Cancer is a complex ecosystem that includes
malignant cells and non-malignant cells in the internal
environment, as well as the stroma, vasculature, and immune,
nervous and endocrine systems. These components of cancer coexist
and collaborate to boost the survival and proliferation of cancer
cells (1). The ecosystem of cancer
possesses various core characteristic capabilities, including
sustaining proliferative signaling, evading growth suppressors, and
avoiding immune destruction (2).
However, these core hallmark capabilities do not explain the
mechanism by which the nervous system regulates cancer growth.
Remarkably, increasing experimental evidence accumulated over the
past decade indicates that the nervous system is crucial to cancer
initiation and progression (3). The
accumulation of data has formed the basis of a new research field
called cancer neuroscience.
As an incurable malignant tumor mainly originating
in the brain, malignant glioma has become a research paradigm in
cancer neuroscience. Malignant gliomas comprise a heterogeneous
group of tumors that mainly include glioblastoma multiforme (GBM)
and other diffuse gliomas, such as grade 3 anaplastic astrocytoma
and oligodendroglioma (4). As the
type of glioma with the greatest malignancy, GBM accounts for 50.9%
of all malignant brain tumors in the Central Brain Tumor Registry
of the United States (5). Patients
with GBM have a dismal prognosis with limited treatment options,
comprising maximally safe surgery, radiotherapy, and concomitant
and maintenance treatment with temozolomide (6–10).
Although research has investigated the molecular profiles, genetic
mutations, epigenetic reprogramming, tumor cell state, and immune
microenvironment of GBM (11–14),
effective treatment methods are lacking. A crucial reason for this
situation is that the brain is dynamically harnessed as malignant
features expand (15), and
clarification of these processes could provide further insights for
the development of therapeutics.
The present review provides an updated framework for
understanding the various aspects of communication between the
nervous system and glioma. Readers will obtain insights into the
construction of neuron-glioma networks, which are constructed from
neurons and glioma cells located at the edge of the tumor, and the
mechanisms by which neurons regulate the malignant phenotype of
glioma. Readers will also understand the construction of
glioma-glioma networks, which are constructed from glioma cells
located inside the glioma by tumor microtubes (TMs), and glioma
cell-induced neural hyperexcitability. Furthermore, the present
review reveals the mechanism by which the complex interconnectivity
among the nervous system, immune system and glioma promotes tumor
growth. Finally, some potential areas of clinical translation and
new research directions were described.
As the host organ of glioma, the brain features
structural and functional networks that are organized according to
complex topological properties. Signaling and information transfer
between neural circuits permeate every facet and spatial scale of
brain function (16,17). As they originate from brain cells,
gliomas naturally exist in neural circuits. However, the specific
type of brain cells from which glioma originates remain
controversial. Because oligodendroglia precursor cells (OPCs) and
neural progenitor stem cells (NPCs) continue to proliferate
throughout life, they are considered the most likely origins of
gliomas (5,18). Liu et al (19) identified notably aberrant growth
prior to malignant tumor in OPCs but not in any other neural stem
cell (NSC)-derived lineages or NSCs themselves. Although OPCs are
not neuronal cells in the brain, they can receive direct synaptic
input from neurons at bona fide synapses. Electrophysiological
analyses confirmed that OPCs express multifarious functional
neurotransmitter receptors and respond physiologically to
presynaptically released neurotransmitters (20–22).
This characteristic is likely to be extended to glioma cells
originating from OPCs. Moreover, gliomas and neural circuits might
interact in a bidirectional manner. In short, higher neural
activity induces faster glioma growth, and the presence of glioma
increases neural activity (23).
Recently, some studies proved that GBM both disrupts neural
circuits (24) and remodels neural
circuits (25) into malignant
networks. These remodeled neural circuits both promote
glioma-genesis and their progression (26–29)
and determine the type of glioma that arises in different brain
tissues. Romero-Garcia et al (30) found that distinct glioma subtypes
exhibited different spatial profiles of occurrence. Lower-grade
gliomas appear to arise more frequently in frontal areas, whereas
GBM is often found in temporoparietal areas (30). Their study further revealed that the
preferential occurrence of low-grade glioma (LGG) versus high-grade
glioma is associated with different regional transcriptomic
characteristics and brain connectomic features in normative
populations (30), that is,
different neural circuits.
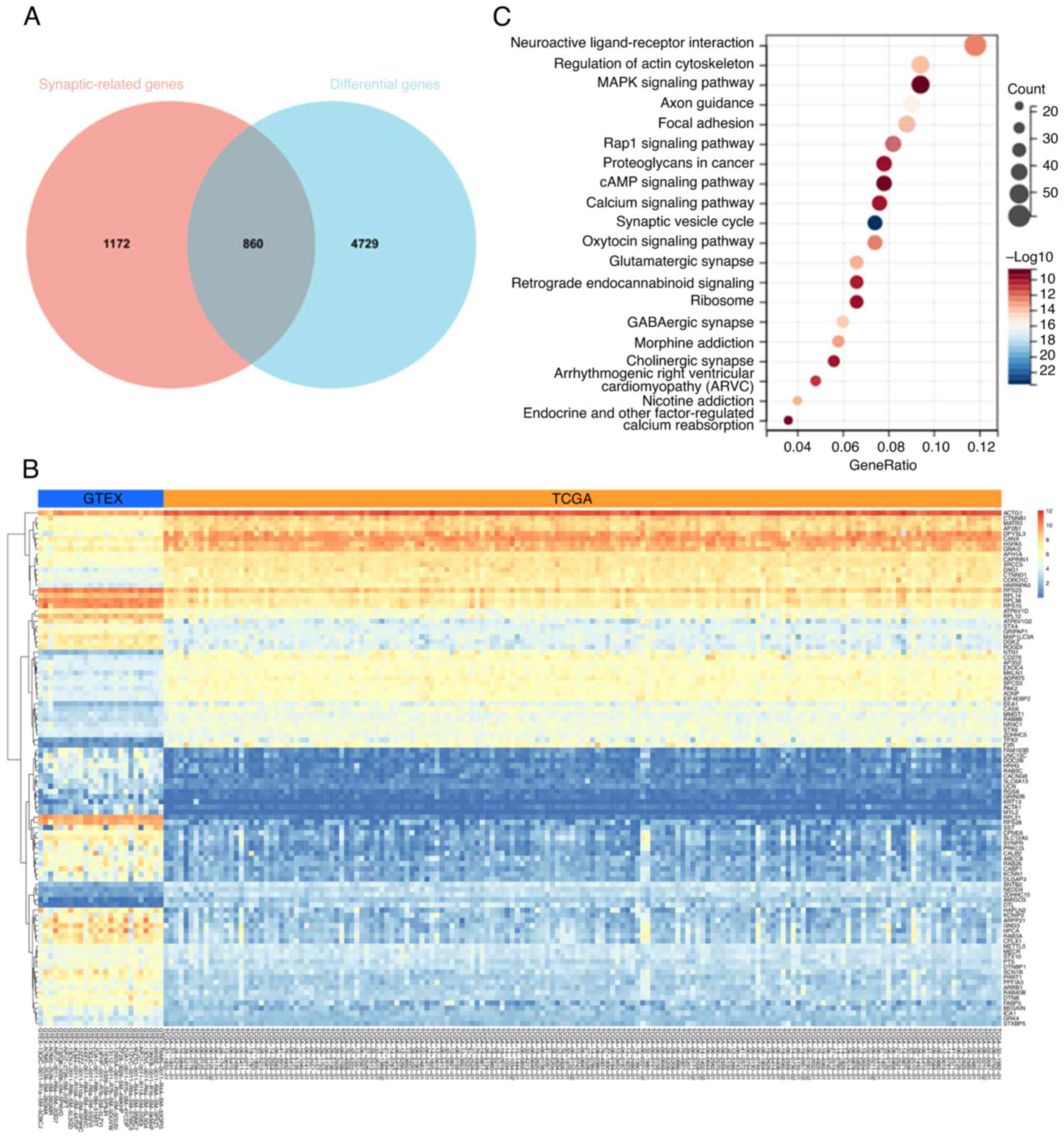
To further clarify the close correlation between GBM
and neural circuits, GBM data from The Cancer Genome Atlas (TCGA)
were analyzed and 4,729 differentially expressed genes in GBM were
identified. By implementing the intersections of genes between
differentially expressed genes in GBM and synaptic genes from
MSigDB, a Venn diagram was generated, demonstrating 860
differentially expressed genes from GBM that are related to
synapses (Fig. 1A). The heatmap in
Fig. 1B displays the expression
changes of differentially expressed genes related to synapses in
GBM. The bubble plots in Fig. 1C
illustrate that these differentially expressed genes in GBM are
significantly involved in neural circuits. From the analysis of
TCGA data, it is evident that the initiation and progression of GBM
are closely related to neural circuits.
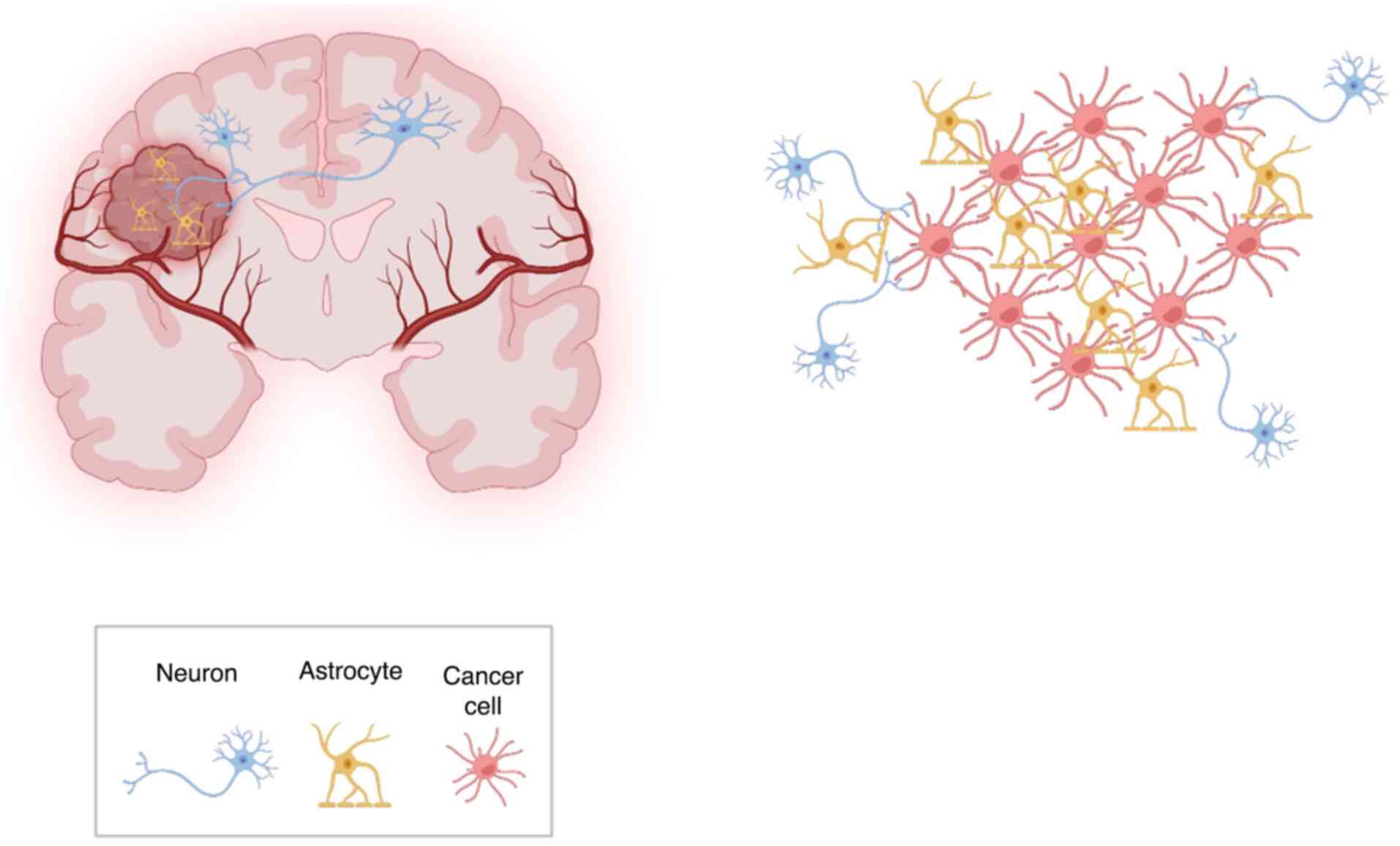
In addition, most glioma cells inside the tumor are
interconnected with each other via TMs, which are ultralong,
cytoskeletal-enriched membrane tubes. These TMs comprise the
anatomical basis of highly functional glioma-glioma networks
coupled by gap junctions (31).
Some studies consistently revealed TMs and their multicellular
networks in incurable gliomas, namely GBMs, WHO grade II–IV
astrocytoma, and K27M mutated midline gliomas (32,33).
Through TMs, glioma cells at the edge of the tumor can transmit
information sent by neurons to every glioma cell inside the tumor
(Fig. 2).
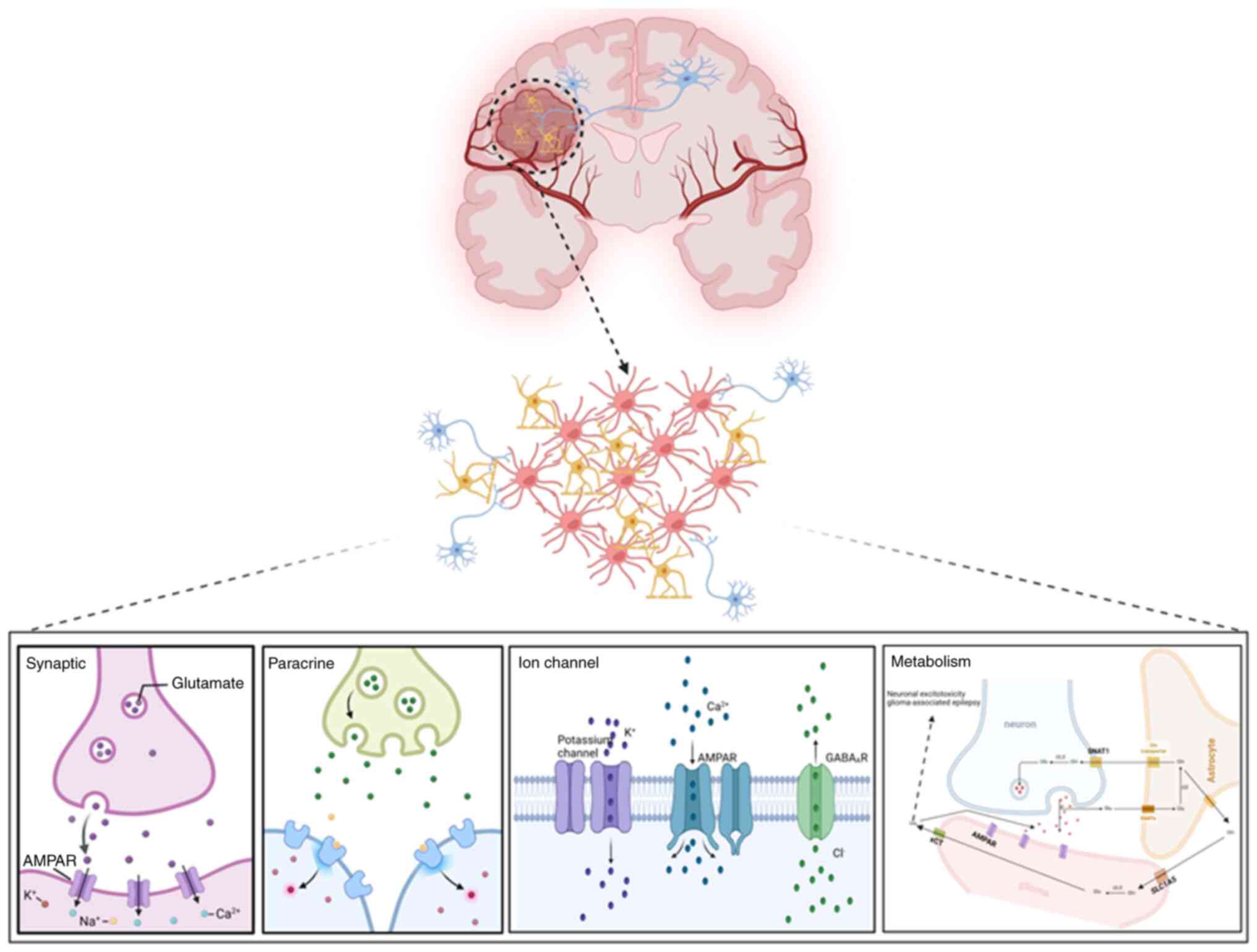
As our understanding of the cancer microenvironment
has increased, neuronal regulation has been deemed to play a
crucial role in glioma biology (34). Some mechanisms by which neurons
regulate the malignant phenotype of glioma cells have been
expounded. Through functional neuron-to-glioma synapses and
paracrine signaling factors, membrane depolarization in glioma
cells drives tumor proliferation and invasion (35). Neural activity-mediated ion channels
also promote the proliferation of glioma cells (3,36). In
addition, GBM hijacks the neuron-astrocyte glutamate-glutamine
cycle to promote tumor proliferation and invasion (37) (Fig.
3).
Although the OPC-like and NPC-like populations of
glioma cells at the rim of the tumor have been found to be enriched
in neuron-glioma synapses (38),
the circuit architecture and neural subtype in neuron-glioma
networks remain to be further elucidated. Sun et al
(39) revealed that GBM cells
rapidly incorporated into brain-wide neural circuits and displayed
different local and long-range connectivity. They also identified
miscellaneous neuro-modulatory inputs across the brain, such as
cholinergic inputs from the basal forebrain excluding glutamatergic
inputs (39). Via comprehensive
whole-brain mapping, Hsieh et al (40) demonstrated that these
glioma-innervating neurons (GINs) constantly arise in brain
regions, including different neuro-modulatory centers and specific
cortical layers, which project to the locations of glioma.
Molecular profiling unveiled that these long-range cortical GINs
are mostly glutamatergic, and subsets express both glutamatergic
and GABAergic markers. Meanwhile, local striatal GINs are mainly
GABAergic. They used electrophysiology to confirm that although
GINs share passive intrinsic properties with cortex-innervating
neurons, GINs possess different action potential waveforms
(40).
As previously mentioned, extensive structural and
functional analyses identified glutamatergic synaptic neurons as
the main neurons involved in neuron-glioma networks. Similarly as
the neuron-OPC synapses that form in the healthy brain,
neuron-glioma synapses are mainly mediated by calcium-permeable
α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)
receptors, which trigger neuronal activity-dependent currents and
membrane depolarization in glioma cells on the postsynaptic side.
Glioma cells upregulate AMPA receptors, and the AMPA receptor
phenotype of glioma cells differs from that in most neurons of the
adult brain, which is not permeable to calcium ions because of the
presence of an edited form of the GluR2 subunit. These bona fide
neuron-glioma synapses promote the growth and invasion of glioma
cells, as evidenced by genetic/pharmacological blockade of AMPA
receptors in neuron-glioma co-culture and in vivo (41). As another receptor associated with
glutamatergic synaptic neurons, N-methyl-d-aspartate receptors
(NMDARs) have been less studied in gliomas. Müller-Längle et
al (42) reported that
treatment with NMDAR antagonists abated the growth and migration of
glutamate-releasing glioma cells and increased their
radiosensitivity by inhibiting double-strand break repair. These
findings suggested that NMDAR activation facilitates the growth and
radio-resistance of glioma (42).
However, the expression of NMDARs on glioma cells and the mechanism
by which neurons regulate the phenotype of glioma cells need to be
further delineated in the future.
In addition to glutamatergic synaptic neurons, other
neurons have been found to participate in the proliferation and
invasion of glioma cells. Sun et al (39) revealed that GBM cells express
multiple types of neurotransmitter receptors, including ionotropic
and metabotropic glutamatergic, GABAergic and cholinergic
receptors, as well as serotonergic, adrenergic and dopaminergic
receptors. Furthermore, they found that acute acetylcholine
stimulation induced uninterrupted calcium oscillation and
long-lasting transcriptional reprogramming of GBM cells into a more
invasive state by the metabotropic CHRM3 receptor. In vitro
and in vivo experiments proved that knockdown of CHRM3 can
inhibit GBM cell invasion, proliferation, and survival (39). These results were confirmed by
Drexler et al (43), who
demonstrated that cholinergic neurons in the midbrain have
long-range projections to midline structures that foster
activity-dependent growth of diffuse midline glioma (DMG) via CHRM1
and CHRM3 cholinergic receptors. Regarding inhibitory neuron
synapses, GABAergic synaptic neurons release GABA, which activates
GABAA receptors in neural precursors to attenuate NSC
proliferation (44). As a modulator
of GABAergic synaptic neurons, diazepam-binding inhibitor (DBI)
suppresses GABAA receptor-mediated currents. Prior
research illustrated that GBM lacks GABAA receptor
expression. The expression of GABAA receptors is
negatively correlated with the tumor grade of glioma, and high
GABAA receptor expression predicts improved prognosis in
different types of gliomas (45).
Recently, Barron et al (46)
found that GABAergic neuron-to-glioma synapses promoted the growth
of DMG by GABAA receptors. Via NKCC1 chloride
transporter function to elevate intracellular chloride
concentrations in DMG malignant cells, GABAergic input has a
depolarizing role on DMG cells. By inducing glioma cell membrane
depolarization, the activity of GABAergic interneurons boosts DMG
proliferation. By contrast, the activity of GABAergic interneurons
did not affect the growth of hemispheric GBM (46). In addition, DBI is overexpressed in
glioma, and it inhibits GABA signaling, thereby promoting glioma
growth. However, DBI is upregulated in GBM, thereby driving tumor
growth through a GABA-independent pathway (47). Therefore, further detailed research
is needed to clarify the mechanism by which GABAergic synaptic
neurons regulate the growth of different glioma subtypes.
In addition to neuron-glioma synaptic communication,
paracrine signaling also mediates neural activity-induced glioma
growth through brain-derived neurotrophic factor (BDNF) and the
soluble synaptic adhesion protein neuroligin-3 (NLGN3) (48–52).
Venkatesh et al (51)
confirmed that neural activity promotes the proliferation of glioma
cells through secreted factors. Via mass spectrometry, they
identified some secreted proteins that increased the proliferation
of glioma cells in an activity-dependent manner. More unexpectedly,
it was found that NLGN3 is an important activity-regulated
paracrine growth factor, and 10 of 11 different glioma models
exhibited increased proliferation in response to NLGN3 (48). This dependance on microenvironmental
NLGN3 was proved in patient-derived xenograft models of diffuse
intrinsic pontine glioma, pediatric GBM and adult GBM. This
phenomenon did not occur in a patient-derived model of brain
metastasis from breast cancer, suggesting specificity for gliomas
(51). However, with prolonged
observation, some xenografted tumors in mice began to grow in the
NLGN3-deficient brain within each experimental cohort (51). The reason for this finding is
unclear, and further research is needed. Mechanistically, neural
activity increases the expression of a disintegrin and
metalloproteinase domain-containing protein 10 (ADAM10), which
cleaves membrane-bound NLGN3 to generate a soluble bioactive
protein, inducing the proliferation of glioma cells. Both Nlgn3
knockdown and pharmacologic ADAM10 inhibition suppress the growth
of glioma (51). Likewise, neural
activity-regulated BDNF also promotes the proliferation of glioma
cells via a paracrine pathway (53). In addition, NLGN3 and BDNF promote
synaptic connectivity between neurons and glioma cells and regulate
the strength of neuron-glioma synapses (35). They play their respective roles in
various types of gliomas. Specific details still need to be
studied.
In addition to synaptic activity-dependent currents,
neural activity also evokes the depolarization of glioma cell
membranes via non-synaptic activity-dependent currents mediated by
ion channels. Accumulating evidence indicates that ion channels act
a pivotal part in the progression of glioma by mediating
communication between neurons and glioma cells. In the central
nervous system, Na+, K+, Ca2+ and
Cl−channels are involved in regulating various
biological behaviors of cells (54). It was recently indicated that ion
channel mediated-electric currents direct the migration and
invasion of glioma cells. Glioma cells regulate their volumes
through ion channels for migration. Through NKCC1, glioma cells
accumulate Cl− intracellularly, whereas Cl−
channel protein 3 (CLC3) regulates Cl− efflux. To
balance Cl− efflux, glioma cells regulate K+
influx via Ca2+ activation by expressing KCa1.1
and KCa3.1 channels (55).
Chlorotoxin, which causes the internalization of CLC family
members, inhibits the invasion of glioma cells (56). Barish et al (57) developed chimeric antigen receptor
(CAR) T cells incorporating chlorotoxin and evaluated the primary
objectives of feasibility and safety in four patients with
MMP-2-expressing recurrent GBM (NCT04214392). The result showed
that three of the four participants exhibited a best response of
stable disease, and the therapy was well tolerated with no
dose-limiting toxicities (57).
Likewise, blockade of KCa1.1 or KCa3.1 channels also inhibits the
invasion of GBM cells (55). A
preclinical study identified that KCa3.1 channel inhibition
sensitized malignant gliomas to temozolomide (58). Recently, Dong et al (59) found that GBM cells predominantly
express EAG2 and Kvβ2 at the GBM-neuron interface. EAG2 and Kvβ2
physically interact to form a K+ channel complex.
Disruption of the EAG2-Kvβ2 interaction mitigates growth and
temozolomide resistance in GBM (59). However, the mechanism by which
neural activity regulates the proliferation of glioma cells by ion
channels remains incompletely understood. The activity of ion
channels involving neuron-mediated electric signals is essential
for downstream pathway signaling, whereas neural activity promotes
glioma cell proliferation. Based on these facts, it was
hypothesized that ion channels act as important bridges between
neural activity and glioma progression. However, these assumptions
need to be further confirmed.
After integrating into neural circuits, GBM cells
also hijack brain metabolism, co-opting neurons and glia to obtain
nutrients. As a dominant anaplerotic carbon source for the TCA
cycle, glutamine is rapidly consumed by GBM cells in vitro.
However, the in vivo glutamine metabolism of GBM differs
from that in cell culture. In vivo, some GBM cells integrate
into the neuron-astrocyte glutamate-glutamine cycle, allowing these
cells exploit astrocytes as an exogenous source of glutamine. It
has been revealed that glutamine catabolism through the
GLS-initiated pathway is absent in IDH1 wild-type GBM in
vivo. GLUL-positive glioma stem cells (GSCs) can produce
glutamine, whereas GLUL-negative GBM cells lack this ability. In
vivo, GLUL-negative GBM cells reside in close proximity to
astrocytes, which are GLUL-positive (37). This phenomenon indicates that
GLUL-positive astrocytes are the primary sources of glutamine for
anabolism in GBM. By expressing the high-affinity uptake
transporter SLC1A5, GBM cells potentially outcompete neurons for
glutamine (37). After depleting
glutamine, astrocyte-derived glutamine is adequate to maintain the
proliferation of GLUL-negative GBM cells in vitro (60). IDH wild-type GBM cells produce and
secrete high levels of glutamate, and consequently, the
extracellular glutamate concentration in the tumor microenvironment
(TME) surpasses that in normal brain tissue. Excessive glutamate
promotes the growth of GBM and triggers epilepsy in patients with
GBM.
In addition to glutamate, GSCs can acquire and
hydrolyze N-acetylaspartate (NAA), which is synthesized and
secreted exclusively by neurons, into acetate and aspartate to
promote proliferative metabolism. In particular, NAA both boosts
GSC proliferation and suppresses GSC differentiation in
vitro (61).
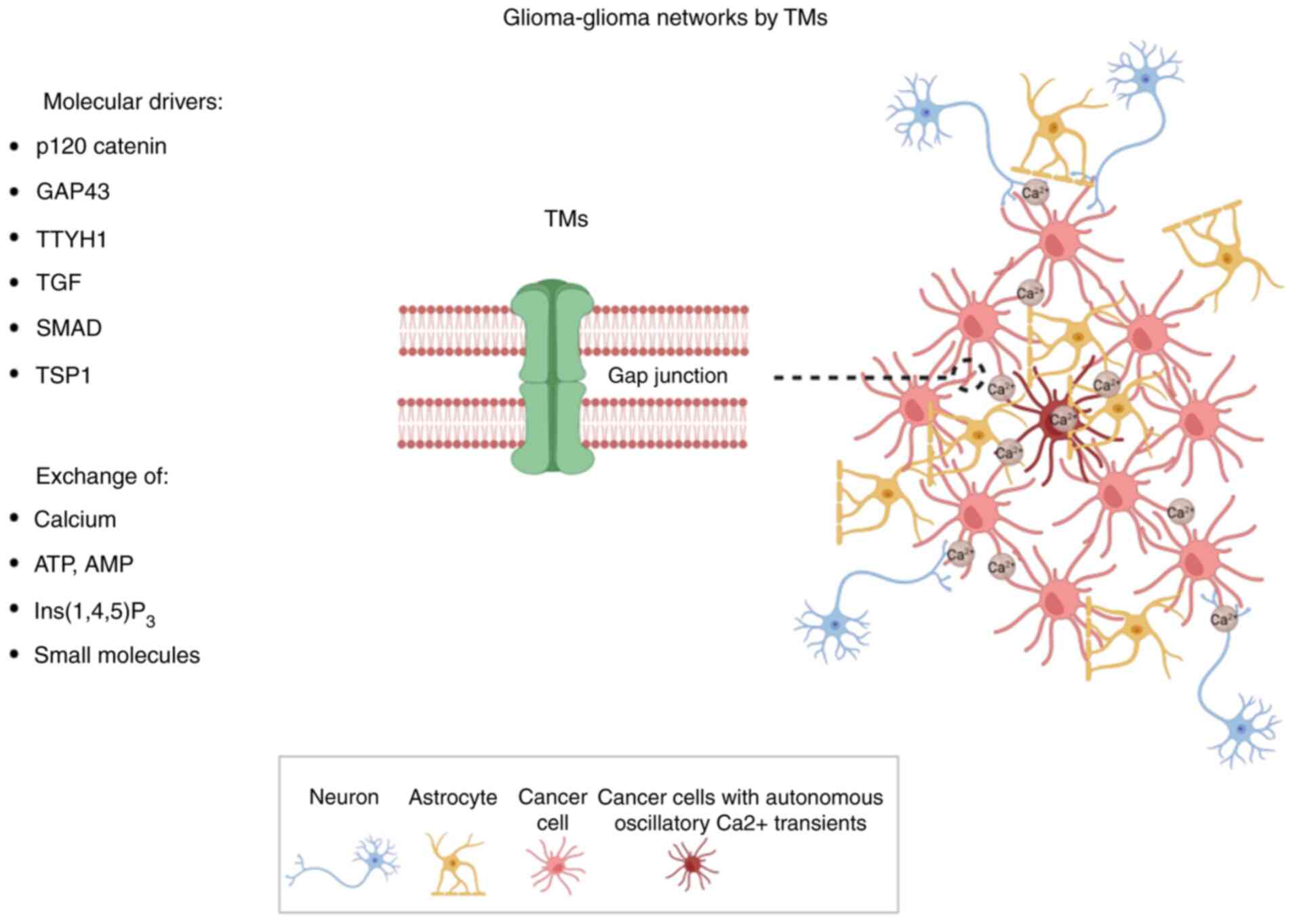
In the tumor core, glioma cells are interconnected
with each other through TMs, thereby forming glioma-glioma networks
(Fig. 4). Astrocyte-like and
MES-like tumor cells enriched in TMs construct the gap
junction-coupled glioma-glioma networks (38). Moreover, most of these cells contain
multiple TMs. Additionally, Venkataramani et al (38) found that connections form between
GBM cells and astrocytes via gap junctional coupling.
TMs are morphologically and molecularly
heterogeneous. Interconnecting TMs represent a continuation of the
membrane of glioma cells, and they extend to other cells while
being separated by gap junctions. Meanwhile, non-connecting TMs are
ultralong membrane protrusions extended by glioma cells. These TMs
hijack numerous characters from neural protrusions. They have blind
endings reminiscent of neurite growth cones, and they facilitate
the exchange of ions and molecules. More importantly, TMs mediating
the functional glioma-glioma networks are predominately resistant
to radiotherapy and standard chemotherapy with temozolomide
(38).
It was recently revealed that a small population of
GBM cells exhibit autonomous oscillatory Ca2+ transients
(Fig. 4). These cells are highly
connected to other glioma cells in glioma-glioma networks, forming
a ‘hub’ within the network. The Ca2+-activated
K+ channel KCa3.1 regulates autonomous and rhythmic
oscillatory Ca2+ transients in these ‘hub cells’ that
propagate through the connected network of gap junction-coupled
tumor cells (62). Furthermore,
Hausmann et al (62) found
that these periodic Ca2+ transients are involved in the
regulation of mitogen-activated protein kinase 2 and nuclear
factor-κB pathways in GBM cells, contributing to their malignant
behaviors. KCa3.1 knockdown abrogates these autonomous
Ca2+ transients, further mitigating glioma growth and
prolonging mouse survival in preclinical models of GBM (62).
At present, several molecules related to neurite
outgrowth and formation, including growth-associated protein
(Gap43), p120 catenin and tweety-homolog 1 (Ttyh1), have been
identified as potent drivers of TM outgrowth (63). Similar to the findings in the
neurodevelopmental program, Gap43 mainly localizes at the tips of
TMs. Gap43 downregulation inhibits TM formation in glioma (64). p120 catenin, an upstream regulator
of genes related to neuronal network formation in glioma, affects
the formation of TMs, further regulating invasion and network
formation by GBM cells (65).
Moreover, p120 catenin also modulates Gap43 expression (32). Ttyh1 is a putative calcium-regulated
chloride channel related to neurito-genesis in the membranes of
axonal growth cones. Ttyh1 downregulation was found to reduce the
invasion and proliferation of GBM cells by inducing abnormal TMs,
whereas tumor network formation between GBM cells was not affected.
These results suggest that molecular functions related to TMs can
be divided into invasive and interconnecting subclasses (66). Moreover, Ttyh1 is localized to
chromosomal arms 1p and 19q, similarly as the neurotrophic factors
nerve growth factor and neurotrophin 4, which upregulates Gap43.
These results suggest that 1p/19q intact astrocytoma has
significantly more and longer TMs than 1p/19q co-deleted
oligodendrogliomas. These data offer a reasonable explanation of
the improved prognosis of 1p/19q co-deleted oligodendrogliomas
(33).
In addition to the aforementioned molecules,
connexin 43 is the most important gap junction protein in TMs. In
prior research, connexin 43 downregulation in glioma cells markedly
decreased the number of glioma cells entering the glioma-glioma
network, reducing communication via intercellular calcium
waves. These changes led to reduced tumor size in vivo
(36). In addition, TGF-β was found
to participate in TM formation via SMAD activation and
thrombospondin 1 (67,68).
As one of the most common symptoms in patients with
gliomas, epilepsy leads to disability and decreases patients'
quality of life. This is closely related to increased neural
activity induced by glioma cells. The involved mechanisms include
the secretion of glutamine by GBM cells via the glutamate-cysteine
exchanger system XC (69), loss of inhibitory GABA interneurons
in the microenvironment (70),
change in the neural response to GABA (70), and glioma cell secretion of
synaptogenic factors such as glypican (71) and TSP-1 (25). Of course, the diversity of somatic
mutations in glioma cells definitely contributes to peritumoral
hyperexcitability and seizures over the course of the disease.
Tobochnik et al (72)
discovered that glioma genetic profiling can reveal diverse somatic
mutations of oncogenes relevant to peritumoral hyperexcitability
that contribute to glioma-related epilepsy. Meanwhile, neural
hyperexcitability and glioma growth engage in a vicious cycle of
mutual promotion. This cycle is a plausible therapeutic target to
manage glioma.
Although numerous studies have confirmed that
bidirectional interactions between glioma cells and neurons
represent a major tumor-promoting factor (83–85),
these related theories have not yet been translated into clinical
applications. Recently, Drexler et al (86) identified an epigenetically defined
neural signature that independently predicts the survival of
patients with GBM. Using the reference signatures of neural cells,
they classified GBM samples into low- or high-neural tumors.
High-neural GBM features hypomethylated CpG sites and increased
expression of genes relevant to synaptic integration. Via
single-cell transcriptomic analysis, they found a high abundance of
malignant stem cell-like cells, primarily of the neural lineage, in
high-neural GBM. Furthermore, these cells are classified as neural
progenitor cell-like, astrocyte-like and oligodendrocyte
progenitor-like, alongside oligodendrocytes and excitatory neurons.
Via survival analysis, researchers revealed a significant survival
benefit of gross total resection (GTR, 100% resection) and near GTR
(≥90% resection) compared with partial resection (<90% contrast
enhancement resection) in low-neural glioblastoma. However, no
survival benefit of near GTR was observed in high-neural GBM. This
suggests that more extensive resection might be necessary to confer
a survival benefit in high-neural GBM. Furthermore, it was also
revealed that BDNF could aid in stratifying patients with GBM based
on their neural subtype (86). The
other study have identified peripheral and CNS BDNF levels as s a
promising biomarker in patients with glioma (87). In order to be clinically applicable,
large-scale multicenter clinical studies is required.
White matter tracts (WMTs) represent one of the main
pathways by which GMB spreads, and these tracts contribute to
treatment failure (88). Using
diffusion-weighted imaging (DWI), Wei et al (89) characterized WMT disruption in a
study of more than 100 patients with GBM. It was found that the
most likely disrupted tracts were associated with fiber pathways
connecting distant cortical brain regions, providing a pathway for
the long-range migration of GBM cells. GBM-induced interruption of
WMTs was linked to distant recurrence and lower overall survival
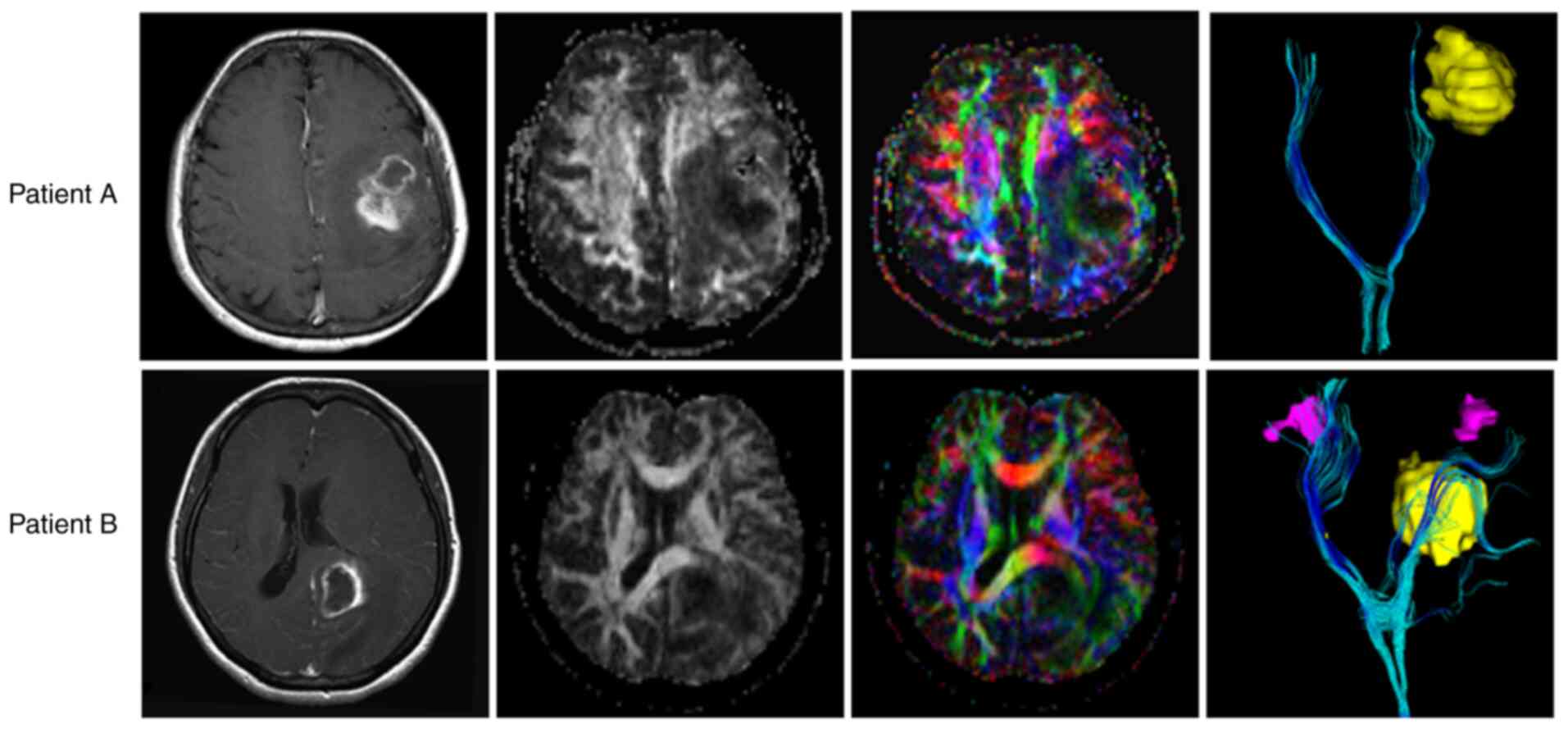
(89). Salvalaggio et al
(90) investigated whether the
local properties of WMTs at the sites of GBM lesions were
predictive of overall survival, revealing that GBM lesions within
regions that contain a higher density of WMTs are related to lower
survival, and vice versa. The correlation between local WMT
characteristics and the survival of patients with GBM. Our result
was similar to that of Salvalaggio et al (90). Two typical DWI of patients with GBM
are presented in Fig. 5. Patient A
had more severe WMT damage than patient B, resulting in shorter
survival for patient A despite the greater possibility of complete
resection in this patient. Consequently, this information from DWI
can improve assistance of surgeons in achieving maximum tumor
resection with minimal impact on patient neurological function.
Moreover, DWI findings can also be used to better predict the
prognosis of patients with GBM. As early as 2015, Abhinav et
al (91) had found that DWI
contributed to surgical planning for patients with glioma (91).
Abnormal molecules identified in glioma neuroscience
can also be used as therapeutic targets to treat gliomas. Targeting
the molecules involved in information communication between neurons
and glioma cells could become a new approach for glioma treatment.
First, inhibiting neuron-glioma synapses has great therapeutic
potential. Venkatesh at al (51) identified soluble neurexins and
ADAM10 inhibitors as favorable treatments for reducing malignant
synaptogenesis. Likewise, by antagonizing the binding of TSP with
its receptor, namely calcium channel auxiliary protein α2δ,
gabapentin and pregabalin suppress excitatory synaptogenesis
(92,93). Recently, Bernstock et al
(94) demonstrated a survival
benefit associated with gabapentin following surgical resection of
newly diagnosed glioblastoma in retrospective research. In
addition, inhibition of synaptic and perisynaptic signal
transmission, such as AMPAR or NMDAR inhibition, is another
therapeutic strategy (95). The
interruption of electric coupling in glioma-glioma networks has
therapeutic potential. Ion channels in neuron-glioma malignant
circuits and neural circuits supplying nutrients could emerge as
treatment targets (95). Finally,
suppressing neural hyperexcitability by anti-epileptics possesses
therapeutic potential in GBM (96).
Although there are numerous targets for neuron-glioma networks, no
target associated with significant inhibition of tumor growth has
been identified. Large numbers of preclinical and clinical trials
are needed in the future to identify effective therapeutic
targets.
The brain features a network of interleaved neural
circuits. Brain connectivity characteristically acts as a network
of nodes and edges, abstracting away the rich biological
information of local neurons (17).
Glioma involves the malignant transformation of a certain node in
the brain network. Consequently, communication between neurons and
glioma cells is inevitable during the initiation and progression of
glioma. Numerous studies have confirmed that glioma and neural
activity mutually regulate each other (97–101).
Although some progress in glioma neuroscience has
been made, knowledge of the specific cellular and molecular
interactions of neurons and glioma cells and the extent of neuron-
and glial cell-specific molecular alterations remains scarce.
Whether there are differences of neuron-glioma synapses at
different grades of glioma or not need to identify. Furthermore,
owing of GBM's heterogeneity, whether neuron-glioma interactions
vary across transcriptional subtypes or not also need to identify.
Guo et al (83) identified
that neuronal activity supported glioblastoma progression through
proneural-to-mesenchymal transition of glioma stem cells. In terms
of the underlying mechanisms of the interactions between neurons
and glioma cells, the identified synaptic inputs onto glioma cells
mainly involve local glutamatergic projections. The role of other
types of neurons in the occurrence and development of gliomas have
been overlooked. In addition, in in vitro glioma
neuroscience studies, neurons are often co-cultured with glioma
cells without other cells of the TME. Organoids are histologically
and functionally similar to human organs, maintaining the
characteristics of glioma and the TME, and organoids have high
sensitivity and high specificity in forecasting the efficacy of
anticancer drugs, thereby improving the accuracy of preclinical
studies (88). Organoids are also
used to study the role of neurons in glioblastoma, as it highly
preserves high fidelity of tumor and the TME. Consequently,
organoids will inevitably become advantageous tools for glioma
neuroscience in future. Conversely, mouse models of glioma do not
always fully recapitulate the human disease. The development of
mouse models that emulate human glioma more faithfully should be a
priority for this field in the future Furthermore, research on the
neuroscience of glioma requires multi-omics research of glioma
relevant to neuroscience, more sophisticated imaging techniques,
and other components. Additionally, some other questions relevant
to clinical translation remain incompletely answered. First, it has
to be investigated which appropriate therapeutic targets can be
successfully targeted with sufficient specificity for clinical
benefit. How can treatment targeting the connections between
neurons and gliomas be synergistically integrated into existing
treatment regimens? Can histopathological or other molecular
biomarkers, which include neural markers (such as NLGN3, BDNF and
Gap43), specify patients who are most likely to benefit from these
therapeutic strategies? Beyond developing novel treatments, the
possibility of repurposing existing drugs that modulate the nervous
system and typically have a tolerable adverse effect profile in
glioma should be investigated.
The nascent field of glioma neuroscience is
increasing rapidly, with parallel efforts aiming to uncover the
mechanistic underpinnings of neuron-glioma interactions and develop
novel therapies. Data obtained from TME studies suggest that
disrupting neuron-glioma crosstalk could eventually become an
important therapeutic strategy of clinical oncology akin to
anti-angiogenic and immunomodulatory therapies.
The authors would like to thank Dr Joe Barber Jr.,
PhD, ELS, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a
draft of this manuscript.
The present study was supported by the Natural Science
Foundation of Tianjin Municipal Science and Technology Commission
(grant no. 23JCYBJC01610) and College Students' Innovation and
Entrepreneurship Training Program Project (grant no.
202513661003).
Not applicable.
CZ and HZ wrote and formatting the manuscript. JC
was responsible for bioinformatic analysis (analysis of sequencing
data from all cited literature). ML provided suggestions on content
related to MRI and participated in writing the manuscript. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
No applicable.
The authors declare that they have no competing
interests.
|
1
|
Chen X and Song E: The theory of tumor
ecosystem. Cancer Commun (Lond). 42:587–608. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Swanton C, Bernard E, Abbosh C, Andre F,
Auwerx J, Balmain A, Bar-Sagi D, Bernards R, Bullman S, DeGregori
J, et al: Embracing cancer complexity: Hallmarks of systemic
disease. Cell. 187:1589–1616. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mancusi R and Monje M: The neuroscience of
cancer. Nature. 618:467–479. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miller KD, Ostrom QT, Kruchko C, Patil N,
Tihan T, Cioffi G, Fuchs HE, Waite KA, Jemal A, Siegel RL, et al:
Brain and other central nervous system tumor statistics, 2021. CA
Cancer J Clin. 71:381–406. 2021.PubMed/NCBI
|
|
5
|
Weller M, Wen PY, Chang SM, Dirven L, Lim
M, Monje M and Reifenberger G: Glioma. Nat Rev Dis Primers.
10:332024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bagley SJ, Logun M, Fraietta JA, Wang X,
Desai AS, Bagley LJ, Nabavizadeh A, Jarocha D, Martins R, Maloney
E, et al: Intrathecal bivalent CAR T cells targeting EGFR and
IL13Ralpha2 in recurrent glioblastoma: Phase 1 trial interim
results. Nat Med. 30:1320–1329. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bagley SJ, Binder ZA, Lamrani L, Marinari
E, Desai AS, Nasrallah MP, Maloney E, Brem S, Lustig RA, Kurtz G,
et al: Repeated peripheral infusions of anti-EGFRvIII CAR T cells
in combination with pembrolizumab show no efficacy in glioblastoma:
A phase 1 trial. Nat Cancer. 5:517–531. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roth P, Gorlia T, Reijneveld JC, de Vos F,
Idbaih A, Frenel JS, Le Rhun E, Sepulveda JM, Perry J, Masucci GL,
et al: Marizomib for patients with newly diagnosed glioblastoma: A
randomized phase 3 trial. Neuro Oncol. 26:1670–1682. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sloan AE, Winter K, Gilbert MR, Aldape K,
Choi S, Wen PY, Butowski N, Iwamoto FM, Raval RR, Voloschin AD, et
al: RG-BN002: Phase I study of ipilimumab, nivolumab, and the
combination in patients with newly diagnosed glioblastoma. Neuro
Oncol. 26:1628–1637. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carpentier A, Stupp R, Sonabend AM, Dufour
H, Chinot O, Mathon B, Ducray F, Guyotat J, Baize N, Menei P, et
al: Repeated blood-brain barrier opening with a nine-emitter
implantable ultrasound device in combination with carboplatin in
recurrent glioblastoma: A phase I/II clinical trial. Nat Commun.
15:16502024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harwood DSL, Pedersen V, Bager NS, Schmidt
AY, Stannius TO, Areskeviciute A, Josefse K, Nørøxe DS, Scheie D,
Rostalski H, et al: Glioblastoma cells increase expression of notch
signaling and synaptic genes within infiltrated brain tissue. Nat
Commun. 15:78572024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alhalabi OT, Fletcher MNC, Hielscher T,
Kessler T, Lokumcu T, Baumgartner U, Wittmann E, Schlue S, Göttmann
M, Rahman S, et al: A novel patient stratification strategy to
enhance the therapeutic efficacy of dasatinib in glioblastoma.
Neuro Oncol. 24:39–51. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hara T, Chanoch-Myers R, Mathewson ND,
Myskiw C, Atta L, Bussema L, Eichhorn SW, Greenwald AC, Kinker GS,
Rodman C, et al: Interactions between cancer cells and immune cells
drive transitions to mesenchymal-like states in glioblastoma.
Cancer Cell. 39:779–792.e11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen L, Qi Q, Jiang X, Wu J, Li Y, Liu Z,
Cai Y, Ran H, Zhang S, Zhang C, et al: Phosphocreatine promotes
epigenetic reprogramming to facilitate glioblastoma growth through
stabilizing BRD2. Cancer Discov. 14:1547–1565. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kloosterman DJ, Erbani J, Boon M, Farber
M, Handgraaf SM, Ando-Kuri M, Sánchez-López E, Fontein B, Mertz M,
Nieuwland M, et al: Macrophage-mediated myelin recycling fuels
brain cancer malignancy. Cell. 187:5336–5356.e30. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seguin C, Sporns O and Zalesky A: Brain
network communication: Concepts, models and applications. Nat Rev
Neurosci. 24:557–574. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bazinet V, Hansen JY and Misic B: Towards
a biologically annotated brain connectome. Nat Rev Neurosci.
24:747–760. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zamler DB and Hu J: Primitive
oligodendrocyte precursor cells are highly susceptible to
gliomagenic transformation. Cancer Res. 83:807–808. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu C, Sage JC, Miller MR, Verhaak RG,
Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L
and Zong H: Mosaic analysis with double markers reveals tumor cell
of origin in glioma. Cell. 146:209–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao Y and Czopka T:
Myelination-independent functions of oligodendrocyte precursor
cells in health and disease. Nat Neurosci. 26:1663–1669. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buchanan J, da Costa NM and Cheadle L:
Emerging roles of oligodendrocyte precursor cells in neural circuit
development and remodeling. Trends Neurosci. 46:628–639. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Miramontes TG, Czopka T and Monk KR:
Synaptic input and Ca2+ activity in zebrafish oligodendrocyte
precursor cells contribute to myelin sheath formation. Nat
Neurosci. 27:219–231. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Douw L, Breedt LC and Zimmermann MLM:
Cancer meets neuroscience: The association between glioma
occurrence and intrinsic brain features. Brain. 146:803–805. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meyer J, Yu K, Luna-Figueroa E, Deneen B
and Noebels J: Glioblastoma disrupts cortical network activity at
multiple spatial and temporal scales. Nat Commun. 15:45032024.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Krishna S, Choudhury A, Keough MB, Seo K,
Ni L, Kakaizada S, Lee A, Aabedi A, Popova G, Lipkin B, et al:
Glioblastoma remodelling of human neural circuits decreases
survival. Nature. 617:599–607. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pan Y, Hysinger JD, Barron T, Schindler
NF, Cobb O, Guo X, Yalçın B, Anastasaki C, Mulinyawe SB, Ponnuswami
A, et al: NF1 mutation drives neuronal activity-dependent
initiation of optic glioma. Nature. 594:277–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Anastasaki C, Chatterjee J, Koleske JP,
Gao Y, Bozeman SL, Kernan CM, Marco Y, Marquez LI, Chen JK, Kelly
CE, et al: NF1 mutation-driven neuronal hyperexcitability sets a
threshold for tumorigenesis and therapeutic targeting of murine
optic glioma. Neuro Oncol. 26:1496–1508. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen P, Wang W, Liu R, Lyu J, Zhang L, Li
B, Qiu B, Tian A, Jiang W, Ying H, et al: Olfactory sensory
experience regulates gliomagenesis via neuronal IGF1. Nature.
606:550–556. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chatterjee J, Koleske JP, Chao A,
Sauerbeck AD, Chen JK, Qi X, Ouyang M, Boggs LG, Idate R, Marco Y,
et al: Brain injury drives optic glioma formation through
neuron-glia signaling. Acta Neuropathol Commun. 12:212024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Romero-Garcia R, Mandal AS, Bethlehem RAI,
Crespo-Facorro B, Hart MG and Suckling J: Transcriptomic and
connectomic correlates of differential spatial patterning among
gliomas. Brain. 146:1200–1211. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hai L, Hoffmann DC, Wagener RJ, Azorin DD,
Hausmann D, Xie R, Huppertz MC, Hiblot J, Sievers P, Heuer S, et
al: A clinically applicable connectivity signature for glioblastoma
includes the tumor network driver CHI3L1. Nat Commun. 15:9682024.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Venkataramani V, Schneider M, Giordano FA,
Kuner T, Wick W, Herrlinger U and Winkler F: Disconnecting
multicellular networks in brain tumours. Nat Rev Cancer.
22:481–491. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Osswald M, Jung E, Sahm F, Solecki G,
Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M,
et al: Brain tumour cells interconnect to a functional and
resistant network. Nature. 528:93–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gillespie S and Monje M: An active role
for neurons in glioma progression: Making sense of Scherer's
structures. Neuro Oncol. 20:1292–1299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Taylor KR, Barron T, Hui A, Spitzer A,
Yalcin B, Ivec AE, Geraghty AC, Hartmann GG, Arzt M, Gillespie SM,
et al: Glioma synapses recruit mechanisms of adaptive plasticity.
Nature. 623:366–374. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Taylor KR and Monje M:
Neuron-oligodendroglial interactions in health and malignant
disease. Nat Rev Neurosci. 24:733–746. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
de Ruiter Swain J, Michalopoulou E, Noch
EK, Lukey MJ and Van Aelst L: Metabolic partitioning in the brain
and its hijacking by glioblastoma. Genes Dev. 37:681–702. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Venkataramani V, Yang Y, Schubert MC,
Reyhan E, Tetzlaff SK, Wißmann N, Botz M, Soyka SJ, Beretta CA,
Pramatarov RL, et al: Glioblastoma hijacks neuronal mechanisms for
brain invasion. Cell. 185:2899–2917.e31. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun Y, Wang X, Zhang DY, Zhang Z,
Bhattarai JP, Wang Y, Park KH, Dong W, Hung YF, Yang Q, et al:
Brain-wide neuronal circuit connectome of human glioblastoma.
Nature. 641:222–231. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsieh AL, Ganesh S, Kula T, Irshad M,
Ferenczi EA, Wang W, Chen YC, Hu SH, Li Z, Joshi S, et al:
Widespread neuroanatomical integration and distinct
electrophysiological properties of glioma-innervating neurons. Proc
Natl Acad Sci USA. 121:e24174201212024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Venkatesh HS, Morishita W, Geraghty AC,
Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami
A, Ni L, et al: Electrical and synaptic integration of glioma into
neural circuits. Nature. 573:539–545. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Muller-Langle A, Lutz H, Hehlgans S, Rodel
F, Rau K and Laube B: NMDA Receptor-mediated signaling pathways
enhance radiation resistance, survival and migration in
glioblastoma Cells-A potential target for adjuvant radiotherapy.
Cancers (Basel). 11:5032019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Drexler R, Drinnenberg A, Gavish A, Yalcin
B, Shamardani K, Rogers A, Mancusi R, Taylor KR, Kim YS, Woo PJ, et
al: Cholinergic neuronal activity promotes diffuse midline glioma
growth through muscarinic signaling. Cell. 188:4640–4657.e30. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stella M, Baiardi G, Pasquariello S, Sacco
F, Dellacasagrande I, Corsaro A, Mattioli F and Barbieri F:
Antitumor potential of antiepileptic drugs in human glioblastoma:
Pharmacological targets and clinical benefits. Biomedicines.
11:5822023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Badalotti R, Dalmolin M, Malafaia O, Ribas
Filho JM, Roesler R, Fernandes MAC and Isolan GR: Gene expression
of GABAA receptor subunits and association with patient survival in
glioma. Brain Sci. 14:2752024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barron T, Yalcin B, Su M, Byun YG, Gavish
A, Shamardani K, Xu H, Ni L, Soni N, Mehta V, et al: GABAergic
neuron-to-glioma synapses in diffuse midline gliomas. Nature.
639:1060–1068. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Duman C, Yaqubi K, Hoffmann A, Acikgoz AA,
Korshunov A, Bendszus M, Herold-Mende C, Liu HK and Alfonso J:
Acyl-CoA-Binding protein drives glioblastoma tumorigenesis by
sustaining fatty acid oxidation. Cell Metab. 30:274–289.e5. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Venkatesh HS, Johung TB, Caretti V, Noll
A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS,
et al: Neuronal activity promotes glioma growth through
Neuroligin-3 secretion. Cell. 161:803–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Colucci-D'Amato L, Speranza L and
Volpicelli F: Neurotrophic factor BDNF, physiological functions and
therapeutic potential in depression, neurodegeneration and brain
cancer. Int J Mol Sci. 21:77772020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li J, Zhang B, Feng Z, An D, Zhou Z, Wan
C, Hu Y, Sun Y, Wang Y, Liu X, et al: Stabilization of KPNB1 by
deubiquitinase USP7 promotes glioblastoma progression through the
YBX1-NLGN3 axis. J Exp Clin Cancer Res. 43:282024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Venkatesh HS, Tam LT, Woo PJ, Lennon J,
Nagaraja S, Gillespie SM, Ni J, Duveau DY, Morris PJ, Zhao JJ, et
al: Targeting neuronal activity-regulated neuroligin-3 dependency
in high-grade glioma. Nature. 549:533–537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yun EJ, Kim D, Kim S, Hsieh JT and Baek
ST: Targeting Wnt/β-catenin-mediated upregulation of oncogenic
NLGN3 suppresses cancer stem cells in glioblastoma. Cell Death Dis.
14:4232023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Guo Z, An P and Hong X: has-miR-134-5p
inhibits the proliferation and migration of glioma cells by
regulating the BDNF/ERK signaling pathway. Aging (Albany NY).
16:6510–6520. 2024.PubMed/NCBI
|
|
54
|
Griffin M, Khan R, Basu S and Smith S: Ion
channels as therapeutic targets in high grade gliomas. Cancers
(Basel). 12:30682020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Elias AF, Lin BC and Piggott BJ: Ion
channels in gliomas-from molecular basis to treatment. Int J Mol
Sci. 24:25302023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sharma G, Braga CB, Chen KE, Jia X,
Ramanujam V, Collins BM, Rittner R and Mobli M: Structural basis
for the binding of the cancer targeting scorpion toxin, ClTx, to
the vascular endothelia growth factor receptor neuropilin-1. Curr
Res Struct Biol. 3:179–186. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Barish ME, Aftabizadeh M, Hibbard J,
Blanchard MS, Ostberg JR, Wagner JR, Manchanda M, Paul J, Stiller
T, Aguilar B, et al: Chlorotoxin-directed CAR T cell therapy for
recurrent glioblastoma: Interim clinical experience demonstrating
feasibility and safety. Cell Rep Med. 6:1023022025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
D'Alessandro G, Grimaldi A, Chece G,
Porzia A, Esposito V, Santoro A, Salvati M, Mainiero F, Ragozzino
D, Di Angelantonio S, et al: KCa3.1 channel inhibition sensitizes
malignant gliomas to temozolomide treatment. Oncotarget.
7:30781–30796. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dong W, Fekete A, Chen X, Liu H, Beilhartz
GL, Chen X, Bahrampour S, Xiong Y, Yang Q, Zhao H, et al: A
designer peptide against the EAG2-Kvβ2 potassium channel targets
the interaction of cancer cells and neurons to treat glioblastoma.
Nat Cancer. 4:1418–1436. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Han L, Zhou J, Li L, Wu X, Shi Y, Cui W,
Zhang S, Hu Q, Wang J, Bai H, et al: SLC1A5 enhances malignant
phenotypes through modulating ferroptosis status and immune
microenvironment in glioma. Cell Death Dis. 13:10712022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Long PM, Moffett JR, Namboodiri AMA,
Viapiano MS, Lawler SE and Jaworski DM: N-acetylaspartate (NAA) and
N-acetylaspartylglutamate (NAAG) promote growth and inhibit
differentiation of glioma stem-like cells. J Biol Chem.
288:26188–26200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hausmann D, Hoffmann DC, Venkataramani V,
Jung E, Horschitz S, Tetzlaff SK, Jabali A, Hai L, Kessler T,
Azoŕin DD, et al: Autonomous rhythmic activity in glioma networks
drives brain tumour growth. Nature. 613:179–186. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jung E, Alfonso J, Monyer H, Wick W and
Winkler F: Neuronal signatures in cancer. Int J Cancer.
147:3281–3291. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Watson DC, Bayik D, Storevik S, Moreino
SS, Sprowls SA, Han J, Augustsson MT, Lauko A, Sravya P, Røsland
GV, et al: GAP43-dependent mitochondria transfer from astrocytes
enhances glioblastoma tumorigenicity. Nat Cancer. 4:648–664. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gritsenko PG, Atlasy N, Dieteren CEJ,
Navis AC, Venhuizen JH, Veelken C, Schubert D, Acker-Palmer A,
Westerman BA, Wurdinger T, et al: p120-catenin-dependent collective
brain infiltration by glioma cell networks. Nat Cell Biol.
22:97–107. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jung E, Osswald M, Blaes J, Wiestler B,
Sahm F, Schmenger T, Solecki G, Deumelandt K, Kurz FT, Xie R, et
al: Tweety-homolog 1 drives brain colonization of gliomas. J
Neurosci. 37:6837–6850. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Joseph JV, Magaut CR, Storevik S, Geraldo
LH, Mathivet T, Latif MA, Rudewicz J, Guyon J, Gambaretti M, Haukas
F, et al: TGF-β promotes microtube formation in glioblastoma
through thrombospondin 1. Neuro Oncol. 24:541–53. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Becker KN and Eisenmann KM: New targets in
the glioblastoma tumor microtube multiverse: Emerging roles for the
TGF-β/TSP1 signaling axis. Neuro Oncol. 24:554–555. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hatcher A, Yu K, Meyer J, Aiba I, Deneen B
and Noebels JL: Pathogenesis of peritumoral hyperexcitability in an
immunocompetent CRISPR-based glioblastoma model. J Clin Invest.
130:2286–2300. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Campbell SL, Robel S, Cuddapah VA, Robert
S, Buckingham SC, Kahle KT and Sontheimer H: GABAergic
disinhibition and impaired KCC2 cotransporter activity underlie
tumor-associated epilepsy. Glia. 63:23–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yu K, Lin CJ, Hatcher A, Lozzi B, Kong K,
Huang-Hobbs E, Cheng YT, Beechar VB, Zhu W, Zhang Y, et al: PIK3CA
variants selectively initiate brain hyperactivity during
gliomagenesis. Nature. 578:166–171. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tobochnik S, Dorotan MKC, Ghosh HS,
Lapinskas E, Vogelzang J, Reardon DA, Ligon KL, Bi WL, Smirnakis SM
and Lee JW: Glioma genetic profiles associated with
electrophysiologic hyperexcitability. Neuro Oncol. 26:323–334.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong
W, Yi M and Xiang B: Regulatory mechanisms of PD-1/PD-L1 in
cancers. Mol Cancer. 23:1082024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xie P, Yu M, Zhang B, Yu Q, Zhao Y, Wu M,
Jin L, Yan J, Zhou B, Liu S, et al: CRKL dictates anti-PD-1
resistance by mediating tumor-associated neutrophil infiltration in
hepatocellular carcinoma. J Hepatol. 81:93–107. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu Q, Guan Y and Li S: Programmed death
receptor (PD-)1/PD-ligand (L)1 in urological cancers: The
‘all-around warrior’ in immunotherapy. Mol Cancer. 23:1832024.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li X, Liu Y, Gui J, Gan L and Xue J: Cell
identity and spatial distribution of PD-1/PD-L1 blockade
responders. Adv Sci (Weinh). 11:e24007022024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs bevacizumab in patients with
recurrent glioblastoma: The CheckMate 143 phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lin H, Liu C, Hu A, Zhang D, Yang H and
Mao Y: Understanding the immunosuppressive microenvironment of
glioma: Mechanistic insights and clinical perspectives. J Hematol
Oncol. 17:312024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nejo T, Krishna S, Yamamichi A,
Lakshmanachetty S, Jimenez C, Lee KY, Baker DL, Young JS, Chen T,
Phyu SSS, et al: Glioma-neuronal circuit remodeling induces
regional immunosuppression. Nat Commun. 16:47702025. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gonzalez-Calvo I, Cizeron M, Bessereau JL
and Selimi F: Synapse formation and function across species:
Ancient roles for CCP, CUB, and TSP-1 structural domains. Front
Neurosci. 16:8664442022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu Z, Wen J, Hu F, Wang J, Hu C and Zhang
W: Thrombospondin-1 induced programmed death-ligand 1-mediated
immunosuppression by activating the STAT3 pathway in osteosarcoma.
Cancer Sci. 113:432–445. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Guo X, Pan Y, Xiong M, Sanapala S,
Anastasaki C, Cobb O, Dahiya S and Gutmann DH: Midkine activation
of CD8+ T cells establishes a neuron-immune-cancer axis
responsible for low-grade glioma growth. Nat Commun. 11:21772020.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Guo X, Qiu W, Wang C, Qi Y, Li B, Wang S,
Zhao R, Cheng B, Han X, Du H, et al: Neuronal activity promotes
glioma progression by inducing Proneural-to-Mesenchymal transition
in glioma stem cells. Cancer Res. 84:372–387. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Guo X, Qiu W, Li B, Qi Y, Wang S, Zhao R,
Cheng B, Han X, Du H, Pan Z, et al: Hypoxia-induced neuronal
activity in glioma patients polarizes microglia by potentiating RNA
m6A demethylation. Clin Cancer Res. 30:1160–1174. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen HC, He P, McDonald M, Williamson MR,
Varadharajan S, Lozzi B, Woo J, Choi DJ, Sardar D, Huang-Hobbs E,
et al: Histone serotonylation regulates ependymoma tumorigenesis.
Nature. 632:903–910. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Drexler R, Khatri R, Sauvigny T, Mohme M,
Maire CL, Ryba A, Zghaibeh Y, Dührsen L, Salviano-Silva A, Lamszus
K, et al: A prognostic neural epigenetic signature in high-grade
glioma. Nat Med. 30:1622–1635. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hasani F, Masrour M, Khamaki S, Jazi K,
Ghoodjani E and Teixeira AL: Brain-derived neurotrophic factor
(BDNF) as a potential biomarker in brain glioma: A systematic
review and Meta-Analysis. Brain Behav. 15:e702662025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Li Y, Wang J, Song SR, Lv SQ, Qin JH and
Yu SC: Models for evaluating glioblastoma invasion along white
matter tracts. Trends Biotechnol. 42:293–309. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wei Y, Li C, Cui Z, Mayrand RC, Zou J,
Wong ALKC, Sinha R, Matys T, Schönlieb CB and Price SJ: Structural
connectome quantifies tumour invasion and predicts survival in
glioblastoma patients. Brain. 146:1714–1727. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Salvalaggio A, Pini L, Gaiola M, Velco A,
Sansone G, Anglani M, Fekonja L, Chioffi F, Picht T, Thiebaut de
Schotten M, et al: White matter tract density index prediction
model of overall survival in glioblastoma. JAMA Neurol.
80:1222–1231. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Abhinav K, Yeh FC, Mansouri A, Zadeh G and
Fernandez-Miranda JC: High-definition fiber tractography for the
evaluation of perilesional white matter tracts in High-grade glioma
surgery. Neuro Oncol. 17:1199–1209. 2015.PubMed/NCBI
|
|
92
|
Mastall M, Roth P, Bink A, Fischer Maranta
A, Laubli H, Hottinger AF, Hundsberger T, Migliorini D, Ochsenbein
A, Seystahl K, et al: A phase Ib/II randomized, open-label drug
repurposing trial of glutamate signaling inhibitors in combination
with chemoradiotherapy in patients with newly diagnosed
glioblastoma: The GLUGLIO trial protocol. BMC Cancer. 24:822024.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yamaguchi K, Kumakura S, Someya A, Iseki
M, Inada E and Nagaoka I: Anti-inflammatory actions of gabapentin
and pregabalin on the substance P-induced mitogen-activated protein
kinase activation in U373 MG human glioblastoma astrocytoma cells.
Mol Med Rep. 16:6109–6115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bernstock JD, Mehari M, E Gerstl JV,
Meredith DM, Valdes PA, Heesen P, Ambati VS, Krishna S, Chen JA,
Arora H, et al: Gabapentinoids confer survival benefit in human
glioblastoma. Nat Commun. 16:44832025. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lan YL, Zou S, Wang W, Chen Q and Zhu Y:
Progress in cancer neuroscience. MedComm (2020). 4:e4312023.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Tobochnik S, Regan MS, Dorotan MKC, Reich
D, Lapinskas E, Hossain MA, Stopka S, Santagata S, Murphy MM,
Arnaout O, et al: Pilot trial of perampanel on peritumoral
hyperexcitability and clinical outcomes in newly diagnosed
high-grade glioma. medRxiv [Preprint]. Apr 18–2024.doi:
10.1101/2024.04.11.24305666. PubMed/NCBI
|
|
97
|
Friess D, Brauer S, Poysti A, Choudhury C
and Harris L: Tools to study neural and glioma stem cell
quiescence. Trends Neurosci. 47:736–748. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Picart T and Hervey-Jumper S: Central
nervous system regulation of diffuse glioma growth and invasion:
From single unit physiology to circuit remodeling. J Neurooncol.
169:1–10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Read RD, Tapp ZM, Rajappa P and
Hambardzumyan D: Glioblastoma microenvironment-from biology to
therapy. Genes Dev. 38:360–379. 2024.PubMed/NCBI
|
|
100
|
Ng S, Duffau H and Herbet G: Perspectives
in human brain plasticity sparked by glioma invasion: From
intraoperative (re)mappings to neural reconfigurations. Neural
Regen Res. 19:947–948. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wei J, Wang M, Li S, Han R, Xu W, Zhao A,
Yu Q, Li H, Li M and Chi G: Reprogramming of astrocytes and glioma
cells into neurons for central nervous system repair and
glioblastoma therapy. Biomed Pharmacother. 176:1168062024.
View Article : Google Scholar : PubMed/NCBI
|