Introduction
Glioblastoma (GBM) is the most common and deadly
type of brain cancer (1). On the
basis of the World Health Organization (WHO) guidelines for central
nervous system tumor classification, GBM is a grade IV diffuse
glioma (2). The GBM cell
characteristic of diffuse infiltration has the ability to invade
the surrounding normal brain tissue, resulting in recurrence even
after complete resection (3). The
current standard of care for GBM involves a combination of surgical
resection with radiation and chemotherapy (4–6). In
recent years, despite advancements in therapeutic efficacy against
GBM, its prognosis remains poor. Therefore, it is necessary to
elucidate the molecular mechanisms governing the pathogenesis and
progression of GBM, while exploring potential therapeutic targets
to increase treatment efficacy and prolong patient survival.
S100B, which is located on chromosome 21, is a
Ca2+-binding protein of the S100 family, and numerous
studies have demonstrated its neurotrophic effects. In the central
nervous system, S100B primarily influences the proliferation and
differentiation of glial cells, as well as the maintenance of
calcium homeostasis (7). Its
carboxyl terminus has a strong affinity for Ca2+; when
it binds to Ca2+, S100B undergoes conformational changes
that expose its target protein-binding site and exerts biological
effects through interactions with these target proteins (8,9).
Melanoma, ovarian and breast cancer, and other malignant tumors
have been observed to express S100B at substantially elevated
levels. Furthermore, it is closely associated with tumor
development, malignancy and prognosis (10–12).
In previous studies, S100B silencing in melanoma cells has been
shown to restore p53-mediated apoptosis, which may restrict
malignant cell proliferation (13,14).
Moreover, intervention with S100B expression in ovarian cancer
stem-like cells has been shown to increase p53 activity and reduce
subcutaneous tumor volume, leading to markedly prolonged survival
time in nude mice (12). In
addition, S100B may drive bevacizumab resistance in ovarian cancer
by promoting angiogenesis. Both in vivo resistant tumors and
in vitro S100B-overexpressing cells were found to enhance
human umbilical vein endothelial cell migration and tube formation,
which can be replicated by exogenous S100B treatment.
Mechanistically, endothelial cells uptake tumor-derived S100B,
which suppresses FOXO1 and releases β-catenin for nuclear
pro-angiogenic signaling (15).
Emerging evidence has implicated S100B in GBM pathogenesis,
although mechanistic insights remain limited. Serum S100B levels
are elevated in patients with glioma compared with those obtained
from control individuals with craniocerebral trauma (16). In addition, S100B concentrations are
positively associated with tumor grade; they are notably higher in
high-grade vs. low-grade glioma, according to the WHO
classification system. However, the tissue-specific expression
patterns of S100B in GBM, its clinical relevance and its functional
roles in tumor progression require further investigation.
The present study identified the upregulated
expression of S100B in GBM and its close association with an
unfavorable prognosis. By contrast, the inhibition of S100B
resulted in suppressed proliferative capacity, and a reduction in
invasion, migration and EMT. The results also revealed that
downregulation of TGF-β2 was observed upon inhibition of S100B.
Consistently, cell invasion, migration and the EMT process were
rescued by exogenous recombinant protein TGF-β2 treatment. These
findings indicate the role of S100B in promoting GBM progression.
The current study elucidates the mechanism by which S100B
facilitates GBM cell invasion and migration via the TGF-β2-induced
EMT process that exhibits an infiltrative growth pattern, thus
providing novel insights for GBM treatment.
Materials and methods
Cell lines and culture
The human GBM cell line LN229 (cat. no. CRL-2611)
was obtained from the American Type Culture Collection. The cell
line was verified through short tandem repeat testing (Guangzhou
Cellcook Biotech Co., Ltd.). The cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; cat. no. 11995065; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(cat. no. PWL001; Dalian Meilun Biology Technology Co., Ltd.) and
1% penicillin-streptomycin (cat. no. ST488S; Beyotime Institute of
Biotechnology) and were incubated at 37°C with 5%
CO2.
Flow cytometric analysis
A total of 2×106 cells were collected and
fixed with 4% paraformaldehyde for 15 min, then washed with PBS.
After incubation with S100B antibody (1:200; cat. no. ab52642;
Abcam) overnight, the cells were washed with PBS and stained with
FITC-Goat Anti-Rabbit IgG secondary antibody (1:200; cat. no.
A22120; Abbkine Scientific Co., Ltd.) for 1.5 h. The final wash was
completed and used for flow cytometry. Flow cytometric analysis was
conducted on a BD FACSCanto™ Flow Cytometer system (10-colour
configuration; BD Biosciences), followed by processing of the data
using BD FACSDiva Software (BD Biosciences).
Lentiviral transduction to knockdown
S100B
LN229 cells were stably transduced with short
hairpin (sh)RNA vectors to knockdown S100B. The shRNA vectors were
purchased from Shanghai GeneChem Co., Ltd. The S100B shRNA vector
(shS100B) sequence was 5′-CTGCCACGAGTTCTTTGAA-3′, and the negative
control (NC) sequence was 5′-TTCTCCGAACGTGTCACGT-3′. For
transduction of the human GBM cell line LN229, 5×104
cells/well in a 12-well plate were transduced with lentiviral
particles (multiplicity of infection, 10) for 12 h. The serum-free
medium was then replaced with medium containing 10% FBS. A total of
48 h after lentivirus transduction, the cells were treated with
puromycin (cat. no. P8230; Beijing Solarbio Science &
Technology Co., Ltd.) working concentration 2.0 µg/ml for 48 h to
screen successfully transduced positive cells. At 72 h
post-infection, GFP expression was examined using a fluorescence
microscope (Nikon Corporation). The validation of shRNA knockdown
efficiency of S100B was verified by reverse
transcription-quantitative PCR (RT-qPCR), western blot analysis and
immunofluorescence (IF) staining. The experimental results were
obtained from three replicate experiments using NC LN229 GBM cells
as a control for relative quantitative analysis. The IF results
were calculated by randomly selecting six fields each time to
analyze their mean fluorescence intensity. The ratio of
S100B-positive cells to all cells in each field was first
calculated separately for the NC and shS100B groups, and then the
relative percentage rate of positive cells was calculated using NC
as the control group. In addition, the shS100B cells were cultured
with recombinant TGF-β2 protein (cat. no. HY-P7119; MedChemExpress)
for 48 h to obtain shS100B + TGF-β2 cells, which were used for
subsequent experimental analyses.
RT-qPCR
Total RNA was isolated from NC and shS100B LN229 GBM
cells using AG RNAex Pro Reagent (cat. no. AG21101; Accurate
Biology). RT-qPCR was performed using the ABScript III RT Master
Mix for qPCR with gDNA Remover (cat. no. RK20429; ABclonal Biotech
Co., Ltd.) and 2X Universal SYBR Green Fast qPCR Mix (cat. no.
RK21203; ABclonal Biotech Co., Ltd.) according to the
manufacturer's protocol. Thermocycling conditions are included in
Tables I and II. GAPDH was used as an internal control
and fold change was determined using the relative quantification
(2−∆∆Cq) method (16).
The sequences of the primers used were as follows: GAPDH forward,
5′-AATGGACAACTGGTCGTGGAC-3′ and reverse,
5′-CCCTCCAGGGGATCTGTTTG-3′; S100B forward,
5′-AGCTGGAGAAGGCCATGGTG-3′ and reverse, 5′-GAACTCGTGGCAGGCAGTAG-3′;
and TGF-β2 forward, 5′-CAGCACACTCGATATGGACCA-3′ and reverse,
5′-CCTCGGGCTCAGGATAGTCT-3′.
 | Table I.Thermocycling conditions of reverse
transcription reaction. |
Table I.
Thermocycling conditions of reverse
transcription reaction.
| Temperature,°C | Time, min |
|---|
| 37 | 2 |
| 55 | 15 |
| 85 | 5 |
| 4 | Hold |
| Table II.Quantitative PCR reaction
conditions. |
Table II.
Quantitative PCR reaction
conditions.
| Step | Temperature,°C | Time | Number of
cycles |
|---|
|
Pre-denaturation | 95 | 3 min | 1 |
| Cyclic
reaction | 95 | 5 sec | 40-45 |
|
| 60 | 30 sec |
|
Western blot analysis
Total protein was extracted from LN229 cells using a
Whole Cell Lysis Assay Kit (cat. no. KGB5303-100; Nanjing KeyGen
Biotech Co., Ltd.) and a BCA assay (cat. no. KGB2101-500; Nanjing
KeyGen Biotech Co., Ltd.) was used to determine the protein
concentration. In total, 30 µg equivalent amounts of protein were
separated by SDS-PAGE on 15% gels and were transferred to PVDF
membranes (cat. no. ISEQ00010; MilliporeSigma). The primary
antibodies used were anti-S100B (1:200; cat. no. ab52642; Abcam),
anti-GAPDH (1:5,000; cat. no. 10494-1-AP; Wuhan Sanying
Biotechnology, Inc.) and anti-TGF-β2 (1:200; cat. no. 19999-1-AP;
Wuhan Sanying Biotechnology, Inc.). The corresponding secondary
antibodies used were horseradish peroxidase (HRP)-conjugated goat
anti-rabbit (1:5,000; cat. no. SA00001-2; Wuhan Sanying
Biotechnology, Inc.) or HRP-conjugated AffiniPure goat anti-mouse
secondary antibodies (1:5,000; cat. no. SA00001-1; Wuhan Sanying
Biotechnology, Inc.). After incubation with secondary antibodies, a
chemiluminescence system (G:box; Syngene Europe) was used to detect
immunoreactive proteins, and the band intensity relative to that of
GAPDH was semi-quantified with Quantity One software (version
4.6.2; Bio-Rad Laboratories, Inc.).
In vivo subcutaneous xenograft
model
Immunodeficient 4-week-old male nude mice weighing
~15 grams from Chongqing Tengxin Biotechnology Co., Ltd., were used
for tumor formation and analysis. Animals were housed in an
specific pathogen free (SPF) environment at 23°C with 30–70%
humidity and 12/12-h light/dark cycle. A total of 1×106
NC cells and shS100B cells in 100 µl DMEM were transplanted
subcutaneously into the nude mice. Tumor size was measured every 3
days during this period. The tumor size was calculated using the
following formula: Volume=width2 × length/2. A total of
six mice were used in the present study. The maximum recorded tumor
volume was 122.301 mm3. At this endpoint, the tumor
length and width measured 6.84 and 5.98 mm, respectively. On day 26
post-injection, the nude mice were placed in a designated
asphyxiator and CO2 is passed through it, with the
CO2 displacing about 70% of the gas in the asphyxiator
per minute, and the animal is removed after it is completely dead.
Death was confirmed by the sustained absence of spontaneous
respiration observed for at least two min, followed by the loss of
all critical reflexes, including pupillary, corneal, and toe-pinch
responses, prior to proceeding with subsequent experiments.
Euthanasia of immunodeficient mice was required if tumor growth
exceeded 10% of body weight, if individual tumors exceeded 17 mm in
diameter, if ulceration, necrosis or infection developed on the
surface of the tumor, or if the mouse lost 15% of its body weight.
The tumor tissues were immediately fixed with 4% paraformaldehyde
for 24 h before being stored in 30% sucrose solution. The fixed and
dehydrated tissues were then embedded in O.C.T. compound (cat. no.
4583; Sakura Finetek USA, Inc.), frozen, sectioned, placed on
slides and subjected to IF staining.
IF staining
For IF staining, NC and shS100B LN229 GBM cells were
fixed with 4% paraformaldehyde, and frozen tissues from NC and
shS100B mice were cut into 15-µm sections. Tumor tissues and GBM
cells were stained with an anti-S100B antibody (1:200; cat. no.
ab52642; Abcam) overnight at 4°C. After being washed three times
with PBS, the samples were incubated with a DyLight 549 goat
anti-rabbit IgG (H+L) secondary antibody (1:200; cat. no. A23320;
Abbkine Scientific Co., Ltd.) for 1.5 h at 4°C. The nuclei were
stained with 4,6-diamidino-2-phenylindole (cat. no. C1002; Beyotime
Institute of Biotechnology) for 15 min. Images were captured using
a laser confocal microscope (Nikon A1R; Nikon Corporation) and were
prepared via Nikon NIS-Elements AR Analysis 5.20.02.64-bit software
for further analysis.
Cell Counting Kit-8 (CCK-8)
proliferation analysis
Cell viability was determined using a CCK-8 assay.
NC and shS100B LN229 GBM cells were cultured in 96-well plates at
1,000 cells/well. According to the manufacturer's instructions, the
cells were incubated with a mixture containing 10 µl CCK solution
(cat. no. M4839; Abmole Bioscience, Inc.) and 90 µl culture medium
for 2 h at 37°C. The OD value was acquired at 450 nm using a
microplate reader (Epoch; BioTek; Agilent Technologies, Inc.).
Notably, OD values were recorded on days 1, 2, 3, 4 and 5.
5-Ethynyl-2′-deoxyuridine (EdU)
proliferation analysis
Another method was also used to evaluate the effect
of S100B on cell proliferation. Briefly, NC and shS100B LN229 GBM
cells were treated with the BeyoClick™ EdU Cell
Proliferation Kit with Alexa Fluor 555 (cat. no. C0075L; Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Subsequently, the cells were stained with
4,6-diamidino-2-phenylindole (cat. no. C1002; Beyotime Institute of
Biotechnology) for 15 min. Images were captured using a laser
confocal microscope (Nikon A1R) and were analyzed with NIS-Elements
AR Analysis 5.20.02.64-bit software.
Colony formation assay
NC and shS100B LN229 cells were seeded into 12-well
plates at a density of 2,000 cells/well and were cultured at 37°C
and 5% CO2 for 8 days. On day 4, the medium was replaced
with fresh medium. On day 8, the medium was removed, the cells were
washed with PBS, and the cells were subsequently fixed with 4%
paraformaldehyde for 15 min. Finally, the cells were stained with
crystal violet staining solution (cat. no. C0121-100 ml; Beyotime
Institute of Biotechnology) for 15 min, and the wells were imaged
by full scanning using a microplate reader (Epoch) to count the
number of visible colonies (≥30 cells).
Transwell migration and invasion
assays
First, NC and shS100B LN229 cells were cultured in
DMEM without FBS for 12 h. Subsequently, 0.5×105 cells
were seeded in the upper chambers of Transwell plates (24 wells;
8-µm pore size; cat. no. 353097; Falcon; Corning, Inc.), which were
precoated with Matrigel (cat. no. HY-K6002; MedChemExpress) at 37°C
for 30 min for the invasion assay or uncoated for the migration
assay. DMEM without FBS was added to the upper chambers, and DMEM
with 10% FBS was added to the lower chambers. After 48 h of
incubation, the migratory/invasive cells in the lower chambers of
the Transwell plate membranes were fixed with 4% paraformaldehyde
and stained with crystal violet solution for 15 min. However, the
non-migratory/invasive cells in the upper chambers were removed.
The number of migratory/invasive cells in the six fields was
randomly determined using a microplate reader at ×100
magnification. The relative proportion of migrating/invasive cells
was calculated using NC as the controls. And the experiment was
repeated three times.
Cross-scratch assay
A scratch assay was used to assess the migratory
properties of the NC and shS100B groups. Briefly, 1×106
cells were seeded in 6-well plates with complete DMEM, and when the
cells formed a confluent monolayer, they were incubated for 24 h at
37°C in a 5% CO2 incubator. A cross-scratch was made
using a sterile 10-µl pipette tip, and the cells were then washed
with sterile PBS to remove nonadherent cells in suspension. The
cells were subsequently cultured in DMEM without FBS, and a
microplate reader (Epoch) was used to capture the center of the
cross-scratch at 0, 24, 48 and 72 h. The areas of the scratches
were analyzed using NIS-Elements AR Analysis 5.20.02.64-bit
software, and the percentage of migrating cells was calculated. The
cell migration rate was calculated as the cell migration area
divided by the cross-scratch area.
Database analysis
The Human Protein Atlas (HPA; http://www.proteinatlas.org/) contains both mRNA and
protein expression data from different human tissues, and
antibodies with different cat. no. have been used to determine the
protein expression level. Thus, using this online database, the
mRNA and protein expression data of S100B in different types of
cancer were obtained. Moreover, the association between gene
expression level and survival time was explored via these
databases.
The Gene Expression Profiling Interactive Analysis
(GEPIA) dataset is an online database used to analyze RNA
sequencing data and Genotype-Tissue Expression projects (http://gepia.cancer-pku.cn/). GEPIA can be used to
perform analyses including tumor and normal differential expression
analysis, patient survival analysis and gene correlation analysis.
For the present study, the mRNA expression of S100B in GBM and
paired normal tissues was evaluated using this database.
The Cancer Genome Atlas (TCGA; http://www.cancer.gov/ccg/research/genome-sequencing/tcga)
is a Cancer Genomics Program, which includes genomic, epigenomic,
transcriptomic and proteomic data. Notably, >20,000 primary
cancer samples and matched normal samples from 33 cancer types have
been molecularly characterized. The database was used to analyze
the expression of S100B in different tumor tissues and normal
tissues, and its expression in association with patient survival
time.
Gene expression profiles of tumor and normal brain
samples generated via chips were obtained from the GSE50161 dataset
from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The online website
SangerBOX (http://sangerbox.com/) was used for
patient survival analysis.
Single-cell data were sourced from Tumor Immune
Single Cell Hub (TISCH; http://tisch.comp-genomics.org/gallery/), which
contains single-cell sequencing data from 17 glioma projects. The
database was used to access the expression profiles of S100B in
different cell types.
Statistical analysis
Statistical analysis was performed using two-tailed
unpaired Student's t-test, One-way ANOVA with GraphPad Prism 9.0
software (Dotmatics). All data are presented as the mean ± standard
error of the mean. Every experiment was repeated at least three
times, and P<0.05 was considered to indicate a statistically
significant difference.
Results
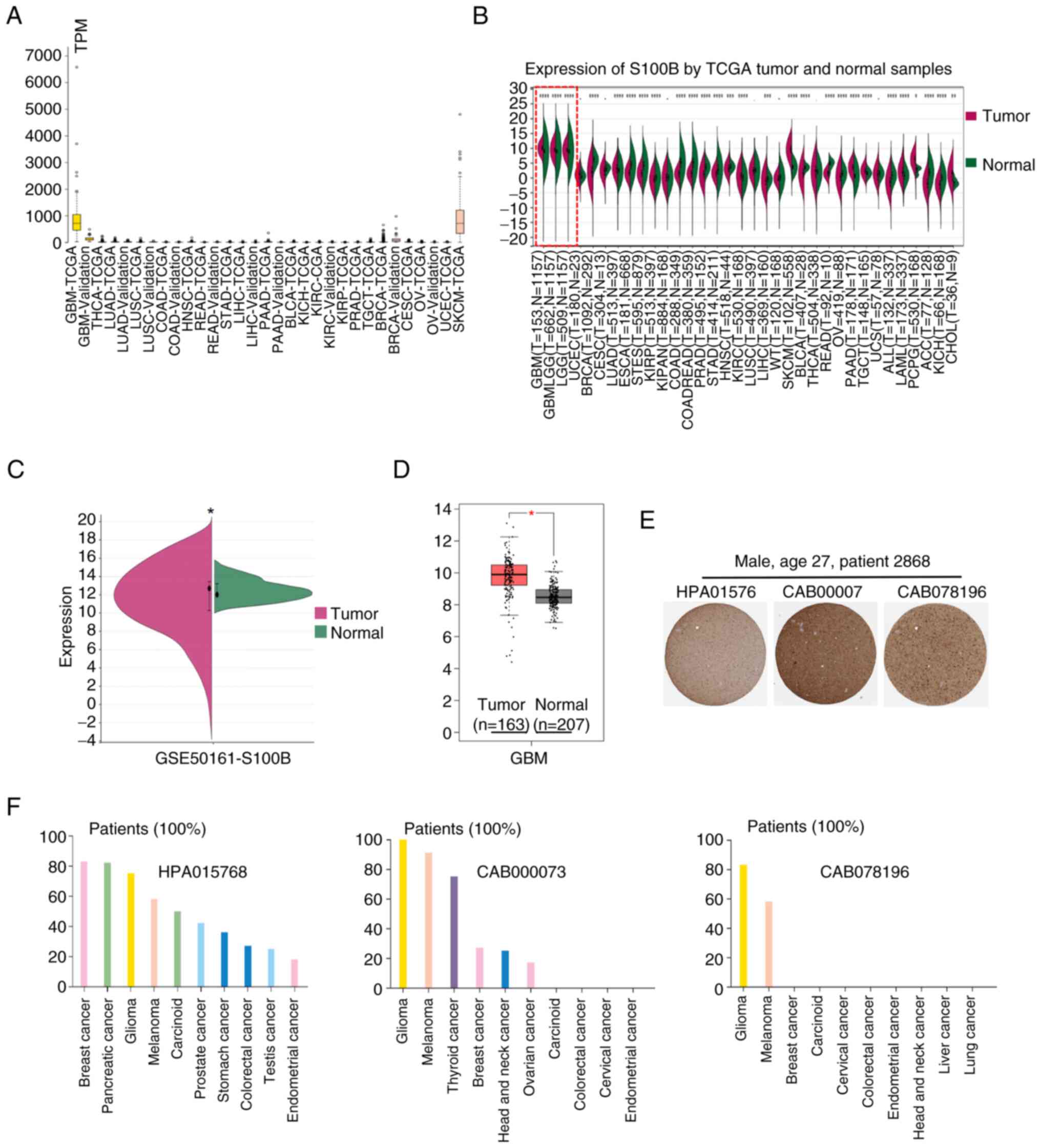
S100B is highly expressed in GBM
tissue
Bioinformatics analyses from two independent
databases consistently revealed elevated S100B expression in GBM.
The HPA demonstrated significantly higher S100B mRNA levels in GBM,
and melanoma compared with other tumor types (Fig. 1A). Notably, this finding was
corroborated by TCGA data, where S100B transcription was markedly
increased in GBM tissues relative to normal controls, a contrast
more pronounced than that detected in other malignancies (Fig. 1B). This phenomenon was subsequently
confirmed in the GSE50161 dataset and the GEPIA database (Fig. 1C and D). Immunohistochemical
examination demonstrated the distribution and expression pattern of
S100B at the protein level in patients with glioma; most samples
displayed moderate to strong nuclear and cytoplasmic positivity,
which was greater than that of melanoma and other types of cancer
(Fig. 1E and F).
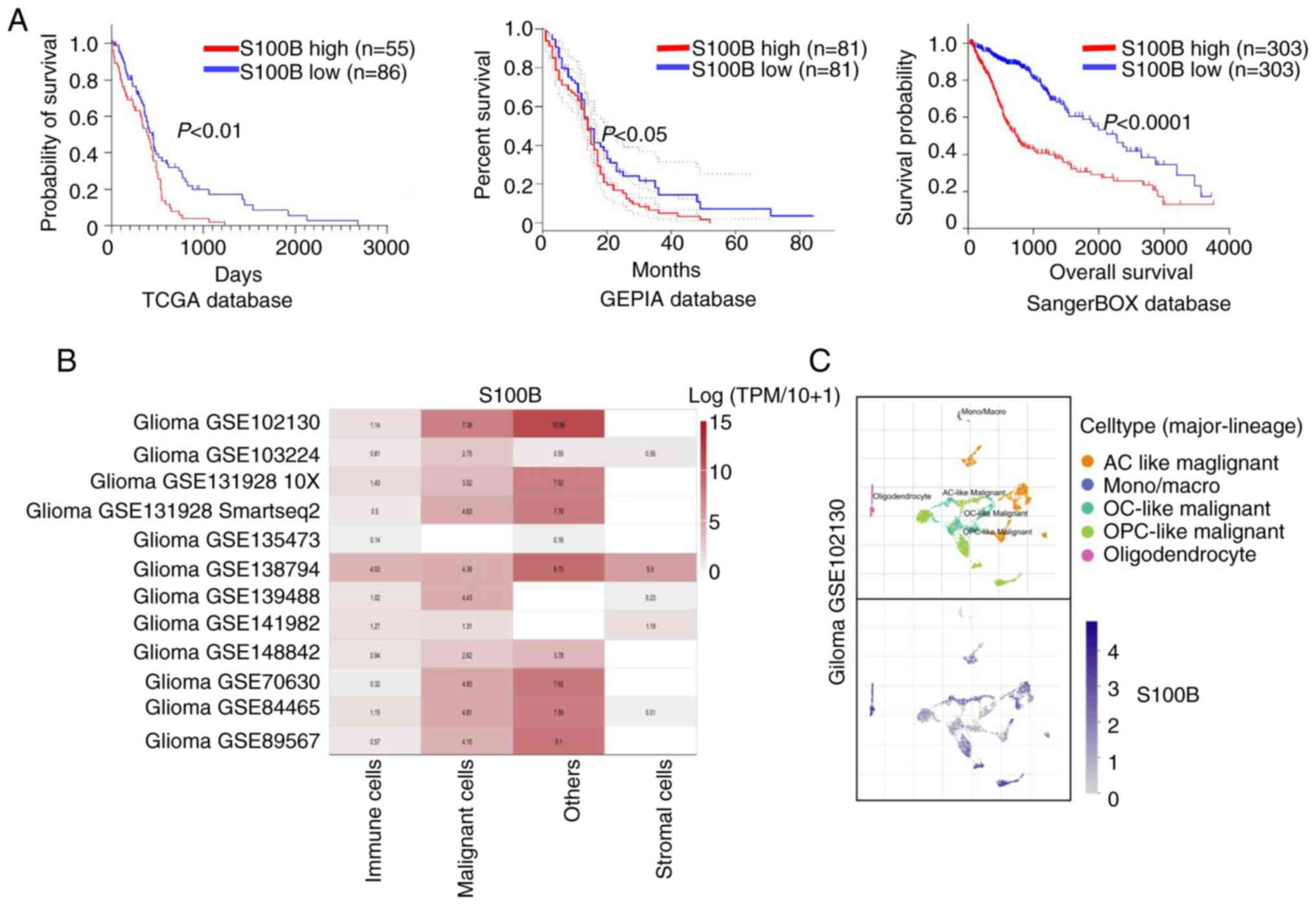
High S100B expression is strongly
associated with adverse outcomes in glioma
To systematically evaluate the prognostic value of
S100B in glioma, multi-omics data from TCGA, GEPIA and SangerBOX
databases were analyzed. Survival analysis revealed that patients
with elevated S100B expression exhibited significantly shorter
overall survival compared with those with lower expression levels
(Fig. 2A), indicating its strong
association with a poor prognosis. The present study also assessed
glioma single-cell transcriptome data from the TISCH database
(17), which confirmed that S100B
was markedly upregulated in malignant cells (Fig. 2B). This result was further supported
by the Synapse (Fig. 2C). These
findings strongly implicate S100B as a potential key regulator and
a promising molecular target worthy of further mechanistic
investigation in glioma therapeutics.
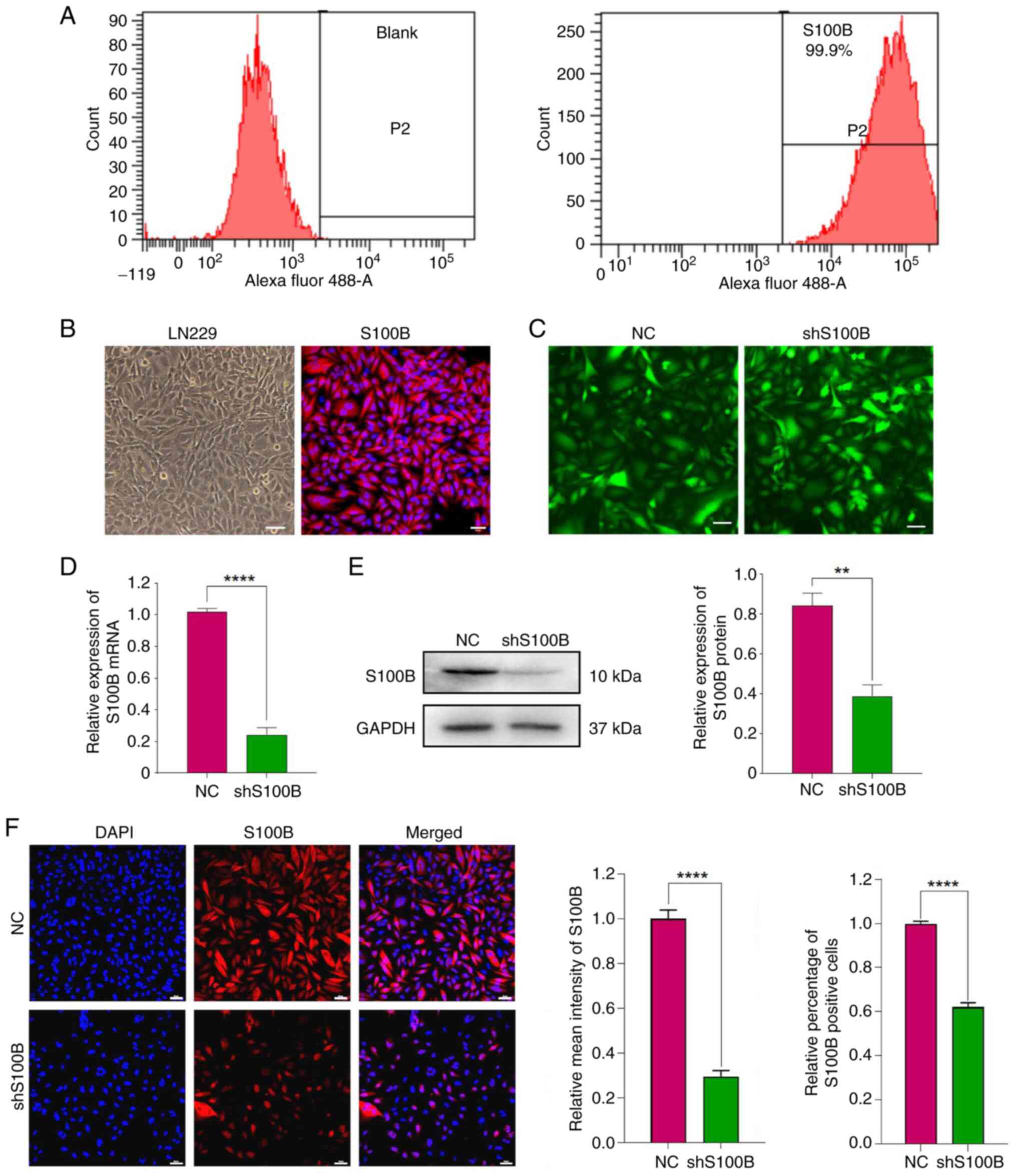
Knockdown of S100B in LN229 cells
Flow cytometric and IF analyses of LN229 GBM cells
stained with S100B revealed that the percentage of positive cells
was as high as 99.9% (Fig. 3A and
B). These results confirmed that S100B is highly expressed in
the GBM cell line LN229. Lentiviral transduction with shS100B
induced intracellular S100B suppression at both transcriptional and
translational levels, as indicated by significantly reduced mRNA
expression and diminished protein abundance compared with that in
the NC group (Fig. 3C-E).
Concurrent IF analysis confirmed this knockdown efficiency at the
cellular level, with both mean intensity and S100B-positive cells
decreased in the shS100B-treated group (Fig. 3F). According to the aforementioned
findings, a stable cell line with low S100B expression was
successfully constructed.
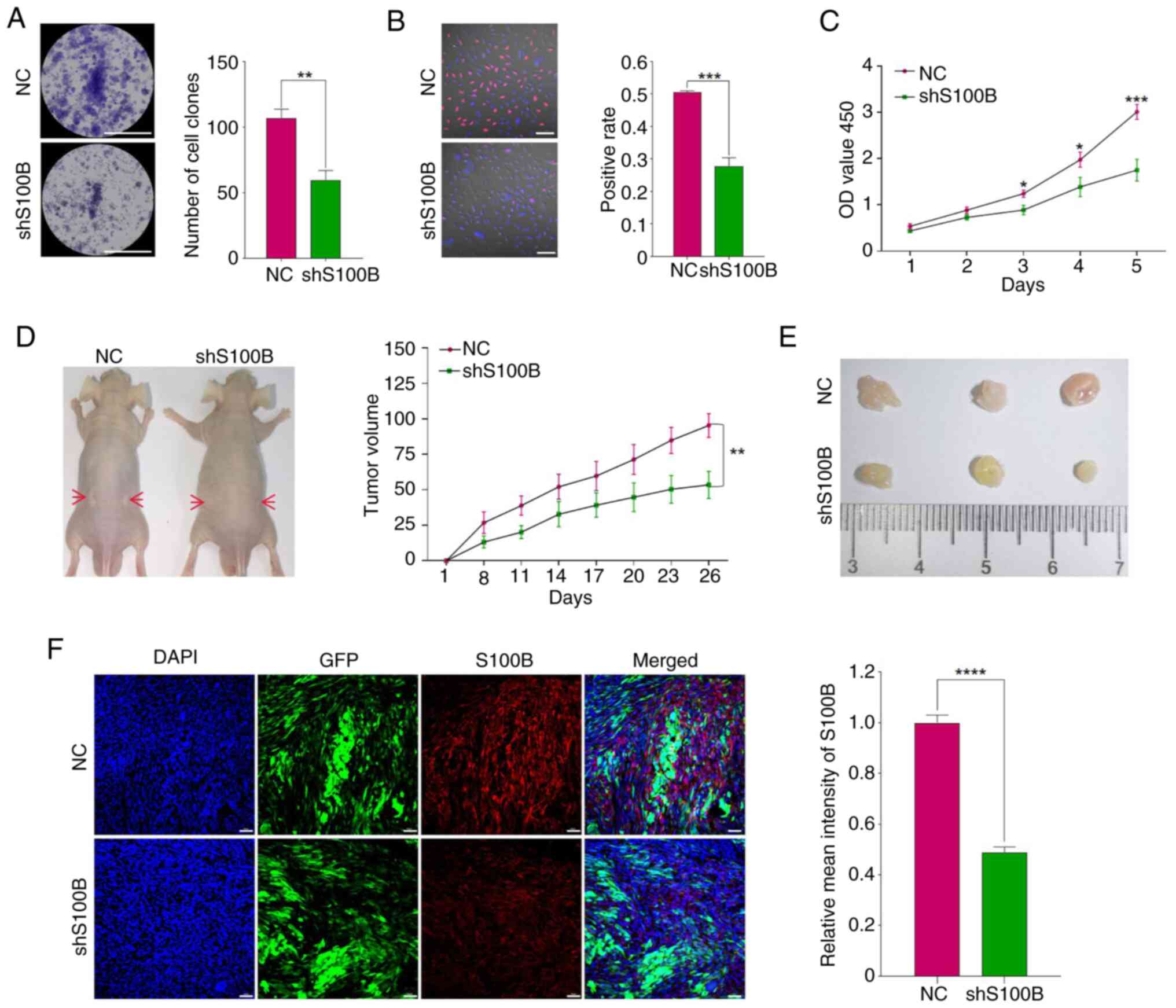
Target-oriented S100B suppresses LN229
proliferative capacity in vitro and tumorigenicity in vivo
Colony formation assays demonstrated that S100B
knockdown significantly impaired clonogenic potential (Fig. 4A), a finding corroborated by EdU
incorporation assays showing significantly decreased DNA synthesis
in shS100B LN229 cells (Fig. 4B).
Furthermore, the results of the CCK-8 assay confirmed that the
inhibition of S100B strongly suppressed the proliferation of LN229
cells (Fig. 4C). To evaluate the
effects of S100B on tumorigenicity, NC and shS100B cells were
transplanted into nude mice. Quantitative analysis revealed that
xenograft tumors in the shS100B group exhibited significantly
slower growth kinetics compared with those in the NC group
(Fig. 4D). Consistently, endpoint
measurements demonstrated a statistically significant reduction in
tumor volume within the shS100B cohort relative to the NC group
(Fig. 4E). Upon termination of the
experiment at day 26 post-implantation, histopathological
examination of resected tumors showed well-circumscribed masses
without evidence of local tissue invasion or metastatic
dissemination. This observation indicated that the subcutaneous
xenograft model, while suitable for evaluating primary tumor growth
parameters, has limitations in assessing invasive and migratory
phenotypes. Subsequently, IF staining of frozen sections
demonstrated that S100B expression in shS100B LN229 tumor tissue
was significantly lower than that in the NC group (Fig. 4F). These collective findings
strongly suggest that S100B depletion attenuates tumorigenic
capacity in this preclinical model.
Cell invasion and migration are
decreased in the S100B-knockdown group via the inhibition of
EMT
The current study further examined the effects of
S100B on GBM cell invasion and migration. Wound healing assays
revealed a significantly reduced migratory capability in the
shS100B group compared with that in the NC group (Fig. 5A). Furthermore, Transwell assays
with or without Matrigel coating demonstrated significantly fewer
invasive/migratory cells in the shS100B group vs. the NC group
(Fig. 5B and C). Collectively,
these results demonstrated that S100B knockdown could attenuate the
invasive and migratory capacities of GBM cells. To elucidate the
molecular mechanism by which S100B affects the invasion and
migration of GBM cells, transcriptome sequencing of NC and shS100B
LN229 cells was performed. RNA sequencing identified 2,294
differentially expressed genes (fold change >2, P<0.05),
comprising 1,130 upregulated and 1,164 downregulated genes between
the two groups (Fig. 5D). Gene
Ontology (GO) analyses revealed that the induction of mesenchyme
morphogenesis, a biological process related to EMT, was among the
20 pathways with the greatest differences in downregulated genes
(Fig. 5E). The expression levels of
the EMT markers E-cadherin, N-cadherin and vimentin were
subsequently analyzed in both the NC and shS100B groups. S100B
downregulation upregulated E-cadherin, whereas it downregulated
N-cadherin and vimentin, suggesting suppression of EMT, which could
consequently impair the invasive and migratory capacities of GBM
cells (Fig. 5F).
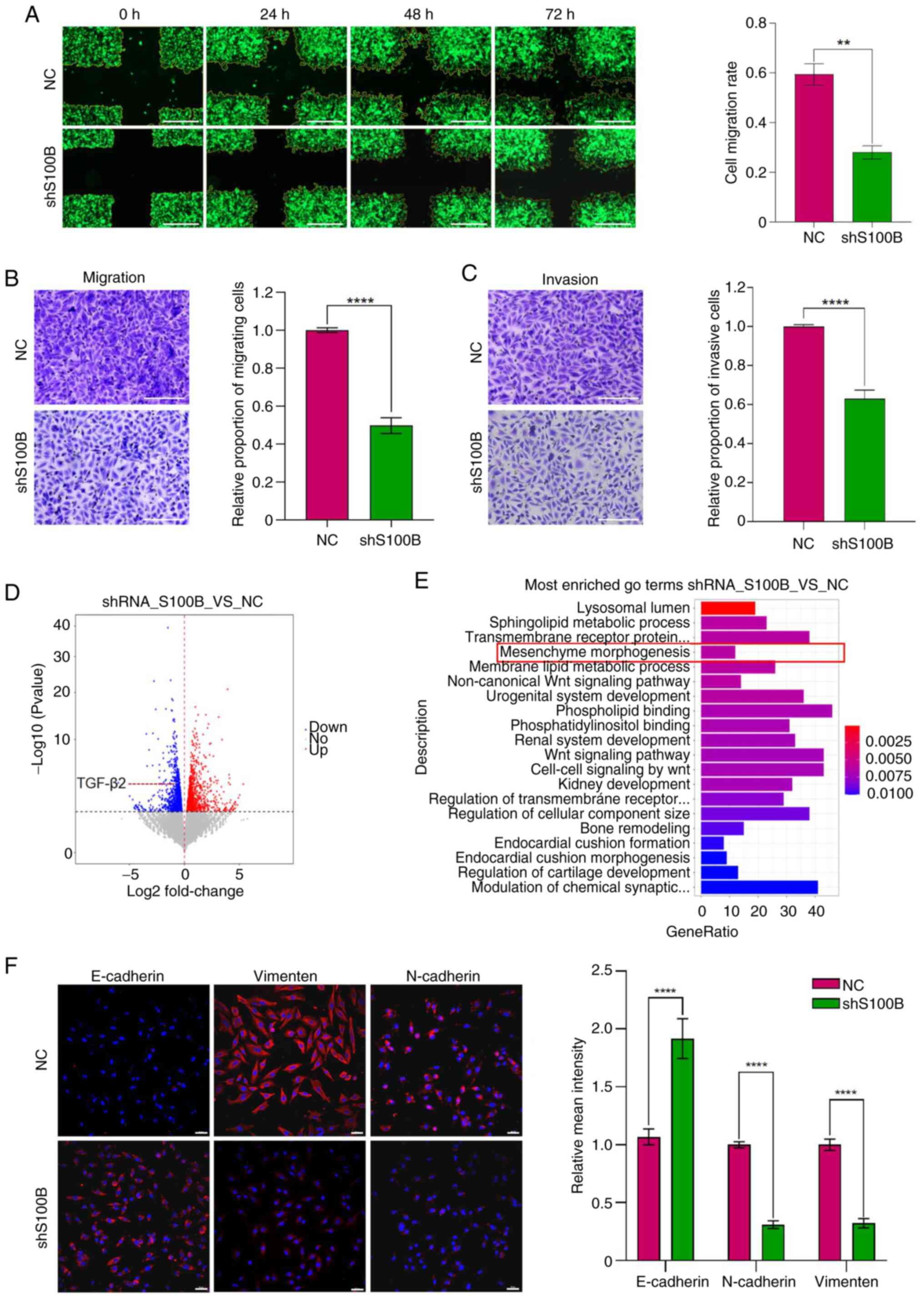 | Figure 5.S100B affects the invasion and
migration of GBM cells via epithelial-mesenchymal transition. (A)
Detection of cell migration ability by cross-scratch assay; Scale
bar, 500 µm. Cell migration rate = Cell migration area/cross
scratch area. (B and C) Transwell assays analyzed the migratory and
invasive capacity of NC and shS100B GBM cells, Scale bar, 200 µm.
Relative proportion of migratory or invasive cells: Number of
migratory or invasive cells in shS100B group/number of migratory or
invasive cells in NC group. (D) Volcano plot showing the DEGs in
the NC and shS100B groups. (E) GO enrichment analysis of the
functions of downregulated DEG. (F) E-cadherin, N-cadherin and
vimentin expression in NC and shS100B LN229 cells were measured by
immunofluorescence staining. Blue represents cell nuclei, red
represents E-cadherin, N-cadherin and vimentin. Statistical
analysis of relative mean fluorescence intensity was performed.
Scale bar, 50 µm. Data are presented as the mean ± SEM. **P<0.01
and ****P<0.0001. GBM, glioblastoma multiforme; shS100B, shRNA
S100B; NC, negative control; DEGs, differentially expressed genes;
GO, Gene Ontology. |
S100B affects the EMT process through
TGF-β2
During mesenchyme morphogenesis pathway activation,
the EMT-associated gene TGF-β2 exhibited significant downregulation
(Fig. 6A). This suppression was
further validated by RT-qPCR and western blotting in
S100B-knockdown cells (Fig. 6B and
C). Accordingly, it was hypothesized that S100B may influence
GBM cell motility through TGF-β2-mediated EMT. To confirm this
hypothesis, GBM cells with downregulated S100B expression were
treated with human recombinant TGF-β2 protein. The experimental
results revealed that compared with those in the shS100B group, the
shS100B + TGF-β2 group had stronger migratory and invasive
properties (Fig. 6D and E).
Furthermore, E-cadherin was reduced, whereas N-cadherin and
vimentin were notably restored in the shS100B + TGF-β2 group
(Fig. 6F). These results indicated
that downregulation of S100B may attenuate TGF-β2-induced EMT, as
well as cell invasion and migration.
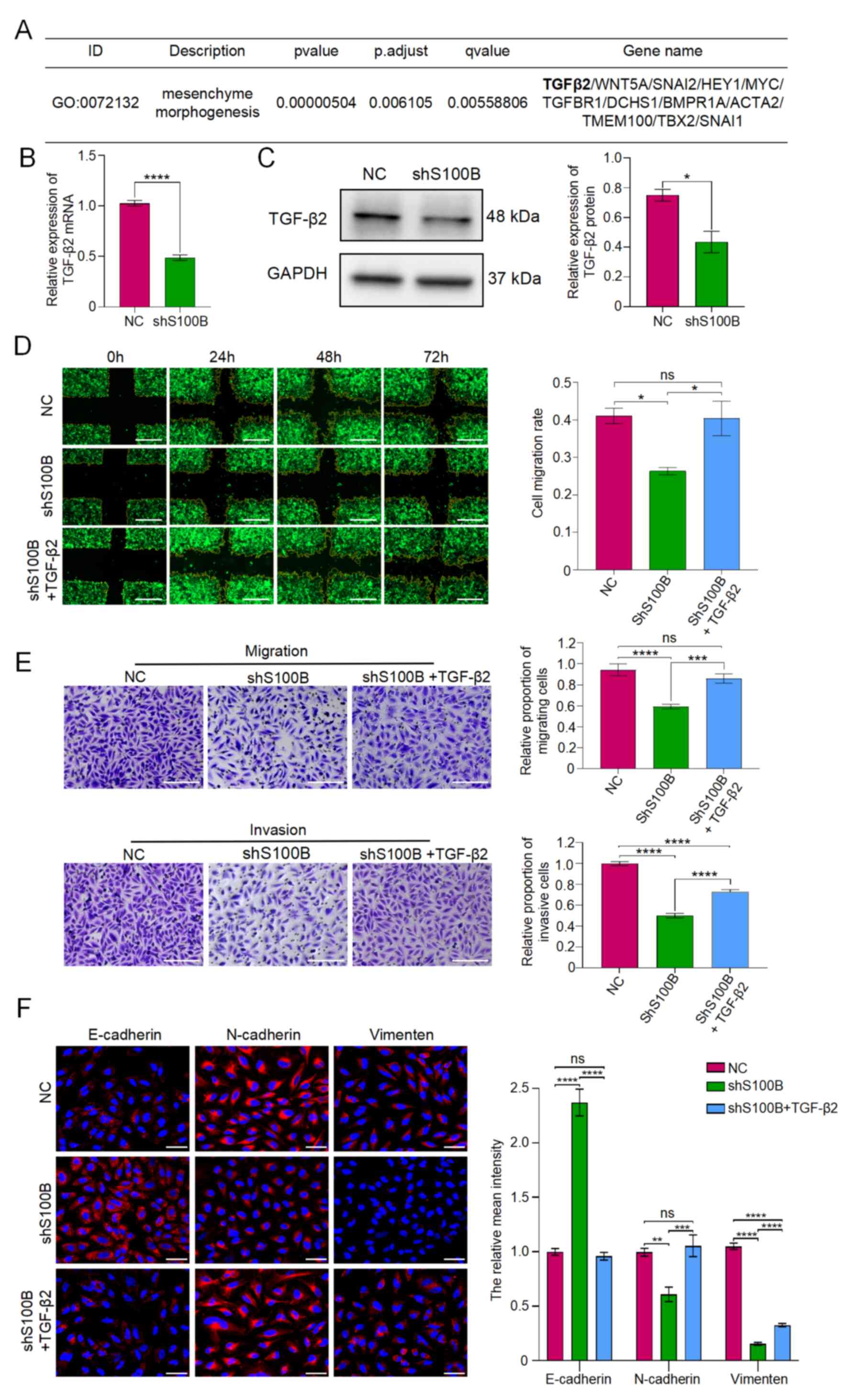 | Figure 6.TGF-β2 induces epithelial-mesenchymal
transition and enhances the invasion and migration of glioblastoma
multiforme cells. (A) Relevant downregulated genes of the
mesenchyme morphogenesis pathway. (B and C) Expression of TGF-β2 in
NC and shS100B LN229 cells was analyzed by reverse
transcription-quantitative PCR and western blotting. (D)
Cross-scratch assay evaluated the migratory capacity of NC, shS100B
and shS100B + TGF-β2 groups, Scale bar, 500 µm. (E) Transwell assay
analyzed the migratory or invasive capacity of NC, shS100B and
shS100B + TGF-β2 groups; Scale bar, 200 µm. Relative proportion of
migratory or invasive cells: Number of migratory or invasive cells
in shS100B group or shS100B + TGF-β2 group/number of migrated or
invaded cells in NC group. (F) E-cadherin, N-cadherin and vimentin
were analyzed by immunofluorescence staining. Blue represents cell
nuclei, and red represents E-cadherin, N-cadherin and vimentin.
Scale bar, 50 µm. Data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001. NC, negative
control; shS100B, shRNA S100B. |
Discussion
GBM is the most aggressive and common type of
malignant brain tumor in the central nervous system. Patients with
GBM have an overall median survival time of 12–16 months (18,19),
and a 5-year survival rate of <5% (20,21).
GBM possesses highly aggressive tumor cells that proliferate in an
infiltrative manner, which results in the absence of a clear
boundary between tumor and healthy areas of brain tissue, making it
difficult to completely remove the tumor during surgery. Although
treatment efficacy has improved with the use of temozolomide and
other combination therapies, the prognosis for patients with GBM
has remained unfavorable for the past 20 years (22). Therefore, identifying the cellular
mechanisms of GBM invasion and preventing its infiltrative growth
by means of molecularly specific therapeutic approaches may be
valuable in enhancing treatment effectiveness and extending patient
survival.
Currently, there are limited findings on the
molecular signaling pathway that underlies the effects of S100B on
the migration and invasion of GBM, and there is little research on
the association between S100B and glioma, especially GBM. S100B is
a member of the multigene family of Ca2+-binding
proteins with EF-hand motifs, which regulate cellular activities
such as metabolism, motility and proliferation. Notably, there is
clear evidence that S100B is elevated in primary malignant melanoma
(13,14). The results of the present study
reported a significantly elevated expression of S100B in both GBM
and melanoma samples, as revealed using the HPA database analysis.
Our previous studies have indicated that S100B is highly expressed
in mouse C6 (23), and in human
U251 and LN229 cell lines. In other databases, S100B was shown to
be negatively associated with patient survival time, and higher
S100B expression was indicated to result in higher mortality in
patients with GBM. In a previous in vitro study, S100B was
shown to promote the proliferation of U251 and T98G GBM cells
(24). Furthermore, overexpression
of S100B has been reported to upregulate CCL2 secretion in G261
glioma cells, leading to increased infiltration of tumor-associated
macrophages into the tumor, increased secretion of inflammatory
factors and tumor angiogenesis, which is favorable to enhance the
growth of malignant tumors (25).
By contrast, decreasing S100B expression in a murine glioma model
has been reported to alter the tumor microenvironment (TME),
inhibit tumor-associated macrophage migration and suppress tumor
progression (26).
The current study demonstrated that targeted
knockdown of S100B using shRNA significantly inhibited GBM
progression both in vitro and in vivo, establishing
S100B as a critical regulator of GBM pathogenesis and a potential
prognostic biomarker. Notably, S100B silencing in LN229 cells
significantly reduced cellular invasion and migration in
vitro, suggesting its pivotal role in tumor metastasis.
However, the precise molecular mechanisms underlying these effects
remain to be elucidated through further investigation. GO pathway
analysis revealed that multiple pathways, including mesenchyme
morphogenesis, were enriched. Despite the fact that neural tissue
does not originate from a traditional epithelial setting, there is
now substantial evidence that the process known as EMT drives
glioma invasion and migration in the brain (27,28).
Cancer cells undergo EMT, allowing them to acquire mesenchymal
characteristics that facilitate invasion and migration. Throughout
this process, cancer cells lose intercellular adhesion and
epithelial cell polarity, which is accompanied by a downregulation
of E-cadherin, ultimately resulting in decreased cell-to-cell or
cell-to-extracellular matrix adhesion. Moreover, N-cadherin and
vimentin expression is upregulated (29). The current investigation revealed
that the process of EMT was reversed following the knockdown of
S100B and resulted in inhibition of tumor cell invasion and
migration, thus indicating that S100B serves an important role in
regulating EMT. The results of the present study highlighted that
TGF-β2 may be a vital signaling molecule that is associated with
mesenchyme morphogenesis and management of the EMT process in GBM.
TGF-β2 is a member of the TGF-β family, which also includes TGF-β1
and TGF-β3. These three subtypes of TGF-β (TGF-β1-3) have the
ability to induce EMT in epithelial cells (30–33).
By contrast, inhibition or deletion of TGF-β expression can trigger
dysregulation of EMT function (34–36).
According to the RNA sequencing results of the present study,
suppression of S100B resulted in significant downregulation of
TGF-β2, but not of TGF-β1 or TGF-β3.
In the present study, the addition of exogenous
recombinant TGF-β2 protein restored the EMT phenotype, which was
inhibited by S100B knockdown, and restored the invasiveness and
migratory ability of the LN229 cell line. It has been demonstrated
that TGF-β mediates the EMT of various tumors, including GBM. For
example, it has been documented that TGF-β causes GBM cells to pass
through a mesenchymal phenotypic transition via enhancing the
expression of ZEB and pSmad2 (37).
Enhydrin hinders EMT, thereby minimizing both migration and
invasion of GBM cells, by mediating the Jun/Smad7/TGF-β1 signaling
process (38). Acidosis adaptation
in cervical and colorectal cancer cells has been shown to lead to
autocrine secretion of TGF-β2, which stimulates the formation of
lipid droplets and facilitates the EMT of tumor cells to support
malignant progression, such as invasion (39). In gastric cancer, HOXA10 mediates
EMT to promote metastasis by regulating the TGF-β2/Smad/METTL3
signaling axis (40). The present
findings suggest a close association between TGF-β2, and tumor
migration and invasion in GBM. However, to the best of the authors'
knowledge, there are no studies at present on the potential role of
the S100B-TGFβ2 axis in promoting GBM cell invasiveness and
migratory ability through the EMT.
The present study revealed that S100B was
predominantly upregulated in GBM tissue, and its elevated
expression levels were associated with shorter patient survival
time, indicating an adverse prognostic impact. Inhibition of S100B
resulted in attenuated cell proliferation, invasion, migration and
EMT. Moreover, S100B knockdown induced downregulation of TGF-β2
expression, and decreased cell invasion and migration, whereas the
EMT process was restored by the addition of recombinant TGF-β2
protein. From the perspective of EMT, the current study elucidated
the mechanism by which S100B promotes the motility of glioma cells
and also revealed that S100B modulates the expression of TGF-β2 to
mediate the occurrence of EMT. These valuable findings indicate
that S100B may be an important marker and potential target site for
GBM therapies. Despite elucidating the pivotal role of S100B in
promoting GBM cell migration and invasion through modulation of the
TGF-β2/EMT axis in vitro, the current study is inherently
limited by the absence of in situ validation, a critical gap
given the profound influence of the complex EMT on metastatic
behavior. To further investigate the role of S100B in GBM
progression, the intracranial GBM xenograft model in
immunocompromised mice is recommended to enable rigorous assessment
of the impact of S100B on invasion via bioluminescence and
histopathological analyses. Furthermore, the transparent zebrafish
system with GFP-labeled tumor cells may offer real-time
visualization of micro-metastasis formation (41,42).
This dual-model approach would not only complement the present
in vitro findings but also provide mechanistic insights into
the role of S100B across different biological contexts. Future
investigations will leverage these platforms to evaluate the
therapeutic potential of S100B through targeted genetic
interventions, potentially paving the way for novel anti-metastatic
strategies in GBM.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Youth Basic Research
Project from the Ministry of Education Key Laboratory of Child
Development and Disorders (grant no. YBRP-202113) and the Science
and Technology Research Program of the Chongqing Municipal
Education Commission (grant no. KJQN202400427).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XL obtained most of the study's outcome data
independently, analyzed and interpreted the data, drafted the
manuscript, and obtained funding. YX made significant contributions
to the acquisition and analysis of part of the data, and provided
valuable assistance in conducting a portion of the experiments. HZ,
QY and SD were involved in obtaining some of the research results.
BT presented the original research concept and design, analyzed and
interpreted the data, revised the contents of the manuscript,
obtained funding and supervised the research. All authors read and
approved the final version of the manuscript. XL and BT confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The animal use protocol was reviewed and approved by
the Ethics Committee of Children's Hospital of Chongqing Medical
University (IACUC approval no: CHCMU-IACUC20210114024; Chongqing,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
EMT
|
epithelial-mesenchymal transition
|
|
WHO
|
World Health Organization
|
|
DMEM
|
Dulbecco's Modified Eagle Medium
|
|
shS100B
|
shRNA S100B LN229 GBM cells
|
|
NC
|
negative control LN229 GBM cells
|
|
GEPIA
|
Gene Expression Profiling Interactive
Analysis
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TISCH
|
Tumor Immune Single Cell Hub
|
|
EdU
|
5-ethynyl-2′-deoxyuridine
|
References
|
1
|
Tan AC, Ashley DM, López GY, Malinzak M,
Friedman HS and Khasraw M: Management of glioblastoma: State of the
art and future directions. CA Cancer J Clin. 70:299–312.
2020.PubMed/NCBI
|
|
2
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sahm F, Capper D, Jeibmann A, Habel A,
Paulus W, Troost D and von Deimling A: Addressing diffuse glioma as
a systemic brain disease with single-cell analysis. Arch Neurol.
69:523–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alexander BM and Cloughesy TF: Adult
glioblastoma. J Clin Oncol. 35:2402–2409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McKinnon C, Nandhabalan M, Murray SA and
Plaha P: Glioblastoma: Clinical presentation, diagnosis, and
management. BMJ. 374:n15602021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Donato R, Cannon BR, Sorci G, Riuzzi F,
Hsu K, Weber DJ and Geczy CL: Functions of S100 proteins. Curr Mol
Med. 13:24–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schafer BW and Heizmann CW: The S100
family of EF-hand calcium-binding proteins: Functions and
pathology. Trends Biochem Sci. 21:134–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Donato R, Sorci G, Riuzzi F, Arcuri C,
Bianchi R, Brozzi F, Tubaro C and Giambanco I: S100B's double life:
intracellular regulator and extracellular signal. Biochim Biophys
Acta. 1793:1008–1022. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiong TF, Pan FQ and Li D: Expression and
clinical significance of S100 family genes in patients with
melanoma. Melanoma Res. 29:23–29. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yen MC, Huang YC, Kan JY, Kuo PL, Hou MF
and Hsu YL: S100B expression in breast cancer as a predictive
marker for cancer metastasis. Int J Oncol. 52:433–440.
2018.PubMed/NCBI
|
|
12
|
Yang T, Cheng J, Yang Y, Qi W, Zhao Y,
Long H, Xie R and Zhu B: S100B mediates stemness of ovarian cancer
stem-like cells through inhibiting p53. Stem Cells. 35:325–336.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin J, Yang Q, Yan Z, Markowitz J, Wilder
PT, Carrier F and Weber DJ: Inhibiting S100B restores p53 levels in
primary malignant melanoma cancer cells. J Biol Chem.
279:34071–34077. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Roy Choudhury S, Heflin B, Taylor E, Koss
B, Avaritt NL and Tackett AJ: CRISPR/dCas9-KRAB-mediated
suppression of S100b restores p53-mediated apoptosis in melanoma
cells. Cells. 12:7302023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu H, Li W, Yue H, Bai Y, Li J, Lu X and
Wang J: S100B induces angiogenesis via the clathrin/FOXO1/β-catenin
signaling pathway and contributes to bevacizumab resistance in
epithelial ovarian cancer. J Adv Res. May 31–2025.(Epub ahead of
print). View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun D, Wang J, Han Y, Dong X, Ge J, Zheng
R, Shi X, Wang B, Li Z, Ren P, et al: TISCH: A comprehensive web
resource enabling interactive single-cell transcriptome
visualization of tumor microenvironment. Nucleic Acids Res. 49(D1):
D1420–D1430. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ostrom QT, Gittleman H, de Blank PM,
Finlay JL, Gurney JG, McKean-Cowdin R, Stearns DS, Wolff JE, Liu M,
Wolinsky Y, et al: American brain tumor association adolescent and
young adult primary brain and central nervous system tumors
diagnosed in the United States in 2008–2012. Neuro Oncol. 18 (Suppl
1):i1–i50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Witthayanuwat S, Pesee M, Supaadirek C,
Supakalin N, Thamronganantasakul K and Krusun S: Survival analysis
of glioblastoma multiforme. Asian Pac J Cancer Prev. 19:2613–2617.
2018.PubMed/NCBI
|
|
23
|
Tan B, Shen L, Yang K, Huang D, Li X, Li
Y, Zhao L, Chen J, Yi Q, Xu H, et al: C6 glioma-conditioned medium
induces malignant transformation of mesenchymal stem cells:
Possible role of S100B/RAGE pathway. Biochem Biophys Res Commun.
495:78–85. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu Y, Song J, Wang Z, Kan J, Ge Y, Wang D,
Zhang R, Zhang W and Liu Y: A novel S100 family-based signature
associated with prognosis and immune microenvironment in glioma. J
Oncol. 2021:35865892021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang H, Zhang L, Zhang IY, Chen X, Da
Fonseca A, Wu S, Ren H, Badie S, Sadeghi S, Ouyang M, et al: S100B
promotes glioma growth through chemoattraction of myeloid-derived
macrophages. Clin Cancer Res. 19:3764–3775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao H, Zhang IY, Zhang L, Song Y, Liu S,
Ren H, Liu H, Zhou H, Su Y, Yang Y and Badie B: S100B suppression
alters polarization of infiltrating myeloid-derived cells in
gliomas and inhibits tumor growth. Cancer Lett. 439:91–100. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kahlert UD, Joseph JV and Kruyt FAE: EMT-
and MET-related processes in nonepithelial tumors: Importance for
disease progression, prognosis, and therapeutic opportunities. Mol
Oncol. 11:860–877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie C, Zhou M, Lin J, Wu Z, Ding S, Luo J,
Zhan Z, Cai Y, Xue S and Song Y: EEF1D promotes glioma
proliferation, migration, and invasion through EMT and PI3K/Akt
pathway. Biomed Res Int. 2020:78047062020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dongre A and Weinberg RA: New insights
into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. Nat Rev Mol Cell Biol. 20:69–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miettinen PJ, Ebner R, Lopez AR and
Derynck R: TGF-beta induced transdifferentiation of mammary
epithelial cells to mesenchymal cells: Involvement of type I
receptors. J Cell Biol. 127:2021–2036. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Piek E, Moustakas A, Kurisaki A, Heldin CH
and ten Dijke P: TGF-(beta) type I receptor/ALK-5 and Smad proteins
mediate epithelial to mesenchymal transdifferentiation in NMuMG
breast epithelial cells. J Cell Sci. 112:4557–4568. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Valcourt U, Kowanetz M, Niimi H, Heldin CH
and Moustakas A: TGF-beta and the Smad signaling pathway support
transcriptomic reprogramming during epithelial-mesenchymal cell
transition. Mol Biol Cell. 16:1987–2002. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao J, Zhu Y, Nilsson M and Sundfeldt K:
TGF-β isoforms induce EMT independent migration of ovarian cancer
cells. Cancer Cell Int. 14:722014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Su J, Morgani SM, David CJ, Wang Q, Er EE,
Huang YH, Basnet H, Zou Y, Shu W, Soni RK, et al: TGF-beta
orchestrates fibrogenic and developmental EMTs via the RAS effector
RREB1. Nature. 577:566–571. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leung DHL, Phon BWS, Sivalingam M,
Radhakrishnan AK and Kamarudin MNA: Regulation of EMT markers,
extracellular matrix, and associated signalling pathways by long
non-coding RNAs in glioblastoma mesenchymal transition: A scoping
review. Biology (Basel). 12:8182023.PubMed/NCBI
|
|
37
|
Joseph JV, Conroy S, Tomar T,
Eggens-Meijer E, Bhat K, Copray S, Walenkamp AM, Boddeke E,
Balasubramanyian V, Wagemakers M, et al: TGF-β is an inducer of
ZEB1-dependent mesenchymal transdifferentiation in glioblastoma
that is associated with tumor invasion. Cell Death Dis.
5:e14432014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen J, Hu J, Li X, Zong S, Zhang G, Guo Z
and Jing Z: Enhydrin suppresses the malignant phenotype of GBM via
Jun/Smad7/TGF-β1 signaling pathway. Biochem Pharmacol.
226:1163802024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Corbet C, Bastien E, Santiago de Jesus JP,
Dierge E, Martherus R, Vander Linden C, Doix B, Degavre C, Guilbaud
C, Petit L, et al: TGFβ2-induced formation of lipid droplets
supports acidosis-driven EMT and the metastatic spreading of cancer
cells. Nat Commun. 11:4542020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song C and Zhou C: HOXA10 mediates
epithelial-mesenchymal transition to promote gastric cancer
metastasis partly via modulation of TGFB2/Smad/METTL3 signaling
axis. J Exp Clin Cancer Res. 40:622021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Almstedt E, Rosén E, Gloger M, Stockgard
R, Hekmati N, Koltowska K, Krona C and Nelander S: Real-time
evaluation of glioblastoma growth in patient-specific zebrafish
xenografts. Neuro Oncol. 24:726–738. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Larsson S, Kettunen P and Carén H:
Orthotopic transplantation of human paediatric high-grade glioma in
zebrafish larvae. Brain Sci. 12:6252022. View Article : Google Scholar : PubMed/NCBI
|