microplate spectrophotometer was employed to measure the
absorbance at a wavelength of 450 nm.
Colony formation assay
To explore the development of cell colonies, cells
were distributed evenly across 6-well culture plates at a density
of 400 cell/.well and were incubated for 10–14 days under standard
culture conditions. The medium was changed and the cell status was
observed every 3 days. Colonies were defined as aggregates of ≥50
cells originating from a single progenitor cell and were manually
counted under a light microscope. The cell colonies were fixed with
4% paraformaldehyde solution at room temperature for 15 min and
stained with 5% crystal violet solution at room temperature for 15
min. After washing with PBS three times, colony quantification and
photographic documentation were performed. Each experimental
condition was independently replicated three times, with
quantitative data normalized to the corresponding control
groups.
EdU staining
Cell proliferation capacity was measured with the
BeyoClick™ EdU-594 Cell Proliferation Assay Kit (cat.
no. C0078S; Beyotime Institute of Biotechnology). Cells were
inoculated into 6-well plates at a density of 2×105
cells/well and were incubated for 24 h in a 37°C incubator.
Subsequently, the cells were treated with pre-warmed (37°C) 2X EdU
working solution (20 µM) at equal volumes, achieving a final 1X EdU
concentration. After 2 h of incubation under standard culture
conditions, cellular samples were subjected to washing with PBS,
followed by 4% paraformaldehyde fixation for 30 min at room
temperature and 0.5% Triton X-100 permeabilization for 10 min at
room temperature. Next, a Click reaction solution was performed by
incubating the cells with 500 µl reaction mixture for 30 min in the
dark at room temperature. Nuclear counterstaining was accomplished
using 1,000 µl 1X Hoechst 33342 solution for 10 min in the dark at
room temperature. Fluorescence imaging was conducted using an
inverted fluorescence microscope, with quantification based on
positive cell counts from five randomly selected fields per
sample.
Xenograft tumor assay
A total of 10 BALB/c-nu female mice (age, 4 weeks;
weight, 16–20 g) from Guangxi Medical University Laboratory Animal
Center (Nanning, China) were selected for the experiment. The mice
were housed in a specific pathogen-free environment with a
temperature of 26–28°C, humidity of 40–60% and under a 12-h
light/dark cycle. The mice were fed an irradiated sterilized high
protein feed ad libitum, and the drinking water was
ultrapure water, with the water bottle changed daily. The Guangxi
Medical University Laboratory Animal Ethics Committee approved the
present animal studies (approval no. 202410013).
BALB/c nude mice were randomly allocated into two
groups (n=5/group): i) Control group receiving MDA-MB-231-NC cells
and ii) experimental group, inoculated with MDA-MB-231-shBUB1B
cells. A suspension containing 1×10⁶ cells in 100 µl PBS was
subcutaneously injected into the right axilla of each mouse. Tumor
growth was monitored every 3 days by measuring orthogonal diameters
with digital calipers, and tumor volume (TV) was calculated using
the ellipsoid formula: TV=1/2 × length × width2. Humane
endpoints were strictly enforced, and experiments were terminated
when mice had a tumor volume ≥1,500 mm3 or lost >20%
of their body weight. The experiment was terminated after 5 weeks
and no mice were sacrificed due to reaching the aforementioned
humane endpoints. Mice were anesthetized via an intraperitoneal
injection of 1.25% tribromoethanol (250 mg/kg body weight) and
euthanized by cervical dislocation. All animals died from
euthanasia. Death was confirmed by continuous observation of
respiratory movements for ≥5 min, including cessation of breathing
and absence of chest rise and fall, and loss of corneal and pain
reflexes.
Immunohistochemistry
For immunohistochemistry, nude mouse tissue
specimens of xenograft tumor origin were incubated in 4%
paraformaldehyde solution for at room temperature 72 h, embedded in
paraffin and sectioned into 4-µm slices. Tissue sections then
underwent sequential processing (deparaffinization in xylene and
rehydration through an alcohol gradient series), and were soaked in
citrate buffer for 3 min for antigen retrieval. Bovine serum
albumin (BSA; cat no. G5001; Wuhan Servicebio Technology Co., Ltd.)
was used to block the sections at room temperature for 30 min.
Subsequently, the sections were incubated with primary antibodies
against Ki67 (cat. no. AF20068; 1:200; Hunan Aifang Biotechnology
Co., Ltd.) overnight at 4°C, and then with a Polymer-HRP anti-mouse
secondary antibody kit (cat. no. AFIHC002; Hunan Aifang
Biotechnology Co., Ltd.) for 30 min at 37°C. DAB (cat. no.
AFIHC004; Hunan Aifang Biotechnology Co., Ltd.) reagent was used
for staining, whereas for counterstaining, the sections were
incubated with hematoxylin at room temperature for 3 min, followed
by sequential dehydration through an ethanol gradient and xylene.
The staining was observed under a light microscope (E100; Nikon
Corporation). Panoramic scanning was performed under a 20X
objective lens using a DS-U3 imaging system (Nikon
Corporation).
TUNEL staining was performed using a TUNEL kit (cat.
no. AFIHC030-C; Hunan Aifang Biotechnology Co., Ltd.) to evaluate
tissue apoptosis. The procedures for tissue fixation, paraffin
embedding, sectioning and antigen retrieval were the same as
aforementioned. Subsequently, a mixture of TDT enzyme and dUTP
(mixed at a ratio of 1:50) was used to cover the tissues, followed
by incubation at 37°C for 1–2 h. Nuclear staining was conducted at
room temperature using 20X DAB staining solution (cat no. AFIHC004;
Hunan Aifang Biotechnology Co., Ltd.). The staining was monitored
under a light microscope (cat no. E100; Nikon Corporation) and the
staining was terminated when brownish-yellow signals appeared. The
stained sections were subjected to panoramic scanning under a 20X
objective using the DS-U3 imaging system (Nikon Corporation).
Gap closure assay
Cell migration was evaluated using the ibidi
Culture-Insert 2 Well (cat no. 81176; ibidi GmbH). The inserts were
placed in 6-well plates, and each well was seeded with 70 µl cell
suspension containing 6×104 cells. After the cell
confluence reached 95%, the inserts were carefully removed. The
cell monolayer was then washed once with PBS and cultured in
serum-free medium. The gap area was captured using a light
microscope immediately after washing in PBS (designated as 0 h) and
was established as the baseline. Images of the same positions were
acquired at 12 and 24 h. The migratory capacity of the cells was
assessed by measuring the changes in the gap area using ImageJ
software (version 1.54g; National Institutes of Health, USA).
Transwell assay
Both Transwell invasion and migration assays were
performed. For the invasion assay, the bottom of the 24-well
Transwell inserts (pore size, 9 µm) were coated with Matrigel
(Corning, Inc.), whereas the migration assay inserts remained
uncoated. The Matrigel was thawed overnight in a refrigerator at
4°C, and the next day it was spread evenly on the bottom of the
chamber and then incubated at 37°C for 1 h to solidify. A total of
200 µl cells resuspended in serum-free DMEM (containing 2×10⁵
cells/well) were seeded into the upper chamber, and 600 µl complete
medium supplemented with 10% FBS was added to the lower chamber.
After 24 h of incubation at 37°C, the Transwell inserts were washed
twice with PBS. The cells were then fixed with 4% paraformaldehyde
for 15 min at room temperature, followed by staining with crystal
violet for 15 min at room temperature. After rinsing the inserts
twice with PBS, the stained cells were visualized and images were
captured using a light microscope.
Immunofluorescence assay
Cells (5×104) were inoculated in 6-well
plates and incubated at 37°C for 24 h. The cells were then treated
with 8 Gy irradiation using a linear accelerator (Elekta Instrument
AB) at a dose rate of 1 Gy/min before the detection of γ-H2AX by
immunofluorescence assay. No irradiation was required for detection
of the other indicators. Upon rinsing with PBS, cells were fixed
using a 4% paraformaldehyde solution for 10 min at room
temperature. After fixation, cell permeability was induced by
treating the cells with a PBS solution containing 0.1% Triton
X-100. Next, to prevent non-specific binding, a blocking procedure
was carried out by incubating the cells with a 5% BSA solution for
30 min at room temperature. Once the blocking step was completed,
the cells were incubated with the following primary antibodies
overnight at 4°C: E-cadherin (cat. no. 60335-1-Ig; 1:200;
Proteintech Group, Inc.), N-cadherin (cat. no. 22018-1-AP; 1:200;
Proteintech Group, Inc.), vimentin (cat. no. R22775; 1:100; Chengdu
Zen-Bioscience Co., Ltd.) and γ-H2AX (cat. no. ab81299; 1:250;
Abcam). After rinsing with PBS, Alexa Fluor (AF)-conjugated Goat
Anti-Mouse lgG H&L (AF594) (cat. no. 550042; Positive Bio) and
Goat Anti-Rabbit lgG H&L (AF594) (cat. no. 550043; Positive
Bio) secondary antibodies were added (dilution, 1:500) and
incubated for 1 h at room temperature. Next, nuclear
counterstaining was carried out using DAPI (cat. no. G1012; Wuhan
Servicebio Technology Co., Ltd.) for 10 min at room temperature.
Finally, fluorescence imaging of the samples was performed using a
Zeiss LSM880 confocal microscope (Zeiss AG).
Neutral comet assay
DNA damage analysis was conducted 2 h after 8 Gy
irradiation using a Comet Assay Kit (cat. no. KGA1302-20; Nanjing
KeyGen Biotech Co., Ltd.). A total of 100 µl 1% normal melting
point agarose was added to the slide, followed by its
solidification at 4°C for 10 min. Next, a cellular suspension
containing 10⁵ cells and 75 µl 0.75% low melting point agarose were
combined, followed by solidification under identical temperature
conditions for 30 min. The slides were then immersed in lysis
buffer for 2 h at 4°C, and the samples were then electrophoresed
for 20 min at 22 V after being equilibrated for 30 min in a neutral
electrophoresis solution. Post-electrophoresis processing included
PBS immersion (pH 7.2–7.4) for 30 min at 4°C and propidium iodide
staining for 10 min at room temperature. Fluorescent images were
obtained with a Zeiss fluorescence microscope (Zeiss AG).
Quantitative analysis was performed using Comet Assay Software
Project (28) (version 1.2.3
beta1), counting 20 cells per group, and the olive tail moments
were shown.
Cell cycle analysis
A commercially available Cell Cycle Staining Kit
[cat. no. CCS012; Multisciences (Lianke) Biotech, Co., Ltd.] was
employed to assess the distribution of the cell cycle. Briefly,
2×10⁵ cells were inoculated in a 6-well dish, placed in an
incubator overnight. On the second day, the cells were treated with
8 Gy irradiation. Trypsin was used to collect cells at
predetermined times post-treatment (0, 12 and 24 h), followed by
washing in PBS and resuspension. Subsequently, 10 µl
permeabilization solution and 1 ml propidium iodide staining
solution (containing RNase A) were incorporated into the mixture
according to the Cell Cycle Staining Kit instructions. The
resulting combination was then incubated at room temperature for 10
min in the dark. Data were acquired using a CytoFLEX flow cytometer
(Beckman Coulter, Inc.). Upon data collection, assessment of the
cell cycle phases was carried out with the aid of FlowJo analysis
software (version 10.8.1; BD Biosciences).
RNA sequencing
Total RNA was extracted from NC and shBUB1B cells
using the MJZol Total RNA Extraction Kit (cat no. T01-200; Shanghai
Majorbio) at 8 h after 8 Gy irradiation. Subsequently, RNA quality
was determined using a 5300 Bioanalyzer (Agilent Technologies,
Inc.) and RNA was quantified using the ND-2000 (NanoDrop; Thermo
Fisher Scientific, Inc.). The molar concentrations of libraries was
determined by fluorescence quantification (Qubit™ 4.0;
Thermo Fisher Scientific, Inc.). The final loading concentration
was adjusted to 2 nM, and 150 bp paired-end sequencing was
performed on the NovaSeq X Plus platform (PE150; Illumina, Inc.)
using the NovaSeq 6000 SP reagent kit (100 cycles; cat. no.
2002746; Illumina Inc.). Bioinformatics processing and initial data
analysis were performed by Shanghai Meiji Biological Co., Ltd.
DESeq2 software (http://bioconductor.org/packages/stats/bioc/DESeq2/)
was used to assess differential gene expression and significant
changes were found using two criteria: P<0.05 and |log2
fold-change|≥1. The Goatools program (https://github.com/tanghaibao/GOatools) was used for
Gene Ontology (GO) enrichment analysis, and the Python SciPy
package (https://scipy.org/install/) was used
for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analysis.
Statistical analysis
All experimental procedures were independently
performed in triplicate, with representative results shown.
GraphPad Prism software version 10.0 (Dotmatics) was utilized to
conduct statistical comparisons. Comparisons between two groups
were performed using the two-tailed unpaired Student's t-test. For
comparisons among multiple groups, one-way ANOVA followed by
Dunnett's post hoc test or two-way ANOVA followed by Bonferroni's
multiple comparisons test were applied. The quantitative outcomes
are presented as the mean ± standard. P<0.05 was considered to
indicate a statistically significant difference.
Results
BUB1B is highly expressed in breast
cancer and is associated with poor prognosis
Analysis of the UALCAN database showed that the
expression levels of BUB1B were significantly higher in breast
cancer tissues compared with in the normal tissues of healthy
controls (Fig. 1A). Among them, the
expression levels of BUB1B were highest in triple-negative breast
cancer, followed by HER2-positive breast cancer, and it was lowest
in luminal breast cancer. Analysis of GEO datasets using the
Mann-Whitney U test revealed that BUB1B expression was markedly
higher in breast cancer tissues vs. normal breast tissues from
healthy controls in the GSE65194 dataset (Fig. 1B). Similarly, BUB1B gene expression
was significantly elevated in breast cancer cells compared with in
normal mammary ductal cells from healthy controls in the GSE38959
dataset (Fig. 1C). Based on the
mRNA expression levels, a ROC curve was generated (Fig. 1D). The area under the ROC curve was
calculated to be 0.963, which suggested that the mRNA levels of
BUB1B may effectively distinguish between patients with breast
cancer and healthy individuals.
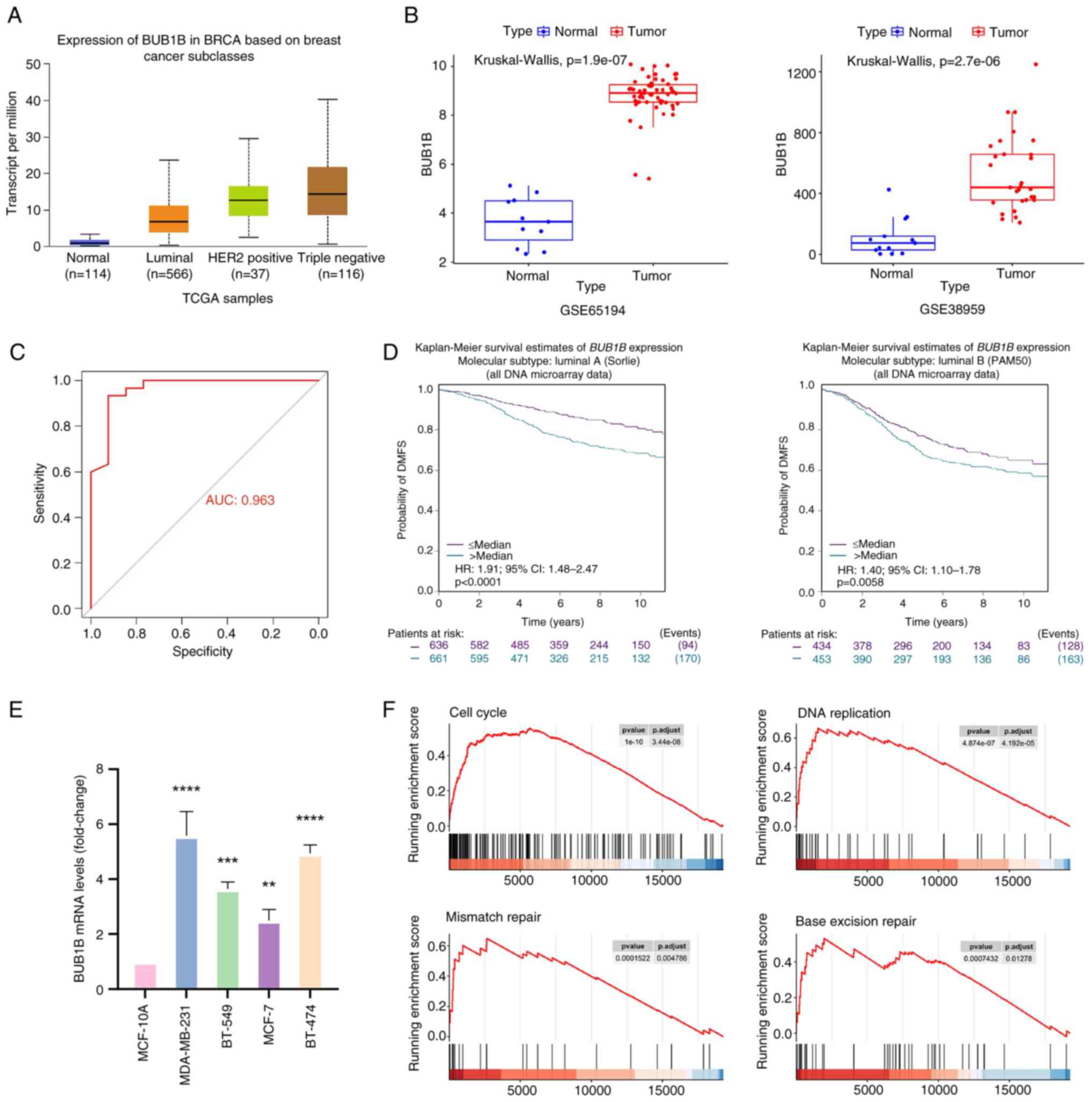 | Figure 1.BUB1B is highly expressed in breast
cancer and is associated with a poor prognosis. (A) BUB1B
expression in healthy and breast cancer tissues in the UALCAN
database. Compared with the Normal group, the P-values for Luminal,
HER2-positive and triple-negative groups were all <0.001. (B)
Expression levels of BUB1B in GSE38959 and GSE65194 datasets. (C)
Receiver operating characteristic curve based on BUB1B mRNA
expression levels. (D) bc-GenExMiner website was used to evaluate
the association between BUB1B and DMFS. (E) mRNA expression levels
of BUB1B in normal mammary epithelial and breast cancer cells. Data
are presented as the mean ± SD, n=3. **P<0.01, ***P<0.001,
****P<0.0001 vs. MCF-10A (one-way ANOVA followed by Dunnett's
post hoc test). (F) Comparative GSEA was conducted using
transcriptomics data from the GSE38959 dataset. Four representative
plots of GSEA enrichment have been demonstrated. AUC, area under
the curve; BUB1B, BUB1 mitotic checkpoint serine/threonine kinase
B; DMFS, distant metastasis-free survival; GSEA, gene set
enrichment analysis; TCGA, The Cancer Genome Atlas. |
The prognostic value of BUB1B was assessed via
Kaplan-Meier survival analysis with log-rank testing using
bc-GenExMiner. The present findings showed that higher BUB1B levels
were associated with a lower probability of distant metastasis-free
survival (DMFS) (Fig. 1D).
Additionally, BUB1B mRNA expression was quantified in normal
mammary epithelial cells (MCF-10A) and in multiple breast cancer
cell lines (MDA-MB-231, BT-549, MCF-7 and BT-474) using RT-qPCR.
The results demonstrated significantly elevated BUB1B mRNA levels
in all breast cancer cell lines compared with in normal mammary
epithelial cells (Fig. 1E).
Furthermore, GSEA of GEO datasets revealed that the expression of
BUB1B was associated with cell cycle, DNA replication, mismatch
repair and base excision repair (Fig.
1F). These results suggested that BUB1B may be associated with
biological processes, such as proliferation, cell cycle regulation
and DNA damage repair in breast cancer cells.
BUB1B induces MDA-MB-231 cell
proliferation in vivo and in vitro
To investigate the role of BUB1B in breast cancer, a
stable knockdown of BUB1B was established in MDA-MB-231 cells
(Fig. 2A and B) and BUB1B knockdown
cell lines constructed using shRNA-2 were selected for cellular
experiments. To assess the impact of BUB1B knockdown on cell
proliferation under in vitro conditions, CCK-8 and colony
formation assays were employed. Knocking down BUB1B significantly
reduced MDA-MB-231 cell proliferation (Fig. 2C and D). EdU staining also
demonstrated that DNA replication was significantly suppressed by
BUB1B knockdown (Fig. 2E). To
evaluate the impact of BUB1B on tumor formation in living
organisms, nude mice were injected with MDA-MB-231 cells that
either exhibited normal BUB1B expression or had BUB1B knockdown.
The findings from the in vivo experiments were consistent
with the aforementioned in vitro findings. Specifically, the
results demonstrated that tumor growth was significantly reduced in
the BUB1B knockdown group compared with that in the control group
(Fig. 2F). Furthermore,
immunohistochemical analysis revealed that BUB1B suppression
resulted in decreased Ki67-positive proliferating cells and
elevated TUNEL-positive apoptotic cell populations (Fig. 2G). These results suggested that
knockdown of BUB1B may affect the growth of transplanted tumors
in vivo by inhibiting cell proliferation and promoting
apoptosis.
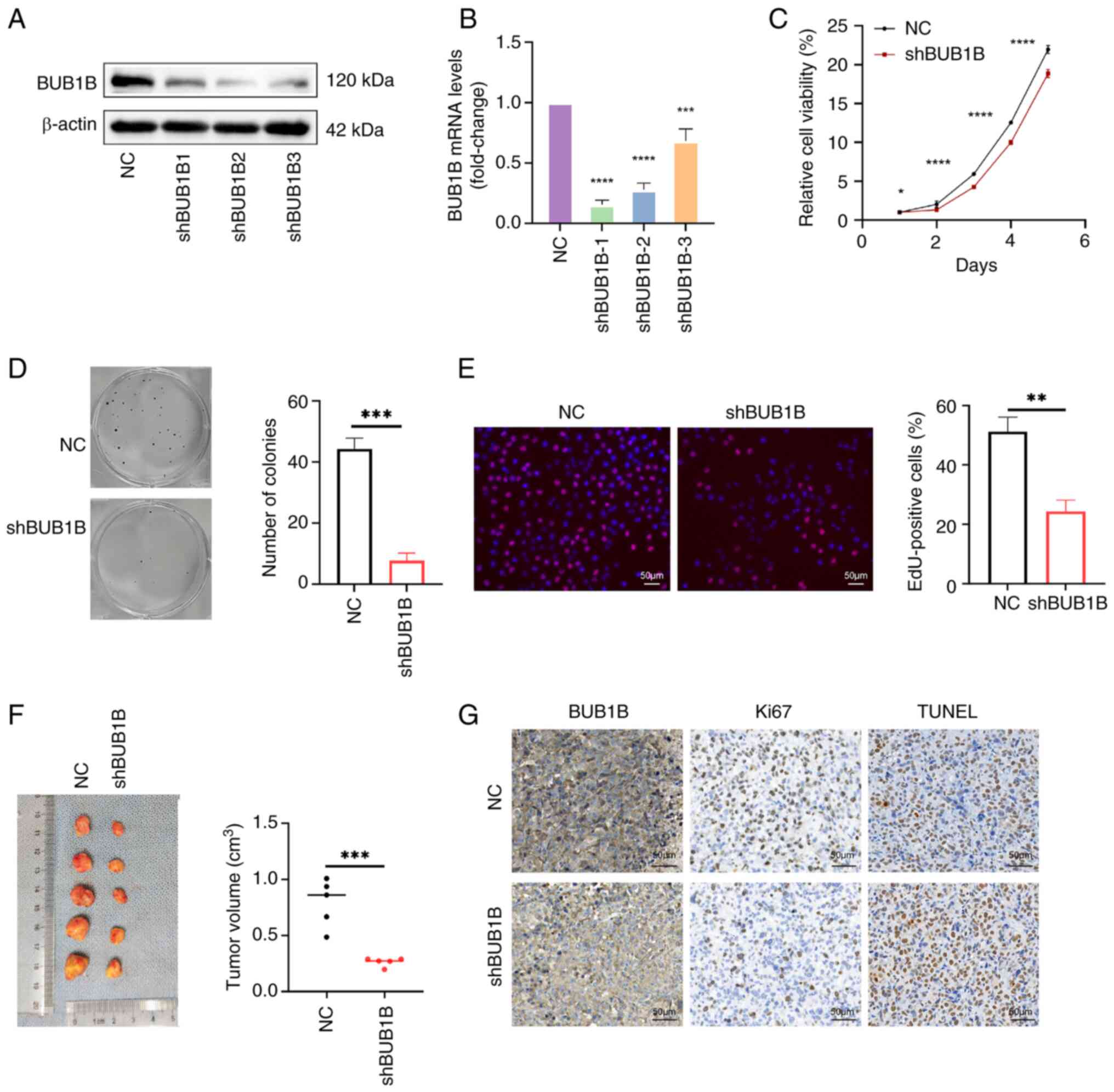 | Figure 2.BUB1B promotes the proliferation of
MDA-MB-231 cells in vivo and in vitro. BUB1B
expression in MDA-MB-231 cells stably infected with NC or shBUB1B
was detected by (A) western blotting and (B) reverse
transcription-quantitative PCR. ***P<0.001, ****P<0.0001 vs.
NC (one-way ANOVA followed by Dunnett's post hoc test). (C) Cell
Counting Kit-8 assay was used to detect the proliferation of
MDA-MB-231 cells. *P<0.05, ****P<0.0001 vs. NC (two-way ANOVA
followed by Bonferroni's multiple comparisons test). (D) Colony
formation assay was used to detect the colony-forming ability of
MDA-MB-231 cells. ***P<0.001 vs. NC (unpaired Student's t-test).
(E) EdU assay of DNA replication in MDA-MB-231 cells. Scale bar, 50
µm; magnification, ×200. **P<0.01 vs. NC (unpaired Student's
t-test). (B-E) Data are presented as the mean ± SD, n=3. (F)
Xenograft tumor assay validated the effect of BUB1B on cell
proliferation in vivo. Data are presented as the mean ± SD,
n=5. ***P<0.001 vs. NC (unpaired Student's t-test). (G) TUNEL
and Ki67 were detected by immunohistochemical staining in tumor
tissues. Scale bar, 50 µm; magnification, ×200. BUB1B, BUB1 mitotic
checkpoint serine/threonine kinase B; NC, negative control; sh,
short hairpin. |
BUB1B promotes the migration and
invasion of MDA-MB-231 cells
Gap closure and Transwell migration assays were used
to assess the influence of BUB1B on the migratory and invasive
capabilities of breast cancer cells. The findings indicated that
downregulation of BUB1B expression suppressed the migration and
invasion of MDA-MB-231 cells (Fig.
3A-D). Immunofluorescence assay was utilized to verify the
influence of BUB1B on the epithelial-mesenchymal transition (EMT)
of breast cancer cells. The findings showed that, when BUB1B was
knocked down in MDA-MB-231 cells, the expression levels of the
epithelial marker E-cadherin were significantly increased, whereas
the levels of mesenchymal markers (N-cadherin and vimentin) were
decreased, compared with those in the NC group (Fig. 3E and F). This result was further
verified by western blot analysis (Fig.
3G and H). These findings suggested that knocking down BUB1B
may inhibit the invasion and metastatic potential of breast cancer
cells through modulating the EMT process.
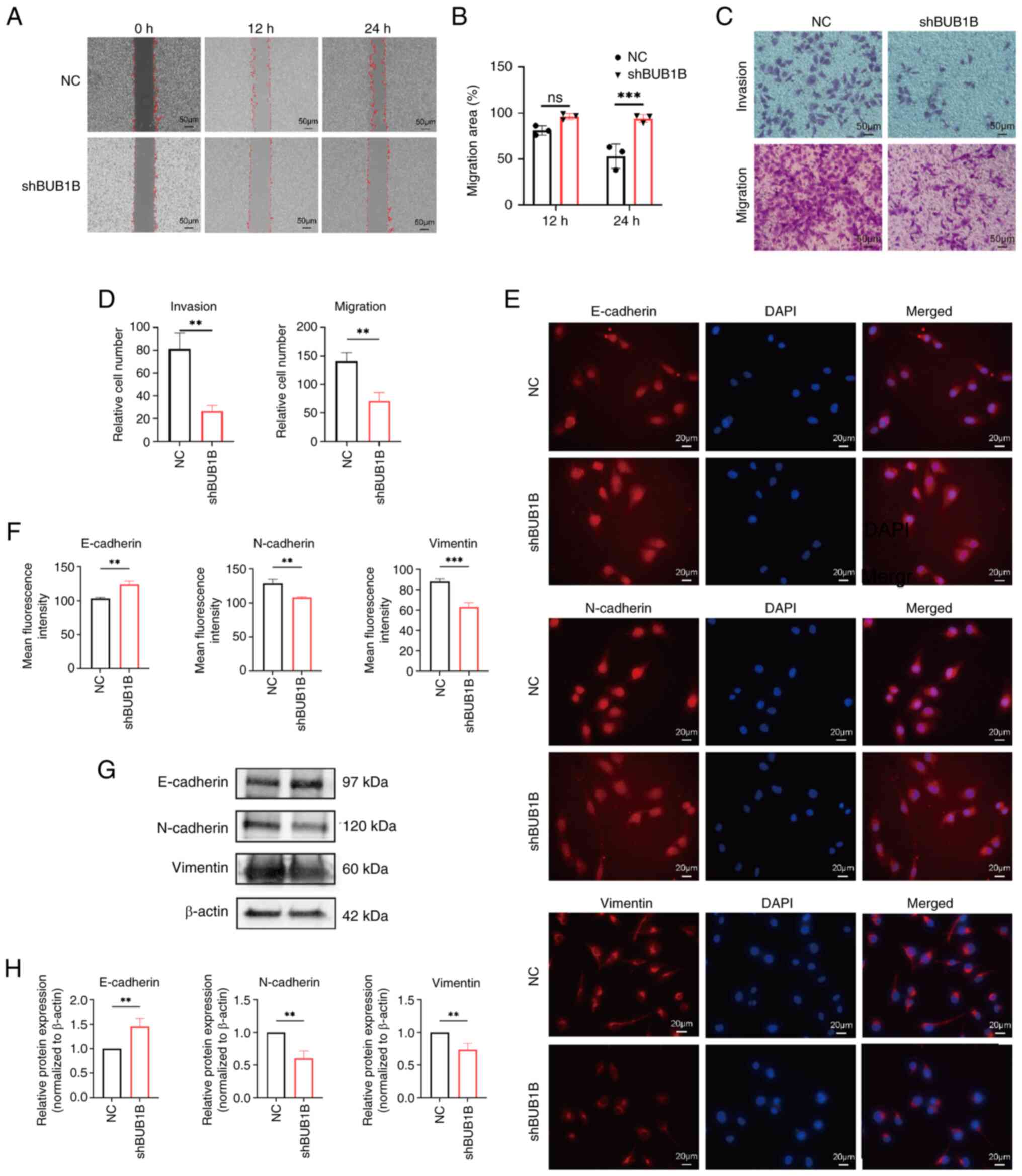 | Figure 3.BUB1B promotes the migration and
invasion of MDA-MB-231 cells. (A) Representative images and (B)
quantitative analysis of cell migration assessed using the gap
closure assay. Scale bar, 50 µm; magnification, ×200. ***P<0.001
vs. NC (two-way ANOVA followed by Bonferroni's multiple comparisons
test). (C) Representative images and (D) quantitative analysis of
migratory and invasive capacity of cells detected by Transwell
assay. Scale bar, 50 µm; magnification, ×200. **P<0.01 vs. NC
(unpaired Student's t-test). (E) Representative immunofluorescence
staining images of the EMT markers E-cadherin, N-cadherin and
vimentin. (F) Semi-quantitative analysis of NC and shBUB1B cells
after irradiation. Scale bar, 20 µm; magnification, ×400.
**P<0.01, ***P<0.001 vs. NC (unpaired Student's t-test). (G)
Representative images and (H) semi-quantitative analysis of the EMT
markers E-cadherin, N-cadherin and vimentin detected by western
blot analysis. **P<0.01 vs. NC (unpaired Students' t-test). All
data are presented as the mean ± SD, n=3. BUB1B, BUB1 mitotic
checkpoint serine/threonine kinase B; NC, negative control; sh,
short hairpin. |
BUB1B promotes cell cycle arrest in
breast cancer cells after irradiation
There is a close association between
radiosensitivity and regulation of the cell cycle (29). Flow cytometry was employed to assess
the impact of BUB1B on the cell cycle after exposure to ionizing
radiation. The results indicated that, upon treatment with ionizing
radiation, in contrast to NC cells, shBUB1B cells exhibited an
increased proportion in the G1/S phase and a concomitant
decrease in the G2/M phase (Fig. 4A and B). These results indicated
that breast cancer cells mainly underwent G2/M phase
cycle arrest when exposed to radiation. Notably, compared with that
in the NC group, the percentage of BUB1B-silenced cells in
G2/M phase was considerably lower. These findings
indicated that silencing BUB1B could impede the G2/M
phase arrest triggered by ionizing radiation.
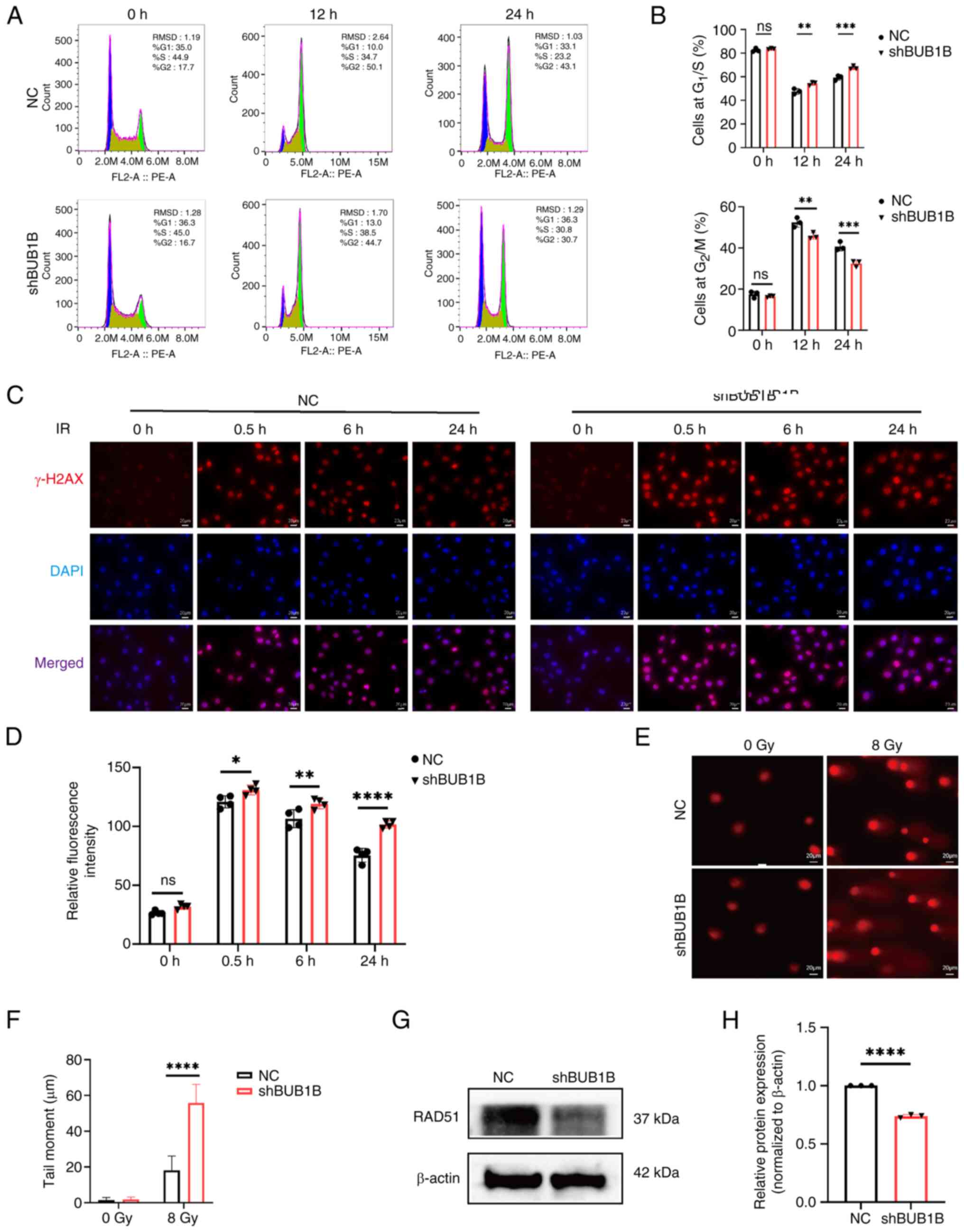 | Figure 4.BUB1B serves an importatn role in the
cell cycle arrest and DNA damage repair after irradiation. (A)
Representative flow cytometry plots of cell cycle analysis and (B)
quantitative analysis of NC and shBUB1B cells after irradiation.
**P<0.01, ***P<0.001 vs. NC (two-way ANOVA followed by
Bonferroni's multiple comparisons test). (C) Representative γ-H2AX
immunofluorescence images and (D) semi-quantitative analysis of NC
and shBUB1B cells after irradiation. Scale bar, 20 µm;
magnification, ×400. *P<0.05, **P<0.01, ****P<0.0001 vs.
NC (two-way ANOVA followed by Bonferroni's multiple comparisons
test). (A-D) Data are presented as the mean ± SD, n=3. (E)
Representative images of the comet assay and (F) tail moment
quantification analysis in MDA-MB-231 cells in the NC and shBUB1B
groups after irradiation. Data are presented as the mean ± SD,
n=20. Scale bar, 20 µm; magnification, ×400. ****P<0.0001 vs. NC
(unpaired Student's t-test). (G) Representative images and (H)
semi-quantitative analysis of RAD51 protein levels detected by
western blot analysis. ****P<0.0001 vs. NC (unpaired Student's
t-test). All data are presented as the mean ± SD, n=3. BUB1B, BUB1
mitotic checkpoint serine/threonine kinase B; IR, ionizing
radiation; NC, negative control; sh, short hairpin. |
BUB1B promotes DNA damage repair in a
HR-biased manner
The primary mechanism of action of radiation therapy
is to cause notable DNA damage (30); therefore, the present study examined
how BUB1B contributes to DNA repair. Immunofluorescence was used to
assess the expression levels of γ-H2AX, a conventional DNA
double-strand break marker, in MDA-MB-231 cells. The results
revealed that cells with BUB1B knockdown had higher levels of
γ-H2AX than NC cells (Fig. 4C and
D). After irradiation with 8 Gy, the comet assay was employed
to detect DSBs. Compared with in the NC cell group, BUB1B-knockdown
cells had a longer olive tail moment (Fig. 4E and F), which indicated that BUB1B
knockdown cells may have more pronounced DNA double-strand breaks.
Together, these results suggested that BUB1B knockdown cells were
more severely DNA damaged than NC cells after irradiation.
HR and NHEJ have been reported to be the two main
pathways of cellular DSB repair. The present study confirmed that
BUB1B knockdown can inhibit irradiation-induced G2/M
phase arrest, which primarily affects cellular HR repair.
Therefore, the study further examined the effect of BUB1B knockdown
on RAD51, a key protein in the HR repair pathway, through western
blot analysis. The results revealed that BUB1B knockdown
significantly suppressed the expression levels of RAD51 (Fig. 4G and H). These results indicated
that BUB1B may promote the repair of DNA damage via HR, which could
offer a novel mechanism for DNA damage repair.
BUB1B regulates the expression of the
PI3K/AKT signaling pathway
To further identify the potential signaling pathways
regulated by BUB1B, differentially expressed genes were screened in
irradiated NC and BUB1B-knockdown cells by RNA sequencing, and GO
and KEGG analyses were performed. The results showed that 297 genes
were upregulated and 76 were downregulated in the BUB1B-knockdown
group compared with in the NC group (Fig. 5C). Further GO functional analysis
revealed that the differentially expressed genes were mainly
involved in functions such as ‘regulation of cellular biological
processes’ and ‘signal transduction’ (Fig. 5A). KEGG pathway enrichment analysis
revealed that the activity of the ‘PI3K-Akt signaling pathway’ was
associated with the BUB1B expression level (Fig. 5B). Several core proteins involved in
the PI3K/AKT signaling pathway were detected by western blotting.
The results showed that the protein levels of PI3K, AKT, p-PI3K and
p-AKT were significantly reduced in MDA-MB-231 cells after
downregulation of BUB1B expression (Fig. 5D and E). These data suggested that
BUB1B regulates the PI3K/AKT signaling pathway.
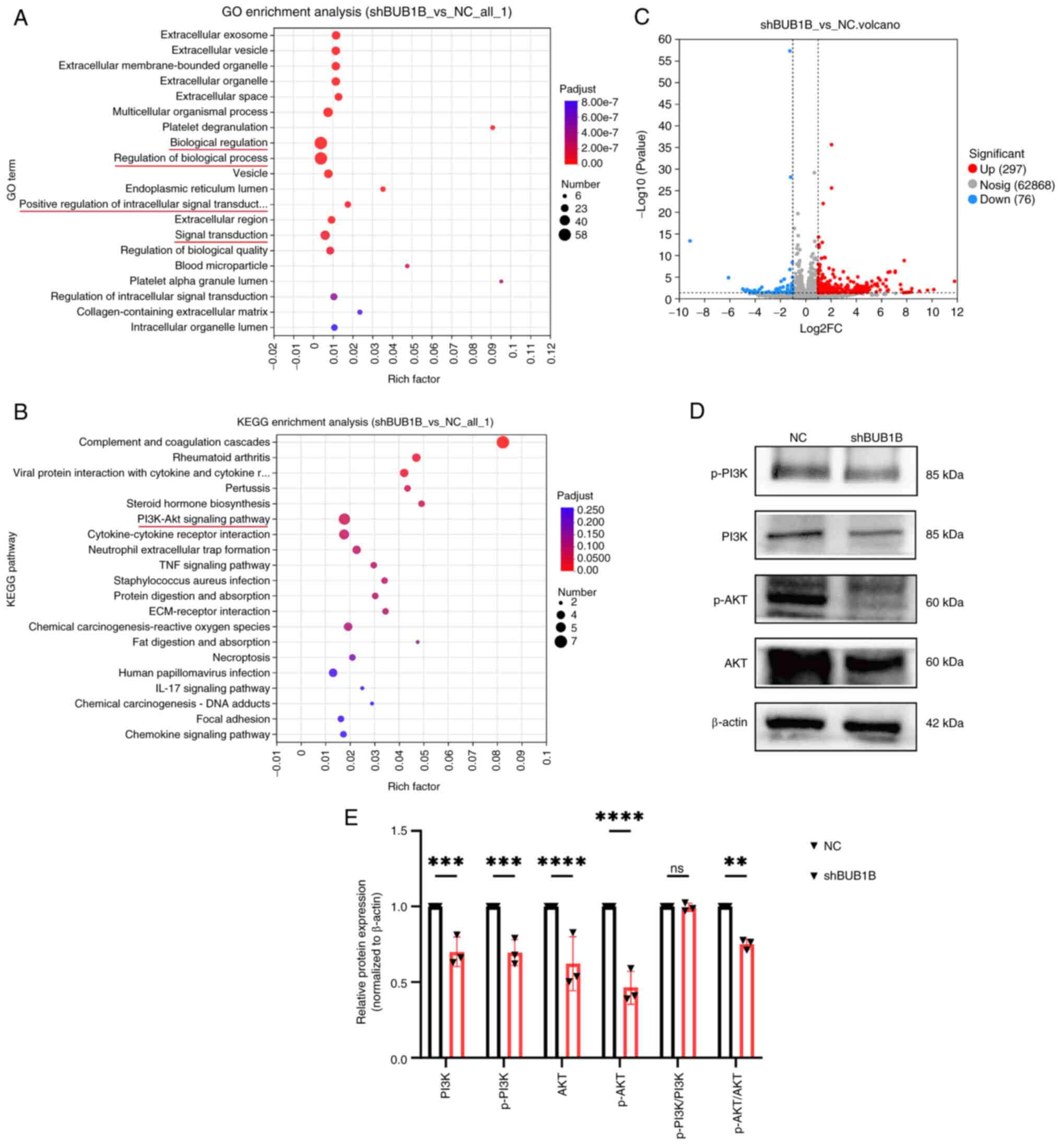 | Figure 5.BUB1B regulates the PI3K/AKT
signaling pathway. (A) GO and (B) KEGG enrichment analyses of
differentially expressed genes in irradiated shBUB1B MDA-MB-231
cells vs. irradiated NC MDA-MB-231 cells. (C) After irradiation, a
volcano plot of the genes that were differentially expressed in
irradiated shBUB1B MDA-MB-231 cells vs. irradiated NC MDA-MB-231
cells was generated (P<0.05, |log2FC|≥1). (D) Representative
images and (E) semi-quantitative analysis of the protein levels in
the PI3K/AKT signaling pathway determined by western blotting. Data
are presented as the mean ± SD, n=3. **P<0.01, ***P<0.001,
****P<0.0001 vs. NC (two-way ANOVA followed by Bonferroni's
multiple comparisons test. BUB1B, BUB1 mitotic checkpoint
serine/threonine kinase B; GO, Gene Ontology; IR, ionizing
radiation; KEGG, Kyoto Encyclopedia of Genes and Genomes; NC,
negative control; ns, not significant; p-, phosphorylated; sh,
short hairpin. |
Discussion
The present results suggested that BUB1B is
upregulated in breast cancer, with higher expression levels
significantly associated with poorer patient survival. Functional
knockdown of BUB1B suppressed breast cancer cell proliferation,
invasion, migration and tumorigenic potential. In addition, BUB1B
was suggested to be a key regulator of radioresistance in breast
cancer: BUB1B knockdown attenuated radiation-induced
G2/M cell cycle arrest and impaired HR-mediated DNA
damage repair following irradiation. Collectively, these findings
indicated that BUB1B may modulate breast cancer radiosensitivity by
orchestrating cell cycle progression and DNA repair capacity,
establishing it as a promising therapeutic target for improving
radiation efficacy in breast cancer treatment.
Increasing evidence has indicated that BUB1B drives
malignant progression in hepatocellular carcinoma, extrahepatic
cholangiocarcinoma, lung adenocarcinoma and renal cell carcinoma by
promoting cell proliferation and oncogenic signaling pathways,
which is associated with poor patient prognosis (19,20,31,32).
For example, in extrahepatic cholangiocarcinoma, aberrant BUB1B
upregulation is significantly associated with a shorter overall
survival and disease-free survival. Mechanistically, BUB1B promotes
tumor cell proliferation, migration and invasion via activation of
the JNK/c-Jun signaling axis (19).
Additionally, knockdown of BUB1B in human breast cancer cells has
been shown to inhibit carcinomatous growth and induce chromosomal
abnormalities (33). These findings
align with the current study, which confirmed that BUB1B was
upregulated in breast cancer and demonstrated a significant
association between high BUB1B expression and shorter DMFS.
Functional analyses further revealed that BUB1B may enhance breast
cancer cell proliferation and invasiveness. Collectively, these
results established BUB1B as a tumor promoter with critical
prognostic significance in breast cancer. The long-term effects of
BUB1B on breast cancer cells, including tumor recurrence and
metastasis, require further validation in the future by
constructing nude mouse metastatic tumor models.
Certain studies have proposed that BUB1B is involved
in radioresistance in malignant tumors, but its biological
mechanism remains controversial. For example, Komura et al
(23) showed that, in bladder
cancer, BUB1B interacts with ATM proteins to enhance cellular DNA
damage repair via mutagenic NHEJ, leading to radioresistance in
bladder cancer. Ma et al (22) demonstrated that, in glioblastoma,
FOXM1 transcriptionally regulates BUB1B expression, thereby
inducing tumor cell radioresistance. By contrast, treatment with a
FOXM1 inhibitor could attenuate tumor radioresistance in
vitro and in vivo (22).
The present findings support the hypothesis that increased BUB1B
expression is associated with the development of radioresistance.
In the current study, GSEA indicated that BUB1B may modulate cell
cycle progression and DNA damage repair. Molecular biology
experiments validated that BUB1B knockdown reduced
irradiation-induced G2/M phase arrest and exacerbated
DNA damage in cells. DNA damage repair is dependent on HR and NHEJ
(34,35). G2/M phase arrest provides
a temporal window for HR repair, such that inhibiting
G2/M arrest predominantly impairs HR repair efficiency.
As RAD51 is a major component of HR-mediated DNA repair, the
present study demonstrated that BUB1B knockdown downregulated
IR-induced RAD51 protein expression, thus suggesting that BUB1B
knockdown may modulate breast cancer radiosensitivity by
suppressing HR-mediated DNA damage repair. However, the impact of
BUB1B knockdown on DNA damage repair efficiency currently lacks
more direct experimental validation, such as verification using the
direct repeat-green fluorescent protein reporter assay system.
The PI3K/AKT signaling pathway serves an important
role in regulating tumor proliferation, invasion and apoptosis
(36–38). Previous research has shown that
activation of the PI3K/AKT pathway is also associated with tumor
radiotherapy resistance (39).
Inhibition of the PI3K/AKT pathway has been reported to increase
the radiosensitivity of cancer cells in different types of tumor,
including nasopharyngeal carcinoma (40), non-small cell lung cancer (41), oral squamous cell carcinoma
(42) and glioblastoma (43,44).
In breast cancer, No et al (45) showed that inhibition of the
PI3K/AKT/mTOR signaling pathway can enhance the radiosensitivity of
SKBR3 cells by inhibiting DNA damage repair, suggesting that the
PI3K/AKT signaling pathway may be a key pathway influencing
radioresistance in breast cancer. The present results of
transcriptome sequencing analysis suggested that the PI3K/AKT
signaling pathway may be a downstream signaling pathway regulated
by BUB1B. Further studies demonstrated that the expression levels
of PI3K, p-PI3K, AKT and p-AKT were downregulated in
BUB1B-knockdown breast cancer cell lines. The concurrent reduction
in both total and phosphorylated levels of PI3K/AKT upon BUB1B
knockdown may be attributed to the synergistic interplay of
multiple mechanisms. BUB1B may enhance the expression of total
PI3K/AKT proteins through interactions with transcription
regulatory factors, or alternatively, it could modulate their total
protein levels by suppressing ubiquitin-mediated degradation. The
decreased phosphorylated forms (p-PI3K, p-AKT) might either
represent a secondary effect of reduced total protein abundance or
result from the direct regulation of phosphorylation-related
machinery by BUB1B. These results demonstrated that BUB1B may serve
as an upstream regulator of the PI3K/AKT signaling pathway.
Collectively, the findings of the current study lead to a
reasonable speculation that BUB1B may promote cell cycle arrest and
DNA damage repair in breast cancer cells by activating the PI3K/AKT
signaling pathway, thereby contributing to radioresistance.
Therefore, the combined inhibition of BUB1B and targeted
suppression of the PI3K/AKT signaling pathway could represent an
effective strategy for radiosensitization in breast cancer.
PARP inhibitors exert therapeutic effects in tumors
with HR repair deficiencies through synthetic lethality, such as in
BRCA1/2-mutated breast and ovarian cancer (46). However, they exhibit limited
efficacy in tumors with intact HR repair function. The present
study demonstrated that BUB1B knockdown inhibited the HR repair
pathway by downregulating RAD51, a key protein in HR repair. The
current findings highlight the potential of combining BUB1B
inhibition with PARP inhibitors for treating cancer insensitive to
PARP inhibitors alone.
The present study has the following limitations:
Firstly, in terms of clinical validation, the current study lacks
validation of the expression level of BUB1B in clinical samples and
its association with clinical parameters, such as patient prognosis
and treatment response. Secondly, at the experimental model level,
the functional validation in the present study was only performed
in the MDA-MB-231 cell line, and further validation in other breast
cancer cell models is needed. Furthermore, when validating the
effect of BUB1B on DNA damage repair efficiency, a control group
using DNA damage repair inhibitors was lacking. Additionally, since
both immunofluorescence and western blotting are protein-level
verification experiments, only immunofluorescence verification of
γ-H2AX was performed, not western blotting. In addition, at the
level of molecular mechanism, the detailed mechanism underlying the
regulatory effects of BUB1B on the PI3K/AKT pathway remains to be
identified and verified by further experiments. Despite the
aforementioned limitations, the present study contributed to an
increased understanding of the molecular mechanisms of breast
cancer radiosensitivity, and provides novel molecular targets and
experimental bases for the in-depth exploration of the regulatory
network of tumor radiosensitivity.
In conclusion, the present study identified BUB1B as
a gene associated with breast cancer radiotherapy resistance, which
is involved in cellular DNA damage repair, especially HR repair, by
regulating the PI3K/AKT signaling pathway. The current findings
provide a novel mechanism for breast cancer radiotherapy resistance
and suggest that BUB1B may be a potential target for improving the
efficacy of breast cancer radiotherapy.
Acknowledgements
The authors would like to thank Professor Lin Yuan
(Guangxi Medical University) for providing the laboratory.
Funding
The present study was supported by the Scientific Research and
Technology Development Program (grant no. Guike AB24010055) of the
Guangxi Zhuang Autonomous Region. This research was also supported
by the following grants: Guangxi Project for the Development and
Promotion of Appropriate Traditional Chinese Medicine Technologies
(grant no. GZSY22-70); Guangxi Project for the Development and
Application of Appropriate Medical and Health Technologies (grant
no. S2018008); and the Wuming District, Nanning City Scientific
Research and Technology Development Program (grant no.
20220117).
Availability of data and materials
The RNA-sequencing data generated in the present
study may be found in the NCBI BioProject database under accession
number PRJNA1277467 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1277467.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
XL and WZ confirm the authenticity of all the raw
data. XL conceptualized the study, developed the methodology,
conducted the investigations and drafted the original manuscript.
YW performed the investigations, and contributed to visualization
and data curation. HL carried out validation and formal analysis.
NX conducted the investigations and contributed to visualization.
WZ conceptualized the study, provided supervision, secured funding,
and revised and edited the manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The experimental protocols involving animal subjects
were carried out in compliance with ethical guidelines and received
formal approval from the Institutional Animal Care and Use
Committee at Guangxi Medical University (Ethical Approval Number
202410013).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Filho AM, Laversanne M, Ferlay J, Colombet
M, Piñeros M, Znaor A, Parkin DM, Soerjomataram I and Bray F: The
GLOBOCAN 2022 cancer estimates: Data sources, methods, and a
snapshot of the cancer burden worldwide. Int J Cancer.
156:1336–1346. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peng L, Jiang J, Tang B, Nice EC, Zhang YY
and Xie N: Managing therapeutic resistance in breast cancer: From
the lncRNAs perspective. Theranostics. 10:10360–10377. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen S, Paul MR, Sterner CJ, Belka GK,
Wang D, Xu P, Sreekumar A, Pan T, Pant DK, Makhlin I, et al: PAQR8
promotes breast cancer recurrence and confers resistance to
multiple therapies. Breast Cancer Res. 25:12023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Azria D, Brengues M, Gourgou S and
Bourgier C: Personalizing breast cancer irradiation using biology:
From bench to the accelerator. Front Oncol. 8:832018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vaidya JS, Bulsara M, Baum M, Wenz F,
Massarut S, Pigorsch S, Alvarado M, Douek M, Saunders C, Flyger HL,
et al: Long term survival and local control outcomes from single
dose targeted intraoperative radiotherapy during lumpectomy
(TARGIT-IORT) for early breast cancer: TARGIT-A randomised clinical
trial. BMJ. 370:m28362020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sousa C, Cruz M, Neto A, Pereira K,
Peixoto M, Bastos J, Henriques M, Roda D, Marques R, Miranda C, et
al: Neoadjuvant radiotherapy in the approach of locally advanced
breast cancer. ESMO Open. 4 (Suppl 2):e0006402020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang RX and Zhou PK: DNA damage response
signaling pathways and targets for radiotherapy sensitization in
cancer. Signal Transduct Target Ther. 5:602020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu Y, Song Y, Wang R and Wang T: Molecular
mechanisms of tumor resistance to radiotherapy. Mol Cancer.
22:962023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu J, Bi K, Yang R, Li H, Nikitaki Z and
Chang L: Role of DNA damage and repair in radiation cancer therapy:
A current update and a look to the future. Int J Radiat Biol.
96:1329–1338. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Piotto C, Biscontin A, Millino C and
Mognato M: Functional validation of miRNAs targeting genes of DNA
double-strand break repair to radiosensitize non-small lung cancer
cells. Biochim Biophys Acta Gene Regul Mech. 1861:1102–1118. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santivasi WL and Xia F: Ionizing
radiation-induced DNA damage, response, and repair. Antioxid Redox
Signal. 21:251–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Oorschot B, Granata G, Di Franco S,
Ten Cate R, Rodermond HM, Todaro M, Medema JP and Franken NAP:
Targeting DNA double strand break repair with hyperthermia and
DNA-PKcs inhibition to enhance the effect of radiation treatment.
Oncotarget. 7:65504–65513. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dietlein F, Thelen L and Reinhardt HC:
Cancer-specific defects in DNA repair pathways as targets for
personalized therapeutic approaches. Trends Genet. 30:326–339.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mekonnen N, Yang H and Shin YK: Homologous
recombination deficiency in ovarian, breast, colorectal,
pancreatic, non-small cell lung and prostate cancers, and the
mechanisms of resistance to PARP inhibitors. Front Oncol.
12:8806432022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Panier S and Boulton SJ: Double-strand
break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol.
15:7–18. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tian J, Wen M, Gao P, Feng M and Wei G:
RUVBL1 ubiquitination by DTL promotes RUVBL1/2-β-catenin-mediated
transcriptional regulation of NHEJ pathway and enhances radiation
resistance in breast cancer. Cell Death Dis. 15:2592024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan Wah Hak CML, Rullan A, Patin EC,
Pedersen M, Melcher AA and Harrington KJ: Enhancing anti-tumour
innate immunity by targeting the DNA damage response and pattern
recognition receptors in combination with radiotherapy. Front
Oncol. 12:9719592022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Karess RE, Wassmann K and Rahmani Z: New
insights into the role of BubR1 in mitosis and beyond. Int Rev Cell
Mol Biol. 306:223–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiao CY, Feng QC, Li CX, Wang D, Han S,
Zhang YD, Jiang WJ, Chang J, Wang X and Li XC: BUB1B promotes
extrahepatic cholangiocarcinoma progression via JNK/c-Jun pathways.
Cell Death Dis. 12:632021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou X, Yuan Y, Kuang H, Tang B, Zhang H
and Zhang M: BUB1B (BUB1 mitotic checkpoint serine/threonine kinase
B) promotes lung adenocarcinoma by interacting with zinc finger
protein ZNF143 and regulating glycolysis. Bioengineered.
13:2471–2485. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan HC and Xiang C: Aberrant expression of
BUB1B contributes to the progression of thyroid carcinoma and
predicts poor outcomes for patients. J Cancer. 13:2336–2351. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma Q, Liu Y, Shang L, Yu J and Qu Q: The
FOXM1/BUB1B signaling pathway is essential for the tumorigenicity
and radioresistance of glioblastoma. Oncol Rep. 38:3367–3375.
2017.PubMed/NCBI
|
|
23
|
Komura K, Inamoto T, Tsujino T, Matsui Y,
Konuma T, Nishimura K, Uchimoto T, Tsutsumi T, Matsunaga T,
Maenosono R, et al: Increased BUB1B/BUBR1 expression contributes to
aberrant DNA repair activity leading to resistance to DNA-damaging
agents. Oncogene. 40:6210–6222. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang X, Guo M, Ding P, Deng Z, Ke M, Yuan
Y, Zhou Y, Lin Z, Li M, Gu C, et al: BUB1B and circBUB1B_544aa
aggravate multiple myeloma malignancy through evoking chromosomal
instability. Signal Transduct Target Ther. 6:3612021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Komatsu M, Yoshimaru T, Matsuo T, Kiyotani
K, Miyoshi Y, Tanahashi T, Rokutan K, Yamaguchi R, Saito A, Imoto
S, et al: Molecular features of triple negative breast cancer cells
by genome-wide gene expression profiling analysis. Int J Oncol.
42:478–506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maubant S, Tesson B, Maire V, Ye M,
Rigaill G, Gentien D, Cruzalegui F, Tucker GC, Roman-Roman S and
Dubois T: Transcriptome analysis of Wnt3a-treated triple-negative
breast cancer cells. PLoS One. 10:e01223332015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Końca K, Lankoff A, Banasik A, Lisowska H,
Kuszewski T, Góźdź S, Koza Z and Wojcik A: A cross-platform public
domain PC image-analysis program for the comet assay. Mutat Res.
534:15–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ho SY, Wu WS, Lin LC, Wu YH, Chiu HW, Yeh
YL, Huang BM and Wang YJ: Cordycepin enhances radiosensitivity in
oral squamous carcinoma cells by inducing autophagy and apoptosis
through cell cycle arrest. Int J Mol Sci. 20:53662019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu T, Wang H, Chen Y, Wan Z, Du Z, Shen
H, Yu Y, Ma S, Xu Y, Li Z, et al: SENP5 promotes homologous
recombination-mediated DNA damage repair in colorectal cancer cells
through H2AZ deSUMOylation. J Exp Clin Cancer Res. 42:2342023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiu J, Zhang S, Wang P, Wang H, Sha B,
Peng H, Ju Z, Rao J and Lu L: BUB1B promotes hepatocellular
carcinoma progression via activation of the mTORC1 signaling
pathway. Cancer Med. 9:8159–8172. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sekino Y, Han X, Kobayashi G, Babasaki T,
Miyamoto S, Kobatake K, Kitano H, Ikeda K, Goto K, Inoue S, et al:
BUB1B overexpression is an independent prognostic marker and
associated with CD44, p53, and PD-L1 in renal cell carcinoma.
Oncology. 99:240–250. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Koyuncu D, Sharma U, Goka ET and Lippman
ME: Spindle assembly checkpoint gene BUB1B is essential in breast
cancer cell survival. Breast Cancer Res Treat. 185:331–341. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mladenov E, Mladenova V, Stuschke M and
Iliakis G: New facets of DNA double strand break repair: Radiation
dose as key determinant of HR versus c-NHEJ engagement. Int J Mol
Sci. 24:149562023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu C, Bugbee T, Dacus D, Palinski R and
Wallace N: Beta human papillomavirus 8 E6 allows colocalization of
non-homologous end joining and homologous recombination repair
factors. PLoS Pathog. 18:e10102752022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou K, Wu C, Cheng W, Zhang B, Wei R,
Cheng D, Li Y, Cao Y, Zhang W, Yao Z and Zhang X: Transglutaminase
3 regulates cutaneous squamous carcinoma differentiation and
inhibits progression via PI3K-AKT signaling pathway-mediated
Keratin 14 degradation. Cell Death Dis. 15:2522024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng D, Zhu G, Liao S, Yi W, Luo G, He J,
Pei Z, Li G and Zhou Y: Dysregulation of the PI3K/Akt signaling
pathway affects cell cycle and apoptosis of side population cells
in nasopharyngeal carcinoma. Oncol Lett. 10:182–188. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dong J, Ru Y, Zhai L, Gao Y, Guo X, Chen B
and Lv X: LMNB1 deletion in ovarian cancer inhibits the
proliferation and metastasis of tumor cells through PI3K/Akt
pathway. Exp Cell Res. 426:1135732023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dong C, Wu J, Chen Y, Nie J and Chen C:
Activation of PI3K/AKT/mTOR pathway causes drug resistance in
breast cancer. Front Pharmacol. 12:6286902021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen Q, Zheng W, Zhu L, Yao D, Wang C,
Song Y, Hu S, Liu H, Bai Y, Pan Y, et al: ANXA6 contributes to
radioresistance by promoting autophagy via inhibiting the
PI3K/AKT/mTOR signaling pathway in nasopharyngeal carcinoma. Front
Cell Dev Biol. 8:2322020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen K, Shang Z, Dai AL and Dai PL: Novel
PI3K/Akt/mTOR pathway inhibitors plus radiotherapy: Strategy for
non-small cell lung cancer with mutant RAS gene. Life Sci.
255:1178162020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu CC, Hung SK, Lin HY, Chiou WY, Lee MS,
Liao HF, Huang HB, Ho HC and Su YC: Targeting the PI3K/AKT/mTOR
signaling pathway as an effectively radiosensitizing strategy for
treating human oral squamous cell carcinoma in vitro and in vivo.
Oncotarget. 8:68641–68653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gil del Alcazar CR, Hardebeck MC,
Mukherjee B, Tomimatsu N, Gao X, Yan J, Xie XJ, Bachoo R, Li L,
Habib AA and Burma S: Inhibition of DNA double-strand break repair
by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for
radiosensitization of glioblastoma. Clin Cancer Res. 20:1235–1248.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kao GD, Jiang Z, Fernandes AM, Gupta AK
and Maity A: Inhibition of phosphatidylinositol-3-OH kinase/Akt
signaling impairs DNA repair in glioblastoma cells following
ionizing radiation. J Biol Chem. 282:21206–21212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
No M, Choi EJ and Kim IA: Targeting HER2
signaling pathway for radiosensitization: Alternative strategy for
therapeutic resistance. Cancer Biol Ther. 8:2351–2361. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun Y, Dong D, Xia Y, Hao L, Wang W and
Zhao C: YTHDF1 promotes breast cancer cell growth, DNA damage
repair and chemoresistance. Cell Death Dis. 13:2302022. View Article : Google Scholar : PubMed/NCBI
|














