Introduction
Pancreatic cancer is a highly lethal malignant
tumor. Due to the lack of specific symptoms in its early stage and
its highly aggressive nature, patients are usually diagnosed at
later stages of the disease (1,2).
Therefore, the prognosis for pancreatic cancer is poor, with 5-year
survival rates of ≤10% (3). Despite
major advances in oncology in recent years, the treatment of
pancreatic cancer remains a challenge. Currently, the main clinical
treatment methods include surgery, radiotherapy and chemotherapy,
with surgery remaining the most effective option. However, only
15–20% of patients qualify for surgical treatment, and even after
surgery, recurrence and metastasis remain highly prevalent
(4). Gemcitabine (GEM) is a
deoxycytidine analog that inhibits DNA replication and is widely
used for the treatment of various cancer types, including
pancreatic, non-small cell lung, bladder, ovarian and breast cancer
(5,6). For patients with inoperable locally
advanced and metastatic pancreatic cancer, GEM monotherapy or
GEM-based combination chemotherapy remain first-line choices
(7). However, enzymatic
deamination, rapid systemic clearance and the emergence of
chemoresistance all limit GEM efficacy, favoring recurrence and
metastasis (6). Therefore,
exploring the mechanisms underlying GEM resistance, improving the
sensitivity of pancreatic cancer cells to GEM and identifying novel
targeted therapeutic drugs is paramount.
The AKT/mTOR signaling pathway crucially influences
basic cellular processes such as protein synthesis, and cell
growth, proliferation and metabolism, and also participates in the
regulation of autophagy, apoptosis and angiogenesis (8,9).
Over-activation of the AKT/mTOR signaling pathway has been reported
in various tumors, such as skin cancer (10), breast cancer (11), thyroid cancer (12), non-small cell lung cancer (NSCLC)
(13) and prostate cancer (14). Abnormal activation of this pathway
is also closely related to the occurrence and development of
pancreatic cancer (15).
Furthermore, sustained activation of AKT/mTOR signaling enhances
drug resistance in pancreatic cancer cells, and creates favorable
conditions for their survival and proliferation by enhancing
metabolic activity and inhibiting apoptosis (16). Therefore, therapeutic inhibition of
the AKT/mTOR pathway has become an important goal in the treatment
of pancreatic cancer.
At present, strategies for reversing drug resistance
in tumor cells mainly include the following approaches: Targeted
inhibition of drug resistance pathways, combination treatments with
conventional agents, administration of traditional Chinese medicine
(TCM) prescriptions, nano-drug delivery systems and gene editing
(17,18). Among these, TCM compounds are
particularly notable for their ability to regulate multiple
targets. This multi-target action can not only enhance the efficacy
of anticancer drugs but also reduce their associated cytotoxicity
and other side effects (19).
Bupleurum, also known as Radix Bupleuri or ‘Chai Hu’, is a
classical herb within TCM (20).
Saikosaponin D (SSD), one of the major bioactive ingredients of
Bupleurum, possesses anti-inflammatory, antioxidant and
immunoregulatory properties (20).
Our previous study demonstrated that SSD exerted cytotoxic effects
on glioma cells and enhanced their sensitivity to temozolomide
chemotherapy (21). Previous
research has further demonstrated that SSD has growth inhibitory
and pro-apoptotic effects in a variety of solid tumors. For
example, Tang et al (22)
found that SSD enhanced the sensitivity of human NSCLC cells to
gefitinib by inhibiting STAT3/Bcl-2 signaling, thereby inducing
tumor cell apoptosis. Hu et al (23) demonstrated that SSD enhanced the
sensitivity of human gastric cancer cells to cisplatin by
inhibiting the IKKβ/NF-κB pathway. In addition, Zhang et al
(24) found that SSD suppressed the
malignant phenotype of Hep3B hepatoma cells and increased the
sensitivity of these cells to chemotherapy in vitro and
in vivo. However, studies on the combination of SSD and GEM
in pancreatic cancer remain scarce. Thus, it remains unclear
whether SSD can enhance the sensitivity of pancreatic cancer cells
to GEM and effectively overcome GEM resistance. Furthermore,
although the antitumor activity of SSD has been extensively studied
in multiple solid tumors, where they demonstrated strong potential
for apoptosis induction through multiple pathways (25,26),
it is not clear whether combined administration of SSD and
chemotherapy agents induces apoptosis via regulation of the
AKT/mTOR pathway. In addition, to the best of our knowledge, it
remains unknown whether SSD can improve the chemosensitivity of
pancreatic cancer cells to GEM by downregulating AKT/mTOR
signaling. Therefore, the present study aimed to investigate
whether SSD could enhance the sensitivity to GEM chemotherapy,
regulate AKT/mTOR activation, and promote apoptosis and autophagy
in pancreatic cancer cells.
Materials and methods
Reagents and antibodies
SSD (purity ≥98% as determined by high-performance
liquid chromatography) was obtained from Shanghai Yuanye
Biotechnology Co., Ltd. GEM was purchased from GLPBIO Technology
LLC. Stock solutions of each drug (1 mmol/l) were prepared by
dissolution in DMSO and stored protected from light at −20°C. The
Cell Counting Kit-8 (CCK-8), Hoechst 33258 staining kit and
immunocytochemistry (ICC) staining kit were acquired from Wuhan
Boster Biological Technology Co., Ltd. The Autophagy Dual Stain kit
[using monodansylcadaverine (MDC)] and 0.1% crystal violet ammonium
oxalate solution were procured from Beijing Solarbio Science &
Technology Co., Ltd. The BCA protein assay kit was obtained from
the Beyotime Institute of Biotechnology. The TUNEL Apoptosis Assay
kit was purchased from TransGen Biotech Co., Ltd. The following
antibodies were purchased from Proteintech Group, Inc.: Mouse
monoclonal antibodies against mTOR (1:500; cat. no. 66888-1-Ig),
phosphorylated (p-)mTOR (Ser2448) (1:2,000; cat. no. 67778-1-Ig),
p-AKT (Ser473) (1:500; cat. no. 66444-1-Ig), caspase-3 (1:2,000;
cat. no. 66470-2-Ig), cleaved caspase-3 (1:1,000; cat. no.
66470-2-Ig), Bax (1:500; cat. no. 60267-1-Ig), AKT (1:500; cat. no.
60203-2-Ig) and β-actin (1:2,000; cat. no. 66009-1-Ig); as well as
rabbit antibodies, including polyclonal antibodies against Bcl-2
(1:1,000; cat. no. 12789-1-AP), Beclin 1 (1:1,000; cat. no.
11306-1-AP) and LC3 (1:1,000; cat. no. 14600-1-AP). The same
monoclonal antibody (cat. no. 66470-2-Ig) was used for the
detection of both total caspase-3 (at 1:2,000 dilution) and cleaved
caspase-3 (at 1:1,000 dilution), as it recognizes an epitope shared
by both forms of the protein. The higher antibody concentration
(1:1,000) for cleaved caspase-3 was used due to its lower abundance
post-cleavage, ensuring optimal detection sensitivity.
Additionally, a separate rabbit monoclonal anti-Bax antibody
(1:1,000; cat. no. 5023T) was obtained from Cell Signaling
Technology, Inc., for western blotting. Peroxidase-conjugated
anti-mouse (1:5,000; cat. no. SA00001-1) and anti-rabbit (1:5,000;
cat. no. SA00001-2) secondary antibodies were also obtained from
Proteintech Group, Inc.
Chemical structure illustration
The chemical structures of SSD and GEM were
generated based on their canonical representations from the PubChem
database (National Center for Biotechnology Information; http://pubchem.ncbi.nlm.nih.gov/), using their
unique Compound Identifiers (119361 for SSD and 60750 for GEM), and
were subsequently exported in high-resolution TIFF format for
publication.
Cell lines and culture
The MIA PaCa-2 and AsPC-1 human pancreatic
adenocarcinoma cell lines were obtained from the cell bank of the
Yan'an University Medical Research and Experimental Center (Yan'an,
China). The MIA PaCa-2 and AsPC-1 cell lines were originally
acquired from American Type Culture Collection. MIA PaCa-2 and
AsPC-1 cells were cultured in DMEM (cat. no. 11965092; Gibco;
Thermo Fisher Scientific, Inc.) and RPMI-1640 medium (cat. no.
11875093; Gibco; Thermo Fisher Scientific, Inc.), respectively,
supplemented with 10% FBS (cat. no. 10270106; Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin (cat. no.
15140122; Gibco; Thermo Fisher Scientific, Inc.). Both cell lines
were maintained at 37°C in a 5% CO2 humidified
incubator. Cells in the logarithmic growth phase were plated for
downstream experiments.
Cell viability assay
MIA PaCa-2 and AsPC-1 cell suspensions were
prepared, and cells were seeded into 96-well cell culture plates
(100 µl; 2.5×103 cells/well), and treated with SSD (0,
2, 4, 6, 8, 10 or 12 µmol/l) or GEM (0, 0.25, 0.5, 1, 2, 4 or 8
µmol/l) for 24, 48 or 72 h at 37°C. CCK-8 reagent was added
to each well and cells were incubated in the dark for 1 h at 37°C.
Absorbance values were subsequently measured at 450 nm using a
microplate reader. Cell viability was calculated using GraphPad
Prism 9 software (Dotmatics). According to the effects of SSD and
GEM and their respective IC50, a GEM dose of 0.25 µmol/l
(referred to as G0.25) and SSD doses of 4 or 6 µmol/l (referred to
as S4 and S6, respectively) were selected for downstream assays to
treat MIA PaCa-2 and AsPC-1 cells. Control groups were treated with
an equal volume of the drug solvent (DMSO) alone, with the final
DMSO concentration not exceeding 0.1% in all groups.
Synergy analysis
Drug interaction effects were quantitatively
analyzed using the Chou-Talalay method (27). Combination Index (CI) values were
calculated from dose-effect curves generated based on CCK-8 and
TUNEL assays, where a CI of <1, =1 and >1 indicates synergy,
additive effects and antagonism, respectively. CalcuSyn software
(version 2.0; Biosoft) was employed for CI calculation with the
following parameters: Fraction affected range, 0.05–0.95; and
median-effect principle for dose-effect relationship modeling.
Cell morphology evaluation
Cell morphology and structure were evaluated by
H&E staining, whereby MIA PaCa-2 and AsPC-1 cells were seeded
onto sterile glass coverslips in 6-well plates at a density of
5×104 cells/well, allowed to adhere overnight, and
treated with a range of concentrations of SSD (0–12 µmol/l), GEM
(0–8 µmol/l) or the combination of G0.25 + S4 for 48 h at 37°C.
After treatment, all subsequent steps were performed at room
temperature. Briefly, cells were washed with PBS, fixed in 95%
ethanol for 20 min, and stained with Harris's hematoxylin (cat. no.
HHS32; Sigma-Aldrich; Merck KGaA) for 8 min. This was followed by
differentiation in 1% acid alcohol for 30 sec, bluing in 0.2%
ammonia water and counterstaining with eosin Y (cat. no. HT110232;
Sigma-Aldrich; Merck KGaA) for 3 min. The samples were then
dehydrated through an ascending graded ethanol series, cleared in
xylene and mounted with neutral balsam. Morphology was evaluated
under a light microscope (Nikon Eclipse Ti2; Nikon Corporation) by
two independent observers. The vehicle control group (0.1% DMSO)
was processed in parallel under identical conditions.
Cell clone formation assay
The cell clone formation assay was performed by
seeding MIA PaCa-2 and AsPC-1 cells into 6-well plates at a density
of 2×103 cells/well, allowing them to adhere for 24 h at
37°C in complete growth medium without treatment, after which the
medium was replaced with fresh medium containing specified
treatments [GEM (0.25 µmol/l), SSD (4 µmol/l), the combination
G0.25 + S4, or vehicle control (0.1% DMSO)] and cells were cultured
for 7–10 days at 37°C with the medium being refreshed every 3 days.
After incubation, the cells were washed with PBS, fixed with 4%
paraformaldehyde (PFA) for 20 min at room temperature and stained
with 0.1% crystal violet for 20 min at room temperature. The plates
were then gently rinsed with distilled water to remove excess dye,
air-dried and imaged using a light microscope. Colonies, defined as
groups of ≥50 cells, were quantified using ImageJ software (version
1.53t; National Institutes of Health) by measuring the crystal
violet-stained area after applying a consistent threshold. Results
were expressed as a percentage of the control group.
Hoechst 33258 staining
Hoechst 33258 staining was performed by seeding MIA
PaCa-2 and AsPC-1 cells on sterile glass coverslips in 6-well
plates at a density of 5×104 cells/well, allowing
adhesion for 24 h at 37°C in untreated complete medium, and then
treating cells with GEM (0.25 µmol/l), SSD (4 µmol/l), G0.25 + S4
or vehicle (0.1% DMSO) for 48 h at 37°C. After treatment, cells
were fixed in 4% PFA for 20 min at room temperature, washed with
PBS and then stained with Hoechst 33258 (5 µg/ml) for 5 min in the
dark at room temperature. Following another wash with PBS, cells
were mounted with anti-fade medium and imaged using a Nikon Eclipse
Ti2 fluorescence microscope (Nikon Corporation) equipped with a
DS-Qi2 camera (Nikon Corporation). Image analysis was conducted
using NIS-Elements software (v5.30; Nikon Corporation).
Autophagy assay
The autophagy assay was performed by seeding MIA
PaCa-2 and AsPC-1 cells into 12-well plates at a density of
5×104 cells/well, followed by treatment the next day
with GEM (0.25 µmol/l), SSD (4 µmol/l), their combination (G0.25 +
S4) or vehicle control (0.1% DMSO) for 48 h at 37°C in a 5%
CO2 humidified incubator. Subsequently, cells were
processed using the Autophagy Dual Stain kit (MDC) (Beijing
Solarbio Science & Technology Co., Ltd.) according to the
manufacturer's instructions. Briefly, cells were washed with 1X PBS
and fixed with the kit-provided fixative (4% paraformaldehyde; 20
min at 25°C). After three washes with the kit-supplied 1X wash
buffer, cells were incubated with the MDC working solution (200
µl/well; 60 min at 37°C in the dark). Following three additional
washes with 1X wash buffer, fresh 1X PBS was added to prevent
drying. Image acquisition was performed immediately using a Nikon
Eclipse Ti2 fluorescence microscope (Nikon Corporation) equipped
with a DS-Qi2 camera. Quantitative analysis of fluorescence
intensity was conducted using NIS-Elements software (v5.30; Nikon
Corporation). Untreated control cells (0.1% DMSO) were processed in
parallel under identical conditions.
TUNEL
The TUNEL assay was conducted by seeding MIA PaCa-2
and AsPC-1 cells onto coverslips in 24-well plates and allowing
them to reach 70% confluence. Cells were then treated with 0.25
µmol/l GEM, 4 µmol/l SSD, their combination G0.25 + S4 or vehicle
control (0.1% DMSO) for 48 h at 37°C with 5% CO2. After
treatment, cells were washed three times with PBS and fixed with 4%
PFA for 20 min at room temperature. Permeabilization was performed
using 0.1% Triton X-100 for 5 min at room temperature. The TUNEL
reaction mixture containing terminal deoxynucleotidyl transferase
was applied to each sample, followed by incubation in a humidified
dark chamber at 37°C for 1 h. Subsequently, cells were washed with
PBS and counterstained with DAPI at a concentration of 1 µg/ml for
10 min at room temperature. After a final rinse with PBS,
coverslips were mounted using antifade mounting medium. Images from
at least five random fields per coverslip were captured using a
fluorescence microscope. Each assay included both a negative
control without terminal transferase enzyme and a vehicle-treated
DMSO control.
Western blotting
MIA PaCa-2 and AsPC-1 cells in the logarithmic
growth phase were treated with 0.25 µmol/l GEM, 4 µmol/l SSD, their
combination G0.25 + S4 or vehicle control (0.1% DMSO) for 48 h at
37°C. Immediately following treatment, cells were harvested using
EDTA-free trypsin, washed twice with cold phosphate-buffered
saline, resuspended in precooled PBS and centrifuged at 1,000 × g
for 5 min at 4°C. The cell pellets were lysed on ice for 40 min
using RIPA lysis buffer (P0013B; Beyotime Institute of
Biotechnology) supplemented with 1% protease inhibitor cocktail
(P1005; Beyotime Institute of Biotechnology). The lysates were
centrifuged at 16,020 × g for 20 min at 4°C, and the supernatant
protein concentrations were determined using a BCA assay (P0010;
Beyotime Institute of Biotechnology,). Equal amounts of protein (30
µg per lane) were separated on 10–15% SDS-polyacrylamide gels and
transferred onto PVDF membranes. After blocking with 5% skim milk
for 2 h at room temperature, the membranes were incubated overnight
at 4°C with the following primary antibodies: Mouse monoclonal
anti-mTOR (1:500; cat. no. 66888-1-Ig; Proteintech Group, Inc.),
anti-p-mTOR (Ser2448) (1:2,000; cat. no. 67778-1-Ig; Proteintech
Group, Inc.), anti-p-AKT (Ser473) (1:500; cat. no. 66444-1-Ig;
Proteintech Group, Inc.), anti-caspase-3 (1:2,000; cat. no.
66470-2-Ig; Proteintech Group, Inc.), anti-cleaved caspase-3
(1:1,000; cat. no. 66470-2-Ig; Proteintech Group, Inc.), anti-AKT
(1:500; cat. no. 60203-2-Ig; Proteintech Group, Inc.) and
anti-β-actin (1:2,000; cat. no. 66009-1-Ig; Proteintech Group,
Inc.); and rabbit polyclonal anti-Bcl-2 (1:1,000; cat. no.
12789-1-AP; Proteintech Group, Inc.), anti-Beclin 1 (1:1,000; cat.
no. 11306-1-AP; Proteintech Group, Inc.) and anti-LC3 (1:1,000;
cat. no. 14600-1-AP; Proteintech Group, Inc.) antibodies, as well
as an anti-Bax monoclonal antibody (1:1,000; cat. no. 5023T; Cell
Signaling Technology, Inc.). The same monoclonal antibody (cat. no.
66470-2-Ig) was used for the detection of both total caspase-3 (at
1:2,000 dilution) and cleaved caspase-3 (at 1:1,000 dilution), as
it recognizes an epitope shared by both forms of the protein. The
higher antibody concentration (1:1,000) for cleaved caspase-3 was
used due to its lower abundance post-cleavage, ensuring optimal
detection sensitivity. The membranes were then washed three times
with TBS with 0.1% Tween-20 (TBST) and incubated for 1.5 h at room
temperature with HRP-conjugated goat anti-mouse (1:5,000;
SA00001-1; Proteintech Group, Inc.) or goat anti-rabbit (1:5,000;
SA00001-2; Proteintech Group, Inc.) secondary antibodies. Following
three additional washes with TBST, the protein bands were
visualized using an Enhanced Chemiluminescence reagent (P0018FS;
Beyotime Institute of Biotechnology) and images were captured with
a Bio-Rad ChemiDoc imaging system (Bio-Rad Laboratories, Inc.).
Semi-quantitative analysis was performed using ImageJ software
(National Institutes of Health) with β-actin serving as the loading
control for normalization.
ICC
MIA PaCa-2 and AsPC-1 cells were seeded onto
coverslips in 24-well plates at a density of 5×104 cells
per well and cultured for 24 h. Cells were then treated with 0.25
µmol/l GEM, 4 µmol/l SSD, their combination G0.25 + S4 or vehicle
control (0.1% DMSO) for 48 h at 37°C. After treatment, cells were
fixed with 4% PFA at 4°C for 20 min, permeabilized with 0.3% Triton
X-100 for 15 min at room temperature, washed with PBS, and
incubated with 3% H2O2 in deionized water at
37°C for 10 min to quench endogenous peroxidase activity. Blocking
was performed with 5% BSA (product no. A8020; Sigma-Aldrich; Merck
KGaA) for 20 min at room temperature. The following primary
antibodies from Proteintech Group, Inc., were applied overnight at
4°C: Mouse monoclonal anti-Bax (1:500; 60267-1-Ig), rabbit
polyclonal anti-Bcl-2 (1:1,000; 12789-1-AP), mouse monoclonal
anti-cleaved caspase-3 (1:1,000; cat. no. 66470-2-Ig), mouse
monoclonal anti-p-AKT (Ser473) (1:500; 66444-1-Ig) and mouse
monoclonal anti-p-mTOR (Ser2448) (1:2,000; 67778-1-Ig). The next
day, samples were incubated with HRP-conjugated goat anti-mouse
(1:5,000; SA00001-1; Proteintech Group, Inc.) or goat anti-rabbit
(1:5,000; SA00001-2; Proteintech Group, Inc.) secondary antibodies
for 1.5 h at room temperature. After three washes with TBST,
signals were developed using 3,3′-diaminobenzidine (DAB). DAB
development was monitored in real-time under a Nikon Eclipse Ti2
light microscope (Nikon Corporation), and the reaction was stopped
by rinsing with pure water once moderate brown cytoplasmic staining
became visible. Nuclei were counterstained with hematoxylin for 3
min at room temperature, followed by washing and dehydration.
Coverslips were mounted with neutral resin and observed under a
light microscope. Images were captured and analyzed using
NIS-Elements software (v5.30; Nikon Corporation). Each experiment
included a negative control with non-immune IgG and a
vehicle-treated DMSO control.
Statistical analysis
GraphPad Prism 9.0 software (Dotmatics) was used for
all statistical analyses and graphing. Based on an a priori power
analysis using G*Power 3.1 (ANOVA: Effect size f=0.4, α=0.05,
power=0.8), five independent biological replicates (n=5) per group
were performed to ensure adequate statistical power. Prior to
analysis, data normality and variance homogeneity were assessed
using the Shapiro-Wilk test and Brown-Forsythe test, respectively.
To account for multiple comparisons, all P-values were adjusted
using the Benjamini-Hochberg correction (false discovery rate
<0.05). Effect sizes with 95% CIs were calculated through
bootstrap resampling (1,000 iterations). Data are presented as the
mean ± standard deviation. An adjusted P-value <0.05 was
considered to indicate a statistically significant difference.
Specific statistical tests were applied based on the experimental
design: Two-way ANOVA with Tukey's post hoc test was used to
compare cell viability across time points and treatments; one-way
ANOVA with Tukey's post hoc test was used for multiple group
comparisons at a single time point. Effect sizes are reported as
partial η2 for ANOVA. Non-parametric alternatives
(Kruskal-Wallis test with Dunn's post hoc test) were used when
parametric assumptions were violated.
Results
SSD inhibits pancreatic cancer cell
proliferation in a concentration-dependent manner
To determine the sensitivity of MIA PaCa-2 and
AsPC-1 cells to SSD (Fig. 1A), cell
proliferation was examined using a CCK-8 assay. Treatment with a
concentration range of 0-12 µmol/l SSD (where 0 µmol/l represents
the vehicle control group) for 24–72 h significantly inhibited the
proliferation of both cell lines in a concentration-dependent
manner (Fig. 1B and C).
Specifically, significant decreases were observed starting at 2
µmol/l after 48 h, with viability falling to <20% of the control
at 10 µmol/l. The sensitivity to SSD treatment was greater in
AsPC-1 cells than in MIA PaCa-2 cells, as evidenced by their lower
survival rates of 58.65% at 4 µmol/l and 53.60% at 6 µmol/l after
48 h of treatment, compared with 71.37 and 61.48% for MIA PaCa-2
cells at the respective concentrations (Fig. 1B and C). Concurrently, H&E
staining demonstrated concentration-dependent morphological
alterations, showing mild cytoplasmic shrinkage and reduced cell
density at 2–4 µmol/l, and pronounced nuclear condensation,
apoptotic body formation and widespread cell detachment at higher
concentrations (6–12 µmol/l) in both cell lines (Fig. 1D and E).
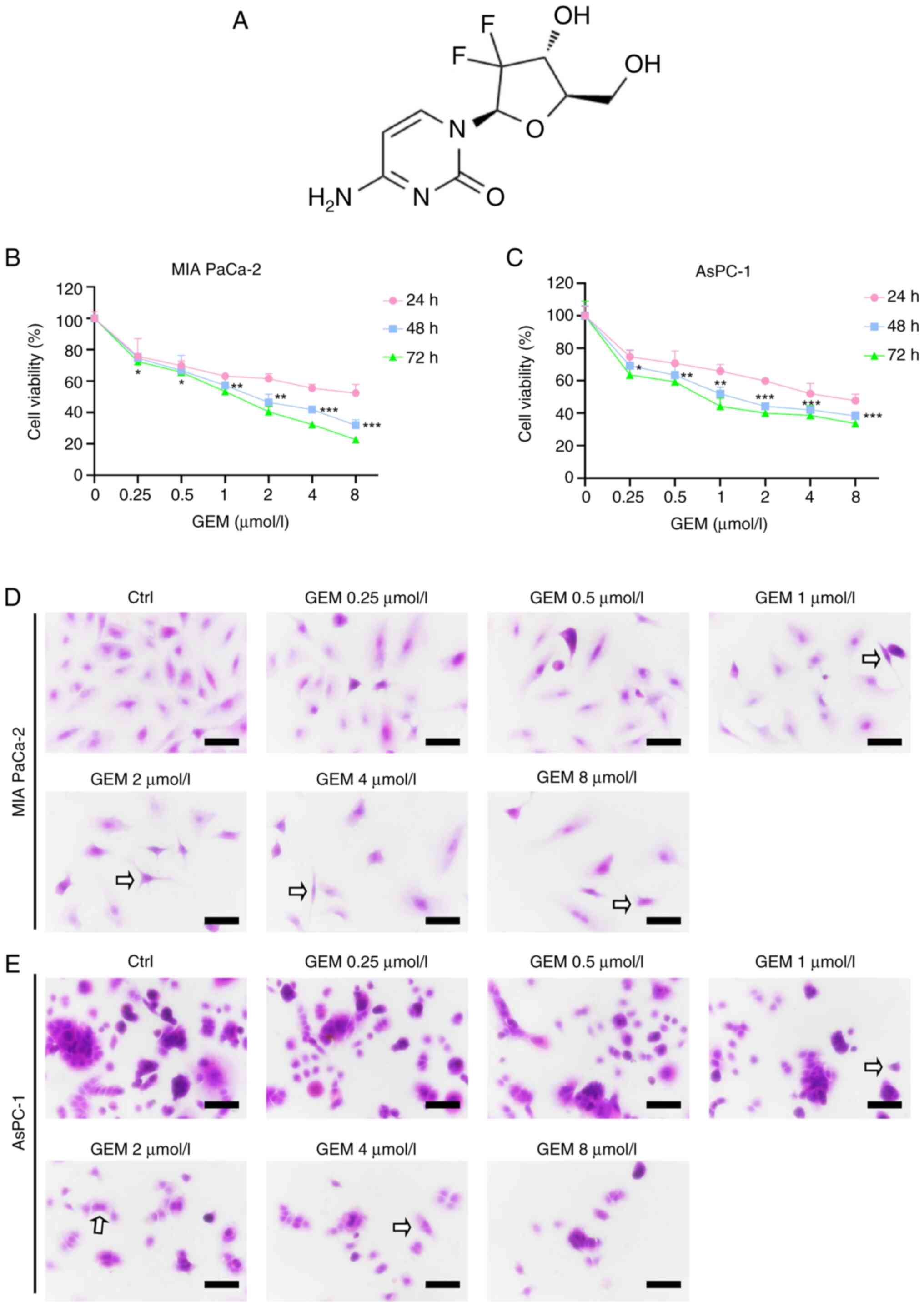
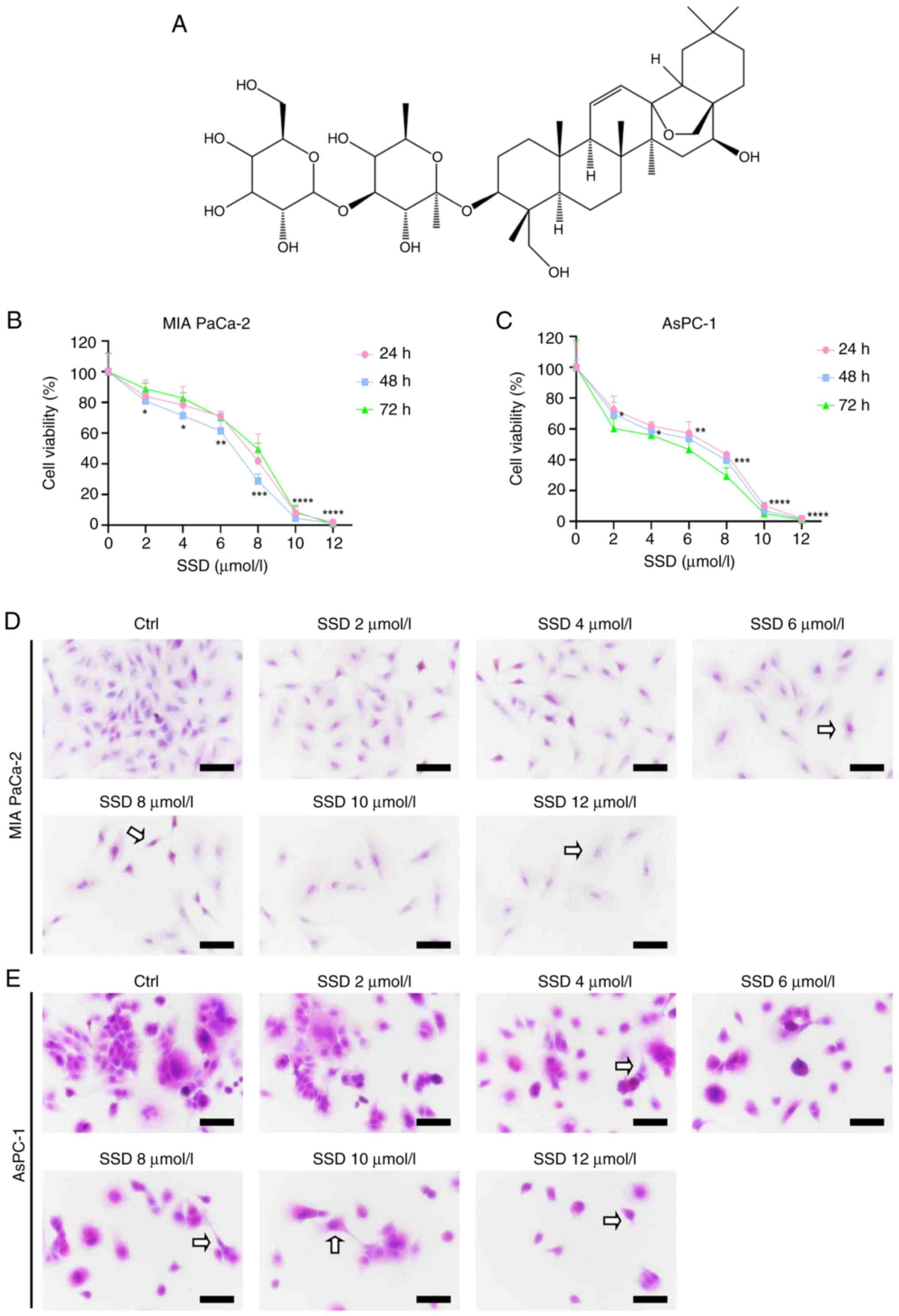 | Figure 1.Dose-dependent effects of SSD on
pancreatic cancer cells. (A) Chemical structure of SSD. (B) MIA
PaCa-2 and (C) AsPC-1 cell viability after treatment with various
concentrations of SSD (0–12 µmol/l; 24–72 h; Cell Counting Kit-8
assay). The Ctrl group was treated with 0 µmol/l SSD. Morphological
changes in (D) MIA PaCa-2 and (E) AsPC-1 cells based on H&E
staining (magnification, ×40). Arrows point to SSD-induced
apoptotic alterations, including nuclear condensation, cytoplasmic
shrinkage and apoptotic bodies. Data are presented as the mean ± SD
(n=5). At the 48-h mark, S12 sharply reduced cell viability in MIA
PaCa-2 cells. The same effect was observed in AsPC-1 cells, where
S12 also significantly inhibited cell proliferation. The
statistical details for S12 are as follows: MIA PaCa-2 cells,
Padj<0.0001; η2=0.73; and AsPC-1 cells,
Padj<0.0001; η2=0.81. All P-values were
adjusted using the Benjamini–Hochberg method. Scale bar, 50 µm. The
significance indicators in (B and C) (*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001) represent the comparison between
each SSD-treated group and the Ctrl group (0 µmol/l). Statistical
analysis was performed using one-way ANOVA followed by Tukey's post
hoc test. Ctrl, control; SSD, Saikosaponin D. |
GEM exhibits time- and
concentration-dependent growth inhibitory effects on pancreatic
cancer cells
To evaluate the sensitivity of MIA PaCa-2 and AsPC-1
cells to GEM (Fig. 2A), cells were
treated with various concentrations of GEM (0, 0.25, 0.5, 1, 2, 4
and 8 µmol/l) for 24, 48 or 72 h. CCK-8 assay results demonstrated
that GEM significantly suppressed the proliferation of both cell
lines in a time- and concentration-dependent manner (Fig. 2B and C), as determined by one-way
ANOVA with Tukey's post hoc test comparing each concentration group
to the vehicle control (0 µmol/l) at each time point. Both cell
lines exhibited reduced proliferation capacity after treatment with
0.25 µmol/l GEM; specifically, after 24, 48 and 72 h of exposure,
the viability of MIA PaCa-2 cells decreased to 75.75, 74.68 and
72.44%, respectively, while that of AsPC-1 cells decreased to
74.80, 69.22 and 63.59%, respectively. Based on this significant
growth inhibition observed at 0.25 µmol/l and to model a sub-lethal
chemotherapeutic pressure, this concentration of GEM (denoted as
G0.25) was selected for subsequent combination experiments.
Similarly, SSD concentrations of 4 and 6 µmol/l (denoted as S4 and
S6, respectively) were chosen for combination treatments as they
induced moderate yet significant growth inhibition (Fig. 1B and C) while maintaining low
cytotoxicity, making them suitable for evaluating synergistic
effects with GEM. H&E staining further confirmed dose-dependent
morphological alterations induced by GEM (0.25–8 µmol/l), including
mild cytoplasmic shrinkage and reduced cell density at lower
concentrations (0.25–1 µmol/l), and marked nuclear condensation,
apoptotic body formation and cell detachment at higher doses (2–8
µmol/l) in both cell lines (Fig. 2D and
E).
SSD significantly enhances the
anti-proliferative effects of GEM in pancreatic cancer cells
through synergistic growth inhibition
The inhibitory effects of 24-, 48- and 72-h
treatments with low-dose SSD and GEM (G0.25 and S4) on the
proliferation of MIA PaCa-2 and AsPC-1 cells were examined. CCK-8
assay results revealed that after 48 h of combined treatment (G0.25
+ S4), the viability of MIA PaCa-2 and AsPC-1 cells was
significantly inhibited compared with the Ctrl group (Fig. 3A and B). The combination therapy
exhibited a stronger inhibitory effect than either monotherapy, as
evidenced by the lower cell viability values. The synergistic
enhancement of the anti-proliferative effect of GEM by SSD is
further quantified in Table I. For
both MIA PaCa-2 and AsPC-1 cells, the addition of SSD (S4 or S6) to
a sub-lethal dose of GEM (G0.25) markedly reduced cell survival
rates beyond the level achieved by GEM alone.
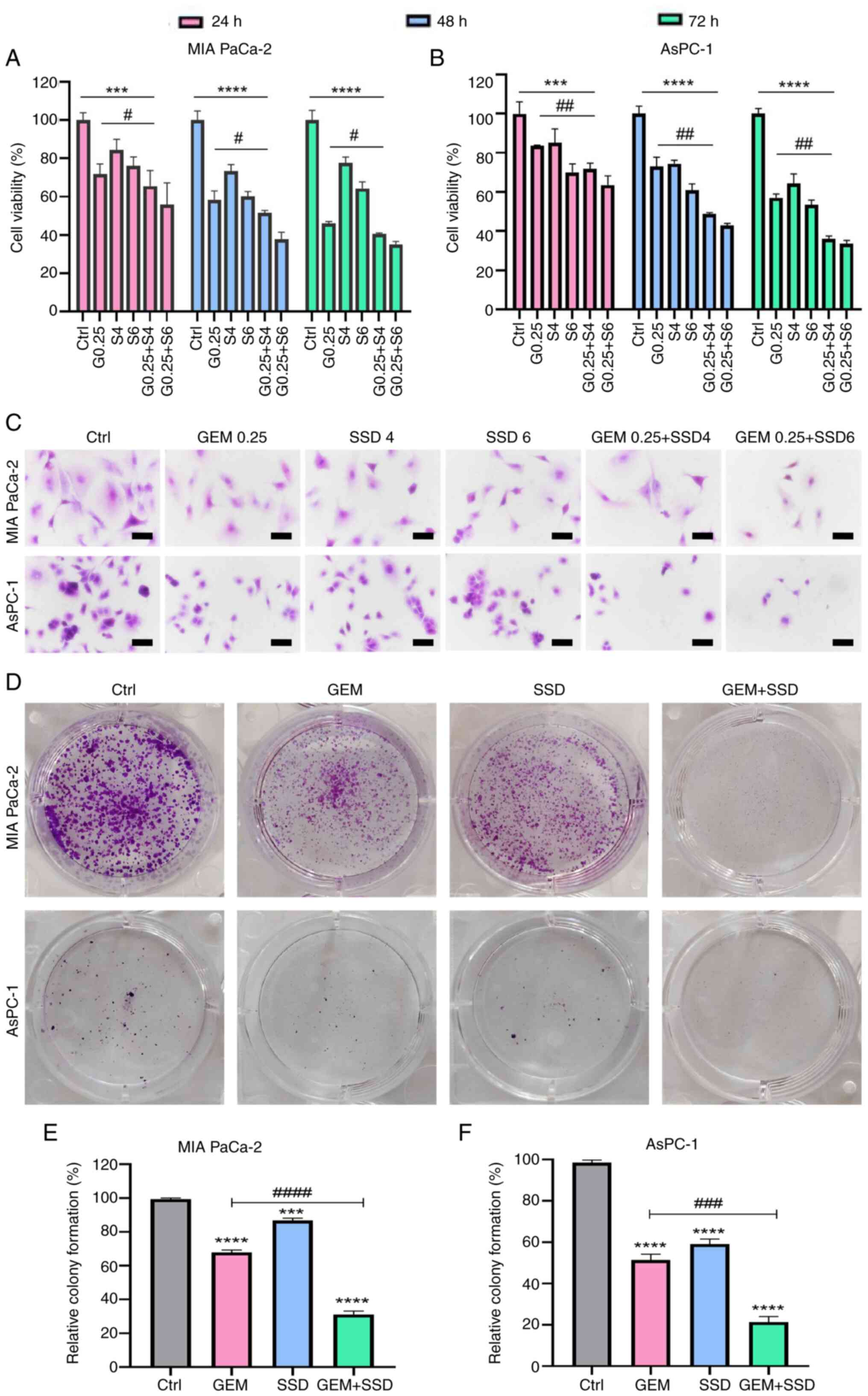 | Figure 3.Combinatorial effects of GEM and SSD
on pancreatic cancer cells. Viability of (A) MIA PaCa-2 and (B)
AsPC-1 cells treated with G0.25, S4, S6 or combinations (G0.25 +
S4/S6) for 24–72 h. (C) H&E staining showing morphological
changes (magnification, ×40). Scale bar, 50 µm. (D-F) Colony
formation assays. (D) Representative images, and quantification in
(E) MIA PaCa-2 and (F) AsPC-1 cells. (D-F) GEM, 0.25 µmol/l and
SSD, 4 µmol/l. The colony formation rate for each group is
expressed as a percentage relative to the untreated control (Ctrl)
group, which was set to 100%. Note that the absolute colony-forming
capacity of the Ctrl group under baseline culture conditions was
defined as the reference point. Data are presented as the mean ± SD
(n=5). The data demonstrate that the combination treatment (G0.25 +
S4) resulted in significantly enhanced antitumor effects compared
with GEM monotherapy (G0.25). With regard to cell viability, the
combination was significantly more effective
(Padj=0.003; effect size: η2=0.62; 95% CI,
0.41–0.79). A strong synergistic effect was also observed in the
colony formation assay, where the combination treatment led to a
marked reduction in clonogenic survival (Padj=0.001;
effect size, η2=0.68; 95% CI, 0.46–0.83). All P-values
are Benjamini-Hochberg-adjusted. ***P<0.001, ****P<0.0001 for
comparisons vs. Ctrl; #P<0.05,
##P<0.01, ###P<0.001,
####P<0.0001 for G0.25 + S4 vs. G0.25. Statistical
analysis was performed using two-way ANOVA with Tukey's post hoc
test (A and B) and one-way ANOVA with Tukey's post hoc test (E and
F). Ctrl, control; GEM, gemcitabine; SSD, Saikosaponin D; G0.25,
0.25 µmol/l GEM; SX, X µmol/l SSD. |
 | Table I.Growth inhibitory effects of combined
GEM + SSD exposure on pancreatic cancer cell lines. |
Table I.
Growth inhibitory effects of combined
GEM + SSD exposure on pancreatic cancer cell lines.
|
| Cell survival rate,
% |
|---|
|
|
|
|---|
| Cell line | G0.25 | G0.25 + S4 | G0.25 + S6 |
|---|
| MIA PaCa-2 | 69.22 | 56.97a | 40.65a |
| AsPC-1 | 74.68 | 45.64b | 42.13b |
To further demonstrate the inhibitory effect of SSD
combined with GEM on the proliferation of pancreatic cancer cell
lines, H&E staining and colony formation assays were performed.
H&E staining was performed to observe morphological changes in
MIA PaCa-2 and AsPC-1 cells treated with SSD, GEM or their
combination. The results showed that the proliferation of both
pancreatic cancer cell lines was inhibited, accompanied by a
reduction in cell number, cell shrinkage, rounding, chromatin
condensation, increased cell debris and formation of apoptotic
bodies (Fig. 3C). Colony formation
assay results showed that the combined treatment group (G0.25 + S4)
exhibited significantly reduced colony formation compared with the
corresponding single-drug GEM group (Fig. 3D). In MIA PaCa-2 and AsPC-1 cells
treated with SSD + GEM (G0.25 + S4), the colony formation rates
decreased by 32.6 and 23.4%, respectively, compared with the
corresponding single-drug GEM groups (Fig. 3E and F).
Synergistic induction of apoptosis by
SSD and GEM in pancreatic cancer cells through modulation of
apoptotic proteins
TUNEL assays were subsequently performed to evaluate
the effect of SSD, GEM and combined SSD + GEM treatment on the
apoptosis of MIA PaCa-2 and AsPC-1 cells. The results showed that
when applied alone, SSD and GEM induced modest, albeit significant,
increases in TUNEL fluorescence intensity in both cell lines
(Fig. 4A). However, the combined
SSD + GEM treatment for 48 h induced a significantly stronger
apoptotic response compared with GEM monotherapy in both MIA PaCa-2
and AsPC-1 cells (Fig. 4B and C;
Padj<0.001), suggesting a synergistic interaction.
This suggested that SSD may act synergistically with GEM to induce
apoptosis in pancreatic cancer cells. Apoptotic features were next
verified by Hoechst 33258 staining, which revealed brightly
stained, irregularly shaped, dense nuclei characteristic of
apoptotic cells (Fig. 4D).
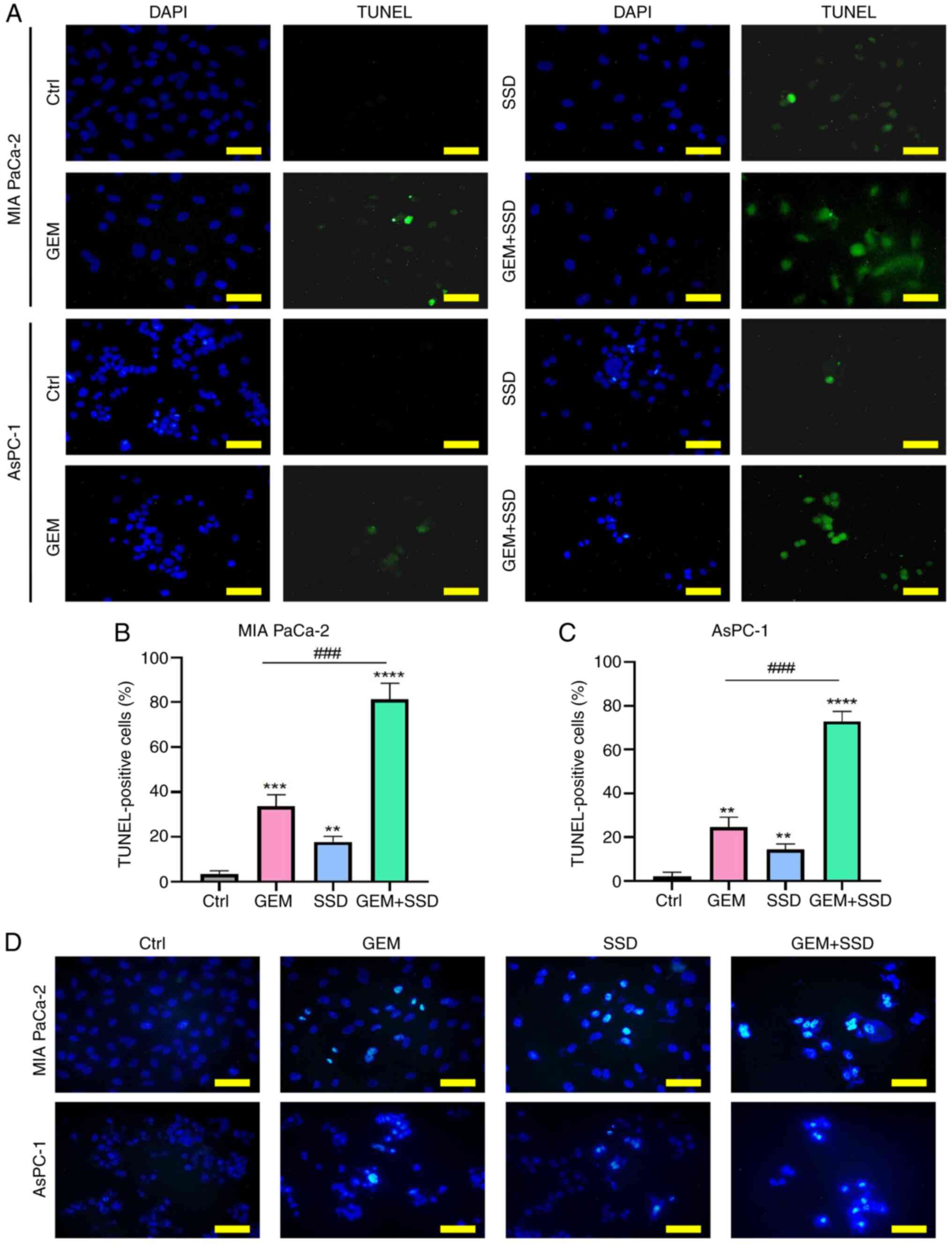 | Figure 4.Apoptosis induction by GEM and SSD
combination therapy. (A) Representative TUNEL/DAPI staining images
(green, apoptotic cells; blue, nuclei) in MIA PaCa-2 (top) and
AsPC-1 (bottom) cells treated with Ctrl, G0.25, S4 or the
combination (G0.25 + S4). Quantification of TUNEL-positive cells in
(B) MIA PaCa-2 and (C) AsPC-1 cells. (D) Hoechst 33258 staining
showing apoptotic nuclei with condensed chromatin. In the figure,
GEM represents 0.25 µmol/l GEM and SSD represents 4 µmol/l SSD.
Data are presented as the mean ± SD (n=5). The data show that the
combination treatment (G0.25 + S4) significantly enhanced the
induction of apoptosis compared with GEM monotherapy (G0.25). The
combination treatment significantly increased apoptosis, evidenced
by a substantial rise in TUNEL-positive cells
(Padj=0.002; η2=0.59; 95% CI, 0.36–0.77) and
a corresponding increase in condensed chromatin, a hallmark of
apoptosis, as shown by Hoechst 33258 staining
(Padj=0.004; η2=0.55; 95% CI, 0.31–0.74). All
P-values are Benjamini-Hochberg-adjusted. Scale bar, 50 µm.
**P<0.01, ***P<0.001, ****P<0.0001 for comparisons vs.
Ctrl; ###P<0.001 for G0.25 + S4 vs. G0.25.
Statistical analysis was performed using one-way ANOVA with Tukey's
post hoc test for multiple comparisons. Ctrl, control; GEM,
gemcitabine; SSD, Saikosaponin D. |
In addition, ICC analysis revealed distinct protein
expression patterns of key apoptotic regulators; specifically,
increased expression of pro-apoptotic Bax and cleaved caspase-3,
alongside decreased expression of anti-apoptotic Bcl-2-in SSD +
GEM-treated pancreatic cancer cells compared with GEM-only treated
controls (Fig. 5A). These ICC
findings were further corroborated by western blot analysis
(Fig. 5B). The combination
treatment potently altered the levels of apoptosis-related proteins
relative to the control. Specifically, it increased the levels of
Bax and cleaved caspase-3, while decreasing the levels of Bcl-2 and
total caspase-3 in both cell lines (Fig. 5C and D). These consistent results
from both ICC and western blot analyses demonstrated that SSD not
only induced apoptosis in pancreatic cancer cells but also enhanced
the pro-apoptotic effect of GEM by modulating key apoptotic
regulators. The observed changes in protein expression levels shown
in Fig. 5C and D are consistent
with a synergistic enhancement of apoptosis by the combination
treatment.
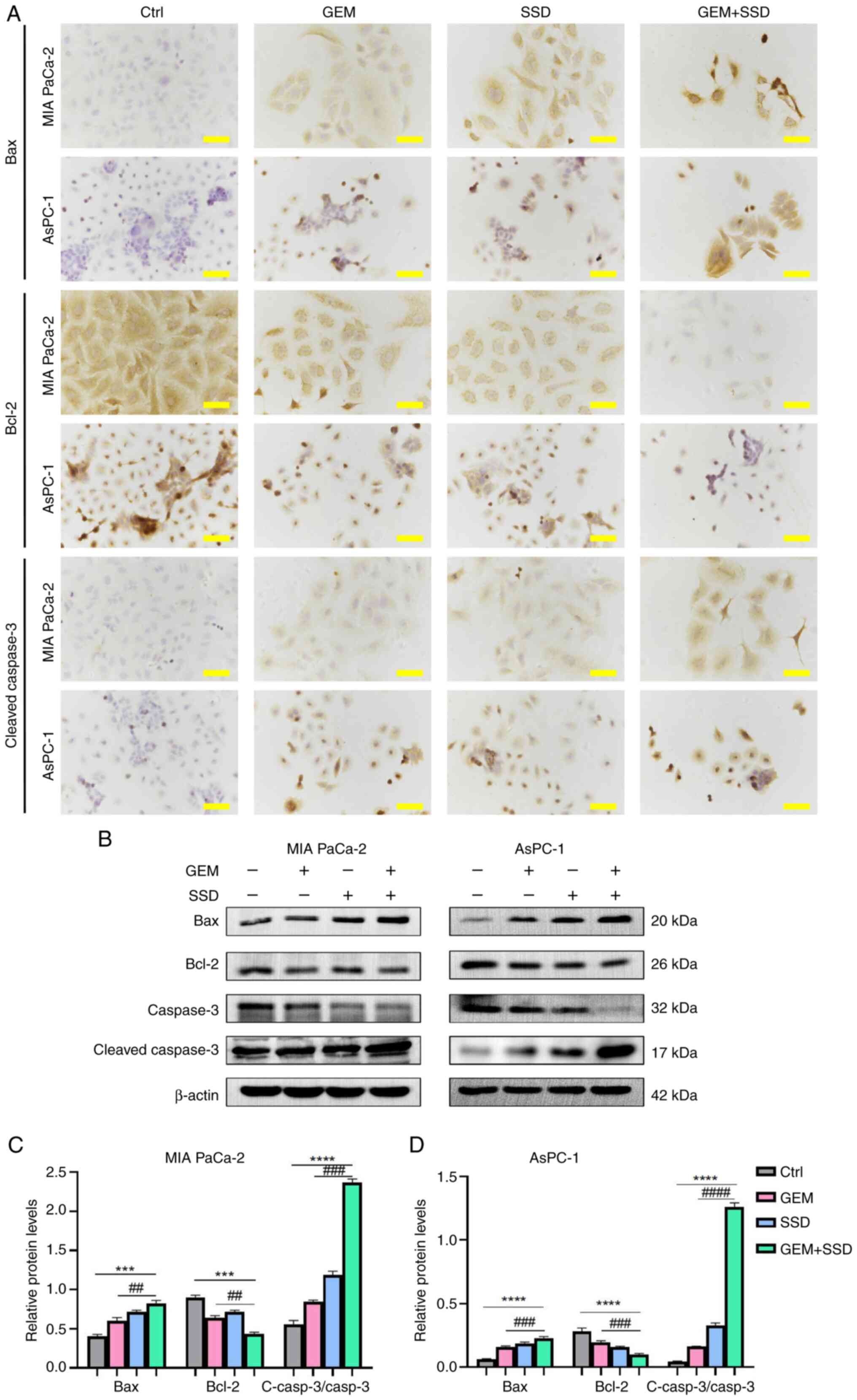 | Figure 5.Molecular mechanisms of apoptosis
induction by GEM and SSD combination treatment. (A)
Immunocytochemistry of MIA PaCa-2 and AsPC-1 cells treated with
Ctrl, G0.25, S4 or the combination (G0.25 + S4; magnification,
×40). Scale bar, 50 µm. (B) Western blot analysis of apoptosis
markers Bax (20 kDa), Bcl-2 (26 kDa), caspase-3 (32 kDa) and
cleaved caspase-3 (17 kDa), and β-actin (42 kDa).
Semi-quantification of immunoblots for (C) MIA PaCa-2 and (D)
AsPC-1 cells. In the figure, GEM represents 0.25 µmol/l GEM and SSD
represents 4 µmol/l SSD. Data are presented as the mean ± SD (n=5).
The combination treatment (G0.25 + S4) increased the expression of
pro-apoptotic Bax and cleaved caspase-3, and elevated the
c-Casp3/Casp3 ratio, while decreasing the expression of
anti-apoptotic Bcl-2 compared with GEM monotherapy (G0.25).
Specifically, Bax expression increased by 2.3-fold in MIA PaCa-2
cells and 3.1-fold in AsPC-1 cells. Conversely, Bcl-2 expression
was reduced to 45 and 38% of GEM monotherapy levels in MIA PaCa-2
and AsPC-1 cells, respectively. The c-Casp3/Casp3 ratio was
significantly enhanced by the combination treatment, indicating
robust activation of the apoptotic executioner pathway. All
P-values are Benjamini-Hochberg-adjusted. ***P<0.001,
****P<0.0001 for G0.25 + S4 vs. Ctrl; ##P<0.01,
###P<0.001, ####P<0.0001 for G0.25 + S4
vs. G0.25. Statistical analysis was performed using one-way ANOVA
with Tukey's post hoc test for multiple comparisons. Ctrl, control;
GEM, gemcitabine; SSD, Saikosaponin D; Casp3, caspase-3; c-Casp3,
cleaved caspase-3. |
To quantitatively validate the mechanistic synergy
between GEM and SSD, drug interaction was analyzed using the
Chou-Talalay method. This analysis confirmed robust synergistic
effects across multiple cellular endpoints (data not shown).
Specifically, the calculated CI values were consistently below the
synergy threshold (CI<1) in both pancreatic cancer cell lines,
indicating pharmacological synergy. These results further support
the conclusion that SSD enhances the efficacy of GEM through
synergistic inhibition of cell viability and induction of
apoptosis.
SSD potentiates the antitumor effects
of GEM through enhanced AKT/mTOR suppression
The involvement of AKT/mTOR signaling in pancreatic
cancer progression is well established (14,21).
To investigate whether SSD mediates its effects through this
pathway, ICC analysis was first performed. As shown in Fig. 6A, ICC staining demonstrated markedly
reduced cytoplasmic levels of p-AKT and p-mTOR in SSD + GEM-treated
cells compared with either single-agent treatment or control
groups, with more pronounced inhibition observed in AsPC-1 cells
than in MIA PaCa-2 cells. These ICC findings were
semi-quantitatively confirmed by western blot analysis (Fig. 6B). SSD treatment alone exhibited
comparable inhibition of p-AKT and p-mTOR levels to GEM
monotherapy. However, the combination treatment exerted
significantly greater suppressive effects compared with GEM alone,
as quantified by western blot analysis (Fig. 6C and D). Densitometric evaluation
revealed a reduction in the p-AKT/AKT ratio by 42.3% in MIA PaCa-2
cells and 72.1% in AsPC-1 cells. Similarly, the p-mTOR/mTOR ratio
was reduced by 51.4 and 80.3% in MIA PaCa-2 and AsPC-1 cells,
respectively. Notably, AsPC-1 cells exhibited more substantial
pathway inhibition than MIA PaCa-2 cells across both detection
methods. These consistent results from complementary techniques
demonstrated that SSD effectively suppressed AKT/mTOR signaling in
pancreatic cancer cells, with enhanced inhibition when combined
with GEM. The combination treatment (G0.25 + S4) resulted in
significantly greater inhibition of both the p-AKT/AKT and
p-mTOR/mTOR ratios compared with GEM alone (G0.25), with these
effects being consistently more pronounced in AsPC-1 cells
(Fig. 6C and D). The inhibition was
statistically significant for both the p-AKT/AKT ratio
(Padj<0.0001; η2=0.73; 95% CI, 0.55–0.86)
and the p-mTOR/mTOR ratio (Padj<0.0001;
η2=0.81; 95% CI, 0.67–0.91).
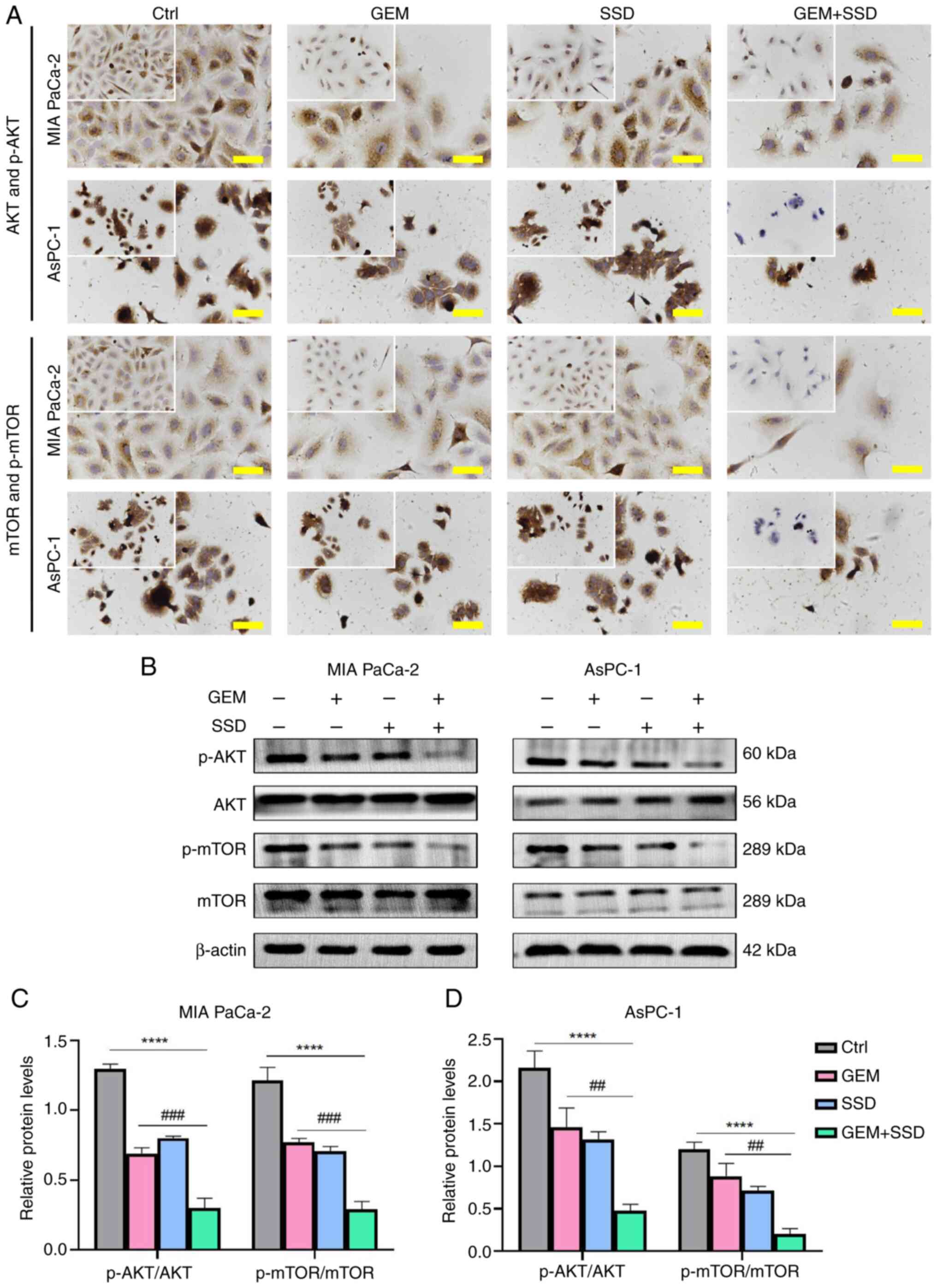 | Figure 6.AKT/mTOR pathway inhibition by GEM
and SSD combination therapy. (A) Immunocytochemistry staining of
AKT/mTOR signaling proteins in MIA PaCa-2 and AsPC-1 cells treated
with Ctrl, G0.25, S4 or the combination (G0.25 + S4; magnification,
×40; insets show phosphoprotein details). Scale bar, 50 µm. (B)
Western blot analysis of p-AKT (60 kDa), AKT (56 kDa), p-mTOR (289
kDa), mTOR (289 kDa) and β-actin (42 kDa). Semi-quantification of
phosphorylation ratios in (C) MIA PaCa-2 and (D) AsPC-1 cells. In
the figure, GEM represents 0.25 µmol/l GEM and SSD represents 4
µmol/l SSD. Data are presented as the mean ± SD (n=5). Compared
with GEM monotherapy, the G0.25 + S4 combination led to a greater
reduction in both p-AKT/AKT and p-mTOR/mTOR ratios (both
Padj<0.0001), with large effect sizes
(η2=0.73 and 0.81, respectively). All P-values are
Benjamini-Hochberg-adjusted. ****P<0.0001 for G0.25 + S4 vs.
Ctrl; ##P<0.01, ###P<0.001 for G0.25 +
S4 vs. G0.25. Statistical analysis was performed using one-way
ANOVA with Tukey's post hoc test. Ctrl, control; GEM, gemcitabine;
SSD, Saikosaponin D; p-, phosphorylated. |
SSD promotes autophagy in pancreatic
cancer cells
The interconnected roles of autophagy and apoptosis
in cancer regulation are well documented (28–32).
Having observed SSD-induced apoptosis in pancreatic cancer cells,
the present study next investigated its potential to induce
autophagy. Initial assessment by MDC staining revealed substantial
accumulation of autophagic vesicles in both MIA PaCa-2 and AsPC-1
cells following treatment. While single-agent SSD or GEM treatment
increased autophagic activity, the combination therapy exerted
markedly greater effects, substantially elevating MDC fluorescence
intensity in both MIA PaCa-2 and AsPC-1 cells compared with GEM
alone (Fig. 7A). ICC analysis
corroborated these findings, demonstrating increased cytoplasmic
expression of autophagy markers Beclin 1 and LC3 in cells treated
with the SSD and GEM combination (G0.25 + S4) (Fig. 7B). The intensity and distribution of
staining were most pronounced in the SSD + GEM combination groups,
consistent with the MDC results. Western blotting
semi-quantification provided precise molecular characterization of
these effects (Fig. 7C). Treatment
with the SSD and GEM combination (G0.25 + S4) elevated both Beclin
1 expression and the LC3II/LC3I ratio compared with GEM alone, with
Beclin 1 expression increasing by 33.5% in MIA PaCa-2 cells and
42.3% in AsPC-1 cells, and the LC3II/LC3I ratio increasing by 48.1%
in MIA PaCa-2 cells and 56.7% in AsPC-1 cells (Fig. 7D and E). The combination treatment
showed additive effects across all autophagy markers, with AsPC-1
cells again exhibiting greater sensitivity than MIA PaCa-2 cells.
These complementary analyses demonstrated that SSD not only induced
autophagy in pancreatic cancer cells but synergistically enhanced
GEM-mediated autophagy activation through inhibition of the
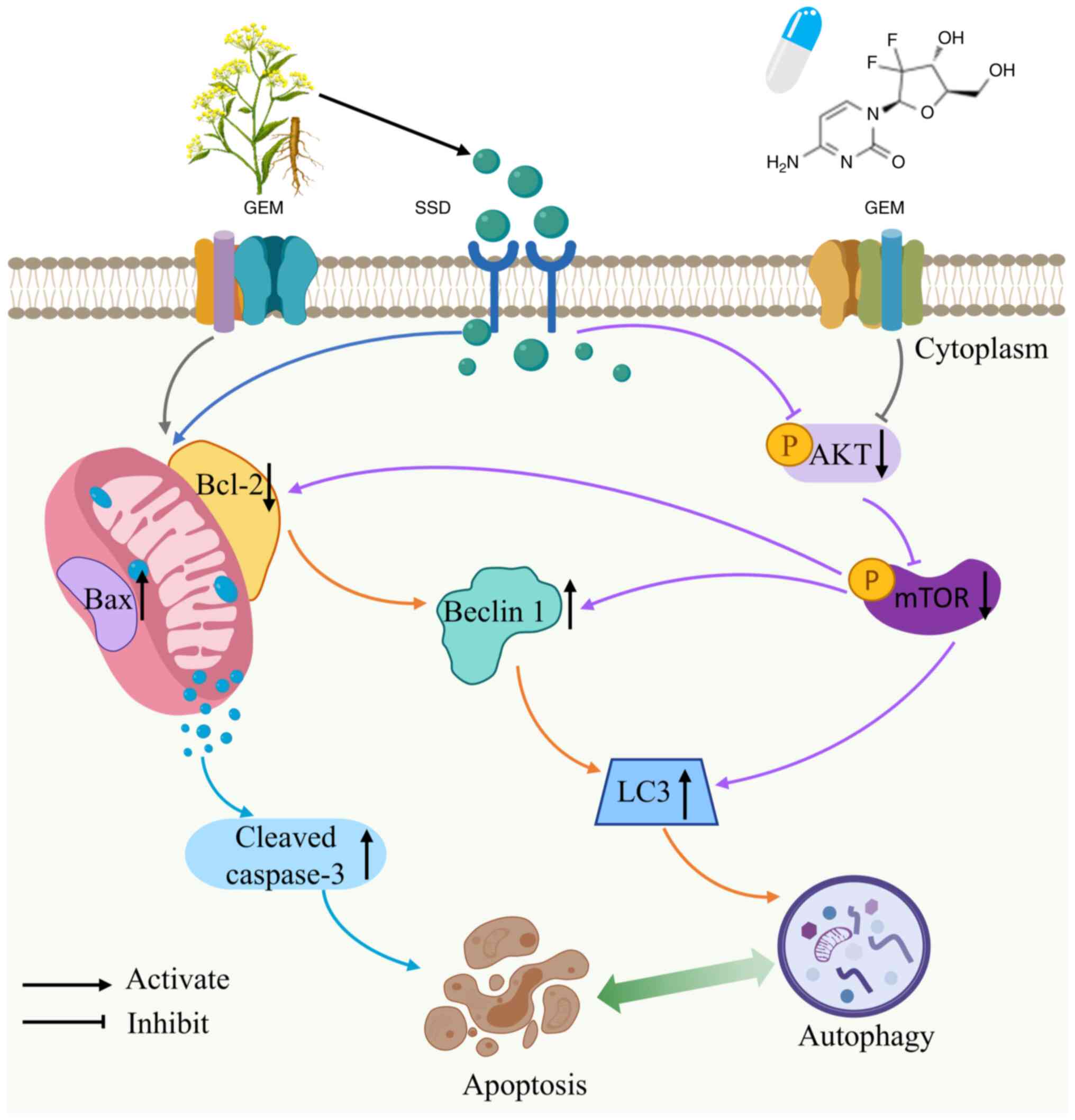
AKT/mTOR pathway (Fig. 8),
potentially contributing to the observed antitumor efficacy of the
combination therapy. The combination treatment (G0.25 + S4)
significantly enhanced autophagic activity compared with GEM
(G0.25) alone. Specifically, it increased the LC3II/LC3I ratio
(Padj=0.001; η2=0.65; 95% CI, 0.43–0.81) and
elevated Beclin 1 expression (Padj=0.002;
η2=0.59; 95% CI, 0.36–0.77).
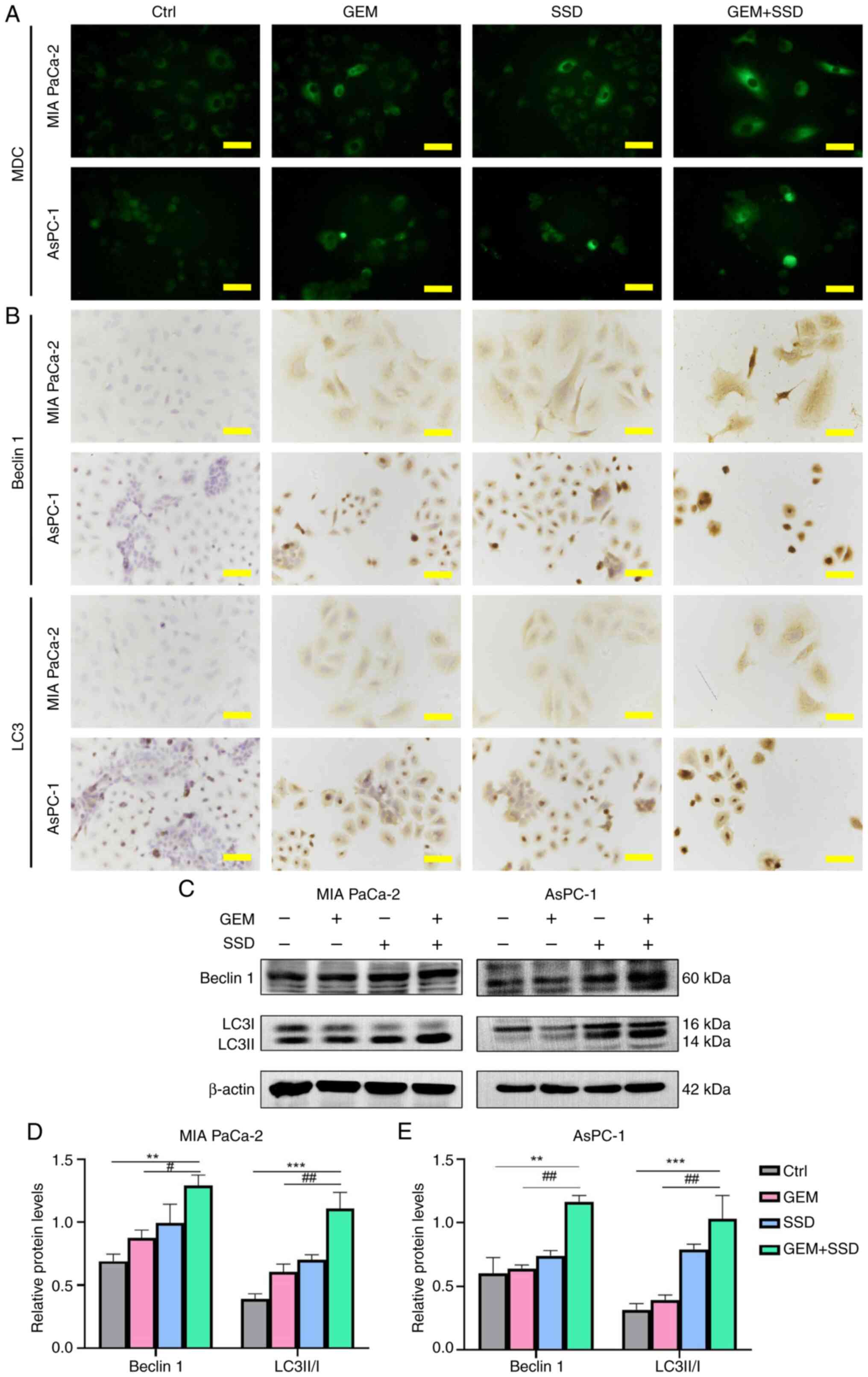 | Figure 7.Autophagy induction by GEM and SSD
combination treatment. (A) MDC staining (green) showing
autophagosome accumulation in MIA PaCa-2 and AsPC-1 cells treated
with Ctrl, G0.25, S4 or the combination (G0.25 + S4; magnification,
×40). (B) Immunocytochemistry of autophagy markers. (C) Western
blot analysis of Beclin 1 (60 kDa), LC3I (16 kDa), LC3II (14 kDa)
and β-actin (42 kDa). Semi-quantification of autophagy markers in
(D) MIA PaCa-2 and (E) AsPC-1 cells. In the figure, GEM represents
0.25 µmol/l GEM and SSD represents 4 µmol/l SSD. Data are presented
as the mean ± SD (n=5). Compared with GEM monotherapy, the G0.25 +
S4 combination significantly increased the LC3-II/I ratio
(Padj=0.001; η2=0.65; 95% CI, 0.43–0.81) and
Beclin 1 levels (Padj=0.002; η2=0.61; 95% CI,
0.38–0.78), indicating strong induction of autophagy. All P-values
are Benjamini-Hochberg-adjusted. Scale bar, 50 µm. **P<0.01,
***P<0.001 for G0.25 + S4 vs. Ctrl; #P<0.05,
##P<0.01 for G0.25 + S4 vs. G0.25. Statistical
analysis was performed using one-way ANOVA with Tukey's post hoc
test. Ctrl, control; GEM, gemcitabine; SSD, Saikosaponin D; MDC,
monodasylcadaverine. |
Discussion
Pancreatic cancer is one of the most lethal
malignant tumors, and is often described as the most aggressive of
all cancers due to its highly metastatic nature, coupled with high
mortality, low early detection and poor survival rates (1,33). In
recent years, the incidence and mortality of pancreatic cancer have
continued to rise worldwide (34).
Although GEM monotherapy or combination therapy with other agents
is widely used as a first-line treatment for pancreatic cancer, the
prognosis remains poor due to the rapid development of drug
resistance in vivo (35).
Therefore, there is an urgent need to identify or develop
anticancer agents with low toxicity and high efficiency to enhance
the sensitivity to GEM and improve treatment outcomes for
pancreatic cancer. TCM has gained increasing attention in cancer
therapy research due to its multi-target pharmacological properties
and favorable safety profiles (19,36,37).
Additionally, TCM formulations contain a variety of bioactive
components, some of which, such as berberine, curcumin and
artemisinin derivatives, have shown potential against pancreatic
cancer in pre-clinical and clinical research (38,39).
SSD is a triterpenoid saponin monomer extracted from
Bupleurum chinense roots (Radix Bupleuri), a classic TCM
formula (40). SSD has antitumor,
anti-inflammatory and antiviral properties, and is considered to be
one of the most bioactive natural saponins (41). Furthermore, SSD has been reported to
be relatively safe in an appropriate dose range (42). In the experimental treatment of
glioma and certain pancreatic cancers, the typical concentration of
SSD is maintained between 5–20 µmol/l, with the lowest effective
dose observed at ~5 µmol/l in in vitro studies (25). The concentration of SSD (4 µmol/l)
selected in the present study therefore falls within this safe
range. The routine clinical dose of GEM is usually 1,000
mg/m2 administered once a week, with a week off after
several weeks of continuous use (43). In the present study, 0.25 µmol/l was
selected as a sub-lethal and biologically relevant concentration of
GEM for combination treatments, based on dose-response curves
demonstrating significant growth inhibition at this dose; the
lowest concentration tested that produced a statistically
significant anti-proliferative effect in both MIA PaCa-2 and AsPC-1
cell lines.
The present study demonstrated that low-dose SSD
exposure inhibited the proliferation of pancreatic cancer cells
in vitro, and also enhanced the antiproliferative effects of
GEM in these cells. While SSD inhibited the proliferation of MIA
PaCa-2 and AsPC-1 cells in a dose-dependent manner, AsPC-1 cells
were more sensitive to SSD than MIA PaCa-2 cells. These effects
were further demonstrated through colony formation assays, which
additionally indicated increased inhibition of tumor cell
clonogenic capacity following combined treatment with SSD and GEM.
TUNEL and Hoechst 33258 staining assays also demonstrated that SSD
induced apoptosis in both cell lines. Notably, TUNEL fluorescence
estimates suggested potential synergistic effects for SSD when
applied in combination with GEM. Furthermore, the combination of
SSD and GEM exhibited synergistic effects on the induction of
autophagy in both MIA PaCa-2 and AsPC-1 cells. These findings
suggest robust potential for SSD as an adjuvant therapy for
pancreatic cancer in patients treated with GEM.
The AKT/mTOR signaling pathway serves an important
role in critical cellular processes, and its dysregulation
contributes to tumor development and progression in numerous cancer
types, as demonstrated in NSCLC (13), breast cancer (11), hepatocellular carcinoma (44) and pancreatic cancer (15). PI3K generates phosphatidylinositol
(3,4,5)-trisphosphate, which binds to and
recruits phosphatidylinositol-dependent protein kinase 1 and AKT to
the cell membrane, thereby activating AKT. Activated AKT regulates
multiple downstream targets, including mTOR and Bad, and through
phosphorylation reactions, regulates a variety of cellular
physiological processes such as proliferation, survival, apoptosis
and metabolism (9). The association
of AKT/mTOR activity with apoptosis and autophagy in cancer cells
has been documented. Wang et al (13) demonstrated that PTPRH promoted NSCLC
progression through glycolysis mediated by the PI3K/AKT/mTOR
signaling pathway. Lai et al (42) showed that SSD inhibited
proliferation and promoted apoptosis in pancreatic cancer cells via
activation of the MKK4-JNK signaling pathway. In line with this
evidence, the present study suggested that inhibition of the
AKT/mTOR pathway underlies SSD + GEM-induced apoptosis and
autophagy in pancreatic cancer cells, as demonstrated by western
blotting and ICC results showing that SSD and GEM inhibited the
phosphorylation of AKT and mTOR. Upon activation, AKT
phosphorylates the pro-apoptotic Bad protein, preventing its
binding to the anti-apoptotic proteins Bcl-2 and Bcl-XL. This
results in inhibition of the pro-apoptotic protein Bax, impeding
the occurrence of apoptosis. Accordingly, when AKT is inhibited,
Bax activation leads to the release of cytochrome c from
mitochondria, which activates caspase-3 (45). Caspase-3 in turn cleaves its
substrate poly (ADP-ribose) polymerase, an enzyme involved in DNA
repair, which accelerates the apoptotic process (46). In the present study, evidence of
SSD-mediated induction of apoptosis was provided by western
blotting and ICC analyses showing downregulated expression of Bcl-2
and total caspase-3, along with upregulated expression of Bax and
cleaved caspase-3, following treatment with SSD, GEM or their
combination. These findings suggested that SSD, both alone and in
combination with GEM, inhibited pancreatic cancer cell
proliferation and induced apoptosis by inhibiting the AKT/mTOR
pathway.
Studies have further highlighted the pivotal role of
AKT/mTOR dysregulation in pancreatic cancer chemoresistance. For
instance, Mortazavi et al (47) highlighted that hyperactivation of
PI3K/AKT/mTOR signaling not only promoted tumor survival but also
mediated adaptive resistance to GEM by upregulating efflux pumps
and DNA repair mechanisms. Similarly, Sheng et al (48) demonstrated that mTOR inhibition
sensitized pancreatic cancer cells to GEM through suppression of
musashi RNA binding protein 2-mediated epithelial-mesenchymal
transition. Notably, Shimia et al (17) reported that dual targeting of AKT
and mTOR synergized with conventional chemotherapy by disrupting
feedback loops that sustain tumor survival. These findings align
with the present results, reinforcing that AKT/mTOR inhibition of
SSD could disrupt multiple resistance mechanisms, including
metabolic reprogramming and anti-apoptotic signaling.
In the present study, combinatorial testing of 4 and
6 µmol/l SSD with sub-lethal GEM (0.25 µmol/l) was conducted. The 4
µmol/l SSD dose (G0.25 + S4) was chosen for detailed mechanistic
analysis to investigate the basis of the synergistic effects under
conditions of minimal cytotoxicity. Specifically, the 4 µmol/l SSD
+ GEM combination achieved proliferation inhibition comparable to
that of higher doses, reducing colony formation by 29.3% in MIA
PaCa-2 cells vs. 15.1% in AsPC-1 cells. Furthermore, this
combination induced marked apoptosis and autophagy in MIA PaCa-2
cells, evidenced by a 61.6% increase in cleaved caspase-3 levels
and a 56.7% rise in LC3-II/I ratios. The favorable safety profile
of this concentration, consistent with prior applications of SSD in
pancreatic models (42), further
supports its suitability for prolonged mechanistic investigation.
Although higher SSD doses (6 µmol/l) further reduced viability (for
instance, resulting in a 31.6% decrease in survival in AsPC-1 cells
treated with G0.25 + S6 at 72 h), the 4 µmol/l dose provided the
best efficacy-toxicity balance, reflecting clinical strategies that
prioritize minimizing chemotherapy doses through the use of
sensitizing adjuvants.
While the present study demonstrated that SSD
synergized with GEM primarily through AKT/mTOR inhibition, the
reported effects of SSD on STAT3/Bcl-2 and IKKβ/NF-κB pathways in
other cancer types (22,23), including NSCLC and gastric cancer,
suggest potential parallel mechanisms in pancreatic cancer.
Although these pathways were not explicitly examined in the present
study, the observed upregulation of Bax and downregulation of Bcl-2
align with STAT3/Bcl-2 axis modulation, as STAT3 often
transcriptionally regulates Bcl-2. Similarly, NF-κB suppression
could further amplify apoptosis via caspase-3 activation (49). Future studies should dissect these
interactions to fully unravel the pleiotropic antitumor effects of
SSD.
Untreated MIA PaCa-2 cells exhibited detectable
cleaved caspase-3 levels, consistent with their known genetic
profile. This cell line carries oncogenic KRASG12J and
mutant TP53R248W (50),
driving constitutive endoplasmic reticulum stress and DNA damage
responses that prime apoptotic pathways even in untreated
conditions. Similar basal caspase activation has been reported in
other KRAS-mutant pancreatic models (51). The present data demonstrated that
SSD + GEM combination therapy further amplified cleaved caspase-3
levels by 47.5% over this elevated baseline, confirming robust
induction of apoptosis beyond intrinsic stress signaling. This
highlights the aggressive biology of pancreatic adenocarcinoma,
where pro-survival and pro-death signals coexist, and underscores
the therapeutic potential of overcoming inherent resistance
mechanisms.
Apoptosis and autophagy are two distinct forms of
cell death. Although there are obvious morphological and molecular
differences between these processes, they are not completely
independent (52,53). Studies have shown that apoptosis and
autophagy affect and restrict each other in different cellular
contexts (28–30). Increasing evidence has shown that a
number of chemotherapies and anticancer drugs, such as temozolomide
(in glioma), cisplatin (in gastric and ovarian cancer) and rapalogs
(such aseverolimus in various solid tumors), can induce autophagy
in a variety of cancer cells (54,55).
Therefore, targeting of autophagy is becoming a promising strategy
for cancer treatment. The unc-51 like autophagy activating kinase 1
(ULK1) complex, which includes ULK1, autophagy-related protein
(ATG)13, focal adhesion kinase family-interacting protein of 200
kDa and ATG101, is a key regulator of autophagy initiation
(56). The AMP-activated protein
kinase (AMPK) and mTOR signaling pathways can initiate autophagy by
regulating the activity of ULK1 (57). In response to nutrient or energy
deprivation, AMPK activates ULK1, while mTOR inhibition promotes
the formation of ULK1 complexes and initiates the autophagic
process. In addition, some upstream signaling molecules such as
Bcl-2 are involved in autophagosome formation by regulating the
activity of Beclin 1 and affecting its binding to vacuolar protein
sorting 34 (58,59). In response to autophagy signals, the
C-terminal amino acid of pro-LC3 is first cleaved by ATG4,
resulting in the formation of LC3I. Subsequently, ATG7 activates
LC3I, which is transferred to ATG3 to be converted into LC3II,
which binds to the autophagosome membrane to promote autophagy
(60). In the present study,
western blotting and ICC assays confirmed that SSD and GEM, both
alone and in combination, increased the expression of Beclin 1 and
the ratio of LC3II/LC3I in pancreatic cancer cells. MDC assays
showed that the accumulation and relative fluorescence intensity of
autophagic vesicles in the cytoplasm of pancreatic cancer cells
were markedly increased upon combined SSD + GEM exposure compared
with either treatment alone, suggesting a potential synergistic
effect.
The Chou-Talalay analysis (27,61)
demonstrated that low-dose SSD (4 µmol/l) synergistically enhanced
GEM efficacy (CI, 0.42–0.72), which mechanistically aligns with the
dual regulation of the AKT/mTOR pathway and autophagic flux. This
synergistic interaction may allow dose reduction of both agents
while maintaining antitumor efficacy, potentially mitigating
GEM-associated toxicity in clinical settings. Particularly
noteworthy is the more pronounced synergy in AsPC-1 cells (CI,
0.42–0.65 vs. 0.58–0.72 in MIA PaCa-2 cells), suggesting cell
line-specific response patterns that warrant further
investigation.
The therapeutic potential of this combination is
further underscored by the favorable safety profile of SSD. The
safety profile of SSD is supported by its selective cytotoxicity,
with reported IC50 values >20 µmol/l in normal
pancreatic cells vs. 4–6 µmol/l in cancer cells (42), suggesting a favorable therapeutic
window. Emerging preclinical evidence from patient-derived
xenograft models has demonstrated the translational potential of
TCM-derived compounds such as SSD in combination with conventional
chemotherapy (19), which aligns
with the present findings. Mechanistically, the observed synergy
may stem from SSD's multifaceted regulation of AKT/mTOR signaling,
a critical node governing both metastatic progression and cellular
metabolism in pancreatic cancer (15), where concurrent pathway inhibition
and autophagy induction create a synthetic lethal effect.
A recognized limitation of the present study is its
focus on two pancreatic cancer cell lines without inclusion of
additional in vitro models [such as normal cell controls or
heterogeneous pancreatic ductal adenocarcinoma (PDAC) subtypes] or
in vivo validation. These aspects are crucial to
comprehensively evaluate the translational relevance and safety of
the combination therapy. Therefore, future studies will prioritize
expanding the biological validation to include toxicity assessments
in non-malignant cells, a broader panel of cell lines representing
the genetic diversity of PDAC and robust in vivo models to
firmly establish the efficacy and therapeutic window of SSD +
GEM.
In the present study, ICC staining utilizing DAB was
employed to evaluate protein expression and subcellular
localization. Although DAB-based methods provide advantages such as
brightfield microscopy compatibility and archival stability, they
are limited by poorer spatial resolution and interpretive
subjectivity. To address these concerns, all ICC findings were
rigorously validated through complementary quantitative approaches:
Western blotting provided objective semi-quantitative measurements
of protein expression, such as the Bax/Bcl-2 ratio, LC3-II/LC3-I
conversion and the p-AKT/AKT ratio, and fluorescence-based assays
(TUNEL, Hoechst and MDC staining) enabled high-resolution
visualization of apoptosis and autophagy, corroborating ICC trends.
The concordance across these orthogonal methods confirmed the
reliability of the ICC data. Future studies would benefit from
multiplex immunofluorescence to enhance spatial resolution for
complex signaling networks.
In conclusion, the present study demonstrated that
SSD inhibited proliferation, and induced autophagy and apoptosis of
pancreatic cancer cells, thereby enhancing the anticancer activity
of GEM. The present data further suggested that negative regulation
of the AKT/mTOR signaling pathway may be, at least in part,
responsible for these effects (Fig.
8). These results provide a strong rationale for the
combination of GEM and SSD in the treatment of pancreatic
cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 82260489 and 82560545), the Shaanxi
Province Youth Talent Lifting Project (grant no. 20240316), the
Shanxi Science and Technology Department Project (grant no.
2025JC-YBQN-1175), the Innovation and Entrepreneurship Training
Program for College Students (grant no. S202210719100), the General
Project of Shaanxi Provincial Department of Education (grant no.
24JK0727), and Yan'an University Doctoral Research Project (grant
no. YDBK2020-29).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YZ and YL conceived and designed the study, and
drafted the original manuscript. RZ, YY, SZ and GL performed the
experiments and analyzed the data. All authors participated in
scientific discussions and data interpretation. YL contributed to
data acquisition and authenticity verification. XL performed
statistical analysis. YZ and YL confirm the authenticity of all the
raw data. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cai J, Chen H, Lu M, Zhang Y, Lu B, You L,
Zhang T, Dai M and Zhao Y: Advances in the epidemiology of
pancreatic cancer: Trends, risk factors, screening, and prognosis.
Cancer Lett. 520:1–11. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klatte DCF, Wallace MB, Löhr M, Bruno MJ
and van Leerdam ME: Hereditary pancreatic cancer. Best Pract Res
Clin Gastroenterol. 58-59:1017832022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Domagała-Haduch M, Gorzelak-Magiera A,
Michalecki Ł and Gisterek-Grocholska I: Radiochemotherapy in
pancreatic cancer. Curr Oncol. 31:3291–3300. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brozos-Vázquez E, Toledano-Fonseca M,
Costa-Fraga N, García-Ortiz MV, Díaz-Lagares Á, Rodríguez-Ariza A,
Aranda E and López-López R: Pancreatic cancer biomarkers: A pathway
to advance in personalized treatment selection. Cancer Treat Rev.
125:1027192024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miao H, Chen X and Luan Y: Small molecular
gemcitabine prodrugs for cancer therapy. Curr Med Chem.
27:5562–5582. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pandit B and Royzen M: Recent development
of prodrugs of gemcitabine. Genes (Basel). 13:4662022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui J, Guo Y, Yin T, Gou S, Xiong J, Liang
X, Lu C and Peng T: USP8 promotes gemcitabine resistance of
pancreatic cancer via deubiquitinating and stabilizing Nrf2. Biomed
Pharmacother. 166:1153592023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shu X, Zhan PP, Sun LX, Yu L, Liu J, Sun
LC, Yang ZH, Ran YL and Sun YM: BCAT1 activates PI3K/AKT/mTOR
pathway and contributes to the angiogenesis and tumorigenicity of
gastric cancer. Front Cell Dev Biol. 9:6592602021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu L, Wei J and Liu P: Attacking the
PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment
in human cancer. Semin Cancer Biol. 85:69–94. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rahaman A, Chaudhuri A, Sarkar A,
Chakraborty S, Bhattacharjee S and Mandal DP: Eucalyptol targets
PI3K/Akt/mTOR pathway to inhibit skin cancer metastasis.
Carcinogenesis. 43:571–583. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou X, Zhao J, Yan T, Ye D, Wang Y, Zhou
B, Liu D, Wang X, Zheng W, Zheng B, et al: ANXA9 facilitates S100A4
and promotes breast cancer progression through modulating STAT3
pathway. Cell Death Dis. 15:2602024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang Q, Guan Y, Zheng J and Lu H: TBK1
promotes thyroid cancer progress by activating the PI3K/Akt/mTOR
signaling pathway. Immun Inflamm Dis. 11:e7962023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Cheng Z, Cui Y, Xu S, Luan Q, Jing
S, Du B, Li X and Li Y: PTPRH promotes the progression of non-small
cell lung cancer via glycolysis mediated by the PI3K/AKT/mTOR
signaling pathway. J Transl Med. 21:8192023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Zhang C, Xu Z, Chen MH, Yu H, Wang
L and Liu R: Differential impact of PI3K/AKT/mTOR signaling on
tumor initiation and progression in animal models of prostate
cancer. Prostate. 83:97–108. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mortazavi M, Moosavi F, Martini M,
Giovannetti E and Firuzi O: Prospects of targeting PI3K/AKT/mTOR
pathway in pancreatic cancer. Crit Rev Oncol Hematol.
176:1037492022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang L, Sun J, Ma Y, Chen H, Tian C and
Dong M: MSI2 regulates NLK-mediated EMT and PI3K/AKT/mTOR pathway
to promote pancreatic cancer progression. Cancer Cell Int.
24:2732024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimia M, Amini M, Ravari AO, Tabnak P,
Valizadeh A, Ghaheri M and Yousefi B: Thymoquinone reversed
doxorubicin resistance in U87 glioblastoma cells via targeting
PI3K/Akt/mTOR signaling. Chem Biol Drug Des. 104:e145872024.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang M, Xue W, Yuan H, Wang Z and Yu L:
Nano-drug delivery systems targeting CAFs: A promising treatment
for pancreatic cancer. Int J Nanomedicine. 19:2823–2849. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Y, Xu H, Li Y, Sun Y and Peng X:
Advances in the treatment of pancreatic cancer with traditional
Chinese medicine. Front Pharmacol. 14:10892452023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Manoharan S, Deivendran B and Perumal E:
Chemotherapeutic potential of saikosaponin D: Experimental
evidence. J Xenobiot. 12:378–405. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu G, Guan Y, Liu Y, Wang Y, Zhang J, Liu
Y and Liu X: Saikosaponin D inducing apoptosis and autophagy
through the activation of endoplasmic reticulum stress in
glioblastoma. Biomed Res Int. 2022:54895532022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang JC, Long F, Zhao J, Hang J, Ren YG,
Chen JY and Mu B: The effects and mechanisms by which
saikosaponin-D enhances the sensitivity of human non-small cell
lung cancer cells to gefitinib. J Cancer. 10:6666–6672. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu J, Li P, Shi B and Tie J: Effects and
mechanisms of saikosaponin d improving the sensitivity of human
gastric cancer cells to cisplatin. ACS Omega. 6:18745–18755. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang CY, Jiang ZM, Ma XF, Li Y, Liu XZ,
Li LL, Wu WH and Wang T: Saikosaponin-d inhibits the hepatoma cells
and enhances chemosensitivity through SENP5-dependent inhibition of
gli1 sumoylation under hypoxia. Front Pharmacol. 10:10392019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang J, Sun J, Liu A, Chen L, Ma X, Liu X
and Zhang C: Saikosaponin D improves chemosensitivity of
glioblastoma by reducing the its stemness maintenance. Biochem
Biophys Rep. 32:1013422022.PubMed/NCBI
|
|
26
|
Tang TT, Jiang L, Zhong Q, Ni ZJ, Thakur
K, Khan MR and Wei ZJ: Saikosaponin D exerts cytotoxicity on human
endometrial cancer ishikawa cells by inducing apoptosis and
inhibiting metastasis through MAPK pathways. Food Chem Toxicol.
177:1138152023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xi H, Wang S, Wang B, Hong X, Liu X, Li M,
Shen R and Dong Q: The role of interaction between autophagy and
apoptosis in tumorigenesis (Review). Oncol Rep. 48:2082022.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sorice M: Crosstalk of autophagy and
apoptosis. Cells. 11:14792022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Niu X, You Q, Hou K, Tian Y, Wei P, Zhu Y,
Gao B, Ashrafizadeh M, Aref AR, Kalbasi A, et al: Autophagy in
cancer development, immune evasion, and drug resistance. Drug
Resist Updat. 78:1011702025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Wu Y, Meng S, Xu P, Li S, Li Y, Hu
X, Ouyang L and Wang G: Selective autophagy in cancer: mechanisms,
therapeutic implications, and future perspectives. Mol Cancer.
23:222024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Y, Tang J, Jiang K, Liu SY, Aicher A
and Heeschen C: Liquid biopsy in pancreatic cancer-current
perspective and future outlook. Biochim Biophys Acta Rev Cancer.
1878:1888682023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhong BH, Ma YT, Sun J, Tang JT and Dong
M: Transcription factor FOXF2 promotes the development and
progression of pancreatic cancer by targeting MSI2. Oncol Rep.
52:932024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen J, Hua Q, Wang H, Zhang D, Zhao L, Yu
D, Pi G, Zhang T and Lin Z: Meta-analysis and indirect treatment
comparison of modified FOLFIRINOX and gemcitabine plus
nab-paclitaxel as first-line chemotherapy in advanced pancreatic
cancer. BMC Cancer. 21:8532021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji J, Wen Q, Yu Y, Xiong F, Zheng X and
Ruan S: Personalized traditional Chinese medicine in oncology:
Bridging the macro state with micro targets. Am J Chin Med.
14:1–34. 2025.
|
|
37
|
Ke Y, Pan Y, Huang X, Bai X, Liu X, Zhang
M, Wei Y, Jiang T and Zhang G: Efficacy and safety of traditional
Chinese medicine (TCM) combined with immune checkpoint inhibitors
(ICIs) for the treatment of cancer: a systematic review and
meta-analysis. Front Pharmacol. 31:16615032025. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Okuno K, Xu C, Pascual-Sabater S, Tokunaga
M, Han H, Fillat C, Kinugas Y and Goel A: Berberine overcomes
gemcitabine-associated chemoresistance through regulation of
Rap1/PI3K-Akt signaling in pancreatic ductal adenocarcinoma.
Pharmaceuticals (Basel). 15:11992022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhattacharyya S, Ghosh H,
Covarrubias-Zambrano O, Jain K, Swamy KV, Kasi A, Hamza A, Anant S,
Van Saun M, Weir SJ, et al: Anticancer activity of novel
difluorinated curcumin analog and its inclusion complex with
2-hydroxypropyl-β-cyclodextrin against pancreatic cancer. Int J Mol
Sci. 24:63362023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ashour ML and Wink M: Genus bupleurum: A
review of its phytochemistry, pharmacology and modes of action. J
Pharm Pharmacol. 63:305–321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheng Y, Liu G, Li Z, Zhou Y and Gao N:
Screening saikosaponin d (SSd)-producing endophytic fungi from
Bupleurum scorzonerifolium Willd. World J Microbiol Biotechnol.
38:2422022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lai M, Ge Y, Chen M, Sun S, Chen J and
Cheng R: Saikosaponin D inhibits proliferation and promotes
apoptosis through activation of MKK4-JNK signaling pathway in
pancreatic cancer cells. Onco Targets Ther. 13:9465–9479. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Conroy T, Castan F, Lopez A, Turpin A, Ben
Abdelghani M, Wei AC, Mitry E, Biagi JJ, Evesque L, Artru P, et al:
Five-year outcomes of FOLFIRINOX vs. gemcitabine as adjuvant
therapy for pancreatic cancer: A randomized clinical trial. JAMA
Oncol. 8:1571–1578. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun EJ, Wankell M, Palamuthusingam P,
McFarlane C and Hebbard L: Targeting the PI3K/Akt/mTOR pathway in
hepatocellular carcinoma. Biomedicines. 9:16392021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kale J, Kutuk O, Brito GC, Andrews TS,
Leber B, Letai A and Andrews DW: Phosphorylation switches Bax from
promoting to inhibiting apoptosis thereby increasing drug
resistance. EMBO Rep. 19:e452352018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li X, He S and Ma B: Autophagy and
autophagy-related proteins in cancer. Mol Cancer. 19:122020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mehra S, Deshpande N and Nagathihalli N:
Targeting PI3K pathway in pancreatic ductal adenocarcinoma:
Rationale and progress. Cancers (Basel). 13:44342021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao
R, Sun J, Wang G, Zhou L and Dong M: Musashi2 promotes EGF-induced
EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin
Cancer Res. 39:162020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deer EL, González-Hernández J, Coursen JD,
Shea JE, Ngatia J, Scaife CL, Firpo MA and Mulvihill SJ: Phenotype
and genotype of pancreatic cancer cell lines. Pancreas. 39:425–435.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fianco G, Mongiardi MP, Levi A, De Luca T,
Desideri M, Trisciuoglio D, Del Bufalo D, Cinà I, Di Benedetto A,
Mottolese M, et al: Caspase-8 contributes to angiogenesis and
chemotherapy resistance in glioblastoma. Elife. 6:e225932017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wei S, Han C, Mo S, Huang H and Luo X:
Advancements in programmed cell death research in antitumor
therapy: A comprehensive overview. Apoptosis. 30:401–421. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Das S, Shukla N, Singh SS, Kushwaha S and
Shrivastava R: Mechanism of interaction between autophagy and
apoptosis in cancer. Apoptosis. 26:512–533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hajdú B, Holczer M, Horváth G, Szederkényi
G and Kapuy O: Fine-tuning of mTORC1-ULK1-PP2A regulatory triangle
is crucial for robust autophagic response upon cellular stress.
Biomolecules. 12:15872022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fujiwara N, Usui T and Nagai H: Regulation
of the Beclin 1/VPS34 complex by post-translational modifications.
Biochem Biophys Res Commun. 566:155–161. 2021.PubMed/NCBI
|
|
56
|
Pareek G and Kundu M: Physiological
functions of ULK1/2. J Mol Biol. 436:1684722024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lu G, Wu Z, Shang J, Xie Z, Chen C and
Zhang C: The effects of metformin on autophagy. Biomed
Pharmacother. 137:1112862021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fujiwara N, Shibutani S, Ohama T and Sato
K: Protein phosphatase 6 dissociates the beclin 1/Vps34 complex and
inhibits autophagy. Biochem Biophys Res Commun. 552:191–195. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bekker M, Abrahams S, Loos B and Bardien
S: Can the interplay between autophagy and apoptosis be targeted as
a novel therapy for Parkinson's disease? Neurobiol Aging.
100:91–105. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Vujić N, Bradić I, Goeritzer M, Kuentzel
KB, Rainer S, Kratky D and Radović B: ATG7 is dispensable for
LC3-PE conjugation in thioglycolate-elicited mouse peritoneal
macrophages. Autophagy. 17:3402–3407. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Foucquier J and Guedj M: Analysis of drug
combinations: Current methodological landscape. Pharmacol Res
Perspect. 3:e001492015. View Article : Google Scholar : PubMed/NCBI
|