Leukemia is a clonal malignant disorder of
hematopoietic precursor cells that present as an excess of one or
more hematologic cell types in the bone marrow and bloodstream. It
is caused by a disruption of the normal processes of cell
proliferation and cell death. There are four major types of
leukemias: Acute lymphoblastic leukemia (ALL), acute myelogenous
leukemia (AML), chronic lymphocytic leukemia (CLL) and chronic
myelogenous leukemia (CML). The classification depends on the
lineage and stage of the presenting leukemia cells and the rate of
leukemia cell proliferation (1). In
2022, there were an estimated 48.7 hundred thousand new cases
(ranked 13th of 33) and 30.5 hundred thousand deaths (ranked 10th
of 33) from leukemia worldwide (2).
Leukemia incidence varies among populations in different age groups
and countries. ALL is most common in children and young adults,
whereas AML, CLL and CML commonly occur in older adults. In adults
aged ≥20 years, AML had the highest incidence and mortality rate
among all leukemia types. A high incidence of leukemia has been
reported in developed regions of the world such as Western Europe,
North America and Australia (3,4).
According to statistics acquired over the last three decades, in
most regions, the cumulative risk of leukemia incidence has
increased faster than the lifetime risk of leukemia mortality
(5,6). The decreasing trends in mortality
rates are potentially a result of significant advancements in
leukemia diagnosis and therapies.
Conventionally, leukemia treatment relies on
chemotherapy, which uses cytotoxic agents to destroy rapidly
dividing leukemia cells. Principal chemotherapy drugs, including
vincristine and an anthracycline, have been used in combination
with other drugs, such as all-trans retinoic acid,
cyclophosphamide, methotrexate and cytarabine, to achieve a
satisfactory complete remission rate (7,8).
However, since these drugs are not selective for cancer cells, they
often cause undesirable side effects, some of which are more severe
than others, such as neurotoxicity (9) and cardiomyopathy (10,11).
These adverse effects not only worsen patient quality of life but
also limit the dose and efficacy of chemotherapy drugs. Moreover,
drug resistance and ineffectiveness in patients with some subtypes
of leukemia have been reported (12,13).
Bone marrow or allogenic hematopoietic stem cell (HSC)
transplantation can be options for the treatment of relapsed or
refractory leukemia. Alternatively, novel therapeutic substances
are needed to simultaneously overcome resistance and alleviate the
adverse effects of chemotherapy. Over the past two decades, an
improved understanding of the pathophysiology of leukemias has
given rise to a rapid progress in leukemia therapeutic research.
Targeted therapy began to play important roles in leukemia
treatment. Cancer cell-selective drugs, including small molecule
inhibitors targeting tyrosine kinases or
phosphatidylinositol-3-kinases and antibodies against specific CD
molecules, have emerged as attractive therapeutic options. To lower
the dose of traditional cytotoxic drugs, such targeted drugs have
often been applied in combination regimens. Cytogenetic and
molecular aberrations are key factors that guide the development of
targeted therapies. Consequently, they increase the complete
cytogenetic response rate and improve patient quality of life in
almost every type of leukemia (14). Excellent reviews discussing the
advancement of leukemia therapy have been published recently
(15,16). However, some leukemia subtypes are
refractory or already resistant to newly developed drugs, making
drug discovery research challenging.
Although genetic alterations of oncogenes play a
significant role in leukemia progression, epigenetic aberrations
have been shown to affect transcriptional regulation and the
development of leukemia cells. In the past few years, several new
therapeutic strategies have been proposed for the treatment of
leukemia on the basis of epigenetic aberrations (17,18).
Among numerous epigenetic modulators, histone deacetylase (HDAC), a
deacetylase that acts on histone and non-histone proteins to alter
chromatin structure and regulate gene expression, is the most
intensively studied therapeutic target in leukemia (19). Overexpression of HDAC has been
demonstrated to stimulate the progression of leukemogenesis and
tumorigenesis by inhibiting tumor suppressor gene expression
(20,21). Blocking HDAC activity reactivates
tumor suppressor genes and directly suppresses tumor cell
proliferation and induces apoptosis. This renders HDAC inhibitors
(HDACis) potential targeted therapeutic agents for the management
of leukemia and other cancers. Over the past decade, novel HDACis
have been discovered and developed from both natural and synthetic
sources (22,23). Among the various compounds that
possess HDAC inhibition activity, peptides derived from diverse
sources, including food, environment and laboratory production, are
potential candidate HDACis that can influence epigenetic modulation
(24). Compared with chemical
drugs, peptide drugs provide advantages such as high specificity,
low toxicity and biological diversity. Additionally, peptides can
be rationally designed, which will be beneficial in targeted
therapy development. The present review provides an overview of
HDAC and HDACis types and functions with an emphasis on the role of
HDACs in leukemogenesis. The development of HDACis and the HDAC
inhibitory effects of potential therapeutic peptides on leukemia in
preclinical and clinical trials are discussed.
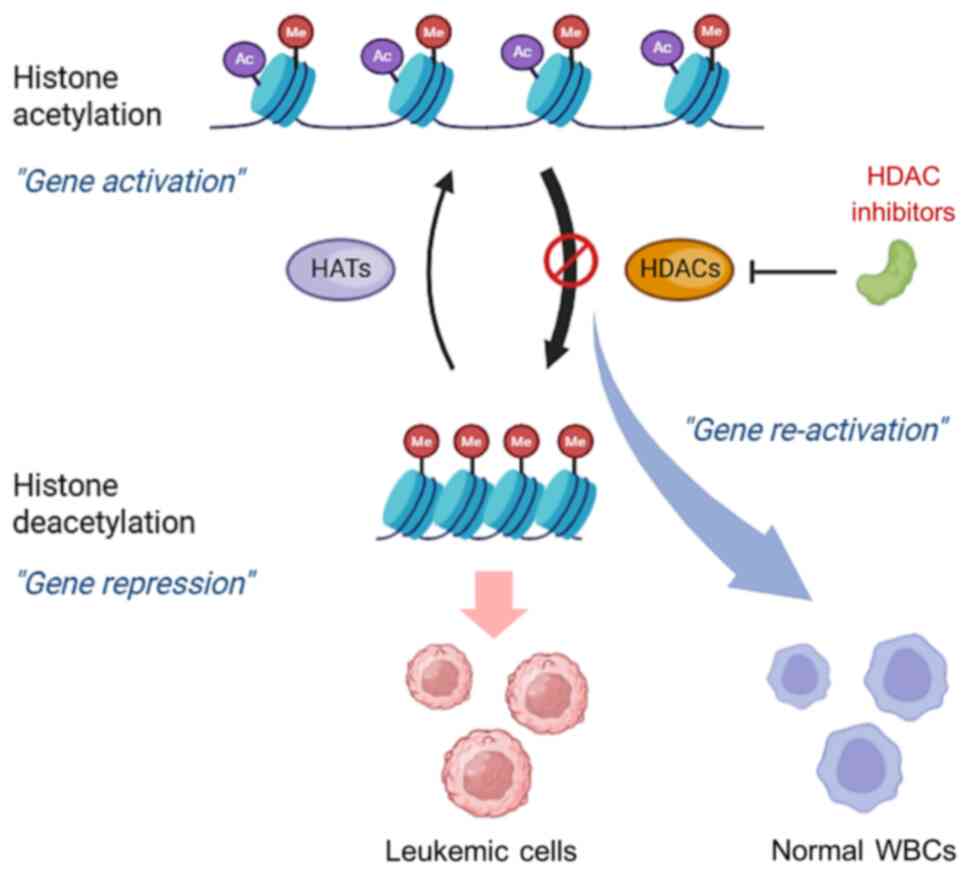
Histone acetylation represents one of the main
mechanisms of epigenetic modifications and is associated with two
different enzymes, namely, histone acetyltransferases (HATs) and
HDACs. HATs mediate the relaxation of nucleosomes to allow opening
of chromatin by adding acetyl groups at the ε-amino group of
N-terminal lysine residues in the histone tail, resulting in the
upregulation of gene expression, whereas deacetylation mediated by
HDACs removes acetyl groups and induces condensation of chromatin,
resulting in the repression of the gene (25). An imbalance in the amount of HAT and
HDAC enzymes interferes with the regulation of gene expression and
induces the progression of several cancers (26).
In humans, HDACs include 18 enzymes categorized into
two families: The classical HDAC family, which is composed of
HDAC1-11, and the silent information regulator (SIR)-2 family,
which is composed of SIRT1-7. These two families can be subdivided
into four classes based on catalytic mechanisms and sequence
homology to yeast deacetylases. HDACs in the same class possess
similar structures and functions (27,28).
HDAC class members and their characteristics are described in
Table I.
Detailed information on the structure and catalytic
mechanisms of each HDAC can be found in the extensive review by
Asmamaw et al (29).
All classes of HDACs participate in hematopoiesis by
regulating multilineage blood cell development. Their functions
involve stemness maintenance of HSCs and the lineage commitment of
progenitor cells. HDACs interact with transcription factors and/or
other cofactor proteins to modulate histone acetylation levels,
which subsequently regulate the expression of several hematopoietic
lineage-related genes (30–32). It has been shown that HDACs play
crucial roles in the cell fate decisions of hematopoietic
progenitors (33,34).
Class I HDACs, especially HDAC1, are major HDACs
expressed in hematopoietic cells. They are found in all
hematopoietic stages, but expression levels differ among the types
of blood cells. HDAC1 is undetectable in early progenitor cells,
moderately expressed in committed progenitor cells, and disappears
in mature granulocytes, reflecting its dose-dependent role in
modulating progenitor cell differentiation (35). A previous study in hematopoietic
cell lines demonstrated that HDAC1 plays essential roles in
maintaining the immature state of committed progenitor cells and
contributes to GATA-1 mediated erythroid differentiation (36). HDAC1 transactivation is driven by
Sp1 and GATA-1, whereas its transcriptional repression is mediated
by C/EBPα and C/EBPβ (34). HDAC1
is accompanied by GATA1-Sp1 complexes during the differentiation of
common myeloid progenitors (CMPs) into megakaryocyte-erythrocyte
progenitor cells. By contrast, HDAC1 is downregulated by
GATA2-C/EBP complexes during CMP differentiation into
granulocyte-monocyte progenitor cells (37,38).
The upregulation of HDAC1 facilitates the movement of
granulocyte-monocyte progenitor cells toward granulocytic cells,
whereas its downregulation induces the development of
granulocyte-monocyte progenitor cells into monocytes, macrophages
and dendritic cells (39). A
previous study in a mouse model indicated that both HDAC1 and HDAC2
in a complex with their corepressor, Sin3A, serve as
cell-autonomous regulators of HSC maintenance (40). They also play a role in the
differentiation of megakaryocytic-erythroid progenitor cells
(41), pre-B cells (42), T lymphocytes (43) and NK cells (44). HDAC1 and HDAC3 are accompanied by
Runx1 as repressor complexes to control the progression and
maturation of granulocytes from progenitor cells, ensuring proper
granulocytic development and function. Class IIa HDAC4 has been
reported to act as a corepressor recruited by BCL6 to repress genes
critical for regulating B-cell development process (45), and class IIa HDAC7 plays a role in
the control of thymic selection during T-cell development (46). In addition, class IIb HDAC6 and
class I HDAC2 stimulate the enucleation process of orthochromatic
normoblasts. Members of class III HDACs have also been implicated
in hematopoiesis. SIRT1 promotes the hematopoietic microenvironment
through CXCL12 upregulation (47),
and influences granulopoiesis through a regulatory loop between
G-CFGR and G-CSF (48). Defects in
hematopoietic progenitor differentiation and the downregulation of
genes associated with hematopoietic development have been
demonstrated in SIRT1-deficient mouse (49). SIRT2, SIRT3 and SIRT7 also play
important roles in HSC maintenance and homeostasis, especially
under stress and in elderly individuals (50–52).
The last HDAC group, class IV HDAC11, is involved in the
development of promyelocytes into neutrophils (53).
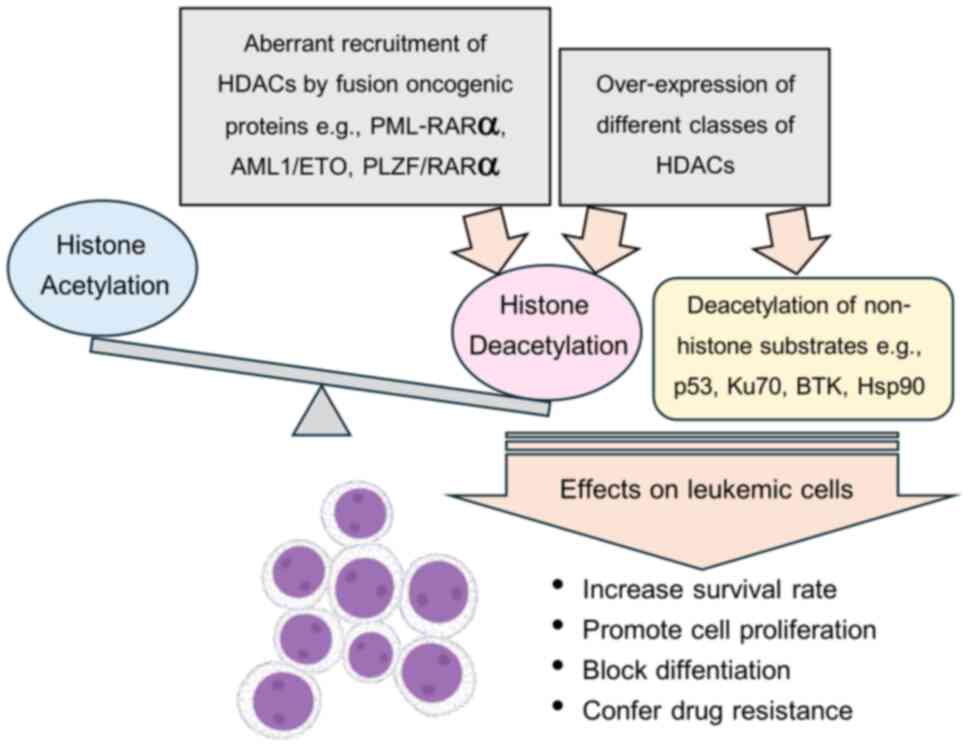
Abnormal histone deacetylation has been implicated
in leukemia initiation and progression via two mechanisms; aberrant
recruitment of an HDAC enzyme by an oncogenic fusion protein and
alteration in the expression of HDACs. Some notable implications of
HDACs in leukemogenesis are described.
It is well-known that one of the most common genetic
rearrangements that causes leukemia is chromosome translocation,
leading to the expression of fusion proteins associated with
oncogenic transformation. In acute promyelocytic leukemia (APL),
the most frequent chromosomal translocation t (15;17) results in
the fusion protein PML-RARα. PML-induced dimerization of RARα
enhances the binding of the corepressor complex NCoR/SMRT and class
I and class II HDACs, thus repressing the expression of retinoic
acid (RA)-target genes which in turn block differentiation of
myeloid precursors (54).
Similarly, other fusion proteins found in AML such as PLZF/RARα and
AML1/EMO have been reported to induce transcriptional repression of
genes critical to the differentiation of granulocytic precursors
through enhanced recruitment of the HDAC-corepressor complex
(55,56). HDAC1 and HDAC3 are recruited by
fusion proteins such as AML1/ETO and subsequently form a complex
that acts as an abnormal TF leading to leukemogenesis. Moreover,
AML1-ETO can recruit HDAC1 and HDAC2 to bind NCoR-mSin3 and
prevents leukemia cells from undergoing TNF-related apoptosis
(57). Overexpression of SETBP1, an
oncogenic protein resulting from t (12;18) in AML, was also
reported to induce leukemia development through transcriptional
repression of the critical hematopoiesis regulator gene Runx1 via
recruitment of HDAC1 (58).
An imbalance in HAT and HDAC enzyme levels also
affects gene transcription and is involved in the development of
hematological malignancies. Cumulative evidence suggests that the
aberrant hypoacetylation of histones associated with the
overexpression of HDACs leads to the repression of tumor suppressor
genes that regulate the cell cycle and DNA repair pathways and
alteration of the cell differentiation program, leading to
leukemogenesis (59,35,60).
The differential overexpression of HDAC isoforms is related to
different subtypes of leukemia. For instance, HDAC1 overexpression
is associated with ALL, CML and AML, whereas the high expression of
HDAC3, 6, and 7 is linked to pathogenesis and prognosis in ALL and
CLL (61). A comprehensive study of
the expression of all 18 HDAC isoforms in 200 CLL patients by Van
Damme et al (62) showed
that there are variations in the expression levels of different
HDACs. HDAC6, HDAC7, HDAC11, SIRT3, SIRT6 and SIRT7 were
upregulated, whereas HDAC2 and SIRT4 were significantly
downregulated in patients with CLL compared with normal B cells.
Correlation analysis revealed that high levels of HDAC6 are
significantly associated with longer treatment-free survival, and
high levels of HDAC3, SIRT2, SIRT3 and SIRT6 are associated with a
longer overall survival. Additionally, the results suggested that
HDAC7 and HDAC10 overexpression and HDAC6 and SIRT3
under-expression are associated with poor prognosis (62). Recently, Verbeek et al
(63) demonstrated that relevancy
between Class IIA HDAC isoforms (HDAC4, HDAC5, HDAC7) shows
critical prognostic relevance in KMT2A-rearranged ALL. Knockdown or
selective inhibition of such HDAC isoforms induced apoptosis and
impacted leukemic infiltration in protective niches such as bone
marrow and CNS, which is crucial for prognosis in infant ALL
(63). HDACs not only act on
histone tails but also affect nonhistone substrates, including
transcription factors and other cofactor proteins. The first
described non-histone substrate of HATs and HDACS is the tumor
suppressor p53 protein, which is the key transcription factor in
cellular stress response pathways (64). P53 plays important roles in
regulating several biological mechanisms, including cell
proliferation, cell differentiation, the cell cycle, the apoptotic
pathway and DNA repair (65).
Normally, p53 activity is modulated by CBP/p300-mediated
acetylation. It has been found that upregulation of HDAC1
potentially block apoptosis induced by the deacetylation of p53,
which results in an increased survival rate of AML cells (66). In AML with inv(16)+, high expression
of HDAC8 was detected in primitive CD34+ cells and it was recruited
by CBFβ-SMMHC fusion proteins to form complexes with p53. These
complexes mediate the aberrant deacetylation of p53 by HDAC8 and
subsequently promote AML progression (67). Furthermore, high expression of class
I HDACs induces aberrant acetylation of p53 and Ku70, resulting in
resistance to imatinib in patients with CML with positive
Philadelphia chromosome (68). It
has been reported that HDAC7 regulates the phagocytic activity of
monocyte-derived macrophages obtained from patients with CLL
through direct modulation of BTK acetylation and phosphorylation.
It has been suggested that HDAC7 contributes to therapeutic
antibody resistance in patients with CLL and CLL who have high HDAC
expression may result in a poor prognosis (69). The proven mechanisms that lead to
abnormal deacetylation, resulting in leukemogenesis and leukemia
progression, are depicted in Fig.
1.
Unlike genetic alterations, aberrant epigenetic
modifications are reversible; therefore, targeting epigenetic
modulators is a promising approach for the treatment of leukemia.
Understanding of HDAC-related leukemogenesis has encouraged the
development of HDACis that aim to restore normal gene expression by
reactivating silenced tumor suppressor genes, inducing
differentiation, and promoting the cessation of leukemia cell
proliferation. In recent years, numerous HDACis have been
extensively investigated in the search for viable therapeutics for
leukemia. Some HDACis can be combined with conventional therapy to
improve treatment efficacy and overcome resistance. Moreover,
precision therapy can be established by rationally designing
selective HDACi-based treatments via the guidance of HDAC
expression profiling or the development of muti-targeted
dual/hybrid inhibitors.
As aforementioned, several lines of evidence suggest
that the aberrant expression and function of HDAC enzymes play
important roles in several solid cancers and hematological
malignancies. HDACs facilitate tumor suppressor gene silencing in
cancer cells, preventing them from undergoing apoptosis. Thus, the
development of a new therapeutic agent that targets the HDAC enzyme
to restore the acetylation of histones is needed (Fig. 2). It has been clearly described that
HDACis can induce cell cycle arrest and cancer cell death. The main
inhibitory mechanism of HDACis involves blocking the substrate
binding of HDAC by interacting with the enzyme catalytic domain.
HDACis have been discovered and identified from both natural and
synthetic sources. HDACis can be classified into 4 groups based on
their chemical structure. The three groups are small-molecule
inhibitors, which include short-chain fatty acids, hydroxamic acids
and benzamides. The other group is cyclic peptide inhibitors.
Small-molecule HDACis are usually pan-HDACis, whereas peptide-based
HDACis are more class-selective or isoform-selective HDACis due to
their larger molecular dimensions and conformational flexibility.
This feature enables them to mimic natural protein interactions
more precisely. All HDACis share the same core structure consisting
of three domains, which consists of a cap region, a hydrophobic
linker and a zinc binding group (ZBG). The cap region interacts on
the surface at the entrance of the substrate, ZBG is the position
of the functional group that binds and chelates a zinc ion in the
catalytic site, and the hydrophobic linker domain is the region
between the cap region and ZBG (70,71).
Peptide inhibitors typically have a more complex ‘cap group’ that
binds extensively to the enzyme surface (72), triggering disruption of HDAC
interactions with specific corepressor complexes or
chromatin-associated scaffolds in leukemia cells. This allows them
to potentially modulate leukemia-specific epigenetic states more
precisely. On the other hand, small-molecule inhibitors
predominantly rely on zinc-chelation and catalytic site blockade,
thus broadly inhibiting catalytic activity and affecting multiple
HDAC isoforms and complexes (73).
This mechanistic difference highlights the potential of peptides
for refined epigenetic therapy in leukemia. The differences between
small-molecule and peptide-based HDACis are summarized in Table II.
Short-chain fatty acids such as butyrate and
valproic acid (VPA) are inhibitors of class I and IIa HDACs.
Butyrate is produced natively by anaerobic bacterial fermentation
of carbohydrates in the colon and longer-chain fatty acid
metabolism. It has been reported that the possible HDAC inhibition
mechanism of butyrate may be attributed to its hydrophobic
interaction with the HDAC active pocket; hence, it binds
non-specifically (74). Butyrate
induces the hyperacetylation of histones, regulating gene
expression, and has been used for cancer treatment (75,76).
It is characterized by low activity, short half-life and rapid
metabolism, leading to a high effective concentration in
vivo (77,78). VPA, a short chain aliphatic acid,
has been reported to have antileukemic effects, such as
anti-proliferation, induction of differentiation and stimulation of
apoptosis, in AML (79,80). Certain studies have shown that VPA
also has low HDAC inhibition activity; however, the combination of
VPA with another chemotherapy drugs could increase the response in
patients with leukemia (81,82).
Hydroxamic acids are organic compounds that can form
table complexes with a variety of metal ions; therefore, their
mechanism of action involves chelation zinc ions in the HDAC
catalytic site (71). Hydroxamic
acids act as potent pan-HDACis that affect HDAC classes I and II.
Trichostatin A (TSA) is a naturally occurring compound that
consists of an aromatic group as a cap region linked by a diene
region connected to the hydroxamate tail. TSA has been proposed as
an effective drug for the treatment of CLL (83). Since it has been shown that the
mutant HDAC enzyme affects the binding activity between TSA and
HDAC isoforms, TSA is used in clinical treatment with limitations
(84). Suberoylanilide hydroxamic
acid (SAHA; vorinostat) is a synthetic hydroxamic acid-containing
HDACi that is structurally related to TSA. SAHA was approved by the
FDA for the treatment of refractory cutaneous T-cell lymphoma
(85). Other HDACi drugs composed
of hydroxamic acid groups are belinostat and panobinostat (86,87).
These hydroxamate compounds have been recently investigated in
clinical trials for the treatment of other hematological
malignancies.
Benzamides (amino anilides) are synthetic compounds
that display selective inhibitory activity against class I HDACs.
Benzamides can interact with and chelate a zinc ion in the HDAC
pocket site, but their chelating activity is lower than that of
hydroxamate and cyclic peptide compounds (88). Several benzamide derivatives,
including entinostat and mocetinostat, have been reported to
inhibit malignant cell proliferation and are currently undergoing
clinical trials for the treatment of hematologic malignancies
(89,90).
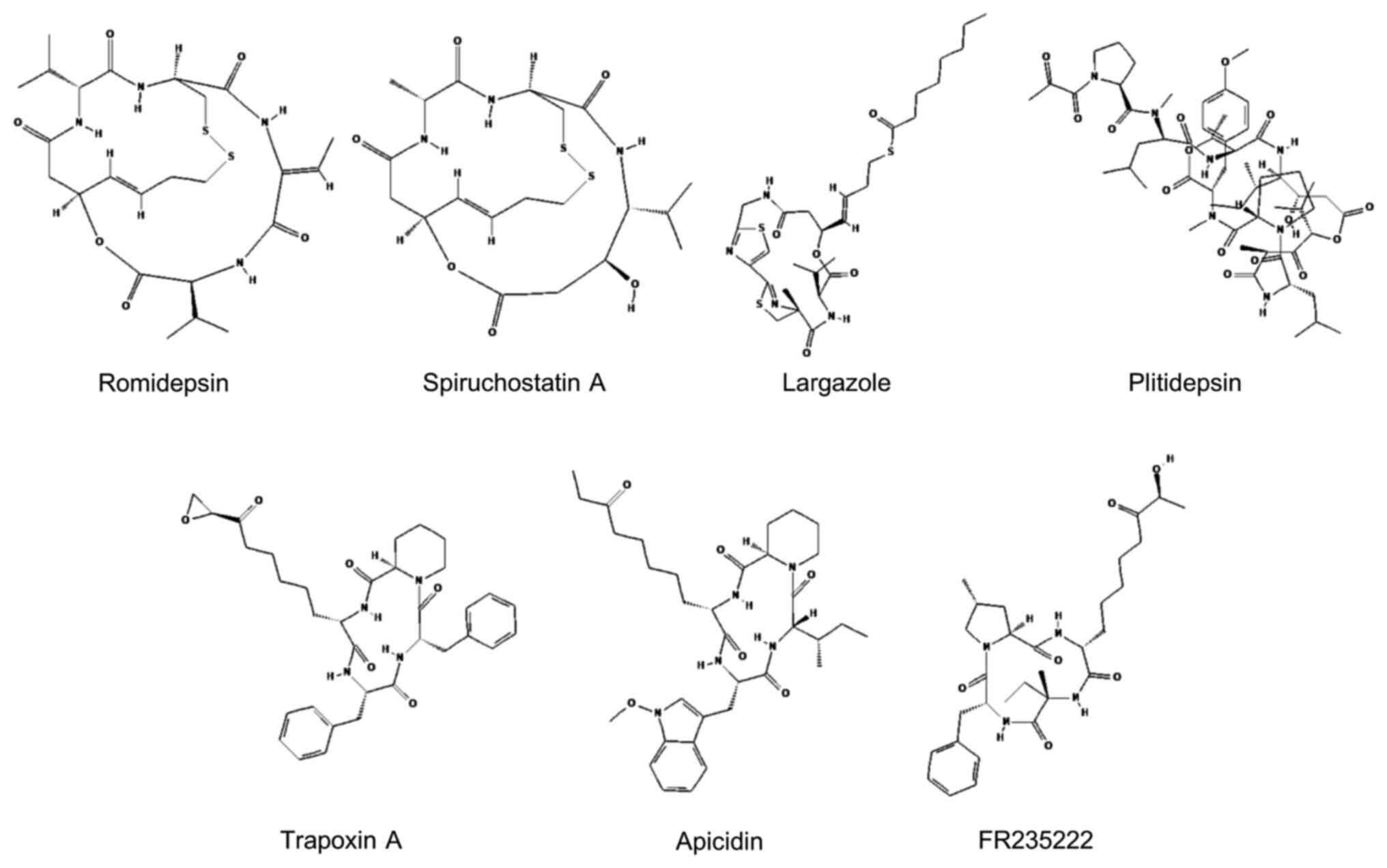
Cyclic peptides are the most structurally diverse
class of HDACis. They can be divided into the following two main
groups: Cyclic tetrapeptides and bicyclic depsipeptides. Cyclic
tetrapeptides contain epoxy ketone groups that interact with amino
acids at the HDAC catalytic site, mainly through covalent bonds.
The bicyclic depsipeptide structure contains disulfide bonds that
attach to a zinc ion in the HDAC active site, leading to inhibition
of HDAC activity. These cyclic peptides play important roles in
targeting different HDAC isoforms depending on variations in cap
regions (72,91). Owing to their strong inhibitory
activity and potential HDAC-isoform selectivity, cyclic peptides
are being intensively studied as prospective candidates for
anticancer therapy development. Insights into cyclic peptide HDACis
that have been investigated for leukemia therapy applications are
elaborated upon in the following section. Their chemical structures
are shown in Fig. 3.
Romidepsin (FK228), a natural bicyclic depsipeptide,
was first isolated from the gram-negative bacterium,
Chromobacterium violaceum, and characterized as an antitumor
substance both in vitro and in vivo (92). Romidepsin possesses HDAC inhibitory
activity by interacting with zinc ions in the active region of HDAC
enzymes. It acts as a prodrug because it is reduced to the active
compound after uptake into cells (93). Romidepsin is mainly responsible for
binding class I HDAC enzymes, including HDAC1, HDAC2, HDAC3 and
HDAC8, but this drug has minimal selectivity for HDAC6 targeting
(94). It is the most extensively
studied among peptide HDACis for its impact and mechanism of action
in a hematological aspect. Since 2009, the FDA has approved
romidepsin for the treatment of numerous hematological
malignancies, including cutaneous T-cell lymphoma (CTCL) and
peripheral T-cell lymphoma (95,96).
Several lines of evidence demonstrate that romidepsin has antitumor
effects in various subtypes of leukemia. Romidepsin demonstrated
antileukemic activity against APL cell lines by inducing APL cell
apoptosis via a mitochondria-dependent pathway and targeting the
NF-κB and p53 transcription factors (97) and increasing cell differentiation
induced by retinoic acid (98).
With respect to CML, romidepsin has been associated with the
induction of apoptosis by inactivation of the BCR-ABL fusion
protein (99). Another study in
AML1/ETO positive leukemia cell supported that romidepsin can exert
both differentiation and cytotoxic activity in AML cells,
regardless of the underlying genomic aberration (100). The combination of a
DNA-methyltransferase inhibitor and romidepsin has been reported to
enhance several biological activities, including histone
hyperacetylation, cytotoxic activity and IL-3 expression, in
leukemia cells (100,101). Additionally, the use of romidepsin
in combination with chemotherapy drugs has shown the potential to
overcome drug resistance in numerous types of leukemia (102,103). Recently, a phase I trial of
romidepsin in combination with gemcitabine, oxaliplatin, and
dexamethasone (Romi-GemOxD), has been conducted in patients with
relapsed or refractory aggressive lymphomas (104). The study showed that Romi-GemOxD
is a well-tolerated and effective treatment option. The overall
response rate was 52%, with a notably high complete response rate
of 43%. Most toxicities were hematologic (thrombocytopenia and
lymphopenia) which are manageable without delays in subsequent
cycles.
Largazole is a cyclic depsipeptide that contains a
4-methylthiazoline unit linearly fused with a thiazole ring. It is
derived from marine cyanobacteria of Symploca spp (105). Largazole shares the same
zinc-binding motif with romidepsin and spiruchostasin. It is also a
prodrug that requires conversion to a thiol derivative in order to
function. Largazole thiol has been shown to be a more effective
HDACi than romidepsin and spiruchostasin (106). In vitro, largazole
demonstrated significant suppressive activity in solid cancer cell
lines (107). Several studies have
synthesized analogs of largazole to improve its activity (108,109). To increase the stability of the
peptide conformation and to enhance HDAC inhibitory activity,
largazole was modified by addition of a C7-benzyl and the
bithiazole analog and capping with an octanoyl group to generate
largazole 4a. A study in the NB4 human leukemia cell line
demonstrated that largazole 4a selectively inhibit the activity of
class I and class II HDAC enzymes (HDAC1 and HDAC4, respectively).
Additionally, it has been shown to upregulatep21 and R-tubulin
acetylation at H3 in the NB4 cell line. However, this compound is
less potent than SAHA (a pan HDAC inhibitor drug that the FDA
approved for the treatment of relapsed CTCL). Previously, an in
vitro study using leukemia cells harvested from patients with
CML demonstrated that largazole induce apoptosis and inhibit CML
cell proliferation (110). It was
suggested that largazole exert its antileukemic effect by
downregulating the expression of the RNA-binding protein Musashi-2,
which subsequently suppresses the mammalian target of rapamycin
signaling pathway.
Apicidin is a cyclic tetrapeptide derived from the
fermenting broth of the fungus Fusarium spp. It was
identified as a hemorrhagic factor and was shown to exhibit
cytotoxic effect on human and mouse leukemia cell lines (134). Like that of trapoxin, its function
is based on an epoxide functional group of Aoe, which mimics the
ε-amino of lysine residues on histones. Apicidin acts as an HDAC
inhibitor with cytotoxic effects on several cancer cell lines
(135). It induced apoptosis in a
Bcr-abl positive leukemia cell line (K562) and primary leukemia
cells from patients with CML. Several reactions have been revealed
to be associated with apoptosis induction in K562 cells, including
increased histone H4 acetylation, disruption of mitochondrial
function, downregulation of Bcr-abl expression, and activation of
the caspase-cascade (136). These
findings indicate that apicidin acts via an intrinsic apoptotic
pathway. In addition, combining apicidin with the tyrosine kinase
inhibitor imatinib, leads to significant enhancement of apoptotic
effect in K562 cells. Apicidin can also induce apoptosis in Bcr-abl
negative leukemia cell lines, namely Jurkat, U937 and HL-60 cells.
However, a synergistic effect of the apicidin/imatinib combination
was not observed (137).
Previously, Ferrante et al (138) demonstrated inhibitory activity of
apicidin against HDAC3, which is shown to be a positive regulator
of the Notch signaling pathway. The study showed that HDAC3
deacetylated the Notch1 intracellular domain (NICD) protein
preventing it from degrading. Apicidin inhibited HDAC3 activity and
destabilized the NICD, which subsequently affected leukemia cell
survival. Correspondently, compared with normal lymphoid cells,
lymphoblastic leukemia cells had higher levels of HDAC3 expression.
Treatment with apicidin resulted in decreased cell viability in
human T-ALL cells and mantle cell lymphoma cells, but the apoptotic
ratio and cell cycle distribution were not altered. These results
suggested that apicidin could be a promising drug for the treatment
of leukemias. However, additional mechanisms underlying
antileukemic activity of apicidin still need to be explored.
Histone modification is an epigenetic regulator that
plays important roles in the development and progression of
leukemia. HDAC overexpression disrupts gene expression homeostasis
associated with cell survival, proliferation and differentiation.
Recent studies have shown that HDACis can be used to counteract
HDAC activity leading to cell cycle arrest and apoptosis (142,143). Therefore, these compounds could be
potential candidates for novel cancer drugs. The present review
describes numerous peptides that act as HDACis as well as their
therapeutic mechanisms and outcomes in cancer studies. Compared
with small molecules, peptide inhibitors exhibit superior
properties, especially high selectivity, offering the potential for
reduced adverse effects and more precise targeting. Personalized
therapy based on HDAC levels can be conducted to avoid pan-HDACis
in favor of isoform-selective inhibitors, thereby improving safety
while retaining antileukemic activity. Combinatorial strategies
employing HDACis with other targeted therapies have also been
explored, aiming to enhance treatment efficacy while overcoming
resistance and minimizing toxicity. Translationally, it can be
suggested that cyclic peptide HDACis can enhance multimodal therapy
efficacy while maintaining acceptable safety profiles.
Notably, in clinical applications, numerous HDACis
fail owing to their low therapeutic effectiveness and risk of
adverse effects. Only two depsipeptides have been approved by the
FDA and are commercially available. Major limitations of peptide
drugs are that, compared with small-molecule inhibitors, they tend
to have higher molecular weights and show lower oral
bioavailability as a result of enzymatic degradation in the
gastrointestinal tract. Their pharmacokinetics are often
characterized by limited membrane permeability, relatively short
half-lives, and challenges in achieving adequate systemic exposure.
The enhancement of the pharmacokinetic properties of peptide drugs
by promising strategies, including encapsulation, drug delivery and
structure alteration, will be beneficial to their application in
leukemia. Nano-delivery technology is widely employed to improve
the bioavailability, antileukemic activity and tolerability of
peptide-based HDACis, as exemplified by developments of
nano-romidepsin (144,145). Furthermore, nanoparticles can be
functionalized with targeting ligands, such as antibodies or
aptamers that specifically bind to proteins presented in abundance
on tumor cells (for example, transferrin receptor, CD20, CD33 and
nucleophosmin). This approach may facilitate selective absorption
of HDACis into target cells, resulting in increased local
concentration. Homotypic targeting using nanoparticles coated with
membranes from leukemia cells or stem cells is also an attractive
option. Structural alteration, such as D-amino acid substitution,
PEGylation, cyclization and cationization, can improve the
stability and permeability of peptide drugs (146). Computational modeling technology
has been used as a tool to modify and optimize structure of HDACis
based on molecular docking and binding interaction analysis to
ensure drug selectivity and stability (147). Additionally, peptidomimetics can
be designed and constructed to generate stabilized peptide HDACis
with improved efficiency and selectivity (148). Future research should also focus
on the design of customized peptide inhibitors enabling precise
treatment for individuals.
Not applicable.
The present study was supported by the Ratchadapisek Sompoch
Endowment Fund (2023), Chulalongkorn University (grant no.
Review_66_005_3700_002).
Not applicable.
PP and YS conceptualized and designed the study and
reviewed the manuscript. PP wrote the manuscript. YS acquired
funding, prepared tables and figures, and revised the manuscript.
Both authors read and approved the final version of the manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Brown G: Introduction and Classification
of Leukemias. Leukemia Stem Cells: Methods and Protocols. Cobaleda
C and Sánchez-García I: Springer; New York, NY: pp. 3–23. 2021
|
|
2
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
3
|
Daltveit DS, Morgan E, Colombet M,
Steliarova-Foucher E, Bendahhou K, Marcos-Gragera R, Rongshou Z,
Smith A, Wei H and Soerjomataram I: Global patterns of leukemia by
subtype, age, and sex in 185 countries in 2022. Leukemia.
39:412–419. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Du M, Chen W, Liu K, Wang L, Hu Y, Mao Y,
Sun X, Luo Y, Shi J, Shao K, et al: The global burden of leukemia
and its attributable factors in 204 countries and territories:
Findings from the Global Burden of Disease 2019 study and
projections to 2030. J Oncol. 2022:16127022022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dong Y, Shi O, Zeng Q, Lu X, Wang W, Li Y
and Wang Q: Leukemia incidence trends at the global, regional, and
national level between 1990 and 2017. Exp Hematol Oncol. 9:142020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharma R and Jani C: Mapping incidence and
mortality of leukemia and its subtypes in 21 world regions in last
three decades and projections to 2030. Ann Hematol. 101:1523–1534.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Briot T, Roger E, Thépot S and Lagarce F:
Advances in treatment formulations for acute myeloid leukemia. Drug
Discov Today. 23:1936–1949. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Škubník J, Pavlíčková VS, Ruml T and
Rimpelová S: Vincristine in combination therapy of cancer: Emerging
trends in clinics. Biology (Basel). 10:8492021.PubMed/NCBI
|
|
9
|
Park SB, Goldstein D, Krishnan AV, Lin CS,
Friedlander ML, Cassidy J, Koltzenburg M and Kiernan MC:
Chemotherapy-induced peripheral neurotoxicity: A critical analysis.
CA Cancer J Clin. 63:419–437. 2013.PubMed/NCBI
|
|
10
|
Mort MK, Sen JM, Morris AL, DeGregory KA,
McLoughlin EM, Mort JF, Dunn SP, Abuannadi M and Keng MK:
Evaluation of cardiomyopathy in acute myeloid leukemia patients
treated with anthracyclines. J Oncol Pharm Pract. 26:680–687. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wallace KB: Doxorubicin-induced cardiac
mitochondrionopathy. Pharmacol Toxicol. 93:105–115. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pogorzala M, Kubicka M, Rafinska B,
Wysocki M and Styczynski J: Drug-resistance profile in
multiple-relapsed childhood acute lymphoblastic leukemia.
Anticancer Res. 35:5667–5670. 2015.PubMed/NCBI
|
|
13
|
Xia CQ and Smith PG: Drug efflux
transporters and multidrug resistance in acute leukemia:
Therapeutic impact and novel approaches to mediation. Mol
Pharmacol. 82:1008–1021. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kantarjian HM, Keating MJ and Freireich
EJ: Toward the potential cure of leukemias in the next decade.
Cancer. 124:4301–4313. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhansali RS, Pratz KW and Lai C: Recent
advances in targeted therapies in acute myeloid leukemia. J Hematol
Oncol. 16:292023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brivio E, Baruchel A, Beishuizen A,
Bourquin JP, Brown PA, Cooper T, Gore L, Kolb EA, Locatelli F,
Maude SL, et al: Targeted inhibitors and antibody immunotherapies:
Novel therapies for paediatric leukaemia and lymphoma. Eur J
Cancer. 164:1–17. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Das AB, Smith-Díaz CC and Vissers MCM:
Emerging epigenetic therapeutics for myeloid leukemia: Modulating
demethylase activity with ascorbate. Haematologica. 106:14–25.
2021.PubMed/NCBI
|
|
18
|
Zhang X, Wang H, Zhang Y and Wang X:
Advances in epigenetic alterations of chronic lymphocytic leukemia:
From pathogenesis to treatment. Clin Exp Med. 24:542024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bian J and Zhang L, Han Y, Wang C and
Zhang L: Histone deacetylase inhibitors: Potent anti-leukemic
agents. Curr Med Chem. 22:2065–2074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gaál Z, Oláh É, Rejtő L, Erdődi F and
Csernoch L: Strong correlation between the expression levels of
HDAC4 and SIRT6 in hematological malignancies of the adults. Pathol
Oncol Res. 23:493–504. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang F, Li Z, Zhou J, Wang G, Zhang W, Xu
J and Liang A: SIRT1 regulates the phosphorylation and degradation
of P27 by deacetylating CDK2 to promote T-cell acute lymphoblastic
leukemia progression. J Exp Clin Cancer Res. 40:2592021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Merarchi M, Sethi G, Shanmugam MK, Fan L,
Arfuso F and Ahn KS: Role of natural products in modulating histone
deacetylases in cancer. Molecules. 24:10472019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Singh AK, Bishayee A and Pandey AK:
Targeting histone deacetylases with natural and synthetic agents:
An emerging anticancer strategy. Nutrients. 10:7312018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Janssens Y, Wynendaele E, Vanden Berghe W
and De Spiegeleer B: Peptides as epigenetic modulators: Therapeutic
implications. Clin Epigenetics. 11:1012019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang XJ and Seto E: HATs and HDACs: From
structure, function and regulation to novel strategies for therapy
and prevention. Oncogene. 26:5310–5318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gray SG and Teh BT: Histone
acetylation/deacetylation and cancer: An ‘open’ and ‘shut’ case?
Curr Mol Med. 1:401–429. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lawson M, Uciechowska U, Schemies J, Rumpf
T, Jung M and Sippl W: Inhibitors to understand molecular
mechanisms of NAD(+)-dependent deacetylases (sirtuins). Biochim
Biophys Acta. 1799:726–739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Seto E and Yoshida M: Erasers of histone
acetylation: The histone deacetylase enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Asmamaw MD, He A, Zhang LR, Liu HM and Gao
Y: Histone deacetylase complexes: Structure, regulation and
function. Biochim Biophys Acta Rev Cancer. 1879:1891502024.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Duan Z, Zarebski A, Montoya-Durango D,
Grimes HL and Horwitz M: Gfi1 coordinates epigenetic repression of
p21 Cip/WAF1 by recruitment of histone lysine methyltransferase G9a
and histone deacetylase 1. Mol Cell biol. 25:10338–10351. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujiwara T, Lee HY, Sanalkumar R and
Bresnick EH: Building multifunctionality into a complex containing
master regulators of hematopoiesis. Proc Natl Acad Sci USA.
107:20429–20434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van Oorschot R, Hansen M, Koornneef JM,
Marneth AE, Bergevoet SM, van Bergen MGJM, van Alphen FPJ, van der
Zwaan C, Martens JHA, Vermeulen M, et al: Molecular mechanisms of
bleeding disorder associated GFI1BQ287* mutation and its affected
pathways in megakaryocytes and platelets. Haematologica.
104:1460–1472. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Calderon A, Mestvirishvili T, Boccalatte
F, Ruggles KV and David G: Chromatin accessibility and cell cycle
progression are controlled by the HDAC-associated Sin3B protein in
murine hematopoietic stem cells. Epigenetics Chromatin. 17:22024.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wada T, Kikuchi J, Nishimura N, Shimizu R,
Kitamura T and Furukawa Y: Expression levels of histone
deacetylases determine the cell fate of hematopoietic progenitors.
J Biol Chem. 284:30673–30683. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang P, Wang Z and Liu J: Role of HDACs in
normal and malignant hematopoiesis. Mol Cancer. 19:52020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yan B, Yang J, Kim MY, Luo H, Cesari N,
Yang T, Strouboulis J, Zhang J, Hardison R, Huang S and Qiu Y:
HDAC1 is required for GATA-1 transcription activity, global
chromatin occupancy and hematopoiesis. Nucleic Acids Res.
49:9783–9798. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Iwasaki H, Mizuno S, Arinobu Y, Ozawa H,
Mori Y, Shigematsu H, Takatsu K, Tenen DG and Akashi K: The order
of expression of transcription factors directs hierarchical
specification of hematopoietic lineages. Genes Dev. 20:3010–3021.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamamura K, Ohishi K, Katayama N, Yu Z,
Kato K, Masuya M, Fujieda A, Sugimoto Y, Miyata E, Shibasaki T, et
al: Pleiotropic role of histone deacetylases in the regulation of
human adult erythropoiesis. Br J Haematol. 135:242–253. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Das Gupta K, Shakespear MR, Iyer A,
Fairlie DP and Sweet MJ: Histone deacetylases in
monocyte/macrophage development, activation and metabolism:
refining HDAC targets for inflammatory and infectious diseases.
Clin Transl Immunol. 5:e622016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Heideman MR, Lancini C, Proost N, Yanover
E, Jacobs H and Dannenberg JH: Sin3a-associated Hdac1 and Hdac2 are
essential for hematopoietic stem cell homeostasis and contribute
differentially to hematopoiesis. Haematologica. 99:1292–1303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu B, Ohishi K, Yamamura K, Suzuki K,
Monma F, Ino K, Nishii K, Masuya M, Sekine T, Heike Y, et al: A
potential activity of valproic acid in the stimulation of
interleukin-3−mediated megakaryopoiesis and erythropoiesis. Exp
Hematol. 38:685–695. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yamaguchi T, Cubizolles F, Zhang Y,
Reichert N, Kohler H, Seiser C and Matthias P: Histone deacetylases
1 and 2 act in concert to promote the G1-to-S progression. Genes
Dev. 24:455–469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boucheron N, Tschismarov R, Göschl L,
Moser MA, Lagger S, Sakaguchi S, Winter M, Lenz F, Vitko D,
Breitwieser FP, et al: CD4+ T cell lineage integrity is controlled
by the histone deacetylases HDAC1 and HDAC2. Nat Immunol.
15:439–448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ni L, Wang L, Yao C, Ni Z, Liu F, Gong C,
Zhu X, Yan X, Watowich SS, Lee DA and Zhu S: The histone
deacetylase inhibitor valproic acid inhibits NKG2D expression in
natural killer cells through suppression of STAT3 and HDAC3. Sci
Rep. 7:452662017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lemercier C, Brocard MP, Puvion-Dutilleul
F, Kao HY, Albagli O and Khochbin S: Class II histone deacetylases
are directly recruited by BCL6 transcriptional repressor. J Biol
Chem. 277:22045–22052. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kasler HG and Verdin E: Histone
deacetylase 7 functions as a key regulator of genes involved in
both positive and negative selection of thymocytes. Mol Cell Biol.
27:5184–5200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li J, Li X, Sun W, Zhang J, Yan Q, Wu J,
Jin J, Lu R and Miao D: Specific overexpression of SIRT1 in
mesenchymal stem cells rescues hematopoiesis niche in BMI1 knockout
mice through promoting CXCL12 expression. Int J Biol Sci.
18:2091–2103. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Skokowa J, Lan D, Thakur BK, Wang F, Gupta
K, Cario G, Brechlin AM, Schambach A, Hinrichsen L, Meyer G, et al:
NAMPT is essential for the G-CSF-induced myeloid differentiation
via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 15:151–158.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ou X, Chae HD, Wang RH, Shelley WC, Cooper
S, Taylor T, Kim YJ, Deng CX, Yoder MC and Broxmeyer HE: SIRT1
deficiency compromises mouse embryonic stem cell hematopoietic
differentiation, and embryonic and adult hematopoiesis in the
mouse. Blood. 117:440–450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brown K, Xie S, Qiu X, Mohrin M, Shin J,
Liu Y, Zhang D, Scadden DT and Chen D: SIRT3 reverses
aging-associated degeneration. Cell Rep. 3:319–327. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kaiser A, Schmidt M, Huber O, Frietsch JJ,
Scholl S, Heidel FH, Hochhaus A, Müller JP and Ernst T: SIRT7: An
influence factor in healthy aging and the development of
age-dependent myeloid stem-cell disorders. Leukemia. 34:2206–2216.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Luo H, Mu WC, Karki R, Chiang HH, Mohrin
M, Shin JJ, Ohkubo R, Ito K, Kanneganti TD and Chen D:
Mitochondrial stress-Initiated aberrant activation of the NLRP3
inflammasome regulates the functional deterioration of
hematopoietic stem cell aging. Cell Rep. 26:945–954.e4. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sahakian E, Chen J, Powers JJ, Chen X,
Maharaj K, Deng SL, Achille AN, Lienlaf M, Wang HW, Cheng F, et al:
Essential role for histone deacetylase 11 (HDAC11) in neutrophil
biology. J Leukoc Biol. 102:475–486. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Grignani F, De Matteis S, Nervi C,
Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara
FF, Zamir I, et al: Fusion proteins of the retinoic acid
receptor-alpha recruit histone deacetylase in promyelocytic
leukaemia. Nature. 391:815–818. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gelmetti V, Zhang J, Fanelli M, Minucci S,
Pelicci PG and Lazar MA: Aberrant recruitment of the nuclear
receptor corepressor-histone deacetylase complex by the acute
myeloid leukemia fusion partner ETO. Mol Cell Biol. 18:7185–7191.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Girard N, Tremblay M, Humbert M, Grondin
B, Haman A, Labrecque J, Chen B, Chen Z, Chen SJ and Hoang T:
RARα-PLZF oncogene inhibits C/EBPα function in myeloid cells. Proc
Natl Acad Sci USA. 110:13522–13527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang J, Hug BA, Huang EY, Chen CW,
Gelmetti V, Maccarana M, Minucci S, Pelicci PG and Lazar MA:
Oligomerization of ETO is obligatory for corepressor interaction.
Mol Cell Biol. 21:156–163. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vishwakarma BA, Nguyen N, Makishima H,
Hosono N, Gudmundsson KO, Negi V, Oakley K, Han Y, Przychodzen B,
Maciejewski JP and Du Y: Runx1 repression by histone deacetylation
is critical for Setbp1-induced mouse myeloid leukemia development.
Leukemia. 30:200–208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nakata S, Yoshida T, Horinaka M, Shiraishi
T, Wakada M and Sakai T: Histone deacetylase inhibitors upregulate
death receptor 5/TRAIL-R2 and sensitize apoptosis induced by
TRAIL/APO2-L in human malignant tumor cells. Oncogene.
23:6261–6271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mehrpouri M, Pourbagheri-Sigaroodi A and
Bashash D: The contributory roles of histone deacetylases (HDACs)
in hematopoiesis regulation and possibilities for pharmacologic
interventions in hematologic malignancies. Int Immunopharmacol.
100:1081142021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Van Damme M, Crompot E, Meuleman N, Mineur
P, Bron D, Lagneaux L and Stamatopoulos B: HDAC isoenzyme
expression is deregulated in chronic lymphocytic leukemia B-cells
and has a complex prognostic significance. Epigenetics.
7:1403–1412. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Verbeek TCAI, Vrenken KS, Arentsen-Peters
STCJM, Castro PG, van de Ven M, van Tellingen O, Pieters R and Stam
RW: Selective inhibition of HDAC class IIA as therapeutic
intervention for KMT2A-rearranged acute lymphoblastic leukemia.
Commun Biol. 7:12572024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Gu W and Roeder RG: Activation of p53
sequence-specific DNA binding by acetylation of the p53 C-terminal
domain. Cell. 90:595–606. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Molica M, Mazzone C, Niscola P and de
Fabritiis P: TP53 mutations in acute myeloid leukemia: Still a
daunting challenge? Front Oncol. 10:6108202021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kuo YH, Qi J and Cook GJ: Regain control
of p53: Targeting leukemia stem cells by isoform-specific HDAC
inhibition. Exp Hematol. 44:315–321. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qi J, Singh S, Hua WK, Cai Q, Chao SW, Li
L, Liu H, Ho Y, McDonald T, Lin A, et al: HDAC8 inhibition
specifically targets inv(16) acute myeloid leukemic stem cells by
restoring p53 acetylation. Cell Stem Cell. 17:597–610. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lee SM, Bae JH, Kim MJ, Lee HS, Lee MK,
Chung BS, Kim DW, Kang CD and Kim SH: Bcr-Abl-independent
imatinib-resistant K562 cells show aberrant protein acetylation and
increased sensitivity to histone deacetylase inhibitors. J
Pharmacol Exp Ther. 322:1084–1092. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Burgess M, Chen YCE, Mapp S, Blumenthal A,
Mollee P, Gill D and Saunders NA: HDAC7 is an actionable driver of
therapeutic antibody resistance by macrophages from CLL patients.
Oncogene. 39:5756–5767. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Micelli C and Rastelli G: Histone
deacetylases: Structural determinants of inhibitor selectivity.
Drug Discov Today. 20:718–735. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang L, Zhang J, Jiang Q, Zhang L and
Song W: Zinc binding groups for histone deacetylase inhibitors. J
Enzyme Inhib Med Chem. 33:714–721. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rajak H, Singh A, Dewangan PK, Patel V,
Jain DK, Tiwari SK, Veerasamy R and Sharma PC: Peptide based
aacrocycles: Selective histone deacetylase inhibitors with
antiproliferative activity. Curr Med Chem. 20:1887–1903. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Curcio A, Rocca R, Alcaro S and Artese A:
The histone deacetylase family: Structural features and application
of combined computational methods. Pharmaceuticals (Basel).
17:6202024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Davie JR: Inhibition of histone
deacetylase activity by butyrate. J Nutr. 133 (7
Suppl):2485S–2493S. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Luu M, Riester Z, Baldrich A, Reichardt N,
Yuille S, Busetti A, Klein M, Wempe A, Leister H, Raifer H, et al:
Microbial short-chain fatty acids modulate CD8(+) T cell responses
and improve adoptive immunotherapy for cancer. Nat Commun.
12:40772021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ozkan AD, Eskiler GG, Kazan N and Turna O:
Histone deacetylase inhibitor sodium butyrate regulates the
activation of toll-like receptor 4/interferon regulatory factor-3
signaling pathways in prostate cancer cells. J Cancer Res Ther.
19:1812–1817. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Sampathkumar SG, Jones MB, Meledeo MA,
Campbell CT, Choi SS, Hida K, Gomutputra P, Sheh A, Gilmartin T,
Head SR and Yarema KJ: Targeting glycosylation pathways and the
cell cycle: Sugar-dependent activity of butyrate-carbohydrate
cancer prodrugs. Chem Biol. 13:1265–1275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Steliou K, Boosalis MS, Perrine SP,
Sangerman J and Faller DV: Butyrate histone deacetylase inhibitors.
Biores Open Access. 1:192–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Tang R, Faussat AM, Majdak P, Perrot JY,
Chaoui D, Legrand O and Marie JP: Valproic acid inhibits
proliferation and induces apoptosis in acute myeloid leukemia cells
expressing P-gp and MRP1. Leukemia. 18:1246–1251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zapotocky M, Mejstrikova E, Smetana K,
Stary J, Trka J and Starkova J: Valproic acid triggers
differentiation and apoptosis in AML1/ETO-positive leukemic cells
specifically. Cancer Lett. 319:144–153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fredly H, Gjertsen BT and Bruserud Ø:
Histone deacetylase inhibition in the treatment of acute myeloid
leukemia: The effects of valproic acid on leukemic cells, and the
clinical and experimental evidence for combining valproic acid with
other antileukemic agents. Clin Epigenetics. 5:122013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Garcia-Manero G, Kantarjian HM,
Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M,
Wierda WG, Ravandi F, Koller C, et al: Phase 1/2 study of the
combination of 5-aza-2′-deoxycytidine with valproic acid in
patients with leukemia. Blood. 108:3271–3279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Peiffer L, Poll-Wolbeck SJ, Flamme H,
Gehrke I, Hallek M and Kreuzer KA: Trichostatin A effectively
induces apoptosis in chronic lymphocytic leukemia cells via
inhibition of Wnt signaling and histone deacetylation. J Cancer Res
Clin Oncol. 140:1283–1293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Khan N, Jeffers M, Kumar S, Hackett C,
Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al:
Determination of the class and isoform selectivity of
small-molecule histone deacetylase inhibitors. Biochem J.
409:581–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Duvic M, Talpur R, Ni X, Zhang C, Hazarika
P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel
SR: Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic
acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood.
109:31–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Campbell P and Thomas CM: Belinostat for
the treatment of relapsed or refractory peripheral T-cell lymphoma.
J Oncol Pharm Pract. 23:143–147. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Laubach JP, Moreau P, San-Miguel JF and
Richardson PG: Panobinostat for the treatment of multiple myeloma.
Clin Cancer Res. 21:4767–4773. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wagner JM, Hackanson B, Lübbert M and Jung
M: Histone deacetylase (HDAC) inhibitors in recent clinical trials
for cancer therapy. Clin Epigenetics. 1:117–136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Batlevi CL, Crump M, Andreadis C, Rizzieri
D, Assouline SE, Fox S, van der Jagt RHC, Copeland A, Potvin D,
Chao R and Younes A: A phase 2 study of mocetinostat, a histone
deacetylase inhibitor, in relapsed or refractory lymphoma. Br J
Haematol. 178:434–441. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Carraway HE, Sawalha Y, Gojo I, Lee MJ,
Lee S, Tomita Y, Yuno A, Greer J, Smith BD, Pratz KW, et al: Phase
1 study of the histone deacetylase inhibitor entinostat plus
clofarabine for poor-risk Philadelphia chromosome-negative (newly
diagnosed older adults or adults with relapsed refractory disease)
acute lymphoblastic leukemia or biphenotypic leukemia. Leuk Res.
110:1067072021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Maolanon AR, Kristensen HM, Leman LJ,
Ghadiri MR and Olsen CA: Natural and synthetic macrocyclic
inhibitors of the histone deacetylase enzymes. Chembiochem.
18:5–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ueda H, Manda T, Matsumoto S, Mukumoto S,
Nishigaki F, Kawamura I and Shimomura K: FR901228, a novel
antitumor bicyclic depsipeptide produced by Chromobacterium
violaceum No. 968. III. Antitumor activities on experimental tumors
in mice. J Antibiot (Tokyo). 47:315–323. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Furumai R, Matsuyama A, Kobashi N, Lee KH,
Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M
and Horinouchi S: FK228 (depsipeptide) as a natural prodrug that
inhibits class I histone deacetylases. Cancer Res. 62:4916–4921.
2002.PubMed/NCBI
|
|
94
|
Murata M, Towatari M, Kosugi H, Tanimoto
M, Ueda R, Saito H and Naoe T: Apoptotic cytotoxic effects of a
histone deacetylase inhibitor, FK228, on malignant lymphoid cells.
Jpn J Cancer Res. 91:1154–1160. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Campas-Moya C: Romidepsin for the
treatment of cutaneous T-cell lymphoma. Drugs Today (Barc).
45:787–795. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Coiffier B, Pro B, Prince HM, Foss F,
Sokol L, Greenwood M, Caballero D, Morschhauser F, Wilhelm M,
Pinter-Brown L, et al: Romidepsin for the treatment of
relapsed/refractory peripheral T-cell lymphoma: Pivotal study
update demonstrates durable responses. J Hematol Oncol. 7:112014.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Savickiene J, Treigyte G, Borutinskaite V,
Navakauskiene R and Magnusson KE: The histone deacetylase inhibitor
FK228 distinctly sensitizes the human leukemia cells to retinoic
acid-induced differentiation. Ann NY Acad Sci. 1091:368–384. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kosugi H, Ito M, Yamamoto Y, Towatari M,
Ito M, Ueda R, Saito H and Naoe T: In vivo effects of a histone
deacetylase inhibitor, FK228, on human acute promyelocytic leukemia
in NOD/Shi-scid/scid mice. Jpn J Cancer Res. 92:529–536. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Okabe S, Tauchi T, Nakajima A, Sashida G,
Gotoh A, Broxmeyer HE, Ohyashiki JH and Ohyashiki K: Depsipeptide
(FK228) preferentially induces apoptosis in BCR/ABL-expressing cell
lines and cells from patients with chronic myelogenous leukemia in
blast crisis. Stem Cells Dev. 16:503–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Klisovic MI, Maghraby EA, Parthun MR,
Guimond M, Sklenar AR, Whitman SP, Chan KK, Murphy T, Anon J,
Archer KJ, et al: Depsipeptide (FR 901228) promotes histone
acetylation, gene transcription, apoptosis and its activity is
enhanced by DNA methyltransferase inhibitors in AML1/ETO-positive
leukemic cells. Leukemia. 17:350–358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shaker S, Bernstein M, Momparler LF and
Momparler RL: Preclinical evaluation of antineoplastic activity of
inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone
deacetylation (trichostatin A, depsipeptide) in combination against
myeloid leukemic cells. Leuk Res. 27:437–444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Brunvand MW and Carson J: Complete
remission with romidepsin in a patient with T-cell acute
lymphoblastic leukemia refractory to induction hyper-CVAD. Hematol
Oncol. 36:340–343. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cox WPJ, Evander N, Van Ingen Schenau DS,
Stoll GR, Anderson N, De Groot L, Grünewald KJT, Hagelaar R, Butler
M, Kuiper RP, et al: Histone deacetylase inhibition sensitizes
p53-deficient B-cell precursor acute lymphoblastic leukemia to
chemotherapy. Haematologica. 109:1755–1765. 2024.PubMed/NCBI
|
|
104
|
Foley N, Riedell PA, Bartlett NL, Cashen
AF, Kahl BS, Fehniger TA, Fischer A, Moreno C, Liu J, Carson KR and
Mehta-Shah N: A phase I study of romidepsin in combination with
gemcitabine, oxaliplatin, and dexamethasone in patients with
relapsed or refractory aggressive lymphomas enriched for T-Cell
lymphomas. Clin Lymphoma Myeloma Leuk. 25:328–336. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Seiser T, Kamena F and Cramer N: Synthesis
and biological activity of largazole and derivatives. Angew Chem
Int Ed Engl. 47:6483–6485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bowers A, West N, Taunton J, Schreiber SL,
Bradner JE and Williams RM: Total synthesis and biological mode of
action of largazole: A potent class I histone deacetylase
inhibitor. J Am Chem Soc. 130:11219–11222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Taori K, Paul VJ and Luesch H: Structure
and activity of largazole, a potent antiproliferative agent from
the floridian marine cyanobacterium symploca sp. J Am Chem Soc.
130:1806–1807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Souto JA, Vaz E, Lepore I, Pöppler AC,
Franci G, Alvarez R, Altucci L and de Lera AR: Synthesis and
biological characterization of the histone deacetylase inhibitor
largazole and C7-modified analogues. J Med Chem. 53:4654–4667.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhang B, Ruan ZW, Luo D, Zhu Y, Ding T,
Sui Q and Lei X: Unexpected enhancement of HDACs inhibition by MeS
substitution at C-2 position of fluoro largazole. Mar Drugs.
18:3442020. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wang M, Sun XY, Zhou YC, Zhang KJ, Lu YZ,
Liu J, Huang YC, Wang GZ, Jiang S and Zhou GB: Suppression of
Musashi-2 by the small compound largazole exerts inhibitory effects
on malignant cells. Int J Oncol. 56:1274–1283. 2020.PubMed/NCBI
|
|
111
|
Yurek-George A, Habens F, Brimmell M,
Packham G and Ganesan A: Total synthesis of spiruchostatin A, a
potent histone deacetylase inhibitor. J Am Chem Soc. 126:1030–1031.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Narita K, Fukui Y, Sano Y, Yamori T, Ito
A, Yoshida M and Katoh T: Total synthesis of bicyclic depsipeptides
spiruchostatins C and D and investigation of their histone
deacetylase inhibitory and antiproliferative activities. Eur J Med
Chem. 60:295–304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kanno S, Maeda N, Tomizawa A, Yomogida S,
Katoh T and Ishikawa M: Involvement of p21waf1/cip1 expression in
the cytotoxicity of the potent histone deacetylase inhibitor
spiruchostatin B towards susceptible NALM-6 human B cell leukemia
cells. Int J Oncol. 40:1391–1396. 2012.PubMed/NCBI
|
|
114
|
Rehman MU, Jawaid P, Yoshihisa Y, Li P,
Zhao QL, Narita K, Katoh T, Kondo T and Shimizu T: Spiruchostatin A
and B, novel histone deacetylase inhibitors, induce apoptosis
through reactive oxygen species-mitochondria pathway in human
lymphoma U937 cells. Chem Biol Interact. 221:24–34. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yao L: Aplidin PharmaMar. IDrugs.
6:246–250. 2003.PubMed/NCBI
|
|
116
|
Erba E, Bassano L, Di Liberti G, Muradore
I, Chiorino G, Ubezio P, Vignati S, Codegoni A, Desiderio MA,
Faircloth G, et al: Cell cycle phase perturbations and apoptosis in
tumour cells induced by aplidine. Br J Cancer. 86:1510–1517. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Erba E, Serafini M, Gaipa G, Tognon G,
Marchini S, Celli N, Rotilio D, Broggini M, Jimeno J, Faircloth GT,
et al: Effect of aplidin in acute lymphoblastic leukaemia cells. Br
J Cancer. 89:763–773. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bresters D, Broekhuizen AJ, Kaaijk P,
Faircloth GT, Jimeno J and Kaspers GJ: In vitro cytotoxicity of
aplidin and crossresistance with other cytotoxic drugs in childhood
leukemic and normal bone marrow and blood samples: A rational basis
for clinical development. Leukemia. 17:1338–1343. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Gómez SG, Bueren JA, Faircloth GT, Jimeno
J and Albella B: In vitro toxicity of three new antitumoral drugs
(trabectedin, aplidin, and kahalalide F) on hematopoietic
progenitors and stem cells. Exp Hematol. 31:1104–1111. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Gajate C, An F and Mollinedo F: Rapid and
selective apoptosis in human leukemic cells induced by Aplidine
through a Fas/CD95- and mitochondrial-mediated mechanism. Clin
Cancer Res. 9:1535–1545. 2003.PubMed/NCBI
|
|
121
|
Mitsiades CS, Ocio EM, Pandiella A, Maiso
P, Gajate C, Garayoa M, Vilanova D, Montero JC, Mitsiades N,
McMullan CJ, et al: Aplidin, a marine organism-derived compound
with potent antimyeloma activity in vitro and in vivo. Cancer Res.
68:5216–5225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Muñoz-Alonso MJ, Álvarez E,
Guillén-Navarro MJ, Pollán M, Avilés P, Galmarini CM and Muñoz A:
c-Jun N-terminal kinase phosphorylation is a biomarker of
plitidepsin activity. Mar Drugs. 11:1677–1692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Broggini M, Marchini SV, Galliera E,
Borsotti P, Taraboletti G, Erba E, Sironi M, Jimeno J, Faircloth
GT, Giavazzi R and D'Incalci M: Aplidine, a new anticancer agent of
marine origin, inhibits vascular endothelial growth factor (VEGF)
secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human
leukemia cells MOLT-4. Leukemia. 17:52–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Morande PE, Zanetti SR, Borge M, Nannini
P, Jancic C, Bezares RF, Bitsmans A, González M, Rodríguez AL,
Galmarini CM, et al: The cytotoxic activity of Aplidin in chronic
lymphocytic leukemia (CLL) is mediated by a direct effect on
leukemic cells and an indirect effect on monocyte-derived cells.
Invest New Drugs. 30:1830–1840. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Barboza NM, Medina DJ, Budak-Alpdogan T,
Aracil M, Jimeno JM, Bertino JR and Banerjee D: Plitidepsin
(Aplidin) is a potent inhibitor of diffuse large cell and Burkitt
lymphoma and is synergistic with rituximab. Cancer Biol Ther.
13:114–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Humeniuk R, Menon LG, Mishra PJ, Saydam G,
Longo-Sorbello GS, Elisseyeff Y, Lewis LD, Aracil M, Jimeno J,
Bertino JR and Banerjee D: Aplidin synergizes with cytosine
arabinoside: Functional relevance of mitochondria in
Aplidin-induced cytotoxicity. Leukemia. 21:2399–2405. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Spicka I, Ocio EM, Oakervee HE, Greil R,
Banh RH, Huang SY, D'Rozario JM, Dimopoulos MA, Martínez S,
Extremera S, et al: Randomized phase III study (ADMYRE) of
plitidepsin in combination with dexamethasone vs. dexamethasone
alone in patients with relapsed/refractory multiple myeloma. Ann
Hematol. 98:2139–2150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mateos MV, Prosper F, Martin Sánchez J,
Ocio EM, Oriol A, Motlló C, Michot JM, Jarque I, Iglesias R, Solé
M, et al: Phase I study of plitidepsin in combination with
bortezomib and dexamethasone in patients with relapsed/refractory
multiple myeloma. Cancer Med. 12:3999–4009. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Itazaki H, Nagashima K, Sugita K, Yoshida
H, Kawamura Y, Yasuda Y, Matsumoto K, Ishii K, Uotani N, Nakai H,
et al: Isolation and structural elucidation of new
cyclotetrapeptides, trapoxins A and B, having detransformation
activities as antitumor agents. J Antibiot (Tokyo). 43:1524–1532.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Furumai R, Komatsu Y, Nishino N, Khochbin
S, Yoshida M and Horinouchi S: Potent histone deacetylase
inhibitors built from trichostatin A and cyclic tetrapeptide
antibiotics including trapoxin. Proc Natl Acad Sci USA. 98:87–92.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kijima M, Yoshida M, Sugita K, Horinouchi
S and Beppu T: Trapoxin, an antitumor cyclic tetrapeptide, is an
irreversible inhibitor of mammalian histone deacetylase. J Biol
Chem. 268:22429–22435. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kosugi H, Towatari M, Hatano S, Kitamura
K, Kiyoi H, Kinoshita T, Tanimoto M, Murate T, Kawashima K, Saito H
and Naoe T: Histone deacetylase inhibitors are the potent
inducer/enhancer of differentiation in acute myeloid leukemia: A
new approach to anti-leukemia therapy. Leukemia. 13:1316–1324.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Maeda T, Towatari M, Kosugi H and Saito H:
Up-regulation of costimulatory/adhesion molecules by histone
deacetylase inhibitors in acute myeloid leukemia cells. Blood.
96:3847–3856. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Park JS, Lee KR, Kim JC, Lim SH, Seo JA
and Lee YW: A hemorrhagic factor (Apicidin) produced by toxic
Fusarium isolates from soybean seeds. Appl Environ Microbiol.
65:126–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Han JW, Ahn SH, Park SH, Wang SY, Bae GU,
Seo DW, Kwon HK, Hong S, Lee HY, Lee YW and Lee HW: Apicidin, a
histone deacetylase inhibitor, inhibits proliferation of tumor
cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res.
60:6068–6074. 2000.PubMed/NCBI
|
|
136
|
Cheong JW, Chong SY, Kim JY, Eom JI, Jeung
HK, Maeng HY, Lee ST and Min YH: Induction of apoptosis by
apicidin, a histone deacetylase inhibitor, via the activation of
mitochondria-dependent caspase cascades in human Bcr-Abl-positive
leukemia cells. Clin Cancer Res. 9:5018–5027. 2003.PubMed/NCBI
|
|
137
|
Kim JS, Jeung HK, Cheong JW, Maeng H, Lee
ST, Hahn JS, Ko YW and Min YH: Apicidin potentiates the
imatinib-induced apoptosis of Bcr-Abl-positive human leukaemia
cells by enhancing the activation of mitochondria-dependent caspase
cascades. Br J Haematol. 124:166–178. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Ferrante F, Giaimo BD, Bartkuhn M,
Zimmermann T, Close V, Mertens D, Nist A, Stiewe T, Meier-Soelch J,
Kracht M, et al: HDAC3 functions as a positive regulator in Notch
signal transduction. Nucleic Acids Res. 48:3496–3512. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Mori H, Urano Y, Abe F, Furukawa S,
Furukawa S, Tsurumi Y, Sakamoto K, Hashimoto M, Takase S, Hino M
and Fujii T: FR235222, a fungal metabolite, is a novel
immunosuppressant that inhibits mammalian histone deacetylase
(HDAC). I. Taxonomy, fermentation, isolation and biological
activities. J Antibiot (Tokyo). 56:72–79. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Petrella A, D'Acunto CW, Rodriquez M,
Festa M, Tosco A, Bruno I, Terracciano S, Taddei M, Paloma LG and
Parente L: Effects of FR235222, a novel HDAC inhibitor, in
proliferation and apoptosis of human leukaemia cell lines: Role of
annexin A1. Eur J Cancer. 44:740–749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
D'Acunto CW, Carratù A, Rodriquez M,
Taddei M, Parente L and Petrella A: LGP1, A histone deacetylase
inhibitor analogue of FR235222, sensitizes promyelocytic leukaemia
U937 cells to TRAIL-mediated apoptosis. Anticancer Res. 30:887–894.
2010.PubMed/NCBI
|
|
142
|
Fuentes-Baile M, García-Morales P,
Pérez-Valenciano E, Mata-Balaguer T, Menéndez-Gutiérrez MP, de Juan
Romero C, Rodríguez-Lescure Á, Martín-Orozco E, Mallavia R, Barberá
VM and Saceda M: Insights into histone deacetylase
inhibitors-induced cell death in cancer cell lines. Biomed
Pharmacother. 191:1185412025. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li Z, Qiu H, Lu W, Duan N, Fan S, Zhou R,
Li X, Zhang H, Liu N and Yang F: Design and synthesis of
thiazole-based hydroxamate histone deacetylase inhibitors with
potent antitumor efficacy by inducing apoptosis, pyroptosis and
cell cycle arrest. Sci Rep. 15:245892025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Aroonthongsawat P, Manocheewa S, Srisawat
C, Punnakitikashem P and Suwanwong Y: Enhancement of the in vitro
anti-leukemic effect of the histone deacetylase inhibitor
romidepsin using Poly-(D, L-lactide-co-glycolide) nanoparticles as
a drug carrier. Eur J Pharm Sci. 207:1070432025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Pal I, Illendula A, Joyner AM, Manavalan
JS, Deddens TM, Sabzevari A, Damera DP, Zuberi S, Marchi E, Fox TE,
et al: Nanoromidepsin, a polymer nanoparticle of the HDAC
inhibitor, improves safety and efficacy in models of T-cell
lymphoma. Blood. Sep 2–2025.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Xiao W, Jiang W, Chen Z, Huang Y, Mao J,
Zheng W, Hu Y and Shi J: Advance in peptide-based drug development:
delivery platforms, therapeutics and vaccines. Signal Transduct
Target Ther. 10:742025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Rizwan A, Aqeel A and Farooqi H: Decoding
HDACs and its inhibitors-artificial intelligence assisted smart
software based super computational modelling technology in
targeting cancer and neurological disorders of the brain. Netw
Modeling Anal Health Inform Bioinform. 14:1042025. View Article : Google Scholar
|
|
148
|
Wang D, Li W, Zhao R, Chen L, Liu N, Tian
Y, Zhao H, Xie M, Lu F, Fang Q, et al: Stabilized peptide HDAC
inhibitors derived from HDAC1 substrate H3K56 for the treatment of
cancer stem-like cells in vivo. Cancer Res. 79:1769–1783. 2019.
View Article : Google Scholar : PubMed/NCBI
|