Introduction
Head and neck squamous cell carcinoma is a highly
malignant cancer with a 5-year survival rate of 30–50% and is the
sixth most common cancer worldwide (1). Surgical excision of this tumor type is
difficult as it occurs in the complex site of the head and neck.
Moreover, even if the tumor is excised, the quality of life of the
patient is significantly reduced. Even after reduction or resection
of head and neck cancer by chemotherapy or surgery, recurrence or
metastasis occurs in 30% of cases (2,3). The
average survival time of metastatic/recurrent cases of head and
neck squamous cell carcinoma treated with chemotherapy alone is 6–9
months, and the 1-year survival rate is as low as 20–40%. Compared
with no treatment, chemotherapy achieves only a slight survival
benefit (4).
Drug resistance is a critical obstacle to the
effective treatment of patients with cancer. Several multidrug
resistance mechanisms have been identified, such as the expression
of drug efflux ATP binding cassette (ABC) transporters (such as
ABCB1 and ABCG2) that discharge anticancer drugs outside of cancer
cells, apoptosis resistance, decreased drug binding as a result of
gene mutations and the activation of survival signals (5). To the best of our knowledge, a
compound that effectively induces cell death in multidrug-resistant
head and neck cancer cells has not been identified.
We previously established a multi-drug resistant
cell line, which we named R HSC-3 (6). R HSC-3 cells were generated by
long-term repeated treatment of the HCS-3 human metastatic oral
squamous cell carcinoma cell line with cisplatin, the first-line
treatment drug for oral squamous cell carcinoma. The cells showed a
maximum 155-fold higher resistance against anticancer drugs such as
SN-38 and docetaxel, which have different mechanisms of action from
cisplatin (7). This cell line
expresses refractory cancer-specific proteins such as the drug
excretion transporter, ABCG2, the cancer stem cell markers, CD44,
SRY-box transcription factor 9 (SOX9) and Notch, and the poor
prognosis factor, fibroblast growth factor 9 (FGF9) (6). This cell line is a useful in
vitro model for multidrug resistance.
The present study investigated compounds that
effectively induce cell death in multidrug-resistant head and neck
cancer cells and explored the mechanism of cell death induction.
These findings may lead to the establishment of a new treatment
strategy for intractable head and neck cancer with acquired drug
resistance.
Materials and methods
Reagents
Dulbecco's Modified Eagle's Medium (DMEM) and
glucose-free DMEM was obtained from Invitrogen (Thermo Fisher
Scientific, Inc.). Fetal bovine serum (FBS; 10%), 1%
antibiotics/antimycotic solution 100X (10,000 U/ml penicillin G,
10,000 µg/ml streptomycin, 25 µg/ml amphotericin B) were purchased
from HyClone (Cytiva). The anti-receptor interacting
serine/threonine kinase 1 (RIP1/RIPK1) mouse monoclonal antibody
was purchased from BD Biosciences (cat. no. 610458).
Anti-calreticulin rabbit monoclonal antibody (cat. no. C4606) and
anti-β-actin mouse monoclonal antibody (cat. no. A5441) were
obtained from Sigma-Aldrich (Merck KGaA). Anti-gasdermin D rabbit
monoclonal antibody (cat. no. 97558) and horseradish peroxidase
(HRP)-labeled goat anti-rabbit IgG (cat. no. 7074) was from Cell
Signaling Technology, Inc. Goat anti-mouse IgG-HRP antibody was
from Santa Cruz Biotechnology, Inc. (cat. no. sc-2005) and rabbit
anti-mouse IgG H&L (HRP) was purchased from Abcam (cat. no.
ab6728). The RIP1 inhibitor,
5-((7-chloro-1H-indol-3-yl)methyl)-3-methylimidazolidine-2,4-dione
(7-Cl-O-Nec1) was obtained from Merck KGaA and the mixed lineage
kinase domain-like protein (MLKL) inhibitor, necrosulfonamide (NSA)
was from R&D Systems, Inc. N-acetyl cysteine (NAC) and
2-deoxy-D-glucose (2DG) were purchased from FUJIFILM Wako Pure
Chemical. Shikonin, acetyl shikonin and β-hydroxyisovaleryl
shikonin were purchased from Nagara Science Co., Ltd. The Pierce
BCA Protein Assay Kit was purchased from Thermo Fisher Scientific,
Inc. and the Cell Proliferation Kit I (MTT) was obtained from Roche
Diagnostics. The JC-1 MitoMP Detection Kit was purchased from
Dojindo Laboratories, Inc. The CellTiter-Glo®
Luminescent Cell Viability Assay and ROS-Glo™ Assay Kit were
obtained from Promega Corporation.
Cell culture
We established the R HSC-3 multi-drug resistant oral
squamous cell carcinoma cell line in a previous study (6). R HSC-3 cells were generated by
long-term repeated treatment of the HSC-3 human metastatic oral
squamous cell carcinoma cell line (Riken Cell Bank; cat. no.
RBC1975) with cisplatin. The R HSC-3 line was cultured in DMEM with
inactivated 10% FBS and 1% antibiotic/antimycotic solution 100X at
37°C with 5% CO2.
Cell viability assays
Two types of cell viability assays were used: The
MTT assay and the ATP-based CellTiter-Glo® Luminescent
Cell Viability Assay, which assess mitochondrial activity and
cellular energy status, respectively. Using both assays provided
complementary and comprehensive assessment of cytotoxicity. R HSC-3
cells were plated in a 96-well plate (TPP Techno Plastic Products
AG) at 1×105 cells/ml and incubated for 24 h.
Naphthoquinones were then added at various concentrations (shikonin
1–20 µM, acetyl shikonin 1–30 µM and β-hydroxyisovaleryl shikonin
1–50 µM) for an additional 48 h. For the MTT assay, MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Cell
Proliferation Kit I] was added, and the insoluble formazan product
was dissolved with 10% SDS in 0.01 M HCl. Absorbance was measured
at a wavelength of 595 nm using TECAN Infinite M Plex (Tecan Group,
Ltd.). For the ATP assay, the ATP extraction reagent
(CellTiter-Glo® Luminescent Cell Viability Assay) was
added according to the manufacturers' instructions. The plates were
incubated for 1 h, then shaken at room temperature for 1 h and
luminescence was measured in relative light units at wavelengths of
485/520 nm using TECAN Infinite M Plex, multimode microplate reader
(Tecan Group, Ltd.). In some experiments, cells were pre-treated
with 5 mM NAC for 24 h, and in some experiments, 2.5 mM NAC was
co-administered with the naphthoquinones.
Measurement of reactive oxygen species
(ROS)
R HSC-3 cells were plated in a 96-well plate at
1×105 cells/ml and incubated for 24 h. The medium was
changed from DMEM with 10% FBS to glucose-free DMEM with 10% FBS
and 10 nM 2DG (2DG-DMEM). After 24 h of incubation, 20 µM shikonin
or 30 µM acetyl shikonin was added to the media. After 6 h of
treatment, ROS was measured using the ROS-Glo™ Assay following the
manufacturer's protocol. In some experiments, 50 µM 7-Cl-O-Nec1, a
RIP1 inhibitor, and 10 µM NSA, a MLKL inhibitor, were first
incubated with the cells for 24 h. Naphthoquinones were then
administered to the pretreated cells and the ROS levels were
measured using the ROS-Glo™ Assay.
Measurement of mitochondrial
toxicity
The Mitochondrial ToxGlo™ Assay (Promega
Corporation) was used to measure mitochondrial toxicity. Cell
membrane integrity was first assessed by measuring the presence or
absence of a distinct protease activity associated with necrosis
using a fluorogenic peptide substrate
(bis-alanyl-alanyl-phenylalanyl-rhodamine 110; bis-AAF-R110) to
measure the dead cell marker protease activity. Next ATP was
measured by adding the ATP extraction reagent, resulting in cell
lysis and the generation of a luminescent signal proportional to
the level of ATP present. R HSC-3 cells were plated in a 96-well
plate at 1×105 cells/ml and cultured in glucose-free
DMEM supplemented with 1 µM galactose and 10% FBS at 37°C for 24 h.
Various concentrations of naphthoquinones were added and the cells
were cultured for 24 h. Next, 5X Cytotoxicity Reagent
(Mitochondrial ToxGlo™ Assay) was added and the cells were shaken
at room temperature for 1 min, then incubated at 37°C for 30 min.
Fluorescence was then measured at wavelengths of 485/520 nm using a
multimode microplate reader. In some experiments, the cells were
pretreated with 2.5 mM NAC for 24 h or 1.25 mM NAC was
co-administered with the naphthoquinones. Oligomycine (FUJIFILM
Wako Pure Chemical Corporation) was used as a positive control
inhibitor of oxidative phosphorylation (OXPHOS).
Lentiviral transduction and
fluorescent live cell imaging
Calreticulin-red fluorescent protein (RFP) cell
lines were generated as follows. R HSC-3 cells were plated in a
6-well plate at 1×105 cells/ml in DMEM with 10% FBS and
incubated at 37°C for 24 h. R HSC-3 cells were transduced with the
pre-packaged LentiBrite™ Calreticulin-RFP-KDEL Lentiviral Biosensor
(Merck KGaA; cat. no. 17-10146) at a multiplicity of infection of
20 and incubated at 37°C with 5% CO2 for 24 h.
Lentivirus-containing medium was removed and replaced with fresh
growth medium. Cells were cultured for another 24–48 h, with medium
changes every 24 h. RFP expression was observed with a fluorescence
microscope (Olympus IX70; Olympus Corporation).
Lentivirus-infected R HSC-3 cells were seeded at
1×105 cells/ml in a 96-well microplate and cultured in
DMEM with 10% FBS at 37°C for 24 h. Hoechst 33342 was added and the
cells were incubated at 37°C for 20 min. Naphthoquinones were then
added, and images were obtained and evaluated using an HS
all-in-one fluorescence microscope (BIOREVO BZ-9000; Keyence
Corporation) and data processing software (BZ-II observation
application, BZ-II image analysis application; version 1.42;
Keyence Corporation).
Immunoblot analyses
R HSC-3 cells were treated with 1, 3, 5, 6 or 8 µM
shikonin in 2DG-DMEM for 5 h. Whole cell extracts were obtained
using 10X RIPA buffer (Cell Signaling Technology, Inc.) with 1 mM
PMSF plus protease inhibitor cocktail (Complete, EDTA-free; Roche
Diagnostics). Total protein concentration in the lysates was
assayed using the PierceTM BCA Protein Assay Kit (Thermo
Fisher Scientific, Inc.). Protein extracts (50 µg) were separated
on 8% SDS-PAGE gels and transferred to a polyvinylidene difluoride
membrane. Membranes were blocked with 10% non-fat milk for 1 h at
room temperature and then incubated with the following primary
antibodies at 4°C overnight: Anti-calreticulin (1:10,000),
anti-RIP1 (1:1,000), anti-gasdermin D (1:1,000) and anti-β-actin
(1:10,000). Following the primary antibody incubation, the
membranes were incubated with secondary HRP-conjugated goat
anti-mouse (1:10,000) or anti-rabbit IgG (1:10,000) antibodies for
1 h at room temperature. The bands were visualized using Clarity
Western ECL Substrate Peroxide solution (Bio-Rad Laboratories,
Inc.). The blots were developed and images analyzed with
Luminescent Image Analyzer LAS-3000 (FUJIFILM Corporation) and
Science Lab 2001 Image Gauge version 4.0 (FUJIFILM
Corporation).
JC-1 mitochondrial membrane potential
assay
Mitochondrial membrane potential (Dj m) was assessed
using the JC-1 MitoMP Detection Kit (Dojindo Laboratories, Inc.)
according to the manufacturer's instructions. Cells were seeded and
treated with naphthoquinones as aforementioned. Cells were
incubated with 1 mM JC-1 dye at 37°C for 30 min, followed by
Hoechst 33342 staining (1:2,000 in PBS) for 1 h. Fluorescence
images were acquired using a BZ-X810 all-in-one fluorescence
microscope (Keyence Corporation) and analyzed with BZ-H4CM software
(Keyence Corporation).
Statistical analysis
All quantitative data are presented as mean ± SD
from at least three independent experiments. The unpaired Student's
t-test was used to analyze statistical differences between two
groups of data, while two-way ANOVA followed by Tukey's post hoc
test was applied for multiple comparisons. P<0.05 was considered
to indicate a statistically significant difference. Statistical
analysis was performed using Mac Ver.3.0 (Esumi Co. Ltd.) and EZR
software (Easy R) version 1.68 (Saitama Medical Center, Jichi
Medical University) (8).
Results
Naphthoquinones induce cell death of
multidrug-resistant oral squamous cell carcinoma cells
We previously generated a multidrug-resistant head
and neck squamous cell line, named R HSC-3, by culturing the HSC-3
human head and neck squamous cell carcinoma cell line in
cisplatin-containing media (6). R
HSC-3 cells were 29 times more resistant to cisplatin than
wild-type HSC-3 cells. In the present study the half maximal
inhibitory concentration (IC50) values of various
anticancer drugs and naphthoquinones in R HSC-3 cells were examined
using MTT assays. The IC50 values of all examined drugs
were higher in R HSC-3 cells compared with parental cells (Table I). Notably, the IC50
values of shikonin and acetylshikonin, which are naphthoquinones,
were ≤3-fold higher in R HSC-3 cells compared with parental cells
and did not show a significant difference between the parental
strain and resistant strain. These data indicated that
naphthoquinones may exhibit potency against multidrug-resistant
cancer cells.
 | Table I.IC50 values in
drug-resistant and parental HSC-3 cells. |
Table I.
IC50 values in
drug-resistant and parental HSC-3 cells.
| Drug | Unit | IC50 in
HSC-3a (µM) | IC50 in
R HSC-3a (µM) | P-value |
|---|
| Cisplatin | µM | 0.68±0.17 | 20.30±6.32 | P<0.001 |
| SN-38 | nM | 7.11±1.68 | 22.18±3.70 | P<0.001 |
| Docetaxel | nM | 0.90±0.20 | 6.57±1.16 | P=0.002 |
| Erlotinib | µM | 0.21±0.04 | 31.90±2.00 | P<0.001 |
| Doxorubicin | nM | 26.57±6.00 | 223.63±15.70 | P=0.028 |
| Shikonin | µM | 4.37±0.53 | 8.10±0.58 | P=0.003 |
| Acetyl
shikonin | µM | 7.83±1.40 | 23.90±6.50 | P<0.001 |
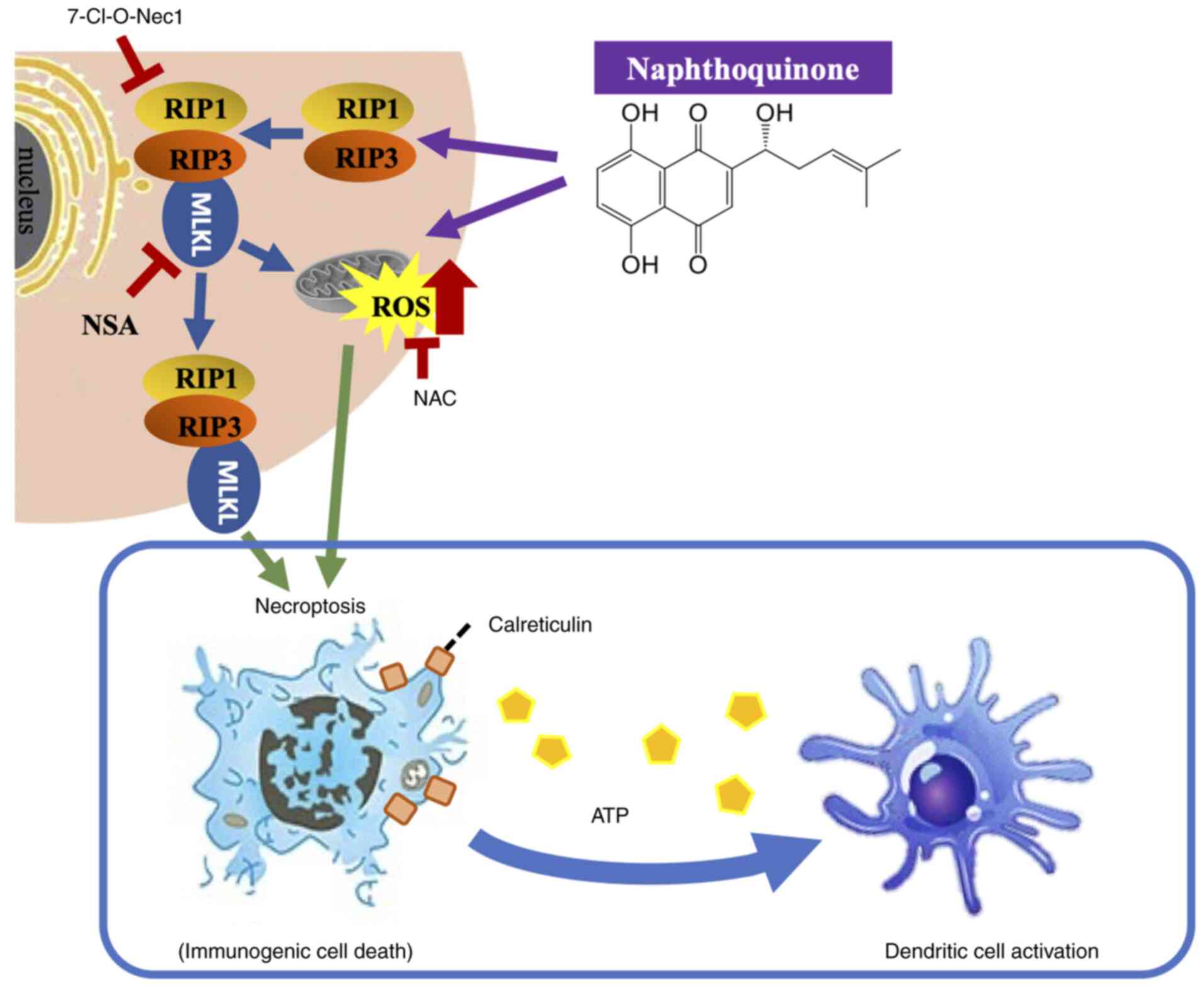
Naphthoquinones induce expression of
the necroptosis-associated protein, RIP1
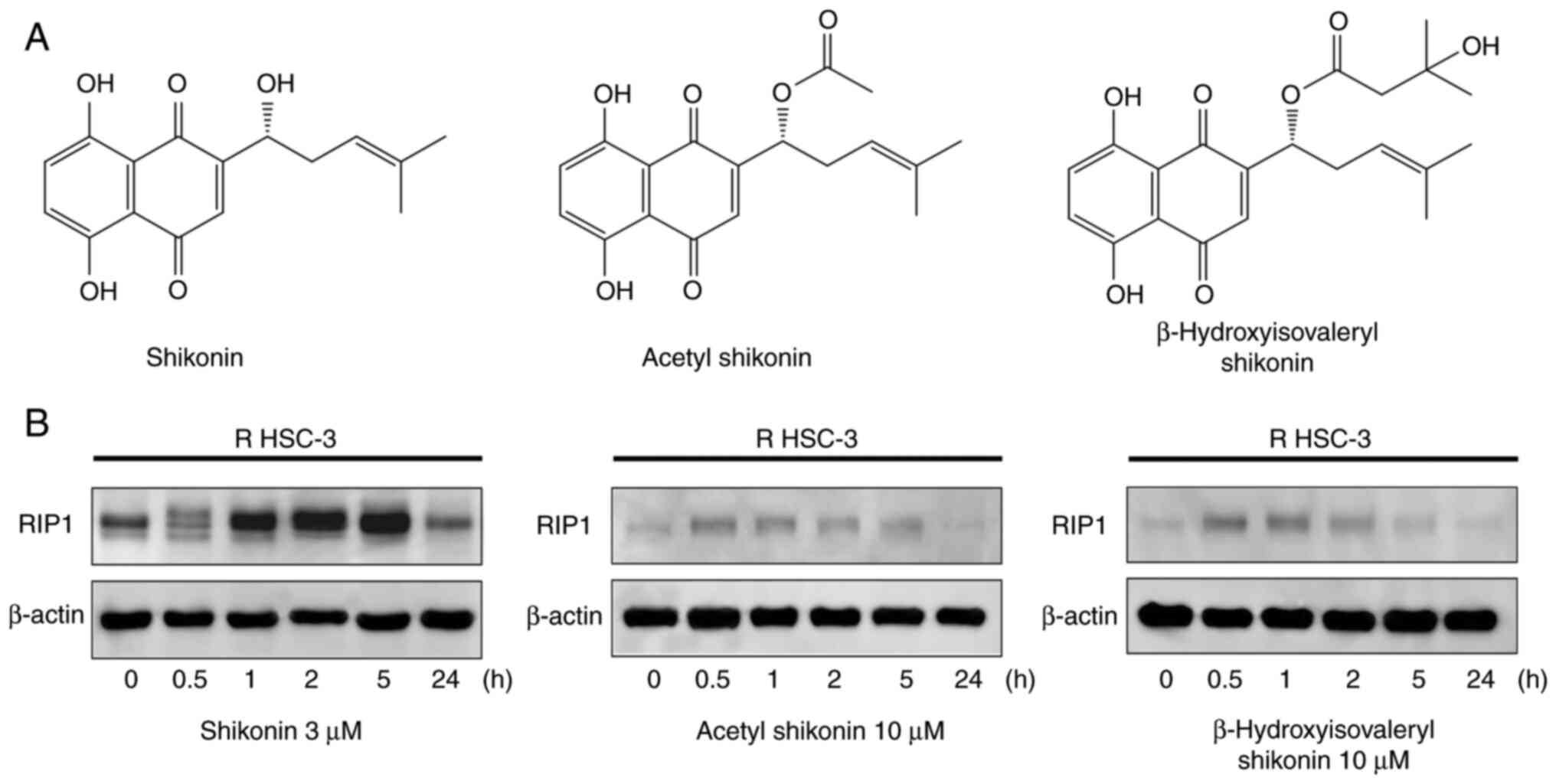
Naphthoquinones are natural red pigment components
isolated from the root of Lithospermum erythrorhizon. The
chemical structures of 1,4-naphthoquinones, including shikonin
(5,8-dihydroxy-2-[(1R)-1-hydroxy-4-methyl-3-penten-1-yl]-1,4-naphthalenedione),
acetylshikonin and β-hydroxyisovaleryl shikonin, are shown in
Fig. 1A. Shikonin is a compound
with an OH group at the β-position of the isoprene side chain of
the naphthoquinone skeleton. Acetyl shikonin and
β-hydroxyisovaleryl shikonin have side chains in which an acetyl
group and a β-hydroxyisovaleryl group, respectively, are
ester-bonded as substituents to the OH group. The mechanism of cell
death of R HSC-3 cells caused by naphthoquinones were next
examined. The induction of cleaved poly(ADP-ribose) polymerase
(PARP), a marker of apoptosis induction, was observed by
immunoblot, but reproducible cleaved PARP expression could not be
obtained (data not shown). Therefore, other forms of cell death
were explored. RIP1 is a kinase involved in necroptosis, one of the
forms of non-apoptotic cell death. Treatment with all
naphthoquinones led to the induction and subsequent decrease of
RIP1 within 0.5 to 5 h (Fig. 1B).
The observed decrease in RIP1 expression after its initial
induction may reflect its degradation through caspase-dependent or
proteasomal pathways, which has been previously reported (9).
Effect of naphthoquinones on cell
viability and the involvement of oxidative stress
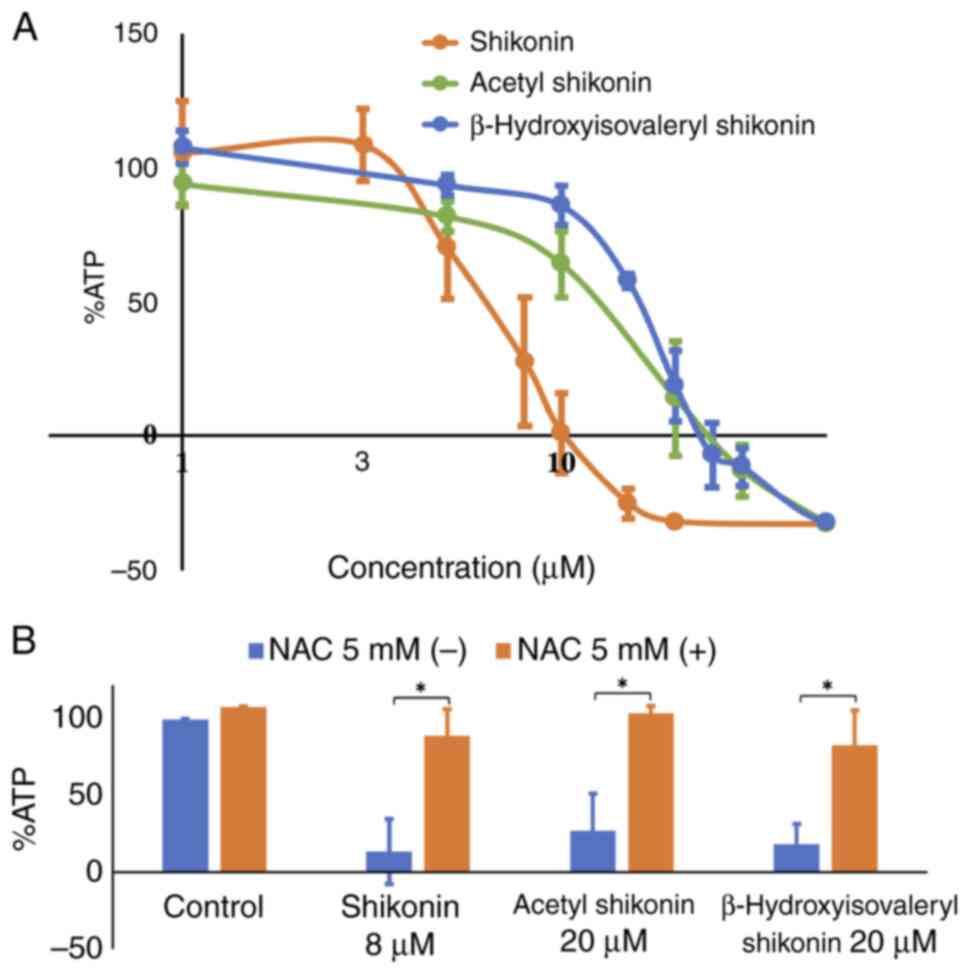
Next, the effects of naphthoquinones on the cell
viability of drug-resistant cells were evaluated using assays that
quantify the ATP levels. The treatment of R HSC-3 cells with all
naphthoquinones reduced the ATP levels in a concentration-dependent
manner, reflecting cell death (Fig.
2A). The IC50 values of shikonin, acetyl shikonin
and β-hydroxyisovaleryl shikonin were 6.0, 12.7 and 15.7 µM,
respectively. These results indicate that the IC50
values of acetyl shikonin and β-hydroxyisovaleryl shikonin, whose
OH group is modified, were 2–2.6 times higher than that for
shikonin, which has a free OH group on its side chain.
Oxidative stress is a significant inducer of
necroptosis, primarily through the production of ROS that activate
necroptosis signaling pathways (10). Thus, whether the
naphthoquinone-induced cell death is related to oxidative stress
was next examined using the antioxidant, NAC. Treatment with NAC
prevented the cytotoxicity induced by naphthoquinones and restored
viability to near control levels (Fig.
2B). Simultaneous treatment of 5 mM NAC significantly increased
survival from 27.4 to 103.8% with shikonin at 8 µM (P=0.018), 13.7
to 89.3% with acetyl shikonin at 20 µM (P=0.037) and 18.6 to 83.1%
with β-hydroxyisovaleryl shikonin at 20 µM (P=0.025). NAC alone did
not cause cytotoxicity. This result indicated that oxidative stress
is involved in the induction of cell death in multidrug-resistant
head and neck cancer cells caused by naphthoquinones.
Changes in the mitochondrial membrane
potential by naphthoquinones
In total, >90% of ROS in cells is produced in
mitochondria (11). Excess ROS
causes oxidative stress damage in cells and mitochondrial
dysfunction (12,13). To examine whether mitochondrial ROS
production is involved in oxidative stress-induced cell death
caused by naphthoquinones, changes in the mitochondrial membrane
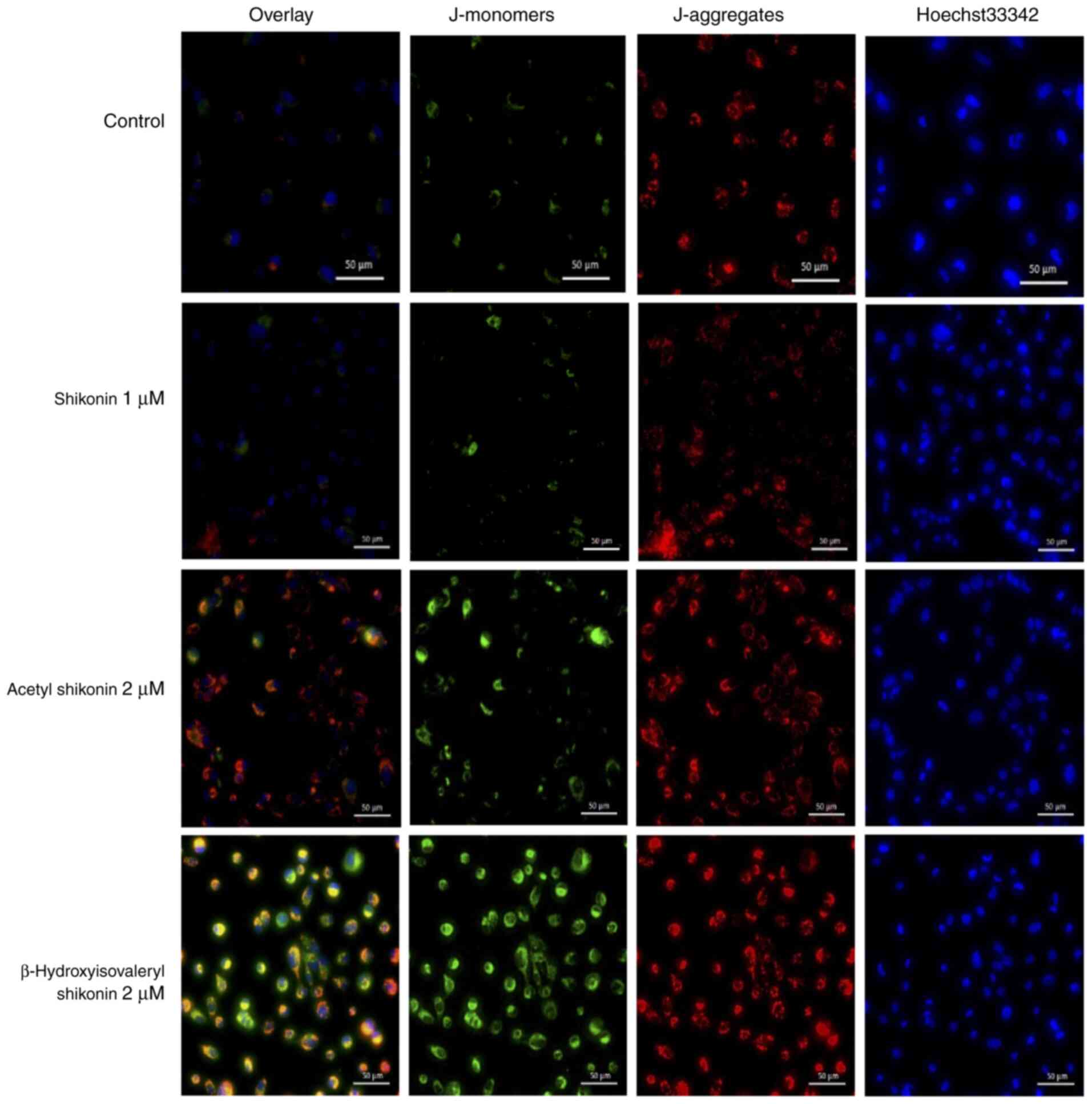
potential were investigated using the JC-1 reagent. When the
mitochondrial membrane potential is normal, JC-1 emits red
fluorescence, and when the membrane potential decreases from
apoptosis or necroptosis, JC-1 emits green fluorescence.
Hoechst33342 was used to stain nuclei and facilitate visualization
of all cells. Treatment of cells with acetyl shikonin and
β-hydroxyisovaleryl shikonin for 2 h resulted in mitochondrial
membrane potential changes and accumulation of JC-1 monomers, and
an increase in red fluorescence and green fluorescence was observed
(Fig. 3). This suggests that the
oxidative stress that induces cell death in multidrug-resistant
head and neck cancer cells by acetyl shikonin and
β-hydroxyisovaleryl shikonin is related to the mitochondrial
electron transport system. Shikonin did not appear to elicit
changes in the mitochondrial membrane potential.
Effects of changes in intracellular
energy metabolism on naphthoquinone-induced cell death
The cell death induced by acetyl shikonin and
β-hydroxyisovaleryl shikonin involves the production of ROS but
shikonin had no effects on the mitochondrial membrane potential
(Fig. 3). One possibility for the
difference in effects is that cancer cells, even under aerobic
conditions, exhibit enhanced ATP production by the glycolytic
pathway rather than mitochondrial OXPHOS (also known as the Warburg
effect) (14). ATP production in
glycolysis is reported to be ~100 times faster than that in
mitochondria (15). Thus, we
speculated that it may be difficult to observe mitochondrial
membrane changes induced by shikonin in drug-resistant head and
neck squamous cells. Therefore, the effect of naphthoquinones on
the mitochondrial respiratory chain of R HSC-3 cells were next
examined. By switching the carbon source of the medium to
galactose, the mitochondrial OXPHOS-dominant state is moderated,
and the effects of mitochondrial toxicity and ATP production can be
examined (16). To confirm that
mitochondrial toxicity is derived from ROS, the antioxidant, NAC,
was used. Mitochondrial toxicity was evaluated by simultaneously
measuring cytotoxicity (mitochondrial-derived dead cell protease)
and ATP production. Cell membrane integrity was first assessed by
measuring the presence or absence of a distinct protease activity
associated with necrosis using a fluorogenic peptide substrate
(bis-AAF-R110) to measure the dead cell marker protease activity.
Fluorescence associated with cytotoxicity was measured, ATP
extraction reagent was added and luminescence associated with the
amount of ATP as a marker of viable cell number was measured
(17). As a positive control,
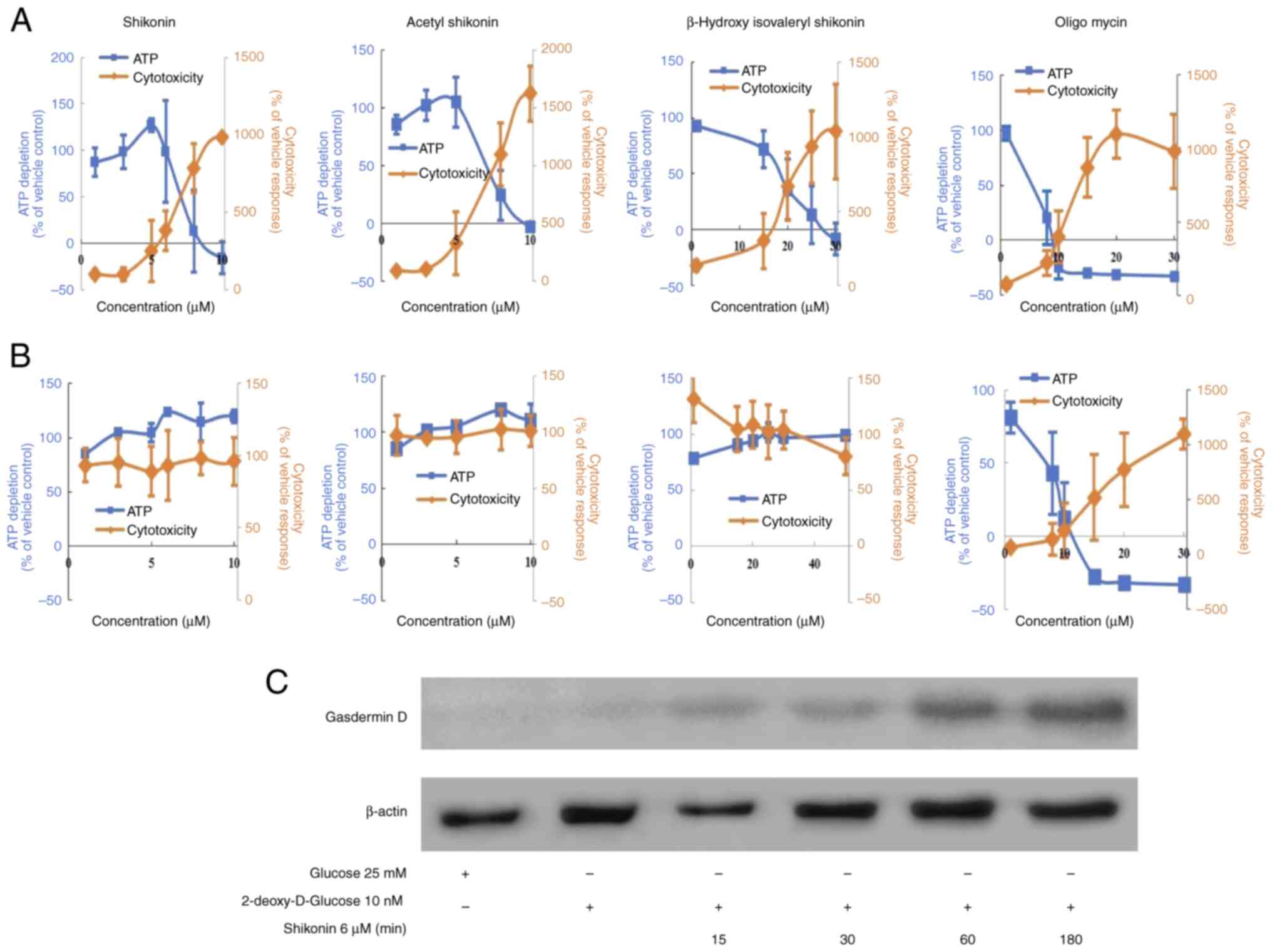
oligomycin, which is an OXPHOS inhibitor (complex V inhibitor), was
used. Under conditions in which the carbon source in the medium was
galactose, the ATP half maximal effective concentration
(EC50) values for shikonin, acetylshikonin and
β-hydroxyisovaleryl shikonin were 7.2, 7.3 and 18.4 µM,
respectively (Fig. 4A and Table II). The cytotoxicity
EC50 values, representing mitochondrial toxicity, were
3.5, 3.4 and 11.8 µM, respectively (Table II).
| Table II.Effects of naphthoquinones under
mitochondrial oxidative phosphorylation conditions was evaluated by
Mitochondrial ToxGlo™ assay. |
Table II.
Effects of naphthoquinones under
mitochondrial oxidative phosphorylation conditions was evaluated by
Mitochondrial ToxGlo™ assay.
| Drug | Cytotoxicity
EC50 (µM) | ATP EC50
(µM) |
|---|
| Shikonin | 3.47 | 7.18 |
| Acetyl
shikonin | 3.43 | 7.27 |
| β-Hydroxy
isovaleryl shikonin | 11.77 | 18.43 |
|
Oligomycina | 6.45 | 3.44 |
The cell viability (%ATP amount) decreased in
response to naphthoquinones and oligomycin in a
concentration-dependent manner; this was accompanied by an increase
in cytotoxicity (mitochondrial-derived dead cell protease)
(Fig. 4A). However, these effects
were almost abolished when cells were co-treated with the
antioxidant NAC (Fig. 4B and
Tables II and III, where Table II shows the results without NAC and
Table III shows the results with
NAC). These results revealed that naphthoquinone-induced cell death
of drug-resistant cancer cells involved mitochondrial toxicity
caused by ROS production. Mitochondrial toxicities that induce
mitochondrial ROS and/or loss of transmembrane potential could
cause pyroptosis (18), which is a
process mediated by the pore-forming cleavage of gasdermin D. To
further verify the properties of shikonin-induced cell death under
OXPHOS, the presence of gasdermin D was analyzed. As a result,
Shikonin induced the expression of gasdermin D under OXPHOS in R
HSC-3 cells (Fig. 4C).
| Table III.Effects of naphthoquinones under
mitochondrial oxidative phosphorylation conditions and in the
presence of the antioxidant, N-acetyl cysteine, was
evaluated by Mitochondrial ToxGlo™ assay. |
Table III.
Effects of naphthoquinones under
mitochondrial oxidative phosphorylation conditions and in the
presence of the antioxidant, N-acetyl cysteine, was
evaluated by Mitochondrial ToxGlo™ assay.
| Drug | Cytotoxicity
EC50 (µM) | ATP EC50
(µM) |
|---|
| Shikonin | >10.00 | >10.00 |
| Acetyl
shikonin | >20.00 | >10.00 |
| β-Hydroxy
isovaleryl shikonin | >50.00 | >50.00 |
|
Oligomycina | 5.57 | 2.42 |
Effects of necroptosis inhibitors on
oxidative stress induced by naphthoquinones
To explore the relationship between
mitochondrial-derived ROS and necroptosis signals, 7-Cl-O-Nec1, a
RIP1 inhibitor, and NSA, an MLKL inhibitor, were used as
necroptosis inhibitors. RIP1 is a signaling molecule for early
necroptosis, and MLKL is a kinase-like molecule that induces pore
formation and destruction of the cell membrane (19–22).
The ROS production induced by naphthoquinones was suppressed by
pretreatment with the RIP1 and MLKL inhibitors (Fig. 5). Under 2DG culture conditions,
shikonin treatment markedly increased ROS production. When compared
with the 2DG + shikonin group, co-treatment with the RIP1 inhibitor
slightly reduced ROS by 21% but this reduction was not
statistically significant (P=0.661). By contrast, co-treatment with
the MLKL inhibitor significantly reduced ROS levels by 59% compared
with the 2DG + shikonin group (P=0.0019). For acetyl shikonin, ROS
levels were reduced by 37% with RIP1 inhibitor (P=0.999) and 71%
with MLKL inhibition (P=0.994) compared with the corresponding 2DG
+ acetyl shikonin group, but neither reduction reached statistical
significance. These results suggest that ROS induction by
naphtoquinones depends more strongly on MLKL than on RIP1, although
the effect of acetyl shikonin was less pronounced.
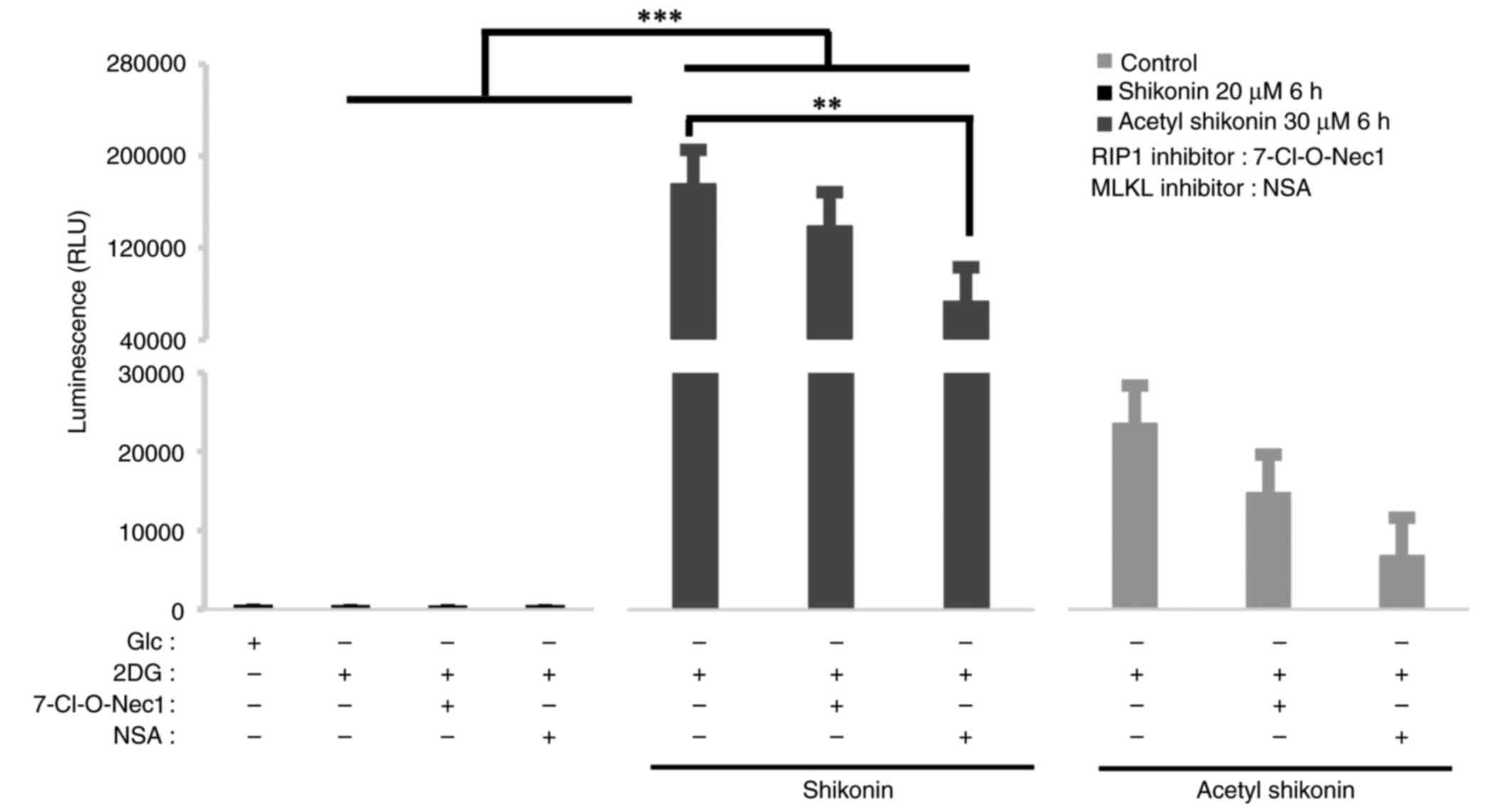 | Figure 5.Effect of naphthoquinone-induced ROS
production in combination with necroptosis inhibitors on R HSC-3
cells. ROS assay of R HSC-3 cells pretreated with RIP1 inhibitor
(50 µM, 7-O-Cl-Nec1) and MLKL inhibitor (10 µM, necrosulfonamide)
before naphthoquinone treatment (20 µM shikonin or 30 µM acetyl
shikonin, 6 h) and cultured in OXPHOS conditions (culture medium
containing 10 nM 2DG) for 24 h. Naphthoquinones (20 µM shikonin or
30 µM acetyl shikonin) were applied to necroptosis inhibitor
pretreated R HSC-3 cells for 6 h, and ROS production was measured.
Data are presented as the mean ± SD (n=3). **P<0.01,
***P<0.0005. ROS, reactive oxygen species; RIP1, receptor
interacting protein 1 kinase; MLKL, mixed lineage kinase-domain
like; OXPHOS, oxidative phosphorylation; 2DG, deoxy-D-glucose; Glc,
glucose; NSA, necrosulfonamide; RLU, relative fluorescence
units. |
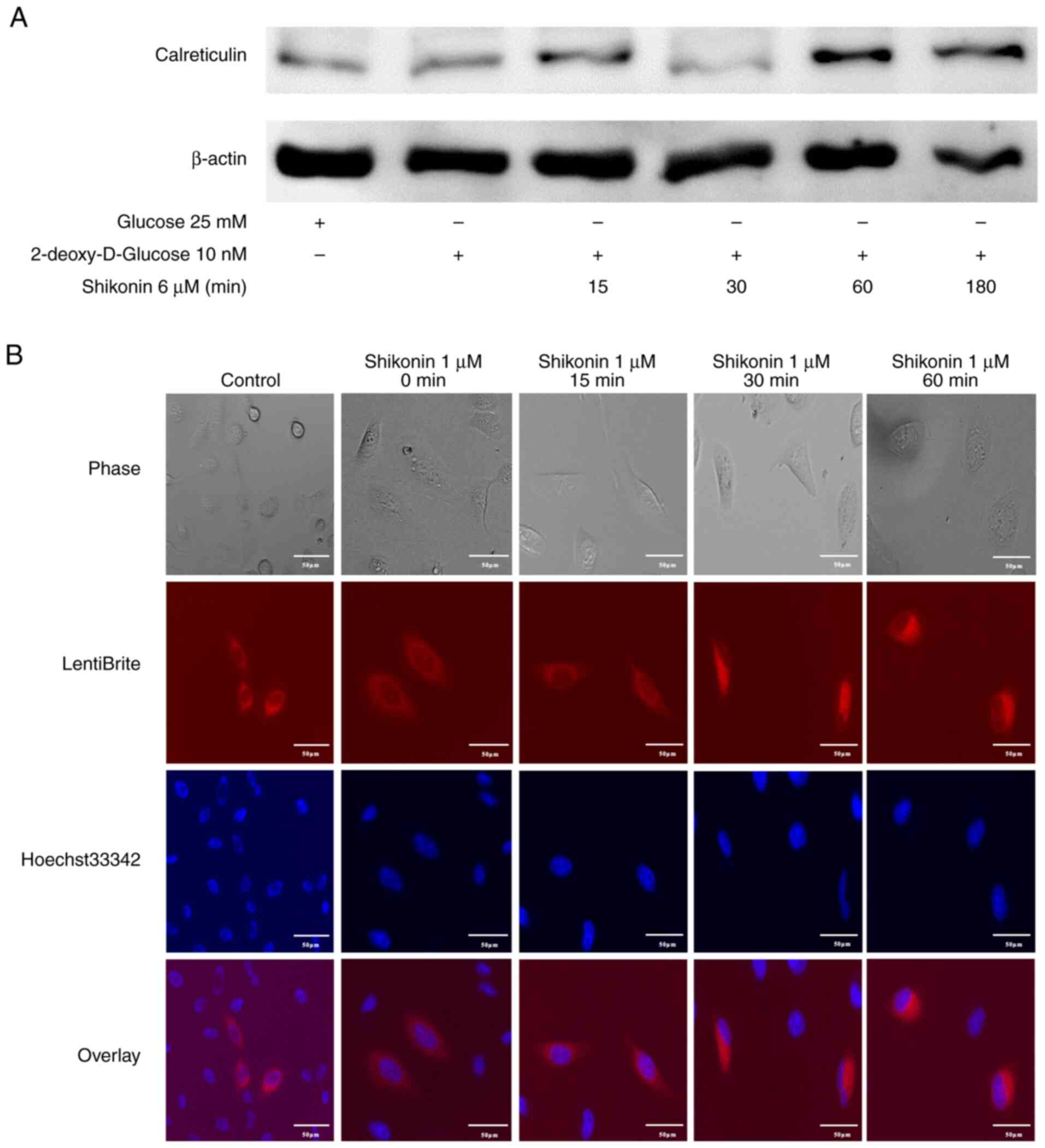
Induction of the damage-associated
molecular pattern (DAMP) molecule calreticulin in R HSC-3 cells by
shikonin
Cancer cells undergoing necroptotic cell death
release DAMPs, which are immunogenic and potentiate cancer
progression (23). DAMPs, such as
extracellular release of ATP and exposure of calreticulin to the
cell membrane surface, are important indicators of immunogenic cell
death (ICD), a form of cell death that induces host immune
responses. Calreticulin has been reported to act as an ‘eat me’
signal for dendritic cells (24,25).
Therefore, it was examined whether necroptosis induced by
naphthoquinones induces the expression of calreticulin, one of the
ICD-related DAMP molecules. R HSC-3 cells cultured for 2 h in
2DG-DMEM, which induces OXPHOS, were treated with shikonin for 5 h
and the expression of calreticulin was examined. The induction of
calreticulin in R HSC-3 cells treated with shikonin was observed
(Fig. 6A). Next, live cell imaging
of intracellular calreticulin in R HSC-3 cells infected with
lentivirus expressing calreticulin-RFP was performed. The
fluorescence intensity in the cytoplasm increased after 30 min of
shikonin treatment (Fig. 6B). This
result indicated that drug-resistant cancer cell death caused by
naphthoquinones may result in ICD, which induces tumor
immunity.
Discussion
In the present study, compounds that induce cell
death in multidrug-resistant head and neck cancer cells were
searched for. The investigation identified naphthoquinones and the
mechanism of their cell death induction activity were further
explored. These results may aid in the development of new
therapeutic agents for multidrug-resistant head and neck cancer
cells. In a cytostatic test using the R HSC-3 multidrug-resistant
head and neck cancer cell line, naphthoquinones, including
shikonin, were effective at reducing cell viability at a low
concentration, with the IC50 values ≤3 times those of
chemotherapeutic drugs. This suggests that naphthoquinones may be
useful for head and neck cancer cells with acquired multidrug
resistance. Shikonin, acetyl shikonin and β-hydroxyisovaleryl
shikonin were analyzed; the latter two are naphthoquinones with
different side chains. All tested naphthoquinones exhibited
cytotoxic effects in cell viability assays. The IC50
results indicated that the side-chain OH group was related to the
strength of cytotoxicity. All three naphthoquinones induced RIP1
expression in the R HSC-3 multidrug-resistant cancer cell line,
suggesting that the common naphthoquinone skeleton was involved in
the necroptosis-inducing activity.
The mechanism by which naphthoquinones induce
necroptosis in multidrug-resistant head and neck cancer cells were
further explored in the present study. Addition of the antioxidant,
NAC, inhibited the cytotoxicity induced by naphthoquinones,
suggesting that ROS, which cause oxidative stress, are involved in
the cell death caused by naphthoquinones. In total, >90% of
intracellular ROS are derived from mitochondria (11). Next, the mitochondrial membrane
potential was measured using the membrane-permeable JC-1 dye.
Treatment with naphthoquinones, except for shikonin, resulted in an
accumulation of JC-1 monomers. These results suggest that
naphthoquinones induce ROS production by altering the mitochondrial
membrane potential. By contrast, another study reported that
shikonin induces ROS production through mitochondrial
depolarization (26). This suggests
that the fluorescence of the mitochondrial membrane potential may
be shorter than the measurement time, which may be why the changes
in the mitochondrial membrane potential induced by shikonin were
not detected in the present study. The absence of detectable
mitochondrial membrane depolarization by shikonin may be due to
transient changes that were not captured at the time of measurement
or to the generation of ROS from non-mitochondrial sources such as
NADPH oxidases (27,28).
Cancer cells predominantly generate ATP produced
through glycolysis rather than mitochondrial OXPHOS, a phenomenon
known as the Warburg effect (14).
In the present study, to investigate the involvement of
mitochondria in ROS induced by naphthoquinones, ROS were measured
under conditions in which energy metabolism was tilted toward
OXPHOS dominance. Naphthoquinones induced the generation of ROS in
R HSC-3 cells in a concentration-dependent manner. ROS production
was stronger with shikonin, showing a difference of >7-fold
compared with acetyl shikonin. When the carbon source was changed
from glycolysis-dependent glucose to mitochondrial-dominant
galactose, treatment with naphthoquinones resulted in
concentration-dependent decreases in ATP levels and increase in
dead cell proteases. This suggests that cell death induced by
naphthoquinones caused mitochondrial dysfunction manifested as a
decrease in ATP levels. The increased mitochondrial toxicity and
decreased ATP levels caused by naphthoquinones were recovered by
the addition of NAC. This indicated that mitochondrial dysfunction
caused by naphthoquinones was due to the production of ROS. The
positive control oligomycin, an OXPHOS inhibitor (complex V
inhibitor) was not affected by NAC. This suggests that
naphthoquinones likely have an effect upstream of mitochondrial
complex V to induce cell death.
To clarify the relationship between
mitochondrial-derived ROS and necroptosis signals, two necroptosis
inhibitors of RIP1 and MLKL were used. RIP1 is an early necroptosis
signaling molecule, and MLKL is a kinase-like molecule that induces
the formation and destruction of membrane pores (29). ROS production induced by
naphthoquinones was suppressed by pretreatment with the necroptosis
inhibitors. MLKL inhibition led to a higher inhibitory effect on
ROS production than RIP1 inhibition. This suggests that
naphthoquinone-induced ROS-induced cell death depends more on the
MLKL execution factor than on RIP1. These results indicate that
naphthoquinones induce necroptotic cell death in
multidrug-resistant cancer cells by oxidative stress damage
mediated by mitochondria-derived ROS.
The present study showed that naphthoquinones induce
necroptosis in multidrug-resistant head and neck cancer cells.
Furthermore, naphthoquinones induced necroptosis signals, including
activation of the RIP1 and MLKL pathways, mediated by
mitochondria-derived oxidative stress. The possibility of
immunogenicity as a result of necroptosis was also suggested. In
recent years, the induction of non-apoptotic cell death has
attracted significant interest in the study of therapy-resistant
and refractory cancer types. Among the cell death mechanisms,
necroptosis has been increasingly recognized as a distinct form of
programmed cell death (30,31). RIP1, RIP3 and MLKL have been
identified as regulators of necroptosis (32–35). A
signal transduction pathway through the RIP1-RIP3-MLKL complex
plays a key role in necroptosis (36–41).
Necroptosis causes cell rupture as a result of morphological
changes such as cell swelling, mitochondrial swelling or rupture
and organelle membrane disruption, and subsequently cell death with
the release of DAMPs (35,42–45).
In cancer cells, DAMPs act on immune cells and are thought to
strengthen their ability to attack cancer (23). ICD is a type of programmed cell
death that stimulates an antitumor immune response. Necroptosis has
emerged as a key contributor to ICD due to its ability to induce
the release of immunostimulatory signals. DAMPs, such as the
exposure of calreticulin to the cell membrane surface and the
extracellular release of ATP, are important indicators of ICD
(46). Calreticulin acts primarily
as an ‘eat me’ signal to dendritic cells (24,25),
and cancer-specific antigens are presented to cytotoxic T cells via
dendritic cells, contributing to the activation of immune cells
with high attack power against cancer (25,47). A
previous transcriptomic analysis showed that patients with head and
neck squamous cell carcinoma with high ICD-related gene signatures
have an improved prognosis and responsiveness to immune checkpoint
inhibitors compared with those with low ICD-related gene signatures
(48) This supports the potential
clinical value of ICD-inducing agents such as shikonin in
combination with immunotherapy. In the present study, it was found
that shikonin induced necroptosis in multidrug-resistant head and
neck cancer cells and induced the expression of calreticulin in a
concentration-dependent manner (Fig.
7).
The present study has several limitations. First,
whether NAC affects the uptake or metabolism of napthoquinones was
not assessed, which could potentially influence the interpretation
of its ROS-scavenging effects. Additionally, NAC is primarily
recognized as a ROS scavenger, and its influence on shikonin
pharmacokinetics is considered minimal but cannot be completely
ruled out (49,50). In the present study, it was also
observed that shikonin induced ROS production but did not alter the
mitochondrial membrane potential at 2 h post-treatment, suggesting
that any membrane depolarization might have been transient or
undetectable at this time point. Moreover, only in vitro
experiments were conducted in the present study. Future studies
using animal models, such as immunodeficient mice bearing
xenografts of drug-resistant head and neck squamous cell carcinoma
cells, are required to validate the antitumor activity of
naphthoquinones in this disease. Further research should also
examine whether naphthoquinones influence tumor immunity.
In conclusion, the present study demonstrated that
mitochondrial-derived ROS-mediated oxidative stress damage by
naphthoquinones caused necroptotic cell death in
multidrug-resistant head and neck cancer cells. Given the high
levels of ROS induced by naphthoquinones, the potential for
systemic toxicity in vivo must be carefully evaluated.
Dose-escalation studies and histopathological analysis of major
organs (such as liver, kidney and heart) should be performed to
assess safety. Co-administration of antioxidants such as NAC may
help mitigate off-target oxidative damage without compromising
antitumor efficacy (49,50). Necroptotic cell death by shikonin
releases DAMPs from multidrug-resistant cancer cells, and these may
contribute to immunogenicity (Fig.
7). This suggests that the ICD of metastatic oral cancer with
acquired multidrug resistance, which is an intractable cancer, may
be a target of the immune system and induce cytotoxic T cells with
higher selectivity. The effect of naphthoquinones on improving the
response rate of head and neck cancer to immunotherapy should be
explored in future studies.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by JSPS KAKENHI (grant
nos. 23K09401 and 20H03785).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SK performed most of the experiments and drafted the
manuscript. NU contributed to the experimental design and drafted
the manuscript. HM performed some experiments and data analysis. MA
performed some experiments and data analysis. HY participated in
the study design, manuscript preparation and critically revised the
manuscript. EO contributed to the experimental conceptualization
and data interpretation and critically revised the manuscript. SK
and NU confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Naoki Umemura, http://orcid.org/0000-0002-3249-782X; Hiromi Miyazaki,
https://orcid.org/0000-0001-7719-4913; Makoto Adachi,
http://orcid.org/0000-0002-9382-7052;
Emika Ohkoshi, http://orcid.org/0000-0001-9700-9973.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI
|
|
2
|
Argiris A, Karamouzis MV, Raben D and
Ferris RL: Head and neck cancer. Lancet. 371:1695–1709. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Price KA and Cohen EE: Current treatment
options for metastatic head and neck cancer. Curr Treat Options
Oncol. 13:35–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chang JH, Wu CC, Yuan KS, Wu ATH and Wu
SY: Locoregionally recurrent head and neck squamous cell carcinoma:
Incidence, survival, prognostic factors, and treatment outcomes.
Oncotarget. 8:55600–55612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sugimoto Y, Tsukahara S, Sato S, Suzuki M,
Nunoi H, Malech HL, Gottesman MM and Tsuruo T: Drug-selected
co-expression of P-glycoprotein and gp91 in vivo from an
MDR1-bicistronic retrovirus vector Ha-MDR-IRES-gp91. J Gene Med.
5:366–376. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murakami K, Umemura N, Adachi M, Motoki M
and Ohkoshi E: ABCG2, CD44 and SOX9 are increased with the
acquisition of drug resistance and involved in cancer stem cell
activities in head and neck squamous cell carcinoma cells. Exp Ther
Med. 24:7222022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith ER, Wang JQ, Yang DH and Xu XX:
Paclitaxel resistance related to nuclear envelope structural
sturdiness. Drug Resist Updat. 65:1008812022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kanda Y: Investigation of the freely
available easy-to-use software ‘EZR’ for medical statistics. Bone
Marrow Transplant. 48:452–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ofengeim D and Yuan J: Regulation of RIP1
kinase signalling at the crossroads of inflammation and cell death.
Nat Rev Mol Cell Biol. 14:727–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie Y, Zhao G, Lei X, Cui N and Wang H:
Advances in the regulatory mechanisms of mTOR in necroptosis. Front
Immunol. 14:12974082023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Butow RA and Avadhani NG: Mitochondrial
signaling: The retrograde response. Mol Cell. 14:1–15. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pfeiffer T, Schuster S and Bonhoeffer S:
Cooperation and competition in the evolution of ATP-producing
pathways. Science. 292:504–507. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marroquin LD, Hynes J, Dykens JA, Jamieson
JD and Will Y: Circumventing the Crabtree effect: Replacing media
glucose with galactose increases susceptibility of HepG2 cells to
mitochondrial toxicants. Toxicol Sci. 97:539–547. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Niles AL, Moravec RA, Eric Hesselberth P,
Scurria MA, Daily WJ and Riss TL: A homogeneous assay to measure
live and dead cells in the same sample by detecting different
protease markers. Anal Biochem. 366:197–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miao R, Jiang C, Chang WY, Zhang H, An J,
Ho F, Chen P, Zhang H, Junqueira C, Amgalan D, et al: Gasdermin D
permeabilization of mitochondrial inner and outer membranes
accelerates and enhances pyroptosis. Immunity. 56:2523–2541.e8.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rickard JA, O'Donnell JA, Evans JM,
Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL,
Anderton H, et al: RIPK1 regulates RIPK3-MLKL-driven systemic
inflammation and emergency hematopoiesis. Cell. 157:1175–1188.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dillon CP, Weinlich R, Rodriguez DA,
Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F,
Gong YN, et al: RIPK1 blocks early postnatal lethality mediated by
caspase-8 and RIPK3. Cell. 157:1189–1202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Z, Zhou T, Sun X, Zheng Y, Cheng B,
Li M, Liu X and He C: Necroptosis in microglia contributes to
neuroinflammation and retinal degeneration through TLR4 activation.
Cell Death Differ. 25:180–189. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Z, Jiang N, Su W and Zhuo Y:
Necroptosis: A novel pathway in neuroinflammation. Front Pharmacol.
12:7015642021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wellenstein MD and de Visser KE:
Cancer-cell-intrinsic mechanisms shaping the tumor immune
landscape. Immunity. 48:399–416. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gardai SJ, McPhillips KA, Frasch SC,
Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg
PA, Michalak M and Henson PM: Cell-surface calreticulin initiates
clearance of viable or apoptotic cells through trans-activation of
LRP on the phagocyte. Cell. 123:321–334. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tesniere A, Apetoh L, Ghiringhelli F, Joza
N, Panaretakis T, Kepp O, Schlemmer F, Zitvogel L and Kroemer G:
Immunogenic cancer cell death: A key-lock paradigm. Curr Opin
Immunol. 20:504–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee MJ, Kao SH, Hunag JE, Sheu GT, Yeh CW,
Hseu YC, Wang CJ and Hsu LS: Shikonin time-dependently induced
necrosis or apoptosis in gastric cancer cells via generation of
reactive oxygen species. Chem Biol Interact. 211:44–53. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Forrester SJ, Kikuchi DS, Hernandes MS, Xu
Q and Griendling KK: Reactive oxygen species in metabolic and
inflammatory signaling. Circ Res. 122:877–902. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Pang J, Liu Z, Ge X, Zhen Y, Jiang
CC, Liu Y, Huo Q, Sun Y and Liu H: Shikonin induces programmed
death of fibroblast synovial cells in rheumatoid arthritis by
inhibiting energy pathways. Sci Rep. 11:182632021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H,
Song Y, Chen X, Shao J and Han J: Distinct roles of RIP1-RIP3
hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis.
Cell Death Differ. 21:1709–1720. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Smith CC and Yellon DM: Necroptosis,
necrostatins and tissue injury. J Cell Mol Med. 15:1797–1806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Linkermann A and Green DR: Necroptosis. N
Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q,
Luo J and Liu ZG: Mixed lineage kinase domain-like is a key
receptor interacting protein 3 downstream component of TNF-induced
necrosis. Proc Natl Acad Sci USA. 109:5322–5327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Sun L, Su L, Rizo J, Liu L, Wang
LF, Wang FS and Wang X: Mixed lineage kinase domain-like protein
MLKL causes necrotic membrane disruption upon phosphorylation by
RIP3. Mol Cell. 54:133–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moreno-Gonzalez G, Vandenabeele P and
Krysko DV: Necroptosis: A novel cell death modality and its
potential relevance for critical care medicine. Am J Respir Crit
Care Med. 194:415–428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Quarato G, Guy CS, Grace CR, Llambi F,
Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T and
Green DR: Sequential engagement of distinct MLKL
Phosphatidylinositol-binding sites executes necroptosis. Mol Cell.
61:589–601. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dhuriya YK and Sharma D: Necroptosis: A
regulated inflammatory mode of cell death. J Neuroinflammation.
15:1992018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Green DR: The coming decade of cell death
research: Five riddles. Cell. 177:1094–1107. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aaes TL, Kaczmarek A, Delvaeye T, De
Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki
B, De Groote P, et al: Vaccination with necroptotic cancer cells
induces efficient anti-tumor immunity. Cell Rep. 15:274–287. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gong YN, Guy C, Crawford JC and Green DR:
Biological events and molecular signaling following MLKL activation
during necroptosis. Cell Cycle. 16:1748–1760. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grootjans S, Vanden Berghe T and
Vandenabeele P: Initiation and execution mechanisms of necroptosis:
An overview. Cell Death Differ. 24:1184–1195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Y, Liu L, Jin L, Yi X, Dang E, Yang
Y, Li C and Gao T: Oxidative stress-induced calreticulin expression
and translocation: New insights into the destruction of
melanocytes. J Invest Dermatol. 134:183–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Obeid M, Tesniere A, Ghiringhelli F, Fimia
GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T,
Casares N, et al: Calreticulin exposure dictates the immunogenicity
of cancer cell death. Nat Med. 13:54–61. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang X, Wu S, Liu F, Ke D, Wang X, Pan D,
Xu W, Zhou L and He W: An Immunogenic cell death-related
classification predicts prognosis and response to immunotherapy in
head and neck squamous cell carcinoma. Front Immunol.
12:7814662021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bauza-Thorbrugge M, Peris E, Zamani S,
Micallef P, Paul A, Bartesaghi S, Benrick A and Wernstedt Asterholm
I: NRF2 is essential for adaptative browning of white adipocytes.
Redox Biol. 68:1029512023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu
Y, Guo M, Ji H, Xu C, Gu C, et al: N-acetyl-L-cysteine protects
against cadmium-induced neuronal apoptosis by inhibiting
ROS-dependent activation of Akt/mTOR pathway in mouse brain.
Neuropathol Appl Neurobiol. 40:759–777. 2014. View Article : Google Scholar : PubMed/NCBI
|