Introduction
Skin cancer remains a major global health burden,
with melanoma representing the most aggressive subtype of cutaneous
malignancy (1), which is
responsible for approximately 80% of skin cancer-associated
fatalities (2–5). Recent advances in post-translational
modification research have identified that acetylation fulfills an
indispensable role in regulating gene expression via dynamic
modulation of chromatin structure (6,7).
Emerging evidence indicates that aberrant acetylation homeostasis
is pathologically associated with various dermatological disorders,
particularly with the initiation, progression and therapeutic
resistance of melanoma (8).
Understanding both the underlying mechanisms and consequences of
acetylation in melanoma is essential for guiding the development of
novel therapeutic strategies targeting these epigenetic
modifications. Notably, acetylation-associated biomarkers have been
shown to have significant clinical relevance in melanoma diagnosis
and prognosis, underscoring the translational importance of this
field.
Melanoma and acetylation
Melanoma
Malignant melanoma originates from melanin-producing
cells derived from the neural crest, and it can be induced by a
range of factors, including physical factors, chemical and
biological mediators, and genetic/molecular determinants (9,10).
According to the World Health Organization classification,
melanomas can be categorized into two major groups, namely those
associated with sun exposure and those that are not, and these
groups are reflected in distinct molecular pathways and different
pathological histories (11). The
sun-associated group is further subclassified based on the extent
of chronic sun damage (CSD) into high-CSD and low-CSD melanomas
(12). The latter group is further
subdivided based on the site of origin into mucosal, acral, uveal
and spitzoid melanomas, melanomas arising in blue or congenital
nevi, as well as rare variants originating in the central nervous
system (13). Ultraviolet radiation
(UVR) has a well-established etiological role in melanoma
pathogenesis; however, its effects on skin physiology are complex,
encompassing both detrimental impacts (for example, DNA damage and
mutagenesis) and beneficial aspects, including vitamin D synthesis
and immunomodulatory functions (14–16).
The process of melanogenesis, regulated by neuroendocrine factors
(for example, α-MSH and ACTH) and enzymatic pathways, not only
determines skin pigmentation, but also modulates melanoma behavior
and therapeutic responses (17).
For example, eumelanin may confer photoprotective effects, whereas
pheomelanin has been shown to promote oxidative stress and
contribute to tumor progression (18,19).
The skin operates as a neuro-immunoendocrine organ, producing a
range of mediators (for example, CRH, β-endorphin and cannabinoids)
that are able to locally modulate melanocyte function and
contribute to melanoma pathogenesis. In advanced stages of the
disease, melanoma-derived factors can disrupt systemic homeostasis,
thereby altering energy balance and immune function beyond the
local microenvironment, which has the effects of facilitating
disease progression and metastatic spread (17).
Acetylation
Acetylation, a dynamic post-translational
modification, serves as an epigenetic rheostat regulating chromatin
accessibility, transcriptional activation and protein functional
states. In cancers, dysregulation of this equilibrium, primarily
mediated by histone acetyltransferases (HATs) and histone
deacetylases (HDACs), drives oncogenic transcription programs and
post-translational rewiring of tumor suppressors (20). HATs, including the p300/CBP
(CREB-binding protein) family and the GNAT/MYST subfamilies,
catalyze the transfer of acetyl groups from acetyl-CoA to histone
tails, thereby neutralizing their positive charges to relax
chromatin structure and facilitate transcriptional activation
(21,22). Beyond histone modification, key
HATs, such as p300, function as transcriptional co-activators that
are able to integrate signaling pathways through acetylating both
histones and non-histone targets, including p53 and STAT3, thereby
modulating apoptosis, cellular differentiation and immune
responses. HDACs have the role of counterbalancing HAT activity,
and they function through the removal of acetyl groups. These
enzymes are divided into four classes of enzymes: Class I HDACs
(HDAC1/2/3/8), which are ubiquitously expressed and predominantly
localized to the nuclear compartment, where they exert their most
prominent HDAC activity (23);
Class II HDACs (HDAC4/5/6/7/9/10), which reside in the cytoplasm
and translocate to the nucleus in response to specific cellular
signaling cues; Class III HDACs, also termed sirtuins (SIRT1-7),
which are NAD+-dependent enzymes that remove acetyl
groups to restore chromatin compaction and silence gene expression
(24); and HDAC11, the sole member
of Class IV HDACs, which exhibits higher defatty-acylase activity
compared with its intrinsic deacetylase activity. Numerous studies
have provided evidence in support of the crucial involvement of
HDAC11 in different types of cancer, immune responses and metabolic
processes (25,26).
The dynamics of acetylation critically influence
melanoma pathogenesis through modulating chromatin accessibility,
transcriptional programs and protein functional states (27). Aberrant histone and non-histone
acetylation, driven by dysregulated HAT/HDAC activity, has been
shown to contribute to melanoma initiation through the silencing of
tumor-suppressive genes and hyperactivation of oncogenic pathways.
During metastatic progression, imbalances in acetylation have the
effect of promoting invasive phenotypes through ‘rewiring’ enhancer
landscapes to favor pro-migratory gene networks, and suppressing
differentiation signals. Clinical studies have shown that
therapeutic strategies targeting acetylation, such as the use of
HDAC inhibitors (HDACi) and sirtuin modulators, demonstrate dual
efficacy in terms of restoring tumor-suppressive transcription and
circumventing immune evasion, thereby underscoring the central role
of this type of epigenetic modification across the melanoma
continuum.
Notably, the acetylation landscape and its
functional consequences may vary significantly across melanoma
subtypes, including cutaneous, acral lentiginous and mucosal
melanoma. Acral melanoma exhibits a distinct molecular profile
compared with other cutaneous melanoma subtypes, characterized by a
lower frequency of canonical driver mutations in the B-Raf
proto-oncogene (BRAF) and NRAS proto-oncogene (NRAS) genes, but a
higher prevalence of alterations, such as KIT mutations and copy
number gains involving cyclin-dependent kinase 4 (CDK4) and cyclin
D1 (CCND1) (28–31). This genomic instability, often
manifested through structural variations and amplifications, is a
hallmark of acral lentiginous melanoma, and may be influenced by
mechanical stress rather than UVR, suggesting that the acetylome
regulating these genomic regions is likely to differ substantially
across the subtypes. Consequently, the differential acetylation
landscape gives rise to varied therapeutic responses; for example,
the reduced frequency of BRAF mutations in acral lentiginous
melanoma has been shown to reduce the efficacy of BRAF/MEK
inhibitors, whereas the activation of alternative pathways (for
example, via KIT or CDK4) suggests a potential role for HDACi in
targeting these non-canonical vulnerabilities either by modulating
oncogene expression or reactivating silenced tumor suppressors.
However, although acetylation dynamics are being increasingly
mapped in cutaneous melanoma, subtype-specific patterns in acral
lentiginous melanoma and mucosal melanoma remain incompletely
defined. Preliminary acetylome profiling, however, has suggested
unique enhancer acetylation landscapes in acral lentiginous
melanoma/mucosal melanoma, potentially reflecting their distinct
mutational spectra and microenvironmental contexts, which may
influence tumor biology and responses to epigenetic therapies,
including HDACi or CBP/p300 inhibitors. Dedicated studies using
patient-derived models and subtype-stratified cohorts are warranted
to elucidate the underlying mechanisms of the acetylation landscape
and to optimize targeted strategies.
Acetylation modifications in melanoma
pathogenesis and progression
Although the key roles of HATs and HDACs in melanoma
are well-established, the specific functions of individual family
members (such as different sirtuins or specific Class I HDACs) and
their heterogeneity across melanoma subtypes require more detailed
characterization. For example, the ‘paradoxical’ role of SIRT6 in
promoting melanoma proliferation, and yet mediating drug
resistance, highlights the complexity of acetylation network
regulation and its potential context-dependence (32,33).
Understanding the effects of nuanced alterations in the acetylation
network is critical in terms of developing effective precision
targeting strategies.
Acetylation-mediated control of cell
fate in melanoma: Regulation of proliferation, autophagy and
apoptosis
Acetylation has been shown to critically regulate
key processes associated with melanoma, including proliferation,
autophagy and apoptosis, through modulating both transcriptional
activators and epigenetic repressors (Fig. 1). Subsequently, the following
section will discuss various factors associated with acetylation
and their underlying mechanisms of action (Table I).
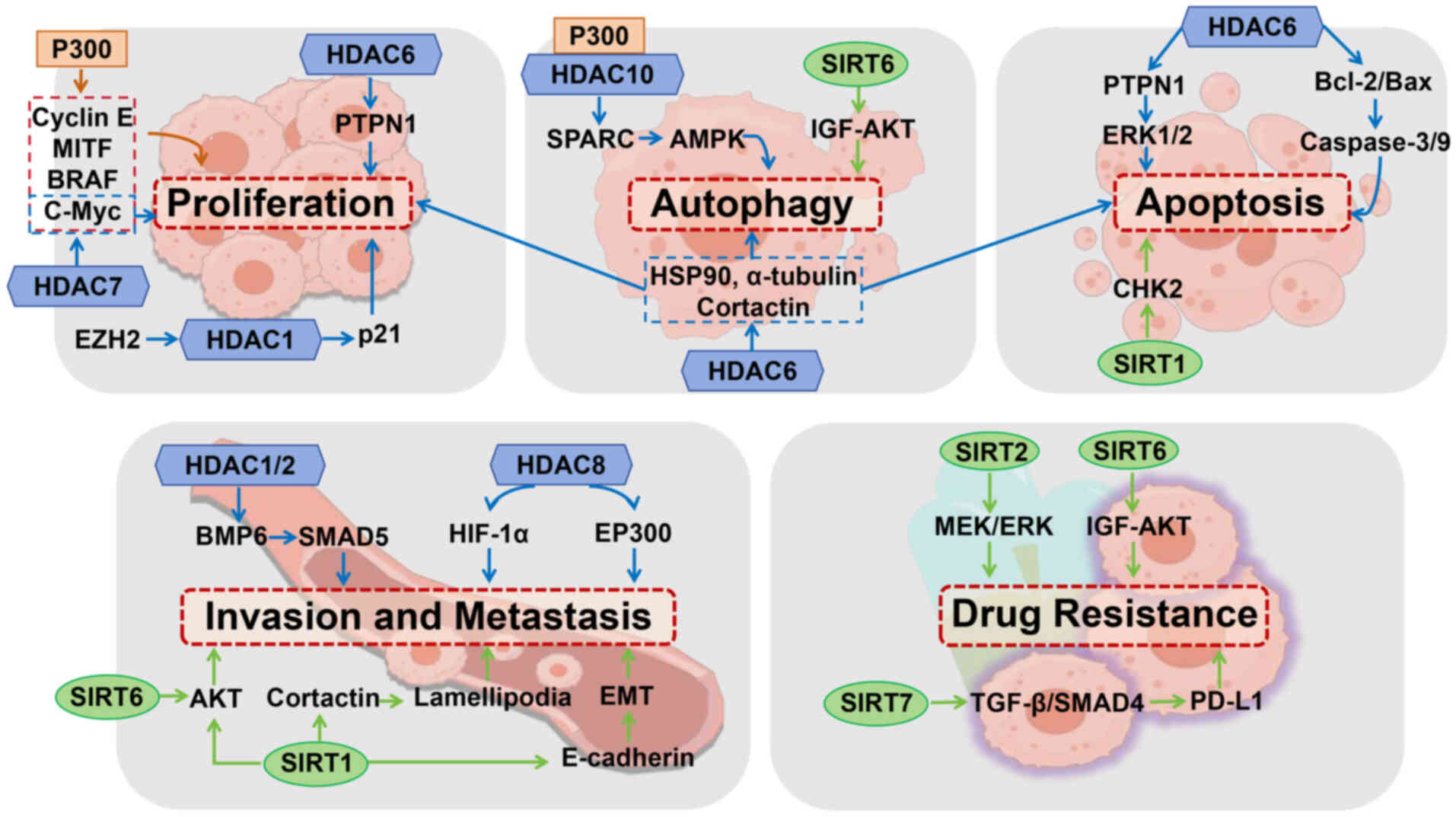 | Figure 1.Acetylation-mediated regulation of
melanoma progression. P300 enhances melanoma cell proliferation via
acetylation of cyclin E, MITF, BRAF and c-Myc. EZH2 represses p21
through HDAC1, disrupting the cell cycle. HDAC6 modulates
proliferation, autophagy, and apoptosis via α-tubulin, HSP90,
cortactin, and the PTPN1-ERK1/2 pathway; its inhibition induces
apoptosis via Bcl-2/Bax regulation. HDAC7 promotes growth via
c-Myc, while P300 and HDAC10 coordinate SPARC-mediated autophagy.
SIRT1 and SIRT6 regulate apoptosis and autophagy through CHK2 and
IGF-AKT signaling, respectively. HDAC1/2 activate BMP6-SMAD5 to
inhibit metastasis, while HDAC8 enhances invasion via deacetylation
of HIF-1α and EP300. SIRT1 promotes migration through EMT,
lamellipodia formation, and AKT deacetylation; SIRT6 antagonizes
this effect. SIRT2 and SIRT6 drive resistance via MEK-ERK and
IGF-AKT signaling. SIRT7 promotes PD-L1-mediated immune evasion by
suppressing TGF-β-SMAD4 signaling. HDAC, histone deacetylase; EMT,
epithelial-mesenchymal transition. |
 | Table I.Summary of HDAC-related function. |
Table I.
Summary of HDAC-related function.
| First author/s,
year | Role of
acetylation | Factor | Target | Mechanism | (Refs.) |
|---|
| Weinert et
al, 2018 | Proliferation,
autophagy and apoptosis | p300 | Cell cycle | Directly activates
the cyclin E promoter | (21) |
| Kim et al,
2019 |
|
| MITF | The MITF is
regulated by p300-mediated histone acetylation. | (35) |
| Dai et al,
2022 |
|
| BRAF | p300 promotes B-Raf
proto-oncogene kinase activity. | (36) |
| Vervoorts et
al, 2003 |
|
| c-Myc | Co-expression of
CBP with c-Myc stimulates acetylation. | (37) |
| Kim et al,
2019; |
| HDAC1 | Cell cycle | HDAC1 is a key
component of EZH2 regulatory axis. | (35,38) |
| Fan et al,
2011 |
|
|
|
|
|
| Kovacs et
al, 2005; |
| HDAC6 | Various | Targeting
substrates such as α-tubulin, HSP90, and cortactin. | (39,40) |
| Pulya et al,
2021 |
|
| cellular
functions |
|
|
| Liu et al,
2018 |
|
| ERK1/2 | Interacting with
the PTPN1; Activating ERK1/2 signaling. | (41) |
| Pulya et al,
2021; |
|
| Bcl-2, Bax, | Inhibiting HDAC6
resulted in a decrease in Bcl-2 and an increase | (40,42) |
| Bai et al,
2015 |
|
| caspase | in
Bax/Caspase-3/9. |
|
| Ling et al,
2023 |
| HDAC10 | SPARC | Depletion of HDAC10
upregulates SPARC and inhibits melanoma growth. | (43) |
| Zhang et al,
2020 |
| SIRT1 | CHK2 | SIRT1 deficiency
induces CHK2 hyperacetylation and results in cell death via mitotic
catastrophe. | (47) |
| Wang et al,
2018 |
| SIRT6 | IGF-AKT | IGF-AKT pathway
mediates the effect of SIRT6 on autophagy in melanoma. | (32) |
| Min et al,
2022 | Invasion and
metastasis | HDAC1/2 | BMP6 | HDAC1/2 inhibits
melanoma metastasis through regulating BMP6/SMAD5 signaling. | (50) |
| Kim et al,
2023 |
| HDAC8 | HIF-1α | HDAC8 deacetylates
HIF-1α, enhancing its protein stability and transcriptional
activity. | (52) |
| Emmons et
al, 2023 |
|
| EP300 | By deacetylating
EP300, HDAC8 inactivates its catalytic function. | (53) |
| Kong et al,
2022 | Drug
resistance | SIRT1 | E-cadherin | SIRT1 suppresses
E-cadherin expression. | (55) |
| Kunimoto et
al, 2014; |
|
| AKT | SIRT1 deacetylates
AKT. | (57–59) |
| Porta et al,
2014; |
|
|
|
|
|
| Yang et al,
2022 |
|
|
|
|
|
| Bajpe et al,
2015; |
| SIRT2 | MEK/ERK | Inhibition of SIRT2
leads to resistance by modulating the MEK/ERK | (60,61) |
| Zhao et al,
2022 |
|
|
| pathway. |
|
| Strub et al,
2018; |
| SIRT6 | IGFBP2 | SIRT6
haploinsufficiency upregulates IGFBP2 expression and confers | (33,62) |
| Wang et al,
2015 |
|
|
| resistance to MAPK
inhibitors. |
|
| Yi et al,
2023 |
| SIRT7 | SMAD4 | SIRT7 deacetylates
SMAD4, thereby antagonizing the TGF-β-SMAD4 pathway. | (63) |
| Yu et al,
2023 |
| HDAC2 | PD-L1 | HDAC2-mediated
deacetylation promotes the nuclear translocation of PD-L1. | (64) |
| Gao et al,
2020 |
| p300 | PD-L1 | p300-mediated
acetylation of PD-L1 prevents its nuclear translocation. | (65) |
Central to the regulatory function mediated via
protein acetylation is the HAT p300, which promotes
melanoma-genesis through multiple interconnected mechanisms. One
key mechanism which involves p300 is the direct activation of the
cyclin E promoter, which thereby facilitates the G1/S
phase transition. Accordingly, inhibition of p300, either by
expressing a dominant negative p300 mutant (DN p300) or using the
pharmacological inhibitor Lys-CoA, was found to markedly reduce
cyclin E transcription, thereby inducing cell cycle arrest
(21). These findings aligned with
its established role in facilitating G1/S progression
through the acetylation of key factors such as E2F1 (34). Furthermore, p300 has been shown to
sustain the pro-proliferative activity of the lineage-survival
oncogene MITF via catalyzing histone acetylation at its proximal
regulatory regions. The inhibitory effect of p300 blockade on the
proliferation of MITFhigh melanoma cells (cells that are
characterized by high levels of the MITF transcription factor)
underscores the significance of this regulatory axis (35). Beyond cell cycle and oncogene
support, p300 also activates the BRAF kinase by promoting BRAF K601
acetylation, thereby enhancing its kinase activity and promoting
melanoma cell proliferation. Significantly, this K601 acetylation
contributes to resistance against BRAFV600E inhibitors
in melanomas harboring the common BRAFV600E mutation, an
effect that is counteracted by the opposing deacetylase activity of
SIRT1 (36). These findings
collectively illustrate the multifaceted roles of p300-mediated
acetylation in melanoma (Table
I).
Building on its multifaceted roles in melanoma,
p300/CBP also acts as a critical positive cofactor for the Myc
family of transcription factors (c-Myc, N-Myc and L-Myc). These
potent regulators drive cell proliferation and suppress
differentiation across diverse cell types. Specifically in the case
of c-Myc, CBP has been shown to acetylate c-Myc in vitro.
Crucially, co-expressing CBP with c-Myc in vivo was shown to
enhance c-Myc acetylation, leading to reduced ubiquitination and
consequently stabilizing the c-Myc protein (37).
Beyond the multifaceted roles of HATs such as p300,
HDACs also have a crucial role in regulating tumor cell
proliferation and apoptosis through epigenetic reprogramming. A key
example is HDAC1, which cooperates with the polycomb group protein,
enhancer of zeste homolog 2 (EZH2), to drive cell cycle
dysregulation in melanoma. EZH2 is highly expressed in metastatic
melanoma cells, where it suppresses the expression of p21 (encoded
by CDKN1A), a cyclin-dependent kinase inhibitor regulated by
histone acetylation. Mechanistically, EZH2 facilitates the
recruitment and retention of HDAC1 at the CDKN1A promoter, thereby
repressing p21 transcription through histone deacetylation.
Consequently, HDAC1 downregulation reactivates p21 expression,
triggering G1/S phase arrest and halting tumor
progression (35,38).
Complementing the multifaceted roles of HATs such as
p300, HDAC6 has emerged as a structurally unique epigenetic
regulator. HDAC6, distinguished by its dual deacetylase domains and
a C-terminal ubiquitin-binding zinc finger domain, targets
substrates such as α-tubulin, heat shock protein 90 and cortactin
(39). These substrates are
involved in various cellular functions, including cell
proliferation, apoptosis, autophagy and DNA repair (40). Functionally, HDAC6 has been shown to
orchestrate a pro-survival signaling network in melanoma. It
directly binds and stabilizes protein tyrosine phosphatase
non-receptor type 1, thereby activating ERK1/2 signaling to drive
cell proliferation, colony formation and metastasis, while
suppressing apoptosis (41). This
axis is further amplified by the regulation of mitochondrial
apoptosis pathways mediated by HDAC6: HDAC6 inhibition has been
shown to trigger reactive oxygen species (ROS)-dependent
mitochondrial depolarization, leading to reduced levels of Bcl-2,
increased levels of Bax, the release of cytochrome c and activation
of caspases-9 and −3, ultimately inducing apoptosis (40,42).
Looking beyond HDAC6′s regulation of pro-survival
signaling networks, the secreted glycoprotein known as secreted
protein acidic and rich in cysteine (SPARC) exemplifies how
acetylation-dependent epigenetic mechanisms converge to control the
cell fate of melanoma. Of crucial importance to this axis is
HDAC10, which acts in concert with HAT p300 to dynamically modulate
H3K27ac, a key epigenetic marker on histone H3, at SPARC regulatory
elements. This epigenetic remodeling facilitates the recruitment of
bromodomain-containing protein 4 (BRD4), a critical transcriptional
co-activator, to SPARC enhancers, thereby repressing SPARC
transcription. Notably, either HDAC10 depletion or its
pharmacological inhibition was shown to reverse this repression,
leading to the robust upregulation of SPARC. The resultant SPARC
overexpression triggers AMPK-dependent autophagy (43), which, in turn, inhibits the activity
of mechanistic target of rapamycin complex 1 (mTORC1), induces
autophagosome formation and activates Unc-51-like kinase 1 (ULK1)
to drive the lysosomal degradation of oncogenic cargo (44). This autophagic flux ultimately
suppresses melanoma proliferation and metastasis. Crucially,
SPARC-mediated autophagy also resensitizes BRAF inhibitor-resistant
melanoma, rendering it susceptible to targeted therapy, thereby
revealing a dual antitumor mechanism (45). Collectively, HDAC10 and SPARC form
an epigenetic-metabolic checkpoint that regulates melanoma
progression, thereby providing a molecular rationale for targeting
this axis in combination therapies.
Beyond the SPARC/HDAC10 axis that couples epigenetic
remodeling with autophagic flux, checkpoint kinase 2 (CHK2) has
emerged as a critical guardian of genomic integrity, orchestrating
DNA damage responses and cell fate decisions in melanoma. Upon
sensing DNA double-strand breaks, CHK2 undergoes ATM-dependent
phosphorylation at Thr-68, triggering dimerization and activation
to enforce either G1/S or G2/M cell cycle
arrest, thereby either enabling DNA repair, or initiating apoptosis
should the damage be irreparable (46). Intriguingly, the
NAD+-dependent deacetylase SIRT1 has been shown to
directly interact with CHK2, deacetylating it at the Lys-520 site
(47). This deacetylation
subsequently antagonizes CHK2 phosphorylation and dimerization,
effectively suppressing its activation. Consequently, SIRT1
deficiency induces CHK2 hyperacetylation, leading to aberrant
kinase activation, mitotic catastrophe and ROS-dependent cell
death, a mechanism exploited by oxidative stress in melanoma
therapy (48). Simultaneously, the
SIRT family member SIRT6 is positively associated with the levels
of autophagy in melanoma. Mechanistically, SIRT6 has been shown to
induce abnormal autophagy in melanoma through inhibiting the
insulin-like growth factor 1 (IGF-1)-AKT signaling pathway
(32). Collectively, the SIRT1-CHK2
and SIRT6-autophagy axes form an integrated network that balances
cell survival and death in response to genomic and metabolic
stresses.
This section of the review has systematically
delineated how multiple acetyl-regulatory factors, including p300,
HDAC1/6/7/10 and SIRT1/6, have been demonstrated to influence the
fate of melanoma cells through modulating key molecules (Cyclin E,
MITF, BRAF, c-Myc, p21, SPARC, CHK2 and IGF-AKT). However, existing
studies have predominantly focused on individual factors, and thus
both a holistic understanding of the dynamic equilibrium within the
acetylation network and knowledge regarding its crosstalk with
other signaling pathways (for example, MAPK and PI3K/AKT) are
currently lacking. Notably, the divergent roles of SIRT1 and SIRT6
in regulating cellular apoptosis/autophagy vs. migration, coupled
with HDAC6′s established function as a multi-pathway hub, suggest
that targeting these molecules may yield pleiotropic effects,
although this would potentially be counterbalanced by a concurrent
increase in off-target risks.
Acetylation-mediated control of cell
fate in melanoma: Influence on invasion and metastasis
The invasion of melanoma cells, which represents a
critical step in metastatic dissemination, relies on the epigenetic
reprogramming of chromatin states that determines phenotypic
plasticity. The epigenetic landscape of acetylation serves as a
critical determinant of melanoma cell invasion and phenotypic
plasticity (Fig. 1). Key evidence
has come from a study by Mendelson et al (49), who stratified primary melanomas into
low-risk (Epgn1, proliferative) and high-risk (Epgn3, invasive)
subtypes based on enhancer landscapes driven by H3K27ac. This
contradiction highlights how acetylated-driven enhancer remodeling
dynamically balances pro- and anti-invasive gene networks, thereby
determining their metastatic potential.
Complementing the epigenetic axis, the bone
morphogenetic protein (BMP)/SMAD signaling pathway, representing a
branch of the TGF-β superfamily, has been shown to orchestrate
melanoma metastasis through dual regulatory modes. Min et al
(50) demonstrated that HDAC1/2
deacetylases suppress metastasis through activating BMP6-dependent
SMAD5 signaling, which leads to an upregulation of adhesion
molecules (for example, E-cadherin) and inhibits matrix
metalloproteinases (MMPs). On the other hand, HDAC1/2 loss triggers
BMP6-SMAD5 axis suppression, which has the effect of enhancing
invasion via epithelial-mesenchymal transition (EMT) transcription
factors (for example, Twist and Slug) and extracellular matrix
(ECM) degradation, a mechanism consistent with earlier findings
reported in a study by Hornig et al (51). Therefore, the HDAC-BMP-SMAD cascade
converges with the dynamics of H3K27ac enhancers to regulate
melanoma invasion.
Beyond the epigenetic reprogramming of
invasion-associated chromatin states, hypoxia-inducible factor 1
(HIF-1) has been found to be pathologically overexpressed, and its
overexpression drives the malignant transformation of melanocytes
through enhancing their proliferation, metastasis and immune
evasion. Critically, acetylation dynamics have the effect of
‘fine-tuning’ the activity of HIF-1α and that of its downstream
regulators. Among these regulators, the Class I HDAC, HDAC8, has
emerged as a pivotal orchestrator. Mechanistically, HDAC8
deacetylates HIF-1α, which has the effect of enhancing its protein
stability and transcriptional activity, thereby promoting the
expression of its target genes and accelerating cellular migration
(52). Moreover, through
deacetylating the HAT EP300, HDAC8 inactivates its catalytic
function, thereby redirecting chromatin accessibility towards
pro-invasive transcription factors such as c-Jun. This modification
has been shown to enhance melanoma cell invasion and resistance to
stress, thereby promoting the development of brain metastasis
(53). Considered altogether, HDAC8
integrates HIF-1α stabilization and EP300 suppression to establish
a feed-forward loop that amplifies melanoma aggressiveness, and
this dual mechanism underscores HDAC8 as a therapeutic node to
disrupt metastatic adaptation in hypoxic and inflammatory
microenvironments.
Building on the HIF-1α-HDAC8 axis that drives
metastatic adaptation, the EMT emerges as a pivotal reprogramming
event, enabling melanoma cells to dissociate from primary tumors
and invade distant tissues. Central to this plasticity is dynamic
lysine acetylation, which fine-tunes the activity of EMT-associated
transcription factors (54,55). Among the multiple acetylation
regulators, the NAD+-dependent deacetylase SIRT1 is
pathologically overexpressed in metastatic melanoma, providing a
signature that correlates with poor prognosis (56). Interestingly, SIRT1 has been shown
to promote cell migration and invasion by inducing EMT through the
suppression of E-cadherin expression (55). Moreover, SIRT1 regulates the
extension of lamellipodia, which are crucial structures for cell
migration, by deacetylating cortactin (56,57).
Previous studies have shown that SIRT1 inhibitors, including
nicotinamide, are able to significantly impair melanoma cell
migration through blocking lamellipodial extension, whereas SIRT1
activation enhances the migratory capability of the cells (56,57).
In addition, phosphoinositide 3-kinase (PI3K) has been shown to
facilitate the formation of membrane protrusions induced by
platelet-derived growth factor (PDGF), a process that is regulated
by SIRT1 through deacetylation of AKT (57–59).
On the other hand, nuclear-localized SIRT6 was found to exert an
antagonistic effect on cytoplasmic SIRT1-AKT signaling through
suppressing AKT activity at the chromatin level, thereby
contributing to the regulation of melanoma cell migration. This
SIRT1-SIRT6 antagonism creates a therapeutic vulnerability: SIRT1
inhibitors (for example, nicotinamide) lead to impairments of
lamellipodial extension and EMT, whereas SIRT6 activation restricts
metastatic dissemination. Targeting this axis may therefore prove
to be an effective means of disrupting the acetylation-dependent
‘migratory plasticity’ of melanoma cells.
The role of acetylation in driving melanoma invasion
and metastasis is highly context-dependent. For example, the
distinct subgroups defined by H3K27ac patterns, the regulation of
HIF-1α and p300 by HDAC8, and the promotion of EMT and cytoskeletal
dynamics by SIRT1 collectively underscore the central importance of
epigenetic reprogramming in determining metastatic potential.
However, it is critical to understand how these processes
dynamically respond to the tumor microenvironment (TME) in
vivo, including hypoxia and immune cell infiltration. Although
current studies have provided robust evidence for the
pro-metastatic roles of HDAC8 and SIRT1, the validation of
effective selective inhibitors through creating metastatic models
has remained limited. Future studies are required that have the
objective of integrating spatial omics technologies to map the
evolution of acetylation modifications across primary tumors,
circulating tumor cells and metastatic niches, which should lead to
the identification of actionable targets. Notably, targeting
metastasis-associated acetylation regulators (for example, HDAC8
and SIRT1) may face significant challenges due to the essential
functions that they perform in normal physiology (for example,
embryonic development and immune regulation), which will
necessitate a rigorous evaluation of the therapeutic window.
Acetylation-mediated control of cell
fate in melanoma: Influence on drug resistance
DNA-damaging agents, including alkylating drugs such
as temozolomide, dacarbazine and fotemustine, are pivotal in the
treatment of metastatic melanoma. However, melanoma cells often
develop resistance to these agents, and this can be attributed, in
part, to epigenetic alterations mediated by acetylation activity.
Overall, the intricate interplay between acetylation modifications
and the cellular response to various anticancer agents underscores
the complexity of melanoma biology (Fig. 1).
Within the regulatory network of melanoma resistance
and immune evasion, the sirtuin family exerts pivotal control
through dynamic acetylation modifications, with individual members
exhibiting both functional heterogeneity and paradoxical roles
across tumor progression stages. For example, SIRT2, a member of
the sirtuin family, drives resistance to BRAF inhibitors (for
example, vemurafenib) through deacetylating and activating the
MEK/ERK pathway (60,61). Interestingly, SIRT6
haploinsufficiency has been found to cause an upregulation of IGF
binding protein 2 (IGFBP2) expression through mechanisms involving
increased chromatin accessibility and H3K56 acetylation at the
IGFBP2 locus, coupled with enhanced IGF-AKT signaling. This
upregulation subsequently confers resistance to MAPK inhibitors in
BRAF-mutant melanoma cells, thereby highlighting its
context-dependent function (33,62).
Furthermore, SIRT7 critically promotes melanoma
progression by enhancing tumor cell survival and facilitating
immune evasion. Deficiency in SIRT7 leads to an increase in tumor
cell death under stress conditions in vitro and a
suppression of tumor growth in vivo. Mechanistically, SIRT7
directly deacetylates SMAD4 protein, which antagonizes the
TGF-β-SMAD4 signaling pathway, ultimately leading to an
upregulation of programmed death-ligand 1 (PD-L1) protein. This
SIRT7-mediated increase in PD-L1 enables immune evasion and
contributes to resistance against immune checkpoint blockade
therapies (63). The regulation of
PD-L1 itself is also subject to acetylation dynamics, as revealed
by contrasting findings: HDAC2-mediated deacetylation was found to
promote PD-L1 nuclear translocation, enabling it to form a complex
with phosphorylated (p-)STAT3 and to activate early growth response
1-mediated tumor angiogenesis (64), whereas p300-mediated acetylation
prevents PD-L1 nuclear translocation, thereby reprogramming
immune-response-associated gene expression and consequently
enhancing the antitumor response to PD-1 blockade (65). Collectively, these findings
underscore the intricate and often context-specific interplay
between sirtuin-mediated acetylation, immune regulation and
therapeutic resistance pathways in melanoma.
The role of acetylation modifications in mediating
melanoma resistance to targeted therapies (for example, BRAF/MEK
inhibitors) and immunotherapies (for example, anti-PD-1) is
increasingly being recognized, especially concerning the
contributions of sirtuins (SIRT2/6/7) and specific HDACs (for
example, HDAC2). The regulation of PD-L1 acetylation status by
p300, and its impact on immune checkpoint inhibitor efficacy,
represent a discovery that has significant clinical translational
potential. Nevertheless, resistance mechanisms are highly complex
and heterogeneous, where alterations in a single acetylation factor
may constitute only one component of an intricate resistance
network. Furthermore, when evaluating strategies targeting
acetylation (for example, combining HDACi or sirtuin inhibitors) to
reverse resistance, it is essential to consider their dual effects
on both tumor cells and immune cells (for example, T-cell
function), as exemplified by SIRT7′s involvement in tumor cell
survival and PD-L1 regulation. Therefore, optimizing the time,
dosage and sequence of combination therapies is critical for
overcoming resistance.
Acetylation modifications in melanoma
diagnosis and prognosis
The prognostic significance of acetylation in
melanoma has become evident, based on the multidimensional
regulatory axis that includes epigenetic reprogramming, metastatic
competence and remodeling of the TME. Numerous studies have shown
that acetylation levels differ across different tumor types, and
that these are correlated with various clinicopathological
parameters and patient survival rates (66–69).
For example, malignant melanoma cells have been shown to exhibit
higher levels of HDAC1/2/3 expression compared with their
non-cancerous counterparts. This positions acetylation status not
only as a potential biomarker for staging, but also as a dynamic
guide for therapeutic intervention.
The HAT Tip60 (Tat interactive protein, 60 kDa)
exemplifies this prognostic value, as its expression is inversely
correlated with primary tumor thickness, serving as an independent
prognostic marker across disease stages; diminished Tip60 levels
were also found to be associated with significantly reduced 5-year
disease-specific survival rates in patients with melanoma (70–72).
Mechanistically, Tip60 promotes apoptosis through p53 acetylation
at K120 (73,74), and its downregulation results in
decreased levels of acetylated DNA methyltransferase 1 (ac-DNMT1).
This Tip60/ac-DNMT1 axis is critically associated with melanoma
progression, and low levels of ac-DNMT1 are correlated with poorer
prognosis in stage IV disease; by contrast, elevated levels of
ac-DNMT1 are a predictor of improved survival (75).
Similarly, the HAT p300 demonstrates significant
stage-dependent prognostic relevance. Clinical analyses have
revealed a redistribution pattern in advanced melanoma,
characterized by nuclear depletion and cytoplasmic accumulation,
and this is strongly correlated with the higher American Joint
Committee on Cancer (AJCC) cancer stages. This redistribution is
mechanistically driven by the BRAF-MAPK/ERK pathway, which targets
nuclear p300 for ubiquitin-proteasomal degradation. Consequently, a
high BRAF/low nuclear p300 profile predicts metastatic transition,
whereas a low BRAF/high nuclear p300 signature aids in
distinguishing nevi from melanoma (36,76).
Critically, Kaplan-Meier survival analysis was employed to confirm
that low nuclear p300 expression, but not low p300 cytoplasmic
expression, is a strong predictor of significantly worse overall
and disease-specific 5-year survival rates (77).
Collectively, these findings have underscored the
complex interplay between specific HATs (Tip60, p300), histone
deacetylases (HDACs), and associated modifiers (for example,
ac-DNMT1) within the acetylation landscape, solidifying their
crucial roles as prognostic indicators and potential therapeutic
targets across the spectrum of melanoma progression. However,
translating these findings into clinical practice faces challenges:
Validating the value of independent prognostic indicators requires
large-scale, multicenter prospective cohorts, and standardized
protocols need to be established for detecting and scoring systems.
Overcoming these challenges will enable acetylation-associated
biomarkers to facilitate clinical risk stratification, thereby
guiding therapeutic decision-making.
Acetylation modifications as therapeutic
targets in melanoma
Recent studies have provided valuable insights into
the mechanisms via which HDAC influences melanoma progression,
which have highlighted the potential of HDACi as a therapeutic
strategy (Fig. 2). The development
of HDACi as therapeutic agents holds promise for the treatment of
melanoma; however, the specific effects of these inhibitors may
differ, according to the HDAC isoform and the cellular context
(Table II).
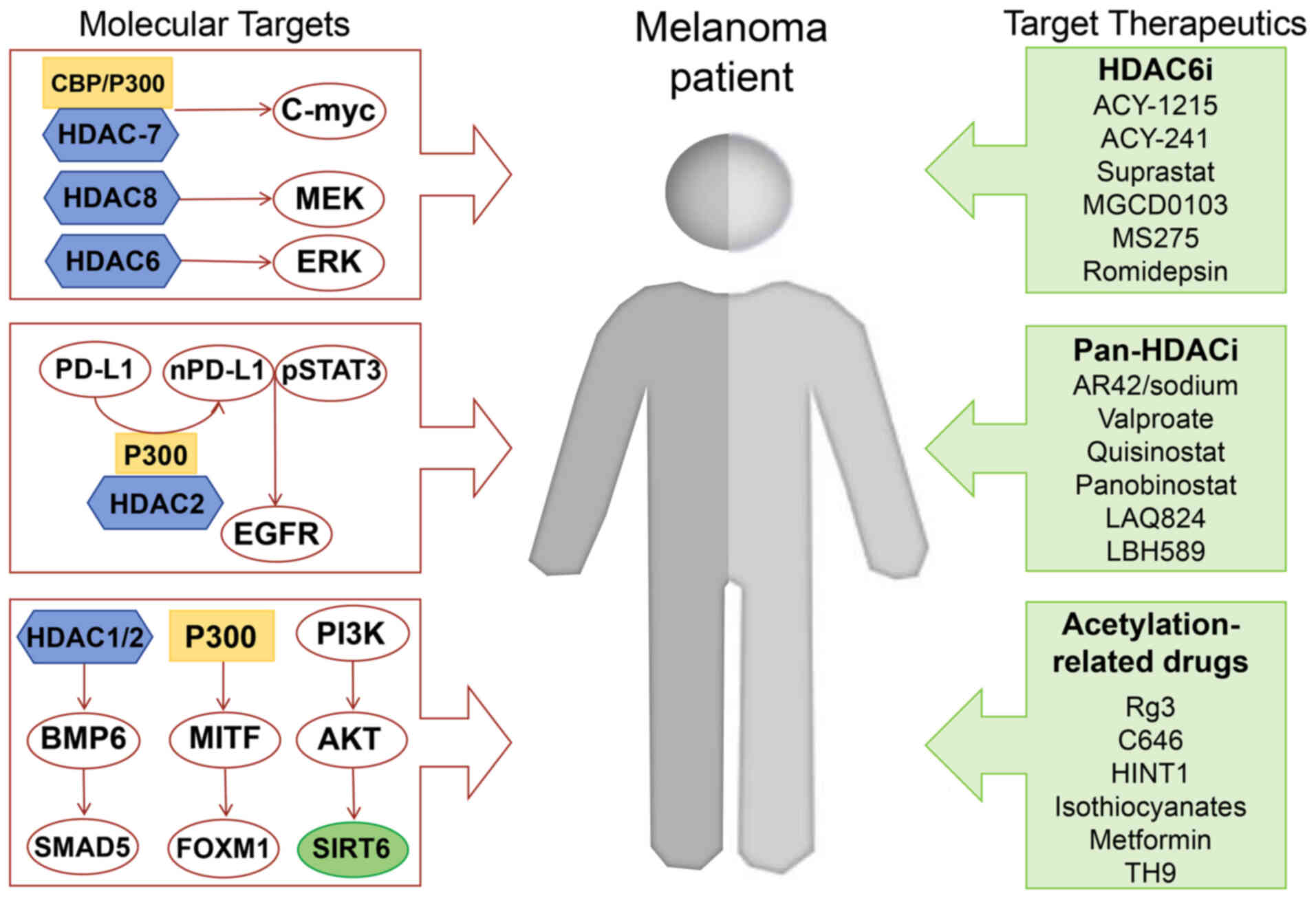 | Figure 2.Integrated signaling pathways and
targeted therapeutic agents in melanoma. Pathways are outlined in
red boxes with directional arrows. Drug categories are color-coded
in light green. Schematic depicts interconnected pathways driving
melanoma pathogenesis: CBP/P300 and HDAC 7/8/6 regulate the
expression of c-Myc, MEK and ERK. SIRT6 is the downstream product
of the PI3K-AKT pathway and P300-MITF-FOXM1 transcriptional network
controlling melanocyte differentiation and proliferation.
HDAC1/2-BMP6-SMAD5 pathway modulating TGF-β signaling.
HDAC6-selective inhibitors (HDAC6i): ACY-1215, ACY-241, Suprastat,
MGCD0103, MS275, and romidepsin. Pan-HDAC inhibitors (Pan-HDACi):
AR42/sodium valproate, quisinostat, panobinostat, LAQ824, and
LBH589. Acetylation-targeting agents: Rg3, C646, HINT1,
isothiocyanates, metformin, and TH9. HDAC, histone deacetylase;
HDACi, HDAC inhibitor. |
| Table II.Summary of HDAC-related therapeutic
agents. |
Table II.
Summary of HDAC-related therapeutic
agents.
| First author/s,
year | Category | Medicine | Target | Mechanism | Model | (Refs.) |
|---|
| Booth et al,
2017 | HDACi | AR42/sodium
valproate | Pan-HDACi | Enhancing of
anti-PD-1 and anti-CTLA4 antibodies | TPF-12-293
cells | (79) |
| Heijkants et
al, 2018 |
| quisinostat | Pan-HDACi | Combining the
treatment with CDK inhibition using flavopiridol | Uveal melanoma cell
lines | (93) |
| Gallagher et
al, 2018 |
| panobinostat | Pan-HDACi | Inducing
caspase-dependent apoptotic cell death | Patient derived
melanoma | (94) |
| Booth et al,
2017 |
| Suprastat | HDAC6i | Increasing the
infiltration of CD8+ effector and memory T-cells | SM1 murine melanoma
model | (80) |
| Vo et al,
2009; |
| LAQ824; | Pan-HDACi | Enhancing the
efficacy of ACT | B16 murine
model | (90,92) |
| Lisiero et
al, 2014 |
| LBH589 |
|
|
|
|
| Noonepalle et
al, 2020 |
| MGCD0103; | Class I-HDACi | Upregulating PD-L1
expression | B16 murine
melanoma | (83) |
|
|
| MS275 |
|
|
|
|
| Badamchi-Zadeh
et al, 2018 |
| Romidepsin |
HDAC1/2-inhibitor | Increasing the
frequency of vaccine-elicited CD8+ T cells | C57BL/6 mice | (95) |
| Shan et al,
2014 | Other
acetylation-related treatments | Rg3 | - | Inducing cell cycle
arrest; decreasing HDAC3 and increasing p53 acetylation | A375; SK-MEL-28;
Xenograft tumor | (96) |
| Yan et al,
2013 |
| C646 | - | Inducing cell cycle
arrest | WM35 cells | (97) |
| Mitsiogianni et
al, 2021 |
|
Isothiocyanates | - | Inducing cell cycle
arrest and cellular senescence in melanoma cells by inhibiting
p300/ CBP | A375, Hs294T, VMM1
and B16F-10 melanoma | (103) |
| Li et al,
2018 |
| Metformin | - | Inhibiting
KAT5-mediated SMAD3 acetylation, transcriptional activity and TRIB3
expression | C57BL/6 mice and
KK-Ay mice | (104) |
HDACi
HDACi hinder the effective repair of DNA damage.
This persistent damage either disrupts or inhibits essential
cellular processes, including transcription and DNA replication,
which ultimately triggers a cell death mechanism in cancer cells
(78).
HDACi and immune checkpoint
therapy
The integration of HDACi with immune checkpoint
therapy represents a paradigm-shifting approach in melanoma
treatment, leveraging epigenetic priming to overcome tumor immune
evasion. Notably, pan-HDACi agents such as AR42 or valproic acid,
when combined with kinase inhibitors such as pazopanib, have
demonstrated significant antitumor activity that extends beyond
growth suppression to the downregulation of the immune checkpoint
molecules, PD-L1/PD-L2. This epigenetic reprogramming leads to a
critical enhancement of tumor responsiveness to subsequent immune
checkpoint blockade, thereby amplifying antitumor immunity
(51,79). The synergy arises through
multifaceted mechanisms; For example, HDACi remodel the TME by
promoting the infiltration of macrophages, natural killer (NK)
cells, neutrophils and activated T cells (80), whereas Class I HDACi also modulate
checkpoint ligand expression, causing an upregulation of PD-L1 and
PD-L2 in melanoma (81,82). For example, the pan-HDACi LBH589
(targeting Class I/II/IV) in combination with PD-1 blockade was
shown to reduce tumor burden and to extend survival in melanoma
models. Similarly, selective HDAC6 inhibitors, such as Suprastat,
exhibit combinatorial efficacy with PD-L1 blockade by reshaping
immune populations, having the effect of reducing the numbers of
pro-tumoral M2 macrophages while enhancing infiltration of
antitumor CD8+ T-cells and memory T-cells (83,84).
This rational combinatorial approach, which is grounded in
overcoming epigenetic-mediated immune resistance, underpins the
clinical success that has already been observed in metastatic
melanoma and other malignancies that target co-inhibitory pathways.
However, the process of clinical translation warrants caution: Even
though preclinical studies (for example, utilizing LBH589,
Suprastat) have demonstrated efficacy, early clinical trial results
have been mixed. Therefore, developing more selective HDACi (for
example, HDAC6 inhibitors) or tumor-targeted HDACi, or optimizing
currently existing dosing regimens (for example, intermittent
administration), may help to improve the therapeutic window and
efficacy of this combination therapy.
Though the combination therapy involving HDACi and
the immune checkpoint cytotoxic T-lymphocyte associated protein 4
(CTLA-4) targeting agent nivolumab has demonstrated improved
efficacy in patients with melanoma (80), there remains a notable scarcity of
studies that have directly investigated the mechanistic links
between acetylation and immune checkpoints such as CTLA-4,
lymphocyte-activation gene 3 (LAG-3) and T cell immunoreceptor with
immunoglobulin and ITIM domain (TIGIT). Limited evidence has
suggested that HDACi is able to modulate the tumor immune
microenvironment, potentially enhancing T cell function and antigen
presentation. For example, Li et al (85) demonstrated that co-inhibitory
receptors such as CTLA-4, LAG-3 and PD-1 provide essential
balancing signals for T cell activation. Their study also revealed
that the intrinsic HDAC activity within the Tcf1 transcription
factor is crucial for preventing the excessive induction of CTLA-4
in T follicular helper (Tfh) cells, thereby protecting their B-cell
helper function. Specifically, mutations in key amino acids within
the HDAC domain of Tcf1 led to the de-repression of CTLA-4 in Tfh
cells. In spite of these insights, however, the direct mechanistic
interplay between HDACi and the regulation of CTLA-4, LAG-3 or
TIGIT expression and function specifically within the context of
melanoma remains poorly understood. The majority of published
studies to date have focused on the phenotypic outcomes of
combination therapies, rather than on the underlying epigenetic
modifications. Therefore, elucidating how HDACi modulate the
acetylation status of histones or non-histone proteins associated
with genes encoding these immune checkpoints in melanoma represents
a critical and promising direction for future research.
HDACi and adoptive cell therapy
(ACT)
ACT represents a transformative approach for
metastatic melanoma, and its efficacy may be significantly enhanced
through strategic epigenetic modulation with HDACi (86,87).
HDACi reprogram the tumor-immune interface via dual mechanisms: i)
through inducing chromatin relaxation to enhance tumor antigen
presentation; and ii) through reversing T-cell exhaustion by
restoring acetylation-dependent transcriptional programs in
CD8+ T cells (88,89).
Crucially, preclinical studies have demonstrated how this
mechanistic synergy is translated into therapeutic enhancement. For
example, the pan-HDACi LAQ824, when combined with ACT using
gp100-specific pmel-1 T cells, was shown to cause a significant
amplification of the cytotoxic function of the transferred cells,
which led to a heightened level of tumor eradication in a B16
melanoma model (90,91). In addition to enhanced tumor cell
eradication, this combination also led to superior antitumor
efficacy. Similarly, combining the pan-HDACi LBH589 with
gp100-specific T-cell therapy led to substantially prolonged T-cell
survival and reduced tumor burden in melanoma models (92). Furthermore, the activity of HDACi
extend beyond modulating immediate effector functions. For example,
previous studies have shown that exposure to HDACi and
interleukin-21 (IL-21) caused a reprogramming of differentiated
human CD8+ T cells into central memory-like T cells.
This dedifferentiation process is initiated through histone H3
acetylation at the CD28 promoter region, which facilitates
IL-21-mediated p-STAT3 binding to the CD28 locus, thereby
generating a highly persistent T cell population (91). These findings collectively served to
position HDACi as essential adjuvants for overcoming the epigenetic
barriers that limit ACT efficacy in immune-cold melanomas.
Utilizing HDACi to enhance ACT, especially through
modifying memory T cell phenotypes and augmenting their
persistence, represents an innovative therapeutic strategy.
However, this approach is currently being validated primarily in
murine models. When translating HDACi such as LAQ824 or LBH589 to
human ACT, critical evaluations must focus on their potential
toxicity towards both the infused T cells and the host immune
system, as well as the impact they may have on T cell receptor
diversity. Furthermore, substantial optimization work needs to be
undertaken to precisely control the HDACi treatment conditions
(concentration, duration) during ex vivo T cell expansion,
which is required in order to maximize the therapeutic benefits
while minimizing functional impairment.
HDACi and other treatments
The combination of HDACi with CDK blockade offers a
paradigm-shifting strategy for overcoming therapeutic resistance in
refractory melanomas. The potential of this as combination therapy
has been robustly demonstrated preclinically: Utilizing BRAF
wild-type cutaneous melanoma tumors as a model, Heijkants et
al (93) reported that the
combination of pan-HDACi quisinostat and pan-CDK inhibitor
flavopiridol significantly reduced tumor volume to a greater extent
than was accomplished via flavopiridol monotherapy. Promising
therapeutic effects were also observed in patient-derived xenograft
models of cutaneous melanoma (93).
Looking beyond CDK inhibition, HDACi further enhance
targeted therapy through distinct molecular mechanisms. Another
study, conducted by Gallagher et al demonstrated the
synergistic effects of combining the BRAF inhibitor encorafenib
with the HDACi panobinostat in melanoma cells (94). This combination induced
caspase-dependent apoptotic cell death, primarily via the
downregulation of c-Myc expression and by decreasing PI3K pathway
activity, suggesting that the combination of HDACi and MAPK
inhibitors may have therapeutic potential in melanoma
treatment.
The therapeutic scope of HDACi combinations extends
significantly into the immunomodulatory landscape. The
combinatorial targeting of epigenetic regulators, especially
through HDACi and the bromodomain and extra-terminal domain (BET)
family, has emerged as a transformative strategy to overcome
therapeutic resistance in melanoma. The HDACi romidepsin (RMD) and
the BET inhibitor IBET151, both individually and in combination,
have been shown to enhance the frequency of vaccine-elicited
CD8+ T cells and to improve therapeutic and prophylactic
protection against B16-OVA melanoma. Additionally, the increased
IL-6 production and pro-apoptotic gene expression following
RMD+ IBET151 treatment are likely to be contributors
towards the enhanced cancer vaccine responses (95). These findings have identified HDACi
as versatile adjuvants whose synergistic interactions with CDK
inhibitors, BRAF inhibitors and BET inhibitors extend beyond simple
growth suppression to encompass targeted elimination and enhanced
immune surveillance. Optimizing these potent combinations
represents a critical frontier for advancing melanoma treatment,
necessitating a focus on clinical translation studies to fully
realize their potential.
Other acetylation-associated
treatments
Emerging pharmacological strategies targeting
acetylation dynamics extend beyond traditional HDACi, demonstrating
multifaceted antitumor potential through epigenetic-metabolic
crosstalk and combinatorial synergy. For example, Rg3, a bioactive
compound extracted from ginseng roots, has demonstrated efficacy in
inhibiting melanoma cell proliferation via downregulating HDAC3
expression and enhancing the level of p53 acetylation on lysine
residues. This dual action not only serves to arrest cell cycle
progression but also potentiates p53-dependent transcriptional
activation; in vivo studies that were performed using A375
×enografts confirmed these significant antiproliferative effects
(96). Complementing these natural
agents, synthetic inhibitors such as C646 have been shown to target
p300/CBP acetyltransferase activity to induce cell cycle arrest and
to synergize with DNA-damaging agents. Notably, C646 enhances
cisplatin-induced apoptosis in melanoma cells via sensitizing cells
to genomic instability (97–100).
Further broadening the therapeutic landscape,
isothiocyanates (ITCs), which are phytochemicals abundant in
cruciferous vegetables, function as pan-HDACi to suppress p300/CBP
activity, thereby triggering G0/G1 arrest and
senescence in melanoma (101).
Mechanistically, ITCs orchestrate a pleiotropic epigenetic
reprogramming landscape through reducing the total HDAC activity,
modulating the expression of histone-modifying enzymes (HDACs, HATs
and methyltransferases), and altering acetylation-methylation
crosstalk on histones H3/H4 (102,103). This multifaceted activity
positions ITCs as ideal partners for kinase inhibitor combinations
in refractory melanoma. Looking beyond their role as direct
acetylation modifiers, targeting stress-responsive nodes, such as
pseudokinase TRIB3, reveals novel metabolic-epigenetic
interdependencies. Li et al (104) found that treatment with metformin
led to melanoma growth and metastasis via reducing TRIB3
expression. Mechanistically, metformin was shown both to suppress
SMAD3 phosphorylation and to weaken the interaction between the
histone acetylase KAT5 and SMAD3, which, in turn, reduces
KAT5-mediated acetylation of SMAD3, leading to a decrease in SMAD3
transcriptional activity and subsequent TRIB3 expression, and
thereby antagonizing melanoma progression.
Beyond HDACi, targeting HATs, utilizing natural
compounds, or employing repurposed drugs are diverse strategies for
modulating acetylation against melanoma. These substances often
exhibit poly-pharmacology (namely, the design and use of a single
drug that simultaneously acts on multiple biological targets to
achieve a therapeutic effect), which may lead to complex biological
effects and potential off-target risks; however, their in
vivo antitumor activity, pharmacokinetic properties,
bioavailability and synergistic potential with standard therapies
require systematic evaluation in models that closely resemble the
clinical setting. Examples such as Rg3 and metformin suggest that
modulating acetylation might represent an essential component of
their known antitumor mechanisms, providing novel insights into
their modes of action, especially in the case of some established
agents (or ‘old drugs’). However, translating these findings into
effective clinical treatment regimens necessitates addressing
challenges that are associated with optimal dosing, routes of
administration, and how best to integrate these non-canonical
epigenetic agents within standard therapeutic frameworks.
Prospects and perspectives
Acetylation, a critical post-translational
modification, has been increasingly recognized for its profound
impact on the progression and metastasis of melanoma. Given the
critical role of acetylation in melanoma pathogenesis, targeting
this modification has emerged as a promising therapeutic strategy.
Various HDACi have been developed, ranging from pan-HDACi that
target multiple HDAC isoforms, to more selective inhibitors
targeting specific HDACs. Despite the promise that they hold, the
clinical application of HDACi in melanoma treatment continues to
face several challenges. The efficacy and safety profiles of these
compounds require further validation through extensive clinical
trials.
First, one of the primary challenges in the clinical
application of HDACi is the heterogeneity of melanoma tumors, and
the variability in acetylation patterns among patients. This
heterogeneity may lead to variable responses to HDACi treatment,
which would necessitate personalized approaches to therapy.
Secondly, the redundancy and context-dependence of acetylation
networks (such as the dual pro-oncogenic/tumor-suppressive roles of
different sirtuins or HDACs) necessitate the development of more
precise targeting strategies. The toxicity and side effects of
pan-inhibitors limit their application, highlighting the urgent
need to develop highly selective inhibitors (targeting specific
HDAC/HAT isoforms or even specific acetylation sites). Thirdly,
numerous preclinical findings to date that hold promise for
clinical applications in the future have yet to achieve widespread
success in clinical trials. Key reasons for this include: i) model
limitations; specifically, that cell lines and genetically
engineered mouse models are generally not suitable for fully
recapitulating human tumor heterogeneity and microenvironment
complexity; ii) toxicity management, due to the fact that the
toxicity of HDACi limit their sufficient dosing within combination
regimens; and iii) lack of patient selection, given that there is
an absence of reliable predictive biomarkers for response to
acetylation-targeted therapies. Finally, emerging evidence
highlights the significance of other lysine acylations,
particularly lactylation, in cancer biology (105). Lactylation involves the transfer
of a lactate-derived lactyl group to lysine residues on histones
and non-histone proteins, which is analogous to the transfer of an
acetyl group in acetylation (106). Histone lactylation has been shown
to regulate gene expression programs that are distinct from
acetylation, influencing multiple physiological and pathological
processes (107). Mechanistically,
the enzymes involved in adding or removing lactyl marks are still
being identified, although evidence already exists to suggest
potential interplay or competition with acetylation pathways, given
that both modifications target lysine residues (108,109). This presents a compelling future
direction-namely, to explore the potential crosstalk and hierarchy
between different types of acylation reactions (for example,
acetylation and lactylation) that shape melanoma pathogenesis.
Therefore, future studies should not only continue to delineate the
acetylation-specific networks but also map the landscape of
lactylation and other novel modifications in melanoma, which will
serve to identify their convergent and unique roles in
oncogenesis.
In conclusion, acetylation modifications fulfill a
crucial role in the pathogenesis and progression of melanoma.
Understanding the molecular mechanisms underlying these
modifications may provide insights into novel therapeutic
strategies. Future studies should focus on a number of different
aspects, including developing isoform-selective HDACi to minimize
toxicity, validating acetylation-based biomarkers in large clinical
cohorts for patient stratification, and elucidating
subtype-specific acetylation patterns in acral and mucosal
melanoma. Through harnessing the power of epigenetics, we will be
able to pave the way for more effective and personalized treatments
for this devastating disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82103726), the Guangdong Basic and
Applied Basic Research Foundation (grant nos. 2023A1515010575 and
2025A1515010947), the Shenzhen Science and Technology Program
(grant nos. JCYJ20210324110008023 and JCYJ20230807095809019), the
Shenzhen Sanming Project (grant no. SZSM202311029), the Shenzhen
Key Medical Discipline Construction Fund (grant no. SZXK040) and
the Shenzhen High-level Hospital Construction Fund and Peking
University Shenzhen Hospital Scientific Research Fund (grant no.
KYQD2024378).
Availability of data and materials
Not applicable.
Authors' contributions
JW and XC wrote the first draft of the manuscript.
JW, XC and CH created the images. ZZ, XL, KZ and CW performed
literature review. CH and BY provided advice in revising the
manuscript and supervised the study. All authors read and approved
the final version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fateeva A, Eddy K and Chen S: Current
state of melanoma therapy and next steps: Battling therapeutic
resistance. Cancers (Basel). 16:15712024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Urban K, Mehrmal S, Uppal P, Giesey RL and
Delost GR: The global burden of skin cancer: A longitudinal
analysis from the global burden of disease study, 1990–2017. JAAD
Int. 2:98–108. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arnold M, Singh D, Laversanne M, Vignat J,
Vaccarella S, Meheus F, Cust AE, de Vries E, Whiteman DC and Bray
F: Global burden of cutaneous melanoma in 2020 and projections to
2040. JAMA Dermatol. 158:495–503. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beasley GM and Terando AM: Articles from
2022 to 2023 to inform your cancer practice: Melanoma. Ann Surg
Oncol. 31:1851–1856. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan J, Li X and Yu S: Global, regional,
and national incidence trend analysis of malignant skin melanoma
between 1990 and 2019, and projections until 2034. Cancer Control.
31:107327482412273402024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gracia-Hernandez M, Munoz Z and Villagra
A: Enhancing therapeutic approaches for melanoma patients targeting
epigenetic modifiers. Cancers (Basel). 13:61802021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang L and Yan Y: Emerging roles of
post-translational modifications in skin diseases: Current
knowledge, challenges and future perspectives. J Inflamm Res.
15:965–975. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reolid A, Muñoz-Aceituno E, Abad-Santos F,
Ovejero-Benito MC and Daudén E: Epigenetics in non-tumor
immune-mediated skin diseases. Mol Diagn Ther. 25:137–161. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Centeno PP, Pavet V and Marais R: The
journey from melanocytes to melanoma. Nat Rev Cancer. 23:372–390.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shain AH and Bastian BC: From melanocytes
to melanomas. Nat Rev Cancer. 16:345–358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elder DE, Bastian BC, Cree IA, Massi D and
Scolyer RA: The 2018 World Health Organization classification of
cutaneous, mucosal, and uveal melanoma: Detailed analysis of 9
distinct subtypes defined by their evolutionary pathway. Arch
Pathol Lab Med. 144:500–522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Long GV, Swetter SM, Menzies AM,
Gershenwald JE and Scolyer RA: Cutaneous melanoma. Lancet.
402:485–502. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gelmi MC, Houtzagers LE, Strub T, Krossa I
and Jager MJ: MITF in normal melanocytes, cutaneous and uveal
melanoma: A delicate balance. Int J Mol Sci. 23:60012022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Slominski RM, Kim TK, Janjetovic Z,
Brożyna AA, Podgorska E, Dixon KM, Mason RS, Tuckey RC, Sharma R,
Crossman DK, et al: Malignant melanoma: An overview, new
perspectives, and vitamin D signaling. Cancers (Basel).
16:22622024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Slominski RM, Chen JY, Raman C and
Slominski AT: Photo-neuro-immuno-endocrinology: How the ultraviolet
radiation regulates the body, brain, and immune system. Proc Natl
Acad Sci USA. 121:e23083741212024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Slominski RM, Raman C, Jetten AM and
Slominski AT: Neuro-immuno-endocrinology of the skin: how
environment regulates body homeostasis. Nat Rev Endocrinol.
21:495–509. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Slominski RM, Sarna T, Płonka PM, Raman C,
Brożyna AA and Slominski AT: Melanoma, melanin, and melanogenesis:
The Yin and Yang RELATIONSHIP. Front Oncol. 12:8424962022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wacker M and Holick MF: Sunlight and
vitamin D: A global perspective for health. Dermatoendocrinol.
5:51–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Menzies KJ, Zhang H, Katsyuba E and Auwerx
J: Protein acetylation in metabolism-metabolites and cofactors. Nat
Rev Endocrinol. 12:43–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weinert BT, Narita T, Satpathy S,
Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang
WW, Liu WR, et al: Time-resolved analysis reveals rapid dynamics
and broad scope of the CBP/p300 acetylome. Cell. 174:231–244.e12.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shvedunova M and Akhtar A: Modulation of
cellular processes by histone and non-histone protein acetylation.
Nat Rev Mol Cell Biol. 23:329–349. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Parveen R, Harihar D and Chatterji BP:
Recent histone deacetylase inhibitors in cancer therapy. Cancer.
129:3372–3380. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee HY, Hsu MJ, Chang HH, Chang WC, Huang
WC and Cho EC: Enhancing anti-cancer capacity: Novel class I/II
HDAC inhibitors modulate EMT, cell cycle, and apoptosis pathways.
Bioorg Med Chem. 109:1177922024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Tong X, Hu W and Chen D: HDAC11: A
novel target for improved cancer therapy. Biomed Pharmacother.
166:1154182023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jia G, Liu J, Hou X, Jiang Y and Li X:
Biological function and small molecule inhibitors of histone
deacetylase 11. Eur J Med Chem. 276:1166342024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Strub T, Ballotti R and Bertolotto C: The
‘ART’ of epigenetics in melanoma: From histone ‘alterations, to
resistance and therapies’. Theranostics. 10:1777–1797. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cintra Lopes Carapeto F, Neves Comodo A,
Germano A, Pereira Guimarães D, Barcelos D, Fernandes M and Landman
G: Marker protein expression combined with expression heterogeneity
is a powerful indicator of malignancy in acral lentiginous
melanomas. Am J Dermatopathol. 39:114–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Comodo-Navarro AN, Fernandes M, Barcelos
D, Carapeto FCL, Guimarães DP, de Sousa Moraes L, Cerutti J,
Iwamura ESM and Landman G: Intratumor heterogeneity of KIT gene
mutations in acral lentiginous melanoma. Am J Dermatopathol.
42:265–271. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng PF: Medical bioinformatics in
melanoma. Curr Opin Oncol. 30:113–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Castillo JJAQ, Silva W, Barcelos D and
Landman G: Molecular landscape of acral melanoma: an integrative
review. Surg Exp Pathol. 8:172025. View Article : Google Scholar
|
|
32
|
Wang L, Guo W, Ma J, Dai W, Liu L, Guo S,
Chen J, Wang H, Yang Y, Yi X, et al: Aberrant SIRT6 expression
contributes to melanoma growth: Role of the autophagy paradox and
IGF-AKT signaling. Autophagy. 14:518–533. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Strub T, Ghiraldini FG, Carcamo S, Li M,
Wroblewska A, Singh R, Goldberg MS, Hasson D, Wang Z, Gallagher SJ,
et al: SIRT6 haploinsufficiency induces BRAFV600E
melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat
Commun. 9:34402018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Manickavinayaham S, Vélez-Cruz R, Biswas
AK, Bedford E, Klein BJ, Kutateladze TG, Liu B, Bedford MT and
Johnson DG: E2F1 acetylation directs p300/CBP-mediated histone
acetylation at DNA double-strand breaks to facilitate repair. Nat
Commun. 10:49512019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim E, Zucconi BE, Wu M, Nocco SE, Meyers
DJ, McGee JS, Venkatesh S, Cohen DL, Gonzalez EC, Ryu B, et al:
MITF expression predicts therapeutic vulnerability to p300
inhibition in human melanoma. Cancer Res. 79:2649–2661. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dai X, Zhang X, Yin Q, Hu J, Guo J, Gao Y,
Snell AH, Inuzuka H, Wan L and Wei W: Acetylation-dependent
regulation of BRAF oncogenic function. Cell Rep. 38:1102502022.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vervoorts J, Lüscher-Firzlaff JM, Rottmann
S, Lilischkis R, Walsemann G, Dohmann K, Austen M and Lüscher B:
Stimulation of c-MYC transcriptional activity and acetylation by
recruitment of the cofactor CBP. EMBO Rep. 4:484–490. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fan T, Jiang S, Chung N, Alikhan A, Ni C,
Lee C-CR and Hornyak TJ: EZH2-dependent suppression of a cellular
senescence phenotype in melanoma cells by inhibition of p21/CDKN1A
expression. Mol Cancer Res. 9:418–429. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kovacs JJ, Murphy PJM, Gaillard S, Zhao X,
Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB and Yao TP: HDAC6
regulates Hsp90 acetylation and chaperone-dependent activation of
glucocorticoid receptor. Mol Cell. 18:601–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pulya S, Amin SA, Adhikari N, Biswas S,
Jha T and Ghosh B: HDAC6 as privileged target in drug discovery: A
perspective. Pharmacol Res. 163:1052742021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu J, Luan W, Zhang Y, Gu J, Shi Y, Yang
Y, Feng Z and Qi F: HDAC6 interacts with PTPN1 to enhance melanoma
cells progression. Biochem Biophys Res Commun. 495:2630–2636. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bai J, Lei Y, An G and He L:
Down-regulation of deacetylase HDAC6 inhibits the melanoma cell
line A375.S2 growth through ROS-dependent mitochondrial pathway.
PLoS One. 10:e01212472015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ling H, Li Y, Peng C, Yang S and Seto E:
HDAC10 blockade upregulates SPARC expression thereby repressing
melanoma cell growth and BRAF inhibitor resistance. bioRxiv
[Preprint]. 2023.12.05.570182. 2023.
|
|
44
|
Yuan W, Fang W, Zhang R, Lyu H, Xiao S,
Guo D, Ali DW, Michalak M, Chen XZ, Zhou C and Tang J: Therapeutic
strategies targeting AMPK-dependent autophagy in cancer cells.
Biochim Biophys Acta Mol Cell Res. 1870:1195372023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ling H, Li Y, Peng C, Yang S and Seto E:
HDAC10 inhibition represses melanoma cell growth and BRAF inhibitor
resistance via upregulating SPARC expression. NAR Cancer.
6:zcae0182024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mustofa MK, Tanoue Y, Tateishi C, Vaziri C
and Tateishi S: Roles of Chk2/CHEK2 in guarding against
environmentally induced DNA damage and replication-stress. Environ
Mol Mutagen. 61:730–735. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang W, Feng Y, Guo Q, Guo W, Xu H, Li X,
Yi F, Guan Y, Geng N, Wang P, et al: SIRT1 modulates cell cycle
progression by regulating CHK2 acetylation-phosphorylation. Cell
Death Differ. 27:482–496. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guo QQ, Wang SS, Zhang SS, Xu HD, Li XM,
Guan Y, Yi F, Zhou TT, Jiang B, Bai N, et al: ATM-CHK2-Beclin 1
axis promotes autophagy to maintain ROS homeostasis under oxidative
stress. EMBO J. 39:e1031112020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mendelson K, Martin TC, Nguyen CB, Hsu M,
Xu J, Lang C, Dummer R, Saenger Y, Messina JL, Sondak VK, et al:
Differential histone acetylation and super-enhancer regulation
underlie melanoma cell dedifferentiation. JCI Insight.
9:e1666112024.PubMed/NCBI
|
|
50
|
Min D, Byun J, Lee EJ, Khan AA, Liu C,
Loudig O, Hu W, Zhao Y, Herlyn M, Tycko B, et al: Epigenetic
silencing of BMP6 by the SIN3A-HDAC1/2 repressor complex drives
melanoma metastasis via FAM83G/PAWS1. Mol Cancer Res. 20:217–230.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hornig E, Heppt MV, Graf SA, Ruzicka T and
Berking C: Inhibition of histone deacetylases in melanoma-a
perspective from bench to bedside. Exp Dermatol. 25:831–838. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kim JY, Cho H, Yoo J, Kim GW, Jeon YH, Lee
SW and Kwon SH: HDAC8 deacetylates HIF-1α and enhances its protein
stability to promote tumor growth and migration in melanoma.
Cancers (Basel). 15:11232023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Emmons MF, Bennett RL, Riva A, Gupta K,
Carvalho LADC, Zhang C, Macaulay R, Dupéré-Richér D, Fang B, Seto
E, et al: HDAC8-mediated inhibition of EP300 drives a
transcriptional state that increases melanoma brain metastasis. Nat
Commun. 14:77592023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kong F, Ma L, Wang X, You H, Zheng K and
Tang R: Regulation of epithelial-mesenchymal transition by protein
lysine acetylation. Cell Commun Signal. 20:572022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sun T, Jiao L, Wang Y, Yu Y and Ming L:
SIRT1 induces epithelial-mesenchymal transition by promoting
autophagic degradation of E-cadherin in melanoma cells. Cell Death
Dis. 9:1362018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kunimoto R, Jimbow K, Tanimura A, Sato M,
Horimoto K, Hayashi T, Hisahara S, Sugino T, Hirobe T, Yamashita T
and Horio Y: SIRT1 regulates lamellipodium extension and migration
of melanoma cells. J Invest Dermatol. 134:1693–1700. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Porta C, Paglino C and Mosca A: Targeting
PI3K/Akt/mTOR signaling in cancer. Front Oncol. 4:642014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang Y, Liu Y, Wang Y, Chao Y, Zhang J,
Jia Y, Tie J and Hu D: Regulation of SIRT1 and its roles in
inflammation. Front Immunol. 13:8311682022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bajpe PK, Prahallad A, Horlings H,
Nagtegaal I, Beijersbergen R and Bernards R: A chromatin modifier
genetic screen identifies SIRT2 as a modulator of response to
targeted therapies through the regulation of MEK kinase activity.
Oncogene. 34:531–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhao N, Guo Y, Liu P, Chen Y and Wang Y:
Sirtuin 2 promotes cell stemness and MEK/ERK signaling pathway
while reduces chemosensitivity in endometrial cancer. Arch Gynecol
Obstet. 305:693–701. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang QL and Guo SJ: Sirtuins function as
the modulators in aging-related diseases in common or respectively.
Chin Med J (Engl). 128:1671–1678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yi X, Wang H, Yang Y, Wang H, Zhang H, Guo
S, Chen J, Du J, Tian Y, Ma J, et al: SIRT7 orchestrates melanoma
progression by simultaneously promoting cell survival and immune
evasion via UPR activation. Signal Transduct Target Ther.
8:1072023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu J, Zhuang A, Gu X, Hua Y, Yang L, Ge S,
Ruan J, Chai P, Jia R and Fan X: Nuclear PD-L1 promotes
EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell
Discov. 9:332023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gao Y, Nihira NT, Bu X, Chu C, Zhang J,
Kolodziejczyk A, Fan Y, Chan NT, Ma L, Liu J, et al:
Acetylation-dependent regulation of PD-L1 nuclear translocation
dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol.
22:1064–1075. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wilmott JS, Colebatch AJ, Kakavand H,
Shang P, Carlino MS, Thompson JF, Long GV, Scolyer RA and Hersey P:
Expression of the class 1 histone deacetylases HDAC8 and 3 are
associated with improved survival of patients with metastatic
melanoma. Mod Pathol. 28:884–894. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
López-Bañuelos L and Vega L: Inhibition of
acetylation, is it enough to fight cancer? Crit Rev Oncol Hematol.
176:1037522022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pan S and Chen R: Pathological implication
of protein post-translational modifications in cancer. Mol Aspects
Med. 86:1010972022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
He W, Li Q and Li X: Acetyl-CoA regulates
lipid metabolism and histone acetylation modification in cancer.
Biochim Biophys Acta Rev Cancer. 1878:1888372023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Krumm A, Barckhausen C, Kücük P,
Tomaszowski KH, Loquai C, Fahrer J, Krämer OH, Kaina B and Roos WP:
Enhanced histone deacetylase activity in malignant melanoma
provokes RAD51 and FANCD2-triggered drug resistance. Cancer Res.
76:3067–3077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hess L, Moos V, Lauber AA, Reiter W,
Schuster M, Hartl N, Lackner D, Boenke T, Koren A, Guzzardo PM, et
al: A toolbox for class I HDACs reveals isoform specific roles in
gene regulation and protein acetylation. PLoS Genet.
18:e10103762022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen G, Cheng Y, Tang Y, Martinka M and Li
G: Role of Tip60 in human melanoma cell migration, metastasis, and
patient survival. J Invest Dermatol. 132:2632–2641. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Karst AM, Dai DL, Martinka M and Li G:
PUMA expression is significantly reduced in human cutaneous
melanomas. Oncogene. 24:1111–1116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang Y, Subbaiah VK, Rajagopalan D, Tham
CY, Abdullah LN, Toh TB, Gong M, Tan TZ, Jadhav SP, Pandey AK, et
al: TIP60 inhibits metastasis by ablating DNMT1-SNAIL2-driven
epithelial-mesenchymal transition program. J Mol Cell Biol.
8:384–399. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang X, Bustos MA, Shoji Y, Ramos RI,
Iida Y, Gentry R, Takeshima TL and Hoon DSB: Acetylated DNMT1
downregulation and related regulatory factors influence metastatic
melanoma patients survival. Cancers (Basel). 13:46912021.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bhandaru M, Ardekani GS, Zhang G, Martinka
M, McElwee KJ, Li G and Rotte A: A combination of p300 and Braf
expression in the diagnosis and prognosis of melanoma. BMC Cancer.
14:3982014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rotte A, Bhandaru M, Cheng Y, Sjoestroem
C, Martinka M and Li G: Decreased expression of nuclear p300 is
associated with disease progression and worse prognosis of melanoma
patients. PLoS One. 8:e754052013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Roos WP and Krumm A: The multifaceted
influence of histone deacetylases on DNA damage signalling and DNA
repair. Nucleic Acids Res. 44:10017–10030. 2016.PubMed/NCBI
|
|
79
|
Booth L, Roberts JL, Sander C, Lee J,
Kirkwood JM, Poklepovic A and Dent P: The HDAC inhibitor AR42
interacts with pazopanib to kill trametinib/dabrafenib-resistant
melanoma cells in vitro and in vivo. Oncotarget. 8:16367–16386.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Booth L, Roberts JL, Poklepovic A,
Kirkwood J and Dent P: HDAC inhibitors enhance the immunotherapy
response of melanoma cells. Oncotarget. 8:83155–83170. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Woods DM, Sodré AL, Villagra A, Sarnaik A,
Sotomayor EM and Weber J: HDAC inhibition upregulates PD-1 ligands
in melanoma and augments immunotherapy with PD-1 blockade. Cancer
Immunol Res. 3:1375–1385. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Beg AA and Gray JE: HDAC inhibitors with
PD-1 blockade: A promising strategy for treatment of multiple
cancer types? Epigenomics. 8:1015–1017. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Noonepalle S, Shen S, Ptáček J, Tavares
MT, Zhang G, Stránský J, Pavlíček J, Ferreira GM, Hadley M, Pelaez
G, et al: Rational design of suprastat: A novel selective histone
deacetylase 6 inhibitor with the ability to potentiate
immunotherapy in melanoma models. J Med Chem. 63:10246–10262. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Peng X, Yu Z, Surineni G, Deng B, Zhang M,
Li C, Sun Z, Pan W, Liu Y, Liu S, et al: Discovery of novel
benzohydroxamate-based histone deacetylase 6 (HDAC6) inhibitors
with the ability to potentiate anti-PD-L1 immunotherapy in
melanoma. J Enzyme Inhib Med Chem. 38:22014082023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li F, Zhao X, Zhang Y, Shao P, Ma X,
Paradee WJ, Liu C, Wang J and Xue HH: TFH cells depend
on Tcf1-intrinsic HDAC activity to suppress CTLA4 and guard B-cell
help function. Proc Natl Acad Sci USA. 118:e20145621182021.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
van der Waart AB, van de Weem NMP, Maas F,
Kramer CSM, Kester MGD, Falkenburg JHF, Schaap N, Jansen JH, van
der Voort R, Gattinoni L, et al: Inhibition of Akt signaling
promotes the generation of superior tumor-reactive T cells for
adoptive immunotherapy. Blood. 124:3490–3500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kreidieh F and Wong MK: New standards in
the treatment of advanced metastatic melanoma: Immunotherapy and
BRAF-targeted therapies as emerging paradigms. Curr Pharm Des. May
26–2025.(Epub ahead of print). PubMed/NCBI
|
|
88
|
Zhang F, Zhou X, DiSpirito JR, Wang C,
Wang Y and Shen H: Epigenetic manipulation restores functions of
defective CD8+ T cells from chronic viral infection. Mol
Ther. 22:1698–1706. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Blake MK, O'Connell P and Aldhamen YA:
Fundamentals to therapeutics: Epigenetic modulation of
CD8+ T Cell exhaustion in the tumor microenvironment.
Front Cell Dev Biol. 10:10821952023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Vo DD, Prins RM, Begley JL, Donahue TR,
Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, et
al: Enhanced antitumor activity induced by adoptive T-cell transfer
and adjunctive use of the histone deacetylase inhibitor LAQ824.
Cancer Res. 69:8693–8699. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang J, Hasan F, Frey AC, Li HS, Park J,
Pan K, Haymaker C, Bernatchez C, Lee DA, Watowich SS and Yee C:
Histone deacetylase inhibitors and IL21 cooperate to reprogram
human effector CD8+ T cells to memory T cells. Cancer Immunol Res.
8:794–805. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lisiero DN, Soto H, Everson RG, Liau LM
and Prins RM: The histone deacetylase inhibitor, LBH589, promotes
the systemic cytokine and effector responses of adoptively
transferred CD8+ T cells. J Immunother Cancer. 2:82014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Heijkants R, Willekens K, Schoonderwoerd
M, Teunisse A, Nieveen M, Radaelli E, Hawinkels L, Marine JC and
Jochemsen A: Combined inhibition of CDK and HDAC as a promising
therapeutic strategy for both cutaneous and uveal metastatic
melanoma. Oncotarget. 9:6174–6187. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gallagher SJ, Gunatilake D, Beaumont KA,
Sharp DM, Tiffen JC, Heinemann A, Weninger W, Haass NK, Wilmott JS,
Madore J, et al: HDAC inhibitors restore BRAF-inhibitor sensitivity
by altering PI3K and survival signalling in a subset of melanoma.
Int J Cancer. 142:1926–1937. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Badamchi-Zadeh A, Moynihan KD, Larocca RA,
Aid M, Provine NM, Iampietro MJ, Kinnear E, Penaloza-MacMaster P,
Abbink P, Blass E, et al: Combined HDAC and BET inhibition enhances
melanoma vaccine immunogenicity and efficacy. J Immunol.
201:2744–2752. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Shan X, Fu YS, Aziz F, Wang XQ, Yan Q and
Liu JW: Ginsenoside Rg3 inhibits melanoma cell proliferation
through down-regulation of histone deacetylase 3 (HDAC3) and
increase of p53 acetylation. PLoS One. 9:e1154012014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yan G, Eller MS, Elm C, Larocca CA, Ryu B,
Panova IP, Dancy BM, Bowers EM, Meyers D, Lareau L, et al:
Selective inhibition of p300 HAT blocks cell cycle progression,
induces cellular senescence, and inhibits the DNA damage response
in melanoma cells. J Invest Dermatol. 133:2444–2452. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
van den Bosch T, Boichenko A, Leus NGJ,
Ourailidou ME, Wapenaar H, Rotili D, Mai A, Imhof A, Bischoff R,
Haisma HJ and Dekker FJ: The histone acetyltransferase p300
inhibitor C646 reduces pro-inflammatory gene expression and
inhibits histone deacetylases. Biochem Pharmacol. 102:130–140.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chen YF, Rahman A, Sax JL, Atala
Pleshinger MJ, Friedrich RM and Adams DJ: C646 degrades exportin-1
to modulate p300 chromatin occupancy and function. Cell Chem Biol.
31:1363–1372.e8. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Mitsiogianni M, Anestopoulos I, Kyriakou
S, Trafalis DT, Franco R, Pappa A and Panayiotidis MI: Benzyl and
phenethyl isothiocyanates as promising epigenetic drug compounds by
modulating histone acetylation and methylation marks in malignant
melanoma. Invest New Drugs. 39:1460–1468. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Okonkwo A, Mitra J, Johnson GS, Li L,
Dashwood WM, Hegde ML, Yue C, Dashwood RH and Rajendran P:
Heterocyclic analogs of sulforaphane trigger DNA damage and impede
DNA repair in colon cancer cells: Interplay of HATs and HDACs. Mol
Nutr Food Res. 62:e18002282018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mitsiogianni M, Mantso T, Trafalis DT,
Vasantha Rupasinghe HP, Zoumpourlis V, Franco R, Botaitis S, Pappa
A and Panayiotidis MI: Allyl isothiocyanate regulates lysine
acetylation and methylation marks in an experimental model of
malignant melanoma. Eur J Nutr. 59:557–569. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mitsiogianni M, Trafalis DT, Franco R,
Zoumpourlis V, Pappa A and Panayiotidis MI: Sulforaphane and iberin
are potent epigenetic modulators of histone acetylation and
methylation in malignant melanoma. Eur J Nutr. 60:147–158. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Li K, Zhang TT, Wang F, Cui B, Zhao CX, Yu
JJ, Lv XX, Zhang XW, Yang ZN, Huang B, et al: Metformin suppresses
melanoma progression by inhibiting KAT5-mediated SMAD3 acetylation,
transcriptional activity and TRIB3 expression. Oncogene.
37:2967–2981. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chen J, Huang Z, Chen Y, Tian H, Chai P,
Shen Y, Yao Y, Xu S, Ge S and Jia R: Lactate and lactylation in
cancer. Signal Transduct Target Ther. 10:382025. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang D, Tang Z, Huang H, Zhou G, Cui C,
Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al: Metabolic
regulation of gene expression by histone lactylation. Nature.
574:575–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhao L, Qi H, Lv H, Liu W, Zhang R and
Yang A: Lactylation in health and disease: Physiological or
pathological? Theranostics. 15:1787–1821. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Dai X, Lv X, Thompson EW and Ostrikov KK:
Histone lactylation: Epigenetic mark of glycolytic switch. Trends
Genet. 38:124–127. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F,
Gill PS, Ha T, Liu L, Williams DL and Li C: Lactate promotes
macrophage HMGB1 lactylation, acetylation, and exosomal release in
polymicrobial sepsis. Cell Death Differ. 29:133–146. 2022.
View Article : Google Scholar : PubMed/NCBI
|














