Acute myeloid leukemia (AML) is a highly aggressive
hematologic malignancy in adults, characterized by a high relapse
rate and persistent treatment resistance, which translates to a
5-year overall survival of <10% (1,2).
Although advances in molecular typing and targeted therapies have
improved the prognosis of patients with specific subtypes, the
heterogeneity of AML, its driving mechanisms, and complex
interactions between leukemia stem cells (LSCs) and the bone marrow
microenvironment remain unclear (3). High-intensity chemotherapy and
allogeneic hematopoietic stem cell transplantation are the current
standard of care; however, due to their severe toxic side effects,
harm to normal hematopoiesis, and limitations in eliminating LSCs
and overcoming drug resistance, novel therapeutic approaches that
are more effective and selective are urgently required (4,5).
Targeting of regulated cell death (RCD) has
attracted increasing attention from researchers in recent years
(6–9). The primary benefit of this method is
its ability to specifically target LSCs while reshaping the
immunosuppressive bone marrow microenvironment to enhance
anti-leukemic efficacy and minimize damage to normal tissues
(10). Studies have shown that
selective modulation of RCD pathways, such as autophagy, apoptosis,
pyroptosis, necroptosis, ferroptosis and cuproptosis, can
effectively target and eliminate leukemic clones resistant to
traditional therapies (11–16). Furthermore, these RCD pathways form
a highly interconnected regulatory network that operates through
dynamic interactions rather than functioning in isolation (17). This complexity suggests that
combinatorial interventions targeting multiple death pathway nodes
synergistically may more effectively disrupt the death-resistant
barriers of leukemia cells, thereby opening multidimensional attack
pathways to overcome therapeutic bottlenecks caused by AML clonal
evolution and microenvironmental sheltering (18).
The present review aims to achieve three objectives:
i) To assess the latest developments and challenges in targeting
RCD in AML treatment by systematically integrating the
understanding of six major RCD pathways; ii) to explore the
crosstalk and synergistic potential between these pathways, a
critical yet underexplored area; and iii) to apply these insights
in developing a framework for future combinatorial therapies. To
provide novel concepts and strategies for overcoming AML treatment
resistance and improving patient outcomes, the present review
examines the RCD modes of action, their key functions, and
potential synergistic effects in maintaining LSCs, chemoresistance
and bone marrow microenvironment remodeling. The present review
also explores the potential for developing a customized therapeutic
system through precise targeting of the death pathway network. The
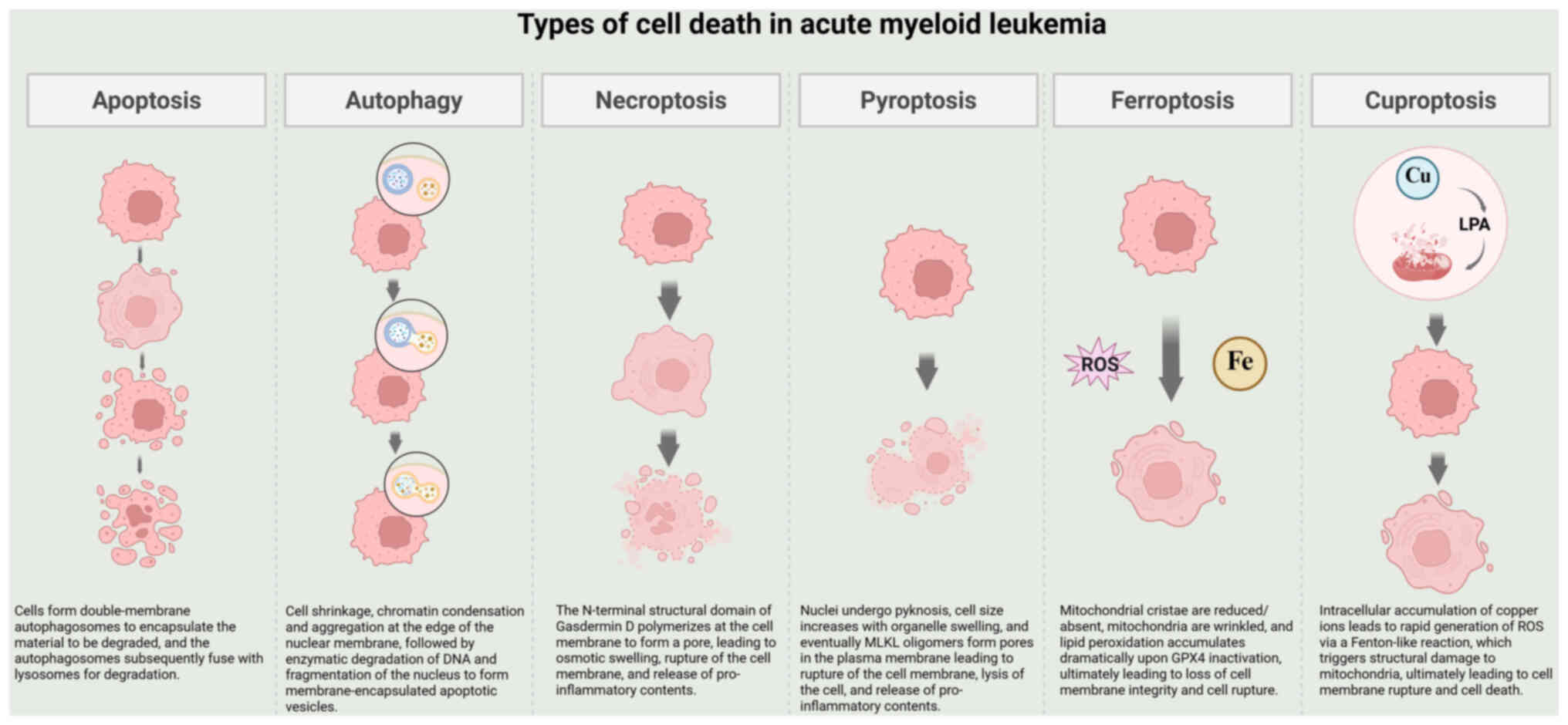
profiles of each type of cell death are illustrated in Fig. 1 (8,19).
Overview of autophagy. Autophagy is a highly
conserved cellular process that involves the formation of
autophagosomal vesicles. These vesicles encapsulate damaged
organelles, misfolded proteins and other cytoplasmic components
within double-membrane structures. Cellular component recycling
occurs when autophagosomes fuse with lysosomes, where the enclosed
contents are enzymatically degraded into smaller molecules
(20).
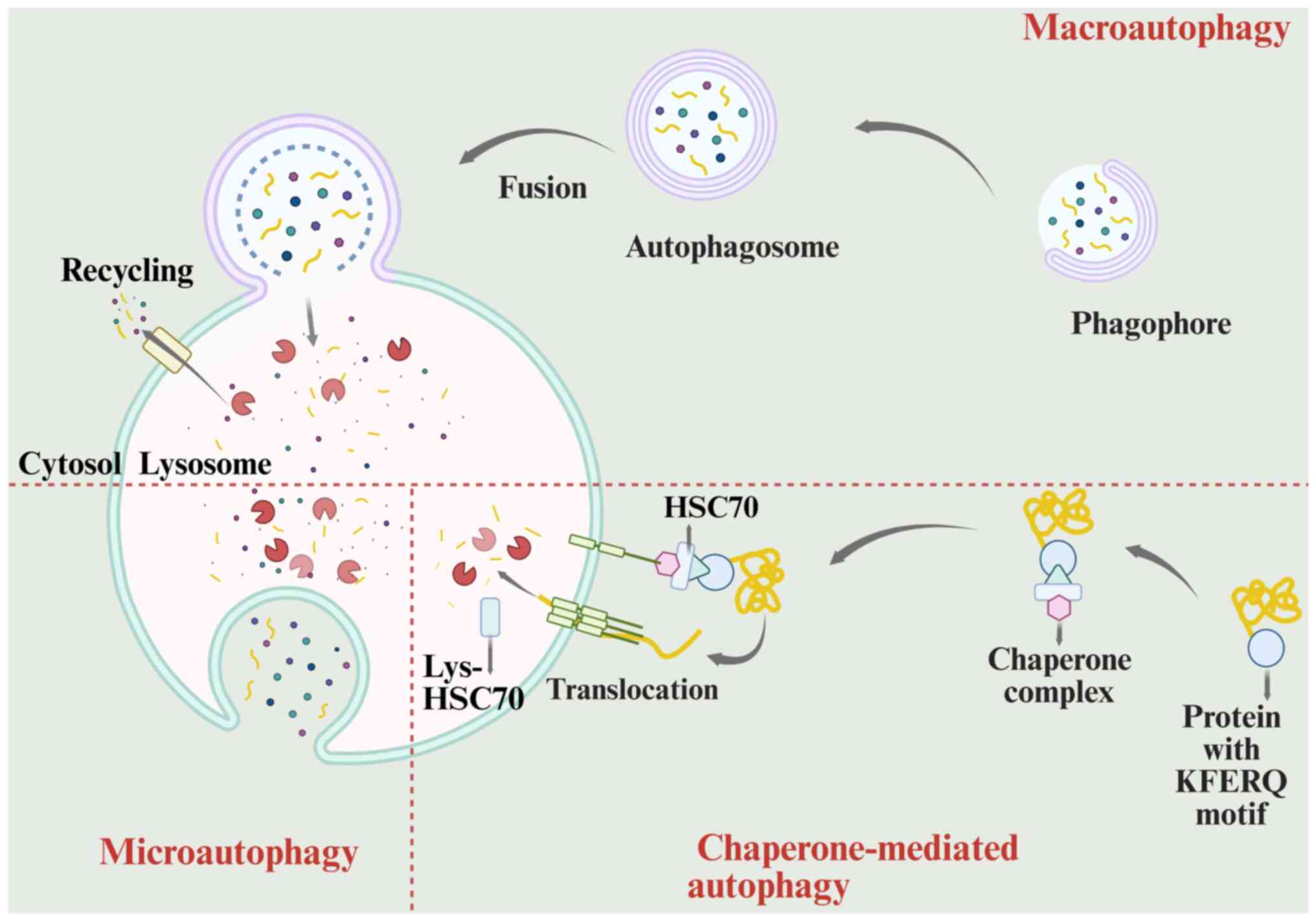
Autophagy can be classified into three types based
on its mechanism: Microautophagy, chaperone-mediated autophagy and
macroautophagy. Macroautophagy is the most widely studied type of
autophagy and involves the encapsulation of damaged organelles or
denatured proteins by double-membrane autophagic vesicles. These
vesicles then transport the contents to fuse with lysosomes, where
enzymatic degradation occurs (21).
Lysosomal-associated membrane protein 2 mediates the highly
specific pathway of chaperone-mediated autophagy, in which heat
shock protein 70 recognizes KFERQ motifs in target proteins and
unidirectionally translocates them into the lysosomal lumen for
selective degradation (21). Unlike
the first two autophagic pathways, microautophagy does not involve
the formation of distinct autophagosomal structures. Instead, it
involves creation of phagocytic vesicles through lysosomal membrane
invagination, directly engulfing cytoplasmic components or
organelles and delivering them to the lysosomal lumen for enzymatic
degradation via membrane fusion (22).
Autophagy is a five-stage molecular process
involving initiation, nucleation, membrane extension, fusion and
degradation (23). When cells
experience nutrient deprivation, the activity of mTOR, a key
energy-sensing kinase, is suppressed, triggering the autophagy
signaling cascade (24). The UNC-51
like kinase 1 (ULK1) complex, which consists of essential
components including ULK1, ATG13 and FIP200, is phosphorylated by
AMP-activated protein kinase. This phosphorylation reverses the
inhibitory effect of mTOR on the complex (25). Upon activation, the complex migrates
towards the autophagy initiation site, where ULK1 catalyzes the
phosphorylation of FIP200 and ATG13 (25). This phosphorylation event recruits
downstream ATG proteins to initiate the formation of the
pre-autophagosome complex (26).
The nucleation phase of autophagy begins with the activation of the
phosphatidylinositol-3-kinase class III complex, which catalyzes
the formation of a lipid-signaling platform at the membrane surface
through phosphatidylinositol 3-phosphate (27). This platform recruits effector
proteins containing the FYVE/PX domain, which promote the assembly
of pre-autophagosomal structures (28). Membrane expansion during autophagy
is driven by two ubiquitin-like modification processes: Atg7
functions as an E1-like enzyme in the Atg5-Atg12 coupling system,
facilitating the covalent binding of Atg12 to Atg5 (29). This complex then associates with
autophagy related 16-like 1 to regulate membrane curvature and
promote the growth of autophagic vesicles (29). In the LC3 lipidation pathway, Atg4
cleaves the LC3 precursor to generate LC3-I, which is then
covalently linked by Atg7 to Atg3, forming membrane-anchored LC3-II
that integrates into the autophagosome membrane, promoting membrane
extension and serving as a marker for substrate recognition,
thereby facilitating targeted transport (30). The two pathways collaborate to
complete autophagosome maturation (31). Ultimately, the autophagic vesicle
membrane closes, enabling fusion with the lysosome to form an
autophagic lysosome. During this process, the autophagosomes are
degraded, and the resulting molecules are recycled for use by the
cell (32,33). Fig.
2 illustrates the three types of autophagy and their respective
processes (24).
Autophagy in AML. AML development is marked by the
dynamic bidirectional regulation of autophagy (34). By eliminating oxidatively damaged
DNA, autophagy maintains the metabolic balance and protects the
genomic integrity of hematopoietic stem cells, preventing their
malignant transformation under normal conditions (35,36).
Autophagy undergoes a dynamic shift during leukemia progression: In
the early stages, it promotes oncogenesis by limiting genomic
instability, while in the later stages, it becomes an adaptive
mechanism that supports tumor cell survival and helps leukemia
cells endure stressful environments through metabolic remodeling
(37,38). This dichotomy suggests that
autophagy dysfunction may contribute to leukemia development, with
an imbalance in its regulatory network accelerating the emergence
of malignant phenotypes (39).
Research has elucidated the mechanisms by which
autophagy contributes to AML development. Targeting the
ATF4-dependent autophagy pathway could offer a therapeutic approach
for patients with Fms-like tyrosine kinase 3 (FLT3) mutations, as
Fms-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD)
expression increases basal autophagy, which is essential for AML
cell survival (40). KIT mutations
increase the likelihood of AML relapse, which is directly linked to
STAT3-mediated autophagy (41,42).
Nucleophosmin 1 (NPM1) mutations are common in AML, where NPM1
induces autophagy through the oncogene PML, thereby promoting AML
cell proliferation (43). By
inhibiting autophagy, the TP53 mutation, which is less common in
hematological cancers than in solid tumors, increases p53
expression and triggers apoptotic responses dependent on
p53-upregulated modulator of apoptosis and BAX, leading to AML cell
destruction (44). Glycolysis, a
key metabolic pathway in AML, influences the disease process by
promoting abnormal invasion, proliferation and drug resistance
(45). Research has demonstrated
that combination of autophagy inducers with glycolysis inhibitors
enhanced leukemia cell sensitivity to chemotherapy (46,47).
In relapsed cases, LSCs promote mitochondrial oxidative
phosphorylation by stimulating fatty acid β-oxidation, creating a
distinct metabolism-dependent drug resistance mechanism (48). Fatty acid metabolism is also
directly linked to AML progression (49–51).
The interconnections among these metabolic pathways create a
complex regulatory network that supports AML cell survival and
treatment resistance. Some of the aforementioned research has
translated into clinical practice, with studies showing that
combination of autophagy inducers with chemotherapeutic drugs
enhanced their effectiveness (52–54).
For instance, the mTOR inhibitor rapamycin, when combined with
chemotherapy drugs, can increase AML cell sensitivity to treatment
and promote apoptosis, providing a novel approach to enhance
chemotherapy efficacy (55).
Combining autophagy inhibitors with targeted therapies has also
improved efficacy. Inhibition of autophagy enhances the
anti-leukemic effect of FLT3 inhibitors in FLT3-ITD-positive AML,
offering a novel strategy to overcome resistance to targeted
therapies (56). Furthermore,
combination of multiple autophagy regulators can more effectively
control the autophagy process in AML cells. For example,
co-treatment with an mTOR inhibitor and a Beclin-1 activator
increases autophagy levels, promoting cell differentiation and
apoptosis, suggesting the potential therapeutic benefits of using
multiple regulators simultaneously (57). Further research on the mechanisms of
AML autophagy, as well as on the clinical efficacy and specific
applicability of related therapies, is required.
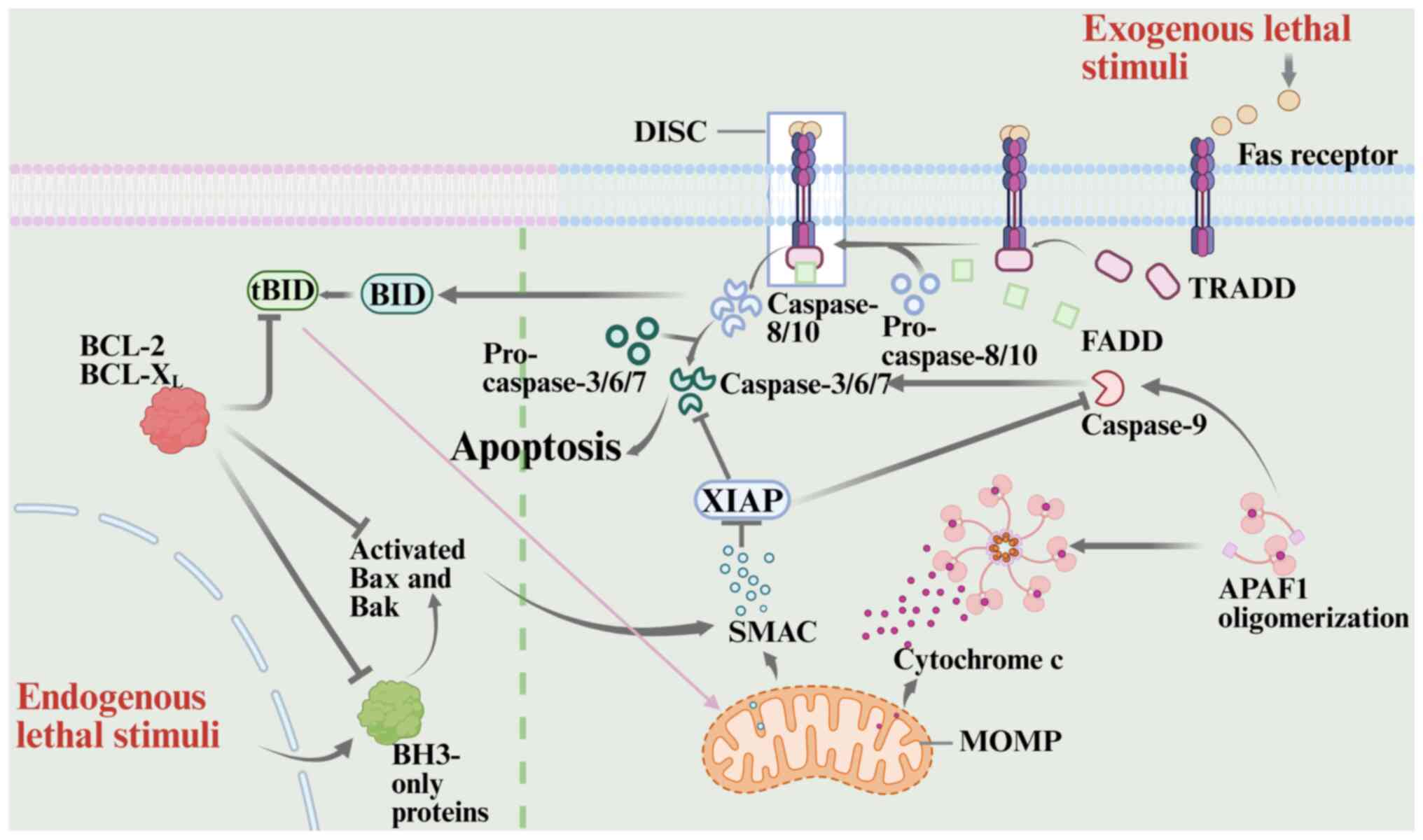
Overview of apoptosis. Apoptosis is a genetically
regulated process of programmed cell death that requires ATP to
execute a series of typical events (58). Endogenous and exogenous apoptosis
are two distinct pathways, and both are regulated by caspases
(59).
Proteins from the Bcl-2 and caspase-9 families are
crucial to the endogenous, or mitochondrial, apoptotic pathway. The
members are classified into pro-apoptotic proteins, which contain
only the BH3 domain, and anti-apoptotic proteins, based on their
function (60). Anti-apoptotic
proteins form heterodimers with pro-apoptotic proteins through the
BH1-3 domains, thereby inhibiting their pro-apoptotic activity
(61). Bax, a pro-apoptotic
protein, is upregulated and undergoes a conformational change upon
DNA damage or oxidative stress, triggering apoptotic signaling
(62). Bax then translocates from
the cytoplasm to the outer mitochondrial membrane, where it
oligomerizes to form channels (63). Meanwhile, BCL2 antagonist/killer,
originally located in the outer mitochondrial membrane, is
activated by BH3-only proteins, triggering a cascade that leads to
oligomerization and the formation of a transmembrane pore (58,64).
The two interact to increase mitochondrial outer membrane
permeability, resulting in the release of cytochrome c (CytC) from
the intermembrane space into the cytoplasm (65). Upon binding to apoptotic protease
activating factor-1, free CytC oligomerizes into heptameric
apoptotic vesicles, driven by deoxyadenosine triphosphate. The
exposed caspase recruitment domain (CARD) then recruits and
activates the initiator caspase-9, which subsequently cleaves the
effector caspase-3, triggering apoptosis (66).
Apoptosis in AML. In AML, the anti-apoptotic protein
BCL-2 is upregulated, enabling leukemia cells to evade apoptosis
and continue their growth (75,76).
Based on the understanding that BCL-2 interacts with pro-apoptotic
proteins, scientists have developed several BCL-2 inhibitors,
including oblimersen, obatoclax mesylate (GX15-070), ABT-737,
ABT-263 (navitoclax), ABT-199 (venetoclax) and S6384559 (77). Venetoclax, one of the most
extensively studied medications, has demonstrated strong clinical
efficacy both as a monotherapy and in combination with other drugs,
particularly low-dose cytarabine or demethylating agents, improving
patient survival and remission rates (78–80).
Venetoclax resistance has become increasingly evident as research
progresses, primarily due to dysfunction in pro-apoptotic
mechanisms and the upregulation of compensatory anti-apoptotic
proteins, such as myeloid cell leukemia-1 (MCL-1). To overcome
venetoclax resistance, appropriate inhibitors are being developed
in parallel (10,81). Despite the use of venetoclax,
additional apoptosis inducers are emerging. For instance, second
mitochondrial-derived activator of caspases (SMAC) analogs
targeting inhibitor of apoptosis protein (IAP) can antagonize IAPs
and promote apoptosis by mimicking endogenous SMAC proteins
(82). A pivotal clinical study has
demonstrated that SMAC analogs, when combined with conventional
chemotherapies or targeted therapies, can enhance the cytotoxic
effects on AML cells (83).
P53, a key pro-apoptotic transcription factor, is
closely associated with complex karyotypes, treatment resistance
and poor survival outcomes in patients with AML. This unique
clinical-molecular feature suggests that dysregulation of the TP53
signaling pathway is not only a key pathogenic mechanism of AML,
but also a potential marker for molecular stratification and a
target for targeted therapies, supporting the optimization of
precision treatment strategies (12). Thus, drugs such as MDM2 inhibitors
and P53 reactivators are being investigated for patients with TP53
mutations who have poor prognoses (84–86).
Numerous studies have demonstrated that leukemia
cells can also undergo apoptosis via activation of the death
receptor pathway, offering a potential basis for immunotherapy in
AML (87,88). AML cell apoptosis is strongly
influenced by the bone marrow microenvironment (89). Components of this microenvironment,
such as the extracellular matrix, regulate the expression of
apoptosis-related proteins in AML cells, affecting their
sensitivity to chemotherapy (90).
Future studies on apoptosis and AML should focus on
enhancing the efficacy and availability of current treatments while
minimizing their side effects to provide more effective AML
therapies.
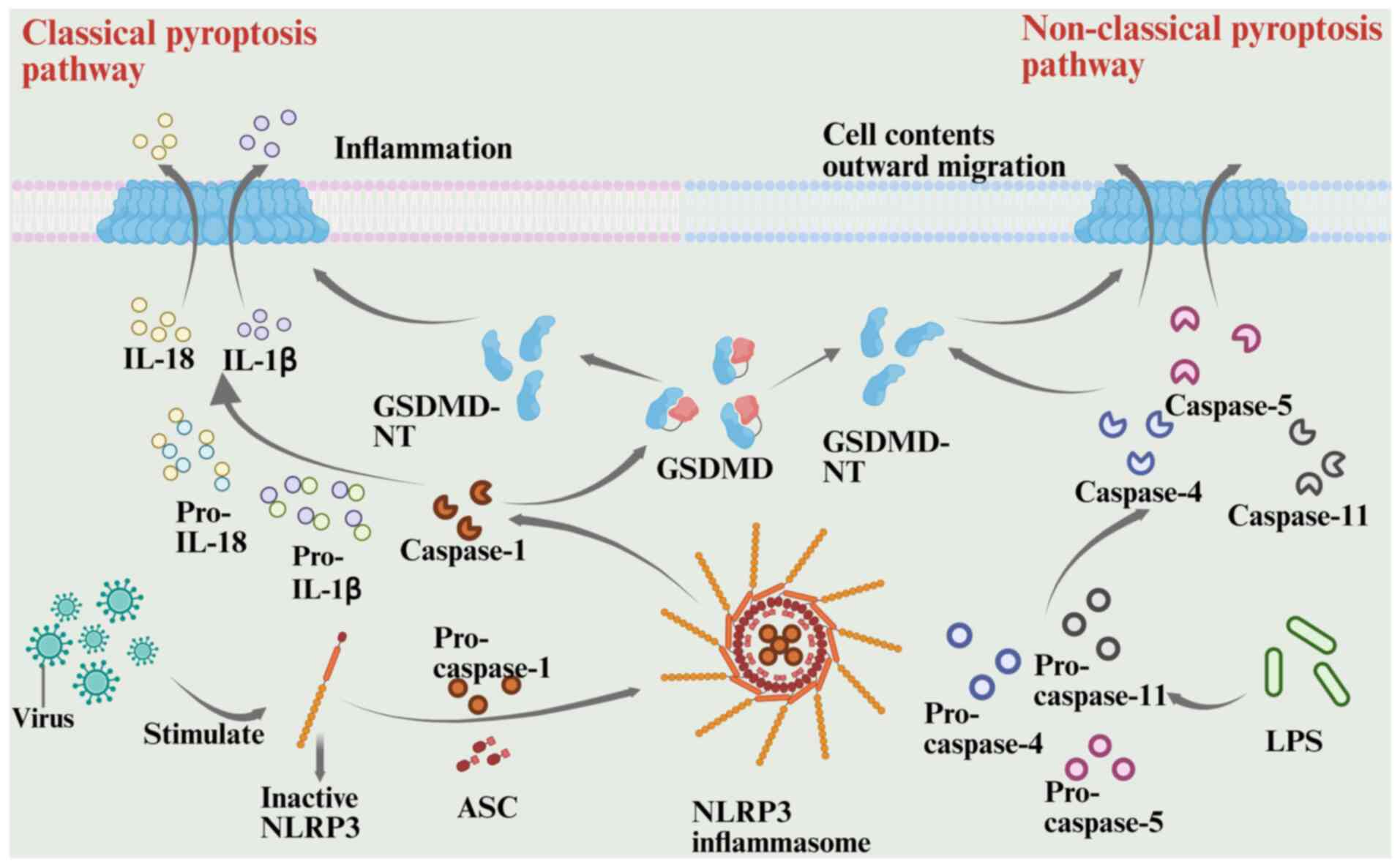
Overview of pyroptosis. The gasdermin (GSDM) family
includes six members (GSDMA, GSDMB, GSDMC, GSDMD, GSDME and
pejvakin), which are primarily expressed in tissues such as the
gastrointestinal tract and skin, and serve a key role in regulating
pyroptosis (91–93). GSDMD-induced pyroptosis can be
categorized into classical and non-classical pathways.
The classical pyroptosis pathway begins with pattern
recognition triggered by pathogen infection and proceeds through
the inflammatory vesicle-GSDMD axis. When intracellular NOD-like
receptors detect pathogen-associated molecular patterns, the pyrin
domain (PYD) recruits the adaptor protein apoptosis-associated
speck-like protein containing a CARD (ASC). This forms a platform
for multimerization and recruits pro-caspase-1, which assembles
into a functional inflammatory vesicle complex (94,95).
When the complex activates caspase-1, it cleaves GSDMD. This
cleavage releases the C-terminal domain of GSDMD, thereby relieving
its inhibition of the N-terminal pore-forming domain. The released
GSDMD N-terminal oligomerizes and embeds in the cell membrane,
forming a nanoscale pore that disrupts cellular osmolality, causes
content leakage and triggers pyroptosis, characterized by cell
swelling (96–98). The primary mechanism of the
anti-infective immunity of the host involves processing the
precursors of IL-1β and IL-18, which are released extracellularly
through the GSDMD pore in their mature forms. This recruits
neutrophils and other inflammatory cells, triggering a cascade
amplification effect (94,99). This is achieved through membrane
pore-mediated ‘inflammatory death’, which eliminates infected cells
while tightly regulating the local inflammatory response (100).
The nonclassical pyroptosis pathway is triggered by
the direct sensing of lipid components, with caspase-4/5/11
detecting intracellular lipopolysaccharide (LPS) (101–103). Unlike the traditional system, this
mechanism does not involve ASC bridging proteins. Caspase-4/5/11
directly binds LPS through its CARD and undergoes oligomerization
for self-activation (102–104). Activated caspase-4/5/11
specifically cleaves GSDMD to release the N-terminal pore domain
(NT), leading to cell membrane perforation and pyroptosis (105). However, since NT does not directly
process IL-1β/IL-18 precursors, the inflammatory response is only
mildly activated (102,106). In non-immune cells, caspase-4
indirectly cleaves pro-IL-18 through conformational changes,
amplifying local inflammatory signals (107) Similarly, by promoting the
activation of inflammatory vesicles containing NACHT, LRR and PYD
domains-containing protein 3 (NLRP3), caspase-11 enhances
caspase-1-mediated IL-1β maturation and secretion, forming a
synergistic network of classical and non-classical regulatory
mechanisms (104).
The pathophysiological relevance of pyroptosis in
infections, tumors and inflammatory diseases is underscored by the
ability of certain GSDM family members (specifically GSDMD, GSDMB
and GSDMC) to induce focal cell death via specific protease
cleavage and link programmed death with the immune response through
a multidimensional regulatory network (108–110). Both the classical and
non-classical pathways of pyroptosis are illustrated in Fig. 4 (100).
Pyroptosis in AML. Research has shown that
inflammasomes, key molecular complexes involved in pyroptosis,
serve a dual role in the occurrence, progression and treatment of
leukemia, with potential clinical applications (111). Evidence suggests that pyroptosis
serves a critical, bidirectional role in both the progression of
AML and its treatment response (111). Mechanistic research has
demonstrated that chemotherapy or targeted therapies enhance
anti-AML efficacy by activating the focal cell death pathway
(18). A study by Ren et al
(12) showed that estrogen receptor
activation enhanced the anti-AML effect of the BCL-2 inhibitor
vincristine, via the pyroptosis pathway. Demethylating agents can
promote venetoclax-induced AML pyroptosis by restoring GSDME
expression, offering a potential strategy to overcome resistance to
BCL-2 inhibitors (14). In addition
to enhancing existing targeted therapies and chemotherapy,
pyroptosis could provide a novel target for AML treatment. Small
molecule inhibitors targeting serine dipeptidyl peptidase
(DPP)8/DPP9 have been shown to induce cellular pyroptosis in a
majority of a panel of human AML cell lines and primary samples,
suggesting that this target may offer a novel approach for AML
treatment (112). Suppression of
reticulocalbin 1 gene expression reduces bone marrow mononuclear
cell activity in patients with AML, further confirming the crucial
role of cellular pyroptosis in AML pathogenesis (113).
The ‘immunogenic death’ feature of pyroptosis, which
releases cytokines that reshape the tumor immune environment and
reduce immunosuppressive cells, underlies its significance in AML
therapy by helping overcome immune escape (12). For example, receptor-interacting
serine/threonine-protein kinase 3 effectively inhibits aberrant
bone marrow proliferation in the FLT3-ITD mutant AML model by
regulating IL-1β production via inflammatory vesicles. However, it
can also modify the bone marrow microenvironment, impairing normal
hematopoietic stem progenitor cell function while promoting LSC
development (114–116). Cytarabine promotes IL-1β secretion
through NLRP3 inflammasomes, offering a novel approach to improve
treatment strategies (117). Ren
et al (12) also
demonstrated that targeted activation of G protein-coupled estrogen
receptor improved vincristine efficacy by enhancing leukemic cell
pyroptosis and CD8+ T-cell immunity in patients with
AML, a process mediated by IL-1β/18.
The challenge lies in the bidirectional regulation
of pyroptosis, where its intensity must be carefully controlled to
prevent inflammatory storms. Despite clinical studies demonstrating
the therapeutic benefit and safety of pyroptosis-inducing therapies
in AML, further research is needed to optimize these treatments and
understand their long-term effects on patients (12,117).
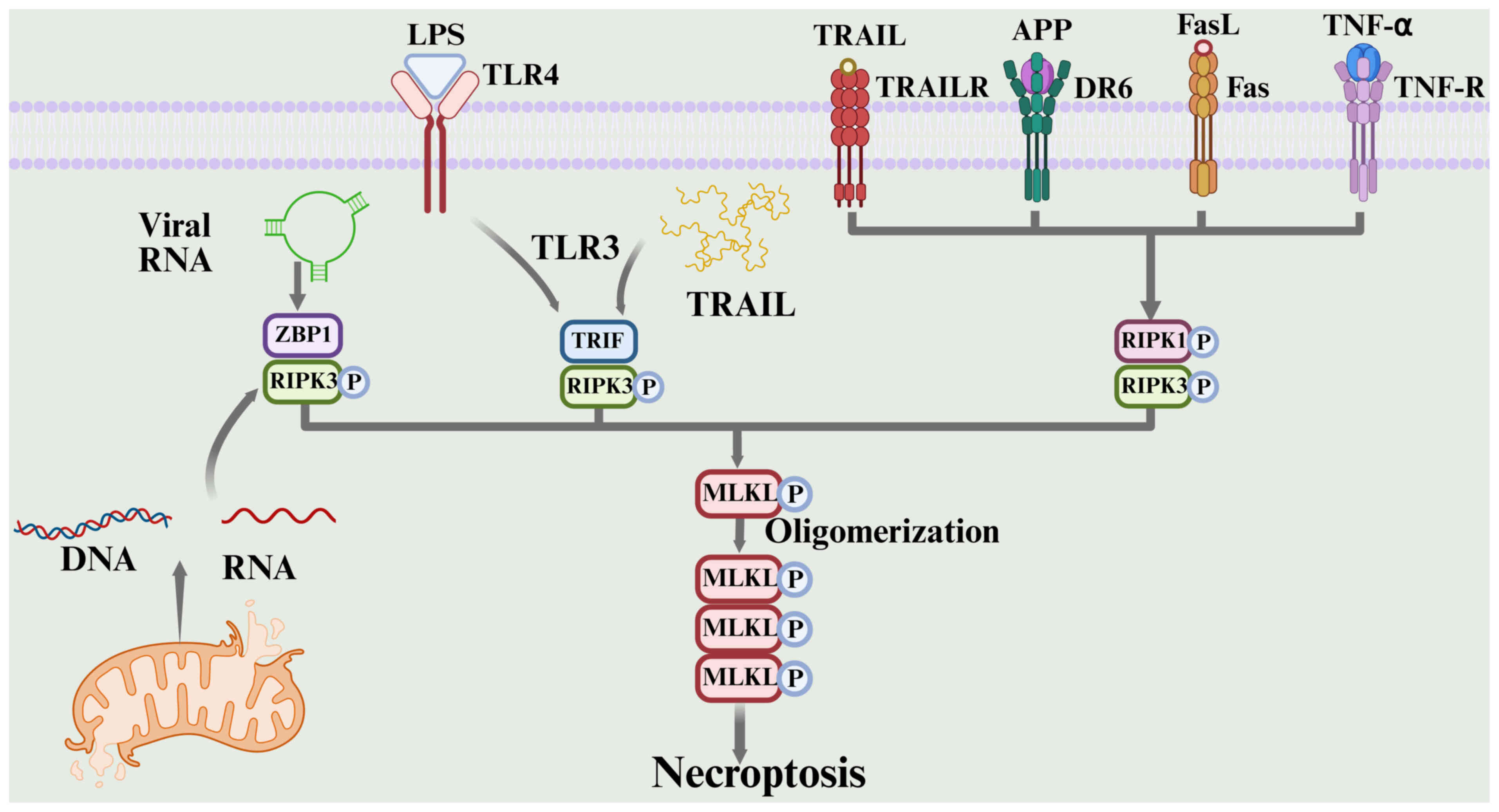
Overview of necroptosis. Necroptosis is a programmed
cell death mechanism independent of cysteine proteases, typically
induced when the apoptotic pathway is blocked by viruses or
medications (118). Its molecular
core involves the receptor interacting serine/threonine kinase 1
(RIPK1)-receptor interacting serine/threonine kinase 3
(RIPK3)-mixed lineage kinase domain like pseudokinase (MLKL)
signaling axis (119). The
activation pathway is complex and diverse. In the classical
pathway, the TNF receptor superfamily recruits RIPK1 through its
death domain when caspase-8, the main executor of apoptosis, is
inhibited genetically or pharmacologically. This results in the
formation of an amyloid-fibril-like signaling complex (necrosome)
with RIPK3, triggering an autophosphorylation cascade of RIPK3
(118,120–122). By contrast, in pattern recognition
receptor-mediated bypass, toll-like receptor 3 directly recruits
RIPK3 via the TIR domain-containing adapter molecule 1 (TRIF),
forming a nonclassical necrosome complex that bypasses RIPK1. This
facilitates signal transduction upon recognition of viral
double-stranded RNA or toll-like receptor 4 in response to
endotoxin LPS (118,123). Additionally, Z-DNA binding protein
1 (ZBP1) acts as a nucleic acid sensor, detecting endogenous
DNA/RNA released from viral replication intermediates or
mitochondrial stress. ZBP1 specifically binds to RIPK3 through its
receptor-interacting protein kinase homotypic interaction motif,
initiating a necroptosis program independent of RIPK1 and TRIF
(118,124). The multi-pathway signals converge,
indicating that RIPK3 phosphorylates the MLKL pseudokinase domain,
triggering a conformational change in MLKL and the formation of
transmembrane pores. This leads to intracellular calcium overload,
osmotic imbalance and loss of membrane integrity, resulting in the
release of cellular contents and a potent pro-inflammatory response
(118,125–129). Notably, the small molecule
necrostatin-1 serves as a molecular tool for precisely modulating
necrotic apoptosis subtypes by blocking RIPK1-dependent necroptosis
through the ATP-binding pocket of the RIPK1 kinase domain. However,
it is ineffective against ZBP1- or TRIF-mediated nonclassical
pathways (130). The flexibility
of this signaling pathway and multiple regulatory mechanisms make
necroptosis potentially bidirectional in tumor microenvironment
remodeling, chemotherapeutic resistance and immune escape, namely,
it can exert both antitumor and pro-tumor effects depending on the
context (131). The process of
necroptosis is shown in Fig. 5
(132).
Necroptosis in AML. The RIPK1/RIPK3/MLKL axis serves
a crucial role in necroptosis, with its dysregulation in AML cells
closely linked to disease progression (13). The role of RIPK3 in AML is complex
and context-dependent. In early leukemogenesis, RIPK3 may act as a
tumor suppressor by inducing necroptosis and promoting the
differentiation of leukemia-initiating cells, thereby preventing
myeloid leukemia progression (13,133).
However, in established AML, the role of RIPK3 is often reversed.
Clinical evidence shows that high RIPK3 expression is associated
with poor prognosis in specific AML subtypes, such as NPM1-mutant
AML (134). Wang et al
(134) specifically reported that
elevated RIPK3 expression was an independent predictor of poor
overall survival in specific AML subtypes, such as FAB-M4/M5,
normal karyotype and NPM1 mutation subtypes, a finding strongly
supported by Kaplan-Meier survival analysis (log-rank P=0.021;
hazard ratio, 1.8; 95% CI, 1.2–2.5). This paradox
(tumor-suppressive role in initiation but oncogenic role in
progression) highlights the dual role of RIPK3 and emphasizes the
need for expression-based patient stratification in future
therapeutic strategies (135). A
study by Zhu et al (136)
identified restoring RIPK3 expression to reverse
R-2-hydroxyglutarate-induced necroptosis in isocitrate
dehydrogenase-mutant AML cells as a potential therapeutic strategy
for patients with AML. However, Hillert et al (137) suggested that targeting the RIPK1
pathway offered a more promising therapeutic approach for patients
with FLT3-ITD-mutant AML. Targeting necroptosis pathways in
combination with traditional chemotherapy or targeted therapies
holds potential for effective treatment (138,139). Li et al (138) found that combination of the RIPK1
inhibitor 22b with cidabenamide enhanced its antileukemic effect in
FLT3-ITD-positive AML cell lines and primary samples. Combining the
RIPK1 inhibitor with the BCL-2 inhibitor venetoclax promotes
apoptosis in AML cells and overcomes resistance to single-drug
treatment (139). A study has
shown that A20 deletion in AML restored sensitivity to
anthracycline by inducing necroptosis (140). Although the aforementioned studies
have demonstrated that targeting the necroptosis pathway is a
viable strategy in AML, the clinical relevance of the
RIPK1/RIPK3/MLKL axis remains unclear (135–137). Future research should focus on
developing biomarkers to predict patient response to therapies
targeting the necroptosis pathway, aiding in precision
medicine.
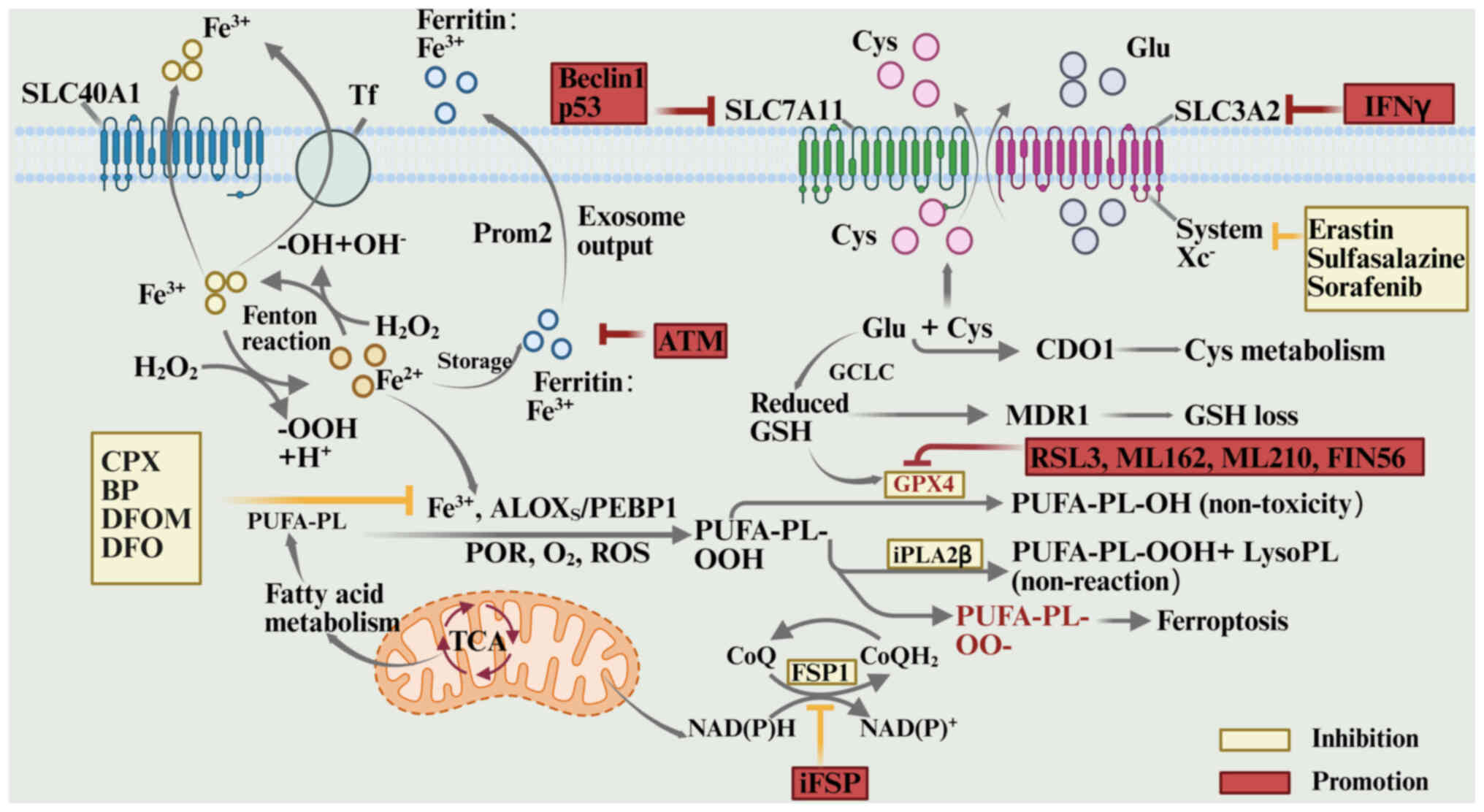
Overview of ferroptosis. Ferroptosis is a form of
programmed cell death driven by iron-dependent lipid peroxidation
(141). Ferroptosis results from
metabolic disruption caused by the imbalance of intracellular redox
homeostasis (142). At
physiological levels, iron serves as a key cofactor for redox
enzymes, contributing to electron transport and energy metabolism
(143). Excess free iron
(Fe2+) generates reactive oxygen species (ROS) through
the Fenton reaction, causing lipid peroxidation of the membrane
(144). This disturbs cellular
membrane integrity and organelle function (145).
The primary mechanism of ferroptosis is a dynamic
imbalance of lipid peroxides, regulated by the glutathione (GSH)
metabolic pathway and antioxidant defenses (146–149). Polyunsaturated fatty acid
phospholipid hydroperoxide (PUFA-PL-OOH) is the key effector in
this process, produced through the combined action of
Fe2+, lipoxygenases (arachidonate lipoxygenases),
phosphatidylethanolamine-binding protein 1 complexes, cytochrome
P450 reductase and ROS. These molecules collaborate to convert
PUFA-PL into the harmful PUFA-PL-OOH (147). Ferroptosis suppressor protein 1
(FSP1) and GSH peroxidase 4 (GPX4) are the main mechanisms
maintaining PUFA-PL-OOH at steady state. GPX4 reduces PUFA-PL-OOH
via a GSH-dependent process, converting it to its non-toxic form
(147). By contrast, FSP1
scavenges lipophilic free radicals and inhibits the lipid
peroxidation chain reaction by producing the reduced form of
coenzyme Q10 (CoQH2) (148,149). In the absence of GPX4 inhibition
or FSP1-CoQH2 system dysfunction, lipid peroxides
accumulate due to impaired scavenging. Furthermore, Fe2+
catalyzes the conversion of hydrogen peroxide into hydroxyl
radicals via the Fenton reaction, intensifying lipid peroxidation
and ROS bursts, and ultimately causing enzyme system collapse and
cellular structural damage (146,147–152).
The ferroptosis regulatory network is multilayered.
At the metabolic input level, the System Xc−
transporter, composed of solute carrier family 7 member 11
(SLC7A11) and solute carrier family 3 member 2, mediates cystine
uptake, providing essential precursors for GSH synthesis (147,153–155). Inhibitors of this system reduce
intracellular GSH levels by blocking cystine uptake, impairing GPX4
function and triggering ferroptosis (156). At the enzymatic regulatory level,
GPX4 activity can be directly inhibited by small molecules such as
RAS-selective lethal 3 and ML162, while GSH production is blocked
by buthionine sulfoximine (157).
Iron homeostasis is crucial, with divalent iron either removed by
chelating agents such as desferrioxamine and bipyridine or expelled
via the exosomal pathway after being stored as Fe3+ in
ferritin (158,159). In addition, external ROS
stimulation or disruption of lipid peroxidation detoxification can
disrupt the redox balance, leading to the accumulation of toxic
lipid products and triggering ferroptosis (160). These multidimensional regulatory
mechanisms collectively shape the complex biological effects of
iron death in cell fate decisions (145). The process of ferroptosis, with
its inhibitory factors and promoting factors, is illustrated in
Fig. 6 (145).
Ferroptosis, an iron-dependent form of non-apoptotic
cell death, offers unique potential for pathological modulation and
therapeutic intervention in AML (15). AML cells are more susceptible to
ferroptosis due to metabolic reprogramming and disrupted redox
homeostasis (161). This
susceptibility is further supported by significantly lower GPX4
expression in primary AML samples compared with normal bone marrow
(P=1.3×10−6), along with reduced GSH levels and
increased mitochondrial lipid peroxidation (162).
Ferroptosis holds significant potential in targeted
therapy for AML. Research suggests that several natural products
possess the potential to induce ferroptosis in cancer treatment
(163). Crotonoside, a key
constituent of Crotonus sativus listed in the Chinese
Pharmacopoeia, induces ferroptosis in AML cells by stimulating
lipid peroxidation and increasing autophagy, offering a novel
approach for AML treatment (164).
Birsen et al (165)
demonstrated that APR-246-induced AML cell death was inhibited by
iron chelators, lipophilic antioxidants and lipid peroxidation
inhibitors, suggesting that APR-246 induced ferroptosis in AML
cells. Lin et al (166)
demonstrated that biomimetic high-density lipoprotein nanoparticles
could disrupt intracellular cholesterol homeostasis by targeting
the scavenger receptor class B type 1 receptor, which was
upregulated in AML cells. This inhibited the GPX4-mediated
antioxidant pathway and induced ferroptosis in AML cells at
nanomolar concentrations, representing a major innovation in AML
treatment.
Research indicates that M2 macrophage-derived growth
differentiation factor 15 confers ferroptosis resistance in the
tumor microenvironment, identifying a key drug resistance mechanism
(167). AML cells are more
resistant to mitoxantrone when co-cultured with M2 macrophages,
providing a novel approach for overcoming AML
microenvironment-mediated drug resistance (168). As a potent ferroptosis inducer and
chemotherapeutic drug carrier for AML treatment, Yu et al
(169) showed that the
ferroptosis-inducing nanomedicine glutathione-capped ferrite
nanoparticles successfully overcame AML resistance. Inhibition of
nuclear factor erythroid 2-related factor 2 (NRF2) expression
enhanced the anti-AML effect of vinpocetine by inducing AML cell
death via the ferroptosis pathway (170). Pardieu et al (171) determined that SLC7A11 promoted AML
cell survival by encoding the xCT cystine importer. The authors
also demonstrated that combining daunorubicin with xCT inhibition
achieved optimal anti-AML efficacy, suggesting that xCT inhibition
combined with chemotherapy could be a promising strategy for AML
treatment.
While ferroptosis has shown considerable promise in
AML treatment, several challenges remain (172). These include the lack of
techniques for monitoring ferroptosis dynamics, tumor
heterogeneity-related differences in response and unclear
mechanisms of microenvironment regulation. Addressing these issues
is crucial for improving the long-term survival of patients with
AML.
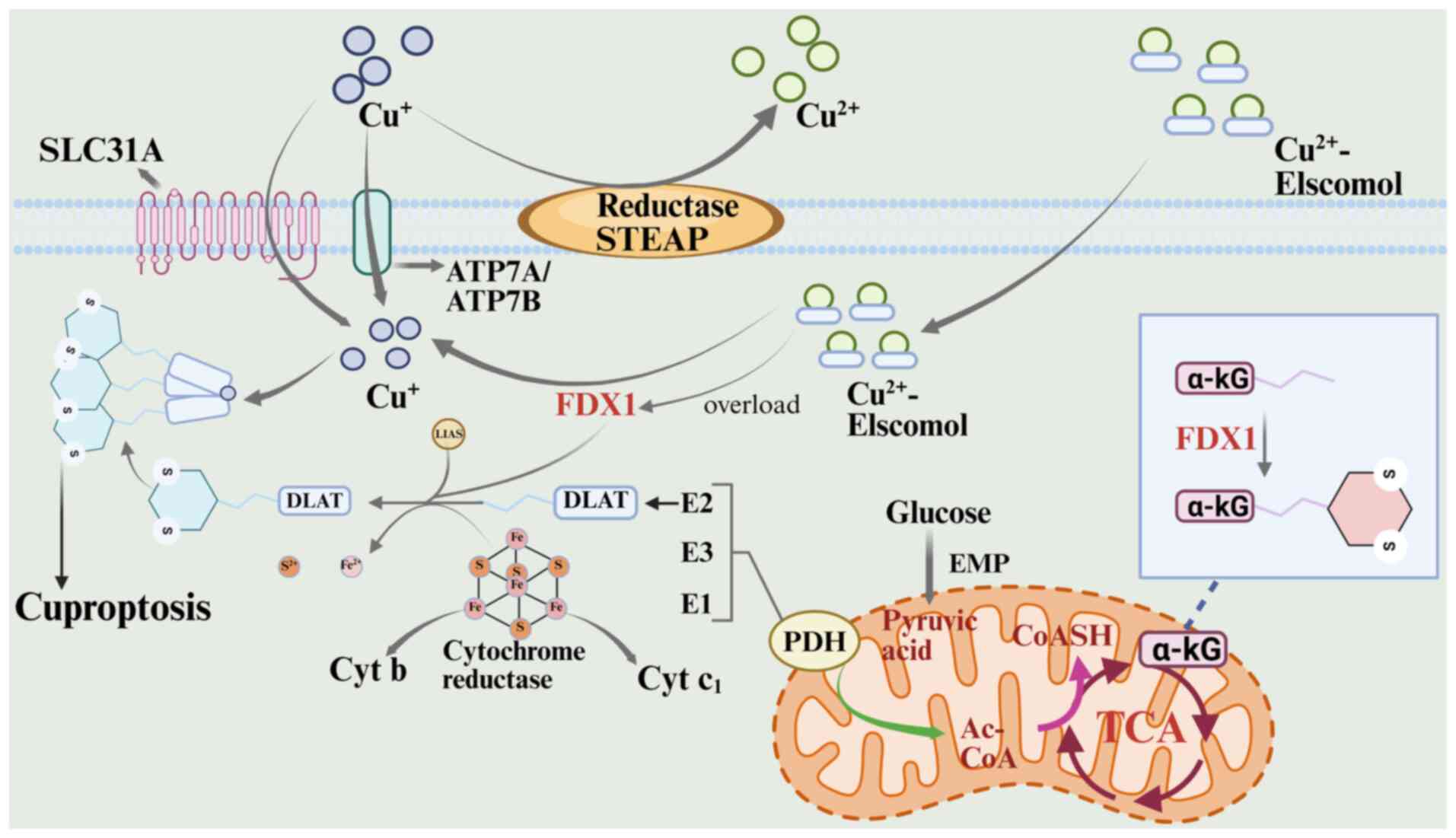
Overview of cuproptosis. Cuproptosis, a
copper-dependent form of RCD, was first proposed by Tsvetkov et
al (173) in 2022. The unique
mechanism distinguishes cuproptosis from other death pathways, such
as apoptosis, pyroptosis, necroptosis and ferroptosis. Cuproptosis
is based on the direct binding of copper ions to lipoylase proteins
in the tricarboxylic acid cycle, leading to abnormal aggregation
(174). This copper-mediated
interaction destabilizes iron-sulfur cluster proteins, disrupting
key components of the mitochondrial respiratory chain and inducing
protein homeostatic imbalance and oxidative stress, ultimately
driving cell death via the proteotoxic stress pathway (175).
A key breakthrough in cuproptosis research was the
2019 discovery by Massachusetts Institute of Technology and Harvard
teams of the copper ion carriers disulfiram and ilithromycin (ES),
two small molecules that can specifically mediate copper ion
transport across membranes, providing a central tool for studying
copper-dependent cell death (176). As an anticancer treatment
targeting mitochondrial metabolism, ES not only increases oxidative
stress and ROS formation, but also triggers a novel cell death
pathway through the accumulation of copper ions, according to
recent findings (173). Tsvetkov
et al (173) demonstrated
that ES-induced cell death occurred without caspase-3 activation
and was resistant to both classical apoptosis inhibitors and other
death pathway blockers, confirming its independence from known
death mechanisms. In-depth mechanistic studies revealed that ES
penetrates the cell membrane by forming a complex with
extracellular Cu2+, reducing Cu2+ to
Cu+, and releasing highly reactive ROS in the
mitochondria. Free ES then re-enters the circulatory system to
transport Cu2+, ultimately leading to copper overloading
in mitochondria through the ‘ion shuttle’ mechanism (177–179). The lethal effects of ES were
reduced by electron transport chain complexes and the mitochondrial
pyruvate carrier, but not by mitochondrial uncouplers. Metabolomics
analyses revealed a time-dependent increase in tricarboxylic acid
cycle intermediates during ES treatment, suggesting that copper
toxicity primarily disrupts the tricarboxylic acid cycle rather
than oxidative phosphorylation (180,181).
Genetic evidence has indicated that ferredoxin 1
(FDX1)-knockout cells were resistant to cuproptosis, confirming
that loss of FDX1, a key regulator of copper toxicity signaling,
prevents the lipoylated protein aggregation cascade triggered by
copper accumulation. This established that FDX1-mediated metabolic
disruption is a core molecular pathway for cuproptosis execution
(182). FDX1-mediated toxicity in
cuproptosis occurs through the specific interaction of
Cu+ with the mitochondrial lipoylase system, which
triggers deleterious protein aggregation (173). Cu+ binds to the fatty
acylated pyruvate dehydrogenase complex, inducing abnormal
oligomerization and inactivation of the enzyme, which prevents
pyruvate conversion to acetyl-CoA and disrupts metabolic flow via
the tricarboxylic acid cycle (173,183). FDX1 serves multiple roles in this
process: It catalyzes the lipid acylation of pyruvate dehydrogenase
and α-ketoglutarate dehydrogenase, and enhances copper ion toxicity
by converting Cu2+ to the more toxic Cu+
(184). Lipoic acid synthase,
meanwhile, facilitates lipid acylation by synthesizing lipoic acid
and aids the assembly of Fe-S cluster proteins in collaboration
with FDX1, together forming the molecular network underlying copper
ion toxicity (184). In this
process, depletion of large Fe-S clusters disrupts the electron
transport chain, while the blockage of the tricarboxylic acid cycle
further impairs mitochondrial energy metabolism, ultimately leading
to cell death through proteotoxic stress and metabolic disruption
(173). This pathway aligns
closely with the previously identified ES-induced cuproptosis
pathway (173). Loss of FDX1
function inhibits both copper ion-mediated lipoylated protein
aggregation and metabolic reprogramming, highlighting its critical
role in cuproptosis signaling (185). The entire process of the
cuproptosis pathway is illustrated in Fig. 7 (174).
The copper-disulfide complex (Cu-DSF) binding
system selectively targets cancer cells and cancer stem cells
(186). The Cu-DSF exhibits
dose-dependent cytotoxicity in LSCs, with minimal effects on normal
hematopoietic progenitor cells. This selective toxicity is likely
due to the enhanced copper ion enrichment in cancer cells (16). A study has demonstrated that
patients with AML had reduced serum levels of zinc and selenium,
accompanied by elevated copper levels, suggesting that disruptions
in copper metabolism contribute to disease progression (187). Since LSCs contain higher copper
levels than normal tissues, researchers have suggested using copper
ion carriers to selectively transport copper ions into tumor cell
mitochondria. This induces intracellular copper overload,
triggering lethal effects such as oxidative stress, proteotoxicity
and mitochondrial dysfunction, ultimately achieving the specific
killing of tumor cells (188).
Current copper-targeted therapies for AML focus on
the antitumor effects of copper chelators in hematologic
malignancies (189–191). Disulfiram-copper complexes
demonstrate anti-AML activity in both in vitro and in
vivo assays, and partially overcome drug resistance in AML
cells (192,193). Xu et al (16) demonstrated that disulfiram, either
alone or in combination with copper, inhibited AML cell
proliferation and induced apoptosis through mechanisms involving
inactivation of the NRF2-NF-κB signaling pathway and activation of
the ROS-JNK stress pathway due to copper accumulation. Furthermore,
in the non-obese diabetic/severe combined immune deficiency mouse
model, disulfiram also suppressed the growth of human
CD34+/CD38+ AML cell xenograft tumors,
further confirming its therapeutic potential in vivo. The
clinical translation of copper-targeted therapies faces challenges,
including off-target effects, systemic toxicity and drug resistance
(194). For instance, copper
chelators damage normal cells due to nonspecific binding, while
long-term use of copper transporter inhibitors may lead to
compensatory metabolic adaptation (195). A growing body of evidence
underscores a strong association between cuproptosis and the
pathogenesis of AML (196–198). This includes the dynamic
expression of copper transporter proteins, the regulation of copper
storage proteins, and the interplay between copper metabolism and
the tumor microenvironment (188,194). Further research is needed to
optimize copper carrier targeting with nanodelivery technologies
and explore the synergistic effects of copper overload on cell
death pathways, aiming to enhance therapeutic specificity and
minimize the toxicity risk (194).
Cell death pathways are intricately interlinked,
forming a complex signaling network that collectively governs cell
fate (199). Autophagy also
mitigates pyroptosis and its associated inflammation by removing
inflammatory vesicle components, inflammatory factor precursors and
damaged mitochondria (200).
Autophagy serves a critical and bidirectional role in ferroptosis:
Lipophagy scavenges lipid peroxides, while ferritin phagocytosis, a
selective form of autophagy, releases ferric ions and drives
ferroptosis (201). In the context
of cuproptosis, autophagy may regulate the process by removing
copper-induced aggregation of proteins or damaged mitochondria
(173). Apoptosis, a conventional
form of programmed cell death, is reciprocally suppressed by
necroptosis: The apoptotic executor caspase-8 cleaves and
inactivates RIPK1/RIPK3, key kinases involved in necroptosis
(202). Necroptosis often serves
as an alternative pathway when apoptosis is inhibited (203). By contrast, apoptosis and
pyroptosis are linked by GSDM proteins; caspase-8 cleavage of GSDME
converts apoptosis into an inflammatory form of cell death
characterized by pyroptosis (203). Apoptosis and ferroptosis share
common regulators, such as p53, which initiate both processes.
Additionally, anti-apoptotic proteins can suppress ferroptosis,
while mitochondrial damage induced by ferroptosis may also trigger
apoptosis (204,205). Oxidative or endoplasmic reticulum
stress caused by copper ion accumulation may indirectly activate
apoptotic pathways (178).
Necroptosis and pyroptosis are forms of inflammatory programmed
necrosis that share the upstream signal RIPK1 (206). At the execution level, their
terminal effectors, MLKL and GSDMD, can further interact through
synergistic or competitive mechanisms during pore formation
(207). Both necroptosis and
pyroptosis share ROS triggers with ferroptosis. TNF produced by
necroptosis or inflammatory factors released during pyroptosis can
create a pro-oxidative environment that indirectly promotes
ferroptosis (208). Additionally,
damage-associated molecular patterns released by ferroptosis can
activate inflammatory vesicles, triggering pyroptosis through a
feedback mechanism (209).
Although ferroptosis and cuproptosis have distinct core mechanisms,
both involve metal ions and oxidative stress. Iron and copper
metabolism are interconnected, with key regulatory nodes, such as
mitochondrial function, ROS levels, inflammatory signaling and the
balance of iron/copper/cystine, serving as pivotal points in the
crosstalk between these death pathways (210). Understanding this complex network
is crucial for explaining cell fate regulation during cancer
pathogenesis and for developing precision treatments that target
specific death pathways, as inhibition of one may trigger
compensatory activation of another.
Although preclinical evidence strongly supports
targeting multiple RCD pathways in AML, clinical translation faces
challenges due to variable patient responses (18). The most successful approach to date
focuses on apoptosis, where the combination of the BCL-2 inhibitor
venetoclax and hypomethylating agents has improved outcomes for
elderly patients with AML or those ineligible for intensive
chemotherapy. This strategy has been validated in phase III trials
and has received regulatory approval, providing an effective means
to overcome apoptosis escape (204). Current research is focused on
overcoming venetoclax resistance, particularly by targeting
complementary anti-apoptotic proteins such as MCL-1 or by combining
it with other targeted therapies, such as FLT3 inhibitors. Several
clinical trials are actively exploring these approaches (204,211,212).
By contrast, direct modulation of other RCD
pathways, such as autophagy, necroptosis, pyroptosis and
ferroptosis, is still in its early stages. Current clinical
evidence primarily comes from observations where these death
pathways are incidentally induced by conventional chemotherapies or
targeted therapies. Targeting autophagy has been particularly
challenging due to its context-dependent dual role, with
non-specific inhibitors such as chloroquine showing limited
efficacy, highlighting the need for more precise agents (213). For example, hypomethylating agents
promote GSDME-mediated pyroptosis, while the p53 reactivator
APR-246 induces ferroptosis, suggesting that the partial
therapeutic efficacy of these agents may stem from non-apoptotic
death mechanisms (14,158). However, first-in-class drugs
designed to activate these pathways, such as DPP8/9 inhibitors for
pyroptosis, RIPK1 activators for necroptosis and GPX4 inhibitors
for ferroptosis, have yet to enter clinical trials for AML,
highlighting a translational gap and the need for further research
(13,103,159).
The copper ionophore disulfiram has been used for
decades with sporadic anticancer activity, while its systematic
evaluation in AML is still confined to an early-stage trial,
highlighting a significant translational challenge for the
cuproptosis pathway (214). The
novel copper ionophore electrochlorol is currently under
early-stage investigation for solid tumors, and its potential to
target mitochondria-rich LSCs in AML remains a hypothesis awaiting
clinical validation (188).
Advancing this field requires overcoming several
key challenges: The lack of reliable biomarkers for predicting
treatment response and enabling precise patient stratification;
managing the complex and often severe immune-related toxicities
triggered by inflammatory cell death; the biological complexity and
potential for overlapping toxicities when designing combination
therapies to exploit synergistic effects across pathways; and the
difficulty in creating targeted delivery systems that enhance
efficacy while minimizing off-target effects. In summary,
translation of mechanistic insights into clinical applications
remains a dynamic frontier. The success of venetoclax demonstrates
the potential of targeting cell death pathways. Future efforts
should focus on translating insights from other RCD pathways into
safe and effective clinical strategies, overcoming resistance and
improving AML outcomes by expanding the therapeutic arsenal. The
clinical development targeting the RCD pathway in AML is summarized
in Table I (13,14,103,158,159,188,204,211–214).
AML is a highly aggressive hematologic malignancy
in adults, characterized by a high recurrence rate and treatment
resistance, both of which threaten patient survival (215). Despite advancements in molecular
typing and targeted therapies that have improved the prognosis of
some subtypes, the mechanisms driving AML heterogeneity and its
microenvironmental interactions remain poorly understood (215). The limitations of chemotherapy and
hematopoietic stem cell transplantation have prompted researchers
to explore novel strategies targeting programmed cell death. These
approaches aim to enhance treatment efficacy while reducing damage
to the normal hematopoietic system, either by selectively inducing
LSC clearance or remodeling the bone marrow immune
microenvironment. Studies have shown that regulation of cell death
processes, including autophagy, apoptosis, necroptosis, pyroptosis,
ferroptosis and cuproptosis, can successfully target drug-resistant
clones (17,216,217). Notably, autophagy can exhibit a
context-dependent dual role in this process. In LSCs, autophagy
helps maintain chemoresistance through homeostatic functions;
however, its overactivation may trigger type II programmed death,
eliminating metabolically compromised cells (53). The BCL-2/MCL-1 balance in the
apoptotic pathway is disrupted in drug-resistant clones, and BH3
mimics can alter mitochondrial apoptotic thresholds, thereby
overcoming anti-apoptotic defenses. In the AML microenvironment,
the RIPK1/RIPK3/MLKL necroptosis cascade is suppressed; however,
its activation can bypass apoptosis resistance and induce
inflammatory cell death. Pyroptosis-induced pore formation by GSDM
family proteins and activation of inflammatory vesicles result in a
dual mechanism that eliminates leukemia cells and reshapes the
immunosuppressive microenvironment. Ferroptosis regulates lipid
peroxidation via the GPX4/acyl-CoA synthetase long chain family
member 4 axis, and targeting of GSH metabolism selectively
eliminates resistant subpopulations that are dependent on
antioxidant defenses. The emerging cuproptosis mechanism induces
proteotoxic stress via FDX1-mediated copper ion overload, providing
a novel strategy to target recalcitrant LSCs with abnormal
mitochondrial metabolism. These RCD processes form a networked
system of spatiotemporal regulation. A combinatorial intervention
targeting multiple nodes can synergistically overcome the
death-resistance barrier of leukemia cells, offering a
multidimensional approach to address therapeutic challenges posed
by AML clonal evolution and microenvironmental sheltering.
Combination therapies targeting RCD are being clinically validated,
but their long-term effects on clonal evolution, immune homeostasis
and metabolic reprogramming remain to be fully explored. Future
research should investigate the synergistic impact of different RCD
modalities on AML stem cell maintenance, chemoresistance and
microenvironmental remodeling in order to enable the development of
personalized therapies that precisely target death pathways.
Not applicable.
The present study was supported by the Tianjin Health
Commission, Research Project of TCM and Western Integrative
Medicine (grant no. 2023130), the Tianjin Education Commission,
Tianjin Education Commission Research Project (General Project)
(grant no. 2021KJ145), and the Shi Zhexin Tianjin Famous
Traditional Chinese Medicine Inheritance Studio (grant no.
tjmzy2406).
Not applicable.
RJ, HC and XY contributed to the conception of the
review, conducted the literature search and screening, and wrote
the original manuscript. LY, YG, CF, XH and GZ assisted with
literature search, analysis of the published data and proofreading
of the drafts. ZS, as the corresponding author, contributed to the
design of the review framework, provided critical revisions for
important intellectual content, and supervised the entire project.
RJ and ZS confirm the accuracy and comprehensive nature of the
literature review presented in this manuscript. Data authentication
is not applicable. All authors have read and approved the final
version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Pollyea DA, Altman JK, Assi R, Bixby D,
Fathi AT, Foran JM, Gojo I, Hall AC, Jonas BA, Kishtagari A, et al:
Acute myeloid leukemia, version 3.2023, NCCN clinical practice
guidelines in oncology. J Natl Compr Canc Netw. 21:503–513. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moore CG, Stein A, Fathi AT and Pullarkat
V: Treatment of Relapsed/Refractory AML-Novel treatment options
including immunotherapy. Am J Hematol. 100:23–37. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sasaki K, Ravandi F, Kadia TM, DiNardo CD,
Short NJ, Borthakur G, Jabbour E and Kantarjian HM: De novo acute
myeloid leukemia: A population-based study of outcome in the United
States based on the Surveillance, Epidemiology, and End Results
(SEER) database, 1980 to 2017. Cancer. 127:2049–2061. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Halik A, Tilgner M, Silva P, Estrada N,
Altwasser R, Jahn E, Heuser M, Hou HA, Pratcorona M, Hills RK, et
al: Genomic characterization of AML with aberrations of chromosome
7: A multinational cohort of 519 patients. J Hematol Oncol.
17:702024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kantarjian H, Borthakur G, Daver N,
DiNardo CD, Issa G, Jabbour E, Kadia T, Sasaki K, Short NJ, Yilmaz
M and Ravandi F: Current status and research directions in acute
myeloid leukemia. Blood Cancer J. 14:1632024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee E, Song CH, Bae SJ, Ha KT and Karki R:
Regulated cell death pathways and their roles in homeostasis,
infection, inflammation, and tumorigenesis. Exp Mol Med.
55:1632–1643. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng J, Zou M, Zhang Q, Liu D, Chen S,
Fang R, Gao Y, Yan X and Hao L: Symphony of regulated cell death:
Unveiling therapeutic horizons in sarcopenia. Metabolism.
172:1563592025. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen C, Wang J, Zhang S, Zhu X, Hu J, Liu
C and Liu L: Epigenetic regulation of diverse regulated cell death
modalities in cardiovascular disease: Insights into necroptosis,
pyroptosis, ferroptosis, and cuproptosis. Redox Biol.
76:1033212024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sorokin O, Hause F, Wedler A, Alakhras T,
Bauchspiess T, Dietrich A, Günther WF, Guha C, Obika KB, Kraft J,
et al: Comprehensive analysis of regulated cell death pathways:
Intrinsic disorder, protein-protein interactions, and cross-pathway
communication. Apoptosis. 30:2110–2162. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng F, Liao M, Qin R, Zhu S, Peng C, Fu
L, Chen Y and Han B: Regulated cell death (RCD) in cancer: Key
pathways and targeted therapies. Signal Transduct Target Ther.
7:2862022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seo W, Silwal P, Song IC and Jo EK: The
dual role of autophagy in acute myeloid leukemia. J Hematol Oncol.
15:512022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ren J, Tao Y, Peng M, Xiao Q, Jing Y,
Huang J, Yang J, Lin C, Sun M, Lei L, et al: Targeted activation of
GPER enhances the efficacy of venetoclax by boosting leukemic
pyroptosis and CD8+ T cell immune function in acute myeloid
leukemia. Cell Death Dis. 13:9152022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan FK: RIPK3 Slams the Brake on
Leukemogenesis. Cancer Cell. 30:7–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye F, Zhang W, Fan C, Dong J, Peng M, Deng
W, Zhang H and Yang L: Antileukemic effect of venetoclax and
hypomethylating agents via caspase-3/GSDME-mediated pyroptosis. J
Transl Med. 21:6062023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An Iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu B, Wang S, Li R, Chen K, He L, Deng M,
Kannappan V, Zha J, Dong H and Wang W: Disulfiram/copper
selectively eradicates AML leukemia stem cells in vitro and in vivo
by simultaneous induction of ROS-JNK and inhibition of NF-κB and
Nrf2. Cell Death Dis. 8:e27972017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo Z, Liu Y, Chen D, Sun Y, Li D, Meng Y,
Zhou Q, Zeng F, Deng G and Chen X: Targeting regulated cell death:
Apoptosis, necroptosis, pyroptosis, ferroptosis, and cuproptosis in
anticancer immunity. J Transl Int Med. 13:10–32. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garciaz S, Miller T, Collette Y and Vey N:
Targeting regulated cell death pathways in acute myeloid leukemia.
Cancer Drug Resist. 6:151–168. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He R, Liu Y, Fu W, He X, Liu S, Xiao D and
Tao Y: Mechanisms and Cross-talk of regulated cell death and their
epigenetic modifications in tumor progression. Mol Cancer.
23:2672024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dikic I and Elazar Z: Mechanism and
medical implications of mammalian autophagy. Nat Rev Mol Cell Biol.
19:349–364. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaushik S and Cuervo AM: The coming of age
of Chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 19:365–381.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamamoto H and Matsui T: Molecular
mechanisms of macroautophagy, microautophagy, and
Chaperone-mediated autophagy. J Nippon Med Sch. 91:2–9. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Niu X, You Q, Hou K, Tian Y, Wei P, Zhu Y,
Gao B, Ashrafizadeh M, Aref AR, Kalbasi A, et al: Autophagy in
cancer development, immune evasion, and drug resistance. Drug
Resist Updat. 78:1011702025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sankar DS, Kaeser-Pebernard S, Vionnet C,
Favre S, de Oliveira Marchioro L, Pillet B, Zhou J, Stumpe M,
Kovacs WJ, Kressler D, et al: The ULK1 effector BAG2 regulates
autophagy initiation by modulating AMBRA1 localization. Cell Rep.
43:1146892024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xi H, Wang S, Wang B, Hong X, Liu X, Li M,
Shen R and Dong Q: The role of interaction between autophagy and
apoptosis in tumorigenesis (Review). Oncol Reps. 48:2082022.
View Article : Google Scholar
|
|
27
|
Cook ASI, Chen M, Nguyen TN, Cabezudo AC,
Khuu G, Rao S, Garcia SN, Yang M, Iavarone AT, Ren X, et al:
Structural pathway for PI3-kinase regulation by VPS15 in autophagy.
Science. 388:eadl37872025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nascimbeni AC, Codogno P and Morel E:
Local detection of PtdIns3P at autophagosome biogenesis membrane
platforms. Autophagy. 13:1602–1612. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wani WY, Boyer-Guittaut M, Dodson M,
Chatham J, Darley-Usmar V and Zhang J: Regulation of autophagy by
protein post-translational modification. Lab Invest. 95:14–25.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang R, Xu Y, Wan W, Shou X, Qian J, You
Z, Liu B, Chang C, Zhou T, Lippincott-Schwartz J and Liu W:
Deacetylation of nuclear LC3 drives autophagy initiation under
starvation. Mol Cell. 57:456–466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ichimura Y, Kirisako T, Takao T, Satomi Y,
Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi
M, et al: A ubiquitin-like system mediates protein lipidation.
Nature. 408:488–492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rogov V, Dötsch V, Johansen T and Kirkin
V: Interactions between autophagy receptors and ubiquitin-like
proteins form the molecular basis for selective autophagy. Mol
Cell. 53:167–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kimmelman AC and White E: Autophagy and
tumor metabolism. Cell Metab. 25:1037–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Santos de Macedo BG, Albuquerque de Melo
M, Pereira-Martins DA, Machado-Neto JA and Traina F: An updated
outlook on autophagy mechanism and how it supports acute myeloid
leukemia maintenance. Biochim Biophys Acta Rev Cancer.
1879:1892142024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nomura N, Ito C, Ooshio T, Tadokoro Y,
Kohno S, Ueno M, Kobayashi M, Kasahara A, Takase Y, Kurayoshi K, et
al: Essential role of autophagy in protecting neonatal
haematopoietic stem cells from oxidative stress in a
p62-independent manner. Sci Rep. 11:16662021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y, Luo X, Zou Z and Liang Y: The role
of reactive oxygen species in tumor treatment and its impact on
bone marrow hematopoiesis. Curr Drug Targets. 21:477–498. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sharma P, Piya S, Ma H, Baran N, Anna Zal
M, Hindley CJ, Dao K, Sims M, Zal T, Ruvolo V, et al: ERK1/2
inhibition overcomes resistance in acute myeloid leukemia (AML) and
alters mitochondrial dynamics. Blood. 138:33382021. View Article : Google Scholar
|
|
38
|
Folkerts H, Wierenga AT, van den Heuvel
FA, Woldhuis RR, Kluit DS, Jaques J, Schuringa JJ and Vellenga E:
Elevated VMP1 expression in acute myeloid leukemia amplifies
autophagy and is protective against venetoclax-induced apoptosis.
Cell Death Dis. 10:4212019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khan A, Singh VK, Thakral D and Gupta R:
Autophagy in acute myeloid leukemia: A paradoxical role in
chemoresistance. Clin Transl Oncol. 24:1459–1469. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heydt Q, Larrue C, Saland E, Bertoli S,
Sarry JE, Besson A, Manenti S, Joffre C and Mansat-De Mas V:
Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid
leukemia. Oncogene. 37:787–797. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Omori I, Yamaguchi H, Miyake K, Miyake N,
Kitano T and Inokuchi K: D816V mutation in the KIT gene activation
loop has greater cell-proliferative and anti-apoptotic ability than
N822K mutation in core-binding factor acute myeloid leukemia. Exp
Hematol. 52:56–64.e54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Larrue C, Heydt Q, Saland E, Boutzen H,
Kaoma T, Sarry JE, Joffre C and Récher C: Oncogenic KIT mutations
induce STAT3-dependent autophagy to support cell proliferation in
acute myeloid leukemia. Oncogenesis. 8:392019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zou Q, Tan S, Yang Z, Zhan Q, Jin H, Xian
J, Zhang S, Yang L, Wang L and Zhang L: NPM1 mutant mediated PML
delocalization and stabilization enhances autophagy and cell
survival in leukemic cells. Theranostics. 7:2289–2304. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Folkerts H, Hilgendorf S, Wierenga ATJ,
Jaques J, Mulder AB, Coffer PJ, Schuringa JJ and Vellenga E:
Inhibition of autophagy as a treatment strategy for p53 wild-type
acute myeloid leukemia. Cell Death Dis. 8:e29272017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang Y, Pu J and Yang Y: Glycolysis and
chemoresistance in acute myeloid leukemia. Heliyon. 10:e357212024.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang YH, Israelsen WJ, Lee D, Yu VWC,
Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG and Scadden DT:
Cell-state-specific metabolic dependency in hematopoiesis and
leukemogenesis. Cell. 158:1309–1323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen S, Tao Y, Wang Q, Ren J, Jing Y,
Huang J, Zhang L and Li R: Glucose Induced-AKT/mTOR activation
accelerates glycolysis and promotes cell survival in acute myeloid
leukemia. Leuk Res. 128:1070592023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Stevens BM, Jones CL, Pollyea DA,
Culp-Hill R, D'Alessandro A, Winters A, Krug A, Abbott D, Goosman
M, Pei S, et al: Fatty acid metabolism underlies venetoclax
resistance in acute myeloid leukemia stem cells. Nat Cancer.
1:1176–1187. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carter JL, Su Y, Qiao X, Zhao J, Wang G,
Howard M, Edwards H, Bao X, Li J, Hüttemann M, et al: Acquired
resistance to venetoclax plus azacitidine in acute myeloid
leukemia: In vitro models and mechanisms. Biochem Pharmacol.
216:1157592023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang HB, Sun ZK, Zhong FM, Yao FY, Liu J,
Zhang J, Zhang N, Lin J, Li SQ, Li MY, et al: A novel fatty acid
metabolism-related signature identifies features of the tumor
microenvironment and predicts clinical outcome in acute myeloid
leukemia. Lipids Health Dis. 21:792022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tcheng M, Roma A, Ahmed N, Smith RW,
Jayanth P, Minden MD, Schimmer AD, Hess DA, Hope K, Rea KA, et al:
Very long chain fatty acid metabolism is required in acute myeloid
leukemia. Blood. 137:3518–3532. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao Y, Guo H, Niu L and Zhao J: Metabolic
pathways and chemotherapy resistance in acute myeloid leukemia
(AML): Insights into Enoyl-CoA hydratase domain-containing protein
3 (ECHDC3) as a potential therapeutic target. Cancer Pathogenesis
and Therapy; 2025, View Article : Google Scholar
|
|
53
|
Du W, Xu A, Huang Y, Cao J, Zhu H, Yang B,
Shao X, He Q and Ying M: The role of autophagy in targeted therapy
for acute myeloid leukemia. Autophagy. 17:2665–2679. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li X, Qian Q, Li J, Zhang L, Wang L, Huang
D, Xu Q and Chen W: The RNA-binding protein CELF1 targets ATG5 to
regulate autophagy and promote drug resistance in acute myeloid
leukemia. Cell Death Dis. 16:5992025. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Herschbein L and Liesveld JL: Dueling for
dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR
inhibitors in AML. Blood Rev. 32:235–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Koschade SE, Klann K, Shaid S, Vick B,
Stratmann JA, Thölken M, Meyer LM, Nguyen TD, Campe J, Moser LM, et
al: Translatome proteomics identifies autophagy as a resistance
mechanism to on-target FLT3 inhibitors in acute myeloid leukemia.
Leukemia. 36:2396–2407. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao Y, Wang Y, Chen Y, Zhang X, Zuo Y, Ge
X, Sun C, Ren B, Liu Y, Wang M and Lu J: Voacamine initiates the
PI3K/mTOR/Beclin1 pathway to induce autophagy and potentiate
apoptosis in acute myeloid leukemia. Phytomedicine. 143:1568592025.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nössing C and Ryan KM: 50 years on and
still very much alive: ‘Apoptosis: A basic biological phenomenon
with wide-ranging implications in tissue kinetics’. Br J Cancer.
128:426–431. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yoon JH and Gores GJ: Death
receptor-mediated apoptosis and the liver. J Hepatol. 37:400–410.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
O'Neill KL, Huang K, Zhang J, Chen Y and
Luo X: Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak
through the outer mitochondrial membrane. Genes Dev. 30:973–988.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiong S, Mu T, Wang G and Jiang X:
Mitochondria-mediated apoptosis in mammals. Protein Cell.
5:737–749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Croce CM, Vaux D, Strasser A, Opferman JT,
Czabotar PE and Fesik SW: The BCL-2 protein family: from discovery
to drug development. Cell Death Differ. 32:1369–1381. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Reyna DE, Garner TP, Lopez A, Kopp F,
Choudhary GS, Sridharan A, Narayanagari SR, Mitchell K, Dong B,
Bartholdy BA, et al: Direct activation of BAX by BTSA1 overcomes
apoptosis resistance in acute myeloid leukemia. Cancer Cell.
32:490–505.e10. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Singh G, Guibao CD, Seetharaman J,
Aggarwal A, Grace CR, McNamara DE, Vaithiyalingam S, Waddell MB and
Moldoveanu T: Structural basis of BAK activation in mitochondrial
apoptosis initiation. Nat Commun. 13:2502022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Cain K, Bratton SB and Cohen GM: The
Apaf-1 apoptosome: A large caspase-activating complex. Biochimie.
84:203–214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kashyap D, Garg VK and Goel N: Intrinsic
and extrinsic pathways of apoptosis: Role in cancer development and
prognosis. Adv Protein Chem Struct Biol. 125:73–120. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zheng L, Yao Y and Lenardo MJ: Death
receptor 5 rises to the occasion. Cell Res. 33:199–200. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Helmke C, Raab M, Rödel F, Matthess Y,
Oellerich T, Mandal R, Sanhaji M, Urlaub H, Rödel C, Becker S and
Strebhardt K: Ligand stimulation of CD95 induces activation of Plk3
followed by phosphorylation of caspase-8. Cell Res. 26:914–934.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Huang K, Zhang J, O'Neill KL, Gurumurthy
CB, Quadros RM, Tu Y and Luo X: Cleavage by Caspase 8 and
mitochondrial membrane association activate the BH3-only protein
bid during TRAIL-induced apoptosis. J Biol Chem. 291:11843–11851.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kantari C and Walczak H: Caspase-8 and
bid: Caught in the act between death receptors and mitochondria.
Biochim Biophys Acta. 1813:558–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kim WS, Lee KS, Kim JH, Kim CK, Lee G,
Choe J, Won MH, Kim TH, Jeoung D, Lee H, et al: The
caspase-8/Bid/cytochrome c axis links signals from death receptors
to mitochondrial reactive oxygen species production. Free Radic
Biol Med. 112:567–577. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Moyer A, Tanaka K and Cheng EH: Apoptosis
in cancer biology and therapy. Annu Rev Pathol. 20:303–328. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Pfeffer CM and Singh ATK: Apoptosis: A
target for anticancer therapy. Int J Mol Sci. 19:4482018.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Campbell KJ and Tait SWG: Targeting BCL-2
regulated apoptosis in cancer. Open Biol. 8:1800022018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ashkenazi A, Fairbrother WJ, Leverson JD
and Souers AJ: From basic apoptosis discoveries to advanced
selective BCL-2 family inhibitors. Nat Rev Drug Discov. 16:273–284.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Konopleva M, Pollyea DA, Potluri J, Chyla
B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, et
al: Efficacy and biological correlates of response in a phase II
study of venetoclax monotherapy in patients with acute myelogenous
leukemia. Cancer Discov. 6:1106–1117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wei AH, Strickland SA Jr, Hou JZ, Fiedler
W, Lin TL, Walter RB, Enjeti A, Tiong IS, Savona M, Lee S, et al:
Venetoclax combined with Low-dose cytarabine for previously
untreated patients with acute myeloid leukemia: Results from a
Phase Ib/II study. J Clin Oncol. 37:1277–1284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
DiNardo CD, Jonas BA, Pullarkat V, Thirman
MJ, Garcia JS, Wei AH, Konopleva M, Döhner H, Letai A, Fenaux P, et
al: Azacitidine and venetoclax in previously untreated acute
myeloid leukemia. N Engl J Med. 383:617–629. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiao CQ, Hu C, Sun MH, Li Y, Wu C, Xu F,
Zhang L, Huang FR, Zhou JJ, Dai JF, et al: Targeting METTL3
mitigates venetoclax resistance via proteasome-mediated modulation
of MCL1 in acute myeloid leukemia. Cell Death Dis. 16:2332025.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chai J, Du C, Wu JW, Kyin S, Wang X and
Shi Y: Structural and biochemical basis of apoptotic activation by
Smac/DIABLO. Nature. 406:855–862. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Carter BZ, Mak PY, Mak DH, Shi Y, Qiu Y,
Bogenberger JM, Mu H, Tibes R, Yao H, Coombes KR, et al:
Synergistic targeting of AML stem/progenitor cells with IAP
antagonist birinapant and demethylating agents. J Natl Cancer Inst.
106:djt4402014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gencel-Augusto J and Lozano G: Targeted
degradation of mutant p53 reverses the Pro-oncogenic
Dominant-negative effect. Cancer Res. 85:1955–1956. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tuval A, Strandgren C, Heldin A,
Palomar-Siles M and Wiman KG: Pharmacological reactivation of p53
in the era of precision anticancer medicine. Nat Rev Clin Oncol.
21:106–120. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Napolitano R, De Matteis S, Carloni S,
Bruno S, Abbati G, Capelli L, Ghetti M, Bochicchio MT, Liverani C,
Mercatali L, et al: Kevetrin induces apoptosis in TP53 wild-type
and mutant acute myeloid leukemia cells. Oncol Rep. 44:1561–1573.
2020.PubMed/NCBI
|
|
87
|
Fulda S: Targeting apoptosis for
anticancer therapy. Semin Cancer Biol. 31:84–88. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chow LQ, Eckhardt SG, Gustafson DL, Langer
CJ, Camidge DR, Padavic K, Gore L, Smith M, Chow LQ, von Mehren M,
et al: HGS-ETR1, an antibody targeting TRAIL-R1, in combination
with paclitaxel and carboplatin in patients with advanced solid
malignancies: Results of a phase 1 and PK study. J Clin Oncol.
24:25152006. View Article : Google Scholar
|
|
89
|
You R, Hou D, Wang B, Liu J, Wang X, Xiao
Q, Pan Z, Li D, Feng X, Kang L, et al: Bone marrow microenvironment
drives AML cell OXPHOS addiction and AMPK inhibition to resist
chemotherapy. J Leukoc Biol. 112:299–311. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cheung HL, Wong YH, Li YY, Yang X, Ko LH,
Tan Kabigting JE, Chan KC, Leung AYH and Chan BP: Microenvironment
matters: In vitro 3D bone marrow niches differentially modulate
survival, phenotype and drug responses of acute myeloid leukemia
(AML) cells. Biomaterials. 312:1227192025. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zou J, Zheng Y, Huang Y, Tang D, Kang R
and Chen R: The versatile gasdermin family: Their function and
roles in diseases. Front Immunol. 12:7515332021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Broz P, Pelegrín P and Shao F: The
gasdermins, a protein family executing cell death and inflammation.
Nat Rev Immunol. 20:143–157. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Tamura M, Tanaka S, Fujii T, Aoki A,
Komiyama H, Ezawa K, Sumiyama K, Sagai T and Shiroishi T: Members
of a novel gene family, Gsdm, are expressed exclusively in the
epithelium of the skin and gastrointestinal tract in a highly
Tissue-specific manner. Genomics. 89:618–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang S, Yuan YH, Chen NH and Wang HB: The
mechanisms of NLRP3 inflammasome/pyroptosis activation and their
role in Parkinson's disease. Int Immunopharmacol. 67:458–464. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yu P, Zhang X, Liu N, Tang L, Peng C and
Chen X: Pyroptosis: Mechanisms and diseases. Signal Transduct
Target Ther. 6:1282021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Garlanda C, Dinarello CA and Mantovani A:
The interleukin-1 family: Back to the future. Immunity.
39:1003–1018. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rao Z, Zhu Y, Yang P, Chen Z, Xia Y, Qiao
C, Liu W, Deng H, Li J, Ning P and Wang Z: Pyroptosis in
inflammatory diseases and cancer. Theranostics. 12:4310–4329. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yang J, Zhao Y and Shao F: Non-canonical
activation of inflammatory caspases by cytosolic LPS in innate
immunity. Curr Opin Immunol. 32:78–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li
P, Hu L and Shao F: Inflammatory caspases are innate immune
receptors for intracellular LPS. Nature. 514:187–192. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hagar JA, Powell DA, Aachoui Y, Ernst RK
and Miao EA: Cytoplasmic LPS activates caspase-11: implications in
TLR4-independent endotoxic shock. Science. 341:1250–1253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liu Y, Pan R, Ouyang Y, Gu W, Xiao T, Yang
H, Tang L, Wang H, Xiang B and Chen P: Pyroptosis in health and
disease: Mechanisms, regulation and clinical perspective. Signal
Transduct Target Ther. 9:2452024. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kayagaki N, Warming S, Lamkanfi M, Vande
Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al:
Non-canonical inflammasome activation targets caspase-11. Nature.
479:117–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Shi X, Sun Q, Hou Y, Zeng H, Cao Y, Dong
M, Ding J and Shao F: Recognition and maturation of IL-18 by
caspase-4 noncanonical inflammasome. Nature. 624:442–450. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Oltra SS, Colomo S, Sin L, Pérez-López M,
Lázaro S, Molina-Crespo A, Choi KH, Ros-Pardo D, Martínez L,
Morales S, et al: Distinct GSDMB protein isoforms and protease
cleavage processes differentially control pyroptotic cell death and
mitochondrial damage in cancer cells. Cell Death Differ.
30:1366–1381. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang S, Chang CW, Huang J, Zeng S, Zhang
X, Hung MC and Hou J: Gasdermin C sensitizes tumor cells to PARP
inhibitor therapy in cancer models. J Clin Invest. 134:e1668412024.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Alves-Hanna FS, Crespo-Neto JA, Nogueira
GM, Pereira DS, Lima AB, Ribeiro TLP, Santos VGR, Fonseca JRF,
Magalhães-Gama F, Sadahiro A and Costa AG: Insights regarding the
role of inflammasomes in leukemia: What Do We Know? J Immunol Res.
2023:55844922023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Johnson DC, Taabazuing CY, Okondo MC, Chui
AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A
and Bachovchin DA: DPP8/DPP9 inhibitor-induced pyroptosis for
treatment of acute myeloid leukemia. Nat Med. 24:1151–1156. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Deng S, Pan Y, An N, Chen F, Chen H, Wang
H, Xu X, Liu R, Yang L, Wang X, et al: Downregulation of RCN1
promotes pyroptosis in acute myeloid leukemia cells. Mol Oncol.
17:2584–2602. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Askmyr M, Ågerstam H, Hansen N, Gordon S,
Arvanitakis A, Rissler M, Juliusson G, Richter J, Järås M and
Fioretos T: Selective killing of candidate AML stem cells by
antibody targeting of IL1RAP. Blood. 121:3709–3713. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ågerstam H, Karlsson C, Hansen N, Sandén
C, Askmyr M, von Palffy S, Högberg C, Rissler M, Wunderlich M,
Juliusson G, et al: Antibodies targeting human IL1RAP (IL1R3) show
therapeutic effects in xenograft models of acute myeloid leukemia.
Proc Natl Acad Sci USA. 112:10786–10791. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang Y, Sun X, Yuan S, Hou S, Guo T, Chu
Y, Pang T, Luo HR, Yuan W and Wang X: Interleukin-1β inhibits
normal hematopoietic expansion and promotes acute myeloid leukemia
progression via the bone marrow niche. Cytotherapy. 22:127–134.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wong J, Tran LT, Magun EA, Magun BE and
Wood LJ: Production of IL-1β by bone marrow-derived macrophages in
response to chemotherapeutic drugs: Synergistic effects of
doxorubicin and vincristine. Cancer Biol Ther. 15:1395–1403. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Yan J, Wan P, Choksi S and Liu ZG:
Necroptosis and tumor progression. Trends Cancer. 8:21–27. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Liu X, Miao M, Sun J, Wu J and Qin X:
PANoptosis: A potential new target for programmed cell death in
breast cancer treatment and prognosis. Apoptosis. 29:277–288. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nat Immunol.
1:489–495. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
122
|
Mandal R, Barrón JC, Kostova I, Becker S
and Strebhardt K: Caspase-8: The double-edged sword. Biochim
Biophys Acta Rev Cancer. 1873:1883572020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kaiser WJ, Sridharan H, Huang C, Mandal P,
Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J and Mocarski ES:
Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J
Biol Chem. 288:31268–31279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Upton JW, Kaiser WJ and Mocarski ES:
DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced
programmed necrosis that is targeted by murine cytomegalovirus
vIRA. Cell Host Microbe. 11:290–297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Linkermann A and Green DR: Necroptosis. N
Engl J Med. 370:455–465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Murphy JM, Czabotar PE, Hildebrand JM,
Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D,
Webb AI, et al: The pseudokinase MLKL mediates necroptosis via a
molecular switch mechanism. Immunity. 39:443–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Gong YN, Guy C, Olauson H, Becker JU, Yang
M, Fitzgerald P, Linkermann A and Green DR: ESCRT-III acts
downstream of MLKL to regulate necroptotic cell death and its
consequences. Cell. 169:286–300.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Cai Z, Jitkaew S, Zhao J, Chiang HC,
Choksi S, Liu J, Ward Y, Wu LG and Liu ZG: Plasma membrane
translocation of trimerized MLKL protein is required for
TNF-induced necroptosis. Nat Cell Biol. 16:55–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Khan N, Downey J, Sanz J, Kaufmann E,
Blankenhaus B, Pacis A, Pernet E, Ahmed E, Cardoso S, Nijnik A, et
al: M. tuberculosis reprograms hematopoietic stem cells to limit
myelopoiesis and impair trained immunity. Cell. 183:752–770.e22.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan
K, Cheng H, Jin K, Ni Q, Yu X and Liu C: The role of necroptosis in
cancer biology and therapy. Mol Cancer. 18:1002019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Zhang T, Wang Y, Inuzuka H and Wei W:
Necroptosis pathways in tumorigenesis. Semin Cancer Biol. 86:32–40.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Höckendorf U, Yabal M, Herold T,
Munkhbaatar E, Rott S, Jilg S, Kauschinger J, Magnani G, Reisinger
F, Heuser M, et al: RIPK3 restricts myeloid leukemogenesis by
promoting cell death and differentiation of leukemia initiating
cells. Cancer Cell. 30:75–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang Y, Zhang TJ, Zhang LC, Xu ZJ, Chu MQ,
Zhao YJ, Lin J, Qian J and Zhou JD: Overexpression and oncogenic
role of RIPK3 in acute myeloid leukemia associated with specific
subtypes and treatment outcome. BMC Cancer. 25:2532025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Xin J, You D, Breslin P, Li J, Zhang J,
Wei W, Cannova J, Volk A, Gutierrez R, Xiao Y, et al: Sensitizing
acute myeloid leukemia cells to induced differentiation by
inhibiting the RIP1/RIP3 pathway. Leukemia. 31:1154–1165. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhu S, Luo Y, Li K, Mei C, Wang Y, Jiang
L, Wang W, Zhang Q, Yang W, Lang W, et al: RIPK3 deficiency blocks
R-2-hydroxyglutarate-induced necroptosis in IDH-mutated AML cells.
Sci Adv. 10:eadi17822024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Hillert LK, Bettermann-Bethge K,
Nimmagadda SC, Fischer T, Naumann M and Lavrik IN: Targeting RIPK1
in AML cells carrying FLT3-ITD. Int J Cancer. 145:1558–1569. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Li J, Liao D, Wang F, Wang Z, Li Y, Xiong
Y and Niu T: RIPK1 inhibition enhances the therapeutic efficacy of
chidamide in FLT3-ITD positive AML, both in vitro and in vivo. Leuk
Lymphoma. 63:1167–1179. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Yue X, Chen Q and He J: Combination
strategies to overcome resistance to the BCL2 inhibitor venetoclax
in hematologic malignancies. Leuk Lymphoma. 20:5242020.
|
|
140
|
Culver-Cochran AE, Hassan A, Hueneman K,
Choi K, Ma A, VanCauwenbergh B, O'Brien E, Wunderlich M, Perentesis
JP and Starczynowski DT: Chemotherapy resistance in acute myeloid
leukemia is mediated by A20 suppression of spontaneous necroptosis.
Nat Commun. 15:91892024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Liang D, Minikes AM and Jiang X:
Ferroptosis at the intersection of lipid metabolism and cellular
signaling. Mol Cell. 82:2215–2227. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Chen F, Kang R, Tang D and Liu J:
Ferroptosis: principles and significance in health and disease. J
Hematol Oncol. 17:412024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Koleini N, Shapiro JS, Geier J and
Ardehali H: Ironing out mechanisms of iron homeostasis and
disorders of iron deficiency. J Clin Invest. 131:e1486712021.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Co HKC, Wu CC, Lee YC and Chen SH:
Emergence of large-scale cell death through ferroptotic trigger
waves. Nature. 631:654–662. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF and Clish CB: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Reyhani A, McKenzie TG, Fu Q and Qiao GG:
Fenton-Chemistry-Mediated Radical Polymerization. Macromol Rapid
Commun. 40:19002202019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Dai E, Chen X, Linkermann A, Jiang X, Kang
R, Kagan VE, Bayir H, Yang WS, Garcia-Saez AJ, Ioannou MS, et al: A
guideline on the molecular ecosystem regulating ferroptosis. Nat
Cell Biol. 26:1447–1457. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ma S, Henson ES, Chen Y and Gibson SB:
Ferroptosis is induced following siramesine and lapatinib treatment
of breast cancer cells. Cell Death Dis. 7:e23072016. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Bannai S: Exchange of cystine and
glutamate across plasma membrane of human fibroblasts. J Biol Chem.
261:2256–2263. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Koppula P, Zhang Y, Zhuang L and Gan B:
Amino acid transporter SLC7A11/xCT at the crossroads of regulating
redox homeostasis and nutrient dependency of cancer. Cancer Commun
(Lond). 38:122018.PubMed/NCBI
|
|
155
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Liu MR, Zhu WT and Pei DS: System Xc-: A
key regulatory target of ferroptosis in cancer. Invest New Drugs.
39:1123–1131. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Scarpellini C, Klejborowska G, Lanthier C,
Hassannia B, Vanden Berghe T and Augustyns K: Beyond ferrostatin-1:
A comprehensive review of ferroptosis inhibitors. Trends Pharmacol
Sci. 44:902–916. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Brown CW, Amante JJ, Chhoy P, Elaimy AL,
Liu H, Zhu LJ, Baer CE, Dixon SJ and Mercurio AM: Prominin2 drives
ferroptosis resistance by stimulating iron export. Dev Cell.
51:575–586.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Cao M, Li H, Sun D, He S, Yu Y, Li J, Chen
H, Shi J, Ren J, Li N and Chen W: Cancer screening in China: The
current status, challenges, and suggestions. Cancer Lett.
506:120–127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhang H and Sun C, Sun Q, Li Y, Zhou C and
Sun C: Susceptibility of acute myeloid leukemia cells to
ferroptosis and evasion strategies. Front Mol Biosci.
10:12757742023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Akiyama H, Zhao R, Ostermann LB, Li Z,
Tcheng M, Yazdani SJ, Moayed A, Pryor ML II, Slngh S, Baran N, et
al: Mitochondrial regulation of GPX4 inhibition-mediated
ferroptosis in acute myeloid leukemia. Leukemia. 38:729–740. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Na X, Li L, Liu D, He J, Zhang L and Zhou
Y: Natural products targeting ferroptosis pathways in cancer
therapy (Review). Oncol Rep. 52:1232024. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Ma C, Wang J, Cui S and Xu R: Crotonoside
induces ferroptosis and mitochondrial dysfunction in AML. Eur J
Pharmacol. 1002:1778592025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Birsen R, Larrue C, Decroocq J, Johnson N,
Guiraud N, Gotanegre M, Cantero-Aguilar L, Grignano E, Huynh T,
Fontenay M, et al: APR-246 induces early cell death by ferroptosis
in acute myeloid leukemia. Haematologica. 107:403–416. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Lin AY, Rink JS, Yang E, Small S, Gerber
JJ, Zak TJ, Altman J, Abaza Y, Platanias LC, Gordon LI and Thaxton
CS: Targeting scavenger receptor class B type 1 with a bioinspired
ligand induces apoptosis or ferroptosis in AML. Blood Neoplasia.
2:1001222025. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Jiang F, Yu WJ, Wang XH, Tang YT, Guo L
and Jiao XY: Regulation of hepcidin through GDF-15 in
cancer-related anemia. Clin Chim Acta. 428:14–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Lu QW and Liao Y: GDF-15 upregulates the
SLC7A11/GPX4 signaling axis and promotes mitoxantrone resistance in
AML cells. Eur J Med Res. 30:5042025. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Yu Y, Meng Y, Xu X, Tong T, He C, Wang L,
Wang K, Zhao M, You X, Zhang W, et al: A Ferroptosis-inducing and
leukemic Cell-targeting drug nanocarrier formed by Redox-responsive
cysteine polymer for acute myeloid leukemia therapy. ACS Nano.
17:3334–3345. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yu X, Wang Y, Tan J, Li Y, Yang P, Liu X,
Lai J, Zhang Y, Cai L, Gu Y, et al: Inhibition of NRF2 enhances the
acute myeloid leukemia cell death induced by venetoclax via the
ferroptosis pathway. Cell Death Discov. 10:352024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Pardieu B, Pasanisi J, Ling F, Dal Bello
R, Penneroux J, Su A, Joudinaud R, Chat L, Wu HC, Duchmann M, et
al: Cystine uptake inhibition potentiates front-line therapies in
acute myeloid leukemia. Leukemia. 36:1585–1595. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Lyu T, Li X and Song Y: Ferroptosis in
acute leukemia. Chin Med J (Engl). 136:886–898. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Tsvetkov P, Coy S, Petrova B, Dreishpoon
M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R,
Spangler RD, et al: Copper induces cell death by targeting
lipoylated TCA cycle proteins. Science. 375:1254–1261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Chen L, Min J and Wang F: Copper
homeostasis and cuproptosis in health and disease. Signal Transduct
Target Ther. 7:3782022. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Tian Z, Jiang S, Zhou J and Zhang W:
Copper homeostasis and cuproptosis in mitochondria. Life Sci.
334:1222232023. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Tsvetkov P, Detappe A, Cai K, Keys HR,
Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, et al:
Mitochondrial metabolism promotes adaptation to proteotoxic stress.
Nat Chem Biol. 15:681–689. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Zheng P, Zhou C, Lu L, Liu B and Ding Y:
Elesclomol: A copper ionophore targeting mitochondrial metabolism
for cancer therapy. J Exp Clin Cancer Res. 41:2712022. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Vo TTT, Peng TY, Nguyen TH, Bui TNH, Wang
CS, Lee WJ, Chen YL, Wu YC and Lee IT: The crosstalk between
copper-induced oxidative stress and cuproptosis: A novel potential
anticancer paradigm. Cell Commun Signal. 22:3532024. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Zeng Y, Cao Y, Ren S, Zhang C, Liu J, Liu
K, Wang Y, Chen H, Zhou F, Yang X, et al: Responsive ROS-Augmented
prodrug hybridization nanoassemblies for multidimensionally
synergitic treatment of hepatocellular carcinoma in cascade
assaults. Adv Sci (Weinh). 12:e25014202025. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Cong Y, Li N, Zhang Z, Shang Y and Zhao H:
Cuproptosis: Molecular mechanisms, cancer prognosis, and
therapeutic applications. J Transl Med. 23:1042025. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Feng Y, Yang Z, Wang J and Zhao H:
Cuproptosis: Unveiling a new frontier in cancer biology and
therapeutics. Cell Commun Signal. 22:2492024. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Kahlson MA and Dixon SJ: Copper-induced
cell death. Science. 375:1231–1232. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Wang Y, Zhang L and Zhou F: Cuproptosis: A
new form of programmed cell death. Cell Mol Immunol. 19:867–868.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Dreishpoon MB, Bick NR, Petrova B, Warui
DM, Cameron A, Booker SJ, Kanarek N, Golub TR and Tsvetkov P: FDX1
regulates cellular protein lipoylation through direct binding to
LIAS. J Biol Chem. 299:1050462023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Sun Z, Xu H, Lu G, Yang C, Gao X, Zhang J,
Liu X, Chen Y, Wang K, Guo J and Li J: AKT1 Phosphorylates FDX1 to
promote cuproptosis resistance in Triple-negative breast cancer.
Adv Sci (Weinh). 12:e24081062025. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Yip NC, Fombon IS, Liu P, Brown S,
Kannappan V, Armesilla AL, Xu B, Cassidy J, Darling JL and Wang W:
Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast
cancer cells with cancer stem cell-like properties. Br J Cancer.
104:1564–1574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Valadbeigi S, Javadian S, Ebrahimi-Rad M,
Khatami S and Saghiri R: Assessment of trace elements in serum of
acute lymphoblastic and myeloid leukemia patients. Exp Oncol.
41:69–71. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Singh RP, Jeyaraju DV, Voisin V, Hurren R,
Xu C, Hawley JR, Barghout SH, Khan DH, Gronda M, Wang X, et al:
Disrupting mitochondrial copper distribution inhibits leukemic stem
cell Self-renewal. Cell Stem Cell. 26:926–937.e10. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
McMahon A, Chen W and Li F: Old wine in
new bottles: Advanced drug delivery systems for disulfiram-based
cancer therapy. J Control Release. 319:352–359. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Weiser Drozdkova D and Smesny Trtkova K:
Possible therapeutic potential of disulfiram for multiple myeloma.
Curr Oncol. 28:2087–2096. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Solak K, Mavi A and Yılmaz B:
Disulfiram-loaded functionalized magnetic nanoparticles combined
with copper and sodium nitroprusside in breast cancer cells. Mater
Sci Eng C Mater Biol Appl. 119:1114522021. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Solier S, Müller S, Cañeque T, Versini A,
Mansart A, Sindikubwabo F, Baron L, Emam L, Gestraud P, Pantoș GD,
et al: A druggable copper-signalling pathway that drives
inflammation. Nature. 617:386–394. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Karsa M, Xiao L, Ronca E, Bongers A,
Spurling D, Karsa A, Cantilena S, Mariana A, Failes TW, Arndt GM,
et al: FDA-approved disulfiram as a novel treatment for aggressive
leukemia. J Mol Med (Berl). 102:507–519. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Oliveri V: Selective targeting of cancer
cells by copper ionophores: An overview. Front Mol Biosci.
9:8418142022. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Wang J, Luo C, Shan C, You Q, Lu J, Elf S,
Zhou Y, Wen Y, Vinkenborg JL, Fan J, et al: Inhibition of human
copper trafficking by a small molecule significantly attenuates
cancer cell proliferation. Nat Chem. 7:968–979. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Cao C, Wang T, Luo Y, Zhang Y, Dai YY and
Shen Y: Comprehensive analysis of cuproptosis-associated LncRNAs
predictive value and related CeRNA network in acute myeloid
leukemia. Heliyon. 9:e225322023. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Moison C, Gracias D, Schmitt J, Girard S,
Spinella JF, Fortier S, Boivin I, Mendoza-Sanchez R, Thavonekham B,
MacRae T, et al: SF3B1 mutations provide genetic vulnerability to
copper ionophores in human acute myeloid leukemia. Sci Adv.
10:eadl40182024. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Zhang J, Zheng Y, Liu H and Liu B:
AC024896.1/miR-363-3p axis regulates the malignant progression of
acute myeloid leukemia by Cuproptosis-related gene MYO1B. Blood
Lymphat Cancer. 14:17–30. 2024.PubMed/NCBI
|
|
199
|
Huang X, Yan H, Xu Z, Yang B, Luo P and He
Q: The inducible role of autophagy in cell death: Emerging evidence
and future perspectives. Cell Commun Signal. 23:1512025. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Gao W, Wang X, Zhou Y, Wang X and Yu Y:
Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor
immunotherapy. Signal Transduct Target Ther. 7:1962022. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Liu X, Tuerxun H and Zhao Y, Li Y, Wen S,
Li X and Zhao Y: Crosstalk between ferroptosis and autophagy:
Broaden horizons of cancer therapy. J Transl Med. 23:182025.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Newton K, Wickliffe KE, Maltzman A, Dugger
DL, Webster JD, Guo H and Dixit VM: Caspase cleavage of RIPK3 after
Asp333 is dispensable for mouse embryogenesis. Cell Death Differ.
31:254–262. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Fritsch M, Günther SD, Schwarzer R, Albert
MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H,
Seeger JM, et al: Caspase-8 is the molecular switch for apoptosis,
necroptosis and pyroptosis. Nature. 575:683–687. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Qiu Y, Hüther JA, Wank B, Rath A, Tykwe R,
Aldrovandi M, Henkelmann B, Mergner J, Nakamura T and Laschat S:
Interplay of ferroptotic and apoptotic cell death and its
modulation by BH3-mimetics. Cell Death Differ. Apr 29–2025.doi:
10.1038/s41418-025-01514-7 (Epub ahead of print). View Article : Google Scholar
|
|
205
|
Xu R, Wang W and Zhang W: Ferroptosis and
the bidirectional regulatory factor p53. Cell Death Discov.
9:1972023. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Gao J, Xiong A, Liu J, Li X, Wang J, Zhang
L, Liu Y, Xiong Y, Li G and He X: PANoptosis: Bridging apoptosis,
pyroptosis, and necroptosis in cancer progression and treatment.
Cancer Gene Ther. 31:970–983. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Qu M, Wang Y, Qiu Z, Zhu S, Guo K, Chen W,
Miao C and Zhang H: Necroptosis, pyroptosis, ferroptosis in sepsis
and treatment. Shock. 57:161–171. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Zhang C and Liu N: Ferroptosis,
necroptosis, and pyroptosis in the occurrence and development of
ovarian cancer. Front Immunol. 13:9200592022. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Chen Y, Fang ZM, Yi X, Wei X and Jiang DS:
The interaction between ferroptosis and inflammatory signaling
pathways. Cell Death Dis. 14:2052023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Li Y, Du Y, Zhou Y, Chen Q, Luo Z, Ren Y,
Chen X and Chen G: Iron and copper: Critical executioners of
ferroptosis, cuproptosis and other forms of cell death. Cell Commun
Signal. 21:3272023. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Hu X, Li L, Nkwocha J, Kmieciak M, Shang
S, Cowart LA, Yue Y, Horimoto K, Hawkridge A, Rijal A, et al: Src
inhibition potentiates MCL-1 antagonist activity in acute myeloid
leukemia. Signal Transduct Target Ther. 10:502025. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Ong F, Kim K and Konopleva MY: Venetoclax
resistance: Mechanistic insights and future strategies. Cancer Drug
Resist. 5:380–400. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Lim J and Murthy A: Targeting autophagy to
treat cancer: Challenges and opportunities. Front Pharmacol.
11:5903442020. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Hassani S, Ghaffari P, Chahardouli B,
Alimoghaddam K, Ghavamzadeh A, Alizadeh S and Ghaffari SH:
Disulfiram/copper causes ROS levels alteration, cell cycle
inhibition, and apoptosis in acute myeloid leukaemia cell lines
with modulation in the expression of related genes. Biomed
Pharmacother. 99:561–569. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Song F, Lin S, Xu T, Yang C, Sharavyn B,
Naranmandura H, Zhang Y and Huang P: Targeted therapy in acute
myeloid leukemia: Resistance and overcoming strategy. Drug Resist
Updat. 83:1012862025. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
D'Amico M and De Amicis F: Challenges of
regulated cell death: Implications for therapy resistance in
cancer. Cells. 13:10832024. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Tong X, Tang R, Xiao M, Xu J, Wang W,
Zhang B, Liu J, Yu X and Shi S: Targeting cell death pathways for
cancer therapy: Recent developments in necroptosis, pyroptosis,
ferroptosis, and cuproptosis research. J Hematol Oncol. 15:1742022.
View Article : Google Scholar : PubMed/NCBI
|