Introduction
Triple-negative breast cancer (TNBC) is the most
aggressive subtype of breast cancer, defined by the lack of
estrogen receptor (ER), progesterone receptor (PR), and human
epidermal growth factor receptor 2 (HER2) expression in tumor
cells. It is characterized by a more aggressive growth pattern and
more enhanced metastatic potential than other subtypes. Despite
advancements in breast cancer therapeutics that have contributed to
improved mortality rates, patients with TNBC continue to face
substantial clinical challenges as they are generally ineligible
for existing targeted therapies (1).
HSP90 is frequently upregulated in cancer, with
expression levels reported to be 2- to 10-fold higher than those in
normal tissues (2). In breast
cancer, its overexpression correlates with adverse
clinicopathological features, including larger tumor size, higher
nuclear grade, and poor prognosis, particularly in early stage
disease (3). Functionally, HSP90
acts as a molecular chaperone that supports the folding and
stability of more than 200 target proteins involved in oncogenic
processes such as cellular proliferation, resistance to apoptosis,
angiogenesis and metastatic progression (4). HSP90 drives tumor progression and
therapy resistance by sustaining pro-survival signaling pathways,
such as JAK/STAT3, AKT and MEK (5,6). Given
its central role in sustaining oncogenic signaling, HSP90 has
emerged as a compelling therapeutic target for TNBC. Therefore,
pharmacological inhibition of HSP90 offers a promising strategy for
simultaneously disrupting multiple oncogenic pathways in this
aggressive breast cancer subtype (7).
Emerging evidence has emphasized the key role of
cancer stem cells (CSCs) in tumor initiation, invasion and
metastasis, particularly in TNBC, where they drive chemoresistance
and metastasis, leading to treatment failure (8). Previous studies have reported that
HSP90 and its co-chaperones are highly implicated in the
maintenance of the CSC phenotype, expression of EMT-related genes,
and stability of pluripotent transcription factors such as Nanog
and Oct4 (9–12). Targeting HSP90 not only disrupts
pro-survival signaling pathways but also impairs the maintenance of
the CSC phenotype, thereby reducing chemoresistance and metastatic
potential, and highlighting its dual role as a promising
therapeutic target in TNBC management.
HSP90 comprises three domains: A N-terminal domain
(NTD) containing an ATP-binding pocket, a middle domain (MD), and a
C-terminal domain (CTD) required for dimerization (13). Most HSP90 inhibitors in clinical
trials target the N-terminus, but none have received FDA approval
owing to serious issues arising from the induction of the heat
shock response (HSR), off-target effects, and organ toxicity
(14,15). NCT-58, a C-terminal HSP90 inhibitor
that interferes with the binding of ATP to its CTD, was developed.
NCT-58 is one of 90 synthesized O-substituted analogs of the B- and
C-ring-truncated scaffolds of deguelin, a naturally occurring
rotenoid. It was observed that NCT-58 exerted antitumor activity in
HER2-positive breast cancer cells by downregulating HER2 and
inducing apoptosis (16). In the
present study, the mechanism underlying the novel effects of NCT-58
on apoptosis, breast cancer stem cells (BCSC)-like properties, and
cell migration in TNBC cells, was explored. Furthermore, the
potential of NCT-58 combined with paclitaxel or doxorubicin was
evaluated, and its potential as a multifaceted therapeutic strategy
for TNBC was highlighted.
Materials and methods
Reagents and antibodies
NCT-58 was synthesized according to a previously
established protocol (17). For all
experiments, NCT-58 was prepared as a stock solution (10 mM) in
dimethyl sulfoxide (DMSO) and vehicle controls contained the
corresponding final concentration of DMSO (0.1% v/v). Chemicals,
including propidium iodide (PI), DMSO and Triton X-100, were
purchased from MilliporeSigma. Protease and phosphatase inhibitor
tablets were obtained from Roche Applied Science. The primary
antibodies used for western blotting and immunocytochemistry were
specific to vimentin, STAT3, phospho-STAT3 (Tyr705), AKT,
phospho-AKT (Ser473), MEK, phospho-MEK (Ser218/222), PARP, cleaved
PARP, cleaved caspase-3, and −7, heat shock transcription factor-1
(HSF)-1, HSP70, cyclin D1, survivin, F-actin (Texas
Red™-X Phalloidin) and GAPDH. Secondary antibodies
included horseradish peroxidase (HRP)-conjugated anti-mouse and
anti-rabbit IgG and Alexa Fluor 488/594-labeled anti-mouse and
anti-rabbit IgG.
Cell culture
The TNBC cell lines MDA-MB-231 (PerkinElmer, Inc.),
Hs578T (American Type Culture Collection; ATCC), BT549 and 4T1
(Japanese Collection of Research Bioresources Cell Bank), and the
cell line 293 (Korean Cell line Bank) were maintained in MEM or
RPMI-1640 supplemented with 10% FBS and 100 U/ml
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
The normal human mammary epithelial cell line MCF10A (ATCC) was
cultured in Mammary Epithelial Cell Growth Basal Medium, including
hEGF (20 ng/ml), insulin (10 µg/ml), hydrocortisone (0.5 µg/ml) and
bovine pituitary extract (50 µg/ml) (SingleQuotsTM Kit;
Lonza Group, Ltd.) containing streptomycin-penicillin (100 U/ml).
The cells were then incubated at 37°C in an atmosphere containing
5% CO2.
Cell viability assay
Cell viability was measured using the CellTiter
96® Aqueous One Solution Cell Proliferation Assay [MTS,
3-(4,
5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]
(Promega Corporation) according to the manufacturer's instructions.
The MTS reagent, which is soluble in cell culture medium, was added
directly to the cells. The quantity of purple formazan product was
determined by measuring the absorbance at 490 nm using a microplate
reader (Agilent BioTek 800 TS; Agilent Technologies, Inc.).
Percentage cell viability was calculated relative to that of the
untreated control (set at 100%). IC50 values were
determined by fitting sigmoidal dose-response curves using
nonlinear regression analysis in GraphPad Prism 9.0
(Dotmatics).
Cell cycle and Sub-G1 analysis
Cell cycle distribution and sub-G1 populations were
analyzed using flow cytometry. Cells were harvested at 70–80%
confluence, washed twice with cold phosphate-buffered saline (PBS),
and fixed by dropwise addition of pre-chilled 95% ethanol
containing 0.5% Tween-20. The samples were then incubated at −20°C
overnight to ensure complete fixation. Following fixation, cells
were centrifuged (7,160 × g), resuspended in staining buffer
containing propidium iodide (50 µg/ml) and RNase A (50 µg/ml), and
incubated for 30 min at room temperature in the dark. DNA content
was assessed using a Beckman Coulter Expo flow cytometer (Beckman
Coulter, Inc.) and data were analyzed using FlowJo software v10.10
(BD Biosciences).
Aldefluor-positive assay,
CD44high/CD24low staining
To evaluate ALDH1 enzymatic activity, cells were
treated using the Aldefluor™ assay kit (Stemcell
Technologies, Inc.) in accordance with the supplier's protocol.
Briefly, cells (0.5×106) were suspended in assay buffer
containing 1 µM BODIPY-amino-acet-aldehyde and incubated at 37°C
for 45 min. Diethyl-amino-benzaldehyde (50 mM) was added to the
control sample to inhibit ALDH1 activity and establish gating
parameters for the Aldefluor-positive population during flow
cytometric analysis. To identify stem-like cell populations
exhibiting the CD44high/CD24low phenotype,
cells were incubated at 4°C for 30 min with FITC-labeled anti-CD24
and PE-labeled anti-CD44 antibodies (1:50; BD Biosciences). FITC-
and PE-conjugated isotype-matched mouse IgG antibodies were used as
controls for non-specific staining. The labeled cells were
subsequently subjected to flow cytometry.
Mammosphere formation assay
To assess sphere-forming capacity, 4T1 cells were
seeded at a density of 3×105 cells/ml in ultralow
attachment plates (Corning, Inc.) and cultured in HuMEC basal
serum-free medium (Gibco; Thermo Fisher Scientific, Inc.),
supplemented with B27 (1:50; Invitrogen; Thermo Fisher Scientific,
Inc.), 20 ng/ml basic fibroblast growth factor (bFGF;
MilliporeSigma), 20 ng/ml mouse epidermal growth factor (EGF,
MilliporeSigma), 4 µg/ml heparin, 1% antibiotic-antimycotic
solution, and 15 µg/ml gentamycin. The number and volume of
mammospheres were determined using an Olympus CKX53 inverted
microscope, and the sphere size was quantified using ImageJ
software (v1.54; National Institutes of Health). Mammosphere
volumes were calculated using the formula:
Volume=4/3×3.14×r3.
C-terminal HSP90 inhibition assay
A C-terminal HSP90α Inhibitor Screening Kit (BPS
Bioscience) was used to evaluate inhibition of the interaction
between the C-terminal region of HSP90α and its co-chaperone
peptidylprolyl isomerase D (PPID) by HSP90 inhibitors, as
previously described (11,18). The assay was conducted in 384-well
Optiplates (PerkinElmer, Inc.) and luminescence signals were
detected using the AlphaScreen® technology on a
Varioskan LUX™ microplate reader (Thermo Fisher
Scientific, Inc.).
N-terminal HSP90 binding activity
assay
To assess compound binding affinity to the NTD of
HSP90α, a fluorescence-based competitive binding assay (HSP90α
N-terminal Assay Kit; BPS Bioscience) was conducted in accordance
with the manufacturer's guidelines. Test compounds (ranging from
0–1,000 nM in DMSO) were mixed with a reaction solution comprising
FITC-tagged geldanamycin (100 nM) and recombinant HSP90α protein
(17 ng/µl), followed by incubation at room temperature for 3 h.
Fluorescence intensity was then measured at an excitation
wavelength of 485 nm and emission at 530 nm using a SpectraMax
Gemini EM microplate reader (Molecular Devices, LLC) to assess the
binding activity to the NTD of HSP90.
Wound healing assay
Cells were plated in 96-well ImageLock plates (Essen
Bioscience) and grown until they reached ~90% confluency. A uniform
wound was created using a 96-pin WoundMaker, followed by washing
with PBS to eliminate detached cells. Immediately after scratch
formation, cells were exposed to varying concentrations of NCT-58.
Cell migration was monitored in real time by capturing images
hourly for 25 to 50 h using an IncuCyte ZOOM Live-Cell Imaging
System (Essen Bioscience). The progression of wound closure was
quantified using the IncuCyte™ Scratch Wound Cell Migration
analysis software (Essen BioScience).
Immunoblot analysis
Membranes were incubated overnight at 4°C with
primary antibodies diluted in 5% bovine serum albumin (BSA) (Thermo
Fisher Scientific, Inc.), targeting the following proteins: AKT
(1:2,000), phospho-AKT (1:2,000), MEK (1:2,000), phospho-MEK
(1:2,000), STAT3 (1:2,000), phospho-STAT3 (Tyr705, 1:2,000),
survivin (1:2,000), cyclin D1 (1:2,000), PARP (1:2,000), cleaved
PARP (1:2,000), cleaved caspase-3 (1:1,000), cleaved caspase-7
(1:1,000), and GAPDH (1:3,000). After washing, the membranes were
incubated with HRP-conjugated secondary antibodies (anti-rabbit or
anti-mouse IgG, 1:3,000-1:5,000). Protein bands were visualized
using an enhanced chemiluminescence (ECL) detection system (Thermo
Fisher Scientific, Inc.).
Cells were lysed in radioimmunoprecipitation assay
buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium
deoxycholate and 0.1% SDS) supplemented with protease and
phosphatase inhibitor cocktails (Roche Diagnostics). The lysates
were incubated on ice for 30 min and then centrifuged at 25,200 × g
for 15 min at 4°C to remove insoluble debris. The supernatant was
collected, and protein concentrations were determined using a BCA
assay (Thermo Fisher Scientific, Inc.). Equal amounts of protein
(20 µg per lane) were mixed with the sample buffer, boiled for 10
min, separated using 4–15% gradient SDS-PAGE, and transferred onto
PVDF membranes (MilliporeSigma). Membranes were blocked with 5%
skim milk for 1 h at room temperature and subsequently incubated
overnight at 4°C with primary antibodies diluted in 5% BSA [AKT
(1:2,000), phospho-AKT (1:2,000), MEK (1:2,000), phospho-MEK
(1:2,000), STAT3 (1:2,000), phospho-STAT3 (Y705; 1:2,000), survivin
(1:2,000), cyclin D1 (1:2,000), PARP (1:2,000), cleaved-PARP
(1:2,000), cleaved-caspase-3 (1:1,000), cleaved-caspase-7
(1:1,000), and GAPDH (1:3,000)]. The catalog numbers and suppliers
of antibodies are included in Table
SI. After washing, the membranes were incubated with
HRP-conjugated secondary antibodies (1:3,000-1:5,000) for 1 h at
room temperature. Protein bands were detected using an Enhanced
Chemiluminescence Kit (Thermo Fisher Scientific, Inc.), quantified
using AlphaEaseFC software (Alpha Innotech Corporation), and
normalized to GAPDH as the internal loading control.
Immunocytochemistry
Cells cultured on 8-well chamber slides (BD
Biosciences) were fixed with 4% paraformaldehyde at 4°C for 2 h,
rinsed with PBS, and permeabilized with 0.2% Triton X-100 for 10
min. The cells were then incubated overnight at 4°C with primary
antibodies against HSP70 (1:100), HSF-1 (1:100), vimentin (1:200),
or F-actin (1:200) diluted in an antibody diluent (Dako; Agilent
Technologies, Inc.). After washing, the cells were treated with
fluorescence-conjugated secondary antibodies (Alexa
Fluor® 488 or 594). Nuclei were counterstained with DAPI
(0.4 µg/ml) and mounted using ProLong™ Gold Antifade
Reagent (Thermo Fisher Scientific, Inc.). Fluorescence images were
captured using a Carl Zeiss confocal microscope, and the signal
intensity was quantified using the intensity profile tool.
Immunofluorescence images were acquired under identical sensitivity
settings, including exposure time, laser power, and gain, across
the control and treated groups. For the nuclear protein HSF1,
nuclei were segmented using DAPI staining in ImageJ. The integrated
density of HSF1 within each nucleus was normalized to the
integrated density of the corresponding DAPI signal (HSF1/DAPI
ratio) to correct for nuclear size and DNA content. For cytoplasmic
proteins HSP70, corrected total cell fluorescence (CTCF) was
calculated as: CTCF=Integrated density-(Area × Background mean
intensity) with background values obtained from four cell-free
regions per image. Quantification was performed using at least 10
cells from a minimum of three independent fields per condition.
Synergy assessment
To evaluate whether two compounds interact
synergistically, additively, or antagonistically, the combination
index (CI) was determined using CompuSyn® software
(ComboSyn, Inc.; http://www.combosyn.com). A CI value of less than,
equal to, or greater than 1 denotes synergism, additive effects, or
antagonism, respectively. The fraction affected (Fa) represents the
proportion of phenotypic inhibition resulting from treatment with a
compound. Fa=percent inhibition of cell viability/100. The Fa-CI
plot was generated for each group treated with more than one
compound, and the corresponding heat map illustrated the percentage
inhibition of cell viability under the combination conditions. The
CI values at specific effect levels were then used to construct
isobolograms, where data points below, on, or above the line of
additivity indicated synergistic, additive, or antagonistic
effects, respectively.
Molecular docking analysis
In silico molecular docking was conducted
using open-access AutoDock Vina-based platforms including DockThor
(https://www.dockthor.lncc.br) and
CB-Dock2 (https://cadd.labshare.cn/cb-dock2). Upon completion of
the docking simulations, both 2D and 3D structures of the
protein-ligand complexes were visualized and analyzed using UCSF
Chimera 1.16 and BIOVIA Discovery Studio 2021. The predicted
binding affinity for the interaction between NCT-58 and the CTD of
hHSP90α (PDB ID: 7RY1) was estimated to be-9.5 kcal/mol. The
binding site and key interactions were analyzed to identify
hydrogen bonds, hydrophobic contacts, and the overall binding
pocket conformation.
Statistical analysis
Statistical analyses were performed using the
GraphPad Prism 9.0 (GraphPad Software; Dotmatics). Data are
presented as the mean ± SEM from at least three independent
experiments. Comparisons between groups were conducted using
unpaired Student's t-test or one-/two-way ANOVA, as appropriate.
For two-way ANOVA, Bonferroni's post-hoc test was used to evaluate
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
NCT-58 exerts an anti-proliferative
effect without triggering the HSR
The anti-proliferative effect of NCT-58 in the human
TNBC cell lines MDA-MB-231, BT549 and Hs578T, and the murine
mammary carcinoma line 4T1, were first evaluated. TNBC cell
viability was dose-dependently reduced in the presence of NCT-58
(0–20 µM, 72 h) in vitro (P<0.05; Figs. 1A and S1; Table
SII). The estimated IC50 values for NCT-58 were
6.701, 4.685, 7.041 and 2.922 µM in MDA-MB-231, BT549, Hs578T and
4T1, respectively. Based on IC50 values, a concentration
range of 2–10 µM in MDA-MB-231 and 1–5 µM in 4T1 cells was selected
for further experiments including apoptosis, western blot analysis,
and cancer stem cell-like characterization. Apoptosis was
significantly increased in the presence of NCT-58 at 5 µM in
MDA-MB-231 cells and at 2 µM in 4T1 cells (P<0.05; Fig. 1B) for 72 h, as evidenced by
increased sub-G1 accumulation. Apoptosis induction was confirmed
using caspase-3 and caspase-7 activation, concomitant with
increased cleavage of PARP, a downstream substrate of caspase-3
(P<0.01; Fig. 1C). Further
quantitative analysis confirmed an increased ratio of cleaved to
total forms of caspase-3, caspase-7 and PARP following NCT-58
treatment (P<0.05; Fig.
S2A-C).
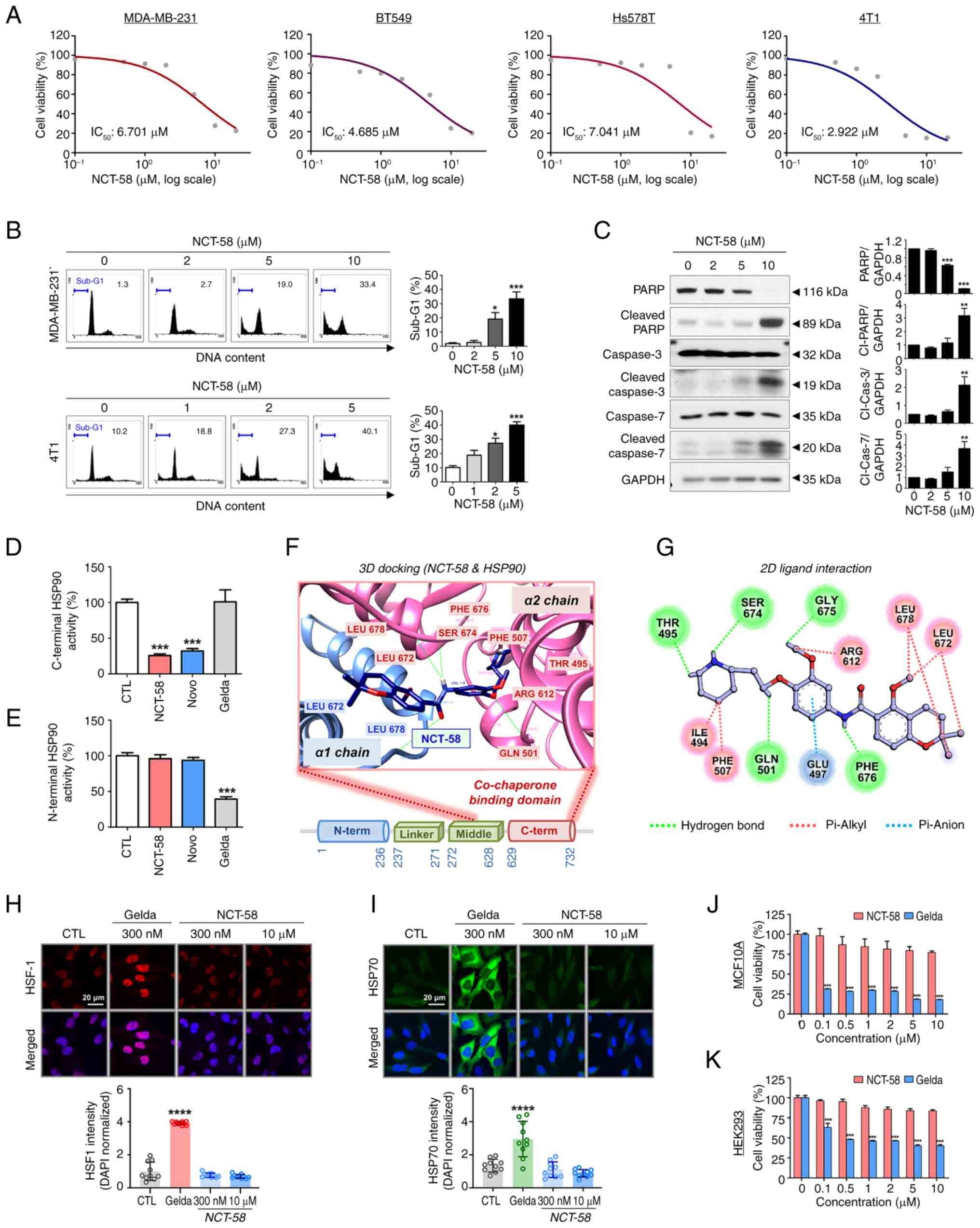 | Figure 1.NCT-58 exerts anti-proliferative
effect in TNBC cells by targeting the C-terminal domain of HSP90.
(A) Four TNBC cell lines, MDA-MB-231, BT549, Hs578T and 4T1 cells
were treated with various concentrations of NCT-58 (0–20 µM) for 72
h. Cell viability was assessed using MTS assay, and IC50
values were calculated using non-linear regression with a sigmoidal
dose-response curve. (B) MDA-MB-231 and 4T1 cells were treated at
the indicated concentrations of NCT-58 (0–10 µM) for 72 h.
Apoptosis was determined through sub-G1-DNA analysis using flow
cytometry. (C) Immunoblot analyses of PARP, cleaved-PARP,
caspase-3, cleaved caspase-3, caspase-7 and cleaved caspase-7
protein expression in MDA-MB-231 cells after treatment with NCT-58
(0–10 µM, 72 h). GAPDH was used as an internal loading control.
Quantitative graphs of these protein levels. The results are
presented as the mean ± SEM of at least three independent
experiments and analyzed using one-way ANOVA followed by
Bonferroni's post hoc test. (D) Effect of NCT-58 on C-terminal
HSP90 binding activity. The inhibitory effect of HSP90 inhibitors
(NCT-58, novobiocin or geldanamycin, 500 µM) on the interaction
between HSP90α (C-terminal) and its co-chaperone peptidylprolyl
isomerase was determined using an HSP90α (C-terminal) inhibitor
screening assay. (E) Influence of NCT-58 on N-terminal HSP90
binding activity. The competitive HSP90α binding activity of HSP90
inhibitors (NCT-58, novobiocin or geldanamycin, 1 µM) with
FITC-labeled geldanamycin was determined using an HSP90α N-terminal
domain assay. (F and G) Molecular docking analysis of NCT-58
binding to the CTD of HSP90 (PDB ID: 7RY1). (F) The binding pose of
NCT-58 at the dimerization interface is displayed as a
space-filling model. The α1 chain of HSP90 is rendered in a blue
ribbon, and the α2 chain in a pink ribbon. Connolly surface
representation of the HSP90 CTD, with NCT-58 modeled within the
binding interface (docking score=−9.5). (G) A 2D interaction
diagram of NCT-58 with key residues in the HSP90 CTD. Hydrogen
bonds and π-anion interactions are indicated by dashed green and
blue lines, respectively. (H and I) Comparison of the effects of
NCT-58 and the N-terminal HSP90 inhibitor geldanamycin on HSF-1 and
HSP70 expression. MDA-MB-231 cells were treated with NCT-58 (300 nM
and 10 µM) or geldanamycin (300 nM) for 24 h. Cells were
immuno-stained for HSF-1 (red, H) or HSP70 (green, I) using DAPI
(nuclei, blue). Images were acquired using a confocal microscope,
and quantification of immunofluorescence intensity was performed
using ImageJ software. Nuclear HSF1 intensity was expressed as the
HSF1/DAPI ratio, and cytoplasmic HSP70 intensity was expressed as
corrected total cell fluorescence normalized to DAPI. (J and K)
Effect of NCT-58 and geldanamycin on cytotoxicity in non-malignant
cells. Normal human mammary epithelial MCF10A (J) and 293 (K) cells
were treated with various concentrations (0.1–10 µM) of NCT-58 or
geldanamycin for 72 h. Cell viability was determined using MTS
assay (***P<0.001). *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. TNBC, triple-negative breast
cancer; Gelda, geldanamycin; Novo, novobiocin; CTD, C-terminal
domain. |
The C-terminal HSP90 binding activity of NCT-58 was
analyzed using the co-chaperone peptidylprolyl isomerase D (PPID),
which exhibits high-affinity ligand binding activity to the
C-terminus of HSP90. NCT-58 significantly inhibited C-terminal
HSP90 activity, as evidenced by the marked inhibition of the
specific interaction between PPID and the C-terminus of HSP90
(P<0.001; Fig. 1D).
Similar results were observed for novobiocin, a potent C-terminal
HSP90 inhibitor (19), whereas
geldanamycin did not affect this interaction. Furthermore, the
competitive HSP90 binding assay with fluorescence-labeled
geldanamycin did not show N-terminal HSP90 binding activity of
NCT-58 (Fig. 1E). Docking studies
using the crystal structure of HSP90 (PDB ID: 7RY1), which includes
the middle and CTD, revealed that NCT-58 precisely fits into the
dimerization interface of the CTD, which is a critical region
required for chaperone function (Fig.
1F). The interaction between NCT-58 and the HSP90 CTD was
extensively stabilized by five hydrogen bonds with the key residues
Thr495, Gln501, Ser674, Gly675 and Phe676. In addition, a π-anion
interaction was observed between NCT-58 and Glu497 of HSP90,
further enhancing binding stability (Fig. 1G). These findings highlight how
NCT-58 specifically occupies the CTD binding pocket to disrupt
dimerization and impair HSP90 function.
Subsequently, it was assessed whether NCT-58 induces
the HSR, a limitation of classical N-terminal HSP90 inhibitors.
Geldanamycin, the first and most extensively studied N-terminal
HSP90 inhibitor, was selected as the reference compound and was
used in all experiments requiring an N-terminal HSP90 inhibitor
(20,21). The N-terminal HSP90 inhibitor
induces the phosphorylation and trimerization of heat shock
factor-1 (HSF-1), promoting its nuclear translocation and
subsequent upregulation of heat shock proteins (HSPs), such as
HSP70 and HSP90, which support pro-survival signaling (15,22).
MDA-MB-231 cells were treated with geldanamycin (300 nM), a potent
N-terminal HSP90 ATP binding inhibitor, and NCT-58 (300 nM and 10
µM), followed by the assessment of HSF-1 and HSP70 expression.
Confocal imaging and intensity analysis revealed no increase in
HSF-1 levels upon exposure to NCT-58, whereas geldanamycin
considerably increased the accumulation of HSF-1 in the nuclei of
MDA-MB-231 cells (Fig. 1H). This
finding was further supported by the fact that geldanamycin induced
the subsequent upregulation of cytoplasmic HSP70, whereas NCT-58
did not elicit this phenomenon (Fig.
1I).
Accumulating preclinical and clinical evidence has
highlighted the significant off-target toxicity of geldanamycin
derivatives, which causes undesirable effects in normal tissues
(23). Notably, NCT-58 exhibited
minimal cytotoxicity at 10 µM in normal mammary gland epithelial
MCF10A cells and 293 cells, while geldanamycin caused significant
cytotoxicity in both normal cell lines at 100 nM, a 1,000-fold
lower concentration (P<0.001; Fig. 1J and K).
NCT-58 simultaneously targets
pro-survival HSP90 client proteins in TNBC cells
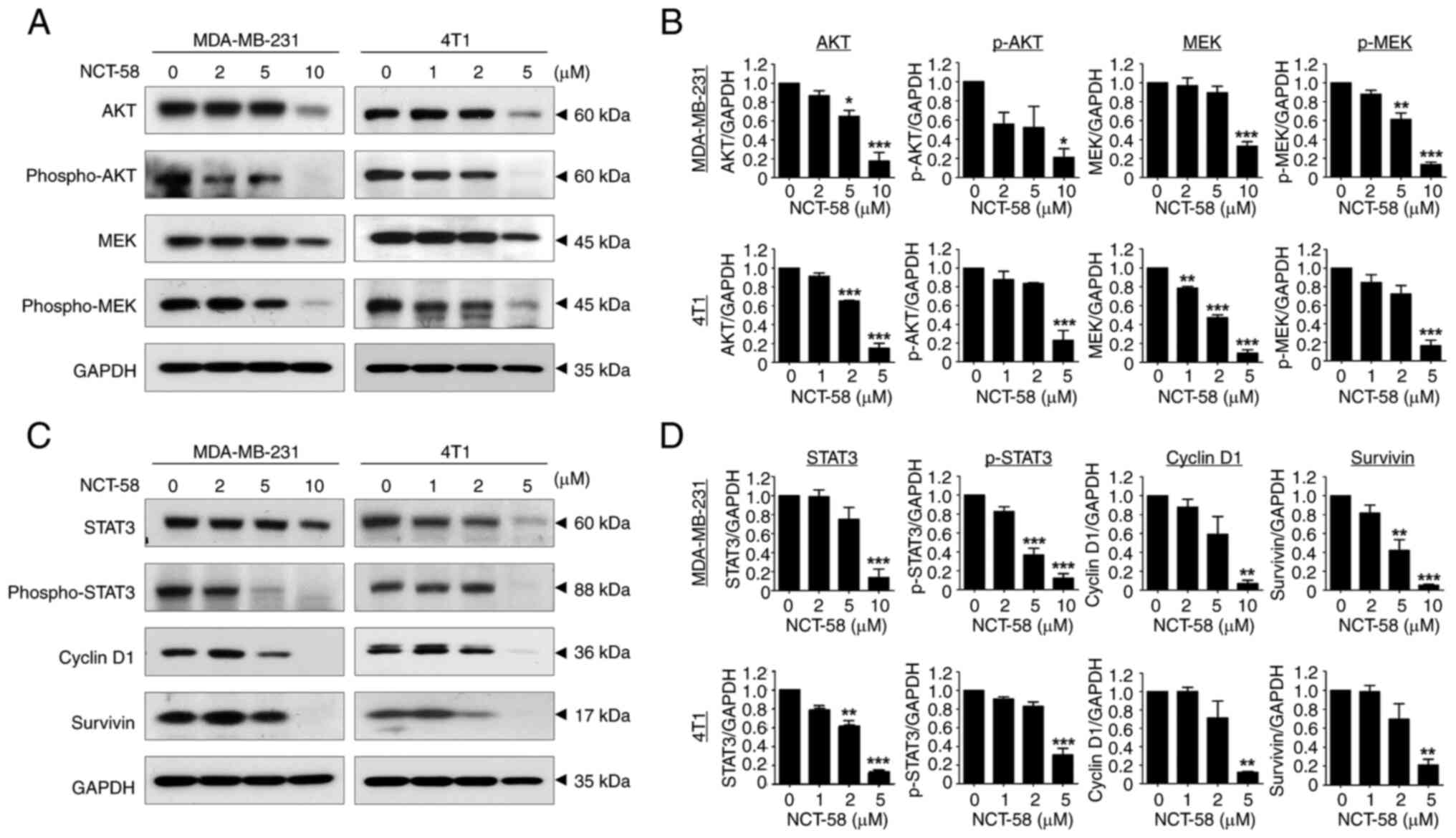
It was observed that NCT-58 significantly
downregulated the expression of AKT and MEK and reduced their
phosphorylation in MDA-MB-231 and 4T1 cells (P<0.05; Fig. 2A and B; Fig. S3A-F), suggesting that the
simultaneous inhibition of these two major survival pathways could
enhance the antiproliferative effect against TNBC cells.
STAT3 is activated by phosphorylation of its
tyrosine residue (Tyr705) in response to stimulation by cytokines
and growth factor receptors, and then translocates to the nucleus
to promote the expression of survival factors such as cyclin D1,
survivin, Bcl-2 and Bcl-xL (24).
Therefore, inhibition of HSP90 may suppress the expression of
several oncoproteins promoted by STAT3. It was observed that NCT-58
significantly impaired the expression and phosphorylation at Tyr705
of STAT3 in MDA-MB-231 and 4T1 cells, accompanied by the
downregulation of survivin and cyclin D1 (P<0.01; Fig. 2C and D).
NCT-58 hampers cancer stem-like
characteristics and migratory ability in TNBC cells
BCSCs confer resistance to chemotherapy during TNBC
treatment and are thought to be a prerequisite for invasion and
metastasis (8). It was examined
whether NCT-58 affects BCSC-like properties, ALDH1 activity,
CD44high/CD24low stem-like populations, and
mammosphere formation. Treatment with NCT-58 significantly
suppressed ALDH1 activity in MDA-MB-231 (P<0.01; Fig. 3A) and 4T1 cells (P<0.01; Fig. 3B). The
CD44high/CD24low phenotype is associated with
a higher incidence of relapse or distant metastasis (25). A significant reduction in the
CD44high/CD24low-population was observed in
MDA-MB-231 cells after NCT-58 challenge (P<0.001; Fig. 3C). Mammosphere 3D culture is a
useful tool for assessing the tumor-like properties of BCSCs
capable of propagating mammary and progenitor cells in vitro
(26,27). The assay revealed that NCT-58
attenuated the mammosphere-forming ability, as evidenced by
significant reductions in the number and volume of mammospheres
derived from 4T1 cells (P<0.001; Fig. 3D).
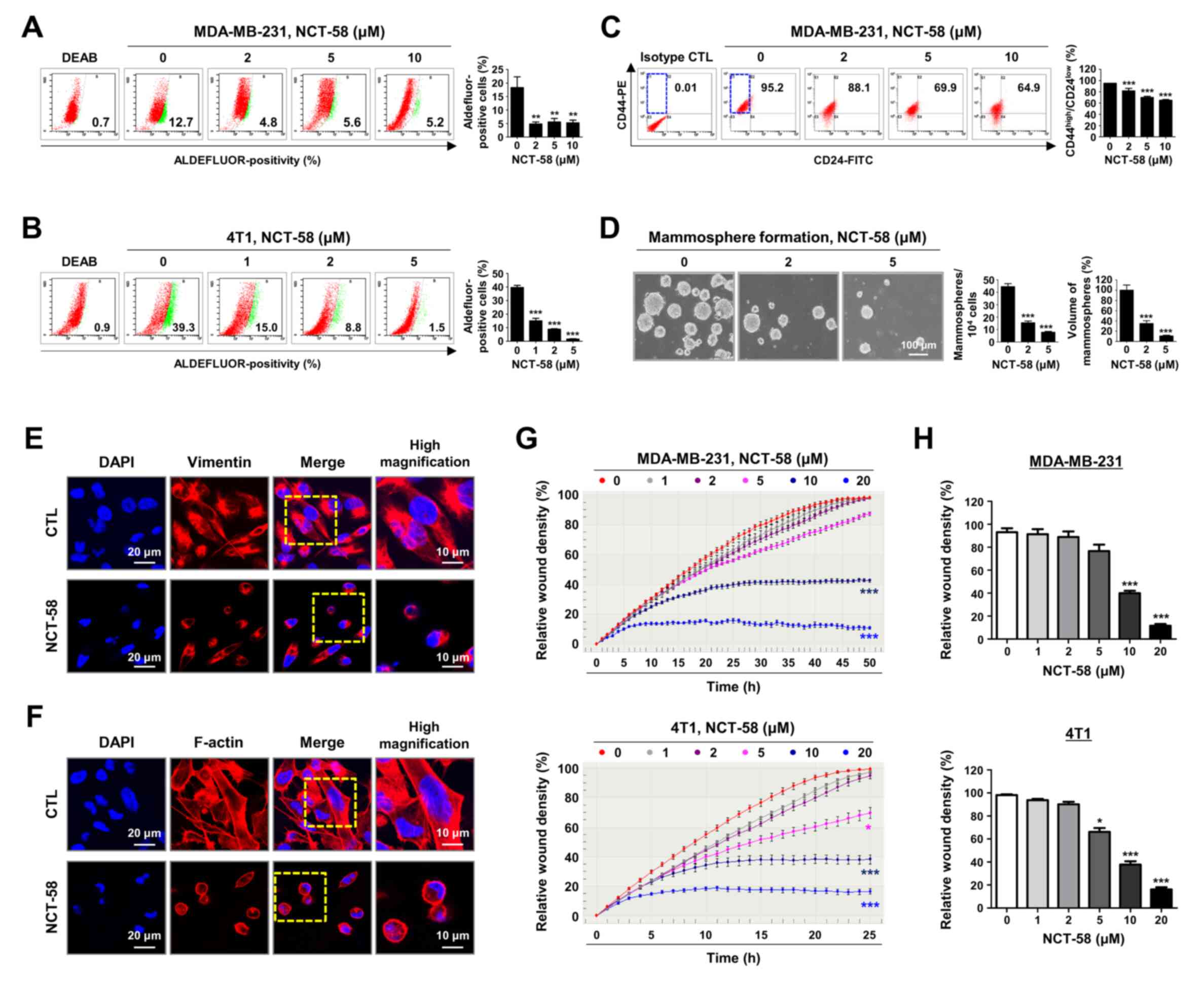 | Figure 3.NCT-58 suppresses cancer stem-like
properties and migratory activity in TNBC cells. (A and B) Effect
of NCT-58 (0–10 µM, 72 h) on ALDH1 activity in (A) MDA-MB-231 and
(B) 4T1 cells. The specific inhibitor diethyl-amino-benzaldehyde
was used to define the Aldefluor-positive population.
Aldefluor-positive cells were quantified using flow cytometry
(right panel). (C) Effect of NCT-58 (0–10 µM, 72 h) on the
CD44high/CD24low stem-like population in
MDA-MB-231 cells. The CD44high/CD24low
population was quantified through flow cytometry. (D) Effect of
NCT-58 on mammosphere formation in vitro. 4T1 cells
(5×104 cells/ml) were cultured in ultralow attachment
plates in the presence or absence of NCT-58 (2–5 µM, 5 days). The
number and volume of mammospheres were quantified using optical
microscopy. (E and F) After exposure to NCT-58 (10 µM) for 48 h,
MDA-MB-231 cells were immuno-stained for vimentin (1:100, E) and
F-actin (1:100, Texas Red-X phalloidin, F) with DAPI nuclear stain
(blue). (G and H) Effect of NCT-58 on TNBC cell migration. (G)
After exposure to NCT-58 (0–20 µM) in MDA-MB-231 and 4T1 cells,
kinetic analysis of cell migration was determined using the
IncuCyte™ Live-Cell Imaging System and quantified for
the indicated time duration. The kinetic graphs of cell migration
represent the relative wound density. (H) Relative wound density
(%) in MDA-MB-231 cells at 50 h and in 4T1 cells at 25 h,
respectively. The results are presented as the mean ± SEM of at
least three independent experiments and analyzed using one- or
two-way ANOVA followed by Bonferroni's post hoc test.
*P<0.05, **P<0.01 and ***P<0.001. TNBC,
triple-negative breast cancer. |
Actin and vimentin are major HSP90 target proteins
and cytoskeletal components that play important roles in cell
interaction, motility, invasiveness and adhesion (13,28).
Vimentin intermediate filament is a hallmark of EMT, and appears to
be involved in TNBC metastasis (29). To explore whether NCT-58 affected
the expression of HSP90 target cytoskeletons, the spatial
distribution of vimentin and filamentous-actin was observed using
immunofluorescence analysis. NCT-58 treatment resulted in vimentin
and F-actin reorganization and the disruption of filament networks,
resulting in concomitant cytoplasmic contraction and cellular
rounding (Fig. 3E and F). Kinetic
analysis of cell migration revealed that NCT-58 significantly and
dose-dependently reduced the migration of both MDA-MB-231 and 4T1
cells in vitro (P<0.05; Fig.
3G and H), indicating that the collapse of filament dynamics
may contribute to the suppression of migratory ability.
NCT-58 significantly enhances the
anti-proliferative effects of paclitaxel and doxorubicin in a
synergistic manner
It was investigated whether NCT-58 could enhance the
sensitivity of cells to the conventional chemotherapeutic agents
paclitaxel and doxorubicin. To assess the synergistic effects of
NCT-58 on paclitaxel- or doxorubicin-induced suppression of cell
viability, MDA-MB-231 cells were treated with NCT-58 (0–20 µM)
combined with paclitaxel (0–2 µM) or doxorubicin (0–3 µM) for 72 h.
The interaction between NCT-58 and paclitaxel or doxorubicin was
evaluated by isobologram analysis, which uses a graphical
representation (isobole) to assess drug effects. Additive
interactions are represented as points along the line connecting
the effective concentration 50 (EC50) values of each
drug, whereas the co-treatment of NCT-58 with paclitaxel or
doxorubicin demonstrated synergistic effects, as indicated by
points positioned below the isobole (Fig. 4A and D). The heat map intensity
represents the relative cell viability of drug combination effects
compared with the control treatment with DMSO alone (Fig. 4B and E). The CI further confirmed
these synergistic interactions, highlighting the potential of
NCT-58 to enhance the efficacy of conventional cytotoxic agents
(Fig. 4C and F).
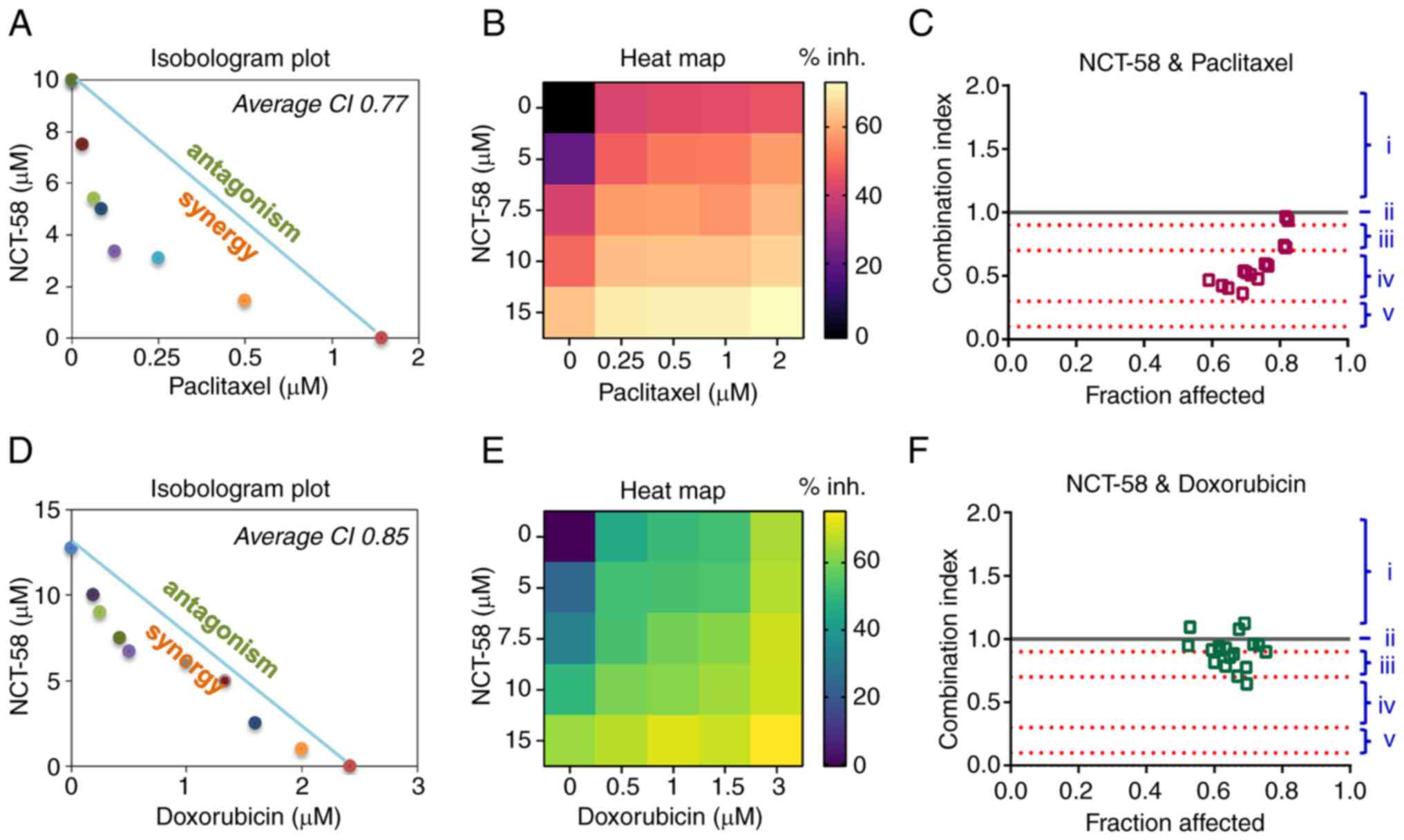 | Figure 4.Synergistic effects of combining
NCT-58 with paclitaxel or doxorubicin in triple-negative breast
cancer cells. (A-F) MDA-MB-231 cells were treated with the
indicated concentrations of NCT-58 (0–15 µM) and paclitaxel (0–2
µM, A-C) or doxorubicin (0–3 µM, D-F) for 72 h and cell viability
was assessed using MTS assay. The isobologram plot, heat map (%
inh., Inhibition rate), and CI were analyzed to assess the
drug-drug synergy of various dose combinations of NCT-58 with
paclitaxel or doxorubicin. Isobologram plot and average CI values
were generated using the CompuSyn software to quantify drug
interactions, where CI<1 indicates synergism, CI=1 indicates an
additive effect, and CI>1 indicates antagonism. (i, antagonism;
ii, addictive effect; iii, moderate synergism; iv, synergism; and
v, strong synergism). The heat map depicts relative cell viability
compared with the DMSO control. CI, combination index. |
Discussion
In the present study, it was demonstrated that
NCT-58, a C-terminal HSP90 inhibitor, exerts multifaceted antitumor
effects in TNBC. Classical HSP90 inhibitors have not been
successful owing to concerns regarding off-target toxicity and the
induction of HSR, which leads to cytoprotective responses (14,15).
N-terminal HSP90 inhibitors trigger HSF-1 activation and
translocation into the nucleus, resulting in elevated transcription
of HSPs such as HSP70 and HSP90, which promote pro-survival
signaling (15,22). NCT-58 effectively suppressed cell
viability and induced apoptosis without triggering the HSR, which
is a major limitation of current HSP90 inhibitors. Although
geldanamycin, a N-terminal HSP90 inhibitor, markedly induced HSF-1
nuclear translocation and the subsequent upregulation of HSP70,
NCT-58 did not produce these effects. This finding suggests that
inhibiting the CTD prevents a key pitfall of numerous existing
HSP90 inhibitors, namely the cytoprotective feedback loop, which
diminishes therapeutic efficacy and enhances cytotoxicity in normal
cells. The present data revealed the minimal cytotoxic effects of
NCT-58 in normal mammary epithelial (MCF10A) and 293 cells, whereas
geldanamycin significantly suppressed cell viability at
considerably lower concentrations.
Mechanistically, NCT-58 simultaneously disrupted the
functions of multiple HSP90 client proteins. By blocking HSP90
C-terminal dimerization, NCT-58 caused pronounced downregulation of
AKT, MEK and STAT3, which are the major pro-survival pathways in
TNBC. Previous genomic profiling revealed STAT3 overexpression and
aberrant activation in TNBC, highlighting its potential as both a
molecular target and biomarker (24,30).
STAT3 inhibition is associated with reduced transcription of
critical survival genes such as survivin and cyclin D1. Consistent
with this observation, NCT-58 treatment decreased STAT3 activation
and its downstream targets, thereby enhancing apoptosis and
diminishing proliferative capacity. A particularly notable finding
was the ability of NCT-58 to target the CSC-like properties of TNBC
cells. The CSC subpopulation, typically characterized by elevated
ALDH1 activity and a CD44high/CD24low
phenotype, plays a pivotal role in tumor initiation and recurrence
(27,31). These cells exhibit robust
self-renewal abilities and multipotency, which contribute to their
resistance to conventional chemotherapy and radiotherapy (32). ALDH is a detoxifying enzyme and its
ability to convert retinol to retinoic acid is implicated in cancer
cell proliferation and CSC differentiation (33). Cyclophosphamide, a commonly used
chemotherapy agent for TNBC, undergoes inactivation through
ALDH-mediated oxidation during its metabolic process (34,35).
Consequently, patients with elevated ALDH expression often
experience relatively poor clinical outcomes and exhibit resistance
to alkylating agents such as cyclophosphamide and ifosfamide
(31,35,36).
Therefore, strategies targeting ALDH activity show significant
promise for the effective eradication of CSCs and the prevention of
chemoresistance and recurrence. Treatment with NCT-58 significantly
reduced ALDH1 activity and decreased the percentage of
CD44high/CD24low cells, ultimately impairing
mammosphere formation in 3D culture. These data align with those of
previous reports implicating HSP90 in the maintenance of CSC
phenotypes and highlight the importance of targeting CSCs to
minimize relapse and metastasis. Accordingly, the 4T1 cell line was
employed, which exhibits highly aggressive and stem cell-like
characteristics, and is widely recognized as a preclinical model
for evaluating cancer stemness and metastatic potential (37–39).
In the in vitro experiments of the present study, 4T1 cells
clearly demonstrated that NCT-58 reduces CSC-related traits such as
ALDH1 activity, thereby supporting the results observed in human
MDA-MB-231 cells. By simultaneously affecting both bulk tumor cells
and CSC-like populations, NCT-58 can potentially overcome the major
mechanisms of resistance in TNBC.
NCT-58 also inhibited cell migration, likely by
modulating the HSP90 client proteins, vimentin and F-actin. During
EMT, vimentin orchestrates cytoskeletal organization and
cell-matrix interactions, enabling cancer cells to remodel the
extracellular matrix and adopt a more flexible and invasive
phenotype (40). Consequently,
vimentin-enriched cells are more elastic and motile, facilitating
tissue invasion, vascular or lymphatic entry, and survival under
mechanical stress (41,42). By disrupting vimentin and F-actin
filament dynamics, NCT-58 induced cytoskeletal reorganization and
collapse, thereby reducing the migratory capacity of TNBC
cells.
In a previous investigation by the authors, it was
demonstrated that NCT-58 overcomes trastuzumab resistance in
HER2-positive breast cancer by destabilizing HER2 family proteins,
including HER2, HER3, Akt and truncated p95HER2. Moreover, NCT-58
downregulated the RAS/RAF/MEK/ERK pathway, thereby impairing
downstream oncogenic signaling (16). These findings established NCT-58 as
a promising therapeutic option for trastuzumab-resistant
HER2-positive disease. In the present study, these observations
were extended to TNBC, and it was shown that NCT-58 suppresses
multiple HSP90 client proteins such as AKT, MEK and STAT3, leading
to impaired pro-survival signaling, reduced cancer stem-like
traits, and diminished migratory potential. Notably, distinct HSP90
client proteins are expressed depending on the breast cancer
subtype (43,44). By targeting HSP90, NCT-58 can
modulate these subtype-specific proteins and thereby block the
oncogenic pathways that drive tumor growth in a subtype-specific
manner. Collectively, the results of the present study highlight
NCT-58 as a versatile therapeutic strategy with efficacy across
different breast cancer subtypes.
Conventional treatments for TNBC are typically
dependent on anthracyclines and taxanes. In recent years,
immunotherapies and antibody-drug conjugates have broadened
therapeutic options, prompting the latest National Comprehensive
Cancer Network guidelines to recommend combining pembrolizumab with
chemotherapy in PD-L1-positive cases (combined positive score ≥10).
Sacituzumab-govitecan (SC) is an additional treatment option for
advanced TNBC following first-line treatment failure (45). However, these approaches are limited
by stringent biomarker requirements, suboptimal patient eligibility
rates (~40% only qualify for immunotherapy) (46), and the potential for acquired
resistance or cost-ineffectiveness (47). For instance, TNBC cells exposed to
SC can eventually develop resistance by altering both the antibody
target and toxic payload (48).
Consequently, achieving sustained therapeutic efficacy in TNBC
remains a significant clinical challenge.
Combination regimens are often required to address
the heterogeneity of TNBC and forestall the emergence of drug
resistance. NCT-58 exhibited synergistic anti-proliferative effects
when co-administered with paclitaxel or doxorubicin, two
foundational chemotherapeutic agents frequently used in TNBC. CI
analysis confirmed this synergy, suggesting that the simultaneous
disruption of HSP90 client protein function by NCT-58 may sensitize
TNBC cells to cytotoxic agents.
Overall, the findings of the present study suggested
that NCT-58 is a promising therapeutic candidate for TNBC,
targeting both proliferative tumor cells and cancer stem-like
populations, while avoiding the limitations of classical HSP90
inhibitors. By targeting key oncogenic pathways, including AKT, MEK
and STAT3, NCT-58 significantly compromises cancer cell survival
and induces apoptosis. Notably, NCT-58 exhibited minimal
cytotoxicity in non-malignant cells, highlighting its favorable
therapeutic profile. The present study has certain limitations, as
additional in vivo validation is warranted. In particular,
investigations using a 4T1-derived syngeneic model will be
necessary to further elucidate the effects of NCT-58 on tumor
growth, angiogenesis, metastasis, and the tumor microenvironment.
Furthermore, comprehensive preclinical studies evaluating long-term
toxicity and safety will be essential prior to clinical application
in TNBC patients. Finally, NCT-58 enhances the anticancer efficacy
of standard chemotherapeutic agents such as paclitaxel and
doxorubicin in a synergistic manner, highlighting its potential as
a potent and well-tolerated treatment option for TNBC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Korea Health Technology
R&D Project through the Korea Health Industry Development
Institute, funded by the Ministry of Health and Welfare, Republic
of Korea (grant nos. HA17C0053 and HR20C0021); the National
Research Foundation funded by the Korean government (grant nos.
2021R1A2C2009723, 2023R1A2C3004010, RS-2024-00342677,
RS-2025-02634306 and RS-2025-00558356); the Korea University Guro
Hospital (grant no. O2411391) and the Brain Korea 21 Plus
Program.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
EJ, KL, JL, YJK, JYK and JHS conceived and designed
the experiments. SJ, EJ, SP, KL, EO, DK, YJK and JYK performed the
experiments. EJ, SJ, KL, SP, MP, DK, SK, YKK, KDN, YJK, LF and JYK
analyzed the data. CTN, MTL, and JA synthesized NCT-58. EJ, KL, LF,
YJK and JYK wrote the manuscript. EJ, JYK and YJK confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zagami P and Carey LA: Triple negative
breast cancer: Pitfalls and progress. NPJ Breast Cancer. 8:952022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferrarini M, Heltai S, Zocchi MR and
Rugarli C: Unusual expression and localization of heat-shock
proteins in human tumor cells. Int J Cancer. 51:613–619. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pick E, Kluger Y, Giltnane JM, Moeder C,
Camp RL, Rimm DL and Kluger HM: High HSP90 expression is associated
with decreased survival in breast cancer. Cancer Res. 67:2932–2937.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Isaacs JS, Xu W and Neckers L: Heat shock
protein 90 as a molecular target for cancer therapeutics. Cancer
Cell. 3:213–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bocchini CE, Kasembeli MM, Roh SH and
Tweardy DJ: Contribution of chaperones to STAT pathway signaling.
JAKSTAT. 3:e9704592014.PubMed/NCBI
|
|
6
|
Streicher JM: The role of heat shock
proteins in regulating receptor signal transduction. Mol Pharmacol.
95:468–474. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Edkins AL: Hsp90 co-chaperones as drug
targets in cancer: Current perspectives. Heat Shock Protein
Inhibitors. McAlpine S and Edkins A: Topics in Medicinal Chemistry,
19. Springer; Cham: pp. 21–54. 2016, View Article : Google Scholar
|
|
8
|
Collina F, Di Bonito M, Li Bergolis V, De
Laurentiis M, Vitagliano C, Cerrone M, Nuzzo F, Cantile M and Botti
G: Prognostic value of cancer stem cells markers in triple-negative
breast cancer. Biomed Res Int. 2015:1586822015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kabakov A, Yakimova A and Matchuk O:
Molecular chaperones in cancer stem cells: Determinants of stemness
and potential targets for antitumor therapy. Cells. 9:8922020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cho TM, Kim JY, Kim YJ, Sung D, Oh E, Jang
S, Farrand L, Hoang VH, Nguyen CT, Ann J, et al: C-terminal HSP90
inhibitor L80 elicits anti-metastatic effects in triple-negative
breast cancer via STAT3 inhibition. Cancer Lett. 447:141–153. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park JM, Kim YJ, Park S, Park M, Farrand
L, Nguyen CT, Ann J, Nam G, Park HJ, Lee J, et al: A novel HSP90
inhibitor targeting the C-terminal domain attenuates trastuzumab
resistance in HER2-positive breast cancer. Mol Cancer. 19:1612020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park M, Jung E, Park JM, Park S, Ko D, Seo
J, Kim S, Nam KD, Kang YK, Farrand L, et al: The HSP90 inhibitor
HVH-2930 exhibits potent efficacy against trastuzumab-resistant
HER2-positive breast cancer. Theranostics. 14:2442–2463. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoter A, El-Sabban ME and Naim HY: The
HSP90 family: Structure, regulation, function, and implications in
health and disease. Int J Mol Sci. 19:25602018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Garcia-Carbonero R, Carnero A and Paz-Ares
L: Inhibition of HSP90 molecular chaperones: Moving into the
clinic. Lancet Oncol. 14:e358–e369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y and McAlpine SR: N-terminal and
C-terminal modulation of Hsp90 produce dissimilar phenotypes. Chem
Commun (Camb). 51:1410–1413. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park S, Kim YJ, Park JM, Park M, Nam KD,
Farrand L, Nguyen CT, La MT, Ann J, Lee J, et al: The C-terminal
HSP90 inhibitor NCT-58 kills trastuzumab-resistant breast cancer
stem-like cells. Cell Death Discov. 7:3542021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nguyen CT, Ann J, Sahu R, Byun WS, Lee S,
Nam G, Park HJ, Park S, Kim YJ, Kim JY, et al: Discovery of novel
anti-breast cancer agents derived from deguelin as inhibitors of
heat shock protein 90 (HSP90). Bioorg Med Chem Lett. 30:1273742020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JY, Cho TM, Park JM, Park S, Park M,
Nam KD, Ko D, Seo J, Kim S, Jung E, et al: A novel HSP90 inhibitor
SL-145 suppresses metastatic triple-negative breast cancer without
triggering the heat shock response. Oncogene. 41:3289–3297. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Donnelly A and Blagg BSJ: Novobiocin and
additional inhibitors of the Hsp90 C-terminal nucleotide-binding
pocket. Curr Med Chem. 15:2702–2717. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stebbins CE, Russo AA, Schneider C, Rosen
N, Hartl FU and Pavletich NP: Crystal structure of an
Hsp90-geldanamycin complex: Targeting of a protein chaperone by an
antitumor agent. Cell. 89:239–250. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gooljarsingh LT, Fernandes C, Yan K, Zhang
H, Grooms M, Johanson K, Sinnamon RH, Kirkpatrick RB, Kerrigan J,
Lewis T, et al: A biochemical rationale for the anticancer effects
of Hsp90 inhibitors: Slow, tight binding inhibition by geldanamycin
and its analogues. Proc Natl Acad Sci USA. 103:7625–7630. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu J, Liu T, Rios Z, Mei Q, Lin X and Cao
S: Heat shock proteins and cancer. Trends Pharmacol Sci.
38:226–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neckers L, Blagg B, Haystead T, Trepel JB,
Whitesell L and Picard D: Methods to validate Hsp90 inhibitor
specificity, to identify off-target effects, and to rethink
approaches for further clinical development. Cell Stress
Chaperones. 23:467–482. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qin JJ, Yan L, Zhang J and Zhang WD: STAT3
as a potential therapeutic target in triple negative breast cancer:
A systematic review. J Exp Clin Cancer Res. 38:1952019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang H, Wang L, Song Y, Wang S, Huang X,
Xuan Q, Kang X and Zhang Q: CD44+/CD24−
phenotype predicts a poor prognosis in triple-negative breast
cancer. Oncol Lett. 14:5890–5898. 2017.PubMed/NCBI
|
|
26
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh E, Kim YJ, An H, Sung D, Cho TM,
Farrand L, Jang S, Seo JH and Kim JY: Flubendazole elicits
anti-metastatic effects in triple-negative breast cancer via STAT3
inhibition. Int J Cancer. 143:1978–1993. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang MH, Lee JS, Kim HJ, Jin DI, Kim JI,
Lee KJ and Seo JS: HSP90 protects apoptotic cleavage of vimentin in
geldanamycin-induced apoptosis. Mol Cell Biochem. 281:111–121.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamashita N, Tokunaga E, Kitao H,
Hisamatsu Y, Taketani K, Akiyoshi S, Okada S, Aishima S, Morita M
and Maehara Y: Vimentin as a poor prognostic factor for
triple-negative breast cancer. J Cancer Res Clin Oncol.
139:739–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sirkisoon SR, Carpenter RL, Rimkus T,
Anderson A, Harrison A, Lange AM, Jin G, Watabe K and Lo HW:
Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription
factors promotes the aggressiveness of triple-negative breast
cancers and HER2-enriched breast cancer. Oncogene. 37:2502–2514.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ginestier C, Hur MH, Charafe-Jauffret E,
Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG,
Liu S, et al: ALDH1 is a marker of normal and malignant human
mammary stem cells and a predictor of poor clinical outcome. Cell
Stem Cell. 1:555–567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei Y, Li Y, Chen Y, Liu P, Huang S, Zhang
Y, Sun Y, Wu Z, Hu M, Wu Q, et al: ALDH1: A potential therapeutic
target for cancer stem cells in solid tumors. Front Oncol.
12:10262782022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Clark DW and Palle K: Aldehyde
dehydrogenases in cancer stem cells: Potential as therapeutic
targets. Ann Transl Med. 4:5182016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sládek NE: Aldehyde dehydrogenase-mediated
cellular relative insensitivity to the oxazaphosphorines. Curr
Pharm Des. 5:607–625. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sladek NE: Metabolism of
oxazaphosphorines. Pharmacol Ther. 37:301–355. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moreb JS: Aldehyde dehydrogenase as a
marker for stem cells. Curr Stem Cell Res Ther. 3:237–246. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schrörs B, Boegel S, Albrecht C, Bukur T,
Bukur V, Holtsträter C, Ritzel C, Manninen K, Tadmor AD, Vormehr M,
et al: Multi-omics characterization of the 4T1 murine mammary gland
tumor model. Front Oncol. 10:11952020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herndon ME, Ayers M, Gibson-Corley KN,
Wendt MK, Wallrath LL, Henry MD and Stipp CS: The highly metastatic
4T1 breast carcinoma model possesses features of a hybrid
epithelial/mesenchymal phenotype. Dis Model Mech. 17:dmm0507712024.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tao K, Fang M, Alroy J and Sahagian GG:
Imagable 4T1 model for the study of late stage breast cancer. BMC
Cancer. 8:2282008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lu W and Kang Y: Epithelial-mesenchymal
plasticity in cancer progression and metastasis. Dev Cell.
49:361–374. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zagouri F, Bournakis E, Koutsoukos K and
Papadimitriou CA: Heat shock protein 90 (hsp90) expression and
breast cancer. Pharmaceuticals (Basel). 5:1008–1020. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wei H, Zhang Y, Jia Y, Chen X, Niu T,
Chatterjee A, He P and Hou G: Heat shock protein 90: Biological
functions, diseases, and therapeutic targets. MedComm (2020).
5:e4702024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gradishar WJ, Moran MS, Abraham J,
Abramson V, Aft R, Agnese D, Allison KH, Anderson B, Burstein HJ,
Chew H, et al: NCCN guidelines® insights: Breast cancer,
version 4.2023. J Natl Compr Canc Netw. 21:594–608. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cortes J, Rugo HS, Cescon DW, Im SA, Yusof
MM, Gallardo C, Lipatov O, Barrios CH, Perez-Garcia J, Iwata H, et
al: Pembrolizumab plus chemotherapy in advanced triple-negative
breast cancer. N Engl J Med. 387:217–226. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xie J, Li S, Li Y and Li J:
Cost-effectiveness of sacituzumab govitecan versus chemotherapy in
patients with relapsed or refractory metastatic triple-negative
breast cancer. BMC Health Serv Res. 23:7062023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Coates JT, Sun S, Leshchiner I, Thimmiah
N, Martin EE, McLoughlin D, Danysh BP, Slowik K, Jacobs RA,
Rhrissorrakrai K, et al: Parallel genomic alterations of antigen
and payload targets mediate polyclonal acquired clinical resistance
to sacituzumab govitecan in triple-negative breast cancer. Cancer
Discov. 11:2436–2445. 2021. View Article : Google Scholar : PubMed/NCBI
|