Introduction
Breast cancer is a heterogeneous disease that
comprises multiple intrinsic subtypes, each associated with
distinct clinical and molecular characteristics (1). Based on gene expression profiling,
four major intrinsic subtypes (luminal A, luminal B, HER2-enriched
and basal-like) have traditionally been identified (2,3). These
classifications guide treatment and provide insights into prognosis
(4). However, a novel intrinsic
subtype, termed claudin-low, was identified in 2007 (5). Claudin-low breast cancers are unique
due to their low expression of claudins, which are tight junction
adhesion molecules integral to epithelial cell cohesion and
polarity. As a result, claudin-low tumors exhibit characteristics
more typical of mesenchymal cells, including increased motility and
invasiveness. These properties are associated with high expression
levels of epithelial-mesenchymal transition (EMT) markers,
reflecting an inherently aggressive phenotype (6–8).
Over 70% of claudin-low breast cancers are also
classified as triple-negative, which means that they lack estrogen
receptor, progesterone receptor and HER2 expression. This renders
claudin-low tumors resistant to conventional endocrine or
HER2-targeted therapies, limiting the treatment options for this
subtype. Beyond the distinctive expression of claudins and EMT
markers, claudin-low tumors share features with cancer stem cells,
such as a CD44+/CD24− surface marker profile
(9). This
CD44+/CD24− phenotype is linked to cancer
stem cell-like traits such as self-renewal and chemoresistance,
contributing to poor clinical outcomes (6,7).
The CD44 protein, a cell surface glycoprotein
involved in cell-cell and cell-matrix interactions, has been
implicated in various aspects of cancer biology, including cell
proliferation, invasion and metastasis (10). In CD44-positive breast cancer cells,
studies have reported that CD44 inhibition can suppress cell
proliferation and, in some cases, diminish invasive capabilities
(11–14). However, the results regarding
invasion are varied; while some studies have suggested that CD44
inhibition can limit invasiveness (15,16),
others have reported negligible effects on invasion (17,18).
This variation may stem from the complex involvement of CD44 in
multiple signaling pathways that collectively influence cell
behavior in diverse mechanisms. Despite these inconsistencies, the
evidence highlights the substantial role of CD44 in promoting the
aggressive characteristics of claudin-low breast cancers,
particularly through its regulatory impact on EMT processes
(19,20).
EMT is a cellular program that drives epithelial
cells to acquire mesenchymal properties, enhancing their migratory
and invasive capabilities. EMT inhibition in claudin-low breast
cancer may offer dual benefits: Cell motility reduction and tumor
growth suppression (21,22). By targeting the EMT pathway, certain
inhibitors, such as histone deacetylase inhibitors and DNA
methyltransferase inhibitors, could theoretically control the
proliferation and metastatic potential of claudin-low breast
cancers (23). However, studies
suggest that EMT inhibition alone provides only limited therapeutic
benefits, indicating a need for combination therapies that target
multiple pathways (24,25). This observation has driven further
research into the identification of co-targets that may enhance the
efficacy of EMT inhibitors.
Previous findings have indicated that the TGF-β
receptor (TGFBR), an upstream EMT regulator, can directly interact
with CD44, providing a novel therapeutic axis for claudin-low
breast cancers (26). Given the
interplay between CD44 and TGF-β signaling, the dual inhibition of
CD44 and TGFBRs may amplify the antiproliferative and anti-invasive
effects of single-agent treatments (23). Pharmacological agents that
downregulate CD44 expression and inhibit TGF-β signaling could,
therefore, provide a combined approach, targeting the EMT pathway
while impacting the CD44-mediated stem cell-like properties of
claudin-low breast cancers (27,28).
This combined approach may provide a mechanistic rationale for
co-targeting CD44 and TGF-β signaling, as CD44 interacts with the
TGF-β receptor complex, modulating downstream Smad2 activation.
Dual inhibition could simultaneously suppress CD44-mediated stem
cell-like survival and TGF-β-driven EMT and invasion, thereby
enhancing antiproliferative efficacy in claudin-low breast
cancer.
Materials and methods
Antibodies
The antibodies used in the present study were
anti-CD44 (1:1,000; cat. no. sc-7297; Santa Cruz Biotechnology,
Inc.), anti-Smad2 (1:1,000; cat. no. 3102; Cell Signaling
Technology, Inc.), anti-phosphorylated (p-)Smad2 (Ser465/467)
(1:1,000; cat. no. 3108; Cell Signaling Technology, Inc.),
anti-TGFBR1 (1:1,000; cat. no. sc-101574; Santa Cruz Biotechnology,
Inc.), anti-TGFBR2 (1:1,000; cat. no. sc-17799; Santa Cruz
Biotechnology, Inc.), anti-Bcl-2 (1:1,000; cat. no. sc-7382; Santa
Cruz Biotechnology, Inc.), anti-Snai1 (1:1,000; cat. no. sc-271977;
Santa Cruz Biotechnology, Inc.), anti-IgG (1:10,000; cat. no. 5415;
Cell Signaling Technology, Inc.) and anti-GAPDH (1:1,000; cat. no.
sc-32233; Santa Cruz Biotechnology, Inc.).
Cell culture and lentiviral
transduction
The MDA-MB-231, SUM159 and MDA-MB-468 human breast
cancer cell lines were obtained from stocks maintained at Shizuoka
General Hospital (Shizuoka, Japan). MDA-MB-231 and MDA-MB-468 cells
were maintained in DMEM (Nacalai Tesque, Inc.) supplemented with
10% FBS (Biowest) and 1% penicillin-streptomycin (100 U/ml
penicillin and 100 µg/ml streptomycin). SUM159 cells were
maintained in Ham's F-12 medium (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 5% FBS (Biowest), 5 µg/ml insulin
(Sigma-Aldrich; Merck KGaA) and 1 µg/ml hydrocortisone
(Sigma-Aldrich; Merck KGaA). All cells were cultured at 37°C in a
humidified incubator containing 5% CO2. These cell lines
were confirmed to be free of mycoplasma contamination and exhibited
morphological features consistent with previously reported
characteristics [MDA-MB-231 and MDA-MB-468 cells: American Type
Culture Collection; SUM159 cells: (29)]. SUM159 and MDA-MB-231 have
previously been classified as claudin-low breast cancer cell lines,
while MDA-MB-468 has been classified as a basal-like breast cancer
cell line (6,25).
CD44 knockdown was performed using short hairpin RNA
(shRNA/sh; TRCN0000296191; Sigma-Aldrich; Merck KGaA), which
targeted the sequence 5′-GGACCAATTACCATAACTATT-3′ (sense strand).
The corresponding antisense strand was 5′-AATAGTTATGGTAATTGGTCC-3′.
The shRNA was cloned into the pLKO.1-puro lentiviral vector
(MISSION pLKO.1-puro; Sigma-Aldrich; Merck KGaA), which expressed
the shRNA under the control of the U6 promoter and included a
puromycin resistance gene for stable selection. MISSION pLKO.1-puro
non-target shRNA control plasmid DNA (SHC016; Sigma-Aldrich; Merck
KGaA) was used as an appropriate control. The sense strand of the
control shRNA was 5′-GCGCGATAGCGCTAATAATTT-3′, and the
corresponding antisense strand was 5′-AAATTATTAGCGCTATCGCGC-3′.
Lentiviral particles were produced in 293 cells (institutional
stock; Shizuoka General Hospital, Shizuoka, Japan) using a
second-generation packaging system (MISSION Lentiviral Packaging
Mix; Sigma-Aldrich; Merck KGaA) and FuGENE HD transfection reagent
(Promega Corporation). For each 10-cm dish, 2.6 µg pLKO.1-puro
transfer plasmid, 26 µl MISSION Lentiviral Packaging Mix and 16 µl
FuGENE HD were combined according to the manufacturers'
instructions and added to cells at 37°C. After 24 h, the medium was
replaced with fresh growth medium. Viral supernatants were
harvested at 48 and 72 h post-transfection and passed through a
0.45-µm filter. MDA-MB-231 and SUM159 cells were transduced at an
MOI of 5 in the presence of 8 µg/ml polybrene for 24 h. Stable
transductants were selected with puromycin (1.0 µg/ml) for 5 days,
followed by culture in puromycin-free medium for 72 h to allow
recovery and to minimize cytotoxic stress associated with prolonged
antibiotic exposure, after which downstream assays were
performed.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from each cell line using
the RNeasy Mini Kit (Qiagen KK) according to the manufacturer's
protocol. RNA was reverse-transcribed into cDNA using the iScript
cDNA Synthesis Kit (Bio-Rad Laboratories, Inc.) in a 20-µl reaction
according to the manufacturer's protocol. Equal cDNA amounts were
used as templates for RT-qPCR to detect mRNA expression relative to
that of GAPDH (endogenous control). mRNA expression was quantitated
using a Thermal Cycler Dice® Real Time System III
(Takara Bio, Inc.) and SYBR-Green qPCR SuperMix (Invitrogen; Thermo
Fisher Scientific, Inc.). The thermocycling conditions were as
follows: Initial denaturation at 95°C for 30 sec, followed by 40
cycles of 95°C for 15 sec and 60°C for 30 sec. Melting curve
analysis was performed to verify product specificity. The primers
used were: Smad2 forward, 5′-AGTGTGTAAAATTCCACCAG-3′ and
reverse, 5′-ATTCTAGTTAGCTGATAGACGG-3′; GAPDH forward,
5′-TCGGAGTCAACGGATTTG-3′ and reverse,
5′-GCAACAATATCCACTTTACCAGAG-3′; snail family transcriptional
repressor 1 (Snai1) forward, 5′-CTCTAATCCAGAGTTTACCTTC-3′
and reverse, 5′-GACAGAGTCCCAGATGAG-3′; and Twist forward,
5′-CTAGATGTCATTGTTTCCAGAG-3′ and reverse,
5′-CCCTGTTTCTTTGAATTTGG-3′. RT-qPCR reactions were performed in
triplicate, and each experiment was repeated three times. The fold
induction of gene expression was calculated using the
2−ΔΔCq method (30).
Immunoprecipitation and western
blotting
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH
7.4; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS)
supplemented with protease and phosphatase inhibitors (Roche
Diagnostics GmbH). For each immunoprecipitation, 50 µl Dynabeads
Protein G magnetic beads (Invitrogen; Thermo Fisher Scientific,
Inc.) were pre-incubated with 3 µg of the indicated primary
antibody (anti-CD44; cat. no. sc-7297; Santa Cruz Biotechnology,
Inc.) or normal mouse IgG (cat. no. 5415; Cell Signaling
Technology, Inc.) for 30 min at room temperature with gentle
rotation to allow formation of bead-antibody complexes.
Subsequently, 500 µg lysate was added to the bead-antibody
complexes and samples were incubated for 1 h at room temperature
with rotation. The immune complexes were isolated magnetically
using a DynaMag™-2 magnet (Thermo Fisher Scientific, Inc.) and
washed three times with PBS at room temperature. Bound proteins
were eluted by boiling the beads in 2X Laemmli sample buffer
(Bio-Rad Laboratories, Inc.) for 5 min, resolved via SDS-PAGE and
transferred onto nitrocellulose membranes. Western blotting was
performed using specific antibodies as previously described
(31). These conditions were
applied to all western blot experiments performed in the present
study. For the analysis of TGF-β-induced Smad2 phosphorylation,
cells were treated with recombinant human TGF-β1 (10 ng/ml;
PeproTech, Inc.; Thermo Fisher Scientific, Inc.) at 37°C for 2 h
prior to protein extraction, unless otherwise specified.
Cell cycle analysis
Cells (2×105 cells/well) were seeded in
6-well plates and incubated at 37°C in a humidified atmosphere with
5% CO2 for 24 h. Subsequently, the cells were treated
with 10 µM LY2109761 (LY-61; Selleck Chemicals) at 37°C for 48 h.
After trypsinization, the cells were fixed and stained according to
the protocol of the Cell Cycle Phase Determination Kit (item no.
10009349; Cayman Chemical Company). The cell cycle distribution was
analyzed via flow cytometry using a BD FACSMelody™ flow cytometer
(BD Biosciences). Propidium iodide fluorescence was detected in the
BP/700/54 channel with a blue laser, and data were analyzed using
BD FACSChorus software (version 1.3.2; BD Biosciences). Cell cycle
phase distributions (G1, S and G2/M) were
calculated using FlowJo software (version 10.10.0; BD
Biosciences).
Apoptosis assay
Apoptosis was analyzed according to the protocol
from BioLegend, Inc. After a 48-h incubation at 37°C in a
humidified atmosphere with 5% CO2 in either standard
media or media containing 10 µM LY-61, the cells were washed twice
with cold Cell Staining Buffer (cat. no. 420201; BioLegend, Inc.)
and resuspended in Annexin V Binding Buffer (cat. no. 422201;
BioLegend, Inc.) at a concentration of 1×106 cells/ml. A
100-µl aliquot of the cell suspension was transferred into a 5-ml
test tube, and 5 µl FITC-conjugated Annexin V (cat. no. 640906;
BioLegend, Inc.) and 5 µl 7-aminoactinomycin D (7-AAD; cat. no.
420403; BioLegend, Inc.) were added simultaneously. Subsequently,
the cells were incubated in the dark for 15 min at room
temperature. After incubation, 400 µl Annexin V Binding Buffer was
added to each sample prior to flow cytometry analysis. Fluorescence
signals were detected using a BD FACSMelody™ flow cytometer (BD
Biosciences) equipped with a 488-nm blue laser, detecting FITC
fluorescence in the BP/530/40 channel and 7-AAD fluorescence in the
BP/700/54 channel. Data were acquired and analyzed using BD
FACSChorus software (version 1.3.2; BD Biosciences), and data were
processed with FlowJo software (version 10.10.0; BD
Biosciences).
MTT assay
MTT assays were performed using the MTT Cell
Proliferation and Cytotoxicity Assay Kit (AR1156; Boster Bio). A
total of 1,000 cells per well were seeded in 96-well cell culture
plates, and the assay was performed according to the manufacturer's
protocol. After incubation with 10 µl MTT labeling reagent for 4 h,
100 µl Formazan Solubilization Solution (included in the kit;
Boster Bio) was added to each well, and the plate was further
incubated at 37°C for 4–18 h to ensure complete dissolution of the
purple formazan crystals. The absorbance was measured at 560 nm
using a microplate reader. Cell viability was quantified on day 5
post-plating, and the relative viability was calculated as the
ratio of absorbance values on day 5.
Clonogenic assay
A total of 500 cells per well were seeded in 6-well
plates and allowed to attach overnight. MDA-MB-231 and SUM159 cells
were cultured in standard media or media containing 10 µM LY-61 at
37°C with 5% CO2 for 7 days. The colonies were fixed
with 4% paraformaldehyde (FUJIFILM Wako Pure Chemical Corporation)
at room temperature for 15 min, stained with 0.5% crystal violet at
room temperature for 20 min and washed with distilled water.
Colonies (diameter >0.5 mm) were counted manually under a light
microscope.
Invasion assay
The invasiveness of MDA-MB-231 and SUM159 cells was
evaluated using the CytoSelect™ 24-well Cell Invasion Assay Kit
(cat. no. CBA-110; Cell Biolabs, Inc.) according to the
manufacturer's protocols. Transwell inserts with polycarbonate
membranes (8-µm pore size) were precoated with Matrigel at 37°C for
2 h, and the Matrigel layer was rehydrated at room temperature for
1 h before cell seeding. For the invasion assay, MDA-MB-231 cells
were maintained in DMEM (Nacalai Tesque, Inc.) and SUM159 cells
were maintained in Ham's F-12 medium (Nacalai Tesque, Inc.) as
aforementioned. A total of 2×105 cells in 300 µl of the
corresponding serum-free medium were added to the upper chamber,
while 500 µl of the same medium containing 10% FBS (Biowest,
distributed by Funakoshi Co., Ltd.) with or without 10 µM LY-61 was
placed in the lower chamber as a chemoattractant. After incubation
at 37°C in 5% CO2 for 48 h, non-invading cells were
removed, and invaded cells were stained with the dye solution
provided in the kit at room temperature for 10 min. Stained samples
(100 µl) from each well were transferred to a 96-well plate, and
absorbance was measured at 560 nm using a microplate reader.
Representative images of invaded cells were captured using a Nikon
ECLIPSE TS2 light microscope (Nikon Corporation).
Statistical analysis
Statistical analyses were conducted using GraphPad
Prism® version 10.3.1 (Dotmatics) and
Microsoft® Excel Version 2019 (Microsoft Corporation).
All data are presented as the mean ± SD from at least three
independent experiments. Data normality was assessed using the
Shapiro-Wilk test. For normally distributed data, statistical
comparisons among multiple groups were conducted using one-way or
two-way ANOVA followed by Tukey's or Dunnett's multiple comparisons
test, as appropriate. Unpaired two-tailed Student's t-tests were
used for pairwise comparisons. For non-normally distributed data
(such as mutation burden data), statistical comparisons between two
groups were performed using the Mann-Whitney U test. P<0.05 was
considered to indicate a statistically significant difference.
Public dataset analysis
Genomic and clinical data, including copy number
alterations (CNAs) and mRNA expression levels, were obtained from
the METABRIC dataset via cBioPortal (https://www.cbioportal.org/). Frequencies of CNAs for
selected genes were summarized according to breast cancer subtype
using the cBioPortal interface. Kaplan-Meier survival analyses were
performed in GraphPad Prism using mRNA expression z-scores and
associated clinical outcome data from the METABRIC cohort. Patients
were divided into high and low expression groups based on the
median value, and P-values were calculated using the log-rank test.
No additional statistical software or custom computational
pipelines were used. Data from the METABRIC study were originally
published by Curtis et al (32).
Protein-protein interaction (PPI)
network analysis
PPI networks were analyzed using the STRING database
(version 11.0; http://string-db.org) (33). Interactions with a medium confidence
score (>0.4) were included.
Results
Genomic alterations in claudin-low
breast cancer: CNA-driven therapeutic implications of CD44, TGFBR1
and TGFBR2
Breast cancer is a heterogeneous disease that
comprises molecular subtypes with distinct genetic and phenotypic
profiles (2,3). CNAs and somatic mutations are crucial
in promoting oncogenesis, as well as influencing tumor progression
and therapeutic response (9,34).
Among the key genes, CD44, a cancer stem cell marker, and TGFBR1/2,
which mediate TGF-β signaling (24,35),
are implicated in tumor invasion and metastasis (36). The presence of CNAs in these genes
suggests that their dysregulation may create potential therapeutic
vulnerabilities, genetic dependencies that could be exploited for
targeted intervention in claudin-low breast cancer.
CD44, TGFBR1 and TGFBR2 exhibited similar CNA
patterns, characterized by moderate deletion frequencies and rare
amplifications across all subtypes. In the claudin-low subtype,
their deletion frequencies were intermediate, ranking third or
fourth highest among all breast cancer subtypes (generally <10%
for CD44 and 10–15% for TGFBR1/2). By contrast, BRCA1 and BRCA2
displayed higher alteration rates, with basal-like tumors showing
prominent deletion frequencies (~40%) (Fig. 1A). This highlights the unique
genomic roles of CD44, TGFBR1 and TGFBR2 in tumor biology, which
are distinct from BRCA1/2.
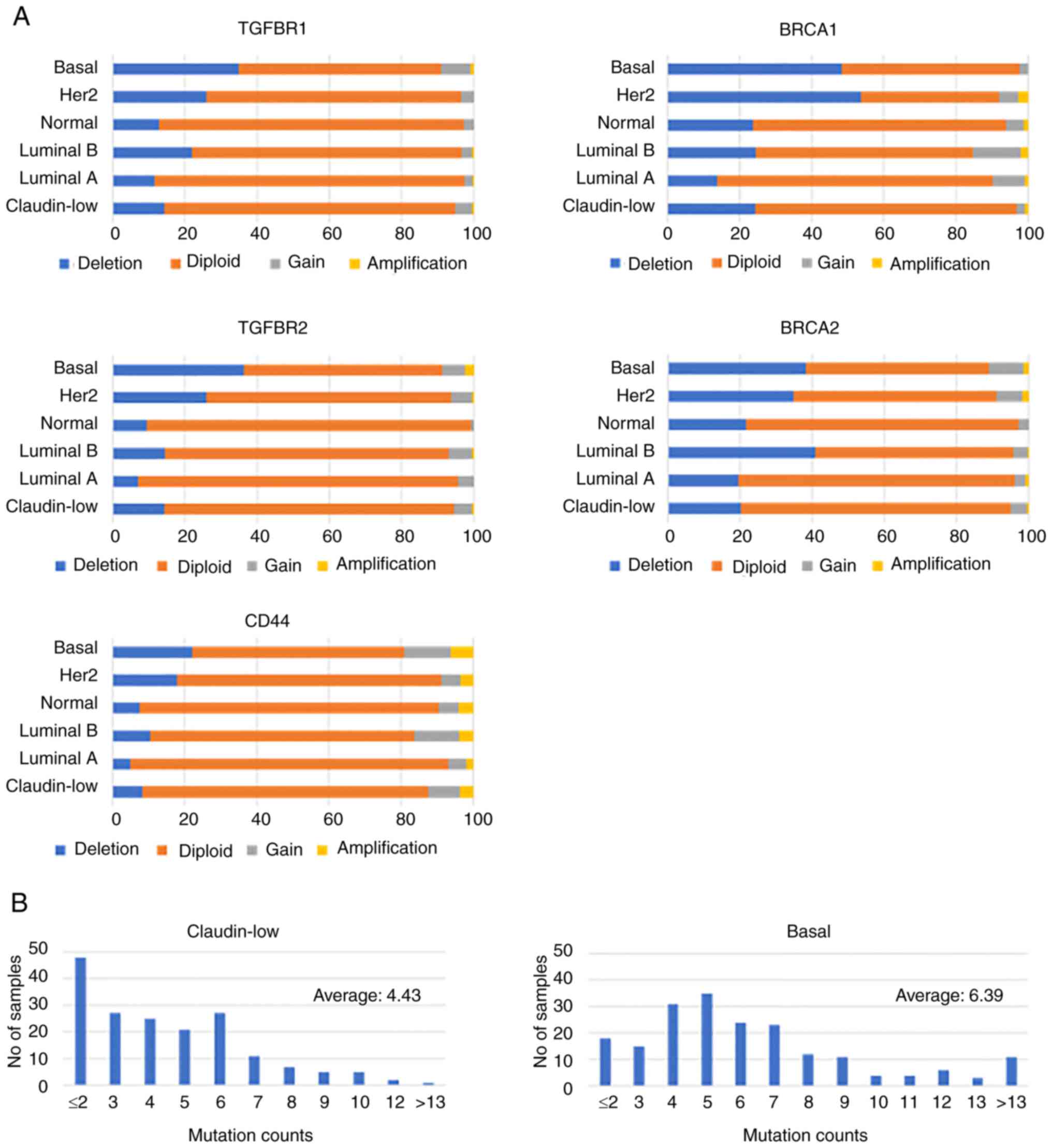 | Figure 1.CNAs and mutation burden across
breast cancer subtypes (METABRIC dataset). (A) Bar plots showing
the distribution of CNAs for TGFBR1, TGFBR2, CD44, BRCA1 and BRCA2
across breast cancer subtypes (claudin-low, luminal A, luminal B,
Her2-enriched, basal and normal-like) based on the METABRIC
dataset. CNAs were categorized as deletions (blue), diploid
(orange), gains (gray) and amplifications (yellow). (B) Histogram
representing the distribution of mutation counts in claudin-low
(n=179) and basal (n=197) breast cancer samples based on the
METABRIC dataset. Claudin-low tumors exhibited a significantly
lower mutational burden (mean ± SD, 4.43±2.70) compared with basal
tumors (mean ± SD, 6.39±3.84) (Mann-Whitney U test; P<0.0001).
CNA, copy number alteration; TGFBR, TGF-β receptor. |
Genome-wide mutation analysis (Fig. 1B) revealed a statistically
significant difference in the mutational burden between claudin-low
and basal subtypes, which are both representatives of
triple-negative breast cancer (TNBC). Claudin-low tumors exhibited
a lower mutational burden (mean, 4.43±2.70; n=179) compared with
basal tumors (mean, 6.39±3.84; n=197), and this difference was
confirmed by a Mann-Whitney U test (P<0.0001). These findings
suggested that claudin-low tumors rely more on CNAs or epigenetic
changes than on mutations.
By contrast, basal tumors exhibited a broader
distribution of mutation counts and higher average burden,
consistent with known genomic instability in this subtype. Such
instability is frequently associated with defects in DNA damage
response pathways, including BRCA1/2 mutations, and may contribute
to the aggressive phenotype characteristic of basal-like TNBC
(9,34).
CD44, TGFBR1 and TGFBR2 displayed negligible
mutation frequencies across all subtypes in the METABRIC dataset
(data not shown), suggesting they are more commonly affected by
CNAs rather than point mutations in claudin-low tumors.
The similar CNA patterns observed in CD44, TGFBR1
and TGFBR2 suggest a potentially coordinated regulatory mechanism,
possibly linked to pathways that drive tumor progression. In
claudin-low tumors, which exhibit a lower overall mutational
burden, such CNAs may serve a more prominent role in shaping tumor
biology. This reliance on CNAs, rather than mutations, highlights a
potential therapeutic vulnerability unique to this subtype
(34,37), and underscores the need for further
investigation into the functional significance and targetability of
these genes.
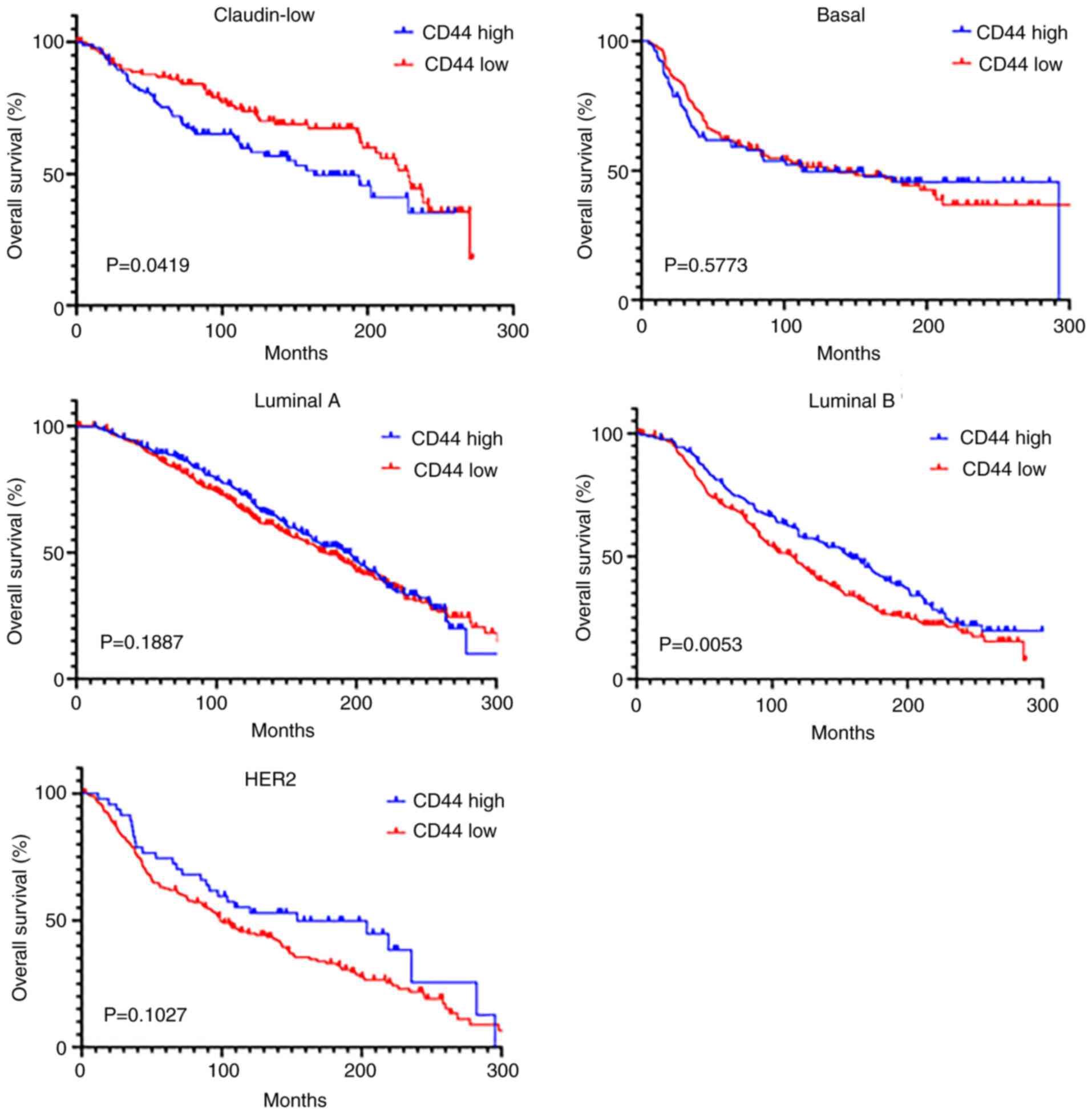
Prognostic role of CD44 expression in
the claudin-low breast cancer subtype
Kaplan-Meier survival curves based on the METABRIC
dataset showed the subtype-specific associations of CD44 expression
with overall survival. In the claudin-low subtype, low CD44
expression was associated with improved survival (P=0.0419),
whereas in the luminal B subtype, low CD44 expression was
associated with poor outcomes (P=0.0053). By contrast, no
significant survival differences were observed between the high and
low CD44 expression groups in patients with the basal, luminal A
and HER2 subtypes (Fig. 2). These
findings suggested that the prognostic value of CD44 was
subtype-specific, highlighting the importance of considering the
breast cancer subtype when evaluating CD44 as a prognostic
biomarker or therapeutic target. This indicated that CD44-targeted
strategies should be tailored to the molecular subtype of the
tumor.
Dual targeting of CD44 and TGF-β
signaling suppresses viability and invasion in claudin-low breast
cancer cells
The effects of multiple TGFBR inhibitors, including
LY-61, SB431542 and Galunisertib, on the viability of various
breast cancer cell lines (SUM159, MDA-MB-231 and MDA-MB-468) were
evaluated. Among these, LY-61 showed the most pronounced inhibitory
effect on viability, particularly in claudin-low subtype cell lines
such as SUM159 and MDA-MB-231 cells, in which its effects were
statistically significant compared with the control. By contrast,
the basal-like cell line MDA-MB-468 exhibited minimal sensitivity
to these inhibitors (Fig. 3A).
These findings demonstrated that LY-61 potently inhibited cell
viability in claudin-low breast cancer cell lines.
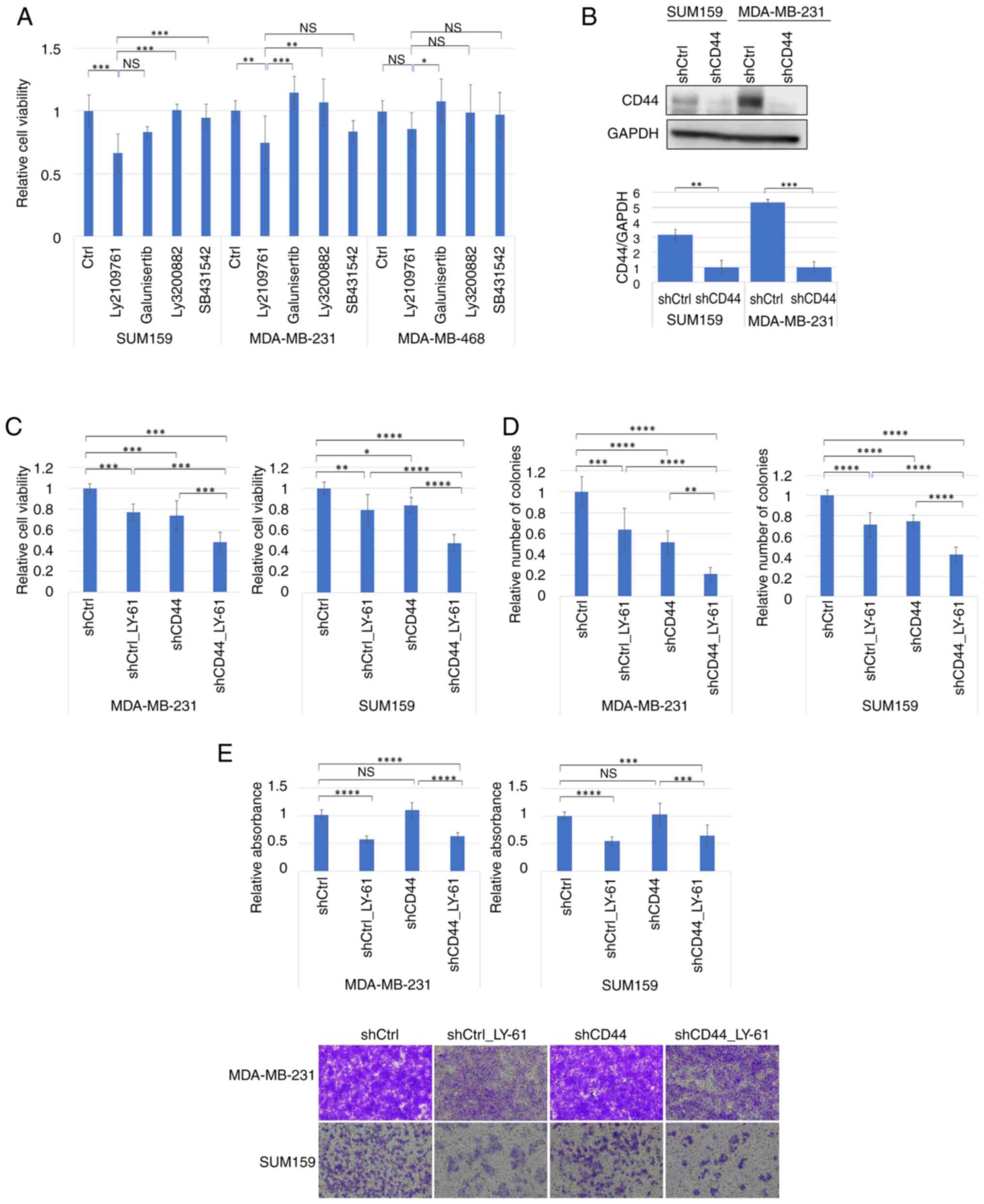 | Figure 3.Additive antitumor effects of LY-61
and CD44 knockdown in claudin-low breast cancer cell lines. (A)
Relative viability of breast cancer cell lines (SUM159, MDA-MB-231
and MDA-MB-468) following treatment with TGF-β receptor inhibitors
(LY-61, Ly3200882, SB431542 and Galunisertib; all at 10 µM). Cells
(1,000 per well) were plated in 96-well plates, and drugs were
added 24 h after plating. Cell viability was measured using the MTT
assay on day 5 after plating. Statistical comparisons were made
using one-way ANOVA followed by Tukey's multiple comparisons test
for each cell line. (B) Western blot analysis of CD44 expression in
SUM159 and MDA-MB-231 cells following shRNA transduction.
Densitometric semi-quantification of CD44 protein levels relative
to GAPDH is shown below the blots (n=3). Data are presented as the
mean ± SD. Statistical comparisons were performed using unpaired
two-tailed t-tests. (C) Relative viability of MDA-MB-231 and SUM159
cells following CD44 knockdown (shCD44) and/or treatment with LY-61
(10 µM). Cells (1,000 per well) were plated in 96-well plates, and
LY-61 was added 24 h after plating. Cell viability was determined
using an MTT assay on day 5 after plating. Statistical significance
was assessed using two-way ANOVA followed by Tukey's multiple
comparisons test. (D) Colony formation assays showing relative
colony numbers in MDA-MB-231 and SUM159 cells following CD44
knockdown and/or LY-61 treatment. Statistical comparisons were
performed using two-way ANOVA followed by Tukey's multiple
comparisons test. (E) Invasion assays in SUM159 and MDA-MB-231
cells showing the relative absorbance and representative microscopy
images of invaded cells stained on the underside of basement
membrane matrix-coated inserts. Cells were treated with LY-61
and/or subjected to CD44 knockdown. Statistical analysis was
performed using two-way ANOVA followed by Tukey's multiple
comparisons test. Representative images are shown at a
magnification of ×100. (A and C-E) Data are presented as the mean ±
SD of three independent experiments (n=3). *P<0.05; **P<0.01;
***P<0.001; ****P<0.0001. Ctrl, control; LY-61, LY2109761;
NS, not significant; sh/shRNA, short hairpin RNA. |
To validate CD44 knockdown, reduced CD44 expression
was confirmed using western blot analysis in SUM159 and MDA-MB-231
cells (Fig. 3B). Further
experiments combining LY-61 treatment with CD44 knockdown
demonstrated an enhanced inhibitory effect on both cell viability
(Fig. 3C) and colony formation
(Fig. 3D) compared with either
treatment alone. Representative images of colony formation in
SUM159 and MDA-MB-231 cells are shown in Fig. S1A. CD44 knockdown and LY-61
treatment individually suppressed viability and colony formation to
a similar extent; however, their combination resulted in a more
pronounced reduction in both parameters. Although individual
comparisons showed significant differences between treatment
groups, no statistically significant interaction was detected in
the two-way ANOVA, suggesting an additive rather than synergistic
effect.
In the invasion assays (Fig. 3E), both SUM159 and MDA-MB-231 cells
were examined. LY-61 treatment markedly reduced invasion in both
cell lines, whereas CD44 knockdown alone had a minimal impact. This
suggested that LY-61 was particularly effective in impairing the
invasive behavior of cancer cells, likely through mechanisms
independent of CD44.
Collectively, these findings indicated that although
CD44 knockdown effectively suppressed cell viability and colony
formation in SUM159 and MDA-MB-231 cells, it had a limited effect
on cell invasion. By contrast, LY-61 treatment consistently
inhibited proliferative and invasive behaviors in both cell lines.
Notably, the combination of CD44 knockdown and LY-61 treatment led
to greater suppression of cell viability and colony formation, and
also reduced the invasive capacity compared with that in the shCtrl
group without LY-61 treatment, the latter effect being primarily
attributable to LY-61. These results highlight the complementary
effects of targeting CD44 and TGF-β signaling in claudin-low breast
cancer cells.
LY-61 attenuates CD44
knockdown-mediated activation of the TGF-β/Smad2 pathway
The STRING network analysis (Fig. S1B) revealed a strong interaction
between CD44 and TGFBR1, suggesting that CD44 serves a potential
regulatory role in TGF-β signaling. This association was confirmed
by co-immunoprecipitation assays (Fig.
4A), as CD44 and TGFBR1 co-precipitated in control cells
transduced with non-targeting shRNA (shCtrl); however, this
interaction was lost following CD44 knockdown. These findings
indicated that CD44 may modulate TGF-β signaling through TGFBR1 by
influencing receptor stability or complex formation.
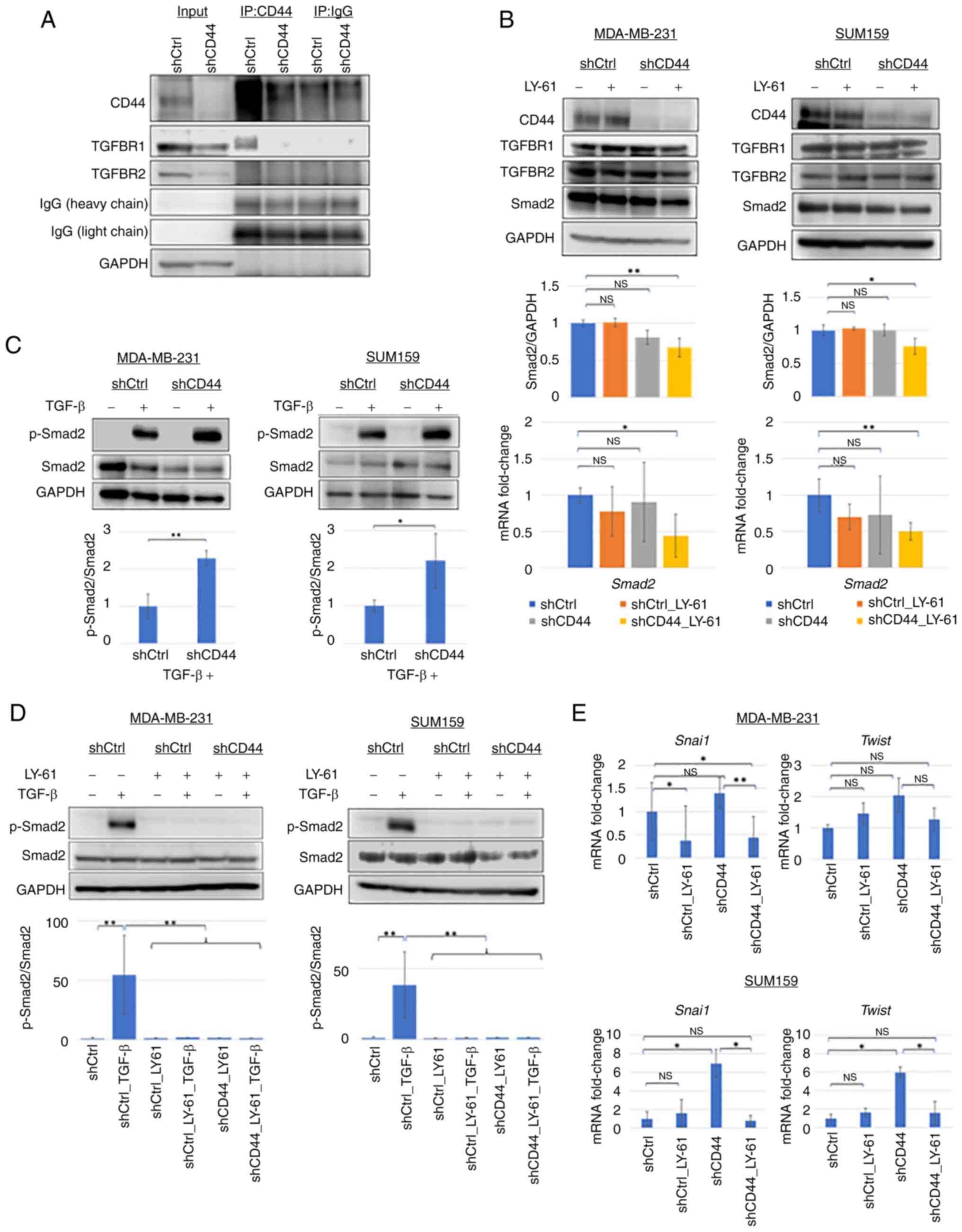 | Figure 4.Effects of LY-61 and CD44 knockdown
on the TGF-β/Smad2 pathway in claudin-low breast cancer cell lines.
(A) Co-immunoprecipitation assays showing that CD44 co-precipitated
with TGFBR1 in MDA-MB-231 cells. The CD44-TGFBR1 interaction signal
was reduced in shCD44 cells. No detectable co-precipitation was
observed between CD44 and TGFBR2. (B) Western blotting and RT-qPCR
showing Smad2 protein and mRNA levels in MDA-MB-231 and SUM159
cells following CD44 knockdown and/or LY-61 treatment.
Representative western blots (top), densitometric
semi-quantification of Smad2 protein levels (middle) and mRNA
expression levels determined by RT-qPCR (bottom) are shown. Data
are presented as the mean ± SD from three independent experiments
(n=3). (C) Western blot analysis of p-Smad2 and total Smad2 protein
levels after TGF-β stimulation in MDA-MB-231 and SUM159 cells with
or without CD44 knockdown. The semi-quantification of Smad2
phosphorylation was performed by calculating the p-Smad2/Smad2
ratio. CD44 knockdown increased TGF-β-induced p-Smad2 levels in
both cell lines. (D) Western blotting showing the effect of LY-61
treatment on p-Smad2 levels in MDA-MB-231 and SUM159 cells.
Densitometric semi-quantification of the p-Smad2/Smad2 ratio from
three independent experiments (n=3) is shown below the blots. (E)
Relative mRNA expression levels of Snai1 and Twist in
SUM159 and MDA-MB-231 cells following CD44 knockdown and/or LY-61
treatment, as determined using RT-qPCR. Data are presented as the
mean ± SD from three independent experiments (n=3). Statistical
significance was assessed using one-way ANOVA followed by (B)
Dunnett's or (D and E) Tukey's multiple comparisons test, or (C)
unpaired two-tailed Student's t-test. *P<0.05; **P<0.01.
Ctrl, control; LY-61, LY2109761; NS, not significant; p-,
phosphorylated; RT-qPCR, reverse transcription-quantitative PCR;
sh, short hairpin RNA; Snai1, snail family transcriptional
repressor 1; TGFBR, TGF-β receptor; IP, immunoprecipitation. |
Western blot analysis (Fig. 4B) showed that Smad2 protein levels
were not notably altered by LY-61 treatment or CD44 knockdown alone
in both MDA-MB-231 and SUM159 cells. However, a modest decrease in
Smad2 protein levels was observed following combined treatment.
Consistently, the results shown in Fig. S1C confirmed efficient CD44
knockdown and showed no marked changes in TGFBR1 or TGFBR2
expression. This pattern was also reflected in the RT-qPCR results,
where Smad2 mRNA levels showed a similar downward trend following
combined treatment. These findings suggested that dual inhibition
exerted a cumulative suppressive effect on Smad2 expression,
although the magnitude of the change remained limited.
A crucial observation was the impact of CD44
knockdown on Smad2 phosphorylation (p-Smad2) in response to TGF-β
stimulation (Fig. 4C). In the
MDA-MB-231 and SUM159 cells, TGF-β treatment markedly increased the
p-Smad2 levels. CD44 knockdown alone did not induce Smad2
phosphorylation; however, it enhanced TGF-β-induced Smad2
phosphorylation compared with that in shCtrl cells, indicating
increased sensitivity to TGF-β signaling. Notably, LY-61 treatment
markedly inhibited TGF-β-induced Smad2 phosphorylation (Fig. 4D), demonstrating its potent
inhibitory effect on Smad2 activation, regardless of CD44
expression.
To assess whether CD44 knockdown affects
TGF-β-induced EMT, the expression levels of the EMT-related
transcription factors Snai1 and Twist were
subsequently examined. As shown in Fig.
4E, CD44 knockdown significantly increased Snai1 and
Twist mRNA expression in SUM159 cells (P<0.05), whereas
only a non-significant upward trend was observed in MDA-MB-231
cells. In MDA-MB-231 cells, LY-61 monotherapy (shCtrl_LY-61)
significantly downregulated Snai1 expression (P<0.05),
while Twist expression remained unchanged. Notably,
treatment with LY-61 significantly reduced Snai1 expression
in CD44-knockdown MDA-MB-231 cells (P<0.01), whereas
Twist showed a non-significant downward trend. In SUM159
cells, LY-61 significantly suppressed both Snai1 and
Twist expression in the context of CD44 knockdown
(P<0.05). These findings suggested that LY-61 counteracted
TGF-β/Smad2-mediated EMT transcriptional activity, even under
conditions of CD44 knockdown, as shown by its suppression of
TGF-β-induced Smad2 phosphorylation (Fig. 4D) and the concomitant reduction of
Snai1 and Twist expression in LY-61-treated
CD44-knockdown cells compared with untreated CD44-knockdown cells
(Fig. 4E). Consistently, LY-61 also
reduced Snai1 protein levels in CD44-knockdown SUM159 cells, with
the combination group (shCD44_LY-61) showing the lowest Snai1/GAPDH
ratio. By contrast, CD44 knockdown alone did not significantly
increase Snai1 expression (Fig.
S1D).
These findings suggested that CD44 knockdown did not
directly enhance Smad2 phosphorylation but increased EMT-related
gene expression, indicating that CD44 may influence TGFBR1-mediated
signaling rather than Smad2 activation itself. LY-61 effectively
inhibited Smad2 phosphorylation and suppressed EMT marker
expression, highlighting its potential to counteract TGF-β-driven
invasive behavior in claudin-low breast cancer.
Cell line-specific effects of LY-61
and CD44 knockdown on the cell cycle and apoptosis in claudin-low
breast cancer cells
In SUM159 cells, LY-61 treatment increased the
proportion of cells in the S phase under both control
(shCtrl_LY-61) and CD44-knockdown conditions (shCD44_LY-61),
suggesting delayed DNA replication and potential checkpoint
activation rather than full cell cycle arrest (38). Furthermore, G2 phase
accumulation was observed following CD44 knockdown in SUM159 cells,
suggesting that CD44 loss may contribute to delayed cell cycle
progression at G2. By contrast, MDA-MB-231 cells showed
only a slight increase in the S phase fraction after LY-61
treatment, and CD44 knockdown exerted a negligible effect on
G2 phase distribution, indicating that the influence of
CD44 on cell cycle regulation differed between these two cell lines
(Fig. 5A and B).
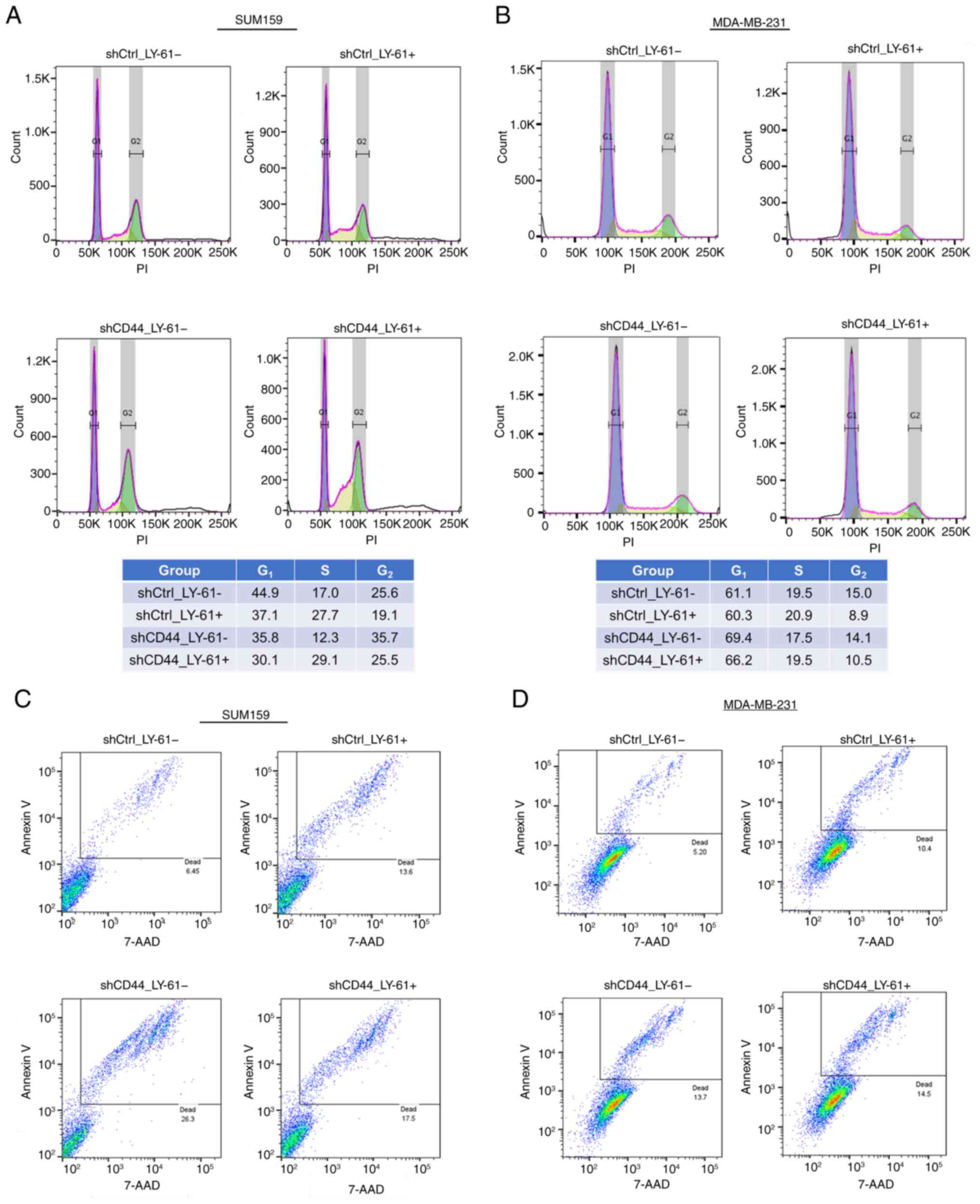 | Figure 5.Effects of LY-61 and CD44 knockdown
on cell cycle progression and apoptosis in claudin-low breast
cancer cells. (A and B) Flow cytometry analysis of cell cycle
distribution in (A) SUM159 and (B) MDA-MB-231 cells treated with
LY-61 and/or CD44 knockdown. LY-61 treatment induced S phase
accumulation, most prominently in SUM159 cells. In SUM159 cells,
CD44 knockdown increased G2 phase accumulation,
suggesting delayed progression through the cell cycle. By contrast,
in MDA-MB-231 cells, the proportion of cells in the G2
phase remained relatively unchanged. The tables summarize the
percentages of cells in each phase. Percentages may not total
exactly 100% due to the exclusion of sub-G1 and debris
populations and rounding errors (values were rounded to one decimal
place after calculation from two decimal places, which may cause
minor deviations such as totals slightly >100%). (C and D)
Apoptosis analysis using annexin V/7-AAD staining in (C) SUM159 and
(D) MDA-MB-231 cells following CD44 knockdown and/or LY-61
treatment. CD44 knockdown increased apoptosis, whereas LY-61
treatment alone had a minimal effect. 7-AAD, 7-aminoactinomycin D;
Ctrl, control; LY-61, LY2109761; sh, short hairpin RNA. |
Apoptosis assays (Fig.
5C and D) revealed that CD44 knockdown increased apoptosis, as
indicated by a higher percentage of annexin V-positive cells
compared with the shCtrl group. This suggested that CD44 knockdown
compromised tumor cell survival by enhancing apoptotic signaling.
Western blotting further showed that CD44 knockdown decreased Bcl-2
protein levels in MDA-MB-231 cells, although the reduction was not
statistically significant (Fig.
S1E), suggesting a role of CD44 in pro-survival signaling via
Bcl-2 regulation.
When LY-61 treatment was combined with CD44
knockdown (shCD44_LY-61), the percentage of apoptotic cells
remained increased compared with that in the shCtrl group but was
slightly reduced compared with that following CD44 knockdown alone
in SUM159 cells, while in MDA-MB-231 cells it was higher than that
in the shCtrl group and similar to that in the CD44 knockdown
group. These findings suggest that LY-61 treatment and CD44
inhibition acted through distinct but complementary mechanisms to
suppress tumor cell survival. LY-61 mainly affected cell cycle
progression by inducing S phase accumulation, particularly in
SUM159 cells, whereas CD44 knockdown increased apoptosis, thereby
reducing the overall proliferative capacity.
These findings highlighted the cell line-specific
effects of LY-61 and CD44 knockdown on cell cycle progression and
apoptosis. LY-61-mediated S phase accumulation was pronounced in
SUM159 cells but only slight in MDA-MB-231 cells, underscoring the
context-dependent nature of cell cycle regulation in claudin-low
breast cancer. Meanwhile, CD44 knockdown consistently increased
apoptosis across both cell lines, demonstrating the role of CD44 in
supporting tumor cell survival. The combined inhibition of CD44 and
TGF-β signaling (via LY-61) is a promising strategy for the
simultaneous disruption of proliferation and survival mechanisms in
claudin-low breast cancer subtypes.
Discussion
The present study provided novel insights into
genomic alterations and potential therapeutic vulnerabilities in
the claudin-low breast cancer subtype, with a particular focus on
CD44, TGFBR1 and TGFBR2. The present results suggested that
claudin-low tumors relied more on CNAs and epigenetic changes than
mutations. This pattern was consistent with the lower overall
mutational burden observed in claudin-low tumors compared with
basal-like tumors, indicating that while mutations are involved,
alternative mechanisms, such as CNAs and epigenetic changes, may
serve a more prominent role in promoting tumor progression in this
subtype (9,34,37).
These findings highlight the unique genomic landscape of
claudin-low breast cancer, distinct from basal-like tumors, which
are characterized by high genomic instability and frequent BRCA1/2
mutations (9,34,37,39).
Understanding these subtype-specific differences is crucial for the
identification of effective therapeutic targets.
One of the key findings of the present study was the
complex role of CD44 in claudin-low breast cancer. While low CD44
expression was associated with improved survival in claudin-low
tumors, CD44 knockdown led to significant upregulation of
Snai1 and Twist expression in SUM159 cells, whereas
only a modest, non-significant increase was observed in MDA-MB-231
cells. This suggests a potential enhancement of EMT-related
transcriptional responsiveness. Although previous studies have
predominantly reported that CD44 promotes EMT and stemness in
various cancer types, including breast cancer (10,28,40–42),
some reports suggest that CD44 may also restrain EMT under specific
conditions (for example, when differences in the cellular context,
microenvironment or signaling network alter its downstream
interactions) (10,12–14).
The present findings differ from numerous previous reports
describing CD44 knockdown as suppressing EMT markers and invasive
potential (10,42,28),
but may reflect a context-specific response in claudin-low cells.
This contradiction indicates that CD44 serves dual roles:
Supporting tumor cell survival while modulating TGF-β signaling in
a way that restrains EMT. The present results demonstrated that
CD44 knockdown increased apoptosis, which may explain the improved
prognosis associated with low CD44 expression regardless of its
pro-EMT effects. These findings underscore the context-dependent
functions of CD44 in claudin-low breast cancer and the importance
of considering subtype-specific differences when evaluating CD44 as
a biomarker or therapeutic target.
From a therapeutic perspective, the present study
demonstrated that LY-61, a TGFBR inhibitor, exerted potent
antitumor effects on claudin-low breast cancer cell lines. However,
its effects on cell cycle regulation differed between SUM159 and
MDA-MB-231 cells. In SUM159 cells, LY-61 induced S phase
accumulation and G2 phase delay, indicating impaired
cell cycle progression. By contrast, MDA-MB-231 cells exhibited a
slight increase in the S-phase fraction accompanied by a modest
reduction in the G2-phase fraction compared with the
control, suggesting that LY-61 did not induce full cell cycle
arrest but rather delayed S phase progression in a cell
line-dependent manner. These differences may reflect variations in
DNA damage response or checkpoint activation (38).
These findings align with the known complexity of
TGF-β signaling in breast cancer. Although TGF-β typically induces
G1 arrest via CDK inhibitors, this mechanism is often
dysregulated in cancer (35,43).
Inhibition of TGF-β signaling may promote S phase entry by
bypassing residual G1 control (24). In the present study, S phase
accumulation was observed in both cell lines following LY-61
treatment, but the effect was more pronounced in SUM159 cells,
whereas only a slight increase was detected in MDA-MB-231 cells. In
both cell lines, LY-61 treatment was accompanied by a comparable
reduction in the G2-phase fraction (from 25.6 to 19.1%
in SUM159 cells and from 15.0 to 8.9% in MDA-MB-231 cells),
consistent with delayed progression from S to G2. These
results underscore the cell line-specific nature of TGF-β pathway
responses and suggest that therapeutic strategies targeting this
pathway may need to account for differences in cell cycle
regulation.
Aside from affecting cell cycle progression, LY-61
also effectively counteracted the modest EMT-related gene
expression changes observed following CD44 knockdown. CD44
knockdown increased Snai1 and Twist expression in
SUM159 cells, while the changes were modest in MDA-MB-231 cells.
However, LY-61 treatment markedly inhibited TGF-β-induced Smad2
phosphorylation, and also suppressed the expression of Snai1
and Twist compared with that in CD44-knockdown cells without
LY-61 treatment, effectively counteracting the pro-invasive signals
induced by TGF-β. These findings suggested that while CD44
knockdown may enhance tumor cell plasticity, LY-61 mitigated this
effect by inhibiting the key drivers of EMT.
These findings are consistent with previous studies
showing that pharmacological inhibition of TGF-β signaling (for
example, using SB431542) suppressed Smad2 phosphorylation and
downregulated EMT-related transcription factors such as
Snai1 and Twist, thereby inhibiting TGF-β-induced
invasion in breast cancer cells (36,44,45).
Although CD44 has been reported to promote EMT and its knockdown
has been reported to suppress EMT markers (28,41,42),
the present data revealed a modest increase in Snai1 and
Twist expression following CD44 knockdown. This discrepancy
may reflect differences in cellular context, such as variations in
TGF-β signaling activity or other regulatory pathways. It is
possible that, under certain conditions, CD44 knockdown sensitizes
cells to TGF-β-induced EMT. This complexity may also help explain
why low CD44 expression was not consistently associated with
improved prognosis across subtypes, showing a favorable association
in claudin-low tumors, but not in basal, luminal A, luminal B or
HER2-enriched subtypes. Even in tumors with low CD44 expression,
EMT-inducing pathways such as TGF-β signaling may remain active,
thereby limiting the survival benefit associated with low CD44
expression (40,46–48).
The present data revealed a functional interplay
between CD44 and TGF-β signaling in claudin-low breast cancer.
Co-immunoprecipitation assays in MDA-MB-231 cells demonstrated a
physical interaction between CD44 and TGFBR1, and this interaction
was lost following CD44 knockdown. These findings suggest that CD44
may exert its role in TGF-β signaling not through the direct
modulation of Smad2 phosphorylation, but rather through its
interaction with TGFBR1, possibly affecting receptor stability or
accessibility to downstream effectors, consistent with a previous
study describing the functional relevance of CD44 in TGF-β receptor
regulation (26). Although SUM159
cells also exhibit claudin-low characteristics, CD44 knockdown and
LY-61 treatment did not induce an apparent reduction in TGFBR1 and
TGFBR2 protein levels, whereas MDA-MB-231 cells showed a tendency
toward decreased TGFBR2 expression, while TGFBR1 levels remained
largely unchanged. This difference may reflect subtle, cell
line-specific variations in the regulation of TGFBR stability or
turnover. It should be noted that the CD44-TGFBR1 interaction was
assessed only in MDA-MB-231 cells; thus, further studies are
required to determine whether similar mechanisms operate in SUM159
cells.
While CD44 knockdown alone markedly increased
apoptosis, the combination of LY-61 and CD44 knockdown resulted in
comparable or slightly lower apoptosis levels than CD44 knockdown
alone; however, apoptosis remained elevated compared with shCtrl
cells. This suggested that LY-61 and CD44 inhibition acted through
distinct but complementary mechanisms. CD44 depletion increased
apoptosis by disrupting survival signaling, whereas LY-61 delayed
cell cycle progression, which may reduce apoptotic stress in a
compensatory manner. Although LY-61 reduced the level of apoptosis
observed compared with CD44 knockdown alone in SUM159 cells, no
such decrease was observed in MDA-MB-231 cells. Nevertheless,
combined treatment with LY-61 and CD44 knockdown effectively
suppressed both proliferation and invasion compared with those in
the shCtrl group without LY-61 treatment, suggesting a
complementary therapeutic mechanism.
These results highlighted the complex and
potentially dual role of CD44 in claudin-low breast cancer. While
CD44 supports tumor survival, it may also restrain EMT by
modulating TGF-β signaling, as suggested by the present findings
and supported by a previous report demonstrating the regulatory
interaction between CD44 and TGFBR1 (26). In the present study, CD44 knockdown
increased apoptosis and was associated with a modest upregulation
of EMT-related transcription factors such as Snai1 and
Twist, possibly reflecting a compensatory response to the
loss of survival signaling. These findings are consistent with
previous reports showing that CD44 promoted cancer stemness and may
suppress EMT under certain conditions (10,11),
whereas other studies have linked CD44 upregulation to enhanced EMT
and metastasis (10,40). It remains to be clarified whether
this EMT-related gene expression change is a direct effect of CD44
loss or a secondary adaptation due to disrupted cell survival
mechanisms.
The effects of TGFBR inhibition observed in the
present study underscore the therapeutic significance of this
pathway in claudin-low breast cancer. Prior studies have
demonstrated that TGF-β serves a role in mediating EMT, immune
evasion and resistance to therapy (36,49).
The present results reinforced the notion that selective inhibition
of TGF-β signaling, particularly when combined with disruption of
CD44-mediated pathways, may represent a promising strategy for
managing claudin-low tumors, which currently lack effective
treatment options.
The findings of the present study suggest a
potential therapeutic strategy for claudin-low breast cancer by
targeting the TGF-β/Smad2 pathway (via LY-61) and CD44-mediated
survival mechanisms. However, the differential effects of LY-61 on
cell cycle progression across claudin-low cell lines underscore the
need for further investigation into the molecular determinants of
its efficacy.
A key limitation of the present study was the lack
of in vivo validation. Although the present in vitro
findings provided strong mechanistic insights into the cooperative
effects of CD44 knockdown and TGFBR inhibition, the therapeutic
implications of this dual-targeting strategy remain hypothetical.
Future studies using xenograft models or patient-derived organoids
will be essential to validate these in vitro observations in
physiologically relevant systems, and to evaluate treatment
efficacy, systemic toxicity and interactions within the tumor
microenvironment.
Although in vivo analyses were beyond the
scope of the present study, the present findings offer a valuable
preclinical framework. The present study demonstrated how CD44 and
TGF-β signaling interacted to regulate proliferation, invasion, EMT
and cell survival in claudin-low breast cancer. These insights
establish a basis for future translational research aimed at
developing combinatorial therapeutic strategies for this aggressive
and poorly understood subtype. Additional investigations should
also explore potential compensatory signaling pathways that might
limit the effectiveness of TGF-β blockade and evaluate the
long-term outcomes of Smad2 inhibition in vivo.
Aside from the CD44-TGF-β axis investigated in the
present study, other EMT-related transcription factors (such as
zinc finger E-box binding homeobox 1, forkhead box C2 and Slug) and
cancer stemness-associated pathways (such as the Wnt/β-catenin and
aldehyde dehydrogenase 1 pathways) may also trigger the malignant
phenotype of claudin-low breast cancer (50–53).
The present findings demonstrated that dual targeting of CD44 and
TGF-β signaling suppressed cell proliferation and invasion, CD44
knockdown increased apoptosis, and LY-61 counteracted EMT-related
transcriptional activation. These results support the therapeutic
potential of this combinatorial approach and provide a mechanistic
basis for further development of more comprehensive treatment
approaches.
Although most comparisons using standard statistical
methods (such as t-tests and ANOVA) yielded significant results,
more advanced multivariate approaches, such as interaction models
or multiple regression, could have yielded deeper insights into the
interplay among CD44 knockdown, TGF-β inhibition and downstream
signaling networks. As such, future studies should incorporate such
analyses to better capture the complex relationships among these
pathways under varying experimental conditions.
The differential responses observed in SUM159 and
MDA-MB-231 cells highlight the need for further mechanistic studies
to understand the cell line-specific effects. These differences
likely reflect underlying biological heterogeneity within
claudin-low breast cancer, including variations in TGFBR
expression, downstream signaling responsiveness or EMT status.
Additional preclinical studies using a broader panel of claudin-low
cell lines and patient-derived xenografts will be required to
validate the generalizability of the present findings and assess
their translational relevance.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Yoshihiro Sowa
(Department of Molecular-Targeting Cancer Prevention, Kyoto
Prefectural University of Medicine, Kyoto, Japan) and Dr Masahiro
Shibata (Department of Breast Surgery, Nagoya Eiseikai Hospital,
Nagoya, Japan) for their valuable advice. This abstract was
presented at the 2024 San Antonio Breast Cancer Symposium, held on
December 10–14, 2024, in San Antonio, TX, USA, and was published as
Abstract no. P2-04-06.
Funding
The present study was supported by JSPS KAKENHI (grant no.
21K08612) and the Medical Research Support Project of Shizuoka
Prefectural Hospital Organization.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
RM designed the study. RM and KK performed the
experiments and confirmed the authenticity of all the raw data. RM,
KK, SI, SS, RH and MT contributed to data analysis and manuscript
preparation. All authors have read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J
and Shi B: Breast cancer intrinsic subtype classification, clinical
use and future trends. Am J Cancer Res. 5:2929–2943.
2015.PubMed/NCBI
|
|
2
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiong X, Zheng LW, Ding Y, Chen YF, Cai
YW, Wang LP, Huang L, Liu CC, Shao ZM and Yu KD: Breast cancer:
Pathogenesis and treatments. Signal Transduct Target Ther.
10:492025. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herschkowitz JI, Simin K, Weigman VJ,
Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S,
Chandrasekharan S, et al: Identification of conserved gene
expression features between murine mammary carcinoma models and
human breast tumors. Genome Biol. 8:R762007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prat A, Parker JS, Karginova O, Fan C,
Livasy C, Herschkowitz JI, He X and Perou CM: Phenotypic and
molecular characterization of the claudin-low intrinsic subtype of
breast cancer. Breast Cancer Res. 12:R682010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sabatier R, Finetti P, Guille A, Adelaide
J, Chaffanet M, Viens P, Birnbaum D and Bertucci F: Claudin-low
breast cancers: Clinical, pathological, molecular and prognostic
characterization. Mol Cancer. 13:2282014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fougner C, Bergholtz H, Norum JH and
Sørlie T: Re-definition of claudin-low as a breast cancer
phenotype. Nat Commun. 11:17872020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garrido-Castro AC, Lin NU and Polyak K:
Insights into molecular classifications of triple-negative breast
cancer: Improving patient selection for treatment. Cancer Discov.
9:176–198. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hassn Mesrati M, Syafruddin SE, Mohtar MA
and Syahir A: CD44: A multifunctional mediator of cancer
progression. Biomolecules. 11:18502021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim KJ, Godarova A, Seedle K, Kim MH, Ince
TA, Wells SI, Driscoll JJ and Godar S: Rb suppresses collective
invasion, circulation and metastasis of breast cancer cells in
CD44-dependent manner. PLoS One. 8:e805902013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dehbokri SG, Noorolyai S, Baghbani E,
Moghaddamneshat N, Javaheri T and Baradaran B: Effects of CD44
siRNA on inhibition, survival, and apoptosis of breast cancer cell
lines (MDA-MB-231 and 4T1). Mol Biol Rep. 51:6462024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nam K, Oh S, Lee KM, Yoo SA and Shin I:
CD44 regulates cell proliferation, migration, and invasion via
modulation of c-Src transcription in human breast cancer cells.
Cell Signal. 27:1882–1894. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Z, Chen D, Nie J, Zhou S, Wang J,
Tang Q and Yang X: MicroRNA-143 targets CD44 to inhibit breast
cancer progression and stem cell-like properties. Mol Med Rep.
13:5193–5199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Montgomery N, Hill A, McFarlane S, Neisen
J, O'Grady A, Conlon S, Jirstrom K, Kay EW and Waugh DJ: CD44
enhances invasion of basal-like breast cancer cells by upregulating
serine protease and collagen-degrading enzymatic expression and
activity. Breast Cancer Res. 14:R842012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ouhtit A, Madani S, Gupta I,
Shanmuganathan S, Abdraboh ME, Al-Riyami H, Al-Farsi YM and Raj MH:
TGF-β2: A novel target of CD44-promoted breast cancer invasion. J
Cancer. 4:566–572. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu H, Niu M, Yuan X, Wu K and Liu A: CD44
as a tumor biomarker and therapeutic target. Exp Hematol Oncol.
9:362020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou L, Sheng D, Deng Q, Wang D and Liu S:
Development of a novel method for rapid cloning of shRNA vectors,
which successfully knocked down CD44 in mesenchymal triple-negative
breast cancer cells. Cancer Commun (Lond). 38:572018.PubMed/NCBI
|
|
19
|
Huang P, Chen A, He W, Li Z, Zhang G, Liu
Z, Liu G, Liu X, He S, Xiao G, et al: BMP-2 induces EMT and breast
cancer stemness through Rb and CD44. Cell Death Discov.
3:170392017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao F, Zhang G, Liu Y, He Y, Sheng Y, Sun
X, Du Y and Yang C: Activation of CD44 signaling in leader cells
induced by tumor-associated macrophages drives collective
detachment in luminal breast carcinomas. Cell Death Dis.
13:5402022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pan C, Xu A, Ma X, Yao Y, Zhao Y, Wang C
and Chen C: Research progress of Claudin-low breast cancer. Front
Oncol. 13:12261182023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pommier RM, Sanlaville A, Tonon L,
Kielbassa J, Thomas E, Ferrari A, Sertier AS, Hollande F, Martinez
P, Tissier A, et al: Comprehensive characterization of claudin-low
breast tumors reflects the impact of the cell-of-origin on cancer
evolution. Nature Commun. 11:34312020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gooding AJ and Schiemann WP:
Epithelial-mesenchymal transition programs and cancer stem cell
phenotypes: Mediators of breast cancer therapy resistance. Mol
Cancer Res. 18:1257–1270. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Revi Drug Discov. 11:790–811.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsunuma R, Chan DW, Kim BJ, Singh P, Han
A, Saltzman AB, Cheng C, Lei JT, Wang J, Roberto da Silva L, et al:
DPYSL3 modulates mitosis, migration, and epithelial-to-mesenchymal
transition in claudin-low breast cancer. Proc Natl Acad Sci USA.
115:E11978–E11987. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Porsch H, Mehić M, Olofsson B, Heldin P
and Heldin CH: Platelet-derived growth factor β-receptor,
transforming growth factor β type I receptor, and CD44 protein
modulate each other's signaling and stability. J Biol Chem.
289:19747–19757. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang K and Yi T: Tumor cell stemness in
gastrointestinal cancer: Regulation and targeted therapy. Front Mol
Biosci. 10:12976112023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen C, Zhao S, Karnad A and Freeman JW:
The biology and role of CD44 in cancer progression: Therapeutic
implications. J Hematol Oncol. 11:642018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Flanagan L, Van Weelden K, Ammerman C,
Ethier SP and Welsh J: SUM-159PT cells: A novel estrogen
independent human breast cancer model system. Breast Cancer Res
Treat. 58:193–204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chan DW, Mody CH, Ting NS and Lees-Miller
SP: Purification and characterization of the double-stranded
DNA-activated protein kinase, DNA-PK, from human placenta. Biochem
Cell Biol. 74:67–73. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Curtis C, Shah SP, Chin SF, Turashvili G,
Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et
al: The genomic and transcriptomic architecture of 2,000 breast
tumours reveals novel subgroups. Nature. 486:346–352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Voutsadakis IA: Molecular characteristics
and therapeutic vulnerabilities of claudin-low breast cancers
derived from cell line models. Cancer Genomics Proteomics.
20:5392023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Donovan J and Slingerland J: Transforming
growth factor-beta and breast cancer: Cell cycle arrest by
transforming growth factor-beta and its disruption in cancer.
Breast Cancer Res. 2:116–124. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X, Eichhorn PJA and Thiery JP: TGF-β,
EMT, and resistance to anti-cancer treatment. Semin Cancer Biol.
97:1–11. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fougner C, Bergholtz H, Kuiper R, Norum JH
and Sørlie T: Claudin-low-like mouse mammary tumors show distinct
transcriptomic patterns uncoupled from genomic drivers. Breast
Cancer Res. 21:852019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Barcellos-Hoff MH and Gulley JL: Molecular
pathways and mechanisms of TGFβ in cancer therapy. Clin Cancer Res.
29:2025–2033. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kwei KA, Kung Y, Salari K, Holcomb IN and
Pollack JR: Genomic instability in breast cancer: Pathogenesis and
clinical implications. Mol Oncol. 4:255–266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu H, Tian Y, Yuan X, Wu H, Liu Q, Pestell
RG and Wu K: The role of CD44 in epithelial-mesenchymal transition
and cancer development. Onco Targets Ther. 8:3783–3792.
2015.PubMed/NCBI
|
|
41
|
Li L, Qi L, Liang Z, Song W, Liu Y, Wang
Y, Sun B, Zhang B and Cao W: Transforming growth factor-β1 induces
EMT by the transactivation of epidermal growth factor signaling
through HA/CD44 in lung and breast cancer cells. Int J Mol Med.
36:113–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao Y, Ruan B, Liu W, Wang J, Yang X,
Zhang Z, Li X, Duan J, Zhang F, Ding R, et al: Knockdown of CD44
inhibits the invasion and metastasis of hepatocellular carcinoma
both in vitro and in vivo by reversing epithelial-mesenchymal
transition. Oncotarget. 6:7828–7837. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Deng Z, Fan T, Xiao C, Tian H, Zheng Y, Li
C and He J: TGF-β signaling in health, disease and therapeutics.
Signal Transduct Target Ther. 9:612024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chanmee T, Ontong P, Mochizuki N,
Kongtawelert P, Konno K and Itano N: Excessive hyaluronan
production promotes acquisition of cancer stem cell signatures
through the coordinated regulation of Twist and the transforming
growth factor β (TGF-β)-Snail signaling axis. J Biol Chem.
289:26038–26056. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang K, Liu X, Hao F, Dong A and Chen D:
Targeting TGF-β1 inhibits invasion of anaplastic thyroid carcinoma
cell through SMAD2-dependent S100A4-MMP-2/9 signalling. Am J Transl
Res. 8:2196–2209. 2016.PubMed/NCBI
|
|
46
|
Katsuno Y, Meyer DS, Zhang Z, Shokat KM,
Akhurst RJ, Miyazono K and Derynck R: Chronic TGF-β exposure drives
stabilized EMT, tumor stemness, and cancer drug resistance with
vulnerability to bitopic mTOR inhibition. Sci Signal.
12:eaau85442019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ouhtit A, Madani S, Gupta I,
Shanmuganathan S, Abdraboh ME, Al-Riyami H, Al-Farsi YM and Raj MH:
TGF-β2: A novel target of CD44-promoted breast cancer invasion. J
Cancer. 4:566–572. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park NR, Cha JH, Jang JW, Bae SH, Jang B,
Kim JH, Hur W, Choi JY and Yoon SK: Synergistic effects of CD44 and
TGF-β1 through AKT/GSK-3β/β-catenin signaling during
epithelial-mesenchymal transition in liver cancer cells. Biochem
Biophys Res Commun. 477:568–574. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hao Y, Baker D and Ten Dijke P:
TGF-β-mediated epithelial-mesenchymal transition and cancer
metastasis. Int J Mol Sci. 20:27672019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Voutsadakis IA: EMT Features in
Claudin-Low versus Claudin-Non-Suppressed Breast Cancers and the
Role of Epigenetic Modifications. Curr Issues Mol Biol.
45:6040–6054. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Werden SJ, Sphyris N, Sarkar TR, Paranjape
AN, LaBaff AM, Taube JH, Hollier BG, Ramirez-Peña EQ, Soundararajan
R, den Hollander P, et al: Phosphorylation of serine 367 of FOXC2
by p38 regulates ZEB1 and breast cancer metastasis, without
impacting primary tumor growth. Oncogene. 35:5977–5988. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Taube JH, Herschkowitz JI, Komurov K, Zhou
AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et
al: Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with claudin-low and
metaplastic breast cancer subtypes. Proc Natl Acad Sci USA.
107:15449–15454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang H, Benard O, Kumar V, Griffen A, Ren
Z, Sivalingam K, Wang J, de Simone Benito E, Zhang X, Zhang J, et
al: Wnt/ERK/CDK4/6 activation in the partial EMT state coordinates
mammary cancer stemness with self-renewal and inhibition of
differentiation. Br J Cancer. 133:986–1002. 2025. View Article : Google Scholar : PubMed/NCBI
|